
Abstract
The sol-gel synthesis represents a versatile platform to fabricate ceramic inorganic membranes. However, it is still a grand challenge to push the boundary of sol-gel chemistry towards high-quality organic membrane construction. Herein, a facile and controlled nanocrystal suturing strategy in sol-gel solutions is developed to afford highly crystalline and free-standing covalent organic framework membranes. The key chemistry design lies in deploying tiny threads (1 mol% dual-NH2-tail linear polymer) to efficiently suture the highly charged covalent organic framework nanocrystals stabilized and confined in sol-gel solutions, creating a continuous and intact membrane surface. A subsequent treatment heals the sutured covalent organic framework nanocrystals, yielding a free-standing membrane with high crystallinity and ordered pores. The structure evolution and role of the thread linker are elucidated via operando spectroscopy and microscopy. The as-afforded covalent organic framework membranes demonstrate attractive proton transport performance in high temperature and anhydrous fuel cell applications.
Similar content being viewed by others
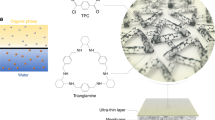
Introduction
Crystalline porous membranes possessing permanent and well-ordered porous channels have garnered remarkable attention1,2,3,4 in research fields related to gas separation, analyte-specific transport, catalysis, and energy transfer/storage5,6,7,8,9,10,11,12, with performance largely surpassing the amorphous and nonporous counterparts. Covalent organic frameworks (COFs) represent an emerging class of crystalline porous materials featuring large surface areas, ordered and customizable pore structures, and tunable chemical properties13,14,15,16,17, making them attractive candidates in membrane-based separation processes. However, similar to most metal-organic framework (MOF) membranes18,19,20,21, the synthesis and fabrication of free-standing COF membranes with strong mechanical properties remain substantial challenges, which mainly arise from the inevitable growth of nano- or micron-sized crystalline powders during the membrane casting process (Supplementary Table 1)22,23,24,25. Moreover, the infusibility of COFs in most solvents and their inherent rigidity further complicate the processability of traditional COF powders26,27,28,29,30. As a result, thin COF active layers are typically grown on various supports to enhance the operability of the resulting membranes to accommodate the practical environments31,32,33. Limited approaches have been developed to fabricate free-standing COF membranes, such as functionalizing building monomers by linear polymer moieties34,35, the self-assembly of a molecular bridge at the aqueous/organic interface36, and the molecular soldering of COF particles via synchronous polymerization of linear polymers37. Nevertheless, the as-afforded ultrathin COF membranes (few nanometers) possessed inferior mechanical robustness, almost non-processibility, small dimensions, and difficulty in achieving large-scale synthesis. The conventional mixed matrix membrane (MMM) method utilizes amorphous and linear polymers, such as polyetherimide (PEI), polybenzimidazole (PBI), poly(vinylamine) (PVAm), and polymers of intrinsic microporosity (PIMs), to mix with COF fillers to enhance the mechanical properties of the resultant MMMs38,39,40,41. However, the loading of COF is typically kept below 50 wt.%, as higher concentrations might lead to the aggregation and non-uniform distribution of COF nanoparticles instead of their individual dispersion within the membrane, ultimately reducing the mechanical properties of the MMMs42. Reduced performance caused by low COF dopant loading, polymer penetration within the porous channels, and segregation of the COF fillers formed large barriers for practical application of the MMMs and remained thistles and thorns.
The sol-gel synthesis procedure represents a versatile platform to fabricate ceramic and glass-based inorganic membranes with high quality and attractive mechanical robustness43,44,45, in which the selection of catalyst/reaction media played critical roles to ensure the formation and stabilization of the highly charged nanosized particles. Our group pioneered and successfully extended the sol-gel approach to fabricate organic membrane counterparts, from which covalent triazine framework (CTF) membranes were fabricated employing a superacid (e.g., CF3SO3H) to dissolve and catalyze the trimerization of aromatic nitrile monomers, resulting in defect-free and flexible CTF membranes46,47,48. The key to this process is the formation of highly concentrated and charged oligomers, which were stably assembled in the reaction media to allow uniform and high molecular weight polymer formation and the subsequent membrane casting. However, the as-synthesized CTF membranes possessed amorphous scaffolds and lacked permanent porosity due to irreversible trimerization and excessive flexibility of the CTF scaffolds. This limitation often necessitates post-synthesis treatments, such as pyrolysis, to create porous carbon molecular sieve membranes for separation applications49. This controlled sol-gel solution-phase polymerization opens a unique avenue to fabricate high quality membranes with organic scaffolds, particularly COF membranes, to mitigate both mechanical property and scaling up issues that plagued traditional ultrathin and non-processable COF membranes. It is crucial to accommodate the reaction media and deploy smart additives to enable highly charged COF nanocrystal formation, avoid COF colloid aggregation, and promote the polymerization and membrane phase creation. To address these challenges, a unique sol-gel synthesis strategy was developed that enables the formation of high-quality, free-standing, crystalline, and porous COF membranes from amine and aldehyde monomers via imine bond formation. The process was governed by the addition of a small amount (as low as 1 mol%) of dual-NH2-tail linear polymer, which can participate in highly charged oligomer formation and self-assembly and act as molecular threads to omnidirectionally suture COF nanocrystals and restrict their growth, thereby promoting the formation of a uniform and stable sol-gel solution ready for membrane casting (Fig. 1). The as-afforded COF membrane precursors were successfully transformed into highly crystallized scaffolds via thermal solution treatment, with well maintained and robust membrane nature and in situ creation of ordered porous channels. Operando spectroscopy and microscopy monitoring revealed the critical role of the additional linker and its dosing amount, in which low quantity (around 1 mol%) was preferred to ensure the high crystallinity and excess amount led to diminished COF nanocrystal formation efficiency and instability of the sol-gel solutions. Notably, unique features of the as-afforded free-standing COF membranes, including the high crystallinity, ordered porous channels, robustness, and extensive hydrogen-bonding networks, provide them with great promise as the next-generation proton exchange membranes (PEMs) in high temperature fuel cell application, as confirmed by the promising proton conductivity and longer-term durability of H3PO4-saturated COF membranes at high temperatures up to 160 °C. The unique sol-gel approach with the ingenious addition of tiny threads being developed herein represented a breakthrough in free-standing, crystalline, thickness tunable, and size scalable COF membrane synthesis and hold great promise to further extend their applications to diverse fields.

Illustration of constructing a thickness-tunable, flexible, and processable free-standing COF membrane via nanocrystal suturing with tiny threads, as demonstrated in this study.
Results
Design concept, membrane fabrication and characterization
The grand challenge in fabricating free-standing and crystalline COF membranes lies in the rigid scaffolds and rapid aggregation and precipitation of the COF nanoparticles during the network connection process. Here, both challenges were successfully addressed using our unique sol-gel approach. The introduction of a small amount of linear and flexible co-crosslinker during COF synthesis enhances the interlamellar interactions between COF nanocrystals, facilitating their counter-diffusion growth and leading to the formation of COF membranes with continuous and integrated surface. Notably, in sol-gel procedure, the solvent selection is critical and should possess the capability to promote the oligomer formation and stabilize the charged intermediates. As a proof of concept, we selected a well-studied 2D Schiff-base COF constructed by triformylphloroglucinol (TP) and p-phenylenediamine (PDA) building blocks to validate our nanocrystal suturing strategy for producing free-standing COF membranes50. Trifluoroacetic acid (TFA) was selected as the solvents and catalysts to promote the imine bond formation, provide a protic media to allow charged intermediates generation, and stabilize the assembly of the nanocrystals (Supplementary Table 2)29,51. TP and PDA were first dissolved in pure TFA, followed by the addition of 10 mol% poly(propylene glycol) bis(2-aminopropyl ether) (PPG-NH2) (average Mn ~2000) as the molecular threads to prepare a homogeneous solution (Fig. 2a). The clear solution was then heated to 50 °C with vigorous stirring and maintained for 4 hours to ensure a thorough reaction between TP, PDA, and the polymer linker (PPG-NH2). As the heating progressed, the solution’s color turned from yellow to orange, and finally red, while the clear solution became turbid and gel-like (Fig. 2b). Finally, the resulting sol-gel solution was degassed for 5 min, then poured into a glass Petri dish to allow slow evaporation of the TFA, forming the dry membrane referred to as TpPa-COF-X% (where X represents the molar ratio of PPG-NH2 added during membrane solution preparation; 10 mol% PPG-NH2 was selected as the model for the following study).

a Synthetic scheme of TpPa-COF-X% membrane. b Photographs showing the transformation of the COF membrane precursor solution during the heating process. c Intensity-weighted particle size distributions of COF precursor sol-gel solutions with the addition of PPG-NH2 subjected to 0-4 hours of heating, and d where the particle size in COF precursor solutions without the addition of PPG-NH2 rapidly grew to the micrometer scale and exceeded the DLS measurement range after 1 hour of heating. e In-situ real-time monitoring of membrane fabrication and surface changes.
Dynamic light scattering (DLS) was employed to monitor the particle sizes of the nanocrystals during the heating process. At room temperature, the monomers reached equilibrium in CF3COOH, exhibiting particle sizes ranging from approximately 310 nm to 360 nm (Fig. 2c, d). The results revealed that with the addition of PPG-NH2, the particle size gradually increased during the reaction period from 0.5 to 4 hours and stabilized at approximately 280 nm when a fully homogeneous sol-gel solution was achieved after 4 hours of reaction (Fig. 2c). In contrast, without PPG-NH2, the COF particle size exceeded 350 nm after just 1 hour of heating, rapidly grew to the micrometer scale, and ultimately surpassed the measurement range of DLS (Fig. 2d). This indicates that PPG-NH2 plays a crucial role in regulating the size of the COF nanocrystals, preventing excessive growth and promoting a more uniform distribution of particle sizes. The controlled particle size is essential for ensuring the stability and homogeneity of the sol-gel solution, laying the groundwork for successful membrane formation. The particle size limitation upon the addition of PPG-NH2 was further confirmed by small-angle X-ray scattering (SAXS) measurements (Supplementary Fig. 1), which indicated that the particle size of the resulting COF nanocrystals is approximately 241 nm, as determined using a spherical model through Guinier analysis (Supplementary Fig. 1b). Additionally, both Porod and Kratky analyses supported the successful formation of the COF precursor sol-gel solution with the incorporation of PPG-NH2 (Supplementary Fig. 1c, d).
To gain deeper insight into the membrane formation process, a high-resolution video was recorded during the in-situ evaporation of TFA. The video revealed that at the initial stage, TFA vapor appeared at the liquid interface. As TFA evaporated, well-defined particles emerged and gradually organized into an integrated, rolling surface. Over time, this rolling surface became increasingly smooth, while larger surface cracks gradually diminished through self-healing. This behavior is attributed to dynamic rearrangement and capillary-driven flow facilitated by the sol-gel state and residual TFA plasticization during membrane formation52,53. Ultimately, a dense, free-standing membrane with a rough surface was formed (Fig. 2e and Supplementary Movie). It is important to note that the observed roughness is not due to macroscopic unevenness or large-scale surface distortion, but rather results from the localized aggregation of COF nanocrystals at the membrane surface during solvent evaporation and gel consolidation. As the solvent evaporates, COF nanocrystals tend to cluster at the surface due to capillary forces, leading to the formation of microscale surface features.
The resulting membrane easily floated up from the bottom of the Petri dish when immersed in a mixture of ethanol and water, then washed with ethanol to remove soluble residues and treated with an aqueous solution of triethylamine (TEA) to neutralize any unreacted TFA in sequence. The membrane obtained at this stage was amorphous and lacked pores, as verified by powder X-ray diffraction (PXRD) (Supplementary Fig. 2a) and N2 sorption isotherms (Supplementary Fig. 2b). To enhance the crystallinity and porosity of the obtained membrane, conventional solvothermal processing of COF powders was employed to recrystallize the TpPa-COF-10% membrane. This was achieved by treating the membrane in a mixture of o-dichlorobenzene, n-butanol, and a 6 M acetic acid aqueous solution (v/v/v = 5/5/1) at 120 °C for three days. PXRD analysis of the free-standing TpPa-COF-10% membranes revealed crystalline structures, characterized by a prominent peak at 4.9° and a relatively broad peak at 27.3° (Fig. 3a), corresponding to the (100) and (001) facets, respectively. N2 sorption isotherms revealed a Brunauer−Emmett−Teller (BET) surface area of 1113 m2 g−1 for the TpPa-COF-10% membrane (Fig. 3b), along with a pore size distribution that indicates an average pore size of approximately 1.3 nm (Supplementary Fig. 3). After aging in a water-ethanol mixture at 60 °C for 24 hours and gradually evaporating the solvent, the BET surface area of the aged TpPa-COF-10% membrane improved to 1504 m2 g−1, attributed to the relaxation of the linear polymer composites and optimization of the arrangement of COF nanocrystals.

a Experimental PXRD patterns of TpPa-COF-10% membrane, TpPa-COF-10% powder, and TpPa-COF powder. b N2 sorption isotherms collected at 77 K for the aged TpPa-COF-10% membrane, TpPa-COF-10% membrane, TpPa-COF-10% powder, and TpPa-COF powder. c Summary of the BET surface areas calculated for the aged TpPa-COF-10% membrane, TpPa-COF-10% membrane, TpPa-COF-10% powder, and TpPa-COF powder. d ATR-IR spectra of TP (pink), PDA (light teal), PPG-NH2 (sky blue), and TpPa-COF-10% membrane (green). e SS 13C NMR spectrum of TpPa-COF-10% membrane.
To investigate the chemical structures of TpPa-COF-10% membrane, powder forms of TpTa-COF and TpPa-COF-10% were synthesized via the conventional solvothermal method for comparison. The attenuated total reflection infrared (ATR-IR) analysis was then conducted to confirm the successful formation of the β-ketoenamine structures in the resulting TpPa-COF-10% membranes, revealing a new peak for C = C at ~1570 cm−1, which was consistent with the IR spectra of TpTa-COF and TpPa-COF-10% powder samples (Fig. 3d and Supplementary Fig. 4). Furthermore, the disappearance of the stretching signals of the primary amine (νN–H = between ~3372 cm−1 to 3174 cm−1) and aldehyde (νC=O = ~1635 cm−1) verified that no unreacted monomers were trapped. The incorporation of the dual-NH2-tail linear polymer was also confirmed by ATR-IR, evidenced by the appearance of a stretching signal of C−O at ~1095 cm−1. Additionally, the solid-state 13C nuclear magnetic resonance (SS 13C NMR) spectrum supported this observation, displaying a strong peak at approximately 17.1 ppm corresponding to the −CH3 groups, and another at 82.2 ppm, attributed to the C−O groups, both originating from the amino polymer additive (Fig. 3e). X-ray photoelectron spectroscopy (XPS) analysis further evidenced the formation of β-ketoenamine structures. In the N 1 s spectrum, a peak at 399.7 eV was attributed to the nitrogen in the β-ketoenamine moiety, while the O 1 s spectrum showed a peak at 530.7 eV corresponding to the ketone group. Additionally, a sharp peak at 532.6 eV was observed, representing the C-O group from the polymer composite (Supplementary Fig. 5). To evaluate the wettability of the TpPa-COF-10%, water contact angle (WCA) were performed on both its membrane and powder forms. The water contact angles of TpPa-COF-10% membrane and TpPa-COF-10% powder were approximately 102.2° and 134.9° respectively, indicating significant hydrophobicity of TpPa-COF-10% (Supplementary Fig. 6). Thermogravimetric analysis (TGA) was carried out to assess the stability of TpPa-COF-10% membrane, demonstrating that the membrane remained thermodynamically stable at temperatures up to 319 °C under nitrogen atmosphere (Supplementary Fig. 7).
The resulting free-standing TpPa-COF-10% membrane was flexible and integrated, as shown in Fig. 4a, allowing it to be bent significantly and easily recovered. Notably, it exhibited considerable thickness, making it easy to hold, transfer, and manipulate by hand, which is rarely achieved in previous COF membranes. The microstructure of the free-standing TpPa-COF-10% membrane was examined by scanning electron microscopy (SEM) images and element mapping, which revealed a rough and continuous membrane surface with a thickness of ~47 µm (Fig. 4b and Supplementary Fig. 8). The SEM images also illustrated that the resulting membrane featured hierarchical porous structures. In addition, the high-resolution transmission electron microscopy (TEM) images revealed strong and well-organized interlamellar interaction in TpPa-COF-10% membrane (Supplementary Fig. 9). The atomic force microscopy (AFM) image reveals the surface morphology of the TpPa-COF-10% membrane, exhibiting a root-mean-square (RMS) roughness of 1.27 µm. Particles of similar sizes are clustered together (Fig. 4c), with individual particle sizes remaining at the nanometer scale (Supplementary Fig. 10).

a Optical photographs of the TpPa-COF-10% membrane, with a quarter dollar coin (24.26 mm in diameter) as a reference. b SEM images of TpPa-COF-10% membrane. c AFM images of TpPa-COF-10% membrane.
Chemistry in Membrane Formation
To investigate the role of PPG-NH2 in the production of free-standing membranes, comprehensive studies using operando proton nuclear magnetic resonance (¹H NMR) were conducted with individual COF precursor combination. First, 2 molar equivalents of TP and 0.3 molar equivalents of PPG-NH2 were completely dissolved in deuterated trifluoroacetic acid (CF3COOD) to form a clear, homogeneous solution, which was then loaded into a high-pressure NMR tube with a screwed cap to prevent solvent evaporation during in-situ heating at 50 °C, allowing for the collection of all ¹H NMR spectra over a time range of 0 to 4 h. Surprisingly, the in-situ 1H NMR spectra showed no changes over the 4-hour heating period, with no consumption of the aldehyde groups from TP (Fig. 5a), indicating that TP does not react with PPG-NH2 when pure TFA is used as the solvent. This finding contradicts previous observation (ATR-IR, SS 13C NMR, and XPS), which indicated successful fusion of this polymer into the resulting TpPa-COF-10% membrane. To understand why this polymer can act as molecular threads during the free-standing membrane production, 2.7 molar equivalents of PDA were added to the NMR solution, followed by an additional 6 hours of in-situ heating to obtain the three-component NMR spectra. As shown in Fig. 5b, a slight peak shift in aldehyde groups was observed after just 15 minutes of in-situ heating, whereas the hydrogen peaks of the methine, methyl, and methoxy groups from PPG-NH2 showed no variation during the 30-minute heating period, implying that TP and PDA dominated the Schiff base reaction initially in this three-component system (Stage I). At this stage, the NMR solution remained clear and transparent, indicating a low degree of polymerization of TP and PDA. Afterwards, the polymer participated in the reaction with TP when plenty of imine bonds formed between TP and PDA. This was confirmed by the downfield shift of all hydrogen peaks from the groups adjacent to the newly formed −C=N− bonds in the polymer component (Stage II), which was consistent with the fact that amines have a stronger electron-donating ability than the resulting imine bonds54. Meanwhile, all aldehyde groups were consumed after a 6-hour heating period, and a muddy sol-gel solution was obtained upon cooling the mixture to room temperature (Stage III).

a In-situ ¹H NMR spectra of the reactions between TP and PPG-NH2 monitored over the 4-hour heating period. b In-situ ¹H NMR spectra of the reactions between TP, PDA, and PPG-NH2 monitored over the 6-hour heating period. c DFT-calculated reaction energies of forming the possible products and reaction routes during the sol-gel solution formation using 1-methoxy-2-propylamine as the alternative of PPG-NH2.
The reaction energetics of forming the possible products during sol-gel solution formation were investigated using density functional theory (DFT) calculations. To simplify the analysis, a repeating unit of PPG-NH2 with a single −NH2 tail (1-methoxy-2-propylamine, MPA) was employed to study the reactions. As expected, MPA is more likely to be protonated by TFA (R2) rather than react with TP to form R1. In contrast, PDA preferentially reacts with TP to form R4 lightly more than being protonated by TFA to form R3. R2 can further react with TP; however, the formation of R5 is an endothermic process (ΔE = 32.1 kJ mol-1), making R5 an unfavorable intermediate product. Interestingly, the formation of R6 is exothermic, as the proton in R3 can be delocalized within the newly formed COF unit. Finally, the reaction of R6 with R2 to form R7 (ΔE = 23.5 kJ mol-1) is endothermic because the excess proton cannot be further delocalized. Only PDA can react with R6 to yield R8, meaning that only one MPA unit can attach to the TP framework. This result is consistent with the NMR analysis of model reactions using TP, excess PPG-NH2, and varying ratios of amine monomers (Supplementary Fig. 11). In summary, the DFT calculations confirm that the likely reaction route begins with the formation of R4, progresses to R6, and ultimately yields R8.
Based on both experimental NMR investigations and computational results, the proposed reactions during the formation of the COF precursor sol-gel solution can be summarized as follows: (1) in the presence of TP, PDA exhibit higher reactivity than PPG-NH2 and can rapidly form imine bonds in pure TFA solvent; (2) as some aldehyde groups in TP are replaced by the newly formed imine bonds, the meta-positioned aldehyde groups become activated to react with the polymer component; (3) oligomers formed from TP and PDA are generated continuously until saturation is reached within the limited volume of TFA; (4) A linear and flexible polymer with dual amine-ended tails (PPG-NH2) serves as molecular threads that covalently link neighboring COF nanocrystals by suturing them together. This covalent suturing not only restricts the overgrowth of COF domains but also reinforces interparticle connectivity, thereby enhancing interlamellar interactions and promoting the formation of a uniform sol-gel network. As a result, a free-standing membrane can be cast from this solution by drying it in a glass Petri dish, with the incorporated flexible molecular threads significantly enhancing the overall robustness and rigidity of the resulting membrane (Supplementary Fig. 12). Moreover, increasing the loading amount of the polymer component is unnecessary for producing the free-standing COF membrane, as indicated by the DFT calculation results and model reaction observations. Excess polymer loading causes only a single amino tail to attach to the COF structure, preventing the polymer from effectively acting as the molecular threads to suture COF nanocrystals. Furthermore, an excessive addition of the polymer may take the opportunity of PDA to connect with TP, thereby inhibiting the formation of crystalline COF structures.
Subsequently, we extended this strategy to produce a COF membrane using 1,3,5-triformylbenzene (TFB) and PDA. However, regardless of the amount of PPG-NH2 added, only powder was obtained on the surface of the glass Petri dish after the evaporation of TFA. We speculated that the ortho-positioned phenol group would interact with the amino group through hydrogen bonding55, facilitating the progression of the reaction. Thus, we first selected benzaldehyde and PPG-NH2 in CF3COOD as a model reaction to explore the chemical reactivity between the neat aldehyde group and amino group in this polymer. As expected, no reaction was observed after heating the solution at 50 °C for 4 hours (Supplementary Fig. 13). Then, a CF3COOD solution of TFB and PPG-NH2 was prepared and heated to 50 °C and maintained for 4 hours to collect the initial and final NMR spectra for comparison, which showed no differences, indicating that no reaction occurred between TFB and the polymer component, as well (Supplementary Fig. 14a). However, a significant split and shift of aldehyde group was observed after adding PDA immediately at room temperature, demonstrating that TFB exhibits higher reactivity with PDA compared to TP. Interestingly, as heating progressed, no additional peak changes were detected, even when the temperature was raised to 70 °C for 8 hours. A slight advancement in the reaction occurred until the temperature was further increased to 100 °C which exceeds the boiling point of TFA (72.4 °C) and maintained for 12 hours; however, a considerable amount of aldehyde groups retained and kept unreacted even at this high temperature (Supplementary Fig. 14b). DFT calculation results demonstrate that the formation of S4 (ΔE = 24.8 kJ mol-1) is endothermic and thermodynamically unfavorable compared to the formation of S5 (ΔE = −7.3 kJ mol-1), which is the condensation product of TFB and PDA (Supplementary Fig. 14c). However, all potential products (S6, S7, and S8) derived from the S5 intermediate and protonated MPA involve endothermic reactions, as shown in Supplementary Fig. 14c. This indicates that the reaction pathway is effectively terminated after the formation of S5. The theoretical findings perfectly explain the unusual experimental observations, demonstrating that no COF membrane can be formed when TP is replaced by TFB, and only the reaction between TFB and PDA in CF3COOD is observed in the 1H NMR spectra.
The above results revealed that pure trifluoroacetic acid hindered the forward progression of the Schiff base reaction, despite being a good solvent capable of easily dissolving both the monomers and the formed oligomers, which was considered an optimal solvent for membrane fabrication to verify the feasibility of our in-situ nanocrystal suturing strategy for producing free-standing TpPa-COF-10% membrane. On the other hand, the results also validated our speculation that the ortho-positioned phenol group facilitated the Schiff base reaction between its neighboring aldehyde group and the amino groups.
Then, we reduced the loading molar amount of PPG-NH2 to 5% and even 1%, successfully obtaining free-standing TpPa-COF-X% (X = 5 or 1) membranes that exhibited outstanding mechanical properties, as well (Fig. 6a, Supplementary Figs. 15–17, and Supplementary Table 3). It is worth noting that the size of the membrane only limited by the dimensions of the glass Petri dish. A free-standing TpPa-COF-10% membrane with a diameter of ~9 cm is displayed in Fig. 6b, produced using the largest Petri dish available in our lab. Regarding another dimension of the membrane, its thickness can be readily controlled by adjusting the feeding amount of the precursor mixtures, as demonstrated by the free-standing membrane with a thickness of 1.01 mm, achieved by enlarging the membrane precursor solution, as illustrated in Fig. 6c.

a Optical photographs of the free-standing TpPa-COF-5% and TpPa-COF-1% membranes. b Photographs showing the TpPa-COF-10% membrane, which has a diameter of up to ~9 cm, with a quarter dollar coin (24.26 mm in diameter) as a reference. c Photographs showing the TpPa-COF-10% membrane with a thickness of up to 1.01 mm. d Schematic illustration of creating TpBZ-COF-10% and TpPa-CO2H-COF-10% membranes. e Experimental PXRD patterns of TpBZ-COF-10% and TpPa-CO2H-COF-10% membranes. f N2 sorption isotherms collected at 77 K for TpBZ-COF-10% and TpPa-CO2H-COF-10% membranes, the BET surface areas were calculated as 821 and 135 m2 g−1, respectively.
To expand the applicability of this method and fabricate function-specific COF membranes, we selected benzidine (BZ) and 2,5-diaminobenzoic acid (Pa-CO2H) as building blocks with TP to successfully produce versatile, free-standing COF membranes. These membranes, designated as TpBZ-COF-10% and TpPa-CO2H-COF-10% respectively, highlight the flexibility of the approach in accommodating diverse functional monomers (Fig. 6d, e, and Supplementary Fig. 18). This achievement further underscores the effectiveness and versatility of the unique strategy of free-standing COF membrane fabrication via nanocrystal suturing with tiny molecular threads. Such advancements not only validate the method’s superiority but also pave the way for its application in developing high-performance COF membranes tailored to specific functionalities.
Performance Investigation of the Free-standing TpPa-COF-10% Membrane
Taking advantage of the highly crystalline scaffolds of the free-standing COF membrane, which features uniformly distributed and well-ordered microporous channels, the membrane was evaluated for its capability in selectively sieving water-soluble dyes. The obtained membrane exhibits not only a consistent pore size across the entire framework, but also an open and accessible internal architecture. This unique structural combination facilitates the rapid permeation of dye-containing aqueous solutions, while simultaneously achieving efficient molecular sieving by excluding larger dye molecules based on pore size. A collection of organic dyes with different sizes were selected to evaluate the rejection performance of the TpPa-COF-10% membrane. As expected, the membrane (pore size: ~13 Å) can successfully reject more than 99% of Rose bengal (Mw = 973.67 Da, 15.4 Å × 12.0 Å), Calcein (Mw = 622.55 Da, 17.6 Å × 8.8 Å), Brilliant blue (Mw = 792.84 Da, 20.6 Å × 17.9 Å), and Methyl blue (Mw = 799.81 Da, 23.6 Å × 17.4 Å), all of which have at least one dimension larger than the membrane’s pore size. For those dyes smaller than the membrane’s pore size, such as Methyl orange (Mw = 327.33 Da, 11.3 Å × 4.2 Å) and Rhodamine 6 G (Mw = 479.02 Da, 11.5 Å × 9.0 Å), rejection rates of 83.4% and 88.1% were achieved respectively (Fig. 7a). demonstrating dye separation performance comparable to that of state-of-the-art membranes (Supplementary Table 4).

a Rejection performance of TpPa-COF-10% membranes towards dyes with varying molecular sizes in water. b Nyquist plots of the H3PO4@TpPa-COF-10% membrane at different temperatures ranging from 100 to 160 °C. c Proton conductivity of the H3PO4@TpPa-COF-10% membrane at different temperatures ranging from 100 to 160 °C. d Arrhenius plots of the proton conductivity of the H3PO4@TpPa-COF-10% membrane. e Performance stability of the H3PO4@TpPa-COF-10% membrane after 48-h of continuous operation at 160 °C.
High proton conductivity is essential for the efficient operation of proton exchange membrane fuel cells (PEMFCs)56,57,58. Currently, state-of-the-art proton exchange membranes operating at elevated temperatures (130–220 °C) are primarily based on sulfonated hydrocarbon polymers (SHPs) and polybenzimidazole (PBI)59. However, these conventional membranes suffer from inherent drawbacks: (1) their linear and flexible backbones result in poor thermal and mechanical stability, leading to significant swelling at high temperatures, reduced durability, and increased gas crossover; (2) their dense structures restrict the uptake of proton carriers such as phosphoric acid (H3PO4), thereby limiting proton transport efficiency. In contrast, COFs with their well-defined porous crystalline architectures and tunable local environments offer compelling advantages for proton-conducting applications. Their inherent porosity and ordered channels enable the formation of extensive hydrogen-bonding networks with various proton carriers, making them attractive candidates for high-performance membranes60,61,62,63. Nonetheless, conventional powdered COFs pose significant challenges in membrane processing, often resulting in non-uniform and unstable films that hinder practical deployment in PEMFCs64,65.
Unlike humidity-dependent proton conduction, anhydrous proton conduction operates without the need for water management, making it ideal for various fuel cell devices, especially those operating at high temperatures66,67,68. The TpPa-COF-10% membrane obtained in this study, cast from a strong acid solvent (TFA), exhibits multiple structural and functional advantages, including excellent thermal stability, a free-standing configuration, large surface area, and a robust framework. Under saturated H3PO4 loading, it demonstrates outstanding anhydrous proton conductivity. Its hierarchical pore architecture—composed of interconnected micro- and mesopores—not only promotes high phosphoric acid uptake by offering abundant anchoring sites and internal free volume, but also ensures uniform acid distribution throughout the framework. In addition, the well-aligned microporous channels serve as efficient and directional pathways for proton hopping. These synergistic structural features enable the COF membrane to overcome the limitations of traditional polymer-based PEMs and deliver high-performance, water-free proton conduction suitable for high-temperature fuel cell applications. Proton conductivity of H3PO4@TpPa-COF-10% membrane was assessed using alternating-current impedance analysis, a well-established electrochemical technique for characterizing the intrinsic ion-conducting properties of materials69,70,71.
Neat H3PO4, serving as the H+ source, was loaded into the TpPa-COF-10% membrane by immersing it in an excess of 85 wt.% aqueous H3PO4 solution for 24 hours at 70 °C, allowing the membrane to reach saturation with H3PO4. The membrane was then taken out from the solution and dried with filter papers to remove any H3PO4 residue on its surface, followed by vacuum drying at 100 °C to eliminate any trapped water molecules within the membrane (Supplementary Figs. 19–21). Figure 7b presents the Nyquist plots of the H3PO4@TpPa-COF-10% membrane under a nitrogen atmosphere at temperatures between 100 and 160 °C in an anhydrous environment. The membrane at 160 °C displays a curve intersecting the x-axis, corresponding to a resistance of 4.83 Ω, from which the anhydrous proton conductivity (σ) is calculated to be as high as 1.21 × 10−2 S cm-1 using the equation
where σ represents the proton conductivity (S cm−1), L is the measured thickness of the membrane (cm), A is the electrode area (cm2), and Z is the impedance (Ω). As shown in Fig. 7c, the H3PO4@TpPa-COF-10% membrane exhibits proton conductivities of 4.83 × 10−3, 5.43 × 10−3, 6.25 × 10−3, 7.23 × 10−3, 8.54 × 10−3, 1.02 × 10−2, 1.21 × 10−2 S cm−1 at 100, 110, 120, 130, 140, 150, and 160 °C, respectively, which are comparable to state-of-the-art H3PO4-doped commercial polymer membranes and Nafion-based membranes (Supplementary Table 5)59. The activation energy (Ea) value of H3PO4@TpPa-COF-10% membrane was calculated using Arrhenius equation of
From the slope, the activation energy (Ea) was determined to be 0.21 eV, indicating a Grotthuss mechanism (Ea < 0.5 eV), in which protons hop within the H3PO4@TpPa-COF-10% membrane (Fig. 7d). The rapid proton transfer path observed in H3PO4@TpPa-COF-10% membrane is attributed to the ordered structure of the TpPa-COF-10% framework. The H3PO4@TpPa-COF-10% membrane also demonstrated robust stability in performance when continuously heated at 160 °C for 48 hours (Fig. 7e). Additionally, membranes fabricated over a large area exhibited performance comparable to their smaller counterparts, underscoring the reproducibility and consistency across different membrane scales (Supplementary Fig. 22).
Discussion
In this study, the sol-gel chemistry was further extended to produce free-standing COF membranes through a nanocrystal suturing strategy, using a dual-amino (NH2) tail linear polymer as a molecular thread to suture COF nanocrystals during the membrane molding. The resulting COF membranes exhibit excellent mechanical properties, allowing them to be significantly bent and easily recovered. Furthermore, the size and the thickness of the membranes can be readily manipulated by varying the feeding amount and ratio of the precursor mixtures, which advances the development of free-standing COF membranes for practical applications. The hierarchical porous structures enable the membrane to encapsulate a substantial amount of H3PO4 molecules, facilitating rapid and efficient proton transfer, thereby resulting in high anhydrous proton conductivity. This strategy, which employs tiny threads to restrict the growth of COF nanocrystals and facilitate the formation of homogenous sol-gel solution for the fabrication of high-quality free-standing COF membranes, opens up new avenues for optimizing COF membranes and expanding their practical applications.
Methods
Materials and measurements
Triformylphloroglucinol and 1,3,5-triformylbenzene were purchased from Ambeed. Solvents and other commercially available reagents were purchased from Sigma-Aldrich and used without further purification. Small-angle X-ray scattering (SAXS) experiments were carried out using Xenocs Xeuss 3.0 equipped with a Cu anode X-ray generator and a Dectris Pilatus 2D detector. The 2D data was then radially averaged to 1D. Careful background correction was performed before data analysis. The particle size distribution was obtained via dynamic light scattering (DLS) at 90° scattering angle using a Malvern/Zetasizer Ultra with 4 mW He-Ne laser at 632.8 nm. The Powder X-ray diffraction (PXRD) data were collected on a PANalytical Empyrean diffractometer (40 kV, 40 mA) using Cu Kα (λ = 1.5406 Å) radiation. Nitrogen sorption isotherms at 77 K were measured using a 3-Flex Micromeritics surface area analyzer with a cryo-station for temperature control. The samples were outgassed for 48 h at 373 K before the measurements. ATR-IR spectra were recorded on a Nicolet Nexus iS50 ATR-IR instrument. In-situ liquid 1H NMR (400 MHz) spectra were recorded on a JEOL-400YH spectrometer. Chemical shifts are expressed in ppm downfield from TMS at δ = 0 ppm, and J values are given in Hz. 13C (125 MHz) cross-polarization magic-angle spinning (CP-MAS) was recorded on a Bruker Avance 500 spectrometer equipped with a magic-angle spin probe in a 4-mm (13C) ZrO2 rotor. Thermogravimetric analysis (TGA) measurements were taken under a nitrogen atmosphere with a ramping rate of 10 °C min−1 from 25 °C to 800 °C by using a TGA Q50 thermogravimetric analyzer. X-ray photoelectron spectroscopy (XPS) spectra were performed on a Thermo ESCALAB 250XI with Al Kα irradiation at θ = 90° for X-ray sources (hv=1468.6 eV), and the binding energies were calibrated using the C1s peak at 284.8 eV. Concentrations of dyes in water were determined by a Thermo Scientific Evolution 300 UV-Vis spectrophotometers. Photographs of water on the surface of the samples were measured with KRUSS DSA100. Scanning electron microscopy (SEM) images and Energy-dispersive X-ray spectroscopy (EDS) studies were performed at a Zeiss Auriga Crossbeam FIB-SEM and FEI Helios NanoLab 600 SEM. Transmission electron microscopy (TEM) images were collected on a Fisher Scientific Spectra 300 electron microscope. Atomic Force Microscopy (AFM) measurements were performed at ambient room temperature using a commercial Park NX20-AFM in non-contact mode. Standard silicon cantilevers with a nominal resonant frequency of approximately 330 kHz were used for topographic imaging. Image analysis was conducted using SPIP software. Electrochemical impedance spectroscopy (EIS) data were collected using a CHI 760E electrochemical workstation with a two-probe electrochemical cell. The in-situ membrane formation and surface changes were recorded using a Phantom v711 camera equipped with a micro-scale lens.
Computational details
The DFT-calculated reaction energies of the possible reaction pathways for COF membrane formation were obtained using the Gaussian 16 package72. The repeating unit of PPG-NH2 was replaced as a single -NH2 tail (1-methoxy-2-propylamine, MPA) to simplify the calculations. Geometry optimizations of the reactants (TP, TFA, PDA, MPA) and products (R1-R8) were performed at the B3LYP/6-311 + G* level of theory with Grimme’s D3 dispersion correction and SMD solvation model73,74,75,76. Formic acid was used as the solvent in the SMD model as an approximation to the TFA solvent used in experiments (there are no available SMD parameters for TFA). Default criteria were selected for SCF convergence and geometry optimization. Normal mode analysis was further performed to ensure that local minima were found in geometry optimization without imaginary frequencies.
Fabrication of free-standing TpPa-COF-X% (X = 1, 5, and 10) membranes
A glass vial was charged with triformylphloroglucinol (TP) (43.3 mg, 0.2 mmol), p-phenylenediamine (PDA) (32.4 mg, 0.3 mmol), and poly(propylene glycol) bis(2-aminopropyl ether) (PPG-NH2) (average Mn ~2000) in varying amounts (6 mg, 0.003 mmol; 30 mg, 0.015 mmol; or 60 mg, 0.03 mmol) in 2.0 mL of trifluoroacetic acid (TFA). The mixture was sonicated for 5 min to ensure complete homogenization and then heated at 50 °C for 4 hours to yield a so-gel solution. Afterwards, the so-gel solution was degassed for 5 minutes, poured into a glass Petri dish, and covered to allow for slow evaporation of the TFA, resulting in a dry membrane which was then washed with ethanol to remove soluble residues and treated with an aqueous solution of triethylamine to neutralize any unreacted TFA. The resultant membrane was transferred to a glass high-pressure reactor containing 11 mL of a 5:5:1 v:v:v solution of o-dichlorobenzene:n-butanol:6 M acetic acid aqueous solution. The membrane was heated at 120 °C for 3 days to afford a crystalline TpPa-COF-X% membrane. Subsequently, the membrane underwent an aging process in a water-ethanol mixture at 60 °C for 1 day to further enhance its porosity.
Synthesis of TpPa-COF-10% powder
A Pyrex tube was charged with triformylphloroglucinol (43.3 mg, 0.2 mmol), p-phenylenediamine (32.4 mg, 0.3 mmol), and poly(propylene glycol) bis(2-aminopropyl ether) (average Mn ~2000) (60 mg, 0.03 mmol) in 1.1 mL of a 5:5:1 v:v:v solution of o-dichlorobenzene:n-butanol:6 M acetic acid aqueous solution. The tube was flash frozen at 77 K (liquid N2 bath), evacuated, and flame sealed. The reaction mixture was heated at 120 °C for 3 days to afford a red precipitate, which was isolated by filtration and washed with THF and acetone to afford TpPa-COF-10% powder.
Synthesis of TpPa-COF powder
A Pyrex tube was charged with triformylphloroglucinol (43.3 mg, 0.2 mmol), p-phenylenediamine (32.4 mg, 0.3 mmol) in 1.1 mL of a 5:5:1 v:v:v solution of o-dichlorobenzene:n-butanol:6 M acetic acid aqueous solution. The tube was flash frozen at 77 K (liquid N2 bath), evacuated, and flame sealed. The reaction mixture was heated at 120 °C for 3 days to afford a red precipitate which was isolated by filtration and washed with THF and acetone to afford TpPa-COF powder.
Synthesis of H3PO4@TpPa-COF-10% membrane
The membrane was placed in a glass Petri dish with an excess addition of 85 wt.% phosphoric acid aqueous solution. The mixture was then heated at 70 °C for 24 hours to ensure sufficient permeation of H3PO4 into the hierarchical porous structures of the membrane. After cooling to room temperature, the membrane was carefully removed from the solution, and any residual H3PO4 on both sides was wiped off using filter paper. Finally, a ready-to-use H3PO4@TpPa-COF-10% membrane was dried at 70 °C under vacuum for 24 hours.
In-situ visualization system
A high-speed and micro-scale visualization system (HMVS) was employed to capture the detailed process of the membrane fabrication on the glass surface. Surface changes of the membrane were recorded over a 10-minute period immediately after pouring the sol-gel solution into the glass Petri dish, using a Phantom v711 camera equipped with a micro-scale lens. The camera, positioned vertically above the dish, captured the video. The video was accelerated by 15x for the first 5 minutes and 30x for the last 5 minutes to enable real-time monitoring of the membrane fabrication and surface changes in situ (Supplementary Movie).
Dye rejection performance measurement
The filtration performance of the membranes was evaluated using a Millipore classic glass filter holder kit (effective permeation area: 17.35 cm2; diameter: 47 mm) under a constant pressure of 1 bar at 25 °C. The optimized free-standing TpPa-COF-10% membrane with a diameter of approximately 5 cm and a thickness of around 20 µm (Supplementary Table 6) was securely placed between the glass funnel and the glass frit membrane support, and firmly sealed using a spring clamp. A 100 mL feed solution containing 100 ppm dye in Milli-Q water was added to the glass funnel, with or without stirring. The concentration of dye in the filtrate was monitored using UV–vis spectroscopy. The rejection rate of dye was calculated as follows:
Where C0 and C1 are the initial concentration of dye and the concentration in the filtrate, respectively.
Anhydrous proton conductivity evaluation
The dried H3PO4@TpPa-COF-10% membrane was cut into a disc with a diameter of 1.5 cm, and its thickness was measured with a vernier caliper, confirming a thickness of 0.46 mm. The electrochemical impedance spectroscopy (EIS) measurements were conducted using a CHI 760E electrochemical workstation with a two-probe electrochemical cell. The cell had an effective contact area of 0.785 cm2 (d = 1 cm), and measurements were taken over a frequency range of 0.1 Hz to 4 MHz with an input voltage amplitude of 10 mV. Before conducting the measurement, the cell equipped with the H3PO4@TpPa-COF-10% membrane was heated at 120 °C under vacuum for 24 hours to eliminate any trapped water molecules, then switched to a nitrogen atmosphere.
Data availability
The authors declare that all the data supporting the findings of this study are available within the article (and Supplementary Information files). Additional data are available from the corresponding author upon request.
References
Yuan, S. et al. Covalent organic frameworks for membrane separation. Chem. Soc. Rev. 48, 2665–2681 (2019).
Lin, Q. et al. Crystalline porous framework-based membrane as a sub-nanometer platform for lithium extraction. Coord. Chem. Rev. 515, 215971 (2024).
Denny, M. S., Moreton, J. C., Benz, L. & Cohen, S. M. Metal-organic frameworks for membrane-based separations. Nat. Rev. Mater. 1, 16078 (2016).
Song, Y., Zhu, C. & Ma, S. Advanced porous organic polymer membranes: Design, fabrication, and energy-saving applications. EnergyChem 4, 100079 (2022).
Meng, X., Wang, H.-N., Song, S.-Y. & Zhang, H.-J. Proton-conducting crystalline porous materials. Chem. Soc. Rev. 46, 464–480 (2017).
Knebel, A. & Caro, J. Metal-organic frameworks and covalent organic frameworks as disruptive membrane materials for energy-efficient gas separation. Nat. Nanotechnol. 17, 911–923 (2022).
Hou, L. et al. Understanding the ion transport behavior across nanofluidic membranes in response to the charge variations. Adv. Funct. Mater. 31, 2009970 (2021).
Guo, Y., Ying, Y., Mao, Y., Peng, X. & Chen, B. Polystyrene sulfonate threaded through a metal-organic framework membrane for fast and selective lithium-ion separation. Angew. Chem. Int. Ed. 55, 15120–15124 (2016).
Zheng, L. et al. Covalent organic framework membrane reactor for boosting catalytic performance. Nat. Commun. 15, 6837 (2024).
Zuo, X. et al. Thermo-osmotic energy conversion enabled by covalent-organic-framework membranes with record output power density. Angew. Chem. Int. Ed. 61, e202116910 (2022).
Hou, S. et al. Free-standing covalent organic framework membrane for high-efficiency salinity gradient energy conversion. Angew. Chem. Int. Ed. 60, 9925–9930 (2021).
Cao, L. et al. Giant osmotic energy conversion through vertical-aligned ion-permselective nanochannels in covalent organic framework membranes. J. Am. Chem. Soc. 144, 12400–12409 (2022).
Geng, K. et al. Covalent organic frameworks: design, synthesis, and functions. Chem. Rev. 120, 8814–8933 (2020).
Diercks, C. S. & Yaghi, O. M. The atom, the molecule, and the covalent organic framework. Science 355, eaal1585 (2017).
Côté, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Song, Y., Sun, Q., Aguila, B. & Ma, S. Opportunities of covalent organic frameworks for advanced applications. Adv. Sci. 6, 1801410 (2019).
Huang, N., Wang, P. & Jiang, D. Covalent organic frameworks: a materials platform for structural and functional designs. Nat. Rev. Mater. 1, 16068 (2016).
Li, X. et al. Metal-organic frameworks based membranes for liquid separation. Chem. Soc. Rev. 46, 7124 (2017).
Cheng, Y. et al. Advances in metal-organic framework-based membranes. Chem. Soc. Rev. 51, 8300 (2022).
Liu, J. & Wöll, C. Surface-supported metal-organic framework thin films: fabrication methods, applications, and challenges. Chem. Soc. Rev. 46, 5730 (2017).
Liu, Y., Ban, Y. & Yang, W. Microstructural engineering and architectural design of metal-organic framework membranes. Adv. Mater. 29, 1606949 (2017).
Wang, R. et al. Ultrathin covalent organic framework membranes prepared by rapid electrophoretic deposition. Adv. Mater. 34, 2204894 (2022).
Yang, Y. et al. A self-standing three-dimensional covalent organic framework film. Nat. Commun. 14, 220 (2023).
Khan, N. A. et al. Assembling covalent organic framework membranes via phase switching for ultrafast molecular transport. Nat. Commun. 13, 3169 (2022).
Hao, Q. et al. Confined synthesis of two-dimensional covalent organic framework thin films within superspreading water layer. J. Am. Chem. Soc. 140, 12152–12158 (2018).
Wang, L. et al. Electrocleavage synthesis of solution-processed, imine-linked, and crystalline covalent organic framework thin films. J. Am. Chem. Soc. 144, 8961–8968 (2022).
Carrington, M. E. et al. Sol-gel processing of a covalent organic framework for the generation of hierarchically porous monolithic adsorbents. Chem 8, 2961–2977 (2022).
Rodriguez-San-Miguel, D., Montoro, C. & Zamora, F. Covalent organic framework nanosheets: preparation, properties and applications. Chem. Soc. Rev. 49, 2291–2302 (2020).
Burke, D. W. et al. Acid exfoliation of imine-linked covalent organic frameworks enables solution processing into crystalline thin films. Angew. Chem. Int. Ed. 59, 5165–5171 (2020).
Rodriguez-San-Miguel, D. & Zamora, F. Processing of covalent organic frameworks: an ingredient for a material to succeed. Chem. Soc. Rev. 48, 4375–4386 (2019).
Meng, Q.-W. et al. Optimizing selectivity via membrane molecular packing manipulation for simultaneous cation and anion screening. Sci. Adv. 10, eado8658 (2024).
Ying, Y., Peh, S. B., Yang, H., Yang, Z. & Zhao, D. Ultrathin covalent organic framework membranes via a multi-interfacial engineering strategy for gas separation. Adv. Mater. 34, 2104946 (2022).
Meng, Q.-W. et al. Enhancing ion selectivity by tuning solvation abilities of covalent-organic-framework membranes. PNAS 121, e2316716121 (2024).
Guo, X. et al. Fabrication of photoresponsive crystalline artificial muscles based on PEGylated covalent organic framework membranes. ACS Cent. Sci. 6, 787–794 (2020).
Wang, Z. et al. PolyCOFs: A new class of freestanding responsive covalent organic framework membranes with high mechanical performance. ACS Cent. Sci. 5, 1352–1359 (2019).
Du, J. et al. A 2D soft covalent organic framework membrane prepared via a molecular bridge. Adv. Mater. 35, 2300975 (2023).
Zhang, Y. et al. Molecularly soldered covalent organic frameworks for ultrafast precision sieving. Sci. Adv. 7, eabe8706 (2021).
Cheng, Y. et al. Highly efficient CO2 capture by mixed matrix membranes containing three-dimensional covalent organic framework fillers. J. Mater. Chem. A 7, 4549–4560 (2019).
Kang, Z. et al. Mixed matrix membranes (MMMs) comprising exfoliated 2D covalent organic frameworks (COFs) for efficient CO2 separation. Chem. Mater. 28, 1277–1285 (2016).
Cao, X., Wang, Z., Qiao, Z., Zhao, S. & Wang, J. Penetrated COF channels: Amino environment and suitable size for CO2 preferential adsorption and transport in mixed matrix membranes. ACS Appl. Mater. Interfaces 11, 5306–5315 (2019).
Wu, X. et al. Mixed matrix membranes comprising polymers of intrinsic microporosity and covalent organic framework for gas separation. J. Membr. Sci. 528, 273–283 (2017).
Biswal, B. P., Chaudhari, H. D., Banerjee, R. & Kharul, U. K. Chemically stable covalent organic framework (COF)-polybenzimidazole hybrid membranes: enhanced gas separation through pore modulation. Chem. Eur. J. 22, 4695–4699 (2016).
Ciriminna, R. et al. The sol-gel route to advanced silica-based materials and recent applications. Chem. Rev. 113, 6592–6620 (2013).
Arachchige, I. U. & Brock, S. L. Sol-gel methods for the assembly of metal chalcogenide quantum dots. Acc. Chem. Res. 40, 801–809 (2007).
Korala, L., Wang, Z., Liu, Y., Maldonado, S. & Brock, S. L. Uniform thin films of CdSe and CdSe(ZnS) Core(Shell) quantum dots by sol-gel assembly: enabling photoelectrochemical characterization and electronic applications. ACS Nano 7, 1215–1223 (2013).
Yang, Z. et al. Fabrication of ionic covalent triazine framework-linked membranes via a facile sol-gel approach. Chem. Mater. 33, 3386–3393 (2021).
Zhu, X. et al. A superacid-catalyzed synthesis of porous membranes based on triazine frameworks for CO2 Separation. J. Am. Chem. Soc. 134, 10478–10484 (2015).
Qiu, L., Yang, Z. & Dai, S. Covalent triazine framework-derived membranes: engineered sol-gel construction and gas separation application. Acc. Mater. Res. 4, 1020–1032 (2023).
Yang, Z. et al. Surpassing Robeson upper limit for CO2/N2 separation with fluorinated carbon molecular sieve membranes. Chem 6, 631–645 (2020).
Kandambeth, S. et al. Construction of crystalline 2D covalent organic frameworks with remarkable chemical (acid/base) stability via a combined reversible and irreversible route. J. Am. Chem. Soc. 134, 19524–19527 (2012).
Senarathna, M. C. et al. Highly flexible dielectric films from solution processable covalent organic fameworks. Angew. Chem. Int. Ed. 62, e202312617 (2023).
Caruso, M. M. et al. Solvent-promoted self-healing epoxy materials. Macromolecules 40, 8830–8832 (2007).
Boulogne, F., Pauchard, L. & Giorgiutti-Dauphiné, F. Effect of a non-volatile cosolvent on crack patterns induced by desiccation of a colloidal gel. Soft Matter 8, 8505–8510 (2012).
Kovaříček, P. & Lehn, J.-M. Merging constitutional and motional covalent dynamics in reversible imine formation and exchange processes. J. Am. Chem. Soc. 134, 9446–9455 (2012).
Güngör, Ö. & Gürkan, P. Synthesis and characterization of higher amino acid Schiff bases, as monosodium salts and neutral forms. Investigation of the intramolecular hydrogen bonding in all Schiff bases, antibacterial and antifungal activities of neutral forms. J. Mol. Struct. 1074, 62–70 (2014).
Jiao, K. et al. Designing the next generation of proton-exchange membrane fuel cells. Nature 595, 361–369 (2021).
Xiao, F. et al. Recent advances in electrocatalysts for proton exchange membrane fuel cells and alkaline membrane fuel cells. Adv. Mater. 33, 2006292 (2021).
Devanathan, R. Recent developments in proton exchange membranes for fuel cells. Energy Environ. Sci. 1, 101–119 (2018).
Song, J. et al. Rational materials and structure design for improving the performance and durability of high temperature proton exchange membranes (HT-PEMs). Adv. Sci. 10, 2303969 (2023).
Cao, L. et al. Weakly humidity-dependent proton-conducting COF membranes. Adv. Mater. 32, 2005565 (2020).
Ma, H. et al. Cationic covalent organic frameworks: A simple platform of anionic exchange for porosity tuning and proton conduction. J. Am. Chem. Soc. 138, 5897–5903 (2016).
Liu, L. et al. Surface-mediated construction of an ultrathin free-standing covalent organic framework membrane for efficient proton conduction. Angew. Chem. Int. Ed. 60, 14875–14880 (2021).
Sasmal, H. S. et al. Superprotonic conductivity in flexible porous covalent organic framework membranes. Angew. Chem. Int. Ed. 57, 10894–10898 (2018).
Tao, S. et al. Confining H3PO4 network in covalent organic frameworks enables proton super flow. Nat. Commun. 11, 1981 (2020).
Yang, Y. et al. Combined intrinsic and extrinsic proton conduction in robust covalent organic frameworks for hydrogen fuel cell applications. Angew. Chem. Int. Ed. 59, 3678–3684 (2020).
Tao, S. & Jiang, D. Exceptional anhydrous proton conduction in covalent organic frameworks. J. Am. Chem. Soc. 146, 18151–18160 (2024).
Wu, X. et al. Perfluoroalkyl-functionalized covalent organic frameworks with superhydrophobicity for anhydrous proton conduction. J. Am. Chem. Soc. 142, 14357–14364 (2020).
Xu, H., Tao, S. & Jiang, D. Proton conduction in crystalline and porous covalent organic frameworks. Nat. Mater. 15, 722–726 (2016).
Yu, B. et al. Linkage conversions in single-crystalline covalent organic frameworks. Nat. Chem. 16, 114–121 (2023).
Yang, F.-F., Wang, X.-L., Tian, J., Yin, Y. & Liang, L. Vitrification-enabled enhancement of proton conductivity in hydrogen-bonded organic frameworks. Nat. Commun. 15, 3930 (2024).
Lu, C. et al. Thiatruxene-based conductive MOF: Harnessing sulfur chemistry for enhanced proton transport. J. Am. Chem. Soc. 146, 26313–26319 (2024).
Frisch, M. J. et al. Gaussian 16 Rev. C.01, Gaussian Inc. Wallingford CT (2016).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Acknowledgements
The research was supported financially by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy. The DLS experiments were performed at the Center for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory. SAXS measurements were enabled by the Major Research Instrumentation program of the National Science Foundation under Award No. DMR-1827474 (E.L.). AFM measurements were performed on a Park NX20-AFM of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704 (X.T.).
Author information
Authors and Affiliations
Contributions
Y.S., Z.Y. and S.D. conceived and designed the research. Y.S., Q.W., E.L., T.W., W.W., J.L., F.-Y.Z., B.L., D-e.J., Y.W., X.T., X.Y. and S.M.M. conducted and analyzed the experiments described in this report. All authors participated in drafting the paper and gave approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shengqian Ma, Fusheng Pan, and Qi Sun for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Y., Wang, Q., Li, E. et al. Creating free standing covalent organic framework membranes by nanocrystal suturing in sol gel solutions. Nat Commun 16, 5712 (2025). https://doi.org/10.1038/s41467-025-61325-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61325-9
