
Abstract
The unstable electrolyte–electrode interface and the trace H2O in commercial organic electrolytes critically limit the cycling life of batteries. Herein, a locking-chain sodium 4,4′-(1,4-phenylenebis(oxy))-bis(butane-1-sulfonate)−15-crown-5 (15PBS) is designed for phase-to-interface electrolyte optimization. In the electrolyte phase, the strong hydrophilic sulfonate groups and 15-crown-5 in 15PBS effectively transform H2O from a reactive aggregated state (strong H-bond) into an inactive state (weak H-bond) through adsorption, effectively suppressing H2O-induced electrolyte decomposition. At the electrolyte–electrode interface, 15PBS preferentially adsorbed onto hard carbon, displacing solvents within the electric double layer to form insoluble phenyl-rich sulfide solid electrolyte interphase with fast Na+ transport. Simultaneously, 15PBS facilitates the formation of stable cathode-electrolyte interphase on Na0.72Ni0.32Mn0.68O2, improving Na+ migration kinetics and cycling reversibility. The hard carbon | |Na0.72Ni0.32Mn0.68O2 full cell with high specific energy of 191.7 Wh kg−1 (based on the total active-material mass) delivers long lifespan of 2000 cycles at 500 mA g−1. Moreover, 15PBS is compatible with ester-based electrolytes in lithium-ion batteries, enabling stable cycling of commercial graphite and Si/C negative electrodes. This work provides an effective approach for durable electrolytes towards safe and high-performance batteries.
Similar content being viewed by others
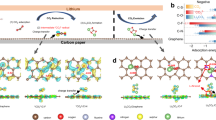


Introduction
Highly stable rechargeable batteries possess great application prospects in portable electronics and electric vehicles. Sodium-ion batteries (SIBs) are considered highly promising large-scale energy storage equipment because of the abundance and low cost of sodium resources1,2. Massive attention has been devoted to the structure optimization of high-capacity positive electrodes (layered oxides (P2-type), Prussian blues, and polyanionics)3 and negative electrodes (hard carbon (HC), alloy compounds, and conversion-type materials)1,4, achieving significant progress for sustainable SIBs. As a “bridge” connecting the positive electrode and negative electrode, the electrolyte is the key to breaking through performance bottlenecks and promoting commercialization, which is worth further exploration5. However, electrolytes suffer from rapid degradation owing to trace H2O-induced corrosion and excessive decomposition caused by the unstable solid electrolyte interphase (SEI), thereby hindering the application of high-energy SIBs.
NaPF6 ethylene carbonate (EC)-based ester electrolytes are extensively used in SIBs because of their high ionic conductivity (9–10 mS cm−1 at 25 °C) and safety6. These electrolytes are extremely sensitive to H2O7. The inevitable trace H2O (less than 20 ppm8) in the electrolytes can cause the hydrolysis of the solvents and salts because of its nucleophile and active proton hydrogen9. The generated HF would accelerate the failure of electrolytes and the dissolution of SEI10,11. Recently, a few strategies have been proposed to address the trace-H2O issue in electrolytes. Molecular sieves are used to adsorb water in nonaqueous liquid electrolytes12 but are applicable only to unused electrolytes, thereby failing to alleviate H2O-catalyzed electrolyte decomposition. Various functional additives have been developed to inhibit the destructive effects of H2O on organic electrolytes. Self-sacrificing additives can react with HF generated from H2O through functional molecular structures (e.g., Si−O and −CN), inhibiting the damage of HF to the electrode and regulating SEI formation13,14. Protective additives can block the direct contact between H2O and electrolytes. However, these continuously consumed self-sacrificing additives during H2O treatment fail to address regenerated H2O from the side interfacial reactions. Additionally, strategies for regulating H2O in SIBs’ electrolytes are few and far from satisfying the requirements of practical applications, especially those concerning interface compatibility with commercial HC negative electrodes and P2-type layered oxide positive electrodes15,16.
The decline in battery capacity is closely related to the electrolyte ingredients because the SEI is generated from the chemical reactions between the electrolyte and the electrode. The unstable organic-rich SEI fails to block the contact between the electrolyte and the electrode, exacerbating the decomposition of the electrolyte. A series of strategies has been proposed to engineer electrolytes, including utilizing new types of salts and solvents17,18, optimizing the proportion of components, and using electrolytes of different concentrations19,20. These strategies are aimed at improving the mechanical properties of SEI by promoting the decomposition of anions to generate inorganic-rich SEI. Unfortunately, the electrolyte optimization through salt or solvent is bound to increase costs and fails to treat the corrosion of trace H2O in the electrolyte. Therefore, simultaneously obtaining the insoluble SEI and suppressing electrolyte hydrolysis are crucial to long-life SIBs.
In this work, we synthesized a locking-chain sodium 4,4’-(1,4-phenylenebis(oxy))-bis(butane-1-sulfonate)−15-crown-5 (15PBS) as the dual-function additive to optimize 1 M NaPF6 EC/diethyl carbonate (DEC) electrolyte from bulk electrolyte to interface. 15PBS trapped trace H2O in the electrolyte phase via polar−group adsorption (−SO3− and 15–C–5), preventing the electrolyte corrosion even at high temperatures and enhancing safety simultaneously. Additionally, the preferentially adsorbed 15PBS at the electrode interface displaced solvents within the electric double layer (EDL) to form insoluble phenyl-rich sulfide SEI. The tough interphase enabled the highly reversible Na+ storage for the HC negative electrode and Na0.72Ni0.32Mn0.68O2 (NNM) positive electrode. Therefore, the HC | | NNM full cell displayed remarkably enhanced cycling stability.
Results and discussion
A locking-chain molecule, 15PBS, was designed (Fig. 1) to realize phase-to-interface electrolyte optimization for sustainable SIBs. The ‘locking chain’ referred to the strong complex structure formed between 15–C–5 molecules and Na+ on long-chain 4,4′-(1,4-phenylenebis(oxy))-bis(butane-1-sulfonate) (PBS) molecules, which interlocked PBS chains with 15–C–5 molecules to form 15PBS additive. 15PBS simultaneously have advantages in inhibiting H2O corrosion in electrolyte phase and modifying the electrode/electrolyte interface for ‘phase-to-interface’ optimization. The structures of 15PBS and PBS in the synthesis process were identified by 1H nuclear magnetic resonance (NMR) spectra (Supplementary Figs. 1, 2). The blue shift of S = O and C–S group in 15PBS compared with that of PBS in FTIR spectra proved the complexation of 15–C–5 and PBS owing to the enhanced electron cloud density (Supplementary Fig. 3 and Supplementary Note 1).

Schematic illustration of locking-chain 15PBS additive synthesis.
The designed 15PBS possessed hydrophilic polar groups (−SO3−, 15–C–5), which can attract H2O from highly active aggregated state (strong H-bond) to inactive H2O (weak H-bond, Fig. 2a). The p-π conjugation between the hydroxyl oxygen in phenol and the benzene ring stabilized 15PBS structure with sp 2 hybridization, which also increased the electrophilicity of the benzene ring by expanding the electron density (Fig. 2b). Meanwhile, the strong coordination amongst sulfonate groups, Na+ and 15–C–5 enhanced the electronegativity of O atoms in 15–C–5, greatly improving their ability to attract H2O and Na+, and thus enabling better performance (Supplementary Fig. 4). Accordingly, density functional theory (DFT) calculations demonstrated that 15PBS provided larger adsorption force (−1.54 eV) than the binding force between H2O molecules (−0.26 eV, Fig. 2c). The commercial 1 M NaPF6 in EC/DEC (1:1 by vol, named as baseline electrolyte, H2O content ~18.5 ± 1.4 ppm, Supplementary Fig. 5)21 and the baseline electrolyte added 1 wt% 15PBS (named as 15PBS electrolyte) was selectively used with high Na+ transference number (0.697) in this work (Supplementary Figs. 6–8 and Supplementary Note 2). The redshift of the O–C–O bond and the _SO3− bond of 15PBS after being added to the baseline electrolyte demonstrated the interaction between the electrolyte and the additive (Supplementary Fig. 9)22.

a Schematic illustration of water-trapping by 15PBS. b Electrostatic potential (ESP) mapping image of 15PBS. c Adsorption energies of H2O-H2O, −SO3−-H2O, 15–C–5-H2O and 15PBS-H2O calculated by DFT calculations. d Fitted Raman spectra of different electrolytes (line: fitted data; dot: pristine data). e 19F NMR spectra of different electrolytes after storage for 7 days at 60 °C. f Radical distribution function and Na+ coordination number of investigated baseline electrolyte and 15PBS electrolyte. g Schematic illustrations of solvation structure. h Raman spectra of baseline electrolyte and 15PBS electrolyte. i EDL capacitance of HC electrodes using different electrolytes by three-electrode systems. Error bars are standard deviations based on three individual tests, and the center metric of the error bars is the average of three tests. j DFT calculations for adsorption energy of H2O, EC, DEC, NaPF6 and 15PBS on graphene sheets with different environments. k, C K-edge synchrotron soft XAS spectra for pristine HC, HC electrodes soaked in different electrolytes, respectively.
Three feature peaks in the Raman spectra of 15PBS electrolyte and baseline electrolyte were attributed to the strong H-bond H2O (~ 3580 cm−1), medium H-bond H2O (~ 3670 cm−1), and weak H-bond H2O (~ 3760 cm−1)23, respectively (Fig. 2d). The high signal of strong H-bond H2O (52.3%) indicated a large amount of highly active H2O in baseline electrolyte. The smaller strong H-bond H2O (13.1%) in 15PBS electrolyte demonstrated that 15PBS downscaled the active H2O in electrolyte. Furthermore, high temperature can accelerate hydrolysis reactions. In the 19F and 1H NMR spectrum, the baseline electrolyte stored at 60 °C for 7 days displayed the signals of HF and sodium ethylene monocarbonate generated from the hydrolysis of NaPF6 and EC, making the electrolyte slightly turbid (Fig. 2e and Supplementary Fig. 10). The absence of electrolyte hydrolysis products in 15PBS electrolyte evidenced the water-trapping ability of 15PBS, stabilizing the electrolyte phase.
The chemical environment of Na+ in the 15PBS electrolyte was characterized by MD calculations at 25 °C and 60 °C, respectively. At 25 °C, H2O molecules occupied the inner solvated structures (2.46 Å) for its strong coordination with Na+ (Supplementary Fig. 11 and Supplementary Table 1). In baseline electrolyte, Na+ had average coordination numbers of 2.62 EC, 1.8 DEC and 1.52 PF6− (Fig. 2f). Meanwhile, the large-sized 15PBS effectively reduced the coordination number of Na+ with solvents and salt (2.35 EC, 1.61 DEC and 1.25 PF6−) owing to its steric-hindrance effect24 and without involving the solvation shell, thereby accelerating desolvation and reducing the accumulation of solvents at the interface (Fig. 2g). The Raman peaks located at about 715.8 cm−1 and 895.2 cm−1 can be attributed to the coordinated PF6− and coordinated EC/DEC in baseline electrolyte. The shift of the PF6− peak from 715.8 cm−1 to 708.9 cm−1 and the shift of EC/DEC peak from 895.2 to 889.3 cm−1 demonstrated the reduced Na+–coordinated PF6– and Na+–coordinated EC/DEC in 15PBS electrolyte than that in baseline electrolyte (Fig. 2h). The downfield trend of the PF6− peaks obtained from the 19F NMR spectrum of the 15PBS electrolyte (−70.67/ − 72.55 ppm) confirmed the decreased electron cloud density around PF6− compared with that of baseline electrolyte (−70.89/ −72.79 ppm), effectively accelerating Na+ transport. The interaction between Na+ and solvents in 15PBS electrolyte was weakened by the enhanced thermal motions of EC (2.19) and DEC (1.29) at 60 °C and disturbed ion−dipole interaction25, which benefited the Na+ transport kinetics (Supplementary Fig. 12).
DFT calculations and synchrotron soft XAS further identified the adsorption of 15PBS at the electrode–electrolyte interface. The calculated EDL capacitance (Supplementary Fig. 13) demonstrated that HC in 15PBS electrolyte had lower double-layer capacitance (CEDL ≈ 5.36 × 10−8 F cm−2, Fig. 2i and Supplementary Table 2) than that of HC in baseline electrolyte (CEDL ≈ 6.79 × 10−8 F cm−2). This indicated that the reduced electrolyte concentration at the interface. DFT calculations with the DFT-D3 corrections and spin polarization26,27 (Supplementary Note 3 and Supplementary Data 1–6) further indicated that the adsorption energy of graphene on 15PBS is much higher than that of electrolyte solvents (Fig. 2j, Supplementary Figs. 14–17 and Supplementary Tables 3–10). Therefore, the preferentially adsorbed 15PBS can replace solvents and H2O in EDL, inhibiting electrolyte decomposition and hydrolysis side reaction during discharge process. The peaks at 285.2 eV and 286.6 eV of HC soaked in 15PBS electrolyte obtained from C K-edge synchrotron soft XAS spectra were assigned to the aromatic π*(C = C) and π*(C − O − C) in 15PBS, proving the adsorption of 15PBS on the HC surface (Fig. 2k). The high-energy shift (from 288.00 to 288.15 eV) of π*(C = O) indicated the electron transfer from HC to 15PBS due to its electronegative groups (−SO3− and 15−C−5), which enhanced the oxidation state π*(C = O) to catalyze rapid decomposition of anions28. HC soaked in baseline electrolyte displayed reduced strength of oxygen-containing functional groups obtained from O K-edge synchrotron soft XAS spectra owing to the more adsorption of solvents and H2O (Supplementary Fig. 18). The loss of oxygen vacancies obtained by electron paramagnetic resonance (EPR, g = 2.004)29,30 further proved that the active sites on HC surface were weakened when more H2O and solvents were adsorbed.
Further research has been conducted on improving SEI stability by the locking-chain 15PBS. The growth rate of SEI is related to the concentration of active molecules at its interface31 according to Fick’s law (Eq. 1)32:
where L is the instantaneous thickness, Di is the chemical diffusion coefficient, ci is the local concentration and the \({\bar{c}}_{i}\) is the mean concentration of the electroactive species i. Reducing the number of solvents on the interface surface can effectively slow down the growth of solvent-induced organic SEI (Supplementary Fig. 19). Moreover, one of the SEI’s functions is to isolate the migration of electrons from the electrode to the electrolyte. The electron tunneling probability (Pr) exponentially decreases with the increased thickness and components of SEI, as explained by the Gamow probability according to quantum mechanics (Fig. 3a, Eq. 2):
where m is the mass of the tunneling particle, L is the interphase thickness, and E and U are the total and potential energies of the tunneling particle, respectively. Therefore, the solvent-induced organic compounds (high Pr) with poor electronic insulation require greater thickness to achieve insulation of electrons, which can be exacerbated by the H2O catalysis. 15PBS reduced the concentration of solvents (ci) at the interface by preferentially adsorbing onto the electrode surface, weakening the contact between the electrolyte and the electrode against solvent decomposition. Meanwhile, theoretical calculations revealed that 15PBS had lower LUMO energy (−1.26 eV, Fig. 3b) than EC and DEC molecules (−0.88 and −0.52 eV) to priorly decompose. A three-electrode cyclic voltammetry (CV) test with glassy carbon33 demonstrated that 15PBS electrolyte had an obvious reduction peak of 15PBS additive at 0.69 V (potential versus Na+/Na) (Supplementary Fig. 20).

a The variation of electron tunneling probability (Pr) with interphase thickness and components. Inset: schematic illustration for the H2O-participated reaction. b Schematic LUMO energy diagram. c In situ DEMS of Na | |HC cell with isotope-labeled 15PBS electrolyte (D2O (2H) was added) for gaseous products analysis at 50 mA g−1, 25 °C. d In situ FTIR of Na | |HC cells using baseline electrolyte and 15PBS electrolyte during initial discharging process at 50 mA g−1, 25 °C. e Cryo-TEM image of the SEI formed on HC negative electrode after initial cycle using 15PBS electrolyte at 50 mA g−1, 25 °C. f Time-dependent mass change (Δm) and energy dissipation (ΔD) of Na | |HC cell at different overtones (n) during CV measurement recorded via in situ EQCM at 25 °C. ΔD is the adsorption of a viscoelastic film (n = 5,7 for the analyzed data). g TOF-SIMS 3D images of F− and S− from SEI of HC negative electrode in 15PBS electrolyte after 100 cycles at 50 mA g−1, 60 °C. h Schematic of SEI derived from different electrolytes.
In situ DEMS and in situ FTIR were used to explore the evolution of electrolyte solvation structure on the electrode surface. HC using 15PBS electrolyte had less CO2 (0.025 μmol) and C2H4 (0.012 μmol) produced by EC decomposition in the five cycles than HC using baseline electrolyte (CO2: 0.069 μmol, C2H4: 0.029 μmol) (Fig. 3c). This evidenced that the SEI formed with 15PBS can effectively inhibit electrolyte decomposition. The reaction pathways of H2O catalysis on HC were also tested with added 20 ppm D2O (D: 2H). The increase in H2 (m/z = 4) from the decomposition of D2O (∼1 V) in baseline electrolyte (Supplementary Fig. 21 and Supplementary Note 4)34 confirmed that H2O participated in electrochemical reactions and caused electrolyte instability. The in situ FTIR peaks at 825 cm−1 and 847 cm−1 corresponded to the P–F of the coordinated PF6− and the free PF6−, respectively (Fig. 3d). The peaks at 1775 and 1820 cm−1 were assigned to the C = O of the coordinated EC and free EC, respectively. The peaks at 1734 and 1747 cm−1 were attributed to the C = O of the coordinated DEC and free DEC, respectively. The 15PBS electrolyte had lower coordinated PF6−, coordinated DEC and EC peaks than baseline electrolyte, proving that 15PBS effectively reduce the interaction between Na+ and solvents and anions. When discharged to 0.72 V, the FTIR peak of coordinated PF6− obviously decreased due to the hydrolysis reaction of PF6−. The blueshift of the coordinated EC (from 1774 cm−1 to 1775 cm−1) and DEC (from 1734 cm−1 to 1735 cm−1) owing to dehydrogenation decomposition indicated that solvated salts and solvents are more likely to contact with the electrode surface and decompose. HC using 15PBS electrolyte had less variation in electrolyte peak, indicating that 15PBS effectively hindered electrolytes decomposition.
The structure and components of SEI formed with 15PBS electrolyte were further investigated. After initial cycling, the widely distributed C 1 s XPS peak at 284.6 eV of HC using 15PBS electrolyte was assigned to the −C = C− of phenyl in the SEI, which improved the insolubility of SEI due to its structural stability (Supplementary Fig. 22). The S 2p peaks at 162.1 eV, 163.8 eV, 168 and 171.4 eV were attributed to Na2S, C–S, Na2SO3 and ROSO2R, therefore enhancing the Na+ transport kinetics and mechanical strength of SEI. The stable 15–C–5 in 15PBS at the surface of SEI was proved by the O 1 s peak at 533.9 eV (C–O–C), strengthening the toughness of SEI. The widely distributed −SO3− groups and 15–C–5 can also promote the decomposition of PF6− to form inorganic NaF in the SEI, which effectively prevented electronic leakage due to its large bandgap35. Consequently, HC formed thin (10 nm, cryo-TEM) and smooth SEI using 15PBS electrolyte (Fig. 3e) and maintained integrity after 200 cycles (Supplementary Fig. 23). Conversely, HC using baseline electrolyte had thick and uneven SEI (8−17 nm), which progressively grew and thickened.
The relationship between the mechanical properties of SEI and the sustainable Na+ storage of HC was investigated with in situ electrochemical quartz crystal microbalance (in situ EQCM). The dissipation factor (ΔD) due to the formed interphase layer and the mass change (Δm) based on the frequency changes were derived from the following equations (Eq. 3 and Eq. 4):
where C is a constant of 17.7 ng Hz−1 cm−2, n is the overtone of the oscillations (n = 3 for the analyzed data).
where \(\frac{\Delta {{{\rm{f}}}}}{n}\) is frequency on multiple harmonics; \(\frac{\Delta {{{\rm{W}}}}}{n}\) is resonance width on multiple harmonics and n is different overtones. HC in 15PBS electrolyte showed high reversible mass (95%) after 100 cycles and displayed highly reversible Na+ storage (Fig. 3f). During cycling, the constantly reduced reversible mass (from 94% to 46%) on HC in baseline electrolyte proved the irreversible electrolyte decomposition and Na+ storage (Supplementary Fig. 24 and Supplementary Note 5). The dissipation factor (ΔD) at different overtones (n = 5, 7) represented the viscoelasticity of SEI on the electrode surface36. The viscoelasticity of the SEI on HC electrode using baseline electrolyte (ΔD ≈ 4.3) and 15PBS electrolyte (ΔD ≈ 4.5) slightly increased in the initial 20 cycles, showing that electrolytes decomposed on the electrode surface, and the viscoelasticity of SEI increased. Subsequently, HC using 15PBS electrolyte delivered viscoelasticity-stable SEI (ΔD ≈ 5.1), which was favorable for highly sustained Na+ storage37. However, the SEI of HC using baseline electrolyte showed dissolution (ΔD ≈ 2.3) and regeneration (ΔD ≈ 12.2), leading to the capacity decline. In situ EQCM tests at 60 °C revealed that the SEI formed with 15PBS maintained regular change in its viscoelasticity during discharge-charge process, evidencing the highly mechanical-strength of its interphase at high temperatures (Supplementary Fig. 25). On the contrary, the continuously dissolved SEI (ΔD reduced 2) formed in baseline electrolyte at high temperatures failed to passivate the interface of the HC and the caused electronic leakage. A broken and uneven SEI (3−8 nm) was seen on HC using baseline electrolyte compared to the integral SEI formed in 15PBS electrolyte after 100 cycles at 60 °C (HRTEM, Supplementary Fig. 26). The poor protection of SEI accelerated interfacial side reactions in baseline electrolyte and ceaselessly produced H2O, and more cycles damaged the electrolyte effectiveness (Supplementary Fig. 27 and Supplementary Note 6). The strong signals of F− and S− obtained by time-of-flight secondary ion mass spectrometry proved that SEI formed with 15PBS remained stable at high temperatures after long cycles (Fig. 3g). The preferential decomposition of 15PBS induced the formation of a thin, mechanically robust SEI enriched with phenyl groups and fluorinated/sulfide components, effectively suppressing the dissolution and regeneration of SEI (Fig. 3h).
The stable interphase chemistry on NNM positive electrodes in 15PBS electrolyte was investigated. DFT calculations demonstrated that the solvents more easily decomposed because of their higher HOMO energy (EC: −6.92 eV; DEC: −6.46 eV) than PF6− (−7.54 eV) in baseline electrolyte (Supplementary Fig. 28). 15PBS had the highest HOMO energy of −4.57 eV, enabling the CEI modification by its decomposition. Meanwhile, 15PBS can catalyze the decomposition of PF6− by breaking P−F bond, realizing its priority decomposition (−6.13 eV, Fig. 4a). S 2p XPS spectra indicated a large number of organic sulfides (RSO3R, RSO2F) and inorganic sulfur salts (Na2SO3, Na2S) in the CEI of NNM, enhancing the Na+ transportation and increasing the toughness of CEI (Fig. 4b). The higher signal of NaF at 685.1 eV on NNM positive electrode using 15PBS electrolyte effectively strengthened the stability of CEI and blocked the contact between electrons and electrolyte. HAADF-STEM exhibited that after initial cycle, NNM positive electrode in baseline electrolyte had thicker CEI of 15 nm (Fig. 4c). While, 15PBS participated in building a thin (8 nm) CEI containing Na2O for NNM positive electrode in 15PBS electrolyte. In situ DEMS demonstrated that NNM positive electrode had higher electrolyte decomposition voltage at 3.5 V (Supplementary Fig. 29) and lower H2 release (0.013 μmol, Fig. 4d) than in baseline electrolyte (3.2 V and 0.024 μmol), indicating the electrolyte decomposition was suppressed.

a Schematic energy diagram of different electrolytes. μC is the cathode electrochemical potential. b S 2p and F 1 s XPS spectra of NNM positive electrode using 15PBS electrolyte after initial cycle at 60 mA g−1, 25 °C with different durations of Ar+ sputtering. c HAADF-STEM images of the CEI formed in different electrolytes after initial cycle at 60 mA g−1, 25 °C. d The H2 release calculated from in situ DEMS of Na | |NNM cells in different electrolytes. e The evolution of Rct fitted from in situ EIS profiles with three-electrode system during initial cycle at 60 mA g−1, 25 °C. f. Time-dependent changes in charge mass retention of different cycles and ΔD at different overtones (n) measured via in situ EQCM using 15PBS electrolyte (n = 5,7 for the analyzed data).
The reversible Na storage of NNM positive electrode with 15PBS was further studied with in situ EIS (Supplementary Figs. 30, 31, and Supplementary Note 7). The charge-transfer impedance (Rct)38 of NNM positive electrode using 15PBS electrolyte (SOC = 0%: 203.5 Ω; SOC = 100%: 70.4 Ω) were lower than that using baseline electrolyte (SOC = 0%: 398.8 Ω; SOC = 100%: 80.0 Ω, Fig. 4e, Supplementary Table 11), evidencing the low impendence of CEI and Na+ transport barrier (Supplementary Figs. 32, 33). The CEI of NNM positive electrode using 15PBS electrolyte showed stable viscoelasticity certificated by in situ EQCM (ΔD: −1.5∼1) (Fig. 4f), enabling the highly average reversible mass of 93%. The CEI generated in baseline electrolyte displayed large change in viscoelasticity (ΔD: −2∼4) during cycling, indicating that the formed CEI was looser, and resulted in the continuous increase of irreversible mass on the electrode and sluggish Na+ transport kinetics (Supplementary Fig. 34). The scanning electron microscopy of NNM positive electrode after 100 cycles in baseline electrolyte displayed obvious cracks, resulting in irreversible sodium storage (Supplementary Fig. 35).
The Na+ storage capability of HC negative electrode was enhanced in the locking-chain 15PBS electrolyte. During charge process, the lower initial voltage (0.012 V) of HC in 15PBS electrolyte demonstrated that HC stored more Na+ in quasi-metallic clusters than HC using baseline electrolyte (0.015 V, Fig. 5a). Owing to the lower polarization, the plateau capacity (16%) and slope capacity (10%) of HC using 15PBS electrolyte increased significantly compared with those in baseline electrolyte (Fig. 5b)34. In situ EQCM was performed to further explore the Na+ storage. The increased mass of HC using baseline electrolyte at 1.22 V was attributed to the electrolyte decomposition catalyzed by H2O (Fig. 5c). HC using 15PBS electrolyte had more mass (Δm = 329 ng cm–2) increased during 0.17−0.01 V due to more Na+ intercalation/ pore-filling in low voltage and high reversible mass ratio (25.6%) than using baseline electrolyte (16.3%, Supplementary Fig. 36). The fitting Rct obtained from in situ EIS with three-electrode measurement demonstrated that the decomposition of electrolyte (0.7−0.3 V) inevitably increased the Rct (Fig. 5d, Supplementary Figs. 37, 38). The thin and sulfide-rich SEI formed in 15PBS electrolyte had the low interface polarization and transmission resistance (25.26 Ω, SOC = 100%), enabling improved Na+ storage kinetics (Supplementary Table 12). Therefore, the HC negative electrode using 15PBS electrolyte delivered reduced desolvation activation energy (Ea) of 57.3 kJ mol−1 than using baseline electrolyte (68.1 kJ mol−1; Supplementary Fig. 39).

a The initial charge curves of HC negative electrodes using different electrolytes at 50 mA g−1, 25 °C. Inset: related charge curves. b The contributions of sloping and plateau capacities obtained from initial charge curves. c Time-dependent changes in Δm of HC using different electrolytes during 4 cycles CV measured via in situ EQCM between 0.01–2.5 V at 25 °C. d Fitting Rct value of HC negative electrodes during the initial discharge/charge process. e Synchrotron radiation in situ XRD of HC using different electrolytes during the initial discharging process at 50 mA g−1. f Solid-state 23Na NMR spectra of HC negative electrodes at various initial discharge states (25 °C, 20 mA g−1). g Schematic illustration of the fast Na+ storage kinetics in HC with 15PBS.
To accurately characterize sodium storage of HC, we used synchrotron radiation in situ XRD and ex-situ solid-state 23Na NMR. The (002) peak of XRD pattern around 22.83° represented that interlayer spacing of HC is 0.388 nm calculated by Bragg law. The (002) peak of HC begins to shift when discharged to 0.5 V, proving that the Na+ inserted into HC layers (Fig. 5e and Supplementary Fig. 40). When discharged to 0.01 V, the (002) peak of HC using 15PBS electrolyte shifted to 22.49° and the interlayer spacing extended to 0.395 nm, which was larger than HC using baseline electrolyte (22.53°) due to more Na+ storage. Furthermore, ex-situ solid-state 23Na NMR revealed that HC using baseline and 15PBS electrolytes both have sharp resonance peaks near −18 ppm when discharged to 0.1 V (Fig. 5f), corresponding to diamagnetic Na+ distributed within the bulk electrode and SEI. A characteristic Knight shift peak at 728 ppm was observed for HC with the 15PBS electrolyte when discharged to 0.01 V, corresponding with the quasi-metallic sodium clusters that formed within HC nanopores. The larger Knight shift of quasi-metallic sodium in HC using 15PBS electrolyte proved that more Na clusters formed in nanopores for larger plateau capacity. To sum up, the lower Rct ensured the highly reversible inserted/pore-filled Na+ for plateau capacity of HC negative electrode (Fig. 5g).
The electrochemical properties tested in half cells manifest the improved compatibility of 15PBS electrolyte with HC negative electrode and NNM positive electrode. HC using 15PBS electrolyte showed improved initial coulombic efficiency (average ICE with three parallel cells: 80.7%) than HC using baseline electrolyte (average ICE with three parallel cells: 78.8%). The increased ICE was due to the suppression of excessive electrolyte decomposition caused by H2O corrosion and the improvement of reversible Na+ storage in HC with 15PBS (Supplementary Fig. 41). Therefore, HC displayed a high charge capacity of 348.7 mAh g−1 at the initial cycle and 48.9%/26.7% plateau capacity retention after 10 and 400 cycles in 15PBS electrolyte at 50 mA g−1. Those values were higher than that using baseline electrolyte (44.1%/10.1%, Fig. 6a-b). HC using 15PBS electrolyte had 85.4% capacity retention (60.5% in baseline electrolyte) after 400 cycles at 50 mA g−1 at 25 °C (Fig. 6c) and still maintained 74.3% (35.6% in baseline electrolyte) after 300 cycles at 60 °C (Fig. 6d). Even at 70 °C, HC using 15PBS electrolyte delivered a reversible capacity retention of 81% (37% in baseline electrolyte) after 100 cycles (Supplementary Fig. 42). Figure 6e showed that increasing temperature can lead to decreased performance of HC in baseline electrolyte (100 cycles, capacity retention reduced from 89% to 37%). HC negative electrode using 15PBS electrolyte displayed improved rate performance (351, 323, 257 and 123 mAh g−1) at 20, 50, 100, and 200 mA g−1, respectively, conforming to the fast Na+ transport kinetics (Supplementary Fig. 43). The mass loading of HC negative electrodes with 2.2, 2.8 and 3.2 mg cm−2 still demonstrated the specific capacities of 307, 300, and 282 mAh g−1 at 50 mA g−1. At 3.2 mg cm−2, the HC negative electrode had 92.8% capacity retention after 80 cycles. Compared with previously reported advanced HC negative electrodes, the HC negative electrode using 15PBS electrolyte also showed improved capacity. The sodium storage kinetics were further supported by the galvanostatic intermittent titration technique (GITT). The Na+ diffusion coefficient of HC using 15PBS electrolyte (10−8.4 cm2 s−1) was higher than HC using baseline electrolyte (Fig. 6f).

a Galvanostatic charge curves from the 10th to 400th cycles of HC negative electrodes at 50 mA g−1. b The plateau-capacities contributions rate of HC obtained from different cycles. c, d Cycling performance of HC negative electrode using 50 mA g-1 at 25 °C (c) and 60 °C (d). e Comparison of the capacity retention of HC at different temperatures after 100 cycles. f Na+-diffusion coefficients calculated from GITT potential profiles for charge process. g, h Voltage hysteresis plots of NNM positive electrodes using baseline electrolyte (g) and 15PBS electrolyte (h). i Cycling performance of NNM positive electrodes at 100 mA g−1, 25 °C.
Even in the 1 M NaPF6 EC/DEC added with 500 ppm H2O electrolyte, HC negative electrode can also steadily work (89.8% capacity retention after 200 cycles, Supplementary Fig. 44) by inhibiting the reactivity of H2O and simultaneously building robust SEI with fast Na+ transport. Furthermore, 15PBS additive can improve the cycling stability of HC in various ester and ether electrolytes (Supplementary Fig. 45). The cycling stability of graphite, Si/C negative electrode, and NCM811 positive electrode could also be improved for lithium-ion batteries with 15PBS additive. (Supplementary Fig. 46 and Supplementary Note 8).
The function of 15PBS on the positive electrode was verified with NNM. The average voltage polarization of NNM positive electrode at high voltage (SOC = 90%) using baseline and 15PBS electrolyte from 2nd to 4th cycles were 0.173 V and 0.151 V, respectively (Fig. 6g, h). After 80 cycles, NNM positive electrode using 15PBS electrolyte maintained smaller voltage polarization of 0.27 V (Supplementary Fig. 47). Therefore, the NNM positive electrode using 15PBS electrolyte had lower desolvation barrier (22.91 kJ mol–1) than using baseline electrolyte (30.67 kJ mol–1, Supplementary Fig. 48), enabling superior capacity retention of 92.8% after 100 cycles (Fig. 6i) and better rate performance of 55.4 mAh g−1 at 600 mA g−1 (Supplementary Fig. 49). Additionally, the 15PBS was applicable to Na3V2(PO4)3 (NVP), and NaxFe4(P2O7)5 x H2O (NFPP) positive electrodes.
To further demonstrate the application potential of 15PBS electrolyte, full cells were fabricated with HC negative electrode and NNM positive electrode. The HC | | NNM full cell using 15PBS electrolyte delivered a high specific energy of 240.7 Wh kg−1 at 50 mA g−1 (based on the total mass of the active materials in positive electrode and negative electrode), better cycling stability (100 cycles, 89.5%) than using baseline electrolyte (100 cycles, 79.9%) (Fig. 7a). At a high specific current of 500 mA g−1, the HC | | NNM full cell showed high capacity of 218 mAh g−1 (Fig. 7b). The HC | | NNM full cell with high NNM loading of ~8.0 mg cm−2 showed high specific energy of 191.7 Wh kg−1 (based on the total mass of the active materials in positive electrode and negative electrode) and impressively low-capacity decay per cycle of 0.017% during 2000 cycles (Fig. 7c) at 500 mA g−1, far exceeding the data of the comparison sample. Further comparison with the electrochemical performance of recently reported coin-type sodium-ion full batteries17,39,40,41,42,43,44,45,46,47,48 revealed that the HC | | NNM coin full cell using 15PBS electrolyte had longest cycle life at specific current of 500 mA g−1, low-capacity decay per cycle and capacity decay per time (1562 h, 0.022% h−1, Fig. 7d, Supplementary Table 13). The capacity decay rate is close to those of previously reported advanced batteries using HC negative electrodes1,49,50,51,52 (Supplementary Fig. 50). The assembled 0.2 Ah pouch cell delivered high specific energy of 115.7 Wh kg−1 (based on the total mass of the active materials in positive electrode and negative electrode) and a capacity retention of 75.8% at 60 mA g−1 after 40 cycles using 15PBS electrolyte (Fig. 7e, Supplementary Fig. 51).

a Cycling performance at 50 mA g−1; Inset: schematic representation of the HC | | NNM full cell. b Rate performance with various specific current ranging from 50 mA g−1 to 500 mA g−1 of the HC | | NNM full cells. c Cycling performance at 500 mA g−1 of the HC | | NNM full cells. d Comparison of the capacity decay per cycle of HC | | NNM full cell in 15PBS electrolyte and other reported SIBs full cells17,39,40,41,42,43,44,45,46,47,48. e Cycling performance of 0.2 Ah HC | | NNM multilayered pouch cell in 15PBS electrolyte at 60 mA g−1.
As for conventional NaPF6-ester electrolyte, the inevitable presence of trace active H2O accelerates the decomposition of anions and solvents, destroying the efficiency and durability of the electrolyte. The solvent-derived SEI/CEI also has high electron tunneling probability (Pr) for its poor electronic insulation, resulting in excessive electrolyte decomposition accompanied by gas release (Fig. 8a). As for advanced 15PBS electrolyte (Fig. 8b), 15PBS served two critical functions compared with conventional NaPF6-ester electrolyte: inhibiting H2O-involved electrolyte decomposition and participating in forming insoluble SEI/CEI with low Pr. The phase-to-interface optimization achieved the stability of batteries. Compared with the reported additives, the dual functional 15PBS additive exhibited better application prospects (Supplementary Table 14).

a NaPF6-ester electrolyte is susceptible to H2O corrosion. b 15PBS additive can effectively inhibit the activity of water and participate in the construction of SEI/CEI. Blue, gray and orange balls represent H2O, gas and 15PBS additive, respectively.
In summary, we designed a locking-chain 15PBS additive to optimize electrolytes from phase to interface. 15PBS trapped trace H2O against electrolyte corrosion and formed stable SEI/CEI for sustainable high-energy-density SIBs. MD simulations revealed that 15PBS lowered the Na⁺−coordination number with EC in the solvation shell, accelerating Na+ desolvation and reducing solvent accumulation at electrode interface. The locking-chain 15PBS adsorbed on HC negative electrodes suppressed parasitic interfacial growth by excluding EC/DEC and H2O in EDL, which lowered local concentration of active molecules (ci), thereby mitigating high-polarity solvent-derived SEI (high Pr) formation. Therefore, preferentially decomposed 15PBS formed thin (10 nm, cryo-TEM) SEI rich in phenyl/sulfide components (RSO3R, Na2SO3, Na2S) to ensure high ion conductivity for enhanced kinetics. The mechanical-tough SEI, confirmed by in situ EQCM and in situ DEMS, exhibited good electronic insulation and effectively suppressed SEI dissolution and regeneration during cycling, even at high temperature. The reduced electrolyte consumption and suppressed gas evolution synergistically improved cycling life and safety. Consequently, the commercialized HC negative electrode achieved extended-plateau capacity due to the accelerated Na+ migration through the low-polarization interphase, enhancing cycling stability. Meanwhile, 15PBS electrolyte formed a thin and robust CEI on NNM positive electrode, mitigating interfacial side reactions. Furthermore, 15PBS effectively attracted water via strong hydrophilic groups (−SO3− and 15–C–5), which broke the strong H-bond of active water to against electrolyte erosion and greatly strengthen the durability of electrolytes. Thus, HC | | NNM full cell achieved a high specific energy of 191.7 Wh kg−1 (based on the total mass of the active materials in positive electrode and negative electrode) and long-term cycling life (2000 cycles) at 500 mA g−1. The stable operation of HC | | NNM pouch cell further certified the practical value. The 15PBS additive was also compatible with graphite and Si/C negative electrodes in lithium-ion batteries, demonstrating broad applicability. This work provides promising opportunities in design of durable electrolytes and stable interphases for advanced batteries.
Methods
Preparation of 15PBS
Firstly, hydroquinone was added to a 250 mL single necked flask and dissolved it in tetrahydrofuran solvent (Aladdin, ≥99.9%), maintaining the reaction at 0 °C. Sodium hydride (Aladdin) was added and maintained the reaction at 0 °C for 1 h. 1,4-butanesulfonic acid lactone (Aladdin, ≥99%) was added to the system, heating to 70 °C and refluxing the overnight. After the reaction was complete, the product precipitated from the tetrahydrofuran solution. The PBS was obtained by filtering the solution with water. Then, PBS was added to a 100 mL single necked bottle and dissolved in 30 mL methanol solvent (Sinopharm Chemical Reagent Co., Ltd, ≥99.9%). By adding a certain amount of 15-crown -5 (15–C–5, Aladdin, ≥97%), the reaction was heat to 80 °C and refluxed for 48 h. After the reaction was completed, the methanol in the reaction system was evaporated and solidified to obtain the 15PBS.
Electrolyte preparation for SIBs
1 M NaPF6 EC/DEC (baseline electrolyte), 1 M NaPF6 EC/DMC, 1 M NaPF6 EC/ DEC/PC, 1 M NaPF6 EC/DMC/PC, 1 M NaClO4 EC/PC, 1 M NaSO3CF3 DGM and 1 M NaPF6 DGM: Battery-grade (≥99.8%) NaPF6, NaClO4 and NaSO3CF3 were purchased from Duoduo chemical reagent Co., Ltd. Battery-grade (≥99.8%) ethylene carbonate (EC), diethyl carbonate (DEC), diethyl carbonate (DMC), propylene carbonate (PC) and diethylene glycol dimethyl ether (DGM) were used as received from Duoduo chemical reagent Co., Ltd. 1 M NaPF6 EC/DEC electrolyte was prepared by dissolving NaPF6 salt into the selected solvent mixture of EC and DEC (1:1 in vol.) 1 M NaPF6 EC/DMC electrolyte was prepared by dissolving NaPF6 salt into the selected solvent mixture of EC and DMC (1:1 in vol.). 1 M NaPF6 EC/DEC/PC electrolyte was prepared by dissolving NaPF6 salt into the selected solvent mixture of EC, DEC and PC (1:1:1 in vol.). 1 M NaPF6 EC/DMC/PC electrolyte was prepared by dissolving NaPF6 salt into the selected solvent mixture of EC, DMC and PC (1:1:1 in vol.). 1 M NaClO4 EC/PC electrolyte was prepared by dissolving NaClO4 salt into the selected solvent mixture of EC and PC (1:1:1 in vol.). 1 M NaSO3CF3 DGM was prepared by dissolving NaSO3CF3 salt into the DGM solvent. 1 M NaPF6 DGM was prepared by dissolving NaPF6 salt into the DGM solvent. All operations were prepared inside an argon-filled glove box with O2, H2O < 1 ppm. The amount of 15PBS additive added to the above electrolytes was 1 wt%.
Electrolyte preparation for LIBs
Battery-grade (≥99.8%) LiPF6, EC, DMC, fluoroethylene carbonate (FEC) and ethyl methyl carbonate (EMC) were used as received from Duoduo chemical reagent Co., Ltd.
1 M LiPF6 EC/ DMC/ EMC was prepared by dissolving LiPF6 salt into the selected solvent mixture of EC, DMC and EMC (1:1:1 in vol.). 1 M LiPF6 EC/DMC/EMC + 3%FEC was prepared by adding 3 wt% FEC into 1 M LiPF6 EC/ DMC/ EMC. 2 M LiPF6 DMC was prepared by dissolving LiPF6 salt into the DMC solvent. The amount of 15PBS additive added to the above electrolytes was 1 wt%. All operations were prepared inside an argon-filled glove box with O2, H2O < 1 ppm.
Electrode preparation for SIBs
For hard carbon (HC) electrodes: HC powder was provided by Kuraray Co., Ltd. and used as received. HC electrode was prepared by coating the slurry on Cu foil (Canrd, ~9-μm-thick), which contained HC: Super P (Canrd, ≥ 99%): polyvinylidene fluoride binder (PVDF) (Canrd, ≥ 99.5%, molecular weight: 1 million) at a mass ratio of 8:1:1 and dried at 80 °C for 24 h, respectively. Methyl-2-pyrrolidone (NMP) (Aladdin, ≥ 99.5%) was used as the solvent. Subsequently, each electrode film was cut into 12 mm disks. The average mass loading was about 1.8–3.2 mg cm–2.
For positive electrodes: Na0.72Ni0.32Mn0.68O2 (NNM) (Paragonage Technology Co., Ltd., ≥ 99%), Na3V2(PO4)3 (NVP) (Canrd, ≥ 99%) and NaxFe4(P2O7)5.xH2O (NFPP) (Canrd, ≥ 99%) electrodes were prepared by mixing the electrode materials, Super P, PVDF with a mass ratio of 8:1:1 in NMP to form a series of slurry mixtures, which were coated onto Al foil (6 μm, Qiandingli electronic technology company) and vacuum dried at 120 °C for 24 h, respectively. Subsequently, each electrode film was cut into 12 mm disks. The mass of each piece of the positive electrode was about 5.0–9.0 mg cm–2.
All the electrodes were coated on one side.
Electrode preparation for LIBs
For graphite electrodes: Graphite powder was provided by Kuraray Co., Ltd. (≥ 99%) and used as received. Graphite electrode was prepared by coating the slurry on Cu foil, which contained graphite: Super P: PVDF (8:1:1 in weight) and dried at 80 °C for 24 h, respectively. Subsequently, each electrode film was cut into 12 mm disks. The average mass loading was about 2.5 mg cm–2.
For Si/C electrodes: Si/C powder was provided by TOPHWA ENERGY Co. (≥ 99%) and used as received. Si/C electrode was prepared by coating the slurry on Cu foil, which contained Si/C: carbon nanotubes: PVDF (8:1:1 in weight) and dried at 80 °C for 24 h, respectively. Subsequently, each electrode film was cut into 12 mm disks. The average mass loading was about 3.1–3.3 mg cm–2.
For LiNi0.8Co0.1Mn0.1O2 (NCM811) electrodes: NCM811 powder was purchased from CANRD Co., LTD ( ≥ 99%) and used as received. NCM811 electrode was prepared by coating the slurry on Al foil, which contained NCM811: Super P: PVDF (8:1:1 in weight) and vacuum dried at 120 °C for 24 h. Subsequently, each electrode film was cut into 12 mm disks. The average mass loading was about 2.5–3 mg cm–2.
All the electrodes were coated on one side.
Materials characterization
Raman spectra was collected with a 532 nm laser under ambient conditions with Raman spectrometer (LabRAM HR Evolution) at room temperature. X-ray photoelectrically spectroscopy (XPS) measurements were measured on an ESCALab MKII X-ray photoelectron spectrometer with Al Ka X-ray radiation as the excitation source. The sample sputtering parameters of Ar+ ions (2 kV; 2 μA; 45° incident angle) were used for the depth profiling investigation. The cycled samples (washed with solvent and dry in argon-filled glovebox) were sealed in an Ar-filled vessel and transferred into the XPS chamber to avoid exposure to air. The sputtering rate is ~ 0.2 nm s–1 based on TaS2. TOF-SIMS (ION-TOF GmbH) was carried out on the cycled electrode (washed with solvent and dry in argon-filled glovebox) at a pulsed Bi3+ (30 eV, 20 ns) ion beam for depth profiling and a Cs+ ion (500 eV) beam for surface analysis. HRTEM (jem-2100, 200kv), cryo-TEM (Glacios, 200 kV) and HAADF-STEM (FEI Titan G2 60–300, 300 kV) were performed to obtained the SEI/CEI. NMR was performed using Bruker Advanced (Bruker). A custom-made electrochemical cell (Beijing Scistar Technology Co., Ltd.) was used for in situ differential electrochemical mass spectrometry experiment and assembled under argon atmosphere. Ultra-high purity dehydrated argon (99.999%) served as the carrier gas, with high-flow purging to evacuate residual atmosphere from gas lines. Operational flow was subsequently regulated at 1 mL min–1 via a mass flow controller. During discharge process, evolved gaseous species were routed through a cryogenic condenser to trap electrolyte volatiles prior to detection by the HPR-20 DEMS system (Hiden Analytical).
SEM was characterized by a scanning electron microscope (ZEISS Merlin Compact). The chemical compositions of electrolyte and electrode were determined by Fourier transform infrared spectroscopy (Bruker, INVENIO S). The electron paramagnetic resonance (EPR) was conducted under vacuum on a Bruker, EMX-9.5/12 instrument with a microwave frequency close to 9.4 GHz. The samples are pristine HC electrode, HC electrode plates soaked in baseline electrolyte and 15PBS electrolyte for five hours, respectively. The solid-state 23Na NMR spectra was carried out on the cycled HC electrode with Bruker 400 M. To avoid the influence of conductive carbon, the HC electrode consists only of HC powder and PVDF binder (mass ratio of 9:1). The test samples come from HC electrode discharged to 0.1 V and 0.01 V at 20 mA g−1 using baseline electrolyte and 15PBS electrolyte, respectively. All the discharged electrodes used in the NMR measurements were cleaned carefully by solvent to eliminate the effect of the electrolyte salt.
In situ FTIR
The in situ FTIR measurements were performed with a Fourier-transformed infrared spectrometer (Bruker, INVENIO S). The HC electrode was fabricated by mixing the HC powder, Super P, and PVDF at a weight ratio of 8:1:1 in NMP to form a slurry. The obtained slurry was coated on copper mesh and dried overnight at 60 °C in a vacuum with an electrode mass loading of 2.0–3.0 mg cm–2.
Synchrotron radiation X-ray absorption spectroscopy (XAS)
Synchrotron radiation X-ray absorption spectroscopy (XAS) measurements were conducted at the wiggler XAS beamline (12ID) at the Australia’s Nuclear Science and Technology Organization (ANSTO) in Melbourne, by using a set of liquid N2-cooled Si (311) monochromator crystals. The samples are pristine HC electrode, HC electrode plates soaked in baseline electrolyte and 15PBS electrolyte for five hours, respectively.
Synchrotron radiation in situ XRD
The synchrotron radiation in situ XRD (beamline BL14B1 of Shanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences) was employed to detect the interlayer spacing changes of HC. The photon beam energy is 18 keV that corresponds to a wavelength of 0.6887 Å with a Huber 5021 hexagonal (4 + 2) diffractometer system and Rayonix MX300 area detector using transmission mode. In situ XRD results were converted to 1.54 Å of Cu radiation wavelength for comparison with XRD in the traditional laboratory. The in situ XRD patterns were collected at a scan rate of 3° min–1 on discharge.
The HC electrode was fabricated by mixing the HC powder, Super P, and PVDF at a weight ratio of 8:1:1 in NMP to form a slurry. The obtained slurry was coated on aluminum foil (one side) and dried overnight at 120 °C in a vacuum with an electrode mass loading of 3.0–4.0 mg cm–2.
In situ electrochemical quartz crystal microbalance analysis (in situ EQCM)
In situ EQCM analysis was applied to track the variation of mass and the SEI viscoelasticity during cyclic voltammetry measurement in Na | |HC and Na | |NNM cells with baseline and 15PBS electrolytes. EQCM measurements were conducted by using a QSense Explorer (Biolin Scientific AB) with gold-coated quartz crystal sensor chips (QSX 313, Biolin Scientific AB). The sensor coated with Au film acted as a working electrode (WE).
The sample preparation is as follows: Firstly, mixing the HC or NNM powders: PVDF: Super P in a ratio of 8: 1: 1, which was subsequently dispersed into NMP solution. Then, the above mixed solution was dropped onto the gold plate and placed it in an 80 °C oven vacuum for drying 12 h.
Electrochemical measurements for SIBs
For half cell: electrochemical examination was conducted via assembling 2025-type cells in a high purify argon-filled glovebox (Mikrouna). The case and spring are made of 304 stainless steel (Canrd). The temperature environment during preparation was carefully controlled and monitored, with an average value of 30 ± 1 °C. The Na disks (300–400 μm thick, Aladdin, ≥99.7%) were used as negative electrodes. Glass fiber separator (Whatman, GF/D, pore size: 2.7 μm, porosity: 40–50%) are used in typical coin-type half cell (assembled with Na-metal negative electrode) and PP separator (Canrd, 2500) are used in pouch-type full cell (assembled with HC negative electrode). The amount of electrolyte added was 115 μL. Cyclic voltammetry (CV) curves of HC negative electrodes were at a scan rate of 0.1 mV s–1 from 0.01 to 2.0 V (potential vs. Na+/Na). Electrochemical impedance spectroscopy (EIS) measurements were performed with amplitude of 5 mV from 100 kHz to 0.005 Hz using three electrode system. All EIS tests were performed in potentiostatic mode and were held at open circuit voltage for 6 h prior to measurement, generating 10 data points per decade frequency. The three-electrode testing system with metallic sodium as the reference electrode was used to eliminate the influence of metallic sodium and achieve accurate measurement of the impedance of the working electrode. Solartron Energylab XM electrochemical workstation was used to measure CV and EIS. Galvanostatic charge-discharge measurements for HC negative electrode were carried out in the voltage range of 0.01 to 2.0 V (vs. Na+/Na) at Neware CT-4008 battery testing system (Shenzhen, China), which is housed in a constant temperature chamber, where the battery test system is maintained at a constant ambient temperature of 25 ± 0.5 °C by forced convection through variable frequency air conditioning. Galvanostatic charge-discharge measurements for NNM positive electrodes were carried out in the voltage range of 2.0 to 4.2 V (vs. Na+/Na) and for NVP and NFPP positive electrodes were carried out in the voltage range of 2.0 to 4.0 V (vs. Na+/Na) at Neware CT-4008 battery testing system (Shenzhen, China). GITT test was carried out in a coin cell after three cycles to reach the thermal equilibrium state and used a pulse current of 0.05 A g−1 for 600 s accompanied with a rest interval of 600 s.
Three-electrode cyclic voltammetry measurements using glassy carbon: Before testing, the glassy carbon electrode (3 mm diameter, LEDONLAB) underwent sequential polishing with alumina slurry, ultrasonic cleaning in deionized water, ethanol rinsing, and drying. A platinum plate served as the counter electrode, while potential values were monitored using a AgCl/Ag reference electrode (saturated KCl electrolyte) and subsequently referred to the Na⁺/Na scale (ENa+/Na = −2.97 V vs AgCl/Ag). The testing device was assembled in the Ar-filled glove box. All CV tests was performed at 25 °C using a Solartron Energylab XM workstation.
For full cell: the full cell was assembled using HC negative electrode and NNM positive electrode at a N/P ratio of 1:1.1. The separator was same to those in half cells. Both negative electrode and positive electrode were electrochemical activated with corresponding electrolytes in half cell at 50 mA g–1 for 2 cycles. Galvanostatic charge-discharge measurements were carried out in the voltage range of 2.0 to 4.0 V at Neware CT-4008 battery testing system (Shenzhen, China) at 25 ± 0.5 °C. Capacity of full cell was calculated based on the active mass of positive electrode and negative electrode.
For pouch cell: Approximately 0.1 MPa of pressure is applied using fixtures throughout the pouch cell testing process. The HC | | NNM pouch cell was made from 20 cm × 4 cm one-sided coated NNM electrodes (mass loading of 12 mg cm–2) and 22 cm × 5 cm HC electrodes (mass loading of 3 mg cm–2). Al lugs were pressure-welded to the NNM electrode, and Ni lugs were clamped to the HC electrode. The prepared cells were encapsulated using an Al-plastic film. The pouch cells were cycled at 60 mA g–1 in the cell voltage range of 1.4–4.2 V at 25 ± 0.5 °C.
Electrochemical measurements for LIBs
For half cell: electrochemical examination was conducted via assembling 2025-type cells in a high purify argon-filled glovebox (Mikrouna). The Li thin disks were used as negative electrodes (300 μm, Canrd Technology Co. Ltd, >99.9%). PP was used as separator. The amount of electrolyte added was 115 μL.
Computational details
The first-principles calculations were carried out within the density functional theory (DFT), utilizing the Vienna ab initio simulation package (VASP). The Perdew-Burke Ernzerhof (PBE) functional of generalized gradient approximation (GGA) was adopted for the exchange correlation energy. The ion-electron interaction was described by the projected augmented wave (PAW) method. A precision energy of 10–5 eV and force criteria of 0.05 eV/Å on each atom are set. Brillouin zone was used by G-centered meshes with fine k-points for the system. The dispersion force correction of DFT-D3 corrections and spin polarization were utilized in the DFT calculations.
To simplify the theoretical calculation model, we used a pure graphene structure with periodic expansion as the HC substrate. In this model, the supercell of graphene is 17 × 17 (39.36 Å × 39.36 Å), containing 578 carbon atoms. The substrate plane size is sufficient to cover 15PBS molecules, with the nearest C–C bond length of 1.42 Å and a vacuum degree of 18 Å set in the z-direction of the box. On this basis, considering the inherent defects and C = O bonds in the HC structure, we constructed graphene models with single vacancy defects and C = O bonds to investigate the effect of complex HC substrates on electrolyte molecule adsorption. It should be clarified that in all three models, the carbon structure are endless graphene sheets (slab configurations with periodic boundary conditions). Therefore, the adsorption results are not affected by the carbon edge structure. Each parallel 15PBS is at least 16 Å away from adjacent 15PBS. The adsorption energies of EC, DEC, H2O, 15PBS, and NaPF6 on graphene substrates are calculated as follows: Eads = Etotal–EA–Esubstrate. Among them, A represents EC, DEC, H2O, 15PBS, and NaPF6, respectively.
The molecular dynamics (MD) simulations: Electrolyte models (50 × 50 × 50 Å3) were constructed to model the Na+, H2O, EC/DEC and 15PBS based electrolytes. The electrolyte model contains 100 NaPF6, 749 EC, 410 DEC and 8 15PBS molecules. The Universal Force Field was applied for all molecular dynamic calculations, with a 1 fs time step implemented53. Electrolyte systems underwent equilibration in the isothermal-isobaric ensemble (constant NPT) using the Berendsen barostat to maintain 0.0001 GPa pressure (decay constant: 0.1 ps) for 2000 ps. Temperature was maintained at 298 K via the Nose thermostat. Subsequently, 2000 ps production runs were conducted in the canonical ensemble (NVT) at 298 K. Equilibrium states were achieved following sufficient simulation duration. The radial distribution function g(r) was obtained by MD simulations of the optimized model and calculated the molecular coordination number by the following formula:
Na+ transference number
Following the Bruce-Vincent method, symmetric cells underwent DC polarization at ΔV = 10 mV until reaching steady-state current. The Na+ transference number (tNa+) was calculated by equation:
where Is and I0 represent the steady−state and initial current, respectively. Rs and R0 are the resistances at steady-state and initial state54, respectively. The polarization voltage ΔV remains at 10 mV.
Double-layer capacitance (CEDL)
where CPE is the constant phase element and Rct is the charge transfer resistance. The relevant parameter values were fitted by ZView software.
Activation energy (Ea) calculation
The charge transfer resistance Rct, obtained from data fitting the temperature-dependent impedance spectra based on the equivalent circuit, is used to determine the activation energies.
The activation energy (Ea) is calculated by Arrhenius equation:
where Rct is the interfacial resistance, A is the frequency factor, R is the gas constant, and T is temperature.
Bragg law:
where d002 is the average interlayer spacing, θ is the diffraction angle of the (002) reflection, λ is the wavelength of the X-ray beam, and n is the order of the reflection.
Na+ diffusion coefficients (DNa+)
where τ (s), mB (g), MB (g mol–1) and VM (mL mol–1) are the pulse duration, the mass, the molar mass and the molar volume of the active material, namely carbon, respectively. In addition, S (cm2) is the active area of the working electrode. ΔES (V) is the potential difference between two adjacent steady states, and ΔEτ (V) is the potential change resulting from the pulse current.
Data availability
The authors declare that the main data supporting our findings of this study are contained within the paper and Supplementary Information. The source data generated in this study are provided in the Source Data file. Source data are provided in this paper. Source data are provided with this paper.
References
Li, Y. et al. Origin of fast charging in hard carbon anodes. Nat. Energy 9, 134–142 (2023).
He, J. et al. Tuning the solvation structure with salts for stable sodium-metal batteries. Nat. Energy 9, 446–456 (2024).
Tang, Y. et al. Sustainable layered cathode with suppressed phase transition for long-life sodium-ion batteries. Nat. Sustain. 7, 348–359 (2024).
Liang, Y. et al. Robust interfacial chemistry induced by B-doping enables rapid, stable sodium storage. Adv. Energy Mater. 13, 2302825 (2023).
Lu, D. et al. Ligand-channel-enabled ultrafast Li-ion conduction. Nature 627, 101–107 (2024).
Cheng, F. et al. Electrolyte salts for sodium-ion batteries: NaPF6 or NaClO4?. ACS Nano 17, 18608 (2023).
Martins, M. et al. Role of catalytic conversions of ethylene carbonate, water, and HF in forming the solid-electrolyte interphase of li-ion batteries. ACS Catal. 13, 9289–9301 (2023).
Cao, X. Important factors for the reliable and reproducible preparation of non-aqueous electrolyte solutions for lithium batteries. Commun. Mater. 4, 10 (2023).
Jiang, H. et al. Electrophilically trapping water for preventing polymerization of cyclic ether towards low-temperature Li metal battery. Angew. Chem. Int. Ed. 62, e202300238 (2023).
Liang, H. et al. Self-purification and silicon-rich interphase achieves high-temperature (70 °C) sodium-ion batteries with nonflammable electrolyte. Energy Storage Mater. 66, 103230 (2024).
Ryu, H. et al. Near-surface reconstruction in Ni-rich layered cathodes for high-performance lithium-ion batteries. Nat. Energy 9, 47–56 (2023).
Zhang, F. et al. Lithium salt dissociation promoted by 18-Crown-6 ether additive toward dilute electrolytes for high-performance lithium oxygen batteries. Angew. Chem. Int. Ed. 135, e202301772 (2023).
Ren, Z. et al. Delicately designed cyano-siloxane as a multifunctional additive enabling high voltage LiNi0.9Co0.05Mn0.05O2/graphite full cell with long cycle life at 50 °C. Adv. Funct. Mater. 33, 2302411 (2023).
Jiang, R. et al. An acetamide additive stabilizing ultra-low concentration electrolyte for long-cycling and high-rate sodium metal battery. Energy Storage Mater. 42, 370–379 (2021).
Xu, K. Electrolytes and interphases in li-ion batteries and beyond. Chem. Rev. 114, 11503–11618 (2014).
Zhang, J. et al. Critical review on cathode electrolyte interphase towards stabilization for sodium-ion batteries. Nano Energy 128, 109814 (2024).
Jin, Y. et al. Low-solvation electrolytes for high-voltage sodium-ion batteries. Nat. Energy 7, 718–725 (2022).
Zhou, X. et al. Regulation of anion–Na+ coordination chemistry in electrolyte solvates for low-temperature sodium-ion batteries. Proc. Natl Acad. Sci. USA 121, e2316914121 (2023).
Zhou, X. et al. Anion-reinforced solvation for a gradient inorganic-rich interphase enables high-rate and stable sodium batteries. Angew. Chem. Int. Ed. 61, e202205045 (2022).
Lin, R. et al. Asymmetric donor-acceptor molecule-regulated core-shell-solvation electrolyte for high-voltage aqueous batteries. Joule 6, 399–417 (2022).
Stich, M. et al. Hydrolysis of LiPF6 in carbonate-based electrolytes for lithium-ion batteries and in aqueous media. J. Phys. Chem. C. 122, 8836–8842 (2018).
Chen, S. et al. Coordination modulation of hydrated zinc ions to enhance redox reversibility of zinc batteries. Nat. Commun. 14, 3526 (2023).
Wang, Y. et al. In situ Raman spectroscopy reveals the structure and dissociation of interfacial water. Nature 600, 81–85 (2021).
Dou, H. et al. Steric-hindrance effect tuned ion solvation enabling high performance aqueous inc ion batteries. Angew. Chem. Int. Ed. 63, e202401974 (2024).
Fang, M. et al. A temperature-dependent solvating electrolyte for wide-temperature and fast-charging lithium metal batteries. Joule 8, 91–103 (2024).
Fan, X. et al. Adsorption and diffusion of Li on pristine and defective graphene. ACS Appl. Mater. Interfaces 4, 2432–2438 (2012).
Vasileiadis, A. et al. Role of defects, pores, and interfaces in deciphering the alkali metal storage mechanism in hard carbon. ACS Appl. Energy Mater. 6, 127–140 (2022).
Xu, X. et al. Establishing exceptional durability in ultralow-temperature organic-sodium batteries via stabilized multiphase conversions. J. Am. Chem. Soc. 146, 1619–1626 (2024).
Chu, Y. et al. Reconfiguring hard carbons with emerging sodium-ion batteries: a perspective. Adv. Mater. 35, 2212186 (2023).
Liu, G. et al. Deciphering electrolyte-dominated Na+ storage mechanisms in hard carbon anodes for sodium-ion batteries. Adv. Sci. 10, 2305414 (2023).
Bruce, P. G. Solid State Electrochemistry. (Cambridge University Press, 1995).
Weppner, W. & Huggins, R. A. Electrochemical studies of parabolic reaction rates and thermodynamic properties. Z. Phys. Chem. 108, 105 (1977).
Cometto, C. et al. Means of using cyclic voltammetry to rapidly design a stable DMC-based electrolyte for Na-ion batteries. J. Electrochem. Soc. 166, A3723–A3730 (2019).
Sui, Y. & Ji, X. Anticatalytic strategies to suppress water electrolysis in aqueous batteries. Chem. Rev. 121, 6654–6695 (2021).
Wang, E. et al. Mitigating electron leakage of solid electrolyte interface for stable sodium-ion batteries. Angew. Chem. Int. Ed. 62, e202216354 (2022).
Niu, L. et al. Understanding the charging of supercapacitors by electrochemical quartz crystal microbalance. Ind. Chem. Mater. 1, 175–187 (2023).
Xia, Y. et al. Designing an asymmetric ether-like lithium salt to enable fast-cycling high-energy lithium metal batteries. Nat. Energy 8, 934–945 (2023).
Keefe, A. S. et al. Temperature-dependent EIS studies separating charge transfer impedance from contact impedance in lithium-ion symmetric cells. J. Electrochem. Soc. 166, A3272–A3279 (2019).
Sun, Y. et al. Molecular engineering enabling high initial coulombic efficiency and rubost solid electrolyte interphase for hard carbon in sodium-ion batteries. Angew. Chem. Int. Ed. 63, e202318960 (2024).
Lu, Z. et al. Consummating ion desolvation in hard carbon anodes for reversible sodium storage. Nat. Commun. 15, 3497 (2024).
Tang, Z. et al. Revealing the closed pore formation of waste wood-derived hard carbon for advanced sodium-ion battery. Nat. Commun. 14, 6024 (2023).
Shi, Q. et al. Niobium-doped layered cathode material for high-power and low-temperature sodium-ion batteries. Nat. Commun. 13, 3205 (2022).
Yin, X. et al. Enabling fast Na+ transfer kinetics in the whole-voltage-region of hard-carbon anodes for ultrahigh-rate sodium storage. Adv. Mater. 34, 2109282 (2022).
Wang, Q. et al. Fast-charge high-voltage layered cathodes for sodium-ion batteries. Nat. Sustain. 7, 338–347 (2024).
Li, W. et al. Binder-induced ultrathin SEI for defect-passivated hard carbon enables highly reversible sodium-ion storage. Adv. Energy Mater. 13, 2300648 (2023).
Liu, Y. et al. Molecular engineering of pore structure/interfacial functional groups toward hard carbon anode in sodium-ion batteries. Energy Storage Mater. 75, 104008 (2025).
Xu, Z. et al. The role of hydrothermal carbonization in sustainable sodium-ion battery anodes. Adv. Energy Mater. 12, 2200208 (2022).
Luo, J. et al. Bifunctional metal-organic coordination interface induced elastic (Cu-F/S, Na-F/S)-rich SEI on hard carbon for durable sodium-ion batteries. Sci. China Chem. 68, 2059–2069 (2024).
Broux, T. et al. High rate performance for carbon-coated Na3V2(PO4)2F3 in Na-ion batteries. Small Methods 3, 1800215 (2018).
Tarascon, J.-M. Na-ion versus Li-ion batteries: complementarity rather than competitiveness. Joule 4, 1616–1620 (2020).
Liu, H. et al. Reviving sodium tunnel oxide cathodes based on structural modulation and sodium compensation strategy toward practical sodium-ion cylindrical battery. Adv. Mater. 36, 2407994 (2024).
Liu, L. et al. Heterogeneous carbon-coated resin-based hard carbon anode material for high-performance sodium ion batteries. Mater. Today Commun. 40, 109854 (2024).
Xie, C. et al. Weak solvent chemistry enables stable aqueous zinc metal batteries over a wide temperature range from –50 to 80 °C. Sci. Bull. 68, 1531–1539 (2023).
Guo, S. et al. Functional separator induced interface potential uniform reformation enabling dendrite-free metal batteries. Adv. Funct. Mater. https://doi.org/10.1002/adfm.202504599 (2025).
Acknowledgments
We acknowledge the funding support from the National Natural Science Foundation of China (U24A20566, 22279121), the Open Project Program of Yaoshan laboratory (2024002), National Key R&D Program of China (No. 2023YFB3809500), Joint Fund of Scientific and Technological Research and Development Program of Henan Province (222301420009), Key Research Development Program of Henan Province (231111241400), and the funding of Zhengzhou University. All theoretical simulations and calculations in this manuscript were performed at National Supercomputing Center in Zhengzhou. The authors thank the support in physical characterizations of the Center of Advanced Analysis & Gene Sequencing of Zhengzhou University. The authors thank Prof. K, Xu from U.S. CCDC Army Research Laboratory, Adelphi, MD, USA. for his valuable advice and guidance on this work. The authors would like to thank Dr. Tadich Anton and Dr. Thomson Lars from ANSTO Australia for their kind assistance in synchrotron SXR data collection and analysis.
Author information
Authors and Affiliations
Contributions
W.C. and Z.Z. conceived and supervised the research. W.L. conducted the experiments, analyzed data, and prepared the manuscript. Y.D. participated in synthesizing samples. S.G. and W.W. conducted electrochemical performances. K.S. and J.Z. performed the theoretical calculations. G.T. conducted the in situ DEMS. L.Z. conducted the synchrotron radiation XAS. P.Y. conducted the HAADF-STEM. E.W., Y.C. and Y.Y. conducted the paper modification. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks Alexandros Vasileiadis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, W., Duan, Y., Ge, S. et al. Locking-chain electrolyte additive enabling moisture-tolerant electrolytes for sodium-ion batteries. Nat Commun 16, 6405 (2025). https://doi.org/10.1038/s41467-025-61603-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61603-6
