
Abstract
Palladium hydride (PdHx) metallenes are efficient electrocatalysts for the oxygen reduction reaction (ORR) due to their high atomic utilization and optimized oxygen binding energies modulated by interstitial hydrogen. However, their practical application is restricted by the highly unstable nature of interstitial hydrogen at working temperatures around 353 K. Here, we report that the use of Mn effectively locks hydrogen atoms within the Pd metallenes lattice, resulting in high alkaline ORR performance across a temperature range of 303–353 K. In contrast, the ORR activity of PdHx metallenes declines sharply with increasing temperature. At 353 K, the mass activity of PdMnHx metallenes at 0.95 V reaches 1.41 A mg−1, which is 14.1 times higher than that of PdHx metallenes. Multiple spectroscopic analyses and theoretical calculations reveal that strong electronic interactions within the immiscible Pd-Mn alloy are critical for locking interstitial hydrogen, thereby enhancing the ORR activity under high temperatures.
Similar content being viewed by others

Introduction
Anion exchange membrane fuel cells (AEMFCs) represent a promising zero-emission energy technology, offering high energy density and environmental sustainability by directly converting the chemical energy of hydrogen into electrical power1,2,3,4. Unfortunately, the inherently slow reaction rate of the oxygen reduction process at the cathode impedes progress towards the full potential of AEMFCs5,6,7. In addition, the operating temperature of AEMFCs has an important impact on their performance and efficiency. At low temperatures, the reaction kinetics of hydrogen and oxygen are poor. To enhance the reaction rate, the operating temperature of AEMFCs is usually in the range 333 K–353 K3,4. However, these high temperatures will lead to the structural collapse and degraded performance of cathodic ORR catalysts. Therefore, the development of heat-resistant and highly efficient ORR catalysts is crucial for AEMFCs.
Pd-based metallenes are a promising class of more efficient ORR electrocatalysts that have gained attention owing to their distinctive two-dimensional architecture with a maximized atomic utilization; high specific surface area; and good electrical conductivity8,9,10,11,12,13,14,15,16,17,18,19,20. Prior research has explored diverse strategies, such as alloying8,9,10,11, defect engineering12,13,14,15,16, and interfacial effects17,18,19,20, to tune the d-band center of Pd metallenes active sites and thereby enhance their performance. Among these approaches, H doping has emerged as the most effective way to enhance the intrinsic ORR mass activity21. The interstitial H atoms existing in the Pd lattice can reduce the binding strength of Pd-O, resulting in Pd metallene catalysts that can counterbalance the relatively strong adsorption energy of oxygen-containing intermediates22,23,24. Unfortunately, the biggest obstacle has been that Pd hydride metallenes may not sufficiently bind H atoms under operating conditions, due to the strong attractive interactions and significant intrinsic compressive strain among surface Pd atoms25. The result is that these insufficiently stable Pd hydride metallenes, while exhibiting high ORR performance at room temperature, do not maintain at the elevated working temperatures of practical membrane electrode assemblies. To this end, designing a strategy for stabilizing the intrinsic structure and enhancing the electrocatalytic ORR activity of PdHx metallenes under practical AEMFCs operating temperatures is important yet remains challenging.
Our prior work demonstrated that interstitial H atoms could be effectively stabilized in Pd metallenes via the Miedema rule of reverse stability by alloying with elements that are immiscible in Pd in the absence of H. We applied this for highly efficient hydrogen evolution reaction and methanol oxidation reaction catalysts26,27. Here, interstitial H atoms are effectively locked within the Pd metallenes lattice via the introduction of Mn, resulting in a good alkaline ORR performance across a temperature range of 303-353 K. At 303 K, PdMnHx metallenes exhibit a high mass activity of 1.08 A mg−1 at 0.95 V vs. RHE, outperforming commercial Pt/C by a factor of 28.4. Importantly, the mass activity value of PdMnHx metallenes can further increase to 1.41 A mg−1 at 353 K. This is in striking contrast with non-alloyed PdHx metallenes, which show a continuing downward trend when the test temperature exceeds 303 K (0.33 A mg−1) and drops to the minimum value with the temperature reaches 353 K (0.10 A mg−1), indicating the rapid deactivation of ORR performance at high working temperatures. Temperature-dependent electrochemical X-ray diffraction (XRD) patterns and operando differential electrochemical mass spectrometry (DEMS) analyses reveal that the deactivation of PdHx metallenes at high working temperature is attributed to the spillover of interstitial H atoms from the Pd lattice. Density functional theory (DFT) calculations demonstrated that interstitial H atoms can be stably confined in PdMnHx metallenes due to the strong electronic interaction within the alloy, resulting in the preservation of hydride nature and exhibit high ORR performance under high temperature.
Results
Morphology characterizations
The Pd-Mn binary phase diagram shows rather asymmetric features and a large isolated Pd phase area at temperatures below 1800 K and Pd content exceeding 70% (Fig. 1a)28. While Pd-Mn alloys can be formed in this range by quenching29, thermodynamic modeling shows that the solubility of Mn in Pd is low below approximately 600 K. In fact, incorporating Mn into the Pd lattice incurs a considerable thermodynamic energy penalty30. Based on our previous research26,27, we hypothesize stabilization of Pd hydride by alloying with the effectively immiscible Mn via the Miedema rule of reverse stability. This is further supported by DFT calculations. We calculated the enthalpy of formation of Pd9Mn1 is 2.30 eV (Fig. 1b and Supplementary Fig. 1), implying that its synthesis demands extra energy input, in agreement with the Pd-Mn phase diagram. Interestingly, the Pd-Mn alloy can produce strong bonding with H atoms, in which each metal binds about 0.3 H atom on average, corresponding to a stoichiometry of Pd9Mn1H3. The enthalpy of formation of Pd9Mn1H3 was calculated based on this stoichiometry to be −0.30 eV, which is much less than the enthalpy of formation of the Mn-Pd alloy, suggesting its good stability under thermodynamic conditions. This is consistent with the reverse Miedema rule as applied to the stability of hydrides31,32,33. This phenomenon implies the obtained PdMn hydride can confine interstitial H atoms with greater stability than those in pure Pd. In the following experimental work, we show that this is the case under electrochemical ORR conditions at elevated working temperatures as illustrated in the schematic Fig. 1c.

a Phase diagram of Pd-Mn alloys. The data is obtained from: Okamoto, H., Schlesinger, M. E. & Mueller, E. M. ASM Handbook, Volume 3: Binary Alloy Phase Diagrams (ASM International, 2016). b The calculated formation enthalpies of and with different reference ground-states. The inset shows the simulation model of Pd9Mn1H3. c Schematic illustration for the structure evolution of PdMnHx and PdHx under ORR conditions. Source data are provided as a Source Data file.
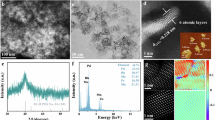
PdMnHx metallenes were prepared through a one-step wet-chemical synthesis using Pd(acac)2 as the palladium source. Mn2(CO)10 provided CO, which acted as a surface-confining agent, while oleylamine functioned simultaneously as the surfactant, solvent, and hydrogen donor (Supplementary Figs. 2–4). The morphology of PdMnHx metallenes was characterized using transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). These show the sheet-like morphology with certain folds (Fig. 2a, b). Elemental mapping by energy-dispersive X-ray spectroscopy (EDS) showed a homogeneous distribution of Pd and Mn in these PdMnHx metallenes, confirming their alloy feature (Fig. 2c). The Pd/Mn atomic ratio, determined based on TEM-EDS results, was found to be 91.2/8.8 (Supplementary Table 1). This was consistent with data obtained from inductively coupled plasma atomic emission spectroscopy (ICP-AES) (Supplementary Fig. 5 and Supplementary Table 2). Atomic force microscopy (AFM) images and corresponding height profiles indicated that our PdMnHx metallenes have a thickness of approximately 2.8 nm (Fig. 2d, e). These PdMnHx metallenes were annealed at 523 K in Ar atmosphere for 1 h to obtain the PdMn alloy (PdMnHx-A metallenes) as a contrast sample for investigating the existence and effect from the confined interstitial H atoms; this contrast sample showed similar composition and identical morphology relative to the PdMnHx metallenes (Supplementary Figs. 6 and 7, Supplementary Tables 1 and 2). Selected area electron diffraction (SAED) patterns of both PdMnHx and PdMnHx-A metallenes are shown in Fig. 2f. The diffraction ring radius of PdMnHx metallenes is smaller than that of PdMnHx-A metallenes, indicating a lattice expansion in PdMnHx metallenes27. In addition, analysis of intensity profiles derived from high-resolution HAADF-STEM imaging further verified the lattice expansion in PdMnHx metallenes, with the (111) lattice spacing measured at 0.230 nm, approximately 3.1% larger than that of PdMnHx-A metallenes (0.223 nm) (Fig. 2g, h, Supplementary Fig. 6). To investigate the hydrogenation state, electron energy loss spectroscopy (EELS) was employed. The PdMnHx-A metallenes exhibit an EELS resonance peak at 7.91 eV, while that of the PdMnHx metallenes appears at 5.71 eV (Fig. 2i, j, Supplementary Fig. 7). This spectral shift is consistent with previously reported hydride characteristics34.

a TEM and b low magnification HAADF-STEM images of PdMnHx metallenes. c TEM-EDS elemental mapping of palladium and manganese in the PdMnHx metallenes. d AFM image and e the relative height profile of PdMnHx metallenes. f SAED images of PdMnHx and PdMnHx-A metallenes. (g) High-resolution HAADF-STEM image of PdMnHx metallenes. h Pixel intensities integrated over the region of PdMnHx metallenes (taken from the red rectangle in Fig. 2g) along the arrow direction ([111] direction). i HAADF-STEM image and j corresponding EELS spectrum of PdMnHx metallenes. Source data are provided as a Source Data file.
Structural and electronic properties
XRD analysis confirms that PdMnHx metallenes possess a face-centered cubic (fcc) crystal structure (Fig. 3a). In comparison to PdMnHx-A metallenes, the diffraction peaks appear at lower angles, indicating a lattice expansion likely caused by hydride formation. To verify the role of Mn elements in the stability of Pd hydride metallenes, XRD analyses were conducted at various temperatures (Supplementary Fig. 8) to investigate the thermal stability of the PdMnHx metallenes. The results revealed that these metallenes retain their structural integrity up to 423 K, but undergo a complete transformation into the metallic phase at 483 K. In comparison, PdHx metallenes lacking Mn exhibit significantly lower thermal stability (Supplementary Fig. 9), preserving their structure only at 303 K and fully converting to metallic Pd at 363 K.

a XRD patterns of PdMnHx-A and PdMnHx metallenes. b Pd 3d and c Mn 2p XPS spectra of PdMnHx-A and PdMnHx metallenes. d1H ssNMR spectra of PdMnHx-A and PdMnHx metallenes. e Pd K-edge XANES of Pd foil, PdMnHx-A, and PdMnHx metallenes. The inset shows the IA/IB values of the three catalysts. f FT-EXAFS and WT-EXAFS spectra of g Pd foil, h PdMnHx-A metallenes, and i PdMnHx metallenes. Source data are provided as a Source Data file.
Based on the established correlation between lattice parameters and composition in palladium hydrides35, the H:(Pd+Mn) ratio is estimated to be 0.32 (Supplementary Fig. 10). The X-ray photoelectron spectroscopy (XPS) result reveals that Pd0 and Mn0 predominates in addition to small fractions of Pd2+ and Mn2+ on the surface of PdMnHx metallenes. Compared to PdMnHx-A metallenes, the Pd 3d and Mn 2p peaks in PdMnHx metallenes are positively shifted 0.35 and 0.15 eV, respectively (Fig. 3b and c), indicating the Pd-H interaction is electronically stronger than the Mn-H interaction27. Furthermore, PdMnHx metallenes exhibited a positive shift in the Pd 3d binding energy relative to the un-hydrided PdMnHx-A metallenes, resulting in a downshift of the d-band center due to electron transfer from Pd to H36. In addition, the galvanic replacement experiment confirmed that Mn atoms are located in the external and internal region of PdMnHx metallenes (Supplementary Fig. 11 and Supplementary Table 3)8,37. To further confirm the presence of interstitial hydrogen atoms, 1H solid-state nuclear magnetic resonance (ssNMR) measurements were conducted. Figure 3d reveals a pronounced peak at 26.2 ppm in the 1H ssNMR spectrum of PdMnHx metallenes, which signifies the formation of Pd-H bonds. Additionally, the interaction between palladium and hydrogen was corroborated by XPS analysis. (Supplementary Fig. 12), which demonstrates that PdMnHx metallenes exhibit a narrower valence band compared to PdMnHx-A metallenes. These observations are consistent with the behavior observed in palladium hydride systems38.
The local coordination and chemical valence states of Pd and Mn in PdMnHx and PdMnHx-A metallenes were elucidated using X-ray absorption spectroscopy (XAS). The Pd K-edge X-ray absorption near-edge structure (XANES) spectra of PdMnHx metallenes are shown in Fig. 3e. The adsorption edge in PdMnHx and PdMnHx-A metallenes is close to that in the Pd foil, demonstrating that Pd atoms in both PdMnHx and PdMnHx-A metallenes predominantly exist in the metallic state. This conclusion aligns with XPS results (Fig. 3b). The first two absorption peaks (denoted as A and B) in the XANES spectra of PdMnHx metallenes, PdMnHx-A metallenes, and Pd foil, labeled as peaks A and B, correspond to electron transitions from 1 s to 5p and 1 s to 4 f orbitals, respectively39. Specifically, the intensity ratio of peak A to peak B (IA/IB) in PdMnHx metallenes (0.91) was found to be slightly higher than that of PdMnHx-A metallenes (0.89) and Pd foil (0.88). This observation is consistent with the characteristic lattice expansion typically associated with hydride formation, further supporting the incorporation of interstitial H atoms into the PdMnHx structure40. While XANES offered general electronic information, EXAFS further resolved the local coordination features, particularly the interatomic distances involving Pd-Pd(Mn) (Fig. 3f, Supplementary Fig. 13, and Supplementary Table 4). After Fourier transformation, the weighted EXAFS spectra (k3) are shown in Fig. 3f. Located approximately at 2.5 Å, the dominant peak reflects the coordination between neighboring Pd atoms in the first shell. Compared to Pd foil and PdMnHx-A metallenes, the incorporation of hydrogen into PdMnHx metallenes induces lattice expansion, as indicated by increased radial distances in the first scattering shell, in agreement with the XRD results. In addition, the Fourier transform (FT) intensities of the samples follow the trend: PdMnHx metallenes<PdMnHx-A metallenes<Pd foil, likely reflecting the lower coordination numbers of PdMnHx and PdMnHx-A metallenes relative to bulk Pd foil. In addition, the incorporation of hydrogen into the lattice induces structural disorder, which further diminishes the intensity of the FT peaks36. Combined with the wavelet transformation (WT) analysis (Figs. 3g–i), the longest radial distance in the first scattering shell of Pd-Pd(Mn) of PdMnHx metallenes and the trend can be seen.
Evaluation of ORR performance
The electrocatalytic performance of PdMnHx metallenes for ORR was evaluated by a rotating disk electrode (RDE) measurement and compared to commercial Pt/C as the benchmark. Before the test, PdMnHx metallenes, PdHx metallenes, and PdMnHx-A metallenes were deposited onto carbon black supports. Cyclic voltammetry (CV) measurements in a 0.1 M KOH solution saturated with N2 were performed for these catalysts. The electrochemical active surface areas (ECSA), estimating the number of active sites available for the ORR, were determined by integrating the reduction peak areas of Pd/Pt oxides. Figure 4a shows that PdMnHx metallenes/C exhibits the highest ECSA value and the most positive Pd-O peak potential compared to the other three catalysts (Supplementary Table 5, Supplementary Figs. 14–16). This observation indicates that PdMnHx metallenes possess a significant number of active sites. ORR polarization curves were measured in an O2-saturated 0.1 M KOH electrolyte at 10 mV s−1. Figure 4b show that PdMnHx metallenes/C catalyst exhibits a half-wave potential (E1/2) of 0.951 V. This is much more positive than that in PdHx metallenes/C (0.913 V), PdMnHx-A metallenes/C (0.901 V), and commercial Pt/C (0.853 V). The Tafel slope of PdMnHx metallenes/C is 36.2 mV dec−1, significantly lower than that of PdHx metallenes/C with 70.5 mV dec−1, PdMnHx-A metallenes/C with 82.6 mV dec−1, and commercial Pt/C with 107.8 mV dec−1 (Fig. 4c), suggesting the faster ORR reaction kinetics on PdMnHx metallenes/C41. The additional analysis of ORR activity under the same precious metal mass loadings for both Pt/C and PdMnHx/C catalysts also demonstrate that PdMnHx/C exhibits the better intrinsic ORR activity (Supplementary Fig. 17). Kinetic currents were extracted from the polarization curves by applying the Koutecky-Levich equation, and subsequently normalized by metal loading and ECSA to calculate the mass activity (MA) and specific activity (SA). Because the universally selected potential of 0.9 V lies near the diffusion-limited region, it cannot reliably capture the dynamic current density owing to the rapid kinetics of PdMnHx metallenes/C. Therefore, the activity was assessed at 0.95 V to remove the effects of O2 diffusion limitation8,42. As shown in Fig. 4d, at 0.95 V, PdMnHx metallenes/C exhibited good MA (1.08 A mg−1Pd) and SA (1.20 mA cm−2), which is 28.4-fold and 19.1-fold enhancements compared to commercial Pt/C, respectively. These results indicate PdMnHx metallenes/C has a competitive performance when compared to the other Pd-based catalysts (Supplementary Table 6). Additionally, optimization experiments for ORR performance with varying H doping amounts revealed that the synthesized PdMnHx catalyst with the H content of 0.32 exhibited the better activity (Supplementary Figs. 18 and 19).

a At a scan rate of 50 mV s-1 and temperature of 298 K, CV curves were collected in N2-saturated 0.1 M KOH solution. b In O2-saturated 0.1 M KOH, LSV curves for ORR were measured at 298 K using a scan rate of 10 mV s−1 and a rotating speed of 1600 rpm. c Corresponding Tafel plots. d The comparisons in MA and SA at 0.95 V. The error bars indicate the standard deviation calculated from three separate experimental replicates. e Durability performance of LSV evolutions, f changes in MA and SA of PdMnHx/C before and after the durability test over multiple potential-scanning cycles. Note that the current density is normalized by the area of the glassy carbon substrate (0.196 cm2). The solution resistance (Rs = 40 ± 5 Ω) was obtained from electrochemical impedance spectroscopy (EIS) measurements. g Schematic illustration of ZABs. h Discharge and power density curves, i The current range of 10 to 100 mA cm−2 was applied during galvanostatic discharge testing of PdMnHx metallenes/C and commercial Pt/C. Source data are provided as a Source Data file.
To assess the electrocatalytic ORR stability of PdMnHx metallenes/C, an accelerated durability test was performed by scanning at 200 mV s−1 within the voltage range of 0.6–1.0 V vs. RHE in 0.1 M KOH saturated with O2 (Fig. 4e)43. The ORR LSV curves of PdMnHx metallenes/C pre- and post-the accelerated durability test exhibit substantial overlap, with only a slight 5 mV drop in E1/2, demonstrating good electrochemical stability. The ECSAs exhibited consistently high values, with negligible change after 30,000 potential cycles (Supplementary Fig. 20). The MA and SA of PdMnHx metallenes/C also remained at 95% and 98% of their initial values after 30,000 cycles, respectively (Fig. 4f). In contrast, the ORR activity of PdHx metallenes/C, PdMnHx-A metallenes/C, and commercial Pt/C decreased notably after 30,000 cycles (Supplementary Fig. 21). Furthermore, the morphology and hydride nature of PdMnHx metallenes were retained after the accelerated stability measurement (Supplementary Figs. 22 and 23), implying the good stability of the hydride. This enhanced durability of PdMnHx metallenes can be attributed to the introduction of Mn atoms and the resulting stabilization of the interstitial H in the Pd lattice. This avoids the structural collapse. To further validate the high ORR performances of PdMnHx metallenes, practical Zn-air batteries (ZABs) tests were conducted with fuel cell devices containing PdMnHx metallenes/C and commercial Pt/C as a cathode material. Compared to commercial Pt/C, PdMnHx metallenes/C exhibit a higher open-circuit voltage of 1.56 V, an elevated power density of 275.7 mW cm−2, and an enhanced specific capacity of 791.4 mAh g−1 at a current density of 10 mA cm-2 (Figs. 4g and h, Supplementary Fig. 24). These values surpass those of Pt/C-based ZABs (1.40 V, 154.1 mW cm−2, and 741.5 mAh g−1, respectively). Furthermore, across a broad range of discharge current densities (10–100 mA cm−2), the PdMnHx metallenes/C-based ZAB consistently demonstrates better performance compared to its Pt/C counterpart (Fig. 4i). These results highlight the strong potential of PdMnHx metallenes/C as an efficient cathode material for metal-air batteries.
Investigation of the temperature-dependent reaction mechanism
We further validate the ORR performance of PdMnHx metallenes at high working temperature. Temperature-dependent ORR polarization measurements were conducted for PdMnHx metallenes and PdHx metallenes over the range of 273-353 K in 0.1 M KOH solution saturated with O2 (Fig. 5a, b, Supplementary Figs. 25 and 26). As shown in Fig. 5a, the limiting current density of PdMnHx metallenes/C increases between 273 and 333 K. This enhancement is primarily attributed to improved oxygen diffusion resulting from reduced electrolyte viscosity at elevated temperatures44. At 353 K, a minor reduction in the limiting current density was observed, likely resulting from reduced oxygen solubility and electrolyte density caused by the substantial rise in water vapor pressure at higher temperatures45. As a strong contrast, for PdHx metallenes/C, the limiting current density showed a declining trend as the temperature increased from 273 K to 303 K and dropped its lowest value when the temperature further increased to 353 K. (Fig. 5b). To more intuitively demonstrate the variation of ORR performance between two catalysts under different working temperatures, the mass-normalized kinetic current densities were extracted from the ORR polarization curves. As shown in Fig. 5c, the mass activity of PdMnHx metallenes/C reached its peak at 333 K. As the temperature continued to rise to 353 K, the mass activity still maintained a high value of 1.41 A mg−1 at 0.95 V, which is 37.1 times higher than that of commercial Pt/C. In contrast, the ORR mass activity of PdHx metallenes/C measured at 0.95 V displayed a similar volcano-type response with increasing temperature, showing a continuing downward trend when the test temperature just exceeds 303 K (0.33 A mg−1) and dropped to the minimum value with the temperature reached 353 K (0.10 A mg−1). In conclusion, the ORR performance of PdHx metallenes is more susceptible to degradation at high operating temperatures compared to PdMnHx metallenes, suggesting that the intrinsic structure of PdHx metallenes may undergo significant changes with increasing temperature. Temperature-dependent electrochemical XRD patterns were performed to explore the changes in the structure of PdMnHx and PdHx metallenes. As shown in Fig. 5d, the XRD peak of PdMnHx metallenes exhibited no significant shift across the temperature range of 303–353 K. In contrast, the XRD peak of PdHx metallenes began shifting toward the Pd phase as early as 313 K and completely transformed into metallic Pd at 353 K (Fig. 5e and f). This suggested that interstitial H atoms escaped from the Pd lattice in PdHx metallenes, whereas they remained stable within the Pd lattice of PdMnHx metallenes. To further illustrate the distinct spill out behavior of interstitial H atoms from the Pd lattice in PdMnHx and PdHx metallenes at the molecular level, operando DEMS of the isotope-labeled ORR process measurements were carried out in the 0.1 M KOD electrolyte with O2-saturated at 353 K on these two catalysts46,47 (Fig. 5g, Supplementary Figs. 27 and 28). For PdMnHx metallenes, no gas signals were detected in the electrolyte during each LSV cycle (Fig. 5h). In contrast, a steady H2 signal was detected in the electrolyte containing PdHx metallenes during two LSV cycles of the ORR process (Fig. 5i). Since no H was present in the reaction solution, the H2 product must have been solely derived from the interstitial H in the PdHx metallenes. The potential difference of the H2 signal observed in two subsequent LSV scans may stem from the progressively reduced stability of H atoms within the PdHx metallenes over time. During the first LSV scan, a relatively higher potential (0.9 V vs. RHE) is required to activate and release the interstitial H atoms from the intact PdHx lattice due to stronger H-metal interactions. However, after the first scan, the original hydride structure is partially disrupted, resulting in a less stable hydrogen environment. Consequently, in the second LSV scan, hydrogen desorption occurs more readily at a lower potential (0.4 V vs. RHE). These results demonstrate that interstitial H atoms could be stably confined within the Pd lattice by introducing immiscible Mn elements, even at high working temperatures.

ORR polarization curves of a PdMnHx/C and b PdHx/C measured from 273 to 353 K in 0.1 M KOH electrolyte saturated with O2 at rotation speed of 1600 rpm and scan rate of 10 mV s−1. c Mass activities of PdMnHx/C and PdHx/C determined at 0.95 V in anodic scan as a function of temperature. Temperature-dependent electrochemical XRD patterns of d PdMnHx/C and e PdHx/C from 303 K to 353 K. f Temperature-dependent lattice parameter of PdMnHx/C and PdHx/C. The solution resistance (Rs = 40 ± 5 Ω) was obtained from EIS measurements. g Schematic illustration of the operando DEMS measurement. The DEMS measurements were conducted using an Ag/AgCl reference electrode (RE), a carbon rod as the counter electrode (CE), and a gold film coated with the catalyst as the working electrode (WE). DEMS signals of H2, HD, and D2 products for h PdMnHx/C and (i) PdHx/C. The labels “off” and “on” in Figs. 5h, i denote cut-off of the first scan and re-start of the second scan during the ORR test program, respectively. Source data are provided as a Source Data file.
DFT calculations
To give a clear description of behavior of interstitial H for PdMnHx and PdHx under ORR conditions, we employed the slow-growth method to sample the energy change during the transfer process of interstitial H atoms from catalysts to electrolyte containing saturated H2O and O2 molecules48. The corresponding potential of the mean force is shown in Supplementary Fig. 29. For both PdMnHx and PdHx metallenes, the migration of H atoms from lattice interstitial positions to the solvent requires overcoming the energy potential barrier, where the energy potential barrier required for H atom migration from PdMnHx (1.14 eV) to the electrolyte is higher than that of PdHx (0.82 eV), indicating that the dissolution of H atoms from PdMnHx is kinetically more difficult than that from PdHx (Fig. 6a, b). This suggests that Mn introduction stabilizes the interstitial H during the ORR process, in agreement with the experimental results. This finding was further confirmed by COHP analysis. As shown in Fig. 6c, d, the metal-H antibonding state near the Fermi energy level on PdMnHx is significantly less than in the case of PdHx, suggesting that the introduction of the Mn element effectively strengthens the electronic interactions between the metal and H, and prevents H atoms from leaving during the ORR process. Both experimental and theoretical results demonstrate that interstitial H atoms cannot be stably confined in PdHx metallenes. Instead, PdHx metallenes will evolve H2, resulting in deactivation of the ORR performance at 353 K. Due to the introduction of Mn into Pd, interstitial H atoms in PdMnHx metallenes are stable in the lattice, which retains the hydride nature and retains high ORR performance at this temperature.

Free energy spectrum of hydrogen precipitated from the a PdMnHx and b PdHx surface to the solution. Green balls = Pd, Purple balls = Mn, Red balls = O, and Grey balls = H. The value in the reaction coordinate represents the distance between the water molecule in the solution and the H atoms in the catalyst. pCOHP between metal and H on c PdMnHx and d PdHx. Source data are provided as a Source Data file.
Discussion
In summary, we report that interstitial H is stabilized in the crystal lattice of Pd metallenes via the introduction of effectively immiscible Mn even under electrocatalytic ORR conditions with high working temperatures. The obtained PdMnHx metallenes exhibit high ORR performance at both 303 K and 353 K, outperforming commercial Pt/C and showing competitive performance with previously reported Pd-based catalysts. In stark contrast, PdHx metallenes show rapid deactivation of ORR performance at 353 K. Temperature-dependent electrochemical XRD patterns and operando DEMS analyses reveal that the deactivation of PdHx metallenes at high working temperature is attributed to the spill out of the interstitial H atoms from the Pd lattice. DFT calculations further demonstrate that interstitial H atoms is able to be stably confined in PdMnHx metallenes due to the strong electronic interaction within immiscible alloy, resulting in the preservation of hydride nature. Our findings uncover the fundamental cause of rapid deactivation in Pd hydride metallenes at elevated temperatures, providing important guidance for developing highly efficient ORR electrocatalysts for practical membrane electrode assemblies.
Methods
Materials
Bis(acetylacetonate) palladium (II) (Pd(acac)2, 98%), commercial Pt/C catalyst, and dimanganese decacarbonyl (Mn2(CO)10, 99%) were purchased from Sigma-Aldrich (USA). Oleylamine (OAm, C18H37N, >90%) was obtained from Aladdin (China). L-ascorbicacid (AA), and potassium hydroxide (KOH, AR) were purchased from Sinopharm Chemical Reagent Co. Ltd. (China). Adamas-beta Chemical Co. (Switzerland) provided the Nafion-ethanol solution. Ultra-pure carbon monoxide was sourced from Xin’guang Gas Co. (China). Throughout the experiments, Milli-Q deionized water (18.2 MΩ·cm) was employed. All reagents were used as received, without additional purification.
Synthesis of PdMnHx metallenes
In a typical preparation of the PdMnHx metallenes, Pd(acac)2 (8 mg), Mn2(CO)10 (8 mg), AA (20 mg), OAm (5 mL) were added into 20 mL glass pressure vessel. After the glass pressure vessel had been capped, the solution was continuously stirred for approximately 20 minutes to achieve a uniform solution. The vessel was then kept at 493 K for 2 h in an oil bath. After the vessel was cooled to 298 K, the resulting black precipitate was isolated by centrifugation and repeatedly washed with cyclohexane and ethanol. The resulting product was re-dispersed in a 4 mL ethanol and cyclohexane mixture (volume ratio 1:1) for subsequent use.
Synthesis of PdMnHx-A metallenes
The above-obtained PdMnHx metallenes were annealed at 523 K under an Ar atmosphere for 1 h to obtain PdMnHx-A metallenes without interstitial H atoms.
Synthesis of PdHx metallenes
In a typical preparation of the PdHx metallenes, Pd(acac)2 (8 mg), AA (20 mg), OAm (5 mL) were added into 20 mL glass pressure vessel. After the glass pressure vessel had been capped, the mixture was stirred at room temperature for approximately 20 minutes to achieve a uniform solution. The homogeneous solution was transferred to a Schlenk flask and purged with CO gas. Then the flask was immediately immersed in an oil bath preheated to 493 K and held for 2 h. Once the mixture reached ambient temperature, the black solid was collected through centrifugation and thoroughly rinsed multiple times using ethanol and cyclohexane. The resulting product was re-dispersed in a 4 mL ethanol and cyclohexane mixture (volume ratio 1:1) for subsequent use.
Material characterizations
TEM and HRTEM characterizations were performed on a JEM-2100F transmission electron microscope (JEOL, Japan). STEM images, along with corresponding EDS mapping and EELS data, were collected using a JEOL ARM200CF instrument (Japan). Note that the dwell time per pixel was increased during TEM-EDS acquisition to enhance the element signal and improve its visibility. AFM images were acquired by depositing the sample onto a mica substrate and analyzing it in air using the tapping mode with ScanAsyst on a Dimension Icon system (Veeco Instruments/Bruker, Germany). XPS measurements were conducted using a monochromatic Al Kα source (1486.6 eV) and a hemispherical analyzer (energy resolution: 0.1 eV) on an ESCALAB 250 system from Thermo Fisher Scientific (USA). ICP-AES analyses were performed on an ELAN 9000/DRC instrument. Noting: the concentration test range of the working sample falls within the standard curve concentration range of the ICP instrument, with no deviation from the standard curve, ensuring the high accuracy of the test results. A Bragg-Brentano diffractometer (D8-tools, Germany) was employed for XRD characterization. Pd K-edge XAS were collected at the Shanghai Synchrotron Radiation Facility (BL14W1). Transmission mode was employed for Pd foil and PdO standards, while fluorescence mode was used for the sample measurements.
Electrochemical measurements
The as-prepared Pd-based metallenes are stored in an ethanol solution, with the concentration of Pd determined by ICP-AES to be 1.0 mgPd/mL. For catalytic evaluation, 5 mL of metallenes solution (containing 5 mg Pd) and 45 mg Ketjen carbon were mixed in 45 mL ethanol and sonicated for 20 minutes to obtain a uniform dispersion. Subsequently, the sonicated mixture is subjected to an additional minute of oscillation. The ultrasonic and oscillating processes were repeated three times to ensure the catalyst was fully loaded on the Ketjen carbon. The solution was then centrifuged, the supernatant discarded, and the residue dried in a vacuum oven at 333 K for 12 h. For electrochemical tests, the catalyst ink was first prepared accordingly. The PdMnHx metallenes/C powder was suspended in a solution composed of 5 μL 5 wt% Nafion solution, 245 μL isopropanol, and 750 μL water. To construct the working electrode, a uniformly dispersed ink served as the coating material, and the benchmark catalyst employed was a commercial 20 wt% Pt/C provided by Johnson-Matthey.
Electrochemical measurements were conducted using a three-electrode setup on an Autolab workstation. The working electrode consisted of a catalyst-coated glassy carbon electrode (5 mm diameter, 0.196 cm2) placed on a rotator, with a Hg/HgO electrode used as the reference and a platinum sheet (1.0 × 1.0 cm2) serving as the counter electrode. All potentials were calibrated against the RHE scale. The 0.1 M KOH electrolyte was prepared by dissolving 2.8 g of KOH in 500 mL of deionized water, followed by ultrasonication to ensure homogeneity. The solution was freshly prepared prior to electrochemical measurements and stored in a sealed container at 298 K. Before catalyst coating, the glassy carbon electrode was polished with alumina polishing slurries, cleaned with DI water, and dried naturally. For catalytic evaluation, 5 mL of the metallenes solution (containing 5 mg of Pd) was fully loaded onto 45 mg of Ketjen carbon, and the final ink solution was configured to 0.1 mgPd/mL. During the ORR measurement, 10 μL of this solution was drop-casted onto the GC electrode, resulting in a Pd loading of 5.2 μg/cm2 for Pd-based metallenes. For Commercial Pt/C, the Pt loading of 10.2 μg cm-2 were added to the GCE to obtain a similar limited diffusion current with other catalysts. The ECSA (m2 g-1PGMs) of the catalysts was calculated according to the equation:
where Q represents the surface charge for oxygen desorption, q represents the charge needed to reduce a PdO monolayer (424 μC cm−2), and m denotes the amount of Pd loading. For ECSA, the cyclic voltammogram was obtained in 0.1 M KOH saturated with N2 at a scan rate of 50 mV s−1. In O2-saturated 0.1 M KOH, ORR polarization measurements were carried out at 10 mV s−1 with a rotation rate of 1600 rpm. After applying iR correction, the Ik was calculated from the ORR curves using the Koutecký-Levich equation:
where Ik is the kinetic current, Id is the limiting current, and I is the measured current at 0.95 V versus RHE. For each catalyst, specific activity and mass activity were calculated by normalizing the kinetic current to the respective ECSA and metal (PGM) mass.
In the accelerated durability tests, the experimental configuration utilized a graphite rod as the counter electrode, which could avoid Pt from the counter electrode dissolves and redeposits on the working electrode during the long hours of test. Accelerated durability tests involved cycling between 0.6 and 1.0 V vs. RHE for 30,000 cycles with a scan rate of 200 mV s−1 applied to the potential. For comparison, the ORR performance of the commercial Pt/C was also measured through similar procedures.
The ORR experiments at different temperatures were conducted by circulating water at controlled temperatures through a constant-temperature water bath. The temperature-regulated water was then circulated through the jacket of the electrochemical cell, enabling precise control of the electrolyte temperature. This setup ensured a stable and uniform temperature environment for the ORR measurements.
At open-circuit potential, EIS analysis was performed using a 10 mV sinusoidal AC signal spanning frequencies from 100 kHz to 10 mHz. Prior to measurement, the OCP was stabilized with a fluctuation of less than 1 mV/min. All potentials were corrected for ohmic drop via iR compensation (Rs = 40 ± 5Ω in 0.1 M KOH).
Zn-air battery test
A polished 0.5 mm-thick Zn foil served as the anode, while a catalyst-coated hydrophobic carbon paper (Spring-P2) was used as the cathode. The electrolyte consisted of 6 M KOH and 0.2 M Zn(AC)2 solution. The catalyst ink was obtained by ultrasonically dispersing 3 mg of catalyst and 1 mg of Vulcan XC 72 carbon was dispersed in 600 μL of a mixed solvent consisting of water, isopropanol, and Nafion in a volume ratio of 44.2:55.2:0.6. The catalyst ink was deposited on carbon paper at a loading of 1.0 mgPd or Pt cm−2 to form the cathode. Discharge polarization curves were recorded using a LAND CT3002A instrument.
XAFS data analysis
The XAFS data were processed in Athena (version 0.9.26) for background correction and edge calibration, followed by Fourier transform fitting using Artemis (version 0.9.26). For Pd fitting, a k3 weighting with a k-range of 3–12 Å-1 and an R-range of 1–3 Å were applied. The four parameters, including coordination number (CN), bond length (R), Debye-Waller factor (σ²), and energy shift (ΔE₀), which were freely fitted without constraints or correlations. For Wavelet Transform analysis, the χ(k) data exported from Athena were processed using the Hama Fortran program. The analysis settings included an R range of 1-4 Å and a k range of 0–13 Å-1 for Pd, with a k-weighting factor of 2. The Morlet wavelet function (κ = 10, σ = 1) was employed as the mother wavelet to capture the overall distribution.
Operando DEMS experiments
DEMS experiments were performed on an operando differential electrochemical mass spectrometer provided by Shpro Instruments (Shanghai) Co. Ltd. The PTFE hydrophobic membrane (pore size = 0.1 μm) provided by Membrane Solutions Company (Shanghai) was employed at the bottom of the electrochemical cell, which facilitates efficient gas diffusion while preventing electrolyte leakage. Electrochemical testing was conducted in a three-electrode configuration at 353 K, employing O2-saturated 0.1 M KOD as the electrolyte. The specific preparation methods of the 0.1 M KOD electrolyte are as follows: The purchased KOD solution (a mixture of KOD and D2O) has a mass fraction of approximately 30% for KOD. To prepare a 0.1 M KOD solution as the electrolyte for operando DEMS measurement, an additional 0.24 mL of D2O was added to 20 mL of the as-received KOD solution. The operando DEMS measurement was carried out by performing LSV tests within a potential range of 0.3–1.1 V (vs. RHE) at a scan rate of 5 mV s−1 on the samples. In this case, the hydrogen molecules are expected to be formed in three ways (i) from interstitial H atoms in PdMnHx and PdHx (H2); (ii) by the combination of hydrogen atoms from KOD solution and interstitial H atoms in PdMnHx and PdHx (HD), and (iii) from two D atoms in KOD solution, without the participation of interstitial H atoms in PdMnHx and PdHx (D2).
Temperature-dependent electrochemical XRD measurement
The catalysts were coated onto carbon paper to serve as the working electrode. A standard LSV measurement was carried out in a three-electrode system with O2-saturated 0.1 M KOH electrolyte across temperatures from 303 to 353 K. Upon completion of the electrochemical measurements, the catalysts were promptly transferred into centrifuge tubes and sealed under an argon atmosphere. XRD measurements were then performed to analyze potential phase transitions that occurred during the electrochemical process.
DFT calculations
All spin-polarized DFT computations were performed using the Vienna ab initio simulation package (VASP)49. The ion-electron interactions were described using the projector augmented-wave (PAW) method50. The exchange-correlation interactions were described using the generalized gradient approximation (GGA) in the form of the Perdew-Burke-Ernzerhof (PBE) functional51. The Brillouin zone was sampled using a 3 × 3 × 1 Monkhorst-Pack k-point mesh for geometry optimization. The DFT-D3 method with Grimme’s empirical dispersion correction was employed to account for van der Waals (vdW) interactions52. All atoms were fully relaxed using a plane-wave cutoff energy of 500 eV until the forces on each atom fell below 0.05 eV/Å. To minimize interactions between periodic images, a 20 Å vacuum spacing was inserted in the Z-axis direction. The formation energy (Eform) of Pd9Mn1H3 was defined as:
The EPd9Mn1H3, EPd, EMn, and EH correspond to the total energies of the respective adsorbed species on the surface, the isolated Pd, Mn and H atom, respectively. Pd9Mn1 exhibit the same formation energy as Pd9Mn1H3, except that H is not involved.
Ab initio molecular dynamics (AIMD) combined with a slow-growth sampling method was employed to simulate the leaching process of H atoms53. A time step of 1 fs was adopted, and the system temperature was maintained at 300 K by applying the Nosé-Hoover thermostat. The collective variable (CV) was incremented by 0.0005 Å, depending on the length of the reaction pathway. The convergence criteria for the electronic step were set to 1 × 10−5, and the POMASS of H is set to 254,55. All AIMD simulations were performed using the Γ-point sampling of the Brillouin zone. (Supplementary Data 1).
Data availability
The data generated in this study are provided in the Supplementary Information and Source Data file. Source data are provided with this paper.
References
Debe, M. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51 (2012).
Yao, Y. et al. Electrocatalysis in alkaline media and alkaline membrane-based energy technologies. Chem. Rev. 122, 6117–6321 (2022).
Xue, J. et al. High-temperature anion-exchange membrane fuel cells with balanced water management and enhanced stability. Joule 8, 1457–1477 (2024).
Douglin, J. et al. High-performance ionomerless cathode anion-exchange membrane fuel cells with ultra-low-loading Ag-Pd alloy electrocatalysts. Nat. Energy 8, 1262–1272 (2023).
Zeng, R. et al. Origins of enhanced oxygen reduction activity of transition metal nitrides. Nat. Mater. 23, 1695–1703 (2024).
Li, M. et al. Spin-polarized PdCu-Fe3O4 in-plane heterostructures with tandem catalytic mechanism for oxygen reduction catalysis. Adv. Mater. 36, 2412004 (2024).
Peng, B. et al. Embedded oxide clusters stabilize sub-2 nm Pt nanoparticles for highly durable fuel cells. Nat. Catal. 7, 818–828 (2024).
Luo, M. et al. PdMo bimetallene for oxygen reduction catalysis. Nature 574, 81–85 (2019).
Xie, L. et al. Modulating the Bader charge transfer in single p-block atoms doped Pd metallene for enhanced oxygen reduction electrocatalysis. Angew. Chem. Int. Ed. 63, e202407658 (2024).
Zhang, K. et al. Interstitial carbon-doped PdMo bimetallene for high-performance oxygen reduction reaction. ACS Energy Lett. 7, 3329–3336 (2022).
Li, Y. et al. Operando elucidation of hydrogen production mechanisms on sub-nanometric high-entropy metallenes. Nat. Commun. 15, 10222 (2024).
Lin, F. et al. Local coordination regulation through tuning atomic-scale cavities of Pd metallene toward efficient oxygen reduction electrocatalysis. Adv. Mater. 34, 2202084 (2022).
Yu, H. et al. Defect-rich porous palladium metallene for enhanced alkaline oxygen reduction electrocatalysis. Angew. Chem. Int. Ed. 60, 12027–12031 (2021).
Guo, J. et al. Template-directed rapid synthesis of Pd-based ultrathin porous intermetallic nanosheets for efficient oxygen reduction. Angew. Chem. Int. Ed. 60, 10942–10949 (2021).
Zeng, T. et al. Control over nitrogen dopant sites in palladium metallene for manipulating catalytic activity and stability in the oxygen reduction reaction. Adv. Funct. Mater. 34, 2408264 (2024).
Tian, J. et al. Greatly enhanced oxygen reduction reaction in anion exchange membrane fuel cell and Zn-air battery via hole inner edge reconstruction of 2D Pd nanomesh. Adv. Mater. 37, 2412051 (2025).
Tao, L. et al. Precise synthetic control of exclusive ligand effect boosts oxygen reduction catalysis. Nat. Commun. 14, 6893 (2023).
Lyu, Z. et al. Amplified interfacial effect in an atomically dispersed RuOx-on-Pd 2D inverse nanocatalyst for high-performance oxygen reduction. Angew. Chem. Int. Ed. 60, 16093–16100 (2021).
Zhao, Y. et al. Atomically engineered defect-rich palladium metallene for high-performance alkaline oxygen reduction electrocatalysis. Adv. Sci. 11, 2405187 (2024).
Prabhu, P. et al. Subnanometric osmium clusters confined on palladium metallenes for enhanced hydrogen evolution and oxygen reduction catalysis. ACS Nano 18, 9942–9957 (2024).
Li, H. et al. Oxidative stability matters: A case study of palladium hydride nanosheets for alkaline fuel cells. J. Am. Chem. Soc. 144, 8106–8114 (2022).
Liu, Y. et al. Exploiting H-induced lattice expansion in β-palladium hydride for enhanced catalytic activities toward oxygen reduction reaction. J. Mater. Sci. Technol. 98, 205–211 (2022).
Liang, J. et al. Gas-balancing adsorption strategy towards noble-metal-based nanowire electrocatalysts. Nat. Catal. 7, 719–732 (2024).
Lu, Y. et al. Highly efficient and durable Pd hydride nanocubes embedded in 2D amorphous NiB nanosheets for oxygen reduction reaction. Adv. Energy Mater. 7, 1700919 (2017).
Fan, J. et al. Spatially confined PdHx metallenes by tensile-strained atomic Ru layers for efficient hydrogen evolution. J. Am. Chem. Soc. 145, 5710–5717 (2023).
Fan, J. et al. Hydrogen stabilized RhPdH 2D bimetallene nanosheets for efficient alkaline hydrogen evolution. J. Am. Chem. Soc. 142, 3645–3651 (2020).
Wu, J. et al. Stable bimetallene hydride boosts anodic CO tolerance of fuel cells. ACS Energy Lett. 6, 1912–1919 (2021).
Okamoto, H., Schlesinger, M. E. & Mueller, E. M. ASM Handbook, Volume 3: Binary Alloy Phase Diagrams (ASM International, 2016).
Saha, D., Ohshima, K., Wey, M., Miida, R. & Kimoto, T. Structure and magnetism of fcc Pd-Mn alloys. Phys. Rev. B 49, 15715–15722 (1994).
Povoden-Karadeniz, E. et al. Thermodynamics of Pd-Mn phases and extension to the Fe-Mn-Pd system. Calphad 51, 314 (2015).
Miedema, A. The electronegativity parameter for transition metals: Heat of formation and charge transfer in alloys. J. Less-Common Met. 32, 117–136 (1973).
Miedema, A., Boom, R. & De Boer, F. On the heat of formation of solid alloys. J. Less-Common Met. 41, 283–298 (1975).
Miedema, A. On the heat of formation of solid alloys. II. J. Less-Common Met. 46, 67–83 (1976).
Fan, J. et al. Interstitial hydrogen atom modulation to boost hydrogen evolution in Pd-based alloy nanoparticles. ACS Nano 13, 12987–12995 (2019).
Schirber, J. & Morosin, B. Lattice constants of β-PdHx and β-PdDx with x near 1.0. Phys. Rev. B. 12, 117–118 (1975).
Povoden-Karadeniz, E. et al. Ligand effect of shape-controlled β‑Palladium Hydride Nanocrystals on liquid-fuel oxidation reactions. Chem. Mater. 31, 5663–5673 (2019).
Huang, S. et al. Sublayer stable Fe dopant in porous Pd metallene boosts oxygen reduction reaction. ACS Nano 16, 522–532 (2022).
Zhao, Z. et al. Synthesis of stable shape-controlled catalytically active β-palladium hydride. J. Am. Chem. Soc. 137, 15672–15675 (2015).
Muller, J. & Jepsen, O. Systematic structure in the K-edge photoabsorption spectra of the 4d transition metals: Theory. Phys. Rev. Lett. 40, 720–722 (1978).
Liu, G. et al. Hydrogen-intercalation-induced lattice expansion of Pd@Pt core-shell nanoparticles for highly efficient electrocatalytic alcohol oxidation. J. Am. Chem. Soc. 143, 11262–11270 (2021).
Sun, K. et al. Co (CN)3 catalysts with well-defined coordination structure for the oxygen reduction reaction. Nat. Catal. 6, 1164–1173 (2023).
Chen, C. et al. Highly crystalline multimetallic nanoframes with three-dimensional electrocatalytic surfaces. Science 343, 1339–1343 (2014).
Chen, J., Jones, C., Linic, S. & Stamenkovic, V. Best practices in pursuit of topics in heterogeneous electrocatalysis. ACS Catal. 7, 6392–6393 (2017).
Wakabayashi, N. et al. Temperature-dependence of oxygen reduction activity at a platinum electrode in an acidic electrolyte solution investigated with a channel flow double electrode. J. Electroanal. Chem. 574, 339–346 (2005).
Khudhayer, W. et al. Oxygen reduction reaction electrocatalytic activity of glancing angle deposited platinum nanorod arrays. J. Electrochem. Soc. 158, B1029–B1041 (2011).
Diaz-Coello, S. et al. Highly active W2C-based composites for the HER in alkaline solution: the role of surface oxide species. ACS Appl. Mater. Interfaces 16, 21877–21884 (2024).
Baltruschat, H. Differential electrochemical mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1693–1706 (2004).
Liu, T. et al. Selective CO2 reduction over γ‑Graphyne supported single-atom catalysts: Crucial role of strain regulation. J. Am. Chem. Soc. 146, 24133–24140 (2024).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B: Condens. Matter Mater. Phys. 47, 558–561 (1993).
Blöchl, P. Projector augmented-wave method. Phys. Rev. B. Condens. Matter Mater. Phys. 50, 17953–17979 (1994).
Perdew, J., Burke, L. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Martyna, G., Klein, M. & Tuckerman, M. Nosé-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 97, 2635–2643 (1992).
Cheng, T., Xiao, H. & Goddard, W. Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu(100) surface at 298 K from quantum mechanics free energy calculations with explicit water. J. Am. Chem. Soc. 138, 13802–13805 (2016).
Liu, X., Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Mechanism of C-N bonds formation in electrocatalytic urea production revealed by ab initio molecular dynamics simulation. Nat. Commun. 13, 5471 (2022).
Acknowledgements
X. Cui and J. Fan acknowledged the support of the National Natural Science Foundation of China (12034002 (X.C.), 22279044 (X.C.), and 22402064 (J.F.)), the Jilin Province Science and Technology Development Program (20210301009GX), and the Fundamental Research Funds for the Central Universities, JLU.
Author information
Authors and Affiliations
Contributions
X.C. and J.F. supervised the execution of the overall project. Y.Q. designed and performed the synthetic and electrochemical experiments, characterized the catalyst, and analyzed the data. D.J. conducted DFT calculations. H.H. assisted with Zn-air battery tests. J.W. assisted with the idea design of catalysts. M.W. assisted with the operando DEMS test. T.G. assisted with the drawing of the schematic diagram. X.Z. assisted with electrochemical ORR tests. X.G. and Wei Z. performed atomic-resolution HAADF-STEM tests. Weitao Z. and D.S. assisted with the paper writing. The results of the manuscript were discussed by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hongjing Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiu, Y., Jiao, D., Huang, H. et al. Locking interstitial hydrogen atoms in Pd metallenes for efficient oxygen reduction reaction. Nat Commun 16, 6103 (2025). https://doi.org/10.1038/s41467-025-61524-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61524-4
