
Abstract
Tumor evolution is one of the major mechanisms responsible for acquiring therapy-resistant and more aggressive cancer clones. Whether the tumor microenvironment through immune-mediated mechanisms might promote the development of more aggressive cancer types is crucial for the identification of additional therapeutic opportunities. Here, we identify a subset of tumor-associated neutrophils, defined as tumor-associated neutrophil precursors (PreNeu). These PreNeu are enriched in highly proliferative hormone-dependent breast cancers and impair DNA repair capacity. Mechanistically, succinate secreted by tumor-associated PreNeu inhibits homologous recombination, promoting error-prone DNA repair through non-homologous end-joining regulated by PARP-1. Consequently, breast cancer cells acquire genomic instability promoting tumor editing and progression. Selective inhibition of these pathways induces increased tumor cell killing in vitro and in vivo. Tumor-associated PreNeu score correlates with copy number alterations in highly proliferative hormone-dependent tumors from breast cancer patients. Treatment with PARP-1 inhibitors counteract the pro-tumoral effect of these neutrophils and synergize with endocrine therapy.
Similar content being viewed by others

Introduction
Hormone-dependent breast cancers, also referred to as estrogen receptor-positive (ER+) or luminal-like tumors, rely on estrogens to promote proliferation and inhibit apoptosis1. Highly proliferative subtype of ER+ breast cancer, associated with Luminal B tumors2, has greater tumor aggressiveness and significantly worse prognoses than low proliferative Luminal A tumors3. Since ERα is the primary oncogenic driver in most ER+ breast cancers, endocrine-based therapeutic options are the current standard of care3. Although endocrine therapies extend overall survival, one-third of all early-stage ER+ breast cancer patients experience treatment resistance, leading to metastasis4,5,6. Genomic alterations in early and advanced breast cancers are proposed to be the cause of emergent therapy-resistant cancer clones7. The well-established dependency of cancer cells on the tumor microenvironment (TME) suggests that the microenvironment might have a role in the development of more aggressive cancer types. ER+ breast cancers are considered to be immunologically cold due to low infiltration of lymphocytes within the TME8. A growing body of evidence suggests that the TME of ER+ breast cancer is highly infiltrated by neutrophils and estrogen signaling contributes to the immunosuppressive nature of breast cancer by driving the recruitment and function of these cells9,10,11. Lectin-type oxidized LDL receptor-1 (LOX-1) expression, which has been associated with higher immunosuppressive activity, defines tumor-infiltrated immunosuppressive neutrophils12,13.
Our laboratory and others have previously demonstrated different pathogenic functions of neutrophils in breast and other hormonally driven cancers and identified different subsets of neutrophils within the TME14,15,16,17. However, a clear picture of neutrophil heterogeneity in ER+ breast cancer is missing, and whether these cells may regulate the pathogenicity of ER+ breast cancers remains unknown. In this work, we identify tumor-associated neutrophil precursors (PreNeu) in ER+ breast cancer. PreNeu inhibits homologous recombination in cancer cells, further promoting error-prone DNA repair and sensitizing to PARP inhibition therapy.
Results
Neutrophil precursors are enriched in the TME of highly proliferative estrogen-positive breast cancers
We performed multiparameter flow cytometry on fresh tumor tissues obtained from 63 women with early-stage ER+ breast cancer (Supplementary Data 1) to define the neutrophil heterogeneity within low and highly proliferative ER+ breast cancers. The patients were classified as low or highly proliferative based on the tumor proliferative capacity. Tumors with Ki67 expression, defined by immunohistochemistry (IHC), higher than 20% were classified as highly proliferative2. Live CD45+ immune cells in the TME were projected onto a 2-dimensional map via the uniform manifold approximation and projection (UMAP)18,19 and clustered using PhenoGraph20, resulting in the identification of three distinct neutrophil clusters (Fig. 1a; Supplementary Fig. 1a, b). The first cluster (Cluster 1) comprised neutrophils (CD45+ CD11b+ CD33+ CD66b+ CD15+ CD163− LOX-1−)21, while the second cluster (Cluster 2) comprised LOX-1+ neutrophils (CD45+ CD11b+ CD33+ CD66b+ CD15+ CD163− LOX-1+)12,13. A third cluster (Cluster 3), that was predominantly found in highly proliferative tumors (Fig. 1a, b), included cells positive for the neutrophil lineage marker CD66b and LOX-1 while lacking maturation markers including CD11b, CD15, CD33, and expression of CD163, a prototypical marker of tumor-associated macrophages22 (Supplementary Fig. 1b).

a UMAP plots derived from PhenoGraph cluster analyses of concatenated low proliferative (n = 3) and highly proliferative (n = 3) breast cancer biopsies gated on CD45+ cells. Cluster 1: CD11b+, CD33+, CD66b+, CD15+, CD163−, LOX-1−; Cluster 2: CD11b+, CD33+, CD66b+, CD15+, CD163−, LOX-1+; Cluster 3: CD11b−, CD33−, CD66b+, CD15−, CD163−, Lox-1+; Cluster 4: CD11b−, CD33−, CD66b−, CD15−, CD163−, LOX-1−. b Bar graph displaying the frequency of the four clusters in normal tissues (NT, n = 3), low proliferative ER+ breast cancer biopsies (Low proliferative ER+ BC; n = 48) and highly proliferative breast cancer biopsies (Highly proliferative ER+ BC; n = 15). c UMAP plot derived from BD Rhapsody scRNA-seq cluster analysis of the neutrophil compartment in a highly proliferative ER+ tumor (n = 1). d Metascape analysis of enriched gene pathways of cluster C neutrophil compared to clusters A and B. Saturation of red color is proportional to p value significance. e Upregulated genes (FDR < 0.01; log2FoldChange > 1) in PreNeu compared to LOX-1+ neutrophils sorted from highly proliferative ER+ breast cancer biopsies resulting from single-cell differential expression analysis (Smartseq-2). Significantly upregulated genes are organized into a protein-protein interaction network generated using String Database. The dimension of the nodes is proportional to the highest statistical significance (represented as −10xlog10FDR). Saturation of red color is proportional to increased gene expression in PreNeu compared to LOX-1+ neutrophils (represented as log2FoldChange). Yellow color indicates the manually selected genes. Highlighted in yellow are genes belonging to the magnified subnetwork shown on the right of the figure panel. f UMAP plot derived from BD Rhapsody scRNA-seq data displaying the enrichment score of the PreNeu signature derived from scSMART-seq analysis based on significantly upregulated genes. g Pseudotime trajectory embedded in the UMAP plot with cluster C as the starting point of the trajectory (green line). h Bar graph showing the percentage of Ki67 positive cells in the different populations of neutrophils (from n = 3 patients). Data are represented as mean ± SEM. Statistical analyses (one-way analyses of variance, ANOVA, multiple comparison test). i Bar graph displaying the percentage of cluster 3 cells within CD45+ cells in normal tissues (NT, n = 3), low proliferative (LP, n = 48), or highly proliferative (HP, n = 15) ER+ breast cancer biopsies. Each symbol represents an individual patient. Data are represented as mean ± SEM. Statistical analyses (one-way analyses of variance, ANOVA, multiple comparison test). j Cluster 3 relative abundance on CD45+ cells stratified based on tumor grade (n = 9 for grade 1, n = 40 for grade 2, n = 13 for grade 3). Data are represented in violin plots with median (bold line) and quartile (regular lines). Statistical analyses (Brown-Forsythe and Welch ANOVA tests). k cluster 3 to cluster 2 ratio in NT (n = 3), LP (n = 48), and HP (n = 15) ER+ breast cancer biopsies. Each symbol represents an individual patient. Data are represented as mean ± SEM. Statistical analyses (one-way analyses of variance, ANOVA, multiple comparison test). l Bar graph displays the percentage of cluster 2 within CD45+ cells in NT (n = 3), LP (n = 48), and HP (n = 15) ER+ breast cancer biopsies. Each symbol represents an individual patient. Data are represented as mean ± SEM. Statistical analyses (one-way analyses of variance, ANOVA, multiple comparison test). Source data are provided as a Source data file.
To characterize the undefined cluster of immune cells (Cluster 3), we performed single-cell (sc)-RNA sequencing using BD Rhapsody known for preserving low-mRNA content cell integrity23, on a highly proliferative ER+ breast biopsy. Applying an unsupervised analysis to these data, we observed three neutrophil clusters (named Clusters A, B, C; Fig. 1c) as previously revealed by flow cytometry (Fig. 1a; Supplementary Fig. 1b). Notably, cells classified in Cluster C possessed strong enrichment in translation, aerobic respiration, and mitosis, as revealed by pathway analyses (Fig. 1d, Supplementary Fig. 1c, Supplementary Data 2). Upon further flow cytometry analysis of the cells in cluster 3, we observed positive expression of CD71, CD117, CD45RA, and CD66b markers, along with negative expression of CD11b, CD33, and CD16 markers (Supplementary Fig. 1d). These phenotypic characteristics are consistent with a population defined as PreNeu, recently identified in the bone marrow of healthy donors24. Next, to cross-validate the definition of the neutrophil subsets, we performed scSMART sequencing to compare the gene expression profile of tumor-sorted cells classified as Cluster 3 to tumor-infiltrating LOX-1+ neutrophils (Cluster 2), which have been reported to be the major tumor-infiltrating, pro-tumoral neutrophil population25,26,27 (Supplementary Fig. 1e, f). Cells classified in Cluster 3 possessed myeloid precursor signatures (Fig. 1e; Supplementary Data 3) that were associated with a subset of human bone marrow PreNeu in a published dataset28 (Supplementary Fig. 1g) and showed a positive enrichment only for a PreNeu signature when analyzed by GSEA enrichment analysis with the other seven validated neutrophil-specific genes sourced from published data28,29 (Supplementary Fig. 1g, h). Consistent with our previous data, overlaying the PreNeu signature generated by scSMART-seq on the Rhapsody dataset, we registered a significant enrichment of the tumor-associated PreNeu signature in cluster 3 compared to the other neutrophil clusters (Fig. 1f; Supplementary Fig. 1i), as well as overlapping with the recently public bone marrow PreNeu signature28 (Supplementary Fig. 1j) and not with a mature neutrophil signature (Supplementary Fig. 1k).
Notably, cluster 3 expressed immunosuppression molecules such as LOX-1, IDO1, PD-L1, and CTLA425 (Fig. 1e; Supplementary Fig. 1d, Supplementary Data 3). Trajectory analysis revealed a transcriptomic continuum connecting cluster 3 with clusters 1 and 2, suggesting that these cells may have the potential to transition into mature neutrophils (Fig. 1g). Cell proliferation is a key biological function that distinguishes PreNeu from mature neutrophils21,30,31. To verify this, we tested Ki67 expression using flow cytometry in tumor-associated neutrophil subsets. We identified cluster 3 cells as the only neutrophil population in breast cancer with proliferative potential (Fig. 1h). Based on these observations, we classified Cluster 3, detected through both flow cytometry and different single-cell sequencing technologies, as tumor-associated PreNeu. We found that tumor-associated PreNeu is enriched in highly proliferative hormone-dependent breast cancers, compared to low proliferative tumors and normal breast tissue (Fig. 1b, i). The abundance of tumor-associated PreNeu was associated with a higher histological tumor grade (Fig. 1j). Of note, no difference in the frequency of LOX-1+ neutrophils was observed (Fig. 1k, l).
In summary, we observed that highly proliferative ER+ breast cancers were specifically infiltrated by tumor-associated PreNeu.
Tumor-associated PreNeu promotes genomic instability by inhibiting high-fidelity homologous recombination DNA repair in cancer cells
To study the role of PreNeu in ER+ breast cancer, we generated PreNeu in vitro from human cord blood (CB) mononuclear cells (CB-derived PreNeu; cbPreNeu; Supplementary Fig. 2a). cbPreNeu clustered together with previously defined neutrophil28 (Supplementary Fig. 2b). Importantly, cbPreNeu have a similar expression profile, phenotypically mimic tumor-infiltrating PreNeu, harbor proliferation capacity, and show immunosuppressive activity (Supplementary Fig. 2c–g). Having demonstrated that the cbPreNeu phenocopies the tumor-associated PreNeu, we used the in vitro differentiated cells as a model to mechanistically study their role in breast cancer progression.
Given that tumor-associated PreNeu was predominantly found in highly proliferative ER+ breast tumors, rather than low proliferative ones, we hypothesized that the presence of these immune cells was associated with the more aggressive phenotype observed in these tumors. We investigated this hypothesis by co-culturing MCF-7 cells, a human ER+ breast cancer cell line32, in the presence of either conditioned medium obtained from PreNeu (cm-PreNeu;), from LOX-1+ neutrophils (cm-LOX-1+), or vehicle (∅; Supplementary Fig. 3a).
Even though the conditioned medium of both cbPreNeu and cbLOX-1+ neutrophils sustained the proliferation and survival of ER+ BC cells cultured under hormone deprivation (Supplementary Fig. 3b), only MCF-7 cells grown in cm-PreNeu showed significant downregulation of DNA Damage Response associated pathways in 4 Gene Set collections tested (Supplementary Fig. 3c) and had lower expression of DNA repair proteins (Supplementary Fig. 3d, e). Of note, the secretome of LOX-1+ neutrophils did not decrease the expression of DNA repair proteins (Supplementary Fig. 3e). To orthogonally validate this finding, we quantified DNA single-strand and double-strand breaks accumulated in MCF-7 cells treated with cm-PreNeu and LOX-1+ neutrophils using a comet assay (Supplementary Fig. 3f). The defects in homologous recombination, induced by cm-PreNeu and not by LOX-1+ neutrophils, led to the accumulation of single-strand and double-strand DNA breaks (Supplementary Fig. 3g, h). To distinguish accumulation of DNA damage per se from indirect effects caused by the accelerated cellular proliferation observed (Supplementary Fig. 3b), we used irradiated MCF-7 cells treated with or without cm-PreNeu in a kinetic experiment (Fig. 2a). MCF-7 cells treated with cm-PreNeu exhibited DNA damage, while untreated cells showed repaired DNA (Fig. 2b). In keeping with this, vehicle and cm-LOX-1+ neutrophils engaged homologous recombination proteins, whereas cells treated with cm-PreNeu failed to do so (Fig. 2c, d; Supplementary Fig. 3i).

a Experimental scheme created using BioRender. Garda, c. (2025) https://BioRender.com/yj15tp2. Briefly, ER+ MCF-7 breast cancer cells were irradiated and co-cultured with conditioned media (cm) from PreNeu for different time points (0 h, 2 h, 6 h, and 24 h) or control (Ø). b Quantification and representative images of the neutral comet assay of MCF-7 cells treated as indicated in a; black for Ø and red for cm-PreNeu). Each dot represents an individual comet from a pool of three biological replicates. Aggregated data from three independent experiments are reported as mean ± SEM (n = 10 for time point). Statistical analyses (two-tailed unpaired Student t test). c Heatmap showing fold change in protein levels using western blot of the reported proteins in MCF-7 cells treated as described in a. Values were normalized to the relative housekeeping gene followed by normalization on t = 0 for both untreated and treated MCF-7 cells upon irradiation (n = 3 biological replicates for time point, except n = 4 biological replicates for pATM/ATM). Statistical analysis (two-tailed unpaired t test). d Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/8prt8a8, Immunofluorescence images (left panel: DAPI blue, TIP60 green, P-53BP1 red; right panel: DAPI blue, p-ATM green, γH2AX red), and quantification of MCF-7 cells treated with either the condition media of the PreNeu or LOX-1+ neutrophil, or control (Ø). Scale bar 5 µm. (P53BP1: n = 91 for Ø and cm-PreNeu, n = 98 for cm-LOX-1+ neutrophils; TIP60: n = 141 for Ø, n = 149 for cm-PreNeu, n = 139 for cm-LOX-1+ neutrophils. γH2AX: n = 34 for Ø, n = 40 for cm-PreNeu, n = 31 for cm-LOX-1+ neutrophils. p-ATM: n = 31 for Ø, n = 50 for cm-PreNeu, n = 33 for cm-LOX-1+ neutrophils. Aggregated data from three biological replicates per condition. Data are reported as mean ± SEM. Statistical analysis (two-tailed unpaired t test). e Correlation analyses of PreNeu score with copy number alterations (R2 = 0.32, p = 9.7e-05 linear regression). RNA sequencing and WES data were collected from 235 breast cancer cases treated with pre-operative chemotherapy. Data are reported with the regression line and confidence interval of 95%. Statistical analyses (simple linear regression). f Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/6n3mu0w. Female NRG mice were surgically implanted with estrogen pellets three weeks before the injection of MCF-7 Luciferase+ cells in the mammary fat pad (FP-MCF-7 Luc+). When the bioluminescence-based total flux reached more than 106 counts, mice were randomly enrolled in the following groups: untreated (Ø; n = 8) or treated with PreNeu (30 × 103, twice a week, i.p.; +PreNeu; n = 5). The mice were sacrificed after 4 months. g Immunofluorescence images (DAPI blue, RAD51 green, γH2AX red) and quantification of untreated vs PreNeu-treated MCF-7 tumors (n = 20 for each group). Scale bar 5 µm. Aggregated data from at least three tumors per condition. Data are reported as mean ± SEM. Statistical analyses (two-tailed unpaired Student t test). h Quantification and representative images of neutral comet assay using cells collected from MCF-7 tumors described in f (n = 20 for each group). Scale bar 5 µm. Data are reported as mean ± SEM. Statistical analyses (two-tailed unpaired Student t test). i Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/014yk4s. Tumors collected from mice treated as described in (f) were processed as single-cell suspensions and EpCaM+ epithelial cells purified to extract DNA. Whole-genome sequencing was performed. Hierarchical clustering of untreated (n = 2) and PreNeu-treated (n = 4) mice based on standardized whole-genome copy number calls (euclidean distance, average linkage), is shown on the left. The violin plot indicates the proportion of the cancer genome affected by aneuploidy. Source data are provided as a Source data file.
Next, we explored the association between the PreNeu signature, which we derived from our single-cell sequencing data (Fig. 1e), and the presence of genomic alterations in breast cancers using data from a published cohort of deeply profiled early breast cancers33. We observed that PreNeu enrichment was associated with increased genomic instability in highly proliferative ER+ breast tumors, with the PreNeu score correlated with the proportion of the genome altered by copy number alterations (Fig. 2e).
To extend our investigation, we tested whether PreNeu promotes genomic instability in ER+ breast cancer in vivo using the MCF-7-FP model34 (MCF-7 Luciferase tumors grafted in the mammary fat pad (FP) treated with PreNeu or vehicle; Fig. 2f). The experimental administration of PreNeu led to a marked increase in tumor volume with a corresponding increase in Ki-67+ cells (Supplementary Fig. 3j, k). Notably, the presence of tumor-associated PreNeu showed a higher percentage of the double-strand break marker γH2AX, accompanied by lower expression of the DNA repair marker RAD51 (Fig. 2g; Supplementary Fig. 3l) and higher DNA damage (Fig. 2h). Strikingly, genomic instability was enhanced in MCF-7-FP tumors grown in mice treated with PreNeu compared to the controls (Fig. 2i). To further validate our findings, we employed three additional allograft models of breast cancer: PyMT-N, PyMT-M, and E0771. The PyMT models are derived from a mammary tumor developed in a transgenic polyoma middle-T (MMTV-PyMT) mouse. MMTV-PyMT is a model of breast cancer metastasis, and early-stage tumors progressively lose ER expression during tumor progression35. PyMT-N and PyMT-M are genetically similar primary tumor lines that are preferentially infiltrated by granulocytic-like or monocytic-like cells, respectively9. PyMT-N allografts were highly infiltrated by neutrophils, which included PreNeu36 compared to PyMT-M allografts (Supplementary Fig. 4a–c). The E0771 allograft has been characterized as highly proliferative, ERα−, ERβ+, PR+, HER2+ mammary cancer cell37 and is infiltrated by tumor-associated PreNeu (Supplementary Fig. 5a). The role of PreNeu in favoring DNA damage by inhibiting the high-fidelity homologous recombination DNA repair in tumor cells was confirmed in all the allografts, in which an external boost of PreNeu (tumor cells co-injected with bone marrow (bm)-derived PreNeu, bmPreNeu) or inhibition of neutrophil recruitment to the tumor using a selective CXCR2 antagonist16,38 (αCXCR2) affected tumor progression, proliferation and accumulation of DNA damage (Supplementary Fig. 4d–i; Supplementary Fig. 5b–f).
Taken together, these data demonstrate that tumor-associated PreNeu promotes breast tumor cell genomic instability by compromising homologous recombination DNA repair in vivo.
Succinate produced by PreNeu drives homologous recombination DNA repair defects in tumor cells
To identify factors secreted by PreNeu which induce defects in homologous recombination DNA repair, we fractioned cm-PreNeu and cm- LOX-1+ neutrophils, testing the ability of the different fractions to induce DNA damage in MCF-7 cells in vitro (Supplementary Fig. 6a). Only the PreNeu secretome fraction containing molecules with a mass lower than 3 kDa induced tumor DNA damage as assessed by comet assay compared to molecules with a mass higher than 3 kDa and LOX-1+ neutrophil conditioned medium (Supplementary Fig. 6b). This finding prompted us to hypothesize that metabolites produced by PreNeu may mediate inhibition of DNA repair. A metabolomic analysis in both murine and human PreNeu (Supplementary Fig. 6c) showed that succinate was the highest-ranked PreNeu metabolite within a cluster of 116 overexpressed metabolites (Fig. 3a; Supplementary Data 4). Succinate is an oncometabolite39 that is synthesized in mitochondria during the tricarboxylic acid cycle and is released extracellularly, where it can modulate cellular gene expression as well as the epigenetic landscape and demonstrate hormone-like signaling40,41. Metabolite quantification by colorimetric assay further confirmed that tumor-associated PreNeu produced succinate in vivo, with tumor-associated PreNeu sorted from highly proliferative ER+ breast cancers and PyMT-N allografts producing even higher levels of succinate compared to other tumor-associated neutrophils (Fig. 3b). Moreover, similar to our results found in vivo, cbPreNeu produces succinate at a higher level compared to cbLOX-1+ neutrophils, additionally validating their functional similarity to cells found in patients or mice (Fig. 3b). Succinate can be converted into fumarate by the enzyme succinate dehydrogenase (SDH). Notably, PreNeu showed significantly decreased SDH activity (Fig. 3c), perhaps explaining the high levels of succinate observed.

a LC-MS/MS quantification of the most upregulated metabolites. b Succinate quantified in sorted neutrophil precursors (PreNeu; n = 1: pool of three independent highly proliferative ER+ breast cancer patients) and LOX-1+ neutrophils (n = 1: pool of three independent highly proliferative ER+ breast cancer patients), cord blood-derived PreNeu (n = 5), cord blood-derived LOX-1+ neutrophils (n = 6), PyMT-N tumor-sorted PreNeu (n = 6) and tumor-associated neutrophils (n = 3). Data are represented as mean ± SEM. Statistical analyses (two-tailed unpaired Student t test). c Succinate dehydrogenase (SDH) activity quantified by SDH Colorimetric Assay in cord-blood derived PreNeu, LOX-1+ neutrophils and PreNeu (n = 4 for neutrophils and PreNeu, n = 5 for LOX-1+ neutrophils). Data are represented as mean ± SEM, one-way analysis of variance (ANOVA multiple comparison test). d Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/8prt8a8. Briefly, MCF-7 cells were irradiated at 10 Gy, then cultured in hormone deprivation and treated or control (Ø) with cm-PreNeu or succinate (2 mM). e Immunofluorescence images (DAPI blue, TIP60 green, P-53BP1 red) and quantification of irradiated MCF-7 cells treated or control (Ø) with condition media of PreNeu (p-53BP1: n = 51 for Ø, n = 50 for both Succinate and cm-PreNeu; TIP60: n = 49 for Ø and cm-PreNeu, n = 48 for Succinate). Data are represented as mean ± SEM. Statistical analysis (two-tailed unpaired Student t test). f Immunofluorescence images (DAPI blue, p-ATM green, γH2AX red) and quantification of irradiated MCF-7 cells treated or control (Ø) with condition media of PreNeu (γH2AX: n = 51 for Ø, n = 50 for Succinate, n = 52 for cm-PreNeu. p-ATM: n = 55 for Ø, n = 53 for Succinate, n = 58 for cm-PreNeu). Aggregated data from three biological replicates per condition. Data are represented as mean ± SEM. Statistical analysis (two-way ANOVA). g Cartoon created using BioRender showing the mechanism of succinate release by PreNeu and uptake/transport by tumor cells. Pharmacological blocking of SCL13a5 prevents succinate transport in tumor cells. h Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/8w9k8zb. Briefly, MCF-7 cells were irradiated at 10 Gy and then cultured in hormone deprivation and treated with either SCL13a5 blocking (αSCL13a5), cm-PreNeu, combinatorial treatment, or control. i Immunofluorescence images (Upper panel: DAPI blue, TIP60 green, P-53BP1 red. Down panel: DAPI blue, pATM green, γH2AX red) and quantification of irradiated MCF-7 cells treated or control (Ø) with cm-PreNeu or cm-PreNeu and αSLC13a5 (P53BP1: n = 48 for Ø and αSLC13a5, n = 45 for cm-PreNeu, n = 51 for cm-PreNeu+ αSLC13a5. TIP60: n = 38 for Ø, n = 30 for αSLC13a5, n = 35 for cm-PreNeu, n = 27 for cm-PreNeu+ αSLC13a5. γH2AX: n = 40 for Ø and αSLC13a5, n = 34 for cm-PreNeu, n = 43 for cm-PreNeu+ αSLC13a5. P-ATM: n = 23 for Ø, n = 35 for αSLC13a5, n = 28 for cm-PreNeu, n = 31 for cm-PreNeu + αSLC13a5). Aggregated data from three biological replicates per condition. Data are represented as mean ± SEM. Statistical analysis (two-way ANOVA). j Quantification and representative images of neutral comet assays of MCF-7 cells treated or control (Ø) with cm-PreNeu or αSLC13a5 (n = 47 for Ø, n = 57 for αSLC13a5, n = 36 for cm-PreNeu, n = 77 for cm-PreNeu+ αSLC13a5). Aggregated data from three biological replicates per condition. Data are represented as mean ± SEM. Statistical analysis (two-way ANOVA). k Cartoon created in BioRender. Garda, c. (2025) https://BioRender.com/svwrqhv showing the mechanism of pharmacological inhibition of succinate release by MCT1 inhibitor and the experimental scheme. Briefly, conditioned media form bone marrow-derived PreNeu (bmPreNeu) was generated in the presence of MCT1 inhibitor (cm-bmPreNeuMCT1i) or control (cm-bmPreNeuØ) and used to pre-treat PyMT-N cells in vitro for 48 hours. Then, cancer cells were implanted in the fat pad (FP) of NRG female mice, and tumors were collected when palpable. l Quantification and representative images of neutral comet assays of tumors whose cancer cells were pre-treated before the experiment with cm-bmPreNeuMCT1i or control (n = 135 for Ø, n = 128 for cm-PreNeuØ, n = 186 for cm-PreNeuMCT1i). Aggregated data from three biological replicates per condition. Data are represented as mean ± SEM. Statistical analysis (one-way ANOVA). m Immunofluorescence images (DAPI blue, RAD51 green, γH2AX red) and quantification of tumors from mice whose cancer cells were pre-treated before the experiment with cm-bmPreNeuMCT1i or control (γH2AX: n = 16 for Ø, n = 16 for cm-PreNeuØ and cm-PreNeuMCT1i. RAD51: n = 31 for Ø, n = 21 for cm-PreNeuØ, n = 21 for cm-PreNeuMCT1i). Aggregated data from three biological replicates per condition. Data are reported as mean ± SEM. Statistical analysis (one-way ANOVA). Source data are provided as a Source data file.
To functionally validate the association between succinate production by PreNeu and homologous recombination deficiency, we have first compared the inhibition of DNA repair proteins to the sites of DNA breaks, finding that both PreNeu secretome and succinate impeded TIP60 and pATM recruitment (Fig. 3d–f). Of note, these findings were also validated in LNCaP cells, a human hormone-dependent prostate cancer cell line (Supplementary Fig. 6d). Secondly, we inhibited succinate uptake by the tumor cells in MCF-7 cells using a selective inhibitor of the succinate transporter SLC13a5 (αSLC13a5)42, which were then cultured in the presence or absence of cm-PreNeu (Fig. 3g, h). While the cm-PreNeu inhibited TIP60 and pATM recruitment, and promoted single and double-strand DNA breaks in untreated MCF-7 cells, MCF-7 cells treated with αSLC13a5 were not affected (Fig. 3i, j). Similar results were also obtained using MCF-7 cells silenced for the succinate receptor (siSUCNR1)42,43 (Supplementary Fig. 6e, f), which were then cultured in the presence or absence of cm-PreNeu (Supplementary Fig. 6g) and validated using a second tumor cell model (Supplementary Fig. 6h–j).
To validate these findings in vivo, we analyzed DNA damage and defects in the homologous recombination pathway in PyMT-N tumor allografts where PreNeu was unable to secrete succinate. The tumor cells were pre-treated with conditioned media from either untreated PreNeu cells (cm-PreNeu∅) or PreNeu cells in which the succinate transporter Monocarboxylate Transporter 1 (MCT1) was pharmacologically inhibited (cm-PreNeuMCT1i; Fig. 3k). Tumors treated with conditioned media from PreNeu that could not secrete succinate (cm-PreNeuMCT1i) showed no defects in DNA damage accumulation and DNA repair capability (Fig. 3l, m). Similarly, when PreNeu was treated with the MCT1 inhibitor were directly administered to NRG mice implanted with PyMT-N cells, DNA damage induced by PreNeu was prevented (Supplementary Fig. 6k–m). Succinate has been shown to suppress DNA repair by inducing inhibition of the lysine demethylase KDM4B, resulting in aberrant hypermethylation and increased expression of histone 3 lysine 9 (H3K9me3)39, leading to the binding of H3K9me3 to genes related to DNA double-strand break repair in MCF-7 cells upon succinate treatment (Supplementary Fig. 7a, b; Supplementary Data 5). We evaluated whether succinate secreted by PreNeu could regulate hypermethylation of H3K9 in breast cancer by acting in a non-cell autonomous manner. Inactivation of SUCNR1 in MCF-7 cells significantly decreased H3K9me3 expression (Supplementary Fig. 7c). In a validation dataset, the degree of PreNeu enrichment was positively associated with SUCNR1 expression and negatively associated with KDM4B expression (Supplementary Fig. 7d). Indeed, succinate modulates the expression of SUCNR1 in a concentration-dependent manner on MCF-7 cells (Supplementary Fig. 7e). While it has been reported that myeloid cells can induce epithelial mutations through the production of reactive oxygen species (ROS)44, we found that PreNeu produced lower amounts of ROS compared to LOX-1+ neutrophils (Supplementary Fig. 8a), and PreNeu did not induce DNA damage through ROS (Supplementary Fig. 8b–e). In summary, these data show that the succinate released by PreNeu into the TME acts by directly impeding homologous recombination DNA repair proteins and promoting DNA damage in tumor cells.
Breast tumor cells affected by tumor-associated PreNeu rely on PARP to survive
Given the induction of homologous recombination deficiency by PreNeu, we interrogated the status of other DNA repair mechanisms in tumor cells challenged with cm-PreNeu, including poly (ADP-ribose) polymerase 1 (PARP-1) and non-homologous end joining (NHEJ) DNA repair. MCF-7 cells challenged with cm-PreNeu showed elevated levels of PARP-1 and DNA-PKcs (Fig. 4a), suggesting a dependence of the cells on these alternative DNA repair mechanisms.

a Quantification and representative images of western blots for PARP1, Ku70, Ku80, and DNA-PKcs levels in MCF-7 cells treated with or without conditioned media obtained from human cord blood-derived neutrophil precursors (cm-PreNeu; n = 4 for all the molecules), except n = 7 for PARP1). Data are reported as mean ± SEM. Statistical analyses (unpaired Student t test). b Quantification and representative image of western blot for PARP1 in MCF-7 cells silenced or not for the succinate receptor (SUCNR1) and treated or not with cm-PreNeu (n = 4 for each group). Data are reported as mean ± SEM. Statistical analysis (one-way ANOVA). c Quantification and representative image of western blot for PARP1 in MCF-7 cells silenced or not for the succinate receptor (SUCNR1) and treated or not with succinate (n = 3 for each group). Data are reported as mean ± SEM. Statistical analysis (unpaired t test). d Graph showing fold change in mRNA level of PARP1 in MCF-7 cells and LNCaP cells. Cells were previously irradiated at 10 Gy and then treated or control (Ø) with cm-PreNeu or succinate (2 mM) for 6 h (n = 3 for each group). Data are reported as mean ± SEM. Statistical analysis (unpaired t test). e Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/uueypah. PARP1-luciferase MCF-7 cells were transfected with pGL3 plasmid, treated or control (Ø) with cm-PreNeu in the presence or absence of αSLC13a5 for 6 hours. Cells were analyzed for luciferase activity using Dual-Glo Luciferase Assay System (Promega, E2920) following manufacturer’s instructions. Expression of PARP1 (Renilla) was normalized to the expression of pGL3 (Firefly) to calculate luciferase activity. The values were further normalized to the condition treated with cm-PreNeu. (n = 7 for each group, each dot is the mean of three independent technical replicates from two independent experiments). Data are reported as mean ± SEM. Statistical analysis (unpaired t test). f Cell proliferation of MCF-7 cells treated or control (Ø) with cm-PreNeu and challenge with PARPi (1 µM) after 24 h. g Annexin V-positive intensity in MCF-7 cells treated or control (Ø) with cm-PreNeu and challenge with PARPi (1 µM) after 24 h. f, g Aggregated data from three independent experiments are reported as mean ± SEM. Statistical analyses (two-way ANOVA and Ordinary one-way ANOVA multiple comparison test, respectively). h–j Tumor growth from the h E0771, i PyMT-N, and j PyMT-M models in female mice treated with PARPi or vehicle (Ø) when tumors were palpable as indicated in the graph (for the E0771 model n = 4 for each group, for the PyMT-N model n = 4 for each group, for the PyMT-M model n = 5 for Ø and n = 4 for PARPi groups). Data are reported as mean ± SEM. Statistical analysis (two-way ANOVA). k Representative histology images and their quantification. H&E and Cleaved-Caspase 3 immunohistochemical staining (Cleaved-Caspase 3 brown; nuclei, blue) of representative E0771, PyMT-N, and PyMT-M tumors from mice at the endpoint, treated with or without PARPi (n = 8 for E0771 + Ø, n = 9 for E0771 + PARPi, n = 30 for both PyMT-N and PyMT-M models; mean of three sections per mouse, ≥3 fields per section). Magnification ×20. Scale bar 200 µm. Quantification of Cleaved-Caspase 3 was reported as a percentage of the total cells within the tumor tissues. Data are represented as mean ± SEM. Statistical analyses (Unpaired Student t test). Source data are provided as a Source data file.
To investigate whether the PreNeu secretome regulates the transcription of PARP-1, we tested levels of PARP-1 in MCF-7 cells silenced for the succinate receptor (siSUCNR1-MCF-7 cells), demonstrating decreased PARP-1 protein level when the cells were challenged with the PreNeu secretome (Fig. 4b) or succinate (Fig. 4c) compared to scrambled MCF-7 cells. Interestingly, PARP-1 is transcriptionally upregulated by the PreNeu secretome or succinate (Fig. 4d, e). Altogether, these data demonstrate that succinate produced by PreNeu induces PARP-1 transcription in tumor cells.
PARP inhibitors (PARPi) are licensed for use in cancer patients harboring BRCA germline mutations45. We hypothesized that tumor-associated PreNeu, by blocking the homologous recombination pathway, may sensitize breast tumor cells to PARPi. We treated MCF-7 cells, pre-conditioned with the PreNeu secretome, with PARPi and observed reduced cellular proliferation (Fig. 4f), the acquisition of apoptotic marker Annexin V (Fig. 4g) and decreased expression of NHEJ DNA repair proteins (Supplementary Fig. 9a). To further validate our results in vivo, we treated E0771, PyMT-N, and PyMT-M allografts with PARPi (Fig. 4h–j). Only E0771 and PyMT-N tumors, enriched in PreNeu (Supplementary Fig. 4b, c; Supplementary Fig. 5a), were sensitive to PARPi, as defined by a reduction in tumor volume (Fig. 4h, i) and higher expression of cleaved caspase 3 (Fig. 4k) upon treatment. To corroborate the role of PreNeu in promoting sensitivity to PARPi in breast cancer cells, we pre-treated PyMT-N and PyMT-M cells for 48 hours with or without the cm-PreNeu and PARPi before tumor cell injection in C57/BL6 mice and assessed tumor growth over time (Supplementary Fig. 9b). In line with our previous results, the pre-treatment of PyMT-N or PyMT-M cells with the cm-PreNeu conferred sensitivity to PARPi (Supplementary Fig. 9c).
Altogether, these data demonstrate that following homologous recombination pathway inhibition induced by PreNeu, breast tumor cells become dependent on error-prone DNA repair mechanisms and are sensitized to PARPi. Our results uncover an unexpected neutrophil feature as a cellular mediator of synthetic lethality.
Olaparib is effective in tumors with PreNeu-induced homologous recombination deficiency
To evaluate the therapeutic relevance of our findings, we next assessed whether olaparib, a safe and widely used PARPi46, could synergize with endocrine therapies used in the treatment of ER+ breast cancer, including tamoxifen (a selective estrogen receptor modulator) and fulvestrant (a selective estrogen receptor degrader)34,47. As expected16, PreNeu promoted tumor progression and decreased the efficacy of endocrine therapy in vitro and in vivo (Supplementary Fig. 9d–f; Fig. 5a–c). Interestingly, olaparib reverted the pro-tumoral activity induced by PreNeu and synergized with tamoxifen, decreasing tumor volume (coefficient of drug interaction = 0.59) and proliferation (Fig. 5b, c).

a Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/uueypah. Female NRG mice were implanted with estrogen pellets 2 weeks before of the injection of 1 × 106 FP-MCF7-Luc+ cells. Starting from this point, every week mice received cbPreNeu (i.p.) and the radiance of the tumors was monitored by luminescence. When the total flux of the tumor was >106, mice were treated as indicated: untreated (Ø, n = 5); Tamoxifen (n = 5), Olaparib (n = 5), neutrophil precursors (PreNeu; n = 9); PreNeu + tamoxifen (n = 6); PreNeu + Olaparib (n = 14); PreNeu + Olaparib + Tamoxifen (n = 16). After 12 weeks of treatment, mice were sacrificed. b Graph showing tumor area (mm2) of mice belonging to the different groups: untreated (Ø, n = 5); Tamoxifen (n = 5), Olaparib (n = 5), PreNeu (n = 9); PreNeu + tamoxifen (n = 6); PreNeu + Olaparib (n = 14); PreNeu + Olaparib + Tamoxifen (n = 16). Synergism was calculated with the coefficient of drug interaction (CDI). CDI (cm-PreNeu + Olaparib + Tamoxifen) = 0.59. Statistical analyses (two-tailed unpaired Student t test). c Quantifications and representative images of immunohistochemistry analysis (H&E, Ki67, γH2AX) of mice treated in the different conditions listed in a. Aggregated data from one tumor per mouse, mean of three sections per mouse, ≥3 fields per section. Statistical analyses (unpaired Student t test). d Experimental scheme created in BioRender. Garda, c. (2025) https://BioRender.com/6n3mu0w. Female NRG mice were injected with either HCC1937 BRCAwt or HCC1937 BRCAmut cells in the mammary fat pad. Starting from when tumors were palpable, mice were injected either with olaparib or PreNeu or PreNeu + olaparib or vehicle (Ø). e, f Tumor growth from mice injected either with e HCC1937 BRCAmut or f HCC1937 BRCAwt and treated as described in d (n = 5 mice for each group). Data are represented as mean ± SEM. Statistical analysis (two-way ANOVA). g, h Quantifications and representative images of immunohistochemistry analysis (H&E, Ki67, γH2AX) of mice injected either with g HCC1937 BRCAmut or h HCC1937 BRCAwt and treated as described in d. g Ki67: n = 10 for each groups, for γH2AX n = 11 for PreNeu and Olaparib groups, n = 10 for Ø and PreNeu + Olaparib. f Ki67: n = 12 for PreNeu, n = 8 for Ø and PreNeu + Olaparib, n = 10 for Olaparib. γH2AX: n = 10 for PreNeu and Ø, n = 9 for Olaparib, n = 8 for PreNeu+Olaparib. Mean of three sections per mouse, ≥3 fields per section. Statistical analyses (one-way ANOVA). Source data are provided as a Source data file.
Since olaparib is approved for treating cancers with BRCA mutations, we sought to enhance the clinical relevance of our findings by testing its effects in the HCC1937 triple-negative breast cancer xenograft model, which harbors a BRCA1 germline mutation (HCC1937 BRCAmut), under experimental administration of PreNeu (Fig. 5d). Interestingly, the efficacy of olaparib was further enhanced in the presence of PreNeu (Fig. 5e, f). These results suggest that in BRCA-mutated tumors, PreNeu may improve the therapeutic effect of olaparib. However, further studies are needed to investigate the presence of this immune cell subset in the TME of BRCA-mutated cancers and its role in modulating olaparib responses in patients affected by BRCA-mutated cancers. Additionally, we validated the therapeutic relevance of our findings by testing olaparib in the BRCA wild-type variant of the HCC1937 xenograft model (HCC1937 BRCAwt), observing an effect only when PreNeu was administered (Fig. 5g, h).
Taken together, these data demonstrate that olaparib treatment can be effective not only in tumors that harbor genetic homologous recombination deficiency per se but also in tumor contexts in which the homologous recombination is altered by the presence of tumor-associated PreNeu.
Discussion
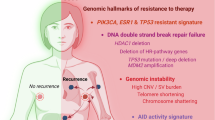
In this study, we described a population of tumor-associated PreNeu as a pro-tumoral subset of neutrophils enriched in highly proliferative ER+ breast cancers. This population can proliferate in the TME and secretes high levels of succinate, an oncometabolite that inhibits high-fidelity homologous recombination DNA repair in tumor cells, resulting in an increased reliance on PARP-1 and error-prone DNA repair mechanisms, leading to increased genomic instability (Supplementary Fig. 10).
During cancer progression, multiple studies have shown that the tumor secretome influences intra-tumoral neutrophil heterogeneity by promoting emergency granulopoiesis30. Recent publications demonstrated that tumor-infiltrating neutrophils may be present in predetermined lineage states but are also incredibly plastic, contributing to multiple neutrophil subsets within the TME15,36,48.
We have observed that neutrophil precursors can infiltrate tumors, where they appear capable of proliferating within the TME and potentially maturing into pro-tumoral neutrophil subsets previously identified in other tumor contexts14,15,16,17. However, this phenomenon is not formally demonstrated in our study, underscoring the need for further research to conclusively establish these dynamics. Our findings open opportunities for investigating the heterogeneity of tumor-associated neutrophils and their functional implications.
Our data sheds light on the heterogeneity of tumor-infiltrating neutrophils and adds to our knowledge of the role of immune cells in cancer editing and progression. Previous data demonstrated that myeloid cells induce mutations in epithelial cells through the production of reactive oxygen species44. We describe a mechanism of tumor editing, wherein PreNeu appears to promote the generation of more aggressive tumor subclones by promoting cancer cell genomic instability.
Nowadays, targeting neutrophils represents a promising anticancer therapy. Our laboratory and others have previously shown that CXCR2 inhibitors prevent the recruitment of immunosuppressive neutrophils in cancers, limiting tumor progression16,17,38,49. Pharmacological inhibition of CXCR2 signaling was largely explored and combined with checkpoint blockers in pancreatic cancer38 and prostate cancer16 or proposed for the treatment of castration-resistant prostate cancer patients in combination with second-line androgen deprivation therapy50. Interestingly, tumor-associated PreNeu infiltrating highly proliferative ER+ breast cancer also express CXCR2 and can be targeted by the CXCR2 inhibitors currently in the clinic.
However, recent insights into neutrophil heterogeneity within the TME have revealed not only the presence of pro-tumorigenic neutrophils with a traditionally short lifespan but also the ability of the TME to extend their survival, enabling these cells to remain functionally active within the tumor15,17. This discovery poses significant challenges for therapeutic strategies, suggesting that simply blocking neutrophil recruitment may be insufficient14.
Our work demonstrates an additional capacity of PreNeu within breast tumors, which supports the previous assertion that CXCR2 inhibition is not enough to combat the neutrophil-related pro-tumoral role. Considering these findings, we propose an alternative strategy: rather than targeting these neutrophils for removal, we aim to harness their functions to sensitize tumor cells to clinically approved cancer therapies. Specifically, our data show that tumor-associated PreNeu induces a dependency in tumor cells on PARP for DNA damage repair, thereby enhancing their sensitivity to PARP inhibitors.
PARP inhibitors, including olaparib, represent groundbreaking therapies approved for the treatment of cancers characterized by germline homologous recombination deficiency. Our findings highlight an additional interplay between tumor-infiltrating immune cells and targeted cancer therapies. The identified mechanism of action played by PreNeu in breast cancer paves the way for the use of PARP inhibitors not only in patients with BRCA-mutated tumors but also in tumors infiltrated by PreNeu, exploiting neutrophils as cellular mediators of synthetic lethality. Additionally, our preclinical work shows that the established PARP-1 inhibitor olaparib synergizes with endocrine therapy in highly proliferative ER+ breast cancers, which are infiltrated by Pre-Neu. While this study lays a compelling foundation and opens avenues for therapeutic innovation, further comprehensive research is crucial to validate these findings and explore their clinical implications in patients. To our knowledge, no current clinical trials are testing PARP inhibitors in tumor contexts without germline homologous recombination deficiencies. This limitation restricts our ability to directly validate our findings in the human setting but highlights the potential for novel clinical trial designs involving PARP inhibitors in broader tumor contexts.
To further investigate the impact of PreNeu and PARP inhibitors in a clinically relevant setting, we evaluated their efficacy using the HCC1937 triple-negative breast cancer xenograft model, which carries a BRCA1 germline mutation, with or without experimental administration of PreNeu. Notably, our results revealed that the therapeutic effect of olaparib was significantly enhanced in the presence of PreNeu. These findings raise the intriguing possibility that PreNeu may potentiate the efficacy of olaparib in BRCA-mutated tumors. Nevertheless, further investigations are required to determine whether this immune cell subset is present in the TME of BRCA-mutated cancers and to elucidate its potential role in modulating olaparib responses in affected patients.
Methods
Statement of ethics
This study complies with all relevant ethical regulations. The umbilical CB was acquired from an anonymous group of pregnant females at Ente Ospedaliero Cantonale di Lugano (Switzerland) who had given their written informed consent to the institutional protocol approved by the Canton Ticino Ethics Committee (reference no. 2020-00373 CE 3602). For breast cancer patients and healthy donors undergoing breast reduction at Ente Ospedaliero Cantonale di Lugano (Switzerland), only females were included in this study following a written informed consent to the institutional protocol approved by the Canton Ticino Ethics Committee (reference no. 2021-00468 CE 3830). Patients were not chosen based on race or ethnicity, and no data regarding these factors were collected. The clinical characteristics of patients and healthy donors are indicated in Supplementary Data 1.
All mice experiments were performed according to state guidelines and approved by the ethics committee “Dipartimento della Sanitá e Socialitá, Esperimenti su animali”, (authorization numbers 34618 and 35145).
Cell lines
MCF-7 cells (obtained from ATCC) were maintained in 75 cm2 flasks in EMEM medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine and 0.25% human-recombinant insulin. PyMT-N and PyMT-M cells (received from the lab of Dr. Xiang H-F Zhang, Baylor College of Medicine) were maintained in 75 cm2 flasks in DMEM medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 2 mM L-glutamine. E0771 cells (obtained from ATCC) were maintained in 75 cm2 flasks in RPMI medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine and 20 mM HEPES (Cat. 15630-080; Thermo Fisher Scientific). HCC1937 BCRAwt and HCC1937 BCRAmut (received from Damia Lab, Mario Negri Institute) were maintained in RPMI medium supplemented with 10% heat inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin. Passaging of the cell lines was performed according to their proliferation frequency. LNCaP cells (obtained from ATCC) were maintained in 75 cm2 flasks in RPMI medium supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin.
Animals
All mice were maintained under specific pathogen-free conditions in the specific pathogen-free (SPF) facility. The mice were housed in individually ventilated cages under standardized conditions (20 ± 2 °C, 55 ± 8% relative humidity, 12 h light/dark cycle) with cage enrichment. Water and food (Kaliba Nafag; Cat. 3432 Maintenance) were provided ad libitum. Only female mice were used in this study. C57BL/6 N (JAX:005304) or NOD-Rag1−/− Il2rγ−/− (NRG; JAX:007799) mice 6 weeks of age were purchased from Charles River Laboratories and acclimated for at least a week before use. Surgical β-estradiol pellet implantation at the neck of immunodeficient ten-week-old NRG mice was performed under anesthesia with isoflurane. Mice were monitored postoperatively for recovery from anesthesia and checked daily with the administration of Bepanthen antiseptic cream (Bayer). Surgical skin tapes were used to close the wound. Mice undergoing treatment were administered control vehicle or therapeutic doses of the appropriate agents. Any mouse suffering distress or greater than 15% weight loss during treatment was euthanized by CO2 asphyxiation. After the study, mice were euthanized by CO2 asphyxiation, and tissue was collected for histology, transcriptomic analysis, genomic analysis, metabolomics, protein analysis, and single-cell suspensions for flow cytometry. For xenograft and allograft experiments, 5 × 105 and 1 × 106 MCF-7 cells, 5 × 105 PyMT-N/PyMT-M cells, or 2.5 × 105 E0771 cells were engrafted to the mammary fat pad of the 4th inguinal mammary gland of six-weeks-old C57BL6/N mice and 3.5 × 106 HCC1937 BRCAwt, or HCC1937 BRCAmut cells were engrafted to the mammary fat pad of the 4th inguinal mammary gland of ten weeks old NRG mice. For allografts, when tumors were palpable at approximately 40–50 mm3, mice were randomized to the treatment groups. Tumor growth was monitored thrice a week by measuring the tumor size with a caliper. The tumor volume was estimated by calculating R1*R2*R3*4/3π, where R1 and R2 are the longitudinal and lateral radii, and R3 is the thickness of the tumor protruding from the surface of normal skin. Animals were sacrificed when the tumor reached approximately 800 mm3. For xenograft experiments, when bioluminescence from tumors reached ~≥106 total flux (p/s/cm²/sr), animals were randomly assigned to different groups (n ≥ 5). Tumor growth was monitored by IVIS technology, and the growth curve was analyzed using GraphPad Prism software 9.3.1.
Human samples
For breast cancer patients, only females were included, with an average age of 31 years for healthy participants, 67.8 years for low proliferative breast cancer patients, and 71.3 years for highly proliferative breast cancer patients. Statistical analysis: one-way ANOVA: p < 0.0001 for age of healthy vs low proliferative and p < 0.0001 for healthy vs highly proliferative.
In vivo treatments
Before treatment, mice were randomized for the treatments. αCXCR2 (AZD5069; AstraZeneca) was administered with daily intraperitoneal injections at a final concentration of 100 mg/kg on a Monday through Friday schedule. Olaparib (Cat. orb61072; Biorbyt) was administered with daily intraperitoneal injections at a final concentration of 50 mg/kg on a Monday through Friday schedule. Anti-mouse PD-L1 (B7-H1) (Cat. BE0101; Bio X Cell) was administered through intraperitoneal injections three times a week at a final concentration of 200 µg/mouse on a Monday, Wednesday, and Friday schedule. Tamoxifen (Cat. 10540-29-1; MedChemExpress) was subcutaneously administered three times a week at a final concentration of 1 mg/mouse on a Monday, Wednesday, and Friday schedule. Control animals received vehicles. Treatment synergism for the experiment shown in Fig. 5b was calculated with the CDI, which indicates a synergistic treatment when CDI < 1.
Immune TME characterization of tumors from patients with breast cancer
Fresh biopsies were analyzed using 28-parameter flow cytometry analyses following the protocol below.
Tumor processing: tumors were disaggregated and digested in collagenase I (1 mg/ml; Cat. 11088858001, Roche) and DNase I (100 μ/ml; Cat. 11284932001, Roche,) for 30 min at 37 °C to obtain single-cell suspension. Single-cell suspension was filtered on a 40µm cell strainer (Cat. 542040, Greiner bio-one), washed with medium containing 10% of FBS, and then stained with specific monoclonal antibodies (primary antibodies directly conjugated) to assess the phenotype or to perform single-cell sorting. Samples were acquired from patients who had given their written informed consent to the institutional protocol approved by the Canton Ticino Ethics Committee (reference no. 2021-00468 CE 3830). Human biological samples were sourced ethically, and their research use was in accordance with the terms of the informed consent provided.
High-dimensional single-cell analysis by flow cytometry: single cell suspensions were incubated with Viability Stain 440UV (Cat. 566332; 1:1000; BD Biosciences) for 10 min at RT to exclude dead cells. Then, washed twice and incubated with FcR blocker purified anti-Human BD Fc Block (Clone Fc1.3216; Cat. 564219; 1:100; BD) for 10 min at 4 °C to block fragment crystallizable (Fc) receptors. Anti-human antibodies were added for 30 min at 37 °C: anti human-CXCR3 PE-Cy5 (Clone 1C6/CXCR3; Cat. 551128, Lot. 1116206; 1:25; BD Biosciences); anti-human CXCR4 BV605 (Clone 12G5; Cat. 306521, Lot. B388999; 1:25, Biolegend); anti-human CXCR2 BV711 (Clone 6C6; Cat. 744199, Lot. 2280642; 1:25, BD Biosciences). Then, after washing using Stain Buffer (Cat 554656; BD Biosciences) the following anti-human antibodies were added for 15 min at 4 °C: anti human-CD45 BUV805 (Clone HI30; Cat. 612892, Lot. 4015948;1:40 BD), anti-human LOX-1 PE (Clone 154 C; Cat. 358604, Lot. B397309; 1:100, Biolegend), anti-human CD15 BV750 (Clone W6D3; Cat. 747426, Lot. 3272135; 1:20, BD), anti-human CD33 BB630- P (Clone WM53; Custom: Cat. 624294; Lot.4012502; 1:80, BD), anti-human CD16 BUV615 (Clone 3G8; Cat. 751572, Lot. 5058361; 1:40, BD), anti-human CD66b BB515 (Clone G10F5; Cat. 564679, Lot. 3156279; 1:100, BD), anti-human CD11b APC (Clone ICRF44; Cat. 561015; 1:25, BD), anti-human HLA-DR APC-R700 (Clone G46-6; Cat. 560743, Lot. 2355906; 1:10, BD), anti-human CD71 BV421 (Clone CY1G4; Cat. 334121, Lot. B353579; 1:100, Biolegend), anti-human CD45RA BV480 (Clone HI100; Cat. 566114, Lot. 2174095; 1:20, BD), anti-human CD117 BV510 (Clone 104D2; Cat. 313220, Lot. B365771; 1:100, Biolegend), anti-human CD226 BV786 (Clone11A8; Cat. 338321; Lot. B327941; 1:16; Biolegend), anti-human PD-L1 PE-Cy7 (Clone MIH3; Cat. 374505; 1:16; Biolegend), anti-human NOTCH2 BV605 (Clone MHN3-25; Cat. 742291; 1:100; BD), anti-human IDO1 APC-R700 (Clone V50-1886; Cat.568017;Lot. 2063442; 1:100; BD), anti-human CD38 BV661 (Clone HIT2; Cat.612969; 1:130; BD).
Then, acquired on a FACS-Symphony A5 flow cytometer (BD Biosciences) equipped with five lasers (UV, 350 nm; violet, 405 nm; blue, 488 nm; yellow/green, 561 nm; red, 640 nm) and capable of detecting 30 parameters.
Computational analysis of flow cytometry data: data were processed as previously described51. Briefly, flow cytometry standard (FCS) 3.1 files were first imported in FlowJo version 10.8 (FlowJo software; Treestar Inc.) to eliminate dead cells by manual gating, and select CD45+ leukocytes, subjected to biexponential transformation, then exported for computational analysis by a custom-made script making use of PhenoGraph (K value set at 30). Here, we modified the Linux-community and the core.py script to fix the seed to “123456” (run in Python version 3.7.3). A unique computational barcode was adopted to label Luminal A and Luminal B samples for subsequent identification. Data were then converted in comma separated (CSV) files and merged into a single file by using the pandas package (v.2.2.3). The obtained data, exported as new CSV file (one for each cluster), were further imported in FlowJo and analyzed to define the percentage of cells positive for each protein as well as their median fluorescent intensity. Data were finally meta-clustered using the gplots R package (v.3.2.0). UMAP was obtained by UMAP Python package (v0.5.7).
High-dimensional single-cell sorting: single cell suspension from fresh biopsies, or CB-derived PreNeu were incubated with FcR blocker purified anti-human BD Fc Block™ (Clone Fc1.3216; Cat. 564219, 1:100, BD) for 10 min at 4 °C to block fragment crystallizable (Fc) receptors, then stained with the following anti-human antibodies: anti-human CD45 BV605 (Clone HI30; Cat. 304042; Lot. B382461; 1:10, Biolegend), anti-human CD11b BV421 (Clone M1/70; Cat.101251; Lot. B394262; 1:25, Biolegend), anti-human CD33 APC (Clone WM53; Cat. 303408; Lot. B2778438; 1:80, Biolegend), anti-human CD15 V500 (Clone HI98; Cat. 561585; Lot. 0027080; 1:20, Biolegend), anti-human CD163 PE-Cy7 (Clone GHI/61; Cat. 333614; Lot. B289916; 1:100, Biolegend), anti-human LOX-1 PE (Clone 154 C; Cat. 358604, Lot. B397309; 1:100, Biolegend), anti-human CD66b BB515 (Clone G10F5; Cat. 564679, Lot. 3156279; 1:100, BD).
Then, after washing with PBS pH 7.2, 0.5% bovine serum albumin (BSA) (A9418, Sigma-Aldrich), and 2 mM EDTA (AM9260G, Thermo Fischer), samples were filtered on a 40 µm cell strainer and sorted to over 98% purity on FACSAria III (BD)after sequential exclusion of doublet and dead cells (7AAD+) using 7AAD viability staining solution; Cat. 00-6993-42; Thermo Fisher Scientific). Within CD45+ CD163− CD33+ CD15+ CD66b+cells, PreNeu and LOX-1+ Neutrophils were selected based on the expression of Lox-1 and negativity or positivity of CD11b, respectively.
Rhapsody single-cell data generation, processing and analysis
Sample processing
Fresh biopsy single-cell suspension was enriched for CD45+ cells using the CD45 MicroBeads, human (Cat. 130-045-801; Miltenyi) following the manufacturer’s protocol. Cells were single cell captured for sequencing using the Rhapsody HT Single-Cell Analysis system and library was generated using the BD Rhapsody WTA Amplification Kit (Cat. 633801, BD Biosciences) with 8 cycles of amplifications, and sequenced using an Illumina NextSeq2000 instrument with a P2 flow cells and chemistry XLEAP obtaining around 8000 reads/cell.
Computational analysis
Tumor sample FASTQ files were aligned, and feature-barcode matrices were generated using the BD Rhapsody™ Sequence Analysis Pipeline on the Seven Bridges Genomics platform, with the GRCh38 genome assembly as the reference. The resulting data was analyzed using Seurat (v.5.1.0)52,53,54 in R (v.4.4.2) [R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL: https://www.R-project.org/]. Quality control measures were applied to each dataset to remove low-quality cells and multiplets. Metrics such as cell counts, UMI counts per cell, genes detected per cell, mitochondrial gene count ratio, and ribosomal gene count ratio were inspected. Data normalization and scaling were performed, regressing out potential confounding factors (UMI counts per cell, genes detected per cell, mitochondrial gene count ratio, and ribosomal gene count ratio). A total of 30 principal components (PCs) was selected for UMAP, which was used for dimensionality reduction and cell clustering. Clustering at a resolution of 0.6 yielded 20 clusters. Marker gene identification utilized a hurdle model designed for scRNA-seq data, implemented in the MAST statistical framework55, with Bonferroni-adjusted p values to correct for multiple testing. Markers were considered significant if they were expressed in at least 70% of cells in a cluster, had an adjusted p value (pval_adj <0.05), and displayed a log2 fold change (log2FC > 1). Major cellular populations were annotated based on marker genes, and the dataset was subsetted to retain only bona fide neutrophils. The subsetted cells underwent further graph-based clustering at a resolution of 0.6, resulting in three clusters. Marker-based annotation was applied for in-depth characterization. Transcriptomic data underwent zero-preserving imputation using the ALRA method56. The Seurat AddModuleScore function was employed to compute and evaluate the enrichment of neutrophil-related signatures. Pathway enrichment analysis was conducted using Metascape57, considering pathways with a Benjamini-Hochberg-adjusted p value < 0.05, involving at least three differentially expressed genes (DEGs), and a minimum enrichment score of 1.5. Finally, trajectory analysis was performed using a custom script based on Monocle358 (v.1.2.7). Cell spatial coordinates were imported from the Seurat object to create the CellDataSet object required by Monocle. Trajectories were constructed using the learn_graph function, and cells were ordered along pseudotime.
Single-cell SMART sequencing (SMART-Seq2)
Sample processing
Fresh biopsy single-cell suspension or CB-derived PreNeu were sorted as described above into 96-well PCR plates containing cell lysis buffer. Samples collected in cell lysis buffer were used for RNA-seq library preparation with the NEBNext Single Cell/Low InputRNA Library Prep Kit for Illumina (NEB, E6420S), following the manufacturer’s protocol for single cells, with the following parameters adjusted: 17 cycles for cDNA amplification PCR, 11 cycles for library enrichment PCR. Libraries were dual-indexed (NEBNext Dual Index Primers Set 1, NEBE7600S), and sequenced on an Illumina NextSeq 500 instrument with 75 cycles reagents. Quality controls and read mapping to the reference genome were performed using the same criteria described for bulk RNA-seq, except that reads having the same start/end coordinates and identical nucleotide sequence were marked and deduplicated to avoid excessive bias due to PCR amplification. Differential expression was performed in R statistical environment using DESeq2 pipeline (v1.28.1). Cells with less than 500 K mapped reads were removed from the analysis. Library-size normalized data were transformed using the variance stabilizing transformation, and the batch effect between tumor-sorted PreNeu or LOX-1+ Neutrophils and CB-derived PreNeu or LOX-1+ Neutrophils was corrected using Combat-Seq. Cell types of origin (PreNeu/PMN-MDSC), which were independently identified in both conditions, were set as covariates to preserve the biological signal.
Computational analysis
Marker genes specific for PreNeu were identified from the single-cell data. We selected all genes that were differentially expressed in PreNeu cells vs LOX-1+ neutrophils (FDR < 0.05). To identify genes being robustly expressed in this setting and reduce the possibility of selecting significant genes expressed at low levels, we restricted the analysis to features according to their mean expression levels (basemean > 50) and then focused on genes showing selective upregulation in PreNeu (log2FoldChange > 1). Filtered elements were used to generate a protein−protein interaction network through String Database. We determined marker genes by identifying a main subnetwork showing a higher degree of connectivity between nodes. We then selected 11 genes within this cluster based on their biological function related to the regulation of immune system processes.
Generation of cord-blood derived PreNeu or LOX-1+ neutrophils
Human cord-blood derived mononuclear cells were collected from umbilical CB by centrifuging the blood with Histopaque−1077 (Cat. 10771; Sigma) at 400 × g for 30 mins followed by washing with PBS twice. CB-derived PreNeu or LOX-1+ neutrophils were differentiated in vitro by seeding 1.5 × 106 ml of cord-blood-derived mononuclear cells with RPMI 1640 containing 10% heat-inactivated FBS in the presence of tumor cell medium (TCM). TCM was prepared, collected the MCF-7 condition medium from a T75 confluent flask, filtered, and centrifuged at 2000 × g using a vivaspin 20-centrifugal concentrator (Cat. Z629464-48EA; Sigma). After 4–5 weeks, the cells were incubated with FcR blocker purified anti-human BD Fc Block (Cat. 564219, Lot. 3299107; 1:20, BD). Then, anti-human antibodies were added for 30 min at 37 °C: anti-human CXCR4 BV605 (Clone 12G5; Cat. 306521, Lot. B388999; 1:25, Biolegend), anti-human CXCR2 BV711 (Clone 6C6; Cat. 744199, Lot. 2280642; 1:25, BD Biosciences). Then, after washing using Stain Buffer (Cat 554656; BD Biosciences) the following anti-human antibodies were added for 15 min at 4 °C: anti-human LOX-1 PE (Clone 154 C; Cat. 358604, Lot. B397309; 1:100, Biolegend), anti-human CD15 BV750 (Clone W6D3; Cat. 747426, Lot. 3272135; 1:20, BD), anti-human CD33 BB630- P (Clone WM53; Custom: Cat. 624294; Lot.4012502; 1:80, BD), anti-human CD16 BUV615 (Clone 3G8; Cat. 751572, Lot. 5058361; 1:40, BD), anti-human CD66b BB515 (Clone G10F5; Cat. 564679, Lot. 3156279; 1:100, BD), anti-human CD11b APC (Clone ICRF44; Cat. 561015; 1:25, BD), anti-human HLA-DR APC-R700 (Clone G46-6; Cat. 560743, Lot. 2355906; 1:10, BD), anti-human CD71 BV421 (Clone CY1G4; Cat. 334121, Lot. B353579; 1:100, Biolegend), anti-human CD45RA BV480 (Clone HI100; Cat. 566114, Lot. 2174095; 1:20, BD), anti-human CD117 BV510 (Clone 104D2; Cat. 313220, Lot. B365771; 1:100, Biolegend). Dead cells were excluded using BD Horizon™ Fixable Viability Stain 440UV (Cat. 566332, 1:1000, BD) or 7AAD (Cat. 559925, 1:100, BD). Then, acquired on a FACS-Symphony A5 flow cytometer (BD Biosciences) equipped with five lasers (UV, 350 nm; violet, 405 nm; blue, 488 nm; yellow/green, 561 nm; red, 640 nm) and capable to detect 30 parameters, and analyzed using FlowJo software (Treestar Inc.). When the cultured cells acquired more than 98% of positivity for CD33, they were sorted as described above.
Cord-blood-derived PreNeu or LOX-1+ neutrophils conditioned medium preparation
The sorted cord-blood derived PreNeu or LOX-1+ neutrophils were resuspended in 1/3rd of their old culture medium and 2/3rd of freshly prepared RPMI 1640 containing 10% heat-inactivated Charcoal Stripped FBS (CS-FBS). Then, stimulated with PMA/ionomycin for 5 hours. The supernatant was then collected, centrifuged at 300 × g for 5 minutes, and directly used or stored at −80 °C.
Differentiation of in vitro mouse neutrophils
Mouse neutrophils were differentiated in vitro as previously described30. Briefly, bone marrow precursors were flushed from the femurs of C57/BL6 mice with RPMI 1640 medium. The cell pellet was resuspended (one femur in 10 ml) in RPMI 1640 containing 10% heat-inactivated charcoal stripped FBS (CS-FBS, Cat. 12676029, Thermo Fisher Scientific), and the cells were cultured in vitro in the presence of 40 ng/ml GM-CSF (Cat. 216-16, Peprotech) and 40 ng/ml IL-6 (Cat. 315-03, Peprotech). After 3 days, the cells were stained following the myeloid panel protocol described above. To impair succinate release by PreNeu, we generated bmPreNeu by differentiating cells in the presence of MCT1 inhibitor AZD3965 (10 μM, Cat. S7339; Selleckchem) for the entire time.
Bone marrow-derived PreNeu conditioned medium preparation
Bone marrow-derived neutrophils were generated as reported above and resuspended in 1/3rd of their old culture medium and 2/3rd of freshly prepared RPMI 1640 containing 10% heat-inactivated Charcoal Stripped FBS (CS-FBS, Cat. 12676029, Thermo Fisher Scientific). Then, stimulated with PMA/ionomycin for 5 hours. The supernatant was then collected, centrifuged at 300 × g for 5 minutes, and directly used or stored at −80 °C. To impede the succinate release by PreNeu in the conditioned medium, bmPreNeu were generated and stimulated in the presence of MCT1 inhibitor AZD3965 (10 μM, Cat. S7339; Selleckchem).
PyMT-N pre-treatment for in vivo injection
PyMT-N cells were seeded in vitro, then after 24 hours media was replaced with fresh media containing 50% of conditioned media and after 48 hours from the seeding olaparib 1 μM (Cat. Orb61072; BioRyb) or control was added to the culture media. Finally, 72 hours from the seeding cancer cells were harvested and injected 0.5 × 106 cells/mouse in the FP.
In vitro T-cell suppression assay
Human cord-blood-derived mononuclear cells were stained with CFSE (Cat. C34570; Invitrogen) according to the manufacturer’s protocol. Then they were activated by anti-human T-Activator CD3/CD28 Dynabeads (Cat. 11161D; ThermoFisher Scientific) and seeded (8 × 104 cells per well of 96-well plate) either alone or in autologous co-culture with cbPreNeu, cbLOX-1+ neutrophils, cbPreNeu, or cord-blood purified CD11b+ CD33+ cells at different ratios. Not activated cord-blood-derived mononuclear cells were used as the negative control. Co-culture was performed for four days, and CFSE dye dilution represents T-cell division. Culture media contained RPMI 1640 + 10% heat-inactivated FBS (Cat. A5670701; Gibco), 1% P/S.
Single-cell 10x data processing and analysis
Neutrophils scRNA-seq raw data from healthy human bone marrow (BM, 2 samples) in FASTQ format were obtained from ArrayExpress under accession no. E-MTAB-1118828. Per sample alignment and generation of feature-barcode matrices were carried out using STARsolo [10.1101/2021.05.05.442755], against GRCh38 genome assembly. Subsequently, all single-cell data were analyzed using Seurat (v.4.0.3)52,54, in R software (v. 4.2.2) [R Core Team (2020). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/]. Quality control measures were applied to remove low-quality cells and multiplets from each sample. This involved inspecting commonly used metrics such as cell counts, UMI counts per cell, genes detected per cell, mitochondrial counts ratio, and ribosomal counts ratio. Samples were merged and normalized using SCTransform (with glmGamPoi method)59,60. A total of 89 PCs were selected for UMAP, which was then used for dimensionality reduction and cell clustering. Clustering at resolution 0.3 was chosen, resulting in 10 clusters. Zero-preserving imputation of transcriptomic data was applied through the ALRA method56. Seurat AddModuleScore function was employed to compute and evaluate the enrichment of the neutrophil precursors signature based on the expression of specific markers identified in the original publication for these cells. Further analyses focused specifically on cells identified as neutrophil precursors in cluster 4, which was subsetted from the original dataset. A subset of cells among neutrophil precursors resulted in a specific enrichment in our custom PreNeu signature, evaluated using the Seurat AddModuleScore function.
Integration of bone marrow and CB datasets
A single Seurat object was created from the CB raw expression matrix. Counts were normalized using SCTransform (with glmGamPoi method). Cells from CB and BM were integrated following the default canonical correlation analysis-based Seurat workflow. A total of nine PCs were selected to generate the UMAP, which was then used to visualize CB neutrophil distribution over the BM dataset. The Seurat AddModuleScore function was employed to evaluate the enrichment of the neutrophil precursors' signature on the integrated dataset.
Proliferation of CB-derived PreNeu or LOX-1+ neutrophils and mouse bone marrow-derived PreNeu or neutrophils
Differentiated cbPreNeu or cbLOX-1+ neutrophils and bmPreNeu or bmNeutrophils were stained by CFSE (Cat. C34570; Invitrogen) according to the manufacturer’s protocol. Then, they were cultured for 6 days, and the proliferation was assessed at days 3 and 6 by measuring CFSE dye dilution.
Proliferation assay
MCF-7 cells were plated in a 96-well plate in RPMI medium supplemented with 10% heat-inactivated CS-FBS, for 24 h. The day after, cells were treated with cm-PreNeu or cm-LOX1+ Neutrophils. Cell proliferation was assessed using IncuCyte (Sartorius) following the manufacturer’s instructions.
ROS detection assay
Oxidation-sensitive dye dichlorodihydrofluorescein diacetate (DCFDA, Cat. C6827; Thermo Fisher Scientific), was used to measure ROS production by the reported immune cells. Cells were incubated at 37 °C in prewarmed PBS in the presence of 2.5 µM DCFDA for 30 min, then washed with PBS and analyzed by flow cytometry.
MCF-7 co-culture experiments
MCF-7 cells were starved in CS-FBS medium for 12 h and then cultured with RPMI 1640 containing 10% heat-inactivated CS-FBS or in the presence of the cbPreNeu or cbPMN-MDSC conditioned medium (ratio 1:1) and/or olaparib. Then, the cells were used for RNA sequencing or protein extraction or comet assay or tested for apoptosis using IncuCyte (Sartorius) following manufacturer’s instructions.
Bulk RNA sequencing
MCF-7 cells, plated in a six-well plate in EMEM medium supplemented with 10% CS-FBS, were treated with or without cm-cbPreNeu in a triplicate for 72 h. After incubation, cells were collected and counted manually with a cell counter followed by RNA extraction using RNeasy Mini Kit (250) (74106, Qiagen), following the manufacturer’s instructions. Prior to library construction, 10–100 ng total RNA was treated with the NEBNext Poly(A) mRNA Magnetic Isolation Module. Library construction was performed with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina®, according to the manufacturer’s protocol. RNA concentration and integrity was assessed using Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA), all hTSC samples had an RNA integrity number equivalent (RINe) value of >9. Quality of sequencing reads was evaluated using FastQC (v.0.11.9) (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequences were mapped to the GRCh38 assembly of the human genome using STAR (v.2.6.1c)61 Alignments were conducted in 2-pass mode and soft-clipping of reads was allowed. PCR duplicates were marked using Picard Tools (v2.18.7) and quantification of expression at gene level was performed using FeatureCounts (v2.0.3, subread package, ignoreDup). Gene annotations were retrieved from Gencode (gencodegenes.org). Downstream analysis was performed in R using DESeq2 (v1.28.1). Raw counts were normalized according to library size and subsequently transformed using the variance stabilizing transformation (VST). Differential expression was conducted by using the Independent Filtering procedure (independentFiltering = TRUE; alpha = 0.1) embedded in DESeq2 to filter out genes that were not expressed at appreciable levels in our setting. Gene set enrichment analysis was performed using Camera function within the limma R package (v.3.46.0). All gene sets collections were retrieved from the Molecular Signature Database (MsigDB).
COMET assay
The intensity of DNA double-strand breaks in cells, harvested both in vitro and in vivo, as determined by a Comet Assay Kit (ab238544, Abcam), following the manufacturer’s instructions. Pictures were taken using a Nikon Eclipse E800 fluorescence microscope, and the tail moment was calculated using the OPENCOMET plugin for Fiji.
Western blot
Tissue and cell lysates were prepared with RIPA buffer (1× PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor cocktail; Cat. 20-188, Roche). Total protein concentration was measured using Bio-Rad Protein Assay Dye Reagent Concentrate (5000006; Bio-Rad). Equal amounts of proteins were separated by SDS-PAGE and western blotted onto a 0.45 μm PVDF membrane. Membranes were blocked in 5% defatted milk or 5% BSA in PBST (PBS and 0.1% Tween-20 (Cat. P1379; Thermo Fisher Scientific), probed with diluted antibodies, and incubated at 4 °C overnight. The following primary antibodies were utilized: rabbit polyclonal anti-HSP90 (4877 s, 1:1000 dilution, Cell Signaling), mouse monoclonal anti-MRE11(18) (sc135992, 1:500 dilution, Santa Cruz Biotechnology), mouse monoclonal anti-TIP60 (C-7) (sc166323, 1:500 dilution, Santa Cruz Biotechnology), rabbit monoclonal anti-RAD51 (ab133534, 1:10000 dilution, abcam), rabbit monoclonal anti-ATM (D2E2) (2873 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-pATM (Ser1981) (D25E5) (13050 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-BRCA1 (9010 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-pChk1 (Ser345) (133D3) (2348 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-Ku70 (D10A7) (4588 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-Ku80 (C48E7) (2180 s, 1:1000 dilution, Cell signaling), rabbit monoclonal anti-DNA- PKcs (Y393) (ab32566, 1:1000 dilution, Abcam), rabbit monoclonal anti-XRCC4 (Y393) (ab32566, 1:1000 dilution, Abcam), rabbit polyclonal anti-XRCC4 (ab97351, 1:1000 dilution, Abcam), rabbit monoclonal anti- Tri-Methyl-Histone H3(Lys9) (D4W1U) (13969 s, 1:1000 dilution, Cell signaling), rabbit polyclonal anti-SUCNR1/GPR91 (NBP1-00861, 1-2ug/ml dilution, Novus bio), mouse monoclonal anti-β-actin (A2228, 1:5000 dilution, Sigma), rabbit monoclonal anti-PARP (9532S, 1:1000 dilution, Cell signaling). After washing in PBST, the membrane was incubated with a secondary antibody conjugated with horseradish peroxidase (HRP) (dilution 1:5000, Promega). The protein bands were visualized using the ECL Western Blotting Substrate (Pierce).
Immunohistochemistry and IHC
For IHC, tissues were fixed in 10% formalin (Thermo Fisher Scientific, 5701) and embedded in paraffin following standard procedures. Preceding immunohistochemical staining, tumor sections (4 µm) were exposed to two washes with OTTIX plus solution (Cat. X0076, Diapath) and subsequent hydration with OTTIX shaper solution (Cat. X0096, Diapath) followed by deionized water. Antigen unmasking was performed by heating sections in the respective pH solutions based on the antibodies used at 98 °C for 20 minutes. Subsequently, the sections were blocked for peroxidases and nonspecific binding of antibodies using 3% H2O2 (VWR chemicals, 23615.248) and Protein-Block solution (DAKO Agilent technologies, X0909), respectively, for 10 mins each, split by 0.5% PBST washing. H&E staining was performed according to standard procedures. Sections were stained for anti-Ki67 (Clone SP6; Cat. MAD-000310QD; Lot. 03100116S; Lab Vision Corporation), anti-gamma-H2AX (Clone S139, Cat. 9718 s; Lot. 47; 1:200 dilution; Cell Signaling), and anti-Cleaved-Caspase 3 (Polyclonal, Cat. 9661; Lot. 21; 1:2000 dilution; Cell Signaling). Images were obtained using ×20 magnification and a pixel image of 200 µm. All the quantifications have been done using the Aperio Image Scope Pathology Slide Viewing Software (Leica Biosystems). For the IF staining, tissue paraffin-embedded sections were stained for 4′,6- Diamidine-2′-phenylindole dihydrochloride (DAPI) (Cat. 70238421, Roche), anti-γH2AX (Cat. 9718 s; Lot. 47; 1:150, Cell Signaling), anti-RAD51 (Clone 14B4; Cat. NB100-148; 1:200 dilution; Novus bio), P-53BP1S1778 (Cat. 2675S; Lot. 3; Cell Signaling), P-ATMS1981 (Cat. 13050S, Lot. 6; 1:1000, Cell Signaling), TIP60 (Cat. sc166323; 1:500, Santa Cruz Biotechnology). Secondary staining was performed using Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 (A-11012; 1:400 dilution; Thermo Fisher Scientific), and Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (A-21202; 1:400 dilution; Thermo Fisher Scientific). Images were obtained with the Leica Thunder Epifluorescence microscope using ×63 or ×100 oil immersion objective.
Whole-genome sequencing
Single-cell suspensions were obtained from MCF-7 xenograft tumors, treated with or without cbPreNeu, by mincing them in collagenase D (Cat. 11088858001, Sigma-Aldrich). After 30 min incubation at 37 °C with continuous revolving, the single-cell suspension was passed through a 40 µm EASYstrainer (542040, Greiner bio-one) and later purified for EpCaM+ cells using human CD326 (EpCAM) microbeads (Cat. 130-061−101; Miltenyi Biotec), following the manufacturer’s instructions. The purified cells were then used to extract DNA with a DNeasy Blood & Tissue Kit (Cat. 69506; Qiagen), following the manufacturer’s instructions. Whole-Genome Sequencing libraries were prepared from 1 to 5 ng eluted DNA using NEBNext Ultra II DNA library Prep Kit (Cat. E76005, New England Biolabs) with 12 cycles of library amplification and sequenced on an Illumina NextSeq500 instrument with High-throughput 300 cycle reagents (2 × 150 bp format). Quality of sequencing reads was evaluated using FastQC (v.0.11.9). Variant-calling and copy-number profiling were performed according to the GATK best practices (v.4.2.0.0). Fastq files were first converted to unmapped BAMs, and read group information (instrument, flowcell, library) was included. Following reconversion to Fastq, files were aligned to the reference human genome (GRCh38) using bwa aligner (bwa-mem2). Subsequent passages strictly adhered to the GATK workflow (MergeBamAlignment, SetNmAndUqTags, MarkDuplicates, BaseRecalibrator, ApplyBQSR). Standardized Copy Number Ratios were assessed from Base Quality Score Recalibrated (BQSR) BAM files according to GATK best practices (CollectReadCounts, DenoiseReadCounts, ModelSegments, CallCopyRatioSegments).
Mutation, copy number and RNA-seq data for validation were obtained from a recently published cohort of breast cancer cases treated with neoadjuvant chemotherapy33. To classify ER+ tumors as high or low proliferation, we performed single-sample gene set enrichment analysis (ssGSEA) on the Genomic Grade Index (GGI) gene set62 using the RNA sequencing data and the GSVA R package (v1.34). High proliferation tumors were defined as those having a GGI ssGSEA score above the median, and low proliferation tumors as those having a GGI ssGSEA score below the median. The PreNeu score was similarly calculated by performing ssGSEA on the RNA-seq data using a set of genes that we showed to be upregulated in PreNeu (NOTCH2, KIT, CD226, IDO1, CTLA4, CD274, CD69, TFRC, IL7R, STAP1, CD36). Genomic metrics, including the proportion of the genome undergoing copy number alterations and mutational signature 3 exposure, were obtained from the original dataset publication33.
Chromatin immunoprecipitation sequencing (ChIP-seq)
MCF-7 cells were irradiated at 10 Gy for 3 minutes with the irradiator RS1800 (Rad Source Technologies). Then cells were plated in RPMI medium supplemented with 10% charcoal-stripped (CS) heat-inactivated FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin. The day after the plating media was replaced with fresh complete RPMI with CS-FBS and Succinate 2 mM (Cat. S9512; Sigma-Aldrich) or control. After 24 hours, cells were treated with 1% formaldehyde (Cat. F8775, Sigma-Aldrich) for 10 min at room temperature (RT). Ten, the reaction was quenched by adding glycine 0.125 M. After spinning and washing with PBS solution, the cross-linked tissue samples were stored at −80 °C. During ChIP experiments, lysis bufer 1 (50 mM Hepes–KOH, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, ddH2O), supplemented with protease inhibitors (Roche) was added to previously fixed-samples; they were homogenized using an ultra-turrax (VWR). After, samples were washed in lysis buffer 2 (10 mM Tris–HCl, 200 mM NaCl, mM EDTA, 0.5 mM EGTA, ddH2O) and lysis buffer 3 (10 mM Tris– HCl, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-deoxycholate, 0.5% N-Lauroylsarcosine, ddH2O), both supplemented with protease inhibitors (Roche). The obtained chromatin was sheared on a Bioruptor sonicator (Diagenode) set at high potency for 30 pulses, each one comprised of 60s ON and 30s OFF. After checking the chromatin smear, 1% of the sample volume was collected and stored at −20 °C to be next used as input sample, while 2 µg of H3K9Me3 antibody (Cat 13969, Lot. 6; Cell Signaling Technology), previously incubated all day at 4 °C in a rotation wheel with Dynabeads (Cat. 10003D; Thermo Fisher Scientific), was added to the remaining sample volume. After overnight incubation in a rotation wheel at 4 °C, immunoprecipitated samples (IPs) were washed with RIPA wash buffer (50 mM Hepes–KOH, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na-deoxycholate, ddH2O) for six times; after spinning, dynabeads were removed in a magnetic stand, samples were eluted in Elution Buffer (TE 1× and 2% SDS) and incubated overnight at 65 °C to remove crosslinks. The recovered material was purified using QIAquick PCR purification kit (Cat. 28104; Qiagen), and DNA was quantified using Qubit dsDNA High Sensitivity Assay Kit (Cat. Q32851; Thermo Fisher Scientific). As a negative control of the ChIP protocol, an IP reaction against rabbit IgG (Cat. 2729S; Lot. 11, Cell Signaling) was performed. ChIP-Seq libraries were created adapting the TruSeq ChIP protocol (Illumina): around 50 ng of IPs and input were used for library preparation. Libraries were run on a NextSeq 500 sequencer (Illumina) using a 1 × 75bp high-output kit (Illumina) with 8 IPs (50M reads per sample) or 4 input samples (100M reads per sample) for the run.
ChIP-seq data were analyzed using the nf-core/chipseq pipeline (version 2.1.0) implemented in Nextflow (version 24.10.4), with Singularity (1.3.6-1.el8) containers to ensure reproducibility. Raw FASTQ files were quality-checked using FastQC (v0.12.1), followed by adapter trimming with Trim Galore (v0.6.7). Reads were aligned to the GRCh38 genome using BWA (v0.7.18) aligner, and PCR duplicates were marked with Picard. Multiple libraries per sample were merged, with duplicates re-marked, and low-quality or problematic reads (e.g., unmapped, multi-mapped, blacklisted, excessive mismatches) were removed using SAMtools (v.1.20), BEDTools (v2.30.0), and BAMTools (v2.5.2). Normalized coverage tracks were generated with BEDTools (v2.30.0), and IP enrichment relative to control was calculated using deepTools (v.3.5.5). Peaks were called with MACS3 (v3.0.1), annotated using HOMER (v4.11), and merged into a consensus peak set for featureCounts-based quantification. Quality metrics, including library complexity and strand cross-correlation, were determined using Preseq (v3.1.2), Picard (v3.2.0) and phantompeakqualtools (v.1.2.2), and then compiled via MultiQC (v.1.23), while downstream analysis of consensus peak counts was performed in R statistical environment using DESeq2 (v.1.28.0).
Mouse immune TME characterization
Mouse allografts were analyzed using 22-parameter flow cytometry analyses following the protocol below.
Tumor processing
Tumors were disaggregated and digested in collagenase I (1 mg/ml, Cat. 11088858001, Roche) and DNase I (100 μ/ml, Cat. 11284932001, Roche) for 30 min at 37 °C to obtain single-cell suspensions. High-dimensional single-cell analysis by flow cytometry: single cell suspensions were incubated with BD Horizon™ Fixable Viability Stain 440UV (Cat. 566332, 1:1000; BD) for 10 min at RT to exclude dead cells. Then, washed twice and incubated with FcR blocker purified anti-mouse CD16/32 antibody (Clone 93; Cat. 101302; Lot. B401727; 1:50; Biolegend) for 10 min at 4 °C to block fragment crystallizable (Fc) receptors. Then, stained with primary antibodies. Myeloid panel: anti-mouse antibodies were added for 30 min at 37 °C: anti mouse-CXCR2 BV711(Clone V48-2310; Cat. 747812; Lot. 4243816; 1:100 dilution; BD Biosciences), anti mouse-CXCR4 PECF594 (Clone L276F12; Cat. 146514; Lot. B337664; 1:100; Biolegend). Then, after washing using Stain Buffer (Cat. 554656; BD Biosciences) the following anti-mouse antibodies were added for 15 min at 4 °C: anti-mouse CD3 BUV395 (Clone 17A2; Ca.t 740268; Lot. 3291839; 1:400; BD Bioscience), anti mouse-CD19 BUV395 (Clone 1D3; Cat. 563557; Lot. 2273150; 1:400 dilution; BD Biosciences), anti-CD16/32 BUV661 (Clone 2.4G2; Cat.741473; 1:100 BD Biosciences), anti-mouse FceRI/alfa BUV737 (Clone MAR-1; Cat. 751766; Lot. 3291878; 1:400, BD Biosciences), anti mouse-CD45 BUV805 (Clone 30-F11; Cat. 748370; Lot. 4354579; 1:400 dilution; BD Biosciences), anti-mouse CD115 BV421 (Clone AFS98; Cat. 750895; Lot. 4243829; 1:400, BD Biosciences), anti-mouse CD162 BV510 (Clone 2PH1; Cat. 563448; Lot. 2126266; 1:100, BD Biosciences), anti-mouse PD-L1 BV650 (Clone MIH5; Cat. 740614; Lot. 5011113; 1:200, BD Biosciences), anti-mouse Ly6G BB700 (Clone 1A8; Cat. 566435; Lot.2203605; 1:400, BD Biosciences), anti-mouse CD71 PE (Clone C2; Cat. 553267; Lot. 2207704; 1:400, BD Biosciences), anti-mouse CD48 PE-Cy7 (Clone HM48-1; Cat. 560731; Lot. 2297610; 1:400, BD Biosciences), anti-mouse CD11a APC (Clone M17/4; Cat. 101120; Lot. B365934; 1:400, Biolegend), anti-mouse CD117 APCR700 (Clone 2B8; Cat. 565476; Lot. 1250593; 1:800, BD Biosciences), anti-mouse LY6A/E BV786 (Clone D7; Cat. 108139; Lot. B354651; 1:800, Biolegend), anti-mouse Ly6B.2 FITC (Clone REA115; Cat. 130-102- 279; Lot. 5220600378; 1:100, Miltenyi), anti-mouse CD36 APCVio770 (Clone REA1184; Cat. 130-122-093; Lot. 5250101206; 1:200, Miltenyi), anti-mouse Ly6C BV605 (Clone AL-21; Cat. 563011; Lot. 3326984; 1:800, BD Biosciences), anti-mouse F4/80 BV480 (Clone T45-2342; Cat. 565635; Lot. 4124279; 1:400, BD Biosciences), anti-mouse CD11b BUV563 (Clone M1/70; Cat. 741242; Lot. 3195081; 1:3200, BD Biosciences). Then, acquired on a FACS-Symphony A5 flow cytometer (BD Biosciences) equipped with five lasers (UV, 350 nm; violet, 405 nm; blue, 488 nm; yellow/green, 561 nm; red, 640 nm) and capable of detecting 30 parameters.
Then, acquired on a FACS-Symphony A5 flow cytometer (BD Biosciences) equipped with five lasers (UV, 350 nm; violet, 405 nm; blue, 488 nm; yellow/green, 561 nm; red, 640 nm) and capable of detecting 30 parameters.
Computational analysis of flow cytometry data
FCS 3.1 files were analyzed by standard gating in FlowJo version 10.8 to remove dead cells and spurious events and identified CD45+CD3−CD19− cells for myeloid analysis and CD45+CD11b− cells for lymphoid analysis. Then, they were concatenated and projected on UMAP plots. Unbiased clustering analysis was performed by X-Shift algorithm. The cluster expression profile was performed to provide clues about the cells constituting the clusters using the Cluster Explorer plugin. The location of the cluster was highlighted on the UMAP plots.
Metabolomic analyses
Sampling and extraction
5 × 105 to 1 × 106 cells were plated out. After growing them (add here growth conditions or refer to them), cells were washed twice with cold PBS. Cells were extracted by adding 500 µl of 80% cold methanol, scraping off the cells and homogenizing by grinding twice. Then, samples were incubated for 15 minutes on ice and centrifuged (13,000 × g, 4 °C, 3 min). The supernatants were stored at −80 °C and used for metabolomic analyses.
LC-MS/MS analysis polar metabolites
Sample preparation for LC-MS/MS analysis was modified from Paglia et al., 2014. Samples were normalized based on cell number. Volume extracts equivalent to 200,000 cells were dried under N2 stream, reconstituted in 20 µl water (MS grade) and incubated on a shaker (800 rpm,15 °C, 10 min). Then, 80 µl injection buffer (90% acetonitrile, 8.8% methanol, 50 mM ammonium acetate) was added, vortexed and centrifuged (10,000 × g, 4 °C, 10 min). A total of 50 µl of the supernatant was transferred to a glass vial with a narrowed bottom (Total Recovery Vials, Waters, Milford, MA, USA) for LC-MS/MS injection. In addition, quality control (QC) standards and pooled samples were prepared in the same way to serve as QC for the measurements.
Metabolites were separated on a nanoAcquity UPLC (Waters, Milford, MA, USA) equipped with a BEH Amide capillary column (150 µm × 100 mm, 1.7 µm particle size, Waters, Milford, MA, USA). Bisolvent system was 5 mM ammonium acetate in water (A) and 5 mM ammonium acetate in 95% acetonitrile (B). The flow rate was adjusted over the gradient from 3 to 2 µl/min. Gradient started with 10% A and increased to 50% A over 10 min. The following 2 min conditions were changed at 98% B, followed by 4 min equilibration to 10% A. Injection volume was 1 µl. The UPLC was coupled to Synapt G2-Si mass spectrometer (Waters, Milford, MA, USA) by a nanoESIsource. MS1 and MS2 data were acquired using negative polarization and MSE over a mass range of 50 to 1200 m/z at a resolution of >20,000. Scan time: 0.3 s, Ramp transfer collision energy: 20 to 40V, Cone voltage: 40V.
Data processing of untargeted metabolomics data
Data were aligned and searched against databases with the Progenesis QI software (Waters). Polar metabolites were searched against the KEGG database with an initial precursor mass tolerance of 20 ppm and fragment mass tolerance of 50 ppm. Quality controls were run on pooled samples and reference compound mixtures to verify technical accuracy and stability.
Identification of differentially abundant features in the metabolome datasets
the Progenesis QI output files were processed further by in-house R scripts. Mass errors were corrected based on known reference compounds, and the most probable identification was assigned to each observed mass.
Succinate and succinate dehydrogenase activity assay
Succinate production was determined by Succinate Colorimetric Assay Kit (Cat. K649-100, Bio Vision), following the manufacturer’s instructions. The SDH activity was determined by a Succinate Dehydrogenase Activity Colorimetric Assay Kit (Cat. K660-100, Bio Vision), following the manufacturer’s instructions.
SUCNR1 silencing
MCF-7 cells were grown in six-well plates to a 60–80% confluence in EMEM medium supplemented with 10% heat-inactivated CS-FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 2 mM L-glutamine and 0.25% human-recombinant insulin. Cells were transfected with Silencer Select siRNA of SUCNR1 (Cat. 4392420, Thermo Fisher Scientific) and negative control (Cat. 4390843, Thermo Fisher Scientific) with the use of RNAiMax (Thermo Fisher). RNAiMax and the siRNA for each gene were diluted in Opti-MEM reduced-Serum Medium (Thermo Fisher Scientific), respectively. The siRNA was then added to the diluted RNAiMax and incubated for 5 min at room temperature. After incubation, the transfection complex was added to the cells in a dropwise manner and maintained in a culture medium for 24–72 h. The silencing of the SUCNR1 gene in transfected cells was confirmed by western blot.
SLC13a5 inhibition
MCF-7 cells were irradiated at 10 Gy for 3 minutes with irradiator RS1800 (Rad Source Technologies). Then cells were plated in six-well plates in RPMI medium supplemented with 10% heat-inactivated CS-FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin. The day after cells were treated with αSLC13a5 (6518/5, TOCRIS), which is a potent and selective inhibitor of human SLC13A5 transporter, with or without cm-PreNeu for 24 hours.
RNA expression and qPCR
RNA isolation (RNeasy minikit, Cat. 74106, Qiagen) and reverse transcription with the go-script reverse transcription system (Cat. A5001, Promega) were performed according to the manufacturer’s instructions. qPCR reactions were performed using TB Green Premix Ex Taq (Cat. RR420L; Takara) and the specific primers reported below. Each expression value was normalized to the HPRT or GADPH level as a reference. The primer sequences (Invitrogen) used were as follows: PARP1 forward, 5′-CGGAGTCTTCGGATAAGCTCT-3′; and reverse, 5′-TTTCCATCAAACATGGGCGAC-3′. GAPDH forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′; and reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′. HSP1 5′-AGTTCCAGACCGTTGATGGG-3′; and reverse, 5′-GTTTGCACCTGGTATGATCTGT-3′.
Luciferase reporter assay for PARP1 gene expression
293 T cells were transfected using JetPRIME reagent (Cat., 101000046; Polyplus) according to the manufacturer’s protocol, at the 1:2 DNA:jetPRIME ratio. After 48 h each PARP1-luciferase reporter lentiviral vector (pLV-Puro-PARP1-Rluc, Vector builder VB230707) containing medium was collected, filtered through 0.45 µL filter and added to 70% confluent MCF-7 cells. After 48 h from transduction, puromycin selection was performed for 48 h. PARP1-luciferase MCF-7 cells were transfected with pGL3 luciferase reporter control plasmid (Cat. E1741; Promega), using jetPRIME reagent following manufacturer’s instructions. After 48h, cells were irradiated at 10 Gy to induce DNA damage and treated with or without cm-PreNeu in the presence or absence of αSLC13a5 for 6hours. The transcription of PARP1 gene is calculated through luminescence assay using Dual-Glo Luciferase Assay System (Cat. E2920; Promega) following manufacturer’s instructions.
Quantification and statistical analysis
GraphPad Prism version 9.3.1 was used to perform statistical analysis. Outliers were detected by Outlier calculator software by setting the significance level at 0.05 and were excluded. Each specific statistical test used and the number of observations (n) are reported for each experiment in the figure legends. p < 0.05 was considered statistically significant. For human samples, no sample size was estimated; we included all the samples collected in the time frame of the study. For mouse and in vitro experiments, we performed a power analysis test (10% significance level and 80% power; 20% foreseen difference between groups). For ethical reasons, the minimum number of animals was used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw single-cell RNA-seq data from human neutrophils produced in this study have been deposited in the European Genome-Phenome Archive (EGA) under study accession EGAS00001008154 and dataset accession EGAD00001015610. The same raw data are available under restricted access on Zenodo (accession 10.5281/zenodo.15492793; https://zenodo.org/records/15492793). Requests for access can be made to the corresponding author and will be reviewed by the relevant institutional committees. Processed data are available on Zenodo (accession 10.5281/zenodo.14790594; https://zenodo.org/records/14790594). Bulk RNA-seq and ChIP-seq datasets are available in the Gene Expression Omnibus (GEO) under accession GSE288570. Whole-genome sequencing data from isolated EpCAM⁺ cells have been deposited in the BioProject database under accession PRJNA1218519. All other data generated throughout this study are included within the article and its Supplementary Files or are available from the corresponding author upon reasonable request. Source data are provided with this manuscript. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
References
Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Falck, A. K., Ferno, M., Bendahl, P. O. & Ryden, L. St Gallen molecular subtypes in primary breast cancer and matched lymph node metastases–aspects on distribution and prognosis for patients with luminal A tumours: results from a prospective randomised trial. BMC Cancer 13, 558 (2013).
DeSantis, C. E. et al. Breast cancer statistics, 2019. CA Cancer J. Clin. 69, 438–451 (2019).
Allred, D. C., Brown, P. & Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 6, 240–245 (2004).
Jeselsohn, R., Buchwalter, G., De Angelis, C., Brown, M. & Schiff, R. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 12, 573–583 (2015).
Haque, R. et al. Impact of breast cancer subtypes and treatment on survival: an analysis spanning two decades. Cancer Epidemiol. Biomark. Prev. 21, 1848–1855 (2012).
McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017).
Burstein, H. J. Systemic therapy for estrogen receptor-positive, HER2-negative breast cancer. N. Engl. J. Med. 383, 2557–2570 (2020).
Kim, I. S. et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 21, 1113–1126 (2019).
Svoronos, N. et al. Tumor cell-independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 7, 72–85 (2017).
Wagner, J. et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell 177, 1330–1345 e1318 (2019).
Condamine, T. et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, aaf8943 (2016).
Si, Y. et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 4, eaaw9159 (2019).
Rys, R. N. & Calcinotto, A. Senescent neutrophils: a hidden role in cancer progression. Trends Cell Biol. 35, 399–411 (2024).
Ng, M. S. F. et al. Deterministic reprogramming of neutrophils within tumors. Science 383, eadf6493 (2024).
Calcinotto, A. et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 559, 363–369 (2018).
Bancaro, N. et al. Apolipoprotein E induces pathogenic senescent-like myeloid cells in prostate cancer. Cancer Cell 41, 602–619.e611 (2023).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38 (2019).
Losurdo, A. et al. Single-cell profiling defines the prognostic benefit of CD39(high) tissue resident memory CD8+ T cells in luminal-like breast cancer. Commun. Biol. 4, 1117 (2021).
Levine, J. H. et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 162, 184–197 (2015).
Quail, D. F. et al. Neutrophil phenotypes and functions in cancer: a consensus statement. J. Exp. Med. 219, e20220011 (2022).
DeNardo, D. G. & Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 19, 369–382 (2019).
Salcher, S. et al. Comparative analysis of 10X Chromium vs. BD Rhapsody whole transcriptome single-cell sequencing technologies in complex human tissues. Heliyon 10, e28358 (2024).
Dinh, H. Q. et al. Coexpression of CD71 and CD117 identifies an early unipotent neutrophil progenitor population in human bone marrow. Immunity 53, 319–334.e316 (2020).
Veglia, F., Sanseviero, E. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 21, 485–498 (2021).
Bronte, V. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 7, 12150 (2016).
Mukherjee, S., Elia, A. R. & Calcinotto, A. Role of myeloid-derived suppressor cells in hormone-dependent cancers. Swiss Med. Wkly 151, w20483 (2021).
Montaldo, E. et al. Cellular and transcriptional dynamics of human neutrophils at steady state and upon stress. Nat. Immunol. 23, 1470–1483 (2022).
Veglia, F. et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med. 218, e20201803 (2021).
Ng, L. G., Ostuni, R. & Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 19, 255–265 (2019).
Hedrick, C. C. & Malanchi, I. Neutrophils in cancer: heterogeneous and multifaceted. Nat. Rev. Immunol. 22, 173–187 (2022).
Levenson, A. S. & Jordan, V. C. MCF-7: the first hormone-responsive breast cancer cell line. Cancer Res. 57, 3071–3078 (1997).
Sammut, S. J. et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 601, 623–629 (2021).
Sflomos, G. et al. A preclinical model for ERalpha-positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell 29, 407–422 (2016).
Maglione, J. E. et al. Transgenic polyoma middle-T mice model premalignant mammary disease. Cancer Res. 61, 8298–8305 (2001).
Zhu, Y. P. et al. Identification of an early unipotent neutrophil progenitor with pro-tumoral activity in mouse and human bone marrow. Cell Rep. 24, 2329–2341.e2328 (2018).
Le Naour, A. et al. EO771, the first luminal B mammary cancer cell line from C57BL/6 mice. Cancer Cell Int. 20, 328 (2020).
Steele, C. W. et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845 (2016).
Sulkowski, P. L. et al. Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591 (2020).
Tretter, L., Patocs, A. & Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta 1857, 1086–1101 (2016).
Yang, M., Soga, T. & Pollard, P. J. Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest. 123, 3652–3658 (2013).
Peruzzotti-Jametti, L. et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell 22, 355–368.e313 (2018).
Trauelsen, M. et al. Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated Gq signaling. Cell Rep. 35, 109246 (2021).
Canli, O. et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell 32, 869–883 e865 (2017).
Tutt, A. N. J. et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl. J. Med. 384, 2394–2405 (2021).
Pusztai, L. et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e985 (2021).
Hanker, A. B., Sudhan, D. R. & Arteaga, C. L. Overcoming endocrine resistance in breast cancer. Cancer Cell 37, 496–513 (2020).
Xue, R. et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature 612, 141–147 (2022).
Jamieson, T. et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 122, 3127–3144 (2012).
Guo, C. et al. Targeting myeloid chemotaxis to reverse prostate cancer therapy resistance. Nature 623, 1053–1061 (2023).
Brummelman, J. et al. Development, application and computational analysis of high-dimensional fluorescent antibody panels for single-cell flow cytometry. Nat. Protoc. 14, 1946–1969 (2019).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e3529 (2021).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Linderman, G. C. et al. Zero-preserving imputation of single-cell RNA-seq data. Nat. Commun. 13, 192 (2022).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Choudhary, S. & Satija, R. Comparison and evaluation of statistical error models for scRNA-seq. Genome Biol. 23, 27 (2022).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Sotiriou, C. & Desmedt, C. Gene expression profiling in breast cancer. Ann. Oncol. 17, x259–x262 (2006).
Acknowledgements
We acknowledge all members of the Calcinotto laboratory and IOR scientists for the scientific discussions. We thank all the members of the IRB animal core facility for their technical assistance and for the animal work. This research was funded by the Fondazione IOR, a Peter Hans Hofschneider Professorship grant, the Pierre Mercier Foundation, San Salvatore Foundation, the Swiss National Science Foundation (SNSF 310030_219673, TMSGI3_218488), and the Ticino Cancer League (Ar.C). Experimental schematic images were generated using BioRender.
Author information
Authors and Affiliations
Contributions
Ar.C., S.Mu., L.B., C.G., and A.R.E. developed the concept and designed the experiments. S.Mu., L.B., C.G., M.T.B., D.B., I.Ce., I.Cr., and A.R.E. performed experiments. C.G., L.B., and A.R.E. performed estrogen pellet implantation and mammary fat pad injection in all the animals. M.B., G.A.C., L.D.R., and S-J.S. performed bioinformatic analyses. S.Mo. performed immunohistochemical experiments, and U.P. performed the analysis. A.R.E., An.C., and G.L. performed flow cytometry analyses and analyzed the results. A.R. performed single-cell RNA sequencing and whole-genome sequencing. D.J. performed cell sorting. A.R.E., C.G., L.B., S.Mu., R.D.S., and I.G. performed in vivo experiments and Ar.C., C.G., L.B., A.R.E., and S.Mu. interpreted the data. S.Mu., L.B., and D.M. carried out fluorescence microscopy. S.Mu. and L.B. performed statistical analyses of IF in mouse tissue sections. P.C. supervised DNA damage experiments. S.B. provided the anti-CXCR2 (αCXCR2) compound. M.L.G., L.R., F.M., Y.H., and S.D.L. provided human samples. E.Lu. and S.P. helped in the interpretation and analysis of flow cytometry data. E.La. and S.S. performed metabolomic analyses and interpretation of data. P.C., S-J.S., and N.H. supervised manuscript preparation. Ar.C., L.B., C.G., M.M., A.R.E., and S.Mu. interpreted the data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare that Dr. Simon Barry is affiliated with IMED Oncology AstraZeneca, Li Ka Shing Center, Cambridge, United Kingdom and provided the αCXCR2 compound. The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jennifer Guerriero and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mukherjee, S., Garda, C., Boffa, L. et al. Tumor-associated neutrophil precursors impair homologous DNA repair and promote sensitivity to PARP inhibition. Nat Commun 16, 6999 (2025). https://doi.org/10.1038/s41467-025-61422-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61422-9
