
Abstract
The alkaline oxygen evolution reaction is a key step in producing green hydrogen through water electrolysis, but its large-scale industrial application remains limited due to challenges with current electrocatalysts—particularly in terms of scalability, efficiency, and long-term stability. Here we show an industrially scalable synthesis of an active NiFe layered double hydroxide (NiFe-LDH) catalyst using a room-temperature, atmospheric-pressure route. The process involves homogeneous alkalinization, where chloride ions nucleophilically attack an epoxide ring, producing a low-dimensional, defect-rich NiFe-LDH with pronounced iron clustering. In-situ spectroscopy and ab-initio calculations reveal that these structural features maximize the conversion of the NiFe-LDH to the catalytic active phase and minimize the energy barrier, improving catalytic efficiency. When used as the anode in an anion exchange membrane water electrolyzer operating at 70 °C, our material delivers 1 A cm⁻² at 1.69 V in a 5 cm2 full-cell setup, with notable durability compared to conventional NiFe-LDHs. This scalable approach could considerably lower the cost of green hydrogen production by enabling more efficient alkaline electrolyzers.
Similar content being viewed by others


Introduction
The oxygen evolution reaction (OER) remains the bottleneck of the electrochemical water-splitting process, suffering from sluggish kinetics and requiring high overpotentials. Among the different water-splitting technologies, anion exchange membrane water electrolysis (AEMWE) has gained increasing attention due to the combination of the low cost of conventional alkaline electrolysis (AWE) and the high current densities, hydrogen purity, and intermittence capabilities of proton exchange membrane water electrolysis (PEMWE)1,2. Despite the advantage of AEMWE in potentially utilizing non-PGM catalysts, the lack of widely available standard materials results in IrO2 still being used as a reference material in the literature, even in alkaline media3,4,5,6,7,8,9. However, the instability of IrO2 at high anodic potentials and particularly the high cost and low abundance of PGM catalysts hinder large scale commercial applications10. Thus, the commercialization of highly active and efficient catalysts based on low-cost earth-abundant elements and non-geolocated is a matter of utmost importance.
Along this front, layered double hydroxides (LDHs)11,12,13,14,15, due to their flexible chemical tunability, can easily satisfy the previously mentioned requirements. Since the first report on NiFe-LDHs exhibiting high OER activity16, a tremendous effort was focused on the optimization of their performance15. Nowadays, state-of-the-art NiFe-LDH catalysts own the best performance for OER in alkaline conditions reported so far, outperforming those based on noble metals and amorphous pure Fe-based oxyhydroxides17. In this line, many different synthetic approaches have been explored to improve their electrochemical performance by increasing the number of the active sites, such as single layer exfoliation18, obtaining sub-nanometric sheets19, the incrementation in the basal space through anion exchange20, or defect engineering21. Moreover, other approximations have been successfully explored beyond increasing the number of centres, including the conductivity enhancement by heteroatom doping22, precious metal single atom doping23, or the change of catalytic mechanism by doping with amorphous FeOOH24,25.
Despite their fascinating performance, the practical application of LDHs in water splitting is restricted by the low efficiency of the available synthetic protocols26, which present high costs and difficulties due to the high temperatures and pressures required in these processes27. As a matter of fact, even the simplest case of NiFe-LDHs containing carbonate anions remains commercially unavailable, alerting about the bottleneck in the implementation of Fe-based LDHs at a large scale. In general, Fe hinders the scalability of NiFe-LDHs due to its high acidity (\({{pK}}_{a}^{{{Fe}}^{{III}}}=2.2\)), low solubility (\({{pK}}_{{sp}}^{{{Fe}\left({OH}\right)}_{3}}=38.6\)) and low water exchange rate (\({k}_{{H}_{2}O}^{{{Fe}}^{{III}}}=1.6\cdot {10}^{2}{S}^{-1}\)), facilitating the formation of less OER active oxide impurity phases when using traditional co-precipitation routes. To avoid this, complex protocols such as topochemical approaches (employing FeII and then a subsequent oxidation step), and/or the use of chelating agents such as triethanolamine (TEA), in hydrothermal conditions, have been carried out during the last years in order to achieve pure NiFe-LDHs20,28,29,30.
Here, we developed a scalable synthetic protocol compatible with industrial needs, that is: straightforward (room temperature conditions, one-pot, aqueous-based), low-cost, and low-time demand, leading to a high-performing NiFe-LDH that even surpasses conventional lab scale materials. The inherent mild conditions of this approach facilitate in situ experiments using synchrotron radiation, allowing to understand the growth mechanism of LDHs. A combination of X-ray spectroscopies, Mössbauer spectroscopy, magnetic measurements, and electron microscopy combined with spectroscopy techniques revealed a low dimensionality, high concentration of defects, and controlled local structure with a pronounced cation clustering. The OER electrochemical performance of this NiFe-LDH has been characterized using a multi-scale testing protocol including (i) rotating disk electrode (RDE), (ii) diffusion electrode cell (DEC), and (iii) full-cell anion-exchange membrane (AEM) water electrolyzer in alkaline media. In this work we have been able to optimize the synthesis of a NiFe-LDH, obtaining a benchmark performance that is at the same time industrially scalable, avoiding the use of precious metals and offering the long-awaited route that allows the transfer of lab-scale Fe-based LDH technologies into commercial water splitting devices.
Results and discussion
For the room temperature synthesis of NiFe-LDHs, we have developed a straightforward homogeneous alkalinization process based on the nucleophilic attack of chloride over an epoxide ring: the so-called epoxide route (Fig. 1a)31. The nucleophilic attack is initiated when chloride ions react with the strained epoxide ring, leading to ring opening and the subsequent generation of hydroxide ions directly in the solution. These hydroxide ions immediately interact with the metal ions to form hydroxides structures. As the reaction proceeds at room temperature, continuous generation of hydroxide ions through the ongoing nucleophilic attacks forces the system to crystallize at low temperatures and atmospheric pressure, opening the doors toward the mass production of highly efficient electrocatalysts. Indeed, achieving kilogram-scale production of highly homogeneous powder in the lab is easily feasible through a one-pot reaction, typically completed in less than 24 h. Further details on the scalability of NiFe-LDH synthesis, including procedural nuances and optimizations, are compiled in patent US20240109787A1, which provides a thorough description of the method’s applicability and efficiency at larger scales32.

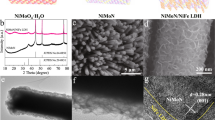
Schematic representation of the NiFe-LDH catalyst synthesis using the nucleophilic attack of chloride over an epoxide ring to obtain a homogeneous alkalinization that allows the room temperature (RT) and atmospheric pressure synthesis of NiFe-LDH (the so-called epoxide route) regardless of the reaction volume used (a). pH profile experiments recorded at room temperature, highlight the plateaus of iron hydroxide precipitation and LDH formation during RT-NiFe-LDH synthesis (b). In-situ X-ray absorption near edge structure (XANES) spectra recorded in fluorescence mode for Fe and Ni K-edges during the first precipitation plateau. The arrow indicates time evolution (c, d). In-situ small angle X-ray scattering (SAXS) patterns for the precipitation process of Ni2+ to Ni(OH)2 (e), Fe3+ to Fe(OH)3 (f) and Ni2+ + Fe3+ to RT-NiFe-LDH (g). Inset: scanning electron microscopy (SEM) (scale bar: 1 µm) and transmission electron microscopy (TEM) (scale bar: 100 nm) inspection of the samples.
In contrast to typical hydrothermal protocols involving high temperature (approximately 120–140 °C) and pressure (1.5–4 atm) using autoclaves20,29,33, the mild synthetic conditions provided by the epoxide route allow the in situ study of the growing processes34,35. Specifically, in-situ pH measurement, X-ray absorption spectroscopy (XAS), and small angle X-ray scattering (SAXS) were carried out during the whole synthetic process to unveil the growth mechanism of NiFe-LDHs. Figure 1b depicts the pH profiles for the kinetic experiments employed to study the precipitation of NiFe-LDHs. As observed, the precipitation pathway can be understood as a two-step mechanism, similar to the case of Al-based LDHs35,36. In the first step, the precipitation of Fe hydroxide takes place reflected by the overshoot centered at pH = 2.7. In the second step, the precipitation of Ni cations precedes onto the already precipitated Fe(OH)3 nanoparticles (NPs), in a plateau around pH = 7.0. This second precipitation step takes place at a lower pH value than with bare Ni (almost 1 unit lower), confirming the higher stability compared to single Ni hydroxide and suggesting the formation of a NiFe-LDH phase.
X-ray absorption near edge structure (XANES) spectra recorded at K-edges during the precipitation of NiFe-LDH confirm, through the position of the absorption edge, the occurrence of NiII and FeIII during the whole process, similar to typical NiFe-LDH catalysts (Fig. 1c,d). Interestingly, while Fe K-edge spectra depict subtle modification attributed to a change in the coordination sphere (i.e.: Fe–H2O by Fe–OH as the pH increases) and subsequent Fe(OH)3 precipitation (Fig. 1c), Ni K-edge spectra remain unaltered during the first plateau, confirming its innocent role (Fig. 1d). Based on SAXS patterns features, the precipitation behaviours of NiII and FeIII exhibit distinct growth mechanisms, influenced by their respective hydrolytic properties. The NiII based hydroxides’ precipitation process initiates with the formation of compact spherical NPs after the overshoot, termed seeds, with an average diameter of approximately 67 ± 1 nm (Fig. 1e). Subsequently, during the plateau phase, these NPs evolve into thin two-dimensional (2D) structures or hierarchical structures containing 2D sheets. These findings align with the characteristic flower-like structures commonly associated with Ni-based hydroxides, corroborated by scanning electron microscopy (SEM) characterization (Fig. 1e – inset). Conversely, FeIII precipitation follows a different pathway; SAXS data indicates the nucleation of small NPs seeds, averaging around 4.1 ± 0.1 nm in diameter. These NPs swiftly aggregate into highly correlated mass fractal structures throughout the precipitation plateau (Fig. 1f). This distinctive behavior of FeIII, typical of a highly acidic cation prone to polymerization into infinite fractal-like structures, resembles the process of hydrogel formation via sol-gel-like mechanisms. Transmission electron microscopy (TEM) imaging confirms the presence of these small NPs, consistent with SAXS observations (Fig. 1f – inset). Notably, when both cations are present, during the initial pH plateau (pH <4), SAXS patterns mirror those of bare FeIII, indicating aggregation into mass fractal structures. However, during the subsequent plateau (pH >7), the SAXS patterns reveal fractal structures persisting even during NiII precipitation, suggesting divalent cations precipitate interacting with Fe(OH)x (Fig. 1g). TEM images depict highly aggregated and smaller flakes compared to those characteristic of Ni-based hydroxides (Fig. 1g – inset). In the supplementary information, two distinct mechanisms during the nucleation and growth of materials are thoroughly examined (Supplementary Fig. 1). Thus, owing to the mild conditions facilitated by the epoxide route, allowing for in situ experiments such as pH kinetics, XAS, and SAXS measurements throughout the entire precipitation process, we present insights confirming the synthesis of NiFe-LDH at room temperature.
Figure 2a depicts the Powder X-ray Diffraction (PXRD) pattern of the NiFe sample synthesized at room temperature (RT-NiFe-LDH) in comparison with a pure NiFe-LDH obtained by using a hydrothermal approach (HT-NiFe-LDH). The RT-NiFe-LDH sample exhibits very low crystallinity (a detailed inspection of the PXRD pattern can be found in Supplementary Fig. 2) with the presence of two very broad signals related to (103) and (110) reflections, confirming the formation of an LDH phase, in agreement with pH profiles. Remarkably, the main interlayer reflection, the (003) peak, remains practically absent, which is characteristic of extremely thin crystals consisting of mono/bilayer structures19,21,37. Supplementary table 1 compiles the structural parameters and the crystallite sizes obtained by the Scherrer equation. In all cases, crystalline domains smaller than 5 nm are obtained suggesting the formation of nano-LDHs.

PXRD pattern for samples synthesized by the epoxide route in comparison with one obtained by hydrothermal synthesis at 120 °C for 12 h (a). Normalized XANES spectra of Ni K-edge (b) and Fe K-edge (c) recorded for NiFe-LDH synthesized at RT and by HT and pure hydroxide as reference. High-resolution XPS spectra for Ni 2p (d), Fe 2p (e), and Cl 2p (f) of the RT-NiFe-LDH sample. Fourier transformation of the EXAFS oscillations (without phase correction) of RT-NiFe-LDH and HT-NiFe-LDH at the Ni K-edge (g) and Fe K-edge (h). Mössbauer spectra taken at 4 K (i). The estimated parameters for these sextets, shown slightly shifted for clarity, are collected in Supplementary Table 5.
XANES measurements confirm the occurrence of NiII and FeIII (see dashed vertical lines in Fig. 2b, c). X-ray photoelectron spectroscopy (XPS) for the RT-NiFe-LDH sample was performed to gain further information about the chemical speciation and the quantitative incorporation of the cations within the layers. The presence of NiII is supported by the main peaks at 855.8 eV (Ni 2p3/2) and 873.3 eV (Ni 2p1/2), and their satellites at 861.5 eV and 879.4 eV, respectively (Fig. 2d). These peaks are very similar to those observed in the HT-NiFe-LDH (Supplementary Fig. 3)38. Analogously, FeIII is confirmed according to both two peaks at 711.8 eV and 723.6 eV, attributable to the Fe 2p3/2 and Fe 2p1/2 signals, respectively (Fig. 2e)38. In the case of Fe, the broad and less defined signals compared to those observed in HT-NiFe-LDH (Supplementary Fig. S3-B) may suggest that the Fe cations are situated in highly defective environments. This is characteristic of nanometric samples with low crystallinity. Additionally, the shift from 711.8 eV to 712.8 eV serves as the first hint of Fe clustering in the RT-NiFe-LDH, as Fe in hydrothermal LDHs typically exhibits higher energies than FeOOH20,39. Additionally, the presence of Cl is confirmed by the main peak around 197.4 eV, with a shoulder at around 199.4 eV, ascribed to Cl 2p3/2 and Cl 2p1/2 respectively (Fig. 2f). Overall, XPS confirms the quantitative incorporation of the cation (Ni:Fe ratio 2:1) and chloride (Cl:Fe ratio 1:1) into the solid phases, in concordance with Energy-dispersive X-ray spectroscopy (EDX) results showing Ni:Fe ratio 2.1:1 and Fe: Cl ratio 1:1, and inductively coupled plasma mass spectrometry (ICP-MS) results showing Ni: Fe ratio 2:1.
Fourier transform of the Extended X-ray Absorption Fine Structure (EXAFS) data, which can be interpreted as a pseudo-radial distribution of the interatomic distances to the absorbing atoms, show for the Ni K-edge two prominent peaks at ca. 1.6 and 2.7 Å (without phase correction), that correspond to the contributions with the first (O atoms), and second neighbours (Ni or Fe atoms), respectively (Fig. 2g). The position of both contributions is practically the same for the β-Ni(OH)2 sample (Supplementary Fig. 4), suggesting a minor effect over the Ni environments by the substitutional doping of Fe in the Ni(OH)2 lattice40, and comparable with the distances observed in the HT-NiFe-LDH. Remarkably, the RT-NiFe-LDH at the Fe K-edge revealed the existence of a third signal at ca. 3.2 Å, which is mainly associated with the second neighbouring Fe-O and Fe-Fe pairs of this phase (Fig. 2h), suggesting a high degree of Fe-clustering in the structure29. This is also supported by the higher Debye-Waller factor (DW) of the RT-NiFe-LDH, which is a measure of the thermal and structural disorder of the surroundings of the absorbing atom (Supplementary Tables 2 and 3).
A thorough magnetic characterization has been carried out to discard the presence of extrinsic magnetic impurities such as Fe spinels, which modify both magnetic29, and electrocatalytic behavior41, resulting in paramagnetic behavior at high temperatures (Supplementary section 5)42. The χ·T versus T plots of the RT-NiFe-LDH reveal the hydroxylated nature of the Fe-clusters instead of FeOx, and the spontaneous magnetization at ca. 12 K confirms the NiFe-LDH phase30. Low-temperature dynamic AC susceptibility measurements exhibit a strong frequency dependence, with a marked spin-glass like behavior, indicative of the presence of clustered Fe–OH–Fe pairs15,29,43,44.
Mössbauer spectroscopy was carried out to determine the degree of Fe-clustering in the RT-NiFe-LDH (Supplementary table 4 and Fig. 2i and Supplementary Fig. 6). As typical for LDHs30,45,46, room temperature Mössbauer spectra show paramagnetic behaviour and confirm the presence of Fe³⁺ (Supplementary Fig. 6). Upon cooling to 4 K, magnetic sextets appear due to slowed relaxation of the Fe magnetic moments, revealing strong magnetic correlations. The presence of multiple sextets and broad quadrupole splitting distributions indicates a partially disordered cation arrangement, in contrast to the ideal case in which all Fe cations would occupy equivalent environments. magnetic hyperfine fields (Bhf) variations among the sextets reflect differences in the number of Fe and Ni nearest neighbours, with stronger Fe–OH–Fe interactions yielding higher Bhf values30. The higher average Bhf observed in RT-NiFe-LDH (~50.1 T) compared to HT-NiFe-LDH (~40.8 T) supports the presence of more Fe-rich local environments in the RT sample. This is further confirmed by the fact that ~70% of Fe atoms in RT-NiFe-LDH are associated with the two highest Bhf components, compared to only 49% in HT-NiFe-LDH, univocally confirming the higher degree of Fe-clustering of the former.
From a morphological perspective, Supplementary Fig. 7 illustrates the morphology of RT-NiFe-LDH. The scanning electron microscope shows the typical microparticle size of agglomerated LDHs (Supplementary Fig. 7A). Transmission electron microscopy (TEM) reveals aggregates approximately 100 nm in size, which aligns with findings from dynamic light scattering (DLS) measurements (Supplementary Fig. 7-B/C). Additionally, a suspension in ethanol exhibits the typical Tyndall effect, indicating notable dispersibility, a crucial attribute for the preparation of OER electrodes. The aggregates consist of highly corrugated and thin layers. High-resolution TEM provides further insight, revealing randomly distributed nanoparticles smaller than 5 nm (Supplementary Fig. 7D). The selected area electron diffraction pattern of the NiFe LDH agglomerates, shown in the inset of Supplementary Fig. 7D, confirms the random distribution and polycrystalline nature of the material, consistent with the PXRD pattern.
Aberration-corrected scanning transmission electron microscopy (STEM) combined with electron energy-loss spectroscopy (EELS) was used to investigate the local structure and chemistry of the samples in the real space. Simultaneously acquired high-resolution annular bright field (ABF) and high-angle annular dark field (HAADF) STEM images (Fig. 3a) of the RT-NiFe-LDH reveal an open structure composed by connected small NPs of ca. 4–5 nm, in perfect agreement with HR-TEM and SAXS information. Remarkably, these NPs exhibit a high degree of crystallinity in the short range (few nm), which is observed in the images and also in the fast Fourier transforms (FFT) in the insets. EELS chemical maps obtained from the analysis of EEL spectrum images containing the Ni L2,3 and Fe L2,3 edges allow direct observation of the local Ni/Fe distribution. Ni/Fe compositional maps reveal the high degree of clustering in our material, clearly revealing a non-uniform distribution of Ni and Fe atoms (Fig. 2a) with Ni/Fe segregation observed again in the few nm length scale. This finding is in stark contrast to what is observed in the case of HT-NiFe-LDH (Fig. 3b), which exhibits a more homogeneous Ni/Fe distribution. From the extensive characterization, including PXRD, XANES, XPS, EXAFS, Mössbauer spectroscopy, and electron microscopy, we depict that the RT-NiFe-LDH synthesized at room temperature exhibits unique structural characteristic that deviate from those of hydrothermally synthesized HT-NiFe-LDH. The distinct structural features such as very low crystallinity, nano-sized crystalline domains, and pronounced Fe clustering are representative of a ‘defective’ structure. In addition to the highly open flower-like structure and low-dimensionality of RT-NiFe-LDH, this notable high clustering is very promising for boosting the OER performance.

ABF and HAADF images (same scale bar) of the RT-NiFe-LDH (a) and HT-NiFe-LDH (b) samples. The insets of the HAADF images show the diffractogram (fast Fourier transform), showing spots derived from the local degree of crystallinity. (Bottom) EELS maps showing the Fe and Ni spatial distribution obtained from EEL spectrum imaging, along with a color overlay of the Fe and Ni maps of the respective samples. DFT structure calculations. RT-NiFe-LDH (110) surface (c) HT-NiFe-LDH (110) surface (d). Gibbs free energy diagram of OER steps in HT and RT NiFe-LDH (110) surface (U = 0 V, pH = 0, T = 273.15 K) (e). Linear sweep voltammetry at 5 mV s−1 in 1 M KOH over glassy carbon rotating disk electrode at RT and 1200 rpm in after 30 activation cycles (50 mV s−1) (f). In situ Raman spectra of NiFe LDHs on carbon paper under different applied potentials. RT-NiFe-LDH (g) HT-NiFe-LDH (h).
Along this front, we carried out first-principles calculations based on density-functional theory (DFT) with the Hubbard U correction (DFT + U) to gain insights into the potential OER performance. Indeed, two different Fe-terminated NiFe-LDH (110) surfaces, namely a (i) highly ordered HT-NiFe-LDH and (ii) a clustered RT-NiFe-LDH, were fully optimized to elucidate the differences in the OER activity between them. The structure of the constructed slabs is shown in Fig. 3c,d. In both cases the relation between Ni and Fe is 2:1. Acidic conditions were considered for simplicity, as they can be considered thermodynamically equivalent to alkaline conditions47,48, focusing on the adsorption of O, OH, and OOH on the NiFe-LDH (110) surface. Molecular Dynamics trajectories are presented in Supplementary Data 1. Supplementary Figs. 8–13 show the relaxed structures of all involved intermediates in the OER process. To obtain the differences in Gibbs free energy (G) for the entire process, we computed the G values for all the species involved in the catalytic process. These results are shown in Supplementary tables 8, 9. As one can see, the differences in energy after the adsorption of the catalytic species are lower in the case of the disordered RT-NiFe-LDH (1.3, 0.4, and 0.2% lower for the adsorption of O, OH, and OOH respectively). This fact can be attributed to a better redistribution of the charge density after adsorption in the case of the Fe-clustered NiFe-LDH, which allows to stabilize the intermediate compounds. Supplementary table 10 shows the ∆G for each step of the OER for both ordered and disordered NiFe-LDH and the Gibbs free energy diagram for the OER process is graphically shown in Fig. 3e. The obtained values are in good agreement with those reported in the literature49,50. We found that in the case of the HT-NiFe-LDH the rate-determining step (RDS) is the fourth (*OOH → O2) with a ∆G = 1.89 eV. On the other hand, the RT-NiFe-LDH shows that the RDS is the second step (*OH → *O) with a ∆G = 1.72 eV. This result points towards a better OER activity as a consequence of the pronounced Fe-clustering due to a faster RDS found in the catalytic mechanism.
In this context, preliminary rotating disk electrode (RDE) experiments demonstrated better catalytic performance for the RT-NiFe-LDH compared to that obtained through a hydrothermal process. A suspension of the samples was drop-cast onto a glassy carbon electrode, and a linear sweep voltammetry (LSV) at 5 mV s⁻¹ was recorded, following a protocol established in the literature20,36. The results indicate that the RT-NiFe-LDH presents an improved performance compared to HT-NiFe-LDHs (Fig. 3f). Notably, RT-NiFe-LDH exhibits a pronounced redox peak at approximately 1.4 V vs RHE, suggesting a more efficient conversion to the oxyhydroxide phase than its HT counterpart, indicative of enhanced catalytic activity.
To better understand the differences in the redox and catalytic behaviour of the two samples, in-situ Raman spectroscopy was conducted at various potentials. The requirement to apply a potential to generate β- and γ-NiOOH phases prohibits the Raman characterization of these pure phases51. In addition, the β-phase is structurally well-defined but the γ-phase is specified as a collection of different crystalline structures. The strong point of Raman spectroscopy is to identify crystal structures by identifying phonon modes originating from spectroscopic unit cells related to the crystalline structures. For the γ-phase, identification lacks certainty due to the undefined nature of this phase. Moreover, several articles try to spectrally identify the NiOOH phase, but it is reported that the synthetic route of the NiOOH precursor has a strong influence on the (purity of the) electrochemical active phase52,53. Therefore, we make a comparative study between the RT-NiFe-LDH and the conventional HT NiFe LDH.
Figure 3g shows the results of the in-situ spectroelectrochemical Raman experiment of RT-NiFe-LDH where at low voltage the typical 450 cm−1 and 520 cm−1 are visible that are related to the A1g mode and a disordered beta Ni(OH)2 respectively51,54. Additionally, there is a weak band around 300 cm−1 that might be a signature of the Eg(T) mode of the same β-Ni(OH)2 phase51,55. The figure shows that at a potential of 1.47 V vs. RHE, both signature NiFe LDH peaks seem to be red-shifted from 450 cm−1 to 472 cm−1 and from 522 cm−1 to 551 cm−1 which are related to NiOOH peaks56,57. Furthermore, a weak shoulder at 596 cm−1 arises that might imply presence of FeOOH, however, due to the lack of studies with 473 nm Raman excitation, this peak remains unassigned57,58. For the conventional HT-NiFe-LDH, we see a similar spectrum at low voltage, solely the position of the two signature NiFe LDH peaks red-shifted which could be related to a bigger lattice constant. After a potential applied 1.57 V vs RHE the spectrum starts undergoes several changes, firstly, the e peak shifts from 457 cm−1 to 472 cm−1. Interestingly, the disordered beta Ni(OH)2 peak at 520 cm−1 remains while a peak appears at 559 cm−1 and the ratio between these, changes with increasing voltage. And lastly a peak at 394 cm−1 which has not been reported to related to NiOOH and most probable will relate to another phase. We will not conclusively assign this peak.
The deconvolution of all spectra is performed by using the mathematical description of several Lorentzian functions. The results of the HT-NiFe-LDH show that there are two peaks in the region at 520 cm−1 and 560 cm−1 which indicate that the shift we see in the two signature NiFe LDH peaks at 450 cm−1 and 520 cm−1 do not shift, but that the material structure changes from the passive Ni(OH)2 to an active phase contrary to what is reported57. Whereas for the RT-NiFe LDH we see full conversion of the peaks at 1.47 V vs RHE while for the HT-NiFe LDH only a partial conversion starts at 1.57 V vs RHE. The results can be correlated with the results of the electrochemical experiment as the RT-NiFe LDH shows conversion an important difference in the redox behaviour in comparison with HT-NiFe-LDH.
These RDE measurements and in-situ Raman characterization give a first hint towards good performance, but are far from operating conditions, therefore a multi-scale electrochemical characterization approach is necessary to confirm the applicability of the RT-NiFe-LDH. Considering some of the previous works on LDHs measured with RDE, it is reported that by applying potential holds, some of the catalysts seem, at first glance, to be unstable, while others can be measured for hours or even days16,59,60,61,62. The changes in current during potential holds are usually related to (i) iron dissolution, (ii) iron adsorption, and (iii) the usage of different conducting powders or polymers. Nevertheless, the effect of bubble formation, catalyst layer (CL) detachment, and the dynamic behavior of LDHs are commonly not taken into account for the electrochemical characterization63,64,65,66. To tackle the current challenges of the field related to poor reproducibility, and to get a more realistic performance compatible with industrial protocols67,68, we designed a systematic multi-scale electrochemical characterization (Fig. 4b), using an automated ultrasonic spray coater for the preparation of the electrodes (Fig. 4a). This way, controlled coating quality and catalyst loading can be ensured for electrodes of increasing dimensions, allowing the progressive characterization at relevant current densities. The prepared inks were sprayed onto glassy carbon (GC) tips for RDE characterization and also on Ni-fiber substrates for diffusion-electrode cell (DEC) experiments and single-cell anion-exchange membrane water electrolysis (AEMWE) measurements. It is important to state that no support, such as conductive carbons, was added to the ink mixture.

Automated ultrasonic spray coating has been used for the multiscale electrode preparation ranging from 0.196 cm2 glassy carbon RDE to 1 cm2 nickel-fiber DEC to finally 5 cm2 AEM water electrolyzer (a). Scheme of electrochemical configuration (b) RDE measurements on GC at RT and 2500 rpm in 1 M KOH after 30 activation cycles (50 mV s−1) and the extended galvanostatic activation protocol (c, d). Slow scan rate CVs (5 mV s−1) of the catalyst after the activation process(c). Polarization curves obtained by galvanostatic steps up to 15 mA cm−2 (d). The plotted potential is corrected with the uncompensated electrolyte resistance (Ru) obtained from impedance measurements at each step. DEC measurements on Ni-fiber substrate at RT and 65 °C in 1 M KOH after 30 activation cycles (50 mV s−1) (e, f). Slow scan rate CVs (5 mV s−1) of the catalyst after the activation process (e). Polarization curves obtained by galvanostatic steps up to 500 mA cm−2 for the RT-NiFe-LDH with three repetitions for each at 25 °C and 65 °C in 1 M KOH. The plotted potential is corrected with Ru obtained from impedance measurements at each step. Polarization curve, high frequency resistance (HFR), and HFR-corrected polarization curve obtained in the AEMWE configuration by galvanostatic steps up to 3000 mA cm−2 after conditioning and break-in at 70 °C in 1 M KOH (g). Polarization curves are always plotted as the mean value from three independent repetitions for each measurement, whereas error bars show the respective standard deviation.
In preliminary RDE experiments it was noticed that by performing the standard reported conditioning cycles (ca. 30 CVs at 50 mV s−1)15,69, the activation of the material was not fully completed, and the OER activity of the LDH continued to increase even after 30 cycles. Nevertheless, the activation cycles were performed to track the evolution of the LDH redox peaks (Supplementary Fig. 14A/B). To fully activate the materials, we implemented a method for RDE evaluation adapting the conditioning applied in AEMWE, with chronopotentiometry (CP) measurements for 16 min at 10 mA cm−2. After the hold, three slow CVs at 5 mV s−1 were measured to compare the activity before and after the extended activation (Supplementary Fig. 15), demonstrating that the OER activity remains constant with the proper activation procedure. To further evaluate the activity of the catalyst in steady state, we implemented a protocol of galvanostatic steps up to 15 mA cm−2 with impedance measurements at each step to correct the uncompensated resistance (Ru). Following, slow scan rate CVs (3 × 5 mV s−1) are measured to compare the charge of the Ni redox peaks and to confirm that the samples are stable during the characterization. The final protocol consists of (i) activation, (ii) slow scan rate CVs, (iii) polarization curve, and (iv) slow scan rate CVs; described in section 9 of the Supplementary Information (Supplementary Fig. 16). For the RDE characterization, the loading of three RT-NiFe-LDH samples was set to 0.2 mg cm−2, delivering a stable potential around 1.52 ± 0.01 V at 10 mA cm−2 and presenting high reproducibility and stability (Supplementary Fig. 17), stressing the high quality of the spray coated samples. The slow scan rate CVs after the activation hold and the polarization curve are shown in Fig. 4c,d, respectively. Comparing the obtained potential during the conditioning and the polarization curve at 10 mA cm−2, the RT-NiFe-LDH catalyst maintains its initial catalytic activity (Supplementary Fig. 18). The RDE results (Supplementary section 10) indicate that using slow scan rate CVs or LSVs to determine electrochemical parameters of dynamic catalysts can result in misleading values. It is worth mentioning that the tip preparation method did not allow to obtain low-loaded samples (<0.2 mg cm−2, catalyst loading in the literature varies between 0.1 and 0.2 mg cm−2)15,70, and high-temperature measurements, due to the thermal expansion of the Teflon holder.
The results observed in RDE (Fig. 4c,d) could be affected by the formation of bubbles at the interface of the glassy carbon and the catalyst layer, which could cause particle detachment64,65,66,71. This effect might be circumvented by improving the interface between the substrate and the catalyst layer, using, for example, coated Ni-fibers measured in DEC configuration. As in RDE, this technique utilizes a reference electrode with a fixed potential, allowing the characterization of the anode overpotential without other contributions. Meanwhile, the electrode structure and catalyst loading can be adjusted to be closer to the application in a real electrolyzer. Additionally, this cell configuration is easily adapted to operate at room temperature (as RDE) and elevated temperature (like AEMWE), offering a valuable tool for bridging the gap between the two techniques. To implement the characterization in the DEC, we decided to deposit the same ink onto Ni-fibers, applying the previously mentioned protocol (Supplementary Fig. 16), but increasing the current density values with respect to RDE. In this case, the loading of three samples was set to 2 mg cm−2, and the conditioning consisted of 30 CVs at 50 mV s−1 and CPs for 16 min at 50 mA cm−2. The slow scan rate CVs after the activation hold and the polarization curve are shown in Fig. 4e,f. In the CVs, the distinct NiII/NiIII catalyst peaks are shifted to higher potentials due to the incorporation of Fe into the catalyst (Supplementary Fig. 20)72. Additionally, a subtle shoulder can be observed, attributed to the contribution of the nickel substrates (Supplementary Fig. 21A)69,73. Thus, Ni-fiber reference samples were also measured following the same protocol (Supplementary Fig. 16). As in RDE, spray coated DEC measurements show a stable potential around 1.50 ± 0.01 V at 50 mA cm−2, presenting high reproducibility and stability (Supplementary Fig. S22). In both techniques, the OER activity is not affected by the increase in the redox peaks suggesting that more material has transitioned to the active phase, but the active sites are not contributing to the OER (Supplementary section 10, 11), limiting the use of CVs or LSVs in RDE & DEC to determine electrochemical parameters.
Although a direct comparison of the activity of RDE and DEC is not possible due to several factors such as (i) loading (0.2 vs 2 mg cm−2), (ii) different catalyst layer formation on substrates (GC and Ni fiber), and (iii) different bubble removal processes (rotation vs stationary), clear trends can be observed for determining the activity of catalysts on both techniques. Due to the uniform distribution and compact nature of the catalyst layer, even a catalyst loading of 0.2 mg cm2 can result in a sufficiently thick catalyst layer (CL) that leads to mass transport problems. Considering that the steady state polarization curve of the RDE measurement presents lower performance than DEC, the activity of the RDE samples measured by applying galvanostatic steps is probably influenced by the low intrinsic conductivity of LDHs and microbubble accumulation (Supplementary Fig. 27). An indirect confirmation of this theory is observed when increasing the loading of the RDE sample from 0.2 to 0.4 mg cm−2, which shows a decrease in the performance (Supplementary Fig. 25). Contrarily, a variation in the loading of the RT-NiFe-LDH samples (from 2 to 0.7 mg cm−2) on Ni-fiber results in the same performance (Supplementary Fig. 26), due to the high surface area of the Ni fibers. In order to understand the contribution of active sites to the OER activity, the RT-NiFe-LDH catalyst and the Ni-fiber reference were also tested in DEC at 65 °C. The RT-NiFe-LDH catalyst shows a decrease in the overpotential from 1.57 to 1.46 V vs. RHE at 500 mA cm−2 (Fig. 4e,f) due to improved kinetics and faster diffusion. The observed large potential shift of 116 mV corresponds well with the potential shift values observed for IrO₂ in the gas diffusion electrode (GDE) at a current density of 1 A cm⁻2 (118 mV) across various temperatures (20 to 60 °C)74. The apparent Tafel slopes obtained from the polarization curves measured by galvanostatic steps in DEC at room temperature and 65 °C are 42.12 ± 2.19 and 36.22 ± 1.50 mV dec−1 (Supplementary Fig. 31). A similar decrease in Tafel slope was also observed in IrO2 GDE samples between 10 and 100 mA cm−2 at 20 and 60 °C74. It is crucial to emphasize the role of measurement parameters and conditioning protocols, as they impact the comparison of these materials (Supplementary Fig. 14, 15). Larrazábal et al.75 highlighted a concerning trend in new catalysts research, often prioritizing notable results from simplified electrochemical methods (LSV, CVs) under unrealistic conditions, like low current densities, room temperature, and non-steady state, leading to misleading outcomes. Therefore, for accurate catalyst screening, experiments should be conducted under realistic conditions. DEC measurements offer the possibility to test at relevant conditions (in our case 1 M KOH, 65 °C, 500 mA cm−2) while providing a defined cathode potential, therefore bridging the gap between RDE and AEMWE measurements.
Next, MEAs were fabricated with the RT-NiFe-LDH OER catalyst and tested at 70 °C in a full-cell AEMWE setup with 5 cm2 active area. In a catalyst coated substrate (CCS) approach, the MEAs were assembled by sandwiching a spray coated Ni-fiber (2 mgNiFe-LDH cm−2, 10 wt.% Aemion + ®) as the anode, together with a commercial membrane (75 µm Aemion + ®)) and spray coated carbon paper (0.5 mgPt cm−2 Pt/C, 10 wt.% Aemion + ®) as the cathode. Three separate MEAs were fabricated and tested, to ensure reproducibility of the results. After assembly, the cell was heated and flushed with 1 M KOH for 1.5 h to reach steady temperature operation. Following a break-in (2 h @ 1.8 V) the initial polarization curve was recorded up to 3 A cm−2 (Fig. 4g). The RT-NiFe-LDH catalyst reaches 1 A cm−2 at 1.69 V and 3 A cm−2 at 2.06 V in this setup. The high frequency resistance (HFR) is determined by fitting an equivalent circuit model to the high frequency intercept of a short impedance sweep at each current density (Supplementary Fig. 32). The polarization curve from the full-cell shows notable deviations compared to DEC measurements across all current densities, with discrepancies increasing at higher currents (Supplementary Fig. 27). These differences likely stem from (i) temperature variations (65 °C vs. 70 °C), (ii) catalyst utilization in the CLs, (iii) the CL-membrane interfaces in the MEA, and (iv) differences in mass transport in each setup. Despite these deviations, the findings align with recent reports76, indicating that cathode overpotential and mass-transport losses contribute to the overall cell voltage in AEMWE. Confirming this would require a detailed analysis of the individual contributions from the anode, cathode, and membrane to the total cell voltage, which is beyond the scope of this study. To sum up, the AEMWE performance of the scalable RT-NiFe-LDH catalyst is notable and remains consistent from laboratory-scale RDE measurements to pilot-scale full-cell MEA, being suitable for operating an electrolyzer at high current densities.
To understand and correlate the electrochemical results with the chemical and structural intrinsic properties of the scalable RT-NiFe-LDH catalyst, a benchmark LDH from the literature was synthesized and denoted as NiFe-LDH22. This reference was chosen because the highly ordered HT-NiFe-LDH cannot be synthesized in quantities necessary for full-cell AEMWE tests. The multiscale electrochemical comparison exhibited higher activities for the RT-NiFe-LDH in all employed characterization methods (Supplementary section S15). Both samples were subjected to a constant current experiment for 100 h at 1 A cm−2 in the full-cell AEMWE setup. During the initial phase of the constant current experiment, from 2 to 14 h, RT-NiFe-LDH exhibited an average potential increase of 1.02 mV/h, compared to the control NiFe-LDH which increased at a rate of 1.84 mV/h. Subsequently, from 85 to 100 h, the potential rise for RT-NiFe-LDH was lower, averaging 0.41 mV/h, while NiFe-LDH showed an increase of 0.80 mV/h (Fig. 5a). The electrodes were analysed at the beginning (BoT) and the end of the tests (EoT) by polarization curves (Fig. 5b). In a complex system such as AEMWE, multiple factors can play a role in the increase of potential over time, such as membrane stability, intrinsic catalyst stability (activity, dissolution, and Pt-Fe poisoning), or mechanical stability of the catalyst layer during operation (influenced by particle size, ionomer used, etc.). As the commercial membrane, ionomer materials, inks, and preparation procedure for all tests were kept constant, the slower potential increase for the RT-NiFe-LDH can likely be ascribed to the higher intrinsic stability of the catalyst77. To shed light on this point, electrochemical impedance spectra (EIS) within the ohmic region of the iV-curve (at 1 A cm−2) were recorded after BoT and EoT polarization curves (Fig. 5c). The BoT as well as EoT impedance characterization shows a qualitatively lower charge transfer resistance for the RT-NiFe-LDH compared to the control material, agreeing with the notable performance shown in the polarization curve and constant current experiment. A slightly higher HFR in the RT-NiFe-LDH sample is observed. Compared to the performance benefit, this drawback seems minor. It is worth mentioning that a quantitative approach to analyze EIS data in AEMWE is not yet established due to the lack of an equivalent circuit model and unknown fundamental parameters (e.g. the specific charge of reaction intermediates to the active sites of the catalyst for calculation of the electrochemical surface area).

Analysis comparing RT-NiFe-LDH with a typical hydrothermal NiFe-LDH at 2 mg/cm2. Constant current operation of the MEAs over 100 h at 1 A cm−2. The first 15 h were conducted in triplicate, with the respective error bars shown to represent the variability in the data (a). AEMWE measurements at 70 °C with 1 M KOH: initial polarization curves obtained by galvanostatic steps after conditioning and break-in (dark blue and red) and polarization curves after 100 h of constant current operation at 1 A cm−2 (light-blue and red) (b). Electrochemical impedance spectra at 1 A cm−2 after the first polarization curve (left, dark colors) and at 1 A cm−2 after 100 h constant current operation at 1 A cm−2 and the second polarization curve (right, light colors) (c). PXRD (interlayer diffraction) pre and post characterization of Ni spray coated substrates. Characterization before (dark colors) and after (light colors) 15 h constant current hold in the MEA of the RT-NiFe-LDH and NiFe-LDH. In order to compare bare Nickel fibers was plotted (gray) (d). In-situ Raman characterization and XANES on carbon paper at 500 mA cm−2. In-situ Raman characterization (e) and normalized XANES spectra of Ni K-edge (f) of RT-NiFe-LDH and NiFe-LDH, respectively.
On the one hand, to further elucidate the differences in activity between both tested catalysts, XRD measurements were performed before and after the full-cell tests (Fig. 5d and Supplementary Fig. 37A). The XRD patterns distinctly differentiated the catalysts. For the RT-NiFe-LDH, the characteristic 8.6 Å peak, indicative of the LDH interlayer spacing, disappears upon electrode formation, transitioning to a 7.2 Å peak associated with the interlayer distance of oxyhydroxide species78. Conversely, in the control NiFe-LDH, the interlayer peak at 7.8 Å remains intact. To confirm the validity of these observations under operational conditions, in-situ Raman spectroscopy and XANES analysis of Ni K-edge at a current density of 500 mA cm−2 on carbon paper were employed (Fig. 5e, f, respectively). Raman spectroscopy confirmed that at a high current density, the behavior of the samples matched the observations made at 1.67 V vs. RHE (approximately 10 mA cm−2). Specifically, there was a complete transformation to an oxyhydroxide phase in RT-NiFe-LDH and only a partial conversion in NiFe-LDH, consistent with the findings from the post-mortem XRD analysis. The XANES analysis highlighted an edge shift of more than 2 eV relative to NiII for the HT sample, suggesting a complete conversion to NiIII. For the RT sample, an additional shift of 0.5 eV to a higher energy level indicates the possibility of environments with Ni valences exceeding (III). This substantial shift in electronic states supports the formation of catalytic centers likely involving NiIV and aligns with the formation of the γ phase of the oxyhydroxide79,80,81,82. Additionally, XANES analysis of the Fe K-edge (Supplementary Fig. 37B) revealed no changes in the oxidation state of Fe.
Thus, the results indicate a complete conversion of the RT-NiFe-LDH to the oxyhydroxide species, but only a partial conversion for the control NiFe-LDH, supporting the higher activity observed in the RDE, DEC, and AEMWE electrochemical measurements. These results make evident, that the unique characteristics of RT-NiFe-LDH (low-dimensionality, highly defective, and clustered nature) maximize the conversion of the brucite layers into highly active oxyhydroxides phases83.
On the other hand, to better understand the improved stability of RT-NiFe-LDH after 100 h of operation and to identify potential degradation mechanisms, FIB-SEM-EDX analyses were conducted on the electrodes both before and after testing (Supplementary Fig. 38A/F and Supplementary Fig. 39A/F). These were complemented by a detailed morphological characterization of the catalytic material recovered from the electrodes using TEM and STEM-EELS (Supplementary Fig. 40A/B). SEM-EDX images show that RT-NiFe-LDH initially forms a cracked catalyst layer (CL), in contrast to the more homogeneous layer produced under similar deposition conditions with hydrothermal NiFe-LDH. Interestingly, post-mortem analysis indicates a lower degree of delamination in the RT-NiFe-LDH sample compared to the hydrothermal counterpart. Moreover, the initial cracks, which tend to propagate perpendicular to the nickel fibers, may serve to alleviate mechanical stress within the porous structure during electrochemical cycling, thereby reducing material detachment. Supporting this observation, cross-sectional FIB-SEM analyses reveal that although both catalyst layers experience compression after operation, the RT-NiFe-LDH layer retains better structural integrity. Nevertheless, high-resolution imaging after 100 h shows that in certain regions—particularly in samples synthesized via urea-based methods—the catalyst layer is considerably delaminated42 or even completely absent, indicating that delamination is the primary degradation pathway. Despite these changes of the CL, TEM analysis of the isolated RT-NiFe-LDH catalyst reveals no notable morphological alterations. However, STEM-EELS analysis shows a slight shift in the Ni/Fe ratio from 2:1 to 3:1, indicative of Fe leaching, which may contribute to the performance degradation. It is also important to consider that this degradation could originate from other components within the single cell, such as the membrane, the cathode, or related interfaces.
As conclusion, we have demonstrated a scalable method for producing relatively large quantities of highly active NiFe-LDHs. This was achieved using the epoxide route, a scalable synthetic approach compatible with industrial needs (room temperature conditions, one-pot, aqueous-based, low-cost, and low time-demand). This is crucial to transfer lab-scale Fe-based LDH technologies into commercial water splitting devices. By in-situ spectroscopic analysis, we unveiled the growth mechanism of NiFe-LDHs, along with a pronounced cation clustering, which turns out to be critical for the notable OER performance of the catalyst. Moreover, we demonstrated the notable electrochemical performance of the RT-NiFe-LDH over a control, state-of-the-art NiFe-LDH in three different setups. The initial catalysts performance evaluation has been carried out in RDE. Afterwards, measurements in DEC with a small active area (1 cm2), providing the overpotential of anode catalysts in steady state conditions and at realistic operating temperatures, allow us bridging the gap between fundamental and applied techniques. Finally, full-cell AEMWE polarization curves demonstrate that we were able to decrease the overpotential of state-of-the-art NiFe-LDHs by 50 mV at 1 A cm−2, from 1.74 to 1.69 V; due to the complete conversion of the RT-NiFe-LDH into its highly active oxyhydroxide phase, and the involvement of catalytic centers likely including NiIV, this aligns with the formation of the γ phase of the oxyhydroxide. The high OER performance of the RT-NiFe-LDH and its industrial scalability pave the way for the development of PGM-free AEMWE electrolyzers operating at high current densities. Moreover, NiFe-LDHs can be applied in other electrolyzers, where commercialization will require the availability of large quantities of highly active material. This makes the demonstration of scalable production methods for NiFe-LDHs critically important. We believe that this work provides a notable step in that direction.
Methods
Catalyst synthesis and characterization
Chemicals
Nickel chloride hexahydrate 99.9% (NiCl2·6H2O), iron chloride hexahydrate 97% (FeCl3·6H2O), nickel nitrate hexahydrate 99.99% (Ni(NO3)2·6H2O), iron nitrate hexahydrate 99.95% (Fe(NO3)3·6H2O), Urea 99.99%, glycidol 96% (Gly), Triethanolamine 99.5% (TEA) and sodium chloride (NaCl) were purchased from Sigma-Aldrich. Ethanol (EtOH) was purchased from Honeywell. All chemicals were used as received. Milli-Q water was obtained from a Millipore Milli-Q equipment. KOH pellets, Normapur®, VWR International LLC >85%, BEK-POR 2N118-0.25 (99.9% Ni), Bekaert.
Synthesis
RT-NiFe sample: Samples were obtained by employing the Epoxide Route. Typically, precipitations were driven by aging for 24–72 h at room temperature aqueous solution adjusted (with HCl solution) at an initial pH around 2 to guarantee the cations’ solubility (especially in the case of FeIII). Total cation concentrations, defined as [NiII]] + [FeIII], were tested from 15 to 750 mM. In all the cases, Ni:Fe ratio of 2:1 was employed, exclusively. Total chloride concentration, [Cl–] = 2[NiII]] + 3[FeIII] + [NaCl], was modulated from 40 to 2000 mM, by adding NaCl solution. Once defined the experimental conditions, Gly is added to the aqueous solution under permanent magnetic stirring. Gly concentrations from 40 to 2000 mM were tested. It is worth mentioning that the addition of Gly starts the reaction. After the precipitation, the solids were obtained either by filtration or centrifugation, washed three times with water (50% of the initial reaction volume), and finally with EtOH. The samples were dried at room temperature and kept in desiccators for further characterization.
HT-NiFe sample: In a typical synthesis of NiFe-LDH30,42, the nitrate salts of the metals were mixed with TEA and dissolved in 50 mL of degassed Milli-Q water to obtain a final solution with a total metal cation concentration of 20 mM, maintaining a fixed Ni to Fe molar ratio of 2:1. The TEA concentration was adjusted based on the amount of Fe present. Next, 50 mL of a 35 mM aqueous urea solution was added, with the amount of urea corresponding to 75% of the total metal concentration. The resulting dark brown solution was transferred to a 125 mL stainless steel autoclave lined with Teflon and heated in a preheated oven at 120 °C for 48 h. After the reaction period, the autoclave was allowed to cool to room temperature on the bench. The obtained yellowish fine powder was collected by filtration, washed thoroughly with Milli-Q water, and dried under vacuum. The pH of the leftover solution was measured to be around 7.5 to 8.0.
NiFe-LDH sample: In a typical procedure22, Ni(NO3)2·6H2O (0.75 mmol), Fe(NO3)3·9H2O (0.25 mmol), and CO(NH2)2 (5 mmol) were dissolved in 36 mL of distilled water and stirred to form a clear solution. The aqueous solution was transferred to a 125 mL Teflon-lined stainless-steel autoclave, which was sealed, maintained at 120 °C for 12 h, and then allowed to cool to room temperature naturally. Then, the final powder was filtered and washed with Milli-Q water and EtOH several times and dried for 24 h under vacuum at room temperature.
Chemical, structural, and physical characterization
Field emission scanning electron microscopy (FESEM) and energy dispersive X-ray spectroscopy (EDS) analyses were carried out using a Hitachi S-4800 microscope operating at an accelerating voltage of 20 kV. Transmission electron microscopy (TEM) was conducted with a JEM-1010 instrument at 80 kV. To prepare the samples, suspensions were deposited onto 300 mesh copper grids coated with lacey Formvar/carbon films.
Scanning transmission electron microscopy combined with electron energy loss spectroscopy (STEM-EELS) was performed on a JEOL ARM200cF microscope located at the Centro Nacional de Microscopía Electrónica (UCM). This equipment is fitted with a spherical aberration corrector and a Gatan Quantum EEL spectrometer. Powder X-ray diffraction (PXRD) data were collected using a PANalytical Empyrean diffractometer equipped with a capillary stage and employing Cu Kα radiation (λ = 1.54 Å), covering a 2θ range from 2° to 70°. X-ray photoelectron spectroscopy (XPS) measurements were obtained in an ESCALAB210 system (Thermo VG Scientific) under ultrahigh vacuum conditions (base pressure of 1.0 × 10⁻¹⁰ mbar), using Al Kα radiation (1486.6 eV) as the excitation source. All spectra were calibrated with reference to the Fermi level. Inductively coupled plasma mass spectrometry (ICP-MS) was performed at the University of Valencia in the Atomic and Molecular Spectrometry Section. Between 3 and 12 mg of each hydroxide sample were digested in ultrapure 69% nitric acid (Scharlab) for trace-level analysis. The digestion was conducted using a Milestone ETHOS EASY high-pressure microwave system, reaching temperatures up to 220 °C.
Magnetic measurements were carried out on the bulk materials using a Quantum Design MPMS-XL-5 SQUID magnetometer. The samples’ magnetic susceptibility was corrected for the diamagnetic contributions of their elemental components, based on Pascal’s constants and the holder’s contribution. DC magnetic data were acquired under applied fields of 100 or 1000 Oe over the 2–300 K temperature range. AC susceptibility data were measured with a 3.95 Oe field at frequencies of 997, 333, 110, 10, and 1 Hz.The Mössbauer spectra were recorded in the transmission mode using a conventional constant-acceleration spectrometer and a 25 mCi 57Co source in an Rh matrix. The velocity scale was calibrated using α-Fe foil. The absorbers were obtained by packing the powdered samples into Perspex holders. The isomer shifts are given relative to metallic α-Fe at room temperature. The spectra at 4.1 K were recorded by using a bath cryostat with the sample immersed in liquid He. The spectra were fitted to Lorentzian lines by using a nonlinear least-squares method84. The distributions of quadrupole splitting were fitted according to the histogram method84. To assess the expected distribution of Fe local environments under a random cation arrangement, the probability P(m) of finding m Ni atoms among the six nearest-neighbor (nnn) cation sites was calculated using the binomial expression:
where y is the Ni/Fe ratio, equal to 2 in both samples. The calculated values of P(m) are provided in Supplementary Table 6. Assuming equivalent local environments and recoil-free fractions, the relative spectral areas I of the sextets observed at 4 K were compared to the corresponding probabilities, as summarized in Supplementary table 7.
In situ Small Angle X-ray Scattering (SAXS) experiments were conducted at the BL11 NCD-SWEET beamline of the ALBA Synchrotron Light Source (Barcelona, Spain), under project IDs 2020024318, 2020094732, and 2021025023.
The experimental setup involved a continuously stirred solution, introduced into a 2 mm glass capillary flow cell optimized for liquid-phase analysis. This configuration helped maintain a homogeneous distribution of nanoparticles during the reaction and reduced X-ray-induced damage to the sample. The X-ray beam was operated at an energy of 12.4 keV, and the distance between the sample and detector was set to 6700 mm. Scattering data were acquired using a Pilatus 1 M detector (Dectris, Switzerland), capturing images with a 5-second exposure time at a rate of four frames per minute. At the beginning of the reaction, the precursor solution had an approximate pH of 2. Data acquisition began immediately after introducing the epoxide into the metal cation solution. Two modeling strategies were applied for interpreting the data. For spherical nanoparticles, a semi-empirical Guinier-Porod function was used85, where the radius of gyration (Rg) served as the primary fitting parameter. The particle diameter was estimated using the relationship with \({{{\rm{d}}}}=2\sqrt{5/3}{{{\rm{Rg}}}}\). For the sol-gel-like process, a mass fractal model was employed86. The fitting parameters included the diameter of the small particles forming the larger aggregate, the fractal dimension (f) related to the aggregate structure, and the fractal cutoff (ε), which corresponds to the characteristic size of the fractal structure. The diameter of the fractal structure was obtained using the relationship87 \({{{\rm{d}}}}=2\sqrt{5/3}\) \(\sqrt{{{{\rm{f}}}}({{{\rm{f}}}}+1)/2}{{{\rm{\varepsilon }}}}\). These models were employed to evaluate the pattern features in order to build a nucleation and growth mechanism during the first steps of the precipitation process.
X-ray Absorption Spectroscopy (XAS) measurements were performed at the BL-22 (CLÆSS) and BL16 – NOTOS beamline from the ALBA synchrotron (Barcelona – Spain). XANES and EXAFS spectra at the Ni and Fe K-edges were measured at room temperature in transmission mode (projects ID: 2020094548, 2021024897 and 2024028425). Absorbent samples were prepared by pressing freshly obtained powders into pellets with a diameter of 1.3 mm, which were then sealed using 50 µm-thick Kapton® tape to protect them from oxidation. The ideal quantity of material required for the measurements was determined using Hephaestus, a software included in the Demeter analysis package88. A Si(111) double-crystal monochromator was used to obtain a monochromatic incident beam over the sample, and the intensities of the incident and transmitted X-rays were measured using two ionization chambers, respectively. XAS spectra were collected from 7000–8000 eV with a reduced step (0.3 eV) in the XANES region (7100 to 7150 eV) for Fe K-edge, and from 8200–9200 eV, with a reduced step of 0.3 eV in the XANES region (8330 to 8360 eV), for the Ni K-edge. The incident photon energy was calibrated using the first inflection point of the Fe K-edge (7112 eV) and Ni K-edge (8333 eV) from reference foils of metallic Fe and Ni, respectively. For each sample, three spectra were taken with exposure times of 4 min each to later be averaged. In situ experiments were carried out using a continuous flow cell available at the CLÆSS beamline. The reaction mixture was continuously circulated from a glass beaker through a borosilicate capillary (3 mm outer diameter) using a peristaltic pump, ensuring a steady flow and constant renewal of the solution within the capillary. XANES spectra were recorded at the Fe and Ni K-edges in fluorescence mode, with a solid-state detector (Amptek silicon drift detector, XR-100SDD) positioned at 90 degrees with respect to the incoming beam. Simultaneously, in situ pH measurements were performed to monitor the progression of the precipitation process and correlate the chemical evolution with the absorption spectra. XANES data treatment was performed by subtracting the pre-edge background followed by normalization, extrapolating a quadratic polynomial fitted at the post-edge region of the spectrum using the ATHENA AUTOBK background removal algorithm89. Quantitative analysis of the EXAFS data was carried out by modeling and fitting the extracted EXAFS oscillations. The χ(k) signals were isolated from the raw experimental data using standard methods implemented in the Athena software. To emphasize features at higher k-values, the χ(k) data were weighted by k³ prior to applying the Fourier transform. The transformation was performed using a Hanning window function. Subsequent EXAFS modeling and fitting were conducted using the Artemis program88. Theoretical scattering path amplitudes and phase shifts for all environments used in the fits were calculated using the FEFF9 code90. The k-range was set from 2.5 to 12 Å−1. The passive reduction factor S02 values were restrained to 0.79 and 0.83 for Fe and Ni, respectively. These values were obtained from the fitting standard foils of metallic Fe and Ni, and constraining the coordination numbers to those corresponding to each structure. Individual point Raman spectra were carried out using a Horiba LabRAM HR evolution, employing a blue laser (473 nm) in the 100–1000 cm−1 range. For the acquisition of all Raman spectra, a 50x Objective with a 600 mm−1 grating was employed.
Computational details
Density functional theory (DFT) calculations were performed as implemented in the Vienna ab initio Simulation Package (VASP)91,92. The plane-wave basis and projector-augmented wave (PAW) method were used93. The generalized gradient approximation (GGA) in combination with the Perdew−Burke−Ernzerhof (PBE) functional was used to describe the exchange-correlation94. Kinetic energy cut-off was set to 500 Ry for expanding the basis set. We used a Grimme’s DFT-D3 scheme to describe van der Waals interactions between the NiFe-LDH (110) surface and the intermediates of the OER95. Self-interaction errors for strongly correlated d electrons were corrected using Hubbard Ueff parameters of 5.3 and 5.5 eV for the Fe and Ni atoms respectively, as implemented by Dudarev et al.96 In our OER calculations, we constructed an undercoordinated Fe-terminated NiFe-LDH (110) surface due to its low surface energy by introducing a vacuum layer of 15 Å to avoid interaction with periodic images along c. Superficial Fe atoms were considered as the active site for the adsorption of the intermediate species as reported in the bibliography97,98. All slab models were fully relaxed until the Hellman-Feynman forces and energies were lower than 0.02 eV A-1 and 1 × 10-6 eV, respectively.
To elucidate the underlying mechanism of the OER activity, we followed the computational methodology developed by Nørskov99. Gibbs free energy (G) for all intermediates involved in OER was calculated using the total energy obtained via DFT calculations and considering the quasi-harmonic vibrational free energy correction term, including the zero-point energy (EZPE) and entropy correction (TS) as:
Acidic conditions were considered for simplicity, as they can be considered thermodynamically equivalent to alkaline conditions47,49.
Electrochemical characterization
Electrolyte preparation
A 1 M KOH solution (pH 14±0.2, Hanna HI1131) was prepared by dissolving 66.01 g of KOH (85% purity) in 1 liter of Milli-Q water using a volumetric flask. The solution was stored in 1 L PTFE bottles, protected from natural light, and kept in a closed cabinet at room temperature.
Initial RDE testing
The initial glassy carbon (GC) rotating disc electrode (RDE) measurements, presented in Fig. 3f of the main text, were carried out by preparing an ink composed of 5 mg of LDH powder, 2.5 mg of graphitized carbon, and 25 μL of 5% Nafion solution D520. These components were dispersed in 1.25 mL of a 1:1 (v/v) ethanol/water mixture and sonicated for 20 min to ensure homogeneity. A 3 μL aliquot of the resulting suspension was drop-cast onto a 3 mm diameter GC RDE, which had been previously polished using alumina powders with particle sizes of 1.0, 0.3, and 0.05 µm. The modified electrode was then dried at room temperature for 30 min. Prior to oxygen evolution reaction (OER) testing, 30 cyclic voltammetry (CV) cycles were performed between 0.9 and 1.7 V versus the reversible hydrogen electrode (RHE) at a scan rate of 50 mV·s⁻¹. Linear sweep voltammetry (LSV) was conducted at 5 mV·s⁻¹ in 1 M KOH aqueous solution previously purged with nitrogen. The RDE was rotated at 1200 rpm during the measurements. Each sample was tested at least three times using independently prepared electrodes.
In-situ Raman characterization under different potential and a high current density
A redox.me cell (model SKU: C-A-RAM_MM_ECFC) with a carbon paper covered with 0.6 mg/cm2 of NiFe-catalysts, a platinum wire counter electrode, and an Ag/AgCl reference electrode were used for EC-Raman measurements. An electrolyte solution of 1 M KOH was applied.
In-situ XANES characterization under high current density
A redox.me cell (model SKU: C-A-MM_FC_GDE_XRD_EC) with a carbon paper covered with 0.6 mg/cm2 of NiFe-catalysts, a platinum wire counter electrode, and a Ag/AgCl reference electrode were used for EC-Raman measurements. An electrolyte solution of 1 M KOH was applied.
Electrode fabrication
The remaining electrodes were fabricated using a catalyst-coated substrate (CCS) method. In this approach, a catalyst ink is first prepared and then deposited onto different substrates—porous transport layers for DEC and MEA testing, and glassy carbon (GC) tips for RDE measurements.
To prepare the anode catalyst ink, an ionomer solution (1 wt.% ionomer in 80/20 wt.% ethanol/acetone) was made by stirring the appropriate amounts of ionomer (AP3-HNN9-00-X, Ionomr), ethanol (EMSURE®, Merck KGaA), and acetone (Merck KGaA) overnight on a hot plate maintained at 60 °C. Simultaneously, the NiFe-LDH catalyst powder was pre-dispersed in Milli-Q water (18.2 MΩ·cm at 25 °C, Merck KGaA) by stirring overnight. The following day, both mixtures were combined, and ethanol was added to achieve the final ink formulation: 3 wt.% total solids (comprising 90 wt.% catalyst and 10 wt.% ionomer) in a 50/50 wt.% mixture of water and ethanol/acetone. The ink was then stirred for one hour at room temperature and further homogenized using an ultrasonic horn (S26d7 sonotrode on a UP200St system, Hielscher Ultrasonics GmbH) in three 20-min intervals at 40 W, while kept cool in an ice bath.
Glassy carbon (GC) disks (5 mm diameter) were thoroughly cleaned using isopropanol and water, followed by polishing with a 3 µm diamond suspension (Struers, DiaPro Mol R). A PTFE mask was applied during spray coating to prevent catalyst overspray onto the RDE tip edges.
The ink was spray-coated using an ultrasonic spray coater (Sonotek ExactaCoat with a 48 kHz AccuMist nozzle) onto GC for RDE, and onto sintered nickel fiber substrates (BEK-POR 2N118-0.25, Bekaert) for DEC and MEA applications. Catalyst loadings were targeted at 0.2 mg/cm2 for RDE and 2 mg/cm2 for DEC and MEA, covering active areas of 0.19 cm2 (RDE), 1 cm2 (DEC), and 5 cm2 (MEA). Loadings were determined gravimetrically using a Satorius Cubis® microscale (MSA66S-000-DH) by weighing the electrodes before and after deposition. The spray coater’s heating plate was set to 120 °C, with a nozzle speed of 170 mm/s and a solution flow rate of 0.45 mL/min.
For MEA cathodes, a similar procedure was followed. The ionomer solution remained the same, but 1-propanol was added to the ink mixture in the morning prior to ultrasonication. The final composition consisted of 1 wt.% total solids (90 wt.% Pt/C with 60 wt.% Pt content, Thermo Fisher Scientific, and 10 wt.% ionomer) in an 80/20 wt.% mixture of water and 1-propanol/ethanol/acetone. This ink was sprayed onto carbon paper (H24C5, Freudenberg) using a 100 °C heated plate, with a nozzle speed of 140 mm/s and a flow rate of 0.33 mL/min.
RDE and DEC test station
The electrochemical characterization of three electrode cells is conducted in an in-house manufactured Teflon cell. The reference electrode is inserted into an isolated compartment connected to the main electrolyte compartment via a Luggin capillary. The counter electrode is directly immersed in the general electrolyte compartment facing the working electrode. The working electrodes are i) a disk insert placed in an RDE tip holder (Pine research, E5TQ ChangeDisk RDE Tip PTFE) for RDE measurements and ii) a 1 cm2 Ni-fiber for DEC measurements (as depicted in Fig. 4a). The cell is connected to a potentiostat (Gamry Reference 600). The main electrolyte compartment is permanently flushed with argon to saturate the 1 M KOH solution. The scheme of the in-house manufactured Teflon cell is described in Fig. 1a) in the following publication (no membrane was used for all the measurements in this work)100. For the CVs, a representative CV from the three reproduced measurements for each sample was selected, and Ru correction was applied. For the polarization curves, impedance was measured at each step, the Ru free curves were calculated and then the mean and the deviation from the three reproduced measurements was represented.
General procedures for RDE and DEC testing
All RDE and DEC measurements were carried out in 1 M KOH (85 wt%) using a custom-built Teflon electrochemical cell. The setup included (i) a platinized mesh counter electrode (Pt-coated Ti mesh, Metakem) and (ii) a reference electrode, which was either a Ag/AgCl electrode (Metrohm) for ambient temperature experiments or a Mini-Hydroflex electrode (Gaskatel) for elevated temperature conditions.
RDE tests were conducted on glassy carbon electrodes rotating at 2500 rpm, while DEC measurements were performed using nickel fiber substrates. RDE experiments were exclusively run at room temperature, whereas DEC experiments were conducted at both room temperature and 65 °C.
To ensure the accuracy of the reference electrode potential, it was cross-checked against platinum rods prior to each measurement by purging the electrolyte with argon. Prior to use, the electrochemical cells were cleaned by boiling in 1 wt.% nitric acid (EMSURE, Merck) followed by three successive boils in ultrapure water. Between experiments, the cell was stored in ultrapure water to prevent contamination. The specific Tafel slope range used in each case is detailed in the corresponding figures.
RDE testing
The electrochemical characterization of the RDE tips is conducted by carefully placing the disk inserts in the RDE tip (Pine research, E5TQ ChangeDisk RDE Tip PTFE). The first electrochemical activation of the LDHs was performed by applying 30 CVs at a scan rate of 50 mV/s between 0.9 and 1.53 V vs RHE. Subsequently, chronopotentiometry at 10 mA/cm2 for 16 min was applied to ensure the complete activation of the materials. Three slow scan rate CVs (5 mV/s) from 0.9 to 1.62 V vs RHE followed the protocol to have a first impression of the catalyst activity. Afterward, chronopotentiometry was performed at current densities ranging from 0.015 to 15 mA/cm2, holding each for 3 min until the voltage response stabilized. The resulting steady-state voltages were then used as DC bias points for subsequent electrochemical impedance spectroscopy measurements to characterize the system under relevant operating conditions. In the end, again three slow scan rate (5 mV/s) CVs from 0.9 to 1.62 V vs RHE were conducted to track any changes in the electrode. To avoid bubble accumulation, all the CVs, impedances, and chronopotentiometry holds were performed at 2500 rpm, purging Ar into the electrolyte. The third CV of the slow scan rate was selected for representation. Impedance measurements were conducted by sweeping a sinusoidal voltage from 100,000 Hz to 1 Hz, using an amplitude of 0.01 V. The uncompensated resistance (Ru) is taken from the high-frequency intercept on the real axis of the Nyquist plot, and it is used to correct the measured electrode potential by subtracting the iR drop (current × Ru) to obtain a more accurate representation of the true electrochemical potential. A more detailed discussion of the protocol is given in supplementary section 9.
DEC testing
The DEC electrochemical characterization was conducted using Ni-fiber substrates (PTL, BEK-POR 2N118-0.25, Bekaert). The first electrochemical activation of the LDHs was performed by applying 30 CVs at a scan rate of 50 mV/s between 0.9 and 1.53 V vs RHE. Subsequently, chronopotentiometry at 50 mA/cm2 for 16 min was applied to ensure the complete activation of the materials. Three slow scan rate CVs (5 mV/s) from 0.9 to 1.62 V vs RHE followed the protocol to have a first impression of the activity of the catalyst. Afterward, chronopotentiometry was performed at current densities ranging from 0.025 mA/cm2 and 500 mA/cm2, holding each for 3 min until the voltage response stabilized. The resulting steady-state voltages were then used as DC bias points for subsequent electrochemical impedance spectroscopy measurements to characterize the system under relevant operating conditions. In the end, again three slow scan rate (5 mV/s) CVs from 0.9 to 1.62 V vs RHE were conducted to track any changes in the electrode. The cell was purged with Ar during the whole measurement time. The third CV of the slow scan rate was selected for representation. No rotation was applied in these experiments. Impedance measurements were conducted by sweeping a sinusoidal voltage from 100,000 Hz to 1 Hz, using an amplitude of 0.01 V. The uncompensated resistance (Ru) is taken from the high-frequency intercept on the real axis of the Nyquist plot, and it is used to correct the measured electrode potential by subtracting the iR drop (current × Ru) to obtain a more accurate representation of the true electrochemical potential. For the elevated temperature measurements, new samples were prepared and the aforementioned protocol was applied first at room temperature, to ensure that the potential measured by the Ag/AgCl electrode (Metrohm) and the Mini-Hydroflex reference electrode (Gaskatel) is the same. Afterward, the temperature was increased to 65 °C. For the final slow scan rate (5 mV/s) CVs, the temperature was decreased to 20 °C. The temperature was controlled by electrolyte recirculation through a heated water bath, and measured by immersing a thermometer in the electrochemical cell.
MEA assembly
The membrane electrode assembly (MEA) is set up by sandwiching the membrane (AF3-HWK9-75, Ionomr, 75 µm) between the two spray coated electrodes. The electrodes are each surrounded by a gasket (225 µm, FIBERFLON®) to ensure tightness for the liquid electrolyte as well as set cell compression and allow for an even pressure distribution. For the final full cell test setup, the MEA is placed between two inhouse designed flow fields (Au-coated Monel, single channel, multi serpentine) with current collectors (Cu), pressure pads (Gylon®) and back plates (Al) on each side. Eight M6 screws are then tightened in two steps (5 Nm, 10 Nm) in a crosswise pattern to establish the cell compression. The Tafel slope range is indicated in the corresponding figures. Voltage data are shown after iR compensation, using the high-frequency resistance (HFR) measured at the corresponding current of the polarization curve.
MEA test station
The electrochemical characterization of the MEA is conducted in a self-built alkaline water electrolysis test station. The cell is connected to a potentiostat (VMP-300, BioLogic) as well as separate liquid electrolyte (1 M KOH) loops for each electrode. The KOH is preheated to 70 °C cell-entry by a heating bath (Hydro H41, LAUDA), while the cell itself is also heated to 70 °C by heating rods. The flow rate of the electrolyte is set to 40 mL/min by a peristaltic pump (Masterflex®, L/S®, Cole-Parmer®), and the electrolyte reservoirs are permanently flushed with nitrogen at a flowrate of 400 mL/min to avoid the formation of an explosive mixture within the test station.
MEA testing protocols
The testing protocol consists of a potential hold of 1 min at 1 V to check for electrical short circuits before starting the actual testing. Then a 2 h break-in procedure with a constant potential at 1.8 V is conducted, in order to exchange the iodine ions (I−) in the storage form of the membrane into hydroxide ions (OH−) from the KOH liquid electrolyte.
After the break-in, a polarization curve is recorded in galvanostatic steps from 2 to 3000 mA/cm2 with a holding time of 3 min at each step. After the 3 min holding time, a short galvanostatic electrochemical impedance spectroscopy (GEIS) sweep is recorded at 10 % amplitude of the respective DC current between 100 kHz to 1 Hz. From this GEIS sweep, the high frequency resistance (HFR) at each point in the polarization curve is approximated by interpolating the zero-crossing within the Nyquist plot of the respective GEIS sweep.
The measurement protocol then continues with impedance measurements between 200 kHz to 100 mHz at three selected regimes of the polarization curve with a 15 min constant current operation beforehand to ensure stable operation. The first electrochemical impedance spectroscopy (EIS) measurement is conducted potentiostatically at 1.5 V cell potential (10 % amplitude) to depict the impedance behavior in the activation region. The second and third EIS measurements are galvanostatically conducted at 1 A/cm2 and 3 A/cm2 (10% amplitude), to characterize the impedance in the Ohmic regime and at high currents, respectively.
An overnight constant current (1 A/cm2) measurement is then conducted for 15 h to characterize the stability of a given MEA. The measurement of the polarization curve and the three EIS measurements are then repeated afterward for evaluation of the stability.
Data availability
The data supporting the findings of this study are included in the published article and its Supplementary Information files. Additionally, all raw data for each graph have been compiled into an Excel file and Molecular Dynamics Trajectories, which is available in https://doi.org/10.5281/zenodo.15466603. Source data are provided with this paper.
References
Li, D. et al. Highly quaternized polystyrene ionomers for high performance anion exchange membrane water electrolysers. Nat. Energy 5, 378–385 (2020).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Chen, Y., Rui, K., Zhu, J., Dou, S. X. & Sun, W. Recent Progress on Nickel-Based Oxide/(Oxy)Hydroxide Electrocatalysts for the Oxygen Evolution Reaction. Chem. – Eur. J. 25, 703–713 (2019).
Thangavel, P. et al. Graphene-nanoplatelets-supported NiFe-MOF: high-efficiency and ultra-stable oxygen electrodes for sustained alkaline anion exchange membrane water electrolysis. Energy Environ. Sci. 13, 3447–3458 (2020).
Du, N. et al. Anion-exchange membrane water electrolyzers. Chem. Rev. 122, 11830–11895 (2022).
Guo, W., Kim, J., Kim, H. & Ahn, S. H. Direct electrodeposition of Ni-Co-S on carbon paper as an efficient cathode for anion exchange membrane water electrolysers. Int. J. Energy Res. 45, 1918–1931 (2021).
Park, J. E. et al. High-performance anion-exchange membrane water electrolysis. Electrochim. Acta 295, 99–106 (2019).
Vinodh, R., Kalanur, S. S., Natarajan, S. K. & Pollet, B. G. Recent advancements of polymeric membranes in anion exchange membrane water electrolyzer (AEMWE): a critical review. Polymers 15, 2144 (2023).
Moreno-González, M. et al. One year operation of an anion exchange membrane water electrolyzer utilizing Aemion+® membrane: Minimal degradation, low H2 crossover and high efficiency. J. Power Sources Adv. 19, 100109 (2023).
Hou, J. et al. Rational design of nanoarray architectures for electrocatalytic water splitting. Adv. Funct. Mater. 29, 1808367 (2019).
Wang, Q. & O’Hare, D. Recent advances in the synthesis and application of layered double hydroxide (LDH) Nanosheets. Chem. Rev. 112, 4124–4155 (2012).
Fan, G., Li, F., Evans, D. G. & Duan, X. Catalytic applications of layered double hydroxides: recent advances and perspectives. Chem. Soc. Rev. 43, 7040–7066 (2014).
Ma, R. & Sasaki, T. Two-dimensional oxide and hydroxide nanosheets: controllable high-quality exfoliation, molecular assembly, and exploration of functionality. Acc. Chem. Res. 48, 136–143 (2015).
He, S., An, Z., Wei, M., Evans, D. G. & Duan, X. Layered double hydroxide-based catalysts: nanostructure design and catalytic performance. Chem. Commun. 49, 5912–5920 (2013).
Shao, M. et al. Layered double hydroxides toward electrochemical energy storage and conversion: design, synthesis and applications. Chem. Commun. 51, 15880–15893 (2015).
Gong, M. et al. An advanced Ni–Fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 135, 8452–8455 (2013).
Zhang, B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Song, F. & Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 5, 1–9 (2014).
Zhao, Y. et al. Sub-3 nm ultrafine monolayer layered double hydroxide nanosheets for electrochemical water oxidation. Adv. Energy Mater. 8, 1703585 (2018).
Carrasco, J. A., Sanchis-Gual, R., Silva, A. S.-D., Abellán, G. & Coronado, E. Influence of the interlayer space on the water oxidation performance in a family of surfactant-intercalated NiFe-layered double hydroxides. Chem. Mater. 31, 6798–6807 (2019).
Zhang, X. et al. A simple synthetic strategy toward defect-rich porous monolayer NiFe-layered double hydroxide nanosheets for efficient electrocatalytic water oxidation. Adv. Energy Mater. 9, 1900881 (2019).
Li, P. et al. Tuning electronic structure of NiFe layered double hydroxides with vanadium doping toward high efficient electrocatalytic water oxidation. Adv. Energy Mater. 8, 1703341 (2018).
Zhai, P. et al. Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting. Nat. Commun. 12, 4587 (2021).
Bai, L., Lee, S. & Hu, X. Spectroscopic and electrokinetic evidence for a bifunctional mechanism of the oxygen evolution reaction. Angew. Chem. Int. Ed. 60, 3095–3103 (2021).
Chen, J. et al. Interfacial interaction between FeOOH and Ni–Fe LDH to modulate the local electronic structure for enhanced OER electrocatalysis. ACS Catal. 8, 11342–11351 (2018).
Zheng, Z. et al. Economizing production of diverse 2D layered metal hydroxides for efficient overall water splitting. Small 14, 1800759 (2018).
Fan, K. et al. Nickel–vanadium monolayer double hydroxide for efficient electrochemical water oxidation. Nat. Commun. 7, 1–9 (2016).
Ma, W. et al. A superlattice of alternately stacked Ni–Fe hydroxide nanosheets and graphene for efficient splitting of water. ACS Nano 9, 1977–1984 (2015).
Abellán, G., Carrasco, J. A. & Coronado, E. Room temperature magnetism in layered double hydroxides due to magnetic nanoparticles. Inorg. Chem. 52, 7828–7830 (2013).
Abellán, G., Coronado, E., Martí-Gastaldo, C., Waerenborgh, J. & Ribera, A. Interplay between chemical composition and cation ordering in the magnetism of Ni/Fe layered double hydroxides. Inorg. Chem. 52, 10147–10157 (2013).
Oestreicher, V. & Jobbágy, M. One pot synthesis of Mg2Al(OH)6Cl·1.5H2O layered double hydroxides: the epoxide route. Langmuir 29, 12104–12109 (2013).
Abellán Sáez, G., Oestreicher, V., Coronado Miralles, E. & Romer Pascual, J. A layered double hydroxide, a process for the synthesis and uses thereof. EP4043404 A1, European Patent Office, 20 Dec 2023.
Carrasco, J. A. et al. Alkoxide-intercalated NiFe-layered double hydroxides magnetic nanosheets as efficient water oxidation electrocatalysts. Inorg. Chem. Front. 3, 478–487 (2016).
Oestreicher, V. et al. Halide-mediated modification of magnetism and electronic structure of α-Co(II) hydroxides: synthesis, characterization, and DFT+U simulations. Inorg. Chem. 58, 9414–9424 (2019).
Oestreicher, V., Fábregas, I. & Jobbágy, M. One-pot epoxide-driven synthesis of M2Al(OH)6Cl·1.5H2O layered double hydroxides: precipitation mechanism and relative stabilities. J. Phys. Chem. C. 118, 30274–30281 (2014).
Oestreicher, V. & Jobbágy, M. On demand one-pot mild preparation of layered double hydroxides and their hybrid forms: advances through the epoxide route. Chem. – Eur. J. 25, 12611–12619 (2019).
Carrasco, J. A. et al. Liquid phase exfoliation of carbonate-intercalated layered double hydroxides. Chem. Commun. 55, 3315–3318 (2019).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and. Ni. Appl. Surf. Sci. 257, 2717–2730 (2011).
McIntyre, N. S. & Zetaruk, D. G. X-ray photoelectron spectroscopic studies of iron oxides. Anal. Chem. 49, 1521–1529 (1977).
Jiang, J. et al. Atomic-level insight into super-efficient electrocatalytic oxygen evolution on iron and vanadium co-doped nickel (oxy)hydroxide. Nat. Commun. 9, 2885 (2018).
Dionigi, F. & Strasser, P. NiFe-based (oxy)hydroxide catalysts for oxygen evolution reaction in non-acidic electrolytes. Adv. Energy Mater. 6, 1600621 (2016).
Abellán, G., Coronado, E., Martí-Gastaldo, C., Pinilla-Cienfuegos, E. & Ribera, A. Hexagonal nanosheets from the exfoliation of Ni2+-Fe3+ LDHs: a route towards layered multifunctional materials. J. Mater. Chem. 20, 7451–7455 (2010).
Carrasco, J. A., Abellán, G. & Coronado, E. Influence of morphology in the magnetic properties of layered double hydroxides. J. Mater. Chem. C. 6, 1187–1198 (2018).
Carrasco, J. A., Oestreicher, V., Silva, A. S.-D. & Abellán, G. Magnetism in two-dimensional layered double hydroxides. Appl. Clay Sci. 243, 107073 (2023).
Carrasco, J. A. et al. Fundamental insights into the covalent silane functionalization of NiFe-layered double hydroxides. Chem. – Eur. J. 26, 6504–6517 (2020).
Seijas-Da et al. Two-dimensional magnetic behaviour in hybrid NiFe-layered double hydroxides by molecular engineering. Dalton Trans. 52, 1219–1228 (2023).
Bajdich, M., García-Mota, M., Vojvodic, A., Nørskov, J. K. & Bell, A. T. Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water. J. Am. Chem. Soc. 135, 13521–13530 (2013).
García-Mota, M. et al. Importance of correlation in determining electrocatalytic oxygen evolution activity on cobalt oxides. J. Phys. Chem. C. 116, 21077–21082 (2012).
Xu, Z. et al. Engineering NiFe layered double hydroxide by valence control and intermediate stabilization toward the oxygen evolution reaction. J. Mater. Chem. A 8, 26130–26138 (2020).
Bi, Y. et al. Understanding the incorporating effect of Co2+/Co3+ in NiFe-layered double hydroxide for electrocatalytic oxygen evolution reaction. J. Catal. 358, 100–107 (2018).
Johnston, C. & Graves, P. R. In situ Raman spectroscopy study of the nickel oxyhydroxide electrode (NOE) system. Appl. Spectrosc. 44, 105–115 (1990).
Hou, C. et al. Rapid large-scale synthesis of ultrathin NiFe-layered double hydroxide nanosheets with tunable structures as robust oxygen evolution electrocatalysts. RSC Adv. 11, 37624–37630 (2021).
Tyndall, D. et al. Demonstrating the source of inherent instability in NiFe LDH-based OER electrocatalysts. J. Mater. Chem. A 11, 4067–4077 (2023).
Bernard, M. C. et al. Structural defects and electrochemical reactivity of β-Ni(OH)2. J. Power Sources 63, 247–254 (1996).
Lu, Z. et al. Three-dimensional NiFe layered double hydroxide film for high-efficiency oxygen evolution reaction. Chem. Commun. 50, 6479–6482 (2014).
Jiang, W. et al. Composition-dependent morphology, structure, and catalytical performance of nickel–iron layered double hydroxide as highly-efficient and stable anode catalyst in anion exchange membrane water electrolysis. Adv. Funct. Mater. 32, 2203520 (2022).
Li, Y. et al. Operando spectroscopies unveil interfacial FeOOH induced highly reactive β-Ni(Fe)OOH for efficient oxygen evolution. Appl. Catal. B Environ. 318, 121825 (2022).
Hedenstedt, K., Bäckström, J. & Ahlberg, E. In-Situ Raman spectroscopy of α- and γ-FeOOH during cathodic load. J. Electrochem. Soc. 164, H621–H627 (2017).
Wilhelm, M., Bastos, A., Neves, C., Martins, R. & Tedim, J. Ni-Fe layered double hydroxides for oxygen evolution Reaction: Impact of Ni/Fe ratio and crystallinity. Mater. Des. 212, 110188 (2021).
Vlamidis, Y., Scavetta, E., Gazzano, M. & Tonelli, D. Iron vs aluminum based layered double hydroxides as water splitting catalysts. Electrochim. Acta 188, 653–660 (2016).
Tang, D. et al. Carbon quantum Dot/NiFe layered double-hydroxide composite as a highly efficient electrocatalyst for water oxidation. ACS Appl. Mater. Interfaces 6, 7918–7925 (2014).
Bhowmik, T., Kundu, M. K. & Barman, S. CoFe layered double hydroxide supported on graphitic carbon nitrides: an efficient and durable bifunctional electrocatalyst for oxygen evolution and hydrogen evolution reactions. ACS Appl. Energy Mater. 1, 1200–1209 (2018).
El-Sayed, H. A., Weiß, A., Olbrich, L. F., Putro, G. P. & Gasteiger, H. A. OER catalyst stability investigation using RDE technique: a stability measure or an artifact?. J. Electrochem. Soc. 166, F458–F464 (2019).
Fathi Tovini, M., Hartig-Weiß, A., Gasteiger, H. A. & El-Sayed, H. A. The discrepancy in oxygen evolution reaction catalyst lifetime explained: RDE vs MEA - dynamicity within the catalyst layer matters. J. Electrochem. Soc. 168, 014512 (2021).
Trogisch, N., Koch, M., El Sawy, E. N. & El-Sayed, H. A. Microscopic bubble accumulation: the missing factor in evaluating oxygen evolution catalyst stability during accelerated stress tests. ACS Catal. 12, 13715–13724 (2022).
Hartig-Weiss, A., Tovini, M. F., Gasteiger, H. A. & El-Sayed, H. A. OER catalyst durability tests using the rotating disk electrode technique: the reason why this leads to erroneous conclusions. ACS Appl. Energy Mater. 3, 10323–10327 (2020).
Thissen, N. et al. Industrially relevant conditions in lab-scale analysis for alkaline water electrolysis. ChemElectroChem 11, e202300432 (2024).
Marquez, R. A. et al. A guide to electrocatalyst stability using lab-scale alkaline water electrolyzers. ACS Energy Lett. 9, 547–555 (2024).
Sanchis-Gual, R. et al. Improving the onset potential and Tafel slope determination of earth-abundant water oxidation electrocatalysts. Electrochim. Acta 388, 138613 (2021).
Weber, R. et al. Meta -kinks are key to binder performance of poly(arylene piperidinium) ionomers for alkaline membrane water electrolysis using non-noble metal catalysts. J. Mater. Chem. A 12, 7826–7836 (2024).
Ehelebe, K. et al. Evaluating electrocatalysts at relevant currents in a half-cell: the impact of Pt loading on oxygen reduction reaction. J. Electrochem. Soc. 166, F1259–F1268 (2019).
Sanchis-Gual, R. et al. Influence of crystallographic structure and metal vacancies on the oxygen evolution reaction performance of Ni-based layered hydroxides. Chem. – Eur. J. 30, e202303146 (2024).
Anantharaj, S., Kundu, S. & Noda, S. “The Fe Effect”: A review unveiling the critical roles of Fe in enhancing OER activity of Ni and Co based catalysts. Nano Energy 80, 105514 (2021).
Geuß, M. et al. Investigation of iridium-based OER catalyst layers in a GDE half-cell setup: opportunities and challenges. J. Electrochem. Soc. 170, 114510 (2023).
Ehlers, J. C., Feidenhans’l, A. A., Therkildsen, K. T. & Larrazábal, G. O. Affordable green hydrogen from alkaline water electrolysis: key research needs from an industrial perspective. ACS Energy Lett. 8, 1502–1509 (2023).
Tricker, A. W. et al. Design and operating principles for high-performing anion exchange membrane water electrolyzers. J. Power Sources 567, 232967 (2023).
Li, H. et al. Poly(vinyl benzyl methylpyrrolidinium) hydroxide derived anion exchange membranes for water electrolysis. J. Mater. Chem. A 7, 17914–17922 (2019).
Dionigi, F. et al. In-situ structure and catalytic mechanism of NiFe and CoFe layered double hydroxides during oxygen evolution. Nat. Commun. 11, 2522 (2020).
Zhang, J., Winkler, J. R., Gray, H. B. & Hunter, B. M. Mechanism of nickel–iron water oxidation electrocatalysts. Energy Fuels 35, 19164–19169 (2021).
Amirzhanova, A. et al. Synthesis and water oxidation electrocatalytic and electrochromic behaviours of mesoporous nickel oxide thin film electrodes. J. Mater. Chem. A 7, 22012–22020 (2019).
Zhou, Y.-N., Li, F.-T., Dong, B. & Chai, Y.-M. Double self-reinforced coordination modulation constructing stable Ni4+ for water oxidation. Energy Environ. Sci. 17, 1468–1481 (2024).
Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
Zhao, J.-W., Shi, Z.-X., Li, C.-F., Gu, L.-F. & Li, G.-R. Boosting the electrocatalytic performance of NiFe layered double hydroxides for the oxygen evolution reaction by exposing the highly active edge plane (012). Chem. Sci. 12, 650–659 (2021).
Meingast, L. et al. Effect of TCNQ layer cover on oxidation dynamics of black phosphorus. Phys. Status Solidi RRL 12, 1800179 (2018).
Hammouda, B. A new Guinier–Porod model. J. Appl. Crystallogr. 43, 716–719 (2010).
Teixeira, J. Small-angle scattering by fractal systems. J. Appl. Crystallogr. 21, 781–785 (1988).
Craievich, A. F. Small-Angle X-ray Scattering by Nanostructured Materials. in Handbook of Sol-Gel Science and Technology (eds. Klein, L., Aparicio, M. & Jitianu, A.) 1–46. https://doi.org/10.1007/978-3-319-19454-7_37-1 (Springer International Publishing, Cham, 2016).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Newville, M. IFEFFIT: interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 8, 322–324 (2001).
Rehr, J. J., Kas, J. J., Vila, F. D., Prange, M. P. & Jorissen, K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 12, 5503 (2010).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Lee, S., Bai, L. & Hu, X. Deciphering iron-dependent activity in oxygen evolution catalyzed by nickel–iron layered double hydroxide. Angew. Chem. 132, 8149–8154 (2020).
Lee, S., Banjac, K., Lingenfelder, M. & Hu, X. Oxygen isotope labeling experiments reveal different reaction sites for the oxygen evolution reaction on nickel and nickel iron oxides. Angew. Chem. 131, 10401–10405 (2019).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Minichová, M. et al. Electrochemical dissolution of PtRu/C: Effect of potential, fuels, and temperature. Electrochim. Acta 502, 144764 (2024).
Acknowledgements
The project SEAL HYDROGEN is supported by the Clean Hydrogen Partnership and its members under the GA 101137915. This work was supported by the European Research Council (ERC Starting Grant No. 2D-PnictoChem 804110, ERC PoC 2D4H2 No. 101101079, ERC-2021-StG-101042680), the Spanish MICINN (CNS2024-154959, PID2022-143297NB-I00, TED2021-131347B-I00, TED2021-131323B-I00, PID2021-122980OB-C51, and Excellence Unit María de Maeztu CEX2019-000919-M), the Generalitat Valenciana (CIDEGENT/2018/001, CIDEXG/2023/1) and Comunidad de Madrid (MAD2D-CM)-UCM3. The authors thank the CELLS-ALBA (Spain) for making all the facilities available for the synchrotron radiation experiment at BL11-NCD SWEET, BL-22 CLÆSS, BL16 - NOTOS beamlines through project numbers 2020024318, 2021025023, 2020094548, 2021024897 and 2024028425. A.S.-D thanks the University of Valencia for an “Atracción del Talento” predoctoral grant. L.J.J.M. acknowledges the FPI fellowship PREP2022-000255 linked to PID2022-143297NB-I00 project funded by MCIN/AEI/10.13039/501100011033 and by FSE+. BV and JCW gratefully acknowledge the FCT support through the UID/04349/2020. M.M. is a research member from CONICET (Argentina) and thanks to the financial support through the RX-EE-1 project of MinCyT Argentina. The authors thank Christian Olivares-Martínez and Federico Juarez for their assistance with the experimental work. Electron microscopy observations carried out at Centro Nacional de Microscopia Electronica ICTS-ELECMI.
Author information
Authors and Affiliations
Contributions
A.S.-D. contributed to the synthesis, design, and oversight of the study’s characterization. A.H. contributed to the electrochemical studies in single cells and the development of inks. V.O., J.R., C.J.-H., and G.T. contributed to the synthesis, scale-up, and material characterization. B.J.C.V. and J.C.W. contributed to the measurements and analysis of Mössbauer spectroscopy. L.J.J.M., G.D., A.F., and S.L. contributed to in situ Raman characterization. A.S.-D., V.O., and C.H.-I. contributed to SAXS data measurement, interpretation, and discussion. A.S.-D., V.O., C.J.-H., and M.M. contributed to XAS experiments, including measurements, interpretation, and discussion. D.L.-A. and J.J.B. contributed to the theoretical part of the study. V.L., A.T.S.F., K.J.J.M., S.C., and S.T. contributed to multi-scale electrochemical characterization, writing of the article, and contextualization of results. M.V. contributed to electron microscopy characterization. V.L. coordinated the interaction between the electrochemical characterization and the synthesis and scaling of layered hydroxides. A.S.-D., A.H., V.O. and V.L. wrote the first draft of the manuscript. G.A. conceived the research and contributed to the funding acquisition, coordinated the research, and wrote the manuscript with the input of all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: G.A., A.S.D.S., J.R. and V.L. has a financial interest in Matteco Team S.L., a spin-off company of the University of Valencia developing new materials for decarbonisation including catalysts for green hydrogen production. G.A., V.O. and J.R. are listed as co-inventors on a patent encompassing the RT-NiFe-LDH referenced herein (EP4043404). The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jungang Hou, Cheng Yang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Seijas-Da Silva, A., Hartert, A., Oestreicher, V. et al. Scalable synthesis of NiFe-layered double hydroxide for efficient anion exchange membrane electrolysis. Nat Commun 16, 6138 (2025). https://doi.org/10.1038/s41467-025-61356-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61356-2
