
Abstract
Monkeypox virus (MPXV) has re-emerged globally since May 2022, posing a significant public health threat. To address this, we develop two multivalent mRNA vaccine candidates—AAL, encoding three MPXV antigens, and AALI, which combines AAL with an immune-enhancing IFN-α protein. Both vaccines are delivered via mannose-modified lipid nanoparticles to target dendritic cells. Here we show that these vaccines elicit strong antibody responses against vaccinia virus and multiple MPXV clades, induce robust memory B-cell and T-cell responses, and promote dendritic cell maturation. In mouse challenge models, both vaccines provide protection against clade IIb MPXV and vaccinia virus, significantly reducing viral loads and preventing lung damage. Immune profiling reveals enhanced B- and T-cell receptor diversity and distinct CDR3 motifs post-vaccination. These findings demonstrate the potential of using mRNA-based multivalent vaccines as an effective strategy for preventing mpox and related Orthopoxvirus infections.
Similar content being viewed by others

Introduction
Mpox (formerly known as monkeypox) is a zoonotic disease caused by the Monkeypox virus (MPXV) of the Orthopoxvirus genus within the Poxviridae family, which also includes variola virus (VARV), vaccinia virus (VACV), and cowpox virus (CPXV)1,2. MPXV has been classified into the Central African clade (MPXV clade I) and the West African clade (MPXV clade II), both of which are currently spreading worldwide. Since early 2022, the number of mpox cases has significantly increased, with over 132,800 confirmed cases and 314 deaths reported across 131 countries by the end of February 2025, demonstrating a substantial increase in mpox infections.
In response to this public health crisis, the World Health Organization (WHO) declared mpox a Public Health Emergency of International Concern (PHEIC) on July 23, 2022, and reiterated the declaration on August 14, 2024, emphasizing the urgent need for global action. This escalation can be partially attributed to the cessation of routine smallpox vaccination globally in 1980, following the successful eradication of smallpox through widespread vaccination with live VACV3. The smallpox vaccination, which provides cross-protection against MPXV due to its highly conserved genome, may have contributed to sustained mpox outbreaks over the past 40 years4. According to guidelines from the Centers for Disease Control and Prevention (CDC), specific treatments for MPXV infection are currently lacking. Standard smallpox vaccines, including ACAM2000 (an attenuated live VACV vaccine) and JYNNEOS (a replication-deficient vaccine)5,6, provide protection against mpox but differ in efficacy and safety profiles7. ACAM2000 provides near-complete protection but poses safety risks, particularly for immunocompromised individuals, owing to its replicative nature. In contrast, JYNNEOS offers robust but incomplete protection with a more favorable safety profile, although its limited availability remains a major challenge8,9,10. Given these limitations of the available vaccines and the resurgence of mpox, development of an improved vaccine with robust immunogenicity, safety, and rapid production capabilities is urgently needed to effectively combat mpox outbreaks.
To address this need, we can leverage existing technology and research advancements. Nucleic acid-based vaccines, as third-generation vaccines, are advantageous because they can be rapidly developed, can be produced in a cell-free manner, exhibit high efficacy and safety, and have low cost11. Notably, the successful development of a COVID-19 mRNA vaccine targeting SARS-CoV-2 provides theoretical guidance and technical support for the development of mpox mRNA vaccines12,13. However, to date, research on MPXV vaccines has not provided detailed insights into the immune landscape, particularly at the single-cell level12,13. Single-cell RNA sequencing (scRNA-seq) provides a relatively high-resolution understanding of immune responses by enabling the identification of rare cell populations, clonal diversity, and functional heterogeneity14. This approach is critical for understanding how different cell types contribute to the immune response to MPXV vaccination. Combining scRNA-seq with challenge studies using clade IIb MPXV strains isolated from clinical cases provides valuable insights into the immune mechanisms underlying vaccine efficacy.
Given that MPXV exists in two forms, the intracellular mature virus (IMV), which mediates systemic transmission, and the extracellular enveloped virus (EEV), which contributes to cell-to-cell transmission14,15,16, we designed MPXV mRNA vaccines to target both forms by selecting appropriate antigens from each of them. According to studies of VACV and other Orthopoxviruses, several proteins likely play key roles in the spread of MPXV infection in host cells, making them potential targets for the development of vaccines against Orthopoxviruses17,18. For example, A27L (homologous to A29L in MPXV) and J1R (homologous to L1R of MPXV) are IMV surface proteins of VACV that play distinct roles: A27L mediates the attachment of VACV to heparan sulfate present on the cell surface, whereas J1R facilitates the formation of a viral assembly complex, which is essential for the morphogenesis of the virus19,20,21,22,23. A33R (homologous to A35R in MPXV), an EEV envelope glycoprotein of VACV, is crucial for cell-to-cell spread during infection and contains conserved B-cell epitopes, making it a key vaccine component21,24. Therefore, an effective vaccine against pandemic mpox should target antigens from both the IMV and EEV forms of the virus25,26,27. Furthermore, we can consider the addition of immunomodulatory factors to increase vaccine efficacy.
Type I interferons (IFN-Is) are key mediators of innate immunity, driving the expression of genes involved in viral clearance and exhibiting potent immunomodulatory functions24,25,26,27,28,29,30,31,32. IFN-α, a member of the IFN-I family, has been shown to promote immune responses to foot-and-mouth disease and acquired immune deficiency syndrome33,34. Therefore, we hypothesize that IFN-α could serve as an adjuvant to increase the efficacy of MPXV vaccines.
On the basis of these considerations, we developed two multivalent mRNA vaccine candidates, AAL and AALI, in this study. These candidates encode the conserved MPXV proteins A35R (analogous to A33R in VACV), an EEV-specific protein, and A29L (homologous to A27L in VACV), along with L1R (homologous to J1R in VACV), both of which are IMV-specific proteins. Additionally, AALI includes the adjuvant-like protein IFN-α. These vaccines are encapsulated in mannose-modified lipid nanoparticles (LNPs) for targeted delivery to antigen-presenting cells (APCs), especially macrophages and dendritic cells (DCs), which exhibit high mannose receptor expression35,36,37. To assess the efficacy of two mRNA vaccines, namely, AALI and AAL, we carried out a comprehensive series of experiments utilizing female BALB/c mice. Mouse models serve as platforms for assessing innate and adaptive immunity, including B/T-cell activation, cytokine responses, and antigen-specific immunity12. They are essential for preclinical evaluation of MPXV vaccines, measuring immunogenicity, efficacy, and viral clearance38. In this study, both vaccines were specifically engineered to elicit robust humoral and cellular immune responses after intramuscular administration. Additionally, to further evaluate their protective capabilities, we isolated a clade IIb MPXV strain from clinical patients and subsequently established a mouse infection model. The detailed immune responses and efficacy of these vaccines are discussed in the results section.
To further explore the immune mechanisms of the vaccines, we employed scRNA-seq to investigate the transcriptional landscape of the immune response to the MPXV mRNA vaccine and elucidate its immune features at the single-cell level. Additionally, we integrated scRNA-seq and V(D)J sequencing analyses to study the immune responses of B and T cells post-vaccination, examining B-cell receptor (BCR) and T-cell receptor (TCR) diversity, gene rearrangement, and predicted complementarity-determining region 3 (CDR3) motifs. These findings offer comprehensive insight into the immune response characteristics elicited by MPXV mRNA vaccines, laying the groundwork for optimizing vaccination strategies, optimizing vaccine design, and devising innovative clinical approaches to combat mpox and Orthopoxvirus outbreaks.
Results
MPXV mRNA vaccine design and fusion protein expression
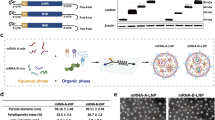
In this study, we developed potent MPXV trivalent mRNA vaccines that target the antigens of both the IMV and EEV forms and characterized their physicochemical properties. First, we generated four codon-optimized mRNAs through the in vitro transcription (IVT) process (Fig. 1A); these mRNAs encode different MPXV antigens derived from the MPXV Zaire-96-I-16 strain (clade I), the IFN-α protein, and red fluorescent protein (RFP). The first mRNA encodes the fusion protein AALI (A35R, A29L, L1R, and IFN-α linked by GSAGSAG), the second encodes AAL (A35R, A29L, and L1R), the third encodes IFN-α as a control, and the fourth encodes RFP as a reporter gene. Notably, we introduced secretion signal peptides (SP) at the N-terminus of the four encoded proteins to increase their immunogenicity. Structural modeling of AALI and AAL with AlphaFold 3 revealed that the domains of A35R, A29L, and L1R exhibited no obvious steric hindrance, and the fused AAL protein was independent of the IFN-α region (Fig. 1B), indicating that the domains of AAL and IFN-α do not affect each other. We subsequently used mannose-conjugated LNPs to encapsulate mRNAs and evaluated their physicochemical properties via transmission electron microscopy (TEM) and dynamic light scattering (DLS). The results displayed in Fig. 1C show that both the LNPs and LNPs-mRNAs (LNPs encapsulating mRNA) exhibit a spherical shape, with particle sizes ranging from 100 to 120 nm. Furthermore, these vaccine particles have a relatively uniform size distribution and are well dispersed in PBS, with a low polydispersity index (PDI) (Fig. 1D). By measuring the concentration of LNPs before and after demulsification of the mRNA in the LNPs, we found that the LNPs presented a high mRNA encapsulation efficacy of ~95 ± 3.1% (Fig. S1A). These results indicate that the MPXV mRNA vaccines AALI and AAL have promising properties for effective vaccine development.

A Two MPXV mRNAs encode fusion proteins: AALI (A35R, A29L, L1R, and IFN-α) and AAL (A35R, A29L, and L1R). mRNAs for IFN-α, RFP, and GFP were used as controls or markers. All sequences were codon optimized, fused with a signal peptide (SP), and inserted into the coding region. B Predicted 3D structures of the AALI and AAL fusion proteins: A29L (light green), A35R (red), L1R (blue), and IFN-α (cyan). C Characterization of LNPs and LNP-mRNA. TEM images of LNPs (left) and LNPs-mRNA (right); scale bar = 200 nm. D LNP (right) and LNPs-mRNA (left) particle sizes were measured via dynamic light scattering (DLS). E HeLa cells transfected with mRFP (RFP mRNA) or LNPs-mRFP were observed under a fluorescence microscope 24 h post-transfection. Images were captured at ×100 magnification. F In vivo bioluminescence imaging of mice 48 h after transfection with LNPs-mRFP. G HeLa cells were transfected with 2 μg of mRNA, and cell lysates collected at 24 h post-transfection were analyzed by SDS-PAGE and Western blotting with anti-A35R and anti-β-actin antibodies. Lane 1: markers; Lanes 2–4: LNPs-, AAL-, and AALI-treated groups. The red box indicates the target protein band. H, Microscopy images (×100) showing the growth status of HeLa cells (top) and RAW264.7 cells (bottom) treated with various LNPs concentrations. I The cell activity induced by LNPs was analyzed at 72 h. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.001; ns, p > 0.05. J, K BMDCs were treated with AALI, AAL, IFN-α, LNPs, or PBS (control) for 48 h, then stained with antibodies against MHC I, MHC II, CD80, and CD86 for flow cytometry analysis. n = 3 biologically independent replicates. Data and error bars represent the means ± SDs. Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, p > 0.05.
Next, the effectiveness of LNPs-mRNAs in expressing target proteins as a vaccine was confirmed through the transfection of LNPs-mRFP (LNPs encapsulating RFP mRNA) into HeLa cells and BALB/c mice. Fluorescence microscopy and in vivo bioluminescence imaging demonstrated successful RFP expression in the LNPs-mRFP group, with no expression observed in the naked mRFP group, at both the cellular and whole-organism levels (Fig. 1E, F). These findings indicate that LNPs effectively protect mRNAs from degradation, increase their delivery into host cells, and enable the mRNA to express the target protein. Moreover, the expression of the fusion proteins AALI and AAL was confirmed through the transfection of an equivalent amount of mRNA into HeLa cells. The cells and their intracellular proteins were harvested and incubated with a primary antibody specific for A35R, followed by the application of either FITC-conjugated or HRP-conjugated secondary antibodies. This process was carried out to facilitate flow cytometry and Western blot analysis. As depicted in Fig. S1B, compared with the control group treated with PBS, the groups treated with AALI and AAL presented a marked increase in the expression of the A35R antigen. Additionally, all proteins of interest were successfully expressed and identified via Western blot analysis, with the mRNAs encoding AALI and AAL yielding robust protein expression levels, as illustrated in Fig. 1G. These findings suggest that the developed MPXV mRNA vaccines are capable of effectively expressing antigenic proteins both in vitro and in vivo.
To verify the ability of the mannose-conjugated LNPs to target DCs, we performed an LNPs-mRNA targeting assay. As shown in Fig. S1C, orange fluorescence indicated colocalization of the green fluorescent protein (GFP) in LNPs-mRNA with red fluorescent DCs, whereas no colocalization was detected in A549 cells. These findings indicate that mannose-modified LNPs can effectively deliver mRNA to DCs and express the target protein, which is consistent with the literature37.
Safety and bioactivity of the MPXV mRNA vaccines
To determine the safety and bioactivity of the mRNA vaccines, we conducted the following experiments. Initially, the cytotoxicity of mannose-modified LNPs at various concentrations was assessed in HeLa and RAW264.7 cells. We found no significant differences in cytotoxicity between the LNPs- and PBS-treated groups at concentrations of 1.25, 12.5, 125, and 1.25 mM (Fig. 1H, I). Additionally, to evaluate the toxicity of LNPs-loaded mRNAs in mice, we conducted histopathological analysis and assessed indicators of liver and kidney function. As expected, no discernible pathological alterations were observed in the heart, liver, spleen, lung, or kidney of the mice in the AALI, AAL or IFN-α groups compared with those in the LNPs and PBS control groups, both in the short term (Fig. S1D, 14 days after the second immunization) and long term (Fig. S2A, 72 days after the third immunization). Furthermore, liver and kidney function indicators, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin (T-BIL), albumin (ALB), alkaline phosphatase (ALP), gamma-glutamyl transferase (GGT), total bile acid (TBA), creatinine (Crea), uric acid (UA), urea, and carbon dioxide (CO₂), remained within the normal range for all groups (Table S1, 14 days after the second immunization; Fig. S2B, 72 days after the third immunization)39,40,41. These results indicate that the MPXV mRNA vaccines are safe.
After establishing the safety and efficacy of the MPXV mRNA vaccine, we investigated its biological activity in vitro. The splenocyte proliferation assay data in Fig. S2C revealed that the AALI, AAL, and IFN-α mRNA vaccines induced clusters of proliferating murine splenocytes. The results from the MTS assay (Fig. S2D) revealed significantly greater proliferation indices (PI) in the AALI, AAL, and IFN-α groups than in the LNPs and PBS control groups. Notably, the AALI group exhibited even greater PIs than the AAL group did (p < 0.5), suggesting that the AALI group had increased immune response capabilities.
DCs, crucial APCs, play pivotal roles in linking innate and adaptive immune responses42. We also treated bone marrow-derived DCs (BMDCs) with the mRNA vaccine and analyzed them using flow cytometry. Compared with treatment with LNPs or PBS, treatment with AALI, AAL, or IFN-α significantly increased the expression levels of the markers CD80, CD86, MHC I, and MHC II in BMDCs, suggesting increased maturation (Fig. 1J). Moreover, the AALI-treated BMDCs presented significantly higher expression levels of CD80 and CD86 than did the AAL-treated BMDCs. Additionally, the expression of MHC I was significantly greater in the AALI and IFN-α groups than in the AAL group (Fig. 1K). Collectively, these findings underscore the potential efficacy of our mRNA vaccines in effectively stimulating the proliferation of mouse splenic lymphocytes and inducing the maturation of BMDCs. Moreover, the combination of IFN-α as an adjuvant with AAL demonstrated promising immunostimulatory effects.
The mRNA vaccines induced antibodies specific to the MPXV and VACV antigens
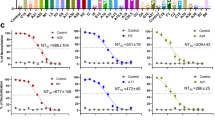
Female BALB/c mice were immunized following a prime-boost-boost schedule, with 2-week intervals between each vaccination, and serum samples were collected 14 days after the final vaccination. To confirm whether the MPXV mRNA vaccine induced the production of MPXV-specific antibodies, we conducted enzyme-linked immunosorbent assays (ELISAs) with six recombinant proteins derived from both clade I MPXV (A29L, A35R, and L1R were derived from the hMpxV/DRC/INRB-0198V/2024 strain) and clade II MPXV (A29L from the MPXV-UK_P2-138 strain, A35R and L1R from the hMpxV/Portugal/INSA-PT0001/2022 strain). As expected, both MPXV mRNA vaccines elicited robust levels of antibodies against those antigens, whereas IFN-α and the LNPs did not induce any effective antibodies (Fig. 2A–F and Table S2). The IgG titers of AALI and AAL against A29L, A35R, and L1R from both clade I and clade II MPXV were >327680, >327680, and >327680, respectively. Interestingly, antisera from the AALI and AAL treatment groups also bound to the antigens A27L and A33R, which were derived from VACV, with high antibody titers (Fig. 2G, H and Table S3). The IgG titers induced by AALI and AAL against the three MPXV antigens were 81920 (A27L) and >327680 (A33R).

A–H The breadth of the total IgG antibody response elicited by the mRNA vaccine was determined by ELISAs of pooled antisera against clade IIb MPXV of A29L (A)-, A35R (B)-, and L1R (C)-, clade Ib MPXV of A29L (D)-, A35R (E)-, L1R (F)-, and VACV of A27L (G)- and A33R (H)-antigen proteins. All the data in (A–H) are shown as the means ± SDs (n = 3 biologically independent replicates). I Fifty percent live-virus neutralizing antibody titers were determined by PRNT for clade II MPXV. n = 3 biologically independent replicates. The data are presented as the means ± SDs. J–M Specific ASC responses to the A29L, AAL, and A35R proteins were measured with an ELISpot assay. J Frequencies of A29L-specific ASCs on days 0, 21, 35, 49 and 100. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, p > 0.05. K, L, and M, Frequencies of AAL-, A29L- and A35R-specific ASCs on day 49. All the data and error bars in (K–M) represent the means ± SDs (n = 3 biologically independent replicates). Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, p > 0.05.
These findings indicate that the mRNA vaccines robustly induced broad antibody binding activity, potentially providing cross-protection. Given the trend observed for the antibody titer, AALI exhibited greater potency in inducing A29L- (from clade II and clade I MPXV), A35R- (from clade I MPXV), L1R- (from clade I MPXV) and A33R-specific antibodies than did AAL (Fig. 2A, D–F, H). Moreover, we measured antigen-specific antibodies of the IgG1 and IgG2a subclasses in antisera, which are essential for understanding B-cell somatic hypermutation and subclass switching43. The results showed that the vaccines induced high levels of A29L-, A35R-, L1R-, A27L-, and A33R-specific IgG1 and IgG2a (Fig. S3A–J), which were consistent with the total IgG results. The elevated levels of the IgG1 and IgG2a subclasses suggest the activation of both Th1 and Th2 immune responses, which are essential for B-cell maturation and subclass switching.
The MPXV mRNA vaccines induce antibodies with high binding affinity for various antigens
We purified the serum antibodies induced by AALI and evaluated their binding affinities for the MPXV and VACV antigens via SPR (Fig. S3K). The results demonstrated that AALI-induced antibodies exhibited strong binding affinities for the clade II MPXV antigens A29L, A35R, and L1R, with KD values of 2.35 × 10−9 M, 8.40 × 10−8 M, and 6.0 × 10−8 M, respectively (Fig. S3L–N). Similarly, for clade I MPXV, the KD values for the A29L, A35R, and L1R antigens were 1.724 × 10−8 M, 6.361 × 10−9 M, and 2.406 × 10−8 M, respectively (Fig. S3O–Q). Additionally, the antibodies bound to the A27L protein derived from VACV, with a KD value of 2.23 × 10−7 M (Fig. S3R). These high-affinity interactions with both the MPXV and VACV antigens suggest that immunization with the MPXV mRNA vaccine induces broad cross-reactive protection against clade II MPXV, clade I MPXV, and VACV.
Neutralizing antibody responses were assessed via live-virus neutralization tests using a clade II MPXV strain. As shown in Fig. 2I, compared with the IFN-α, LNPs, and PBS groups, the AALI and AAL mRNA vaccines induced high neutralizing antibody titers after booster immunization, which prevented MPXV from infecting host cells. Furthermore, the median values of the AALI- and AAL-50% neutralizing antibody titers were 1:900 and 1:300, respectively. The fusion of AAL with the adjuvant-like protein IFN-α may strengthen humoral responses. These results demonstrate that the mRNA vaccines induced potent antibody responses with high binding affinities for both the MPXV and VACV antigens, effectively neutralizing and preventing clade II MPXV infection.
The MPXV mRNA vaccines induced antibody-secreting cell (ASC) responses
ELISpot assays were conducted to measure antigen-specific B-cell responses induced by the mRNA vaccines. The kinetics of MPXV antigen-specific ASCs were tracked before and after immunization, and the results are shown in Fig. S3S. The response strength was positively correlated with vaccination time, peaking on day 49. At this peak, the frequencies of AAL protein (expressed in Escherichia coli), A29L- and A35R-specific ASCs were significantly greater in the AALI- and AAL-immunized groups than in the IFN-α-, LNPs-, and PBS-immunized groups. Additionally, we found a statistically significant difference between the IFN-α group and the LNPs and PBS groups (Fig. 2J). Moreover, at the peak, the frequencies of the AAL-, A29L-, and A35R-specific ASCs induced by AALI were 69 ± 6, 134 ± 8 and 161 ± 17 per million, respectively, which were ~1.4-fold greater than the frequencies induced by AAL (p < 0.05, Fig. 2K–M). Notably, the vaccine-treated groups maintained robust A29L-specific ASC levels at 100 days post-third immunization, which remained significantly elevated compared with the pre-vaccination baseline, as shown in Fig. 2J. This sustained ASC presence beyond the typical plasmablast contraction phase (4–6 weeks post-immunization) strongly indicates the successful generation of memory B-cell pools—a key correlate of long-term immune protection44. In summary, the AALI and AAL mRNA vaccines effectively induced antigen-specific B-cell responses and elicited serological memory. This memory is likely sustained by long-lived plasma cells and bolstered by memory B cells, which rapidly differentiate into ASCs upon re-exposure to specific antigens. In conclusion, these results suggest that MPXV mRNA vaccines induce B cells to differentiate into MPXV-specific antibody-secreting and memory cells, producing high-titer neutralizing antibodies that may provide cross-protection against MPXV and VACV.
The MPXV mRNA vaccines induced T-cell responses
MPXV infection is controlled primarily by antibodies, with T cells assisting in the humoral response, virus clearance, and spread prevention. Here, to investigate the cellular immunity induced by the developed mRNA vaccines, we investigated CD4+ T-cell and CD8+ T-cell immune responses. We isolated splenocytes from vaccinated mice on day 14 post-vaccination to explore the T-cell immune response induced by the vaccines in vivo. As depicted in Fig. 3A, B and Fig. S4A–D, AALI, AAL, and IFN-α, but not LNPs or PBS, significantly induced CD4+ T-cell and CD8+ T-cell proliferation. Furthermore, to investigate CD4+ T-cell differentiation, we used ELISpot assays to quantify the numbers of IFN-γ- and IL-4-secreting cells, which typically represent Th1 and Th2 cells, respectively (Fig. 3C). Compared with those in the LNPs and PBS groups, the numbers of IFN-γ- and IL-4-producing splenocytes in the mRNA vaccine and IFN-α groups were significantly greater (p < 0.05, Fig. 3D). Additionally, the AALI-treated group presented a significantly greater number of these cells than did the AAL-treated group (p < 0.05). Precise measurement of cytokine expression in helper T cells is crucial for understanding the immune responses elicited by vaccines. Thus, we also assessed the serum expression levels of cytokines, including IL-4, IL-6, IL-13, TNF-α, and IFN-γ, following vaccination via cytometric bead assays (Fig. 3E–I). The results were consistent with those obtained in the ELISpot assay, showing that the AALI and AAL mRNA vaccines significantly induced the upregulation of all these cytokines compared with LNPs alone and PBS. Notably, immunization with AALI and IFN-α induced greater expression of the investigated cytokines than did immunization with AAL. Taken together, these results show that immunization with the mRNA vaccines activated Th1 and Th2 cells, which are necessary for immune responses to clear virus infection.

A Splenocytes isolated from immunized mice in each group on day 14 were examined by flow cytometry. The clusters of CD3+CD4+ and CD3+CD8+ cells represented the CD4+ (above) and CD8+ T cells (below). B Summary data for the fractions of CD4+ and CD8+ T cells in different groups. The data and error bars represent the means ± SDs (n = 3 biologically independent replicates). Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, p > 0.05. C Splenocytes were plated in ELISpot wells, and IFN-γ and IL-4 cytokine-producing cells were detected. D Frequencies of IFN-γ- and IL-4 cytokine-producing cells on day 14. The data and error bars represent the means ± SDs (n = 3 biologically independent replicates). Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, p > 0.05. E–I Production of cytokines, including IL-4, IL-6, IL-13, TNF-α, and IFN-γ, in the serum of patients treated with the mRNA vaccines. All the data and error bars in (E–I) represent the means ± SDs (n = 3 biologically independent replicates). Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, p > 0.05. J Equal numbers of CFSEhigh-labeled A35R-pretreated cells and CFSElow-labeled BSA-pretreated splenocytes were mixed and injected into immunized mice. After 12 h, the splenocytes were collected to assess antigen-specific cytolytic responses using flow cytometry. K Infected cell-specific lysis in different groups. The data and error bars represent the means ± SDs (n = 3 biologically independent replicates). Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; ns, p > 0.05.
The cytotoxic effects of CD8+ T cells are crucial for the elimination of infected cells and play an important role in the effective control of MPXV spread. To evaluate whether the developed mRNA vaccines could promote antigen-specific CD8+ T-cell responses, we intravenously administered a mixture of equal numbers of CFSEhigh-labeled A35R-pretreated cells and CFSElow-labeled BSA-pretreated splenocytes into immunized mice. Specific cytotoxic T lymphocyte (CTL) activity was then assessed via flow cytometry after 12 h. As shown in Figs. 3J and S4E, the fluorescence intensity of the A35R-pretreated cells surpassed that of the BSA-pretreated cells, underscoring the discernible difference in labeling. The number of residual A35R-pretreated cells (CFSEhigh) in the mice immunized with AALI or AAL was markedly lower than that in the mice immunized with IFN-α, LNPs, or PBS (Fig. 3K). Furthermore, we found no significant differences between the IFN-α- and LNPs-treated groups (p > 0.05). These findings confirmed that the mRNA vaccines induced strong antigen-specific cytotoxic responses.
Immunization with the developed mRNA vaccines provides in vivo protection against MPXV and VACV challenge
To analyze the protective efficacy of the mRNA vaccines, 3 weeks after the third immunization, the mice were intranasally challenged with 4 × 105 focus-forming units (FFUs) of the clade IIb MPXV hMpxV/China/SZ-SZTH42/2023 or hMpxV/China/SZ-SZTH41/2023 strain or 8 × 104 FFUs of the VACV Tiantan strain45, as shown in Fig. 4A. We separated the lung tissues and measured the viral load of MPXV SZTH42 on days 4 and 7 post-infection via real-time quantitative polymerase chain reaction (qPCR) analysis and a focus formation assay. As expected, the number of infectious viral particles in the mice immunized with AALI or AAL was significantly lower than that in the mice in the IFN-α- and LNPs-treated groups on day 4. Additionally, the lungs of the mice in the IFN-α group had fewer infectious viral particles than those in the LNPs group (Fig. 4B). Similar results were observed via qPCR analysis of the number of viral E9L gene copies in the mice in each group46 (Fig. 4C). The relative viral load in the lungs of the mice in the AALI- and AAL-treated groups was significantly lower than that in the lungs of those in the mice in the IFN-α and LNPs groups at 4 days, indicating that the MPXV-specific antibodies induced by the mRNA vaccines effectively neutralized and inhibited infection in the lung tissue. Additionally, by 7 days post-infection, the viral load sharply decreased in the AALI and AAL groups but continued to increase in the IFN-α and LNPs groups. These findings suggest that the CTLs induced by AALI and AAL rapidly cleared MPXV-infected cells, reducing the spread of the virus. The greater reduction in MPXV load in the AALI group than in the AAL group at 7 days post-infection suggests that AALI offers slightly better protection.

A Mice were infected intranasally with the clade II MPXV SZTH41, SZTH42 strains or VACV Tiantan strain 21 days after the third immunization. Created in BioRender. gN, \. (2025) https://biorender.com/k8huiqq. B The viral load in the lung was detected on days 4 and 7 post-infection via a focus-forming assay. n = 3 biologically independent mice. Data are presented as means ± SD. C The viral copy numbers of MPXV SZTH42 in the lungs, which were derived from the same batch of lung tissue samples as shown in (B), were quantified by qPCR. n = 3 biologically independent mice. The data are presented as the means ± SDs. D (clade II MPXV SZTH42 strain), E (clade II MPXV SZTH41 strain), and F (VACV Tiantan strain) depict the percentage change in body weight monitored 7–14 days post-challenge. For D–F n = 6 biologically independent mice. G (clade II MPXV SZTH42 strain), H (clade II MPXV SZTH41 strain), and I (VACV Tiantan strain) present the survival curves of BALB/c mice following challenge. For G–I n = 6 biologically independent mice. J Histopathology of lung tissue on day 7 post-infection with the MPXV clade II strain SZTH42 (scale bar = 200 μm). The green arrows indicate macrophage, lymphocyte, or granulocyte infiltration in alveolar spaces; the red arrows highlight alveolar hemorrhage; the yellow arrows denote peribronchial infiltration; and the black arrows indicate bronchial epithelial shedding. Untreated and unchallenged naive mice served as controls. K Distribution of MPXV (yellow for A35R), lung-infiltrating CD8+ T cells (green for CD8) and lung-infiltrating macrophages (red for F4/80); scale bar = 100 μm. L Relative quantification of the fluorescence intensity of A35R, CD8, and F4/80. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by one-way ANOVA with Tukey’s multiple comparison tests. *, p < 0.5; **, p < 0.01; ***, p < 0.001; ns, p > 0.05.
The AALI- and AAL-immunized mice experienced a reversible loss of body weight at 2 days and exhibited minimal overall weight loss, whereas the mice in the LNPs- and IFN-α-treated groups continued to lose weight, eventually losing more than 25% of their body weight (Fig. 4D–F). By monitoring the survival of the mice, we found that both AALI and AAL provided 100% cross-protection against the clade II MPXV SZTH42, MPXV SZTH41, and VACV Tiantan strains, as illustrated in Fig. 4G–I. In addition, histopathological analysis revealed the absence of visible pathological changes in the lungs of the AALI- and AAL-immunized mice. In stark contrast, the LNPs- and IFN-α-treated groups displayed severe alveolar damage and interstitial inflammatory infiltration (Fig. 4J) on day 7 post-infection. In conclusion, multiple findings demonstrate that our vaccines offer strong cross-protection against MPXV and VACV challenge in vivo.
To evaluate the inflammatory damage caused by MPXV infection, we examined lymphocyte infiltration into the lungs of the mice via immunohistochemistry (IHC). As expected, the common leukocyte antigen CD45 was widely detected in the lung sections of the IFN-α- and LNPs-treated mice but not in those of the uninfected naive mice (Fig. S4F). Compared with IFN-α and LNPs treatment, immunization with AALI or AAL before MPXV infection substantially reduced the number of lung-infiltrating CD45+ leukocytes (Fig. S4G), suggesting the mitigation of inflammatory lung damage. Additionally, immunofluorescence analysis of lung sections was performed using fluorescent antibodies targeting A35R, F4/80, and CD8 to assess the distribution of MPXV, infiltrating macrophages, and CD8+ T cells (Fig. 4K). As shown in Fig. 4L, the intense yellow fluorescence signals of A35R on day 7 post-infection indicated widespread MPXV distribution in the lungs of the IFN-α- and LNPs-treated mice. In contrast, A35R fluorescence was weaker in the lungs of the AALI- and AAL-vaccinated mice. Compared with the IFN-α and LNPs control groups, the AALI- and AAL-immunized groups presented significantly increased lung CD8+ T-cell distributions (p < 0.01), which was consistent with the CTL assay results. These CD8+ T cells effectively cleared virus-infected cells, leading to reduced viral loads, as indicated by decreased A35R fluorescence. In contrast, the IFN-α- and LNPs-treated mice presented greater macrophage staining intensities than did the AALI- and AAL-immunized mice (p < 0.05). This increased macrophage activity was associated with increased inflammation and delayed viral clearance in the IFN-α and LNPs groups47. These findings underscore the robust protective efficacy of AALI and AAL immunization against MPXV challenge in vivo.
Single-cell transcriptional profiling of splenocytes isolated from mice vaccinated with the MPXV mRNA vaccine
To characterize the immunological features of BALB/c mice vaccinated with the AALI MPXV mRNA vaccine, we conducted droplet-based scRNA-seq using the 10× Genomics platform. Additionally, we performed single-cell TCR and BCR sequencing to comprehensively evaluate the transcriptomic profiles of spleens obtained from three AALI-treated and three LNPs-treated BALB/c mice, which served as controls (Fig. 5A). After employing a unified single-cell analysis pipeline, we sequenced a total of 44333 cells derived from the spleen across all the samples. Among these cells, 20,965 (47.3%) originated from the AALI-treated group, whereas 23,368 (52.7%) originated from the LNPs-treated group. By using graph-based clustering in uniform manifold approximation and projection (UMAP), we delineated the transcriptomes of 11 major cell types or subtypes on the basis of the expression patterns of canonical gene markers (Fig. 5B)48,49. These cells included B cells (Cd79a+Ms4a+), CD4+T cells (Cd3d+Cd3e+Cd4+), CD8+T cells (Cd3d+Cd3e+Cd8a+Cd8b+), DCs (Siglech+Cd83+Cst3+), monocytes (Cd14+Lyz2+Csf1r+Ccr2+), natural killer cells (NK cells: Nkg7+Fcer1g+), plasma cells (Cd38+Mzb1+), hematopoietic stem and progenitor cells (HSPCs: Kit+Sox4+Gata2+Cd34+), erythrocytes (Gypa+Slc4a1+), endothelial cells (Pecam1+Igfbp7+) and neutrophils (Ly6g+S100a9+S100a8+) (Fig. 5C–E). Thus, we effectively delineated the splenic immune cell landscape in BALB/c mice receiving the developed mRNA vaccines.

A A schematic diagram of the overall study design. Splenocytes from AALI-treated (n = 3) and LNPs-treated mice (n = 3) were subjected to scRNA-seq gene expression profiling and TCR and BCR profiling analyses. Created in BioRender. gN, \. (2025) https://biorender.com/ac9szf7. B Cell populations were identified via UMAP projection, which revealed 11 major cell types across six samples. Each dot represents a single cell, colored according to its respective cell type. C Canonical cell markers were utilized to label clusters by cell identity in the UMAP plot, with data colored on the basis of expression levels via a log-scale legend. D Violin plots illustrating the expression distribution of selected canonical cell markers across 11 clusters, with rows representing marker genes and columns representing clusters. E Dot plot depicting cell type marker genes across 11 distinct cell types from spleen samples. F Average proportion of each cell type derived from the two groups. G Distribution of the average percentage of each immune cell type across the two conditions. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by unpaired two-sided Student’s t-tests. *, p < 0.05; ns, p > 0.05.
Next, to gain further insight into the immune cell dynamics in MPXV mRNA-vaccinated mice, we employed scRNA-seq to analyze the distribution of major splenic immune cell populations. By quantifying the percentages of 11 key cell types in the spleens of vaccinated mice, we aimed to characterize the immune cell landscape post-vaccination (Fig. 5F). Our analysis revealed a noteworthy finding: the percentage of plasma cells in the spleens of the mice subjected to AALI was significantly greater (p < 0.05) than that in the LNPs control group on day 7 post-vaccination. This elevation in plasma cell abundance likely reflects increased production of protective neutralizing antibodies directed against MPXV, contributing to the efficacy of the vaccine. While the increases in monocyte and DCs percentages were not statistically significant (p > 0.05), these results should be considered potential trends given their biological context and the mechanisms of vaccine action, providing preliminary insights for future studies (Fig. 5G). These cells play pivotal roles in orchestrating the immune response by presenting antigens and activating other immune effectors. Interestingly, we also observed a postvaccination increase in neutrophils, which play key roles in both innate and adaptive immunity, including in pathogen killing, increasing antigen presentation, and modulating immune cell interactions50. Collectively, these findings provide a comprehensive view of immune cell dynamics post-vaccination, highlighting the specific cell types that may be crucial for mediating protection against MPXV.
Expression profiles of B cells after vaccination
To characterize changes in individual B-cell subsets post-vaccination, we subclustered splenic B cells (23,824) and identified six subsets on the basis of B-cell markers and signature genes (Figs. 6A, B and S5A, B): naive follicular B cells (Ighd+Sell+Fcer2a+), marginal zone B cells (MZ B, Ighd+Cd38+Cr2+), pre-B cells (Ms4a1+Vpreb3+), plasma cells (Mzb1+Jchain+Xbp1+), plasmablast cells (Mzb1+Cd38+Ms4a1+), and debris. Notably, the proportions of active B-cell subsets, including plasma B cells and MZ B cells, were greater in the AALI-treated groups than in the control groups, whereas the proportions of naive follicular B cells were significantly lower (Figs. 6C and S5C).

A UMAP projection of B cells, with each dot representing a single cell and colored by cell type. B Violin plots illustrating the expression distribution of selected canonical cell markers across six clusters, with rows representing marker genes and columns representing clusters. C Distribution of the average percentage of each immune cell type across the two groups. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by unpaired two-sided Student’s t-tests. *, p < 0.05; **, p < 0.001; ns, p > 0.05. D GO terms are labeled with names and IDs and are sorted by −log10 p values. E Volcano plot showing the DEGs between the AALI- and LNPs-treated samples. DEGs refer to genes with a Benjamini‒Hochberg adjusted p value ≤ 0.05. F Bar plot showing the percentages of IGHA, IGHD, IGHG, and IGHM expression in the two groups. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by unpaired two-sided Student’s t-tests. *, p < 0.05; **, p < 0.001; ns, p > 0.05. G Bar plot showing the percentages of IGHG1, IGHG2B, IGHG2C, and IGHG3 expression in the two groups. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by unpaired two-sided Student’s t-tests. *, p < 0.05; **, p < 0.001; ns, p > 0.05. H UMAP plot showing BCR detection. I UMAP plot showing the BCR clone status distribution. J Stacked bar plots showing the multiclonal state of BCR in each sample. K Circos plots showing BCR chain rearrangements in the two groups. Arc lengths indicate VDJ gene family frequencies; colored ribbons represent paired segment usage. L The CDR3 motifs in the IGH, IGK, and IGL chains that are unique to the AALI-treated group were predicted after excluding sequences that were shared with the LNPs-treated group. The label on the x-axis represents the position of the CDR3 amino acid, and the y-axis represents the bits.
To further investigate differential transcriptomic changes in B cells after vaccination, we compared the expression profiles of B cells between the vaccine-treated group and the control group. Initially, we employed Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses to illuminate the functional aspects of B cells (Fisg. 6D and S5D). As anticipated, genes associated with “positive regulation of the MHC class I biosynthetic process” and “antigen processing and presentation” were enriched following MPXV mRNA vaccination, indicating that the presentation and conversion of antigens from the vaccine might be initiated. Additionally, genes related to “protein processing in the endoplasmic reticulum” and “positive regulation of peptide secretion” were enriched in B cells, indicating a heightened demand for protein synthesis and assembly during antibody production. The “Toll-like receptor signaling pathway” and “NOD-like receptor signaling pathway”, which play critical roles in regulating the adaptive immune response, were also enriched after vaccination. Genes associated with the “response to viral infection” were also notably enriched in B cells, suggesting that vaccination triggered a robust antiviral response, which was likely effective against MPXV infection. Furthermore, other biological pathways involved in host immune responses, including the “B-cell receptor signaling pathway” and “positive regulation of the inflammatory response”, were significantly enriched after MPXV mRNA vaccination. Next, we observed significant upregulation of differentially expressed genes (DEGs) in B cells, including Rpl37a, Jchain, Igkv4-91, Igkv8-19, Ighm, and Iglc1 (Fig. 6E), which are involved in antibody production, clonal expansion, and V(D)J gene utilization. These findings suggest that our vaccine activated B cells, potentially leading to distinctive and specific V(D)J rearrangements post-vaccination.
At 7 days after vaccination, B cells expressed genes encoding the constant regions of IgG, IgD, IgA, and IgM (Fig. S5E). Compared with the LNPs-treated samples, the AALI-treated samples presented a significant increase in IgG and a decrease in IgM expression (Fig. 6F), suggesting the occurrence of clonal expansion or a humoral immune response in the MPXV mRNA-vaccinated mice. Further analysis revealed a significant increase in the expression of the Ighg1 and Ighg2c subtypes post-vaccination, which was consistent with the ELISA results (Fig. 6G). Although Ighg2b and Ighg3 expression also tended to increase, the difference was not statistically significant (p > 0.5). The elevated expression of Ighg isotypes suggests B-cell responsiveness to Th1 and Th2 cells, which regulate class switching to IgG1 and IgG2, respectively. In conclusion, our scRNA-seq data revealed high levels of transcriptional specialization in B cells post-vaccination, validating differences in B-cell protective responses between the vaccine- and LNPs-treated groups.
Expansion of B cells and specific rearrangements of V(D)J genes after vaccination
B cells achieve substantial antigen recognition diversity through unique BCR specificity, enabling broad detection of antigens51. Only a minor subset undergoes antigen-driven clonal expansion, transitioning from single-cell clones (clone = 1) to expanded populations (clone ≥2)52. To assess the impact of the MPXV mRNA vaccine on BCR transcripts, we reconstructed BCR sequences and analyzed clonal expansion post-vaccination. Over 77% of the BCRs were detected in each cluster (Fig. 6H, I). To assess the impact of the MPXV mRNA vaccine on BCR transcripts, we reconstructed BCR sequences and analyzed clonal expansion post-vaccination. Notably, comparative analysis of BCR expanded clones between the AALI- and LNPs-treated groups revealed a greater proportion of multiclonal BCRs (clone ≥2) in the vaccine group than in the control group (Fig. 6J). Although there was no statistically significant difference (p > 0.5), the observed trend suggests that the AALI vaccine may have the potential to induce B-cell clonal expansion, as shown in Fig. S5F. Additionally, most of the top 20 clone types were present in the vaccine-treated samples (Fig. S5G). These findings indicate strong activation of B-cell activity and humoral immune responses, which is consistent with the increased antibody titers observed by ELISAs and focus reduction neutralization tests (FRNTs). To delve deeper into the biased V(D)J rearrangements within BCRs in response to specific stimuli, we conducted a comparative analysis of V(D)J gene utilization patterns across two distinct groups, focusing on the immunoglobulin heavy chain variable region (IGHV), immunoglobulin kappa chain variable region (IGKV), and immunoglobulin lambda chain variable region (IGLV) segments (as depicted in Fig. 6K). Our gene usage analysis revealed preferential selection of certain V(D)J genes, notably Ighv1-19, Ighv3-1, and Ighv9-3, among IGHVs; Igkv1-135 and Igkv10-96, among IGKVs; and Iglv1, among IGLVs, which were more prevalent than other germline counterparts. Intriguingly, after vaccination, we observed notable overrepresentation of Ighv1-5 and Igkv1-110, suggesting a potential role in the immune response to vaccination (as evident in Figs. 6K and S5H). Furthermore, our analysis revealed the clonal expansion of specific lineages, including Ighv1-72, Ighv8-4, and Igkv8-34, which was exclusive to mice treated with AALI (as shown in Figs. 6K and S6A). This exclusive expansion suggests that these genes encode antibodies specifically targeted against MPXV, emphasizing their potential importance in the development of an effective immune response against this virus.
CDR3 represents a hypervariable segment of the BCR due to diverse recombination processes and is considered the major determinant of antibody specificity, playing an essential role in the identification of and interaction with distinct antigenic peptides46. To identify CDR3 consensus motifs, we analyzed the amino acid sequences of the IGH, IGK, and IGL chains in the CDR3s of the AALI- and LNPs-treated groups and then predicted the motifs using the motif-based sequence analysis tool Multiple Em for Motif Elicitation (MEME)53. The top five highest-ranked IGH, IGK, and IGL motifs were obtained for each group of BCRs on the basis of sequence conservation and the E value (Table S4), and the results (E value < 0.05) are shown in Fig. S6B, C. We identified the unique CDR3 motifs in the BCRs from the immune and control groups (as shown in Table S4 for the AALI and LNPs groups, respectively). To specifically identify BCR clonotypes induced by the vaccine and minimize interference from the baseline immune repertoire, we excluded the common BCR sequences that were shared between the AALI- and LNPs-treated groups from the AALI-treated group (as shown in Table S4, labeled AALI-LNPs). Motif analysis was then performed solely on the post-immunization BCR sequences, as shown in Fig. 6L. The motifs “YAMDYW”, “CARGGY”, and “GYYYFDY” were found in the IGH chain, “CQQSSSYP” in the IGK chain, and “CALWYSNQ” in the IGL chain. These motifs are likely associated with vaccine-induced B-cell clonal expansion and selection, indicating a specific immune response to the administered antigen.
To gain insight into the BCR response to MPXV antigens, we performed molecular docking simulations of antibody motifs with conserved sequences and the AAL antigen. Specifically, we concatenated two conserved motifs: IGH sequences harboring motifs such as “GYYYFDY” or “CARGGY” and IGK sequences featuring the motif “CQQSSSYP” via a (GGGGS)3 linker. We then predicted the structures of these concatenated motifs using AlphaFold 3 and docked them with the AAL antigen using PDBePISA. The docking results demonstrated that these motifs were able to form hydrogen bonds and salt bridges with the AAL antigen, as illustrated in Fig. S6D, E and Tables S5–S6. These findings indicate that the identified motif sequences have the potential to bind to MPXV antigens.
Expression profiles of T cells in MPXV mRNA vaccine-treated mice
To identify alterations in T-cell subsets after vaccination, we subclustered T cells (11,474) from the spleen and identified 7 subsets on the basis of the expression and distribution of canonical T-cell markers (Figs. 7A, B and S7A, B): naive CD4+ T cells (Cd4+Ccr7+Sell+), Th1 cells (Tbx21+Cd40lg+Ifng+), Th2 cells (Gata3+Maf+Ccr4+Cd44+), Th17 cells (Ccr6+Il23r+Rorc+Il17a+), CD8+ effector T cells (Cd8a+Cd8b1+Prf1+Lamp1+), Treg cells (Foxp3+Ctla4+Ikzf2+) and naive NK-T cells (Ccr7+Sell+Cd8a+Nkg7+Cd8b1+). The abundance of the Th1, Th2, Th17, and CD8 effector subsets increased in the AALI mRNA-vaccinated mice, although the differences were not statistically significant (p > 0.5), suggesting robust induction of the cellular immune response (Figs. 7C and S7C). These T-cell composition changes align with findings from the ELISpot and flow cytometric assays.

A UMAP projection of T cells, with each dot representing a single cell and colored by cell type. B Violin plots depict the expression distribution of selected canonical cell markers across seven clusters, with marker genes in rows and clusters in columns. C Distribution of the average percentage of each immune cell type across the two groups. n = 3 biologically independent replicates. The data and error bars represent the means ± SDs. Statistical significance was calculated by unpaired two-sided Student’s t-tests. *, p < 0.05; **, p < 0.001; ns, p > 0.05. D GO terms are annotated with names and IDs, sorted by decreasing −log10 p values. DEGs denote genes with Benjamini‒Hochberg adjusted p values ≤ 0.05. E Volcano plot showing the DEGs between the AALI- and LNPs-treated samples. F Heatmap illustrating the expression of cytotoxic gene signatures in CD8+ T cells across the AALI- and LNPs-treated groups. The minimum signal is represented in blue, and the maximum signal is depicted in red. G Heatmap of the expression of the IFN response signature in T cells from all samples. The minimum signal is represented in blue, and the maximum signal is depicted in red. H Circos plots depicting the rearrangements of TCR chains in the two groups. Arc lengths reflect the relative frequency of VDJ gene families, whereas colored ribbons indicate the frequency of paired VDJ gene segments. I The CDR3 motifs for TRA and TRB chains that are unique to the AALI-treated group were predicted after sequences that were shared with the LNPs-treated group were excluded. The label on the x-axis represents the position of the CDR3 amino acid, and the y-axis represents the bits.
To characterize transcriptomic changes in T cells between the AALI-vaccinated groups and LNPs-treated groups, we compared the expression profiles of CD4+ and CD8+ T cells at 7 days after vaccination (Figs. 7D, E and S7D, E). As expected, genes associated with “negative regulation by the host of viral transcription”, “defense response”, “regulation of T-cell differentiation” and “positive regulation of cell activation” were significantly enriched in CD4+ and CD8+ T cells after vaccination, indicating an ongoing host immune response against MPXV (Figs. 7E and S7F–H). Genes associated with “regulation of cell motility”, “leukocyte migration involved in the inflammatory response”, and “positive regulation of peptide secretion” were enriched in CD8+ T cells (Fig. S7E). This enrichment suggests that CD8+ T cells may have a targeted killing effect on MPXV-infected cells. At the bulk cell level, we also observed increased expression of several typical cytotoxic genes, such as Nkg7, Gzma, Gzmb, Klrk1, Klrd1, Ctsw and Cst7, in effector-state T cells after vaccination (Fig. 7F). Similar to the GO and KEGG findings for B-cell subsets, genes associated with “NF-kappaB binding”, “antigen processing and presentation of peptide antigen”, and the “JAK-STAT signaling pathway” were enriched, indicating consistent activation of host immune responses after vaccination in splenic immune cells. Moreover, pathways involved in antiviral immune responses, such as “regulation of germinal center formation”, “regulation of B-cell differentiation”, and “positive regulation of the inflammatory response” were significantly enriched post-vaccination. Given the critical role of IFN in viral infection clearance, we compared a panel of proinflammatory and IFN-γ hallmark gene signatures in T cells (Fig. 7G). Our analyses revealed the activation of proinflammatory and IFN-γ response-related genes on day 7 post-vaccination, which was consistent with the enrichment of “cellular response to type II interferon” pathways identified in our GO analyses (Fig. 7D). In conclusion, our scRNA-seq analysis revealed a high degree of transcriptional specialization in both CD4+ and CD8+ T cells following vaccination. This finding corroborates the role of T cells in assisting B cells in producing antibodies and in the clearance of infected cells.
Clonal expansion of T cells and preferred usage of V(D)J genes after vaccination
Like B cells, T cells are also highly diverse, with each bearing a unique TCR that recognizes a specific antigen54. Only a small subset of T cells can recognize antigens and undergo immune expansion, shifting from clone = 1 to clone ≥2. To elucidate the clonal relationships among T cells and analyze the utilization of V(D)J gene segments following vaccination, we performed TCR V(D)J sequencing and conducted a comprehensive analysis by integrating scRNA-seq with V(D)J-seq. In brief, more than 80% of the cells in all subsets had matched TCR information, except for the Cd8 effector and Th17 subsets (Fig. S7I). Notably, evident TCR clonal expansion (clone ≥2) was observed in the AALI-treated groups compared with the LNPs-treated groups (Fig. S7J, K). Moreover, the majority of the top 20 clone types were present in the vaccine-treated samples (Fig. S7L), suggesting that the mRNA vaccine could induce the amplification of AALI-specific T cells. To explore the dynamics and gene preferences of the TCR after vaccination, we compared the usage of V(D)J genes between the two groups. Notably, we observed a decrease in the diversity and distinct usage of V(D)J genes, particularly TRB genes, after vaccination (Fig. S7M). Compared with the LNPs-treated mice, the AALI-treated mice presented overrepresentation of Trav7d-2, Trav12n-2, Trbv13-3, and Trbv29, with Traj15 and Traj31 as the preferred TRAJ genes and Trbj1-1 as the preferred TRBJ gene (Fig. S8A). Additionally, the most distinct paired V‒J frequencies of TRA and TRB in the vaccine groups were Traj18/Trav11 and Trbvj2-4/Trbv12-2 + Trbv13-2, respectively (Fig. 7H). These findings suggest that AALI treatment elicits a unique TCR repertoire, potentially increasing the specificity and magnitude of the immune response to vaccination. To elucidate the differences in the TCR repertoire after immunization, we performed motif analysis of the CDR3 sequences of the TRA and TRB chains, which are critical elements for the identification of antigenic peptides, via MEME. The top five TRA and TRB motifs for each group were identified and ranked according to their sequence conservation and E values, as detailed in Table S7, and the results (E value < 0.05) are shown in Fig. S8B, C. Both the AALI- and LNPs-treated groups presented similar predominant motifs within their TCR repertoires, as shown in Table S7 for the AALI and LNPs groups, respectively. This observation indicates that these motifs may represent a baseline or conserved component of the TCR repertoire across treatment groups.
To specifically identify TCR clonotypes induced by the vaccine and minimize interference from the baseline immune repertoire, we excluded the common TCR sequences that were shared between the AALI- and LNPs-treated groups from the AALI-treated group (as shown in Table S7, labeled AALI-LNPs). This resulted in the unique motifs “CALSEG” and “GGNNKKLTF” in the TRA chain and “SSGGGGNT” in the TRB chain (Fig. 7I). The discovered motifs in the CDR3 sequences of the TCR after immunization suggest that the diversity of the vaccine-induced immune repertoire is increased and that these conserved amino acid regions in CDR3 play important roles in the immune response. Overall, increased T-cell clonality and skewed usage of TCR V(D)J genes post-vaccination suggest that AALI-associated V(D)J rearrangements occur in host T cells. Notably, the selective usage of dominant TRA and TRB genes, as well as the discovery of TRA and TRB motifs, indicate that various immunodominant epitopes may shape the molecular profile of T-cell responses, potentially aiding in vaccine design.
Discussion
Owing to the resurgence of mpox in 2022, which was driven by MPXV infection, efficacious and safe vaccines are urgently needed. In this study, we developed two innovative multivalent mRNA vaccine candidates and systematically examined the resulting cellular and humoral immune responses to MPXV mRNA vaccination. Using single-cell transcriptome sequencing, we obtained detailed insights into the TCR and BCR repertoires, increasing our understanding of antiviral mechanisms at single-cell resolution.
We developed two MPXV mRNA vaccines, AALI and AAL, as a preliminary step toward the development of a mpox vaccine. The AALI vaccine comprises an AAL domain encoding the MPXV antigens A35R, A29L, and L1R as immunogens, along with an IFN-α domain, which serves as an adjuvant-like molecular chaperone. Additionally, we designed an AAL vaccine variant without IFN-α. Furthermore, we modified LNPs, a safe and effective mRNA delivery system, with mannose (Man-LNPs) to target DCs. This modification aims to enhance immune responses, as DCs are potent APCs crucial for linking innate and adaptive immunity. Our study provides evidence for the immunogenicity and efficacy of these mRNA vaccine candidates against MPXV, demonstrating the induction of humoral immune responses and the engagement of cellular immunity in defense against MPXV in mice.
The antisera generated from mice immunized with AALI- and AAL-specific antigens exhibited robust binding to clade Ib MPXV and clade IIb MPXV antigenic fragments A35R, A29L, and L1R, surpassing the antibody titers induced by the BioNTech BNT166 vaccine, which comprises individual mRNAs encoding the MPXV antigens A35, B6, M1, and H338. The detection of high levels of the IgG1 and IgG2a subclasses indicates the induction of both Th1 and Th2 immune responses, which are crucial for B-cell maturation and subclass switching, adding to the complexity and effectiveness of the immune response. The potent induction of VACV-specific antibodies by our vaccines is noteworthy, as it suggests potential cross-neutralization activity within the Orthopoxvirus family. This observation aligns with the high genetic homology between MPXV and VACV, particularly in their central genome regions, and the conservation of key antigenic targets such as A29L, A35R, and L1R. Such cross-reactivity could broaden the protective spectrum of our vaccines, offering protection against not only MPXV but also other Orthopoxviruses, which is a major advancement in the field. Furthermore, the SPR assays confirmed the strong binding affinities of the induced antibodies to both the MPXV and VACV antigens, reinforcing the potential for cross-protection. The ability of the antisera to specifically neutralize clade IIb MPXV, as evidenced by the inhibition of virus replication in host cells and the comparable neutralization titers to those of the BioNTech BNT166 vaccine, underscores the efficacy of our vaccines in eliciting functional antibodies38. Collectively, these discoveries indicate that the antibodies elicited by our vaccines possess a robust capacity to bind to and neutralize MPXV as well as diverse Orthopoxviruses with a high degree of affinity, underscoring their effectiveness.
To further elucidate the B-cell immune responses elicited by our vaccines, we conducted a comprehensive analysis using the ELISpot assay, which assesses splenocyte induction. Our research indicates that mRNA vaccines consistently induce long-lasting antigen-specific ASCs, which is indicative of robust memory B-cell differentiation. This effect is pivotal for a rapid adaptive immune response to MPXV re-exposure. To strengthen our conclusions, a more detailed analysis of the B-cell subsets involved would provide further insights into the durability and quality of the immune response generated by our vaccines.
To delve deeper into the immune mechanisms triggered by our AALI and LNPs vaccines at the single-cell level, we performed scRNA-seq on mouse spleens. In terms of B-cell responses, the observed proliferation of plasma cells underscores robust neutralizing antibody production, which is consistent with the ELISA results and suggests the emergence of vaccine-specific B-cell clones. Comprehensive GO/KEGG analyses provide a panoramic view of B-cell activation, encompassing a broad spectrum of processes from antigen presentation to antibody production.
BCR V(D)J sequencing post-vaccination revealed unique genetic rearrangements characterized by heightened clonality, biased gene utilization, and shifts in immunoglobulin isotype frequencies. To pinpoint vaccine-specific BCRs, we investigated the CDR3 region, the cornerstone of antibody specificity, in sequences from vaccinated and control mice. We excluded common BCR sequences shared between the AALI- and LNPs-treated groups from the AALI-treated group, uncovering distinct CDR3 motifs indicative of vaccine-driven B-cell clonal expansion and selection. Molecular docking demonstrated specific binding between BCR CDR3 motifs and MPXV antigens, highlighting two key functional implications: the critical residues in MPXV antigens define core epitopes for vaccine development, whereas conserved motifs in BCR CDR3 regions provide valuable insights for therapeutic monoclonal antibody discovery. However, docking results alone cannot confirm the protective efficacy against MPXV. Future studies are needed to experimentally validate BCR-MPXV interactions and characterize their immunological role.
With respect to T-cell immune responses, the investigated mRNA vaccines strongly promoted cellular immunity, particularly the functionality of CD4+ and CD8+ T cells. This conclusion was strongly supported by a comprehensive set of experiments, including ELISpot assays and flow cytometric analyses, which together provided a comprehensive picture of vaccine-induced immune activation. Upon activation, CD4+ T cells differentiate into distinct subsets with specialized functions. Specifically, Th1 cells secrete IFN-γ, which plays a pivotal role in promoting IgG2a class switching, thereby bolstering the humoral immune response against pathogens. Conversely, Th2 cells secrete IL-4, facilitating IgG1 class switching and contributing to viral clearance through distinct mechanisms. Our ELISpot analysis conclusively demonstrated that the mRNA vaccines potently promote both Th1 and Th2 immune responses, as indicated by the notable upregulation of IFN-γ- and IL-4-producing CD4+ T cells. Additionally, the high expression of cytokines, specifically IL-4, IL-6, IL-13, TNF-α, and IFN-γ, in helper T cells strengthens our conclusion.
Moreover, CD8+ T cells have emerged as a critical component of the antiviral arsenal, demonstrating their ability to eliminate virus-infected cells and thereby curtail viral dissemination within tissues50. In vivo CTL assays provided compelling evidence for the specific activation of MPXV-targeted CTLs. Notably, mice immunized with the AALI and AAL vaccines displayed good efficacy in killing cells infected with the A35R antigen, whereas they spared cells expressing an unrelated antigen (BSA), indicating the specificity and potency of the induced CTL responses. Collectively, these findings underscore the capacity of mRNA vaccines to elicit robust and well-coordinated T-cell responses, encompassing both helper (CD4+) and killer (CD8+) T-cell functions. This comprehensive immune activation strategy ensures robust defense against viral challenges, with Th1 and Th2 cells coordinating humoral immunity and CD8+ T cells executing direct antiviral effects.
To strengthen our conclusions, an in-depth single-cell analysis of T-cell responses is pivotal. Vaccination with AALI mRNA induced alterations in T-cell subsets, with increased proportions of Th1, Th2, Th17 and CD8 effector T cells, suggesting a robust cellular immune response that aligns with the ELISpot and flow cytometric assay findings. GO analysis revealed enriched pathways linked to T-cell activation, proliferation, differentiation, migration, and defense mechanisms, indicating the potent induction of T-cell cytotoxicity by MPXV mRNA vaccines. TCR V(D)J immune repertoire sequencing provided insights into T-cell clonal dynamics and V(D)J gene utilization post-vaccination. Compared with the LNPs control mice, AALI-treated mice presented enrichment of TCR V genes (Trav7d-2, Trav12n-2, Trbv13-3, and Trbv29), with preferential Traj15, Traj31, and Trbj1-1 usage. The predominant V-J pairs in vaccinated mice were Traj18/Trav11 for TRA and Trbj2-4/Trbv12-2 + Trbv13-2 for TRB, highlighting AALI-induced TCR rearrangements. These findings suggest that the vaccine elicits a targeted and specific T-cell response at the TCR diversity level. Analysis of the CDR3 sequences in the immunized group revealed conserved motifs, including “CALSEG” and “GGNNKKLTF” in TRA and “SSGGGGNT” in TRB, suggesting their functional relevance in T-cell responses. Our study provides a comprehensive immunological profile post-vaccination at the single-cell transcriptome level, offering insights into MPXV mRNA vaccine-induced immunity.
Next, we evaluated the efficacy of our mRNA vaccines by isolating a clade IIb MPXV strain from clinical cases and establishing a robust mouse infection model. Our results demonstrated that both the AALI and AAL vaccines conferred significant cross-protection against clade IIb MPXV and VACV infection, substantially reducing viral loads and mitigating lung inflammation. Within 4 days of vaccination with the mRNA vaccine, the viral loads in the lungs were significantly reduced, demonstrating the effective neutralization capability of the MPXV-specific antibodies induced by these vaccines. By day 7 post-infection, the sharp decline in viral loads observed in the vaccinated groups contrasted with the continued rise in the IFN-α and LNPs groups, indicating that CTLs, which were also induced by the mRNA vaccines, rapidly cleared MPXV-infected cells, thereby limiting viral spread. Notably, both the AALI and AAL vaccines achieved a remarkable 100% protection against both clade IIb MPXV and VACV, underscoring their potential in the development of a universal Orthopoxvirus vaccine. However, the AALI vaccine distinguished itself by comprehensively bolstering immune responses, specifically eliciting increased antibody production and robust T-cell activation, thereby exhibiting a superior protective profile compared with that of the AAL vaccine. ACAM2000 and JYNNEOS, while effective against MPXV, have notable limitations. There are safety concerns associated with ACAM2000, an attenuated virus vaccine, particularly in immunocompromised individuals. Additionally, the limited availability of JYNNEOS underscores the urgent need for a vaccine that can be rapidly developed and widely distributed. Our vaccine candidates represent a promising solution, offering a safe, effective, and rapidly deployable option to address the ongoing mpox threat.
We established a pathogenic BALB/c mouse model for clade IIb MPXV via respiratory infection. Building on this foundation, like other researchers, we utilized the respiratory challenge model to systematically evaluate cross-reactive immune responses and compare vaccine efficacy against both MPXV and VACV12. While mouse models have inherent translational limitations due to species-specific differences in symptom severity and immune responses, including B-cell activation and cytokine profiles, they remain indispensable for foundational research. Integrating them with other model systems will be essential for improving the translatability of vaccines and therapeutic development55. Given the demonstrated safety and cross-protection of our vaccines against both VACV and MPXV, we believe this provides a strong foundation for clinical trials. Since variations in human leukocyte antigen (HLA) alleles can influence immune responses to the vaccine, we recommend that future clinical trials include participants from diverse genetic backgrounds to assess the vaccine’s effectiveness and adaptability across different populations.
In summary (Fig. S9), confronted with the mpox outbreak, we successfully developed two pioneering mRNA vaccines targeting the IMV- and EEV-specific antigens A29L, A35R, and L1R. The vaccines target DCs to deliver mRNA sequences through mannose-modified LNPs, enabling the DCs to present the fusion protein to CD4+ and CD8+ T cells using the MHC II and MHC I pathways. This differentiation of CD4+ T cells into Th1 and Th2 subsets involves collaboration with B cells to produce neutralizing antibodies against MPXV and VACV, while CD8+ T cells become antigen-specific CTLs that eliminate MPXV-infected cells. Furthermore, the application of single-cell transcriptomic sequencing, complemented with detailed TCR- and BCR-based lineage tracing, has offered a comprehensive understanding of the intricate humoral and cellular immune responses triggered by these MPXV mRNA vaccines. These discoveries underscore the potential use of mRNA vaccines as a pioneering clinical approach against mpox, emphasizing their capacity to elicit robust protective responses and providing intricate insights into the comprehensive immune landscape, thereby allowing us to address both present and future Orthopoxvirus outbreaks effectively.
Methods
Research ethics statement
We confirm that the research was conducted following ethical standards for research involving animals. This study was approved by the Ethics Committee of the Shenzhen Third People’s Hospital, China (approval number: 2023-019), and by the Institutional Animal Care and Use Committee of Zhongke Industry Holdings (Shenzhen) Co., Ltd. (Permission ID: 202300181).
Animals
Female BALB/c mice (8–10 weeks old) were purchased from Guangdong Medical Laboratory Animal Center (Foshan, China) and kept at Zhongke Industry Holdings (Shenzhen) Co., Ltd, under controlled conditions (22 ± 1 °C, 50–60% humidity, 15–20 air changes/h) with a 12-h light/dark cycle. The mice were group-housed (5 per cage) in individually ventilated polycarbonate cages (360 × 200 × 150 mm) containing sterile corn cob bedding. Unless stated otherwise, all the mice in the animal facility were provided unrestricted access to sterile food and water, with feeding racks positioned to encourage foraging behavior.
All animal experiments were conducted using protocols approved by the Institutional Animal Care and Use Committee of Zhongke Industry Holdings (Shenzhen) Co., Ltd. (Permission ID: 202300181). During the experiments, the mice were randomly assigned to experimental groups, and the investigators were blinded to group assignments through coding of the experimental groups. The group allocation codes were not revealed until after the completion of data collection and analyses. We adhered to the Guide for the NIH Guidelines for the Care and Use of Laboratory Animals throughout all experimental procedures and ensured compliance with all applicable ethical regulations related to animal experimentation.
Cell lines and virus
The 293T (Cat. No. STCC10301G), A549 (Cat. No. STCC10201G), HeLa (Cat. No. STCC10603P) and RAW264.7 (Cat. No. STCC20020G) cells were provided by Servicebio (Wuhan, China), and Vero E6 cells (Cat. No. CL-0491) were provided by Procell (Wuhan, China). These cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‒streptomycin. The clade IIb MPXV strains SZTH41 (hMpxV/China/SZ-SZTH41/2023, GISAID accession ID: EPI_ISL_ 18213375) and SZTH42 (hMpxV/China/SZ-SZTH42/2023, GISAID accession ID: EPI_ISL_18213374) were isolated from male mpox patients aged 42 and 37 years in Shenzhen China, respectively45,55. The vaccinia virus Tiantan strain was kindly provided by Dr. Shouwen Du from the Guangdong Academy of Agricultural Sciences.
Antigen sequences and plasmids
The recombinant protein AAL consists of A35R (YP_010377142.1), A29L (NP_536566.1), and L1R (AAL40543.1) from MPXV Zaire-96-I-16, which belongs to clade I. AALI consists of AAL at the amino terminus followed by the IFN-α sequence at the carboxyl terminus. A flexible linker peptide (GSAGSAG) was used between each component to prevent steric hindrance. The 3D structures of AALI and AAL were generated using the AlphaFold 3 and PyMOL programs (version 3.1.5.1). The DNA fragments encoding AALI and AAL were optimized according to the codon preference of humans (Table S8) and synthesized by Sangon Biotech (Shanghai, China). These sequences were then cloned and inserted into pmRVac1 vectors (VectorBuilder, Guangzhou, China) with a T7 RNA polymerase promoter and downstream polyA sequences and transformed into E. coli DH5α. Positive clones were confirmed by DNA sequencing.
In vitro transcription
Plasmids linearized with NotI (New England Biolabs, MA, USA) served as DNA templates for IVT using the T7 High Yield RNA Synthesis Kit for Cotranscription (Yeasen Biotechnology, Shanghai, China). N1-Me-Pseudo UTP (Yeasen Biotechnology, Shanghai, China) was substituted for uridine triphosphate (UTP) during this process. The resulting mRNA was capped with Cap1-GAG m7(3’OMeG)(5’)ppp(5’)(2’OMeA)pG (Yeasen Biotechnology, Shanghai, China) and purified using an RNAclean RNA Kit (Biomed, Beijing, China). The mRNA concentration was measured with a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA), and its integrity and purity were verified by agarose gel analysis.
LNPs encapsulation
The LNPs formulation consisted of SM-102 (heptadecan-9-yl 8-((2-hydroxyethyl) (6-oxo-6-(undecyloxy) hexyl) amino) octanoate), DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), DSPE-PEG-Man (mannose-modified1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy(polyethylene glycol)-2000)), and cholesterol at a 50:10:1.5:38.5 molar ratio. These components were sourced from AVT Pharmaceutical Tech Co., Ltd (Shanghai, China). mRNA was encapsulated within the LNPs formulation using a microfluidic device called the Intelligent LNPs Synthesizer (FluidicLab, Shanghai, China). In brief, the LNPs formulation concentration was adjusted to 10 mM, and this formulation was used to encapsulate the mRNA, which was dissolved in citric acid buffer (pH 4.0) at a lipid-to-mRNA ratio of N:P = 6, using an Intelligent LNPs Synthesizer at room temperature. The flow rates of the lipid phase (LNPs formulation) to the water phase (mRNA solution) were set at 5 mL/min and 15 mL/min, respectively. The resulting raw LNPs-mRNA suspension was dialyzed three times using an ultrafiltration tube (100 K) to remove residual citrate salt and ethanol. The purified LNPs-mRNA was then resuspended in Tris buffer (pH 7.4) and stored at −80 °C56.
Transfection experiment
A549 and HeLa cells were seeded in 6-well plates and transfected with 2 μg of RFP-encoded mRNA encapsulated in mannose-modified LNPs per well. At 24–48 h post-transfection, red fluorescence signals were detected using an inverted fluorescence microscope. For the LNPs-mRNA targeting assay, the transfection procedure was similar, with slight modifications. GFP-encoded mRNA was labeled with a Cy3 fluorescent probe, encapsulated in mannose-modified LNPs to form LNPs-mGFP, and transfected into A549 cells and BMDCs in a coculture system at a concentration of 1 μg per well, with a 1:1 mixture of both cell types. After 12 h, BMDCs were specifically labeled with APC-conjugated anti-CD11c fluorescent antibodies, while A549 cells were left untreated. Confocal microscopy was then used to examine the localization of various fluorescent signals to assess the ability of LNPs-mRNAs to target DCs.
Transfection and western blotting
HeLa cells were seeded in 24-well plates and transfected with 2 μg of mRNA per well. At 24 h post-transfection, the cells were collected and lysed in lysis buffer on ice for 10 min to extract the cellular proteins. Equivalent samples (40 μg of protein) were mixed with 5× loading buffer, boiled, and separated on a 12% SDS‒PAGE gel. Proteins were then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore Sigma, Billerica, MA). After being blocked with 5% skim milk, the membranes were split into two sections and incubated overnight at 4 °C with different primary antibodies according to the manufacturer’s specifications: the A35R rabbit polyclonal antibody57 (Antibody System, PVV13101, dilution: 1:1000) on one section and the anti-β-actin antibody58 (Servicebio, GB15003-100, UniProt ID: P60710, dilution: 1:3000) as a loading control on the other. After being washed, the membranes were incubated at room temperature for 60 min with an HRP-conjugated goat anti-rabbit IgG secondary antibody (Proteintech, 20000886) at a 1:5000 dilution. The specificity of all the antibodies was confirmed through previous publications or datasheets provided by the supplier. Detection was performed via enhanced chemiluminescence (ECL), and Western blot images were captured using iBright FL1500 (Bio-Rad Laboratories, Inc., USA) and quantified using ImageJ software (version 1.54 h) to assess the band intensity.
Flow cytometry analysis
HeLa cells, transfected as described above, were fixed, washed, and permeabilized with the appropriate buffers sequentially. The cells were then incubated with A35R rabbit monoclonal antibody (mAb) for 20 min at 4 °C. After two washes with PBS, the cells were incubated with fluorochrome-conjugated IgG-FITC mAbs (Absin, abs20139, dilution 1:100) for 20 min at 4 °C in the dark. Flow cytometry was performed using a FACSARIA II (BD, USA), and the data were analyzed with FlowJo (version 10.0) software.
Freshly isolated splenic cells from immunized mice were stained with fluorochrome-conjugated mAbs. The antibody panel included PE-conjugated anti-mouse CD3 (BioLegend, Cat. No. 100307, 0.25 µg per million cells in 100 µL volume) for T lymphocytes, FITC-conjugated anti-mouse CD4 (BioLegend, Cat. No. 100509, 0.25 µg per million cells in 100 µL volume) for CD4+ T cells, and APC-conjugated anti-mouse CD8 (BioLegend, Cat. No. 100711, 0.25 µg per million cells in 100 µL volume) for CD8+ T cells. The cells (1 × 106 per tube) were resuspended in 100 μL of PBS containing appropriate concentrations of antibodies according to the manufacturer’s instructions and incubated for 20 min at 4 °C in the dark. Flow cytometry was performed, and the data were analyzed with FlowJo (version 10.0) software as described above. Specifically, to define the live, singlet population and exclude debris, aggregates, and doublets in flow cytometry data, we first plotted FSC-A (area) vs. FSC-W (weight) to set a doublet discrimination gate, excluding events outside the expected single-cell population. Next, the FSC vs. SSC plot was used to identify and separate live, singlet cells from debris and aggregates. Specific surface markers, such as CD8+ T cells (CD3+/CD8+) and CD4+ T cells (CD3+/CD4+), were then applied to identify immune cell populations of interest. To ensure accuracy, compensation for spectral overlap between fluorochromes was performed using compensation beads or controls prior to analysis. For each sample, 10,000–20,000 live cells within the defined gate were acquired, and the percentage of each immune population was recorded. Statistical analysis was then conducted to compare population percentages across experimental groups.
In vivo bioluminescence imaging
LNPs-mRNA encoding RFP were administered to BALB/c mice via intranasal and intramuscular routes at a dose of 10 µg per mouse. After 24–48 h, the mice were anesthetized, their hair was clipped, and their lungs were harvested and photographed. Images were captured using the IVIS Spectrum CT system (Perkin-Elmer, MA, USA), and the bioluminescence intensity from the regions of interest was quantified using Living Image software (version 4.4).
Cell survival assay
293T or A549 cells were seeded in 12-well plates and treated with various concentrations of LNPs. After 48 h, 20 µL of MTS reagent was added to each well, and the cells were incubated at 37 °C for 3 h. The absorbance was then measured at 450 nm. The cells treated with PBS served as the control, with the survival rate set at 100%.
For the LNPs-mRNA-mediated splenocyte proliferation assay, splenocytes from naive BALB/c mice were cultured in RPMI 1640 medium supplemented with 10% FBS. The splenocytes were seeded in 96-well plates at a density of 1 × 106 cells per well and stimulated for 72 h with 200 ng of LNPs-AALI, LNPs-AAL, LNPs-IFN-α, or LNPs, with PBS used as a negative control. Cell proliferation was assessed using the MTS assay, and the PI was calculated using the following formula: PI = (Absorbance value in experimental wells - Absorbance value in blank control wells)/(Absorbance value in control wells − Absorbance value in blank control wells) × 10056.
Animal immunization
Female wild-type BALB/c mice (Guangdong Medical Laboratory Animal Center, 8–10 weeks old) were randomly assigned to five groups (n = 15 per group). The sample size for each group was determined via a power analysis (α = 0.05, β = 0.2, effect size = 0.4) conducted using G*Power 3.1 software59,60. Immunization was administered intramuscularly with 10 µg of LNPs-mRNA formulated in 100 µL of PBS following a prime-boost regimen on days 0, 14, and 35. Serum samples were collected via retro-orbital bleeding at 14 days post-immunization (dpi) for each timepoint and stored at −80 °C until analysis. In parallel with serum collection, spleens were aseptically harvested at 14 dpi for isolation of splenocytes for ELISpot assays, flow cytometric analysis and scRNA-seq. Investigators performing immunological assays were blinded to group assignments throughout the data collection and analysis phases.
For the challenge experiments, the mice were randomly divided into five groups (11 mice per group) and intranasally challenged with 4 × 105 FFUs of the clade II MPXV hMpxV/China/SZ-SZTH42/2023 or hMpxV/China/SZ-SZTH41/2023 strain or 8 × 104 FFUs of the VACV Tiantan strain45. Lungs were collected from three mice per group on days 4 and 7 post-infection for viral load quantification and pathological damage assessment. In brief, the right lungs were used for focus formation assays and qPCR, while the left lungs were fixed for histopathological analysis61. The remaining mice were monitored daily for body weight and clinical symptoms for 2 weeks post-challenge. The mice were euthanized at the end of the experiment or earlier if their weight loss exceeded 25% of their initial weight. This study was approved by the Ethics Committee of the Shenzhen Third People’s Hospital, China (approval number: 2023-019). All works with infectious MPXV were carried out in a biosafety level 3 (BSL-3) laboratory.
Enzyme-linked immunosorbent assay
For measurement of antigen-specific IgG, IgG1, and IgG2a levels by ELISA, 96-well microtiter plates were coated overnight with 100 µL of A29L, A35R, L1R, A27L, or A33R protein at a concentration of 2 µg/mL. The plates were washed with PBS containing 0.05% Tween-20 and then blocked with PBS containing 0.05% Tween-20 and 1% BSA for 2 h at room temperature. After washing, 100 µL of each serially diluted serum sample was added to the wells and incubated for 2 h at room temperature. After an additional wash, 100 µL of the following HRP-conjugated secondary antibodies (all from Proteintech) were added to the wells: affinipure goat anti-mouse IgG (H + L) (Cat. No. SA00001-1, 1:10,000 dilution), Fc gamma subclass 1-specific affinipure goat anti-mouse IgG (Cat. No. SA00012-1, 1:10,000 dilution), and Fc gamma subclass 2a-specific affinipure goat anti-mouse (Cat. No. SA00012-2, 1:10,000 dilution). The plates were then incubated at room temperature for 2 h. After being washed, the plates were incubated with TMB substrate for 15 min. The color reaction was stopped with 100 µL of 2 M hydrochloric acid, and the absorbance was measured at 450 nm using an H1MF microplate reader (BioTek, USA).
Binding affinity analysis using SPR
Serum antibodies were purified using the Beaver-Beads Protein A/G Antibody Purification Kit (Beaver Biosciences, Inc., Guangzhou, China) according to the manufacturer’s instructions. The binding affinities of the purified serum antibodies for the A29L, A27L, A35R and L1R proteins of Orthopoxvirus were determined by surface plasmon resonance (SPR) using a Biacore T200 (Cytiva, USA). Purified antibodies (1.25 nM) in PBSP buffer were captured onto a CM5 chip using anti-mouse IgG (H + L) (ImmunoWay, Cat. No. RS0007-100 µl, dilution 1:10000, CA, USA) immobilized by standard amine coupling. An anti-mouse IgG (H + L) sensor channel served as a negative control. Various concentrations of A29L, A27L, A35R, and L1R proteins were flowed over each channel in PBS with 0.05% P20 surfactant (pH 7.4). After each binding event, the sensor surface was regenerated by washing with glycine at pH 2.0. Binding and dissociation were monitored in real time, and binding curves were analyzed after correcting for nonspecific binding. Dissociation constant (KD) values were calculated using a steady-state affinity model with Biacore T200 evaluation software (version 3.1).
Focus reduction neutralization tests
To analyze the neutralization efficacy of the serum antibody induced by the mRNA vaccine against MPXV, Vero E6 cells were meticulously seeded at a density of 1.5 × 104 cells/well into 96-well plates and acclimated overnight for subsequent experimentation. Serum samples were serially 3-fold diluted with DMEM-2 medium containing 2% sterile guinea pig complement, mixed with an equal volume of diluted MPXV, and then incubated at 37 °C for 1 h. The antibody-virus mixture was subsequently added to the plate, which was seeded with Vero E6 cells and allowed to adsorb at 37 °C for 1 h. After this incubation period, the cells were gently rinsed with PBS and incubated with DMEM-2 at 37 °C for ~18 h. Then, the cells were fixed, permeabilized, and stained, and the FFUs were determined as described above for the focus formation assay. The reactions were developed via KPL TrueBlue Peroxidase substrates. The number of MPXV foci was subsequently quantified utilizing an ELISpot reader, and the half-maximal inhibitory dilution (ID50) was determined using GraphPad Prism (version 8.0) software.
ELISpot assays
Splenocytes were freshly isolated from immunized mice, and Orthopoxvirus-specific IgG ASCs were enumerated using an ELISpot assay. MultiScreen HTS 96-well plates (Millipore, MA, USA) were coated overnight at 4 °C with purified AAL, A29L, and A35R proteins at 5 µg/mL in PBS, with PBS-coated wells serving as negative controls. After being blocked with complete medium for 2 h at 37 °C, splenocytes (5 × 105 cells per well) were added to triplicate wells and incubated at 37 °C for 20 h. The plates were then washed and incubated with HRP-conjugated anti-mouse IgG (Proteintech, Cat. No. SA00001-1, dilution 1:10000) at 37 °C for 2 h, after which the spots representing IgG ASCs were counted. Mouse IL-4 and IFN-γ ELISpot kits (MabTech, Stockholm, Sweden) were used to measure the number of IL-4- and IFN-γ-producing splenocytes according to the manufacturer’s instructions.
Cytotoxic lysis assay
An in vivo cytotoxicity assay was carried out to assess antigen-specific killing activity. Briefly, splenocytes freshly isolated from naive BALB/c mice were divided into two parts. One part was pulsed with 10−6 M A35R polypeptide for 1.5 h and then labeled with 5 μΜ CFSE (termed CFSEhigh A35R-pretreated cells), whereas the other part was pulsed with 10−6 M bovine serum albumin (BSA) and labeled with 1 μM CFSE (termed CFSElow BSA-pretreated cells) as a control. The cells from the two groups were mixed at a 1:1 ratio and injected into the immunized recipient mice at 2 × 107 total cells per mouse via the tail vein on day 14 after the third immunization. Twelve hours later, splenocytes were isolated from the recipients, and differential CFSE fluorescence intensities were measured via flow cytometry62. The specific lysis rate was calculated using the following formula:
Cytokine detection by a cytometric bead assay
The cytokine secretion levels were detected with a LEGENDplex™ Mouse Th Cytokine Panel according to the manufacturer’s instructions. In brief, 25 μL of capture beads was mixed with 50 μL of diluted standards or sera, and culture supernatant samples were diluted in assay buffer in tubes before incubation for 15 min at room temperature. After shaking at 800 rpm in the dark for 2 h at room temperature, the tubes were centrifuged at 250 × g for 5 min to remove the supernatant. Then, the beads were washed once, and 25 µL of detection antibody was added. After incubation for 1 h at room temperature in the dark, the beads were not washed, and 25 µL of SA-PE was added directly. After incubation for 15 min at room temperature in the dark, the beads were washed once and detected by flow cytometry. The fluorescence intensity of the APCs represented the cytokine expression level.
Histological analysis
The mice were sacrificed on days 4 and 7 post-infection. Left lung tissues were collected from three mice per group for histological examination. The tissues were fixed in neutral-buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin for microscopic analysis. Additionally, lung sections were prepared for immunohistochemical analysis of CD45 to assess the number of lung-infiltrating leukocytes. The procedure involved dewaxing and rehydrating the paraffin sections using environmentally friendly solutions and alcohols, followed by antigen retrieval and washing with PBS. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide, and nonspecific binding was prevented by serum blocking with 3% BSA. The sections were incubated overnight at 4 °C with a 1:300 dilution of CD45 rabbit antibody (Servicebio, Cat. No. GB113886, dilution 1:300), followed by incubation with an HRP-conjugated goat anti-rabbit IgG secondary antibody (Proteintech, Cat. No. SA00001-1, dilution 1:10000). HRP-mediated color development was performed using 3,3’-diaminobenzidine (DAB), and the sections were counterstained with hematoxylin. The sections were then visualized under a white light microscope with Pannoramic MiDl (3DHISTECH/PerkinElmer, Hungary), and the immunohistochemical images were quantified using ImageJ software (version 1.54 h) to assess the positive signal intensity.
The distributions of MPXV, lung-infiltrating T lymphocytes, and lung-infiltrating macrophages were examined using immunofluorescence analysis of A35R, CD8, and F4/80, respectively. The lung sections were deparaffinized, rehydrated, subjected to antigen repair, blocked with endogenous peroxidase and hydrogen peroxide, and blocked with BSA. The samples were subsequently incubated with rabbit primary antibodies specific for A35R, CD8 (Servicebio, Cat. No. GB15068, dilution 1:400), and F4/80 (Servicebio, Cat. No. GB113373, dilution 1:500), followed by incubation with the corresponding secondary antibodies. Nuclei were stained with DAPI, and autofluorescence was quenched before the slides were sealed. Images were captured using a Pannoramic MiDl fluorescence microscope (3DHISTECH/PerkinElmer, Hungary) with appropriate filters. For each scanned image, three fields of view were randomly chosen for quantitative analysis of the fluorescence signals. ImageJ software (version 1.54 h) was used to measure the intensity of the fluorescence signals for each marker. The data were analyzed by one-way analysis of variance (ANOVA) using SPSS software (version 26.0).
Real-time quantitative polymerase chain reaction
For determination of the virus titers in the lungs of infected mice, total RNA was extracted from lung tissues using the MolPure Cell/Tissue Total RNA Kit (Yeasen, Shanghai, China), and 1 μg of total RNA was reverse transcribed to cDNA using the HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, Jiangsu, China) according to the manufacturer’s instructions. The resulting cDNA was used for quantitative real-time PCR analysis with a Taq Pro Universal SYBR qPCR Master Mix Kit (Vazyme, Jiangsu, China) using an ABI VII7 (Thermo Fisher Scientific, USA). In brief, an equal amount of cDNA was used to prepare the qPCR system. The amplification program typically included denaturation at 95 °C, annealing at 60 °C, and extension at 72 °C for 40 cycles, followed by melting curve analysis at the end of the reaction to assess nonspecific amplification. The MPXV-specific primer sequences were selected from the DNA polymerase gene E9L (forward primer: 5´-TCA ACT GAA AAG GCC ATC TAT GA-3´; reverse primer: 5´-GAG TAT AGA GCA CTA TTT CTA AAT CC CA1-3´)63. The internal reference primer sequences for β-actin were as follows: forward primer: 5´-TGGAATCCTGTGGCATCCATGAAAC-3´; reverse primer: 5´-TAAAACGCAGCTCAGTAACAGTCCG-3´64. Relative transcription levels were determined using the 2-ΔΔCt method64.
Focus formation assay
For determination of the infectious virus titer, the left lung was placed into a homogenization tube containing 1 mL of PBS, homogenized, and subjected to two freeze‒thaw cycles. The mixture was subsequently centrifuged at 11,000 × g for 5 min, and the supernatant was collected for MPXV titration analysis using the following method. Vero E6 cells were seeded in 96-well plates and incubated overnight. Tenfold serial dilutions of MPXV were prepared in DMEM-2. The Vero E6 cells were then inoculated with the serially diluted virus (100 μL/well). After 1 h of adsorption, the virus was removed, and the cells were washed once with PBS. Subsequently, DMEM-2 was added at 100 μL/well. Following an 18-h incubation period, the medium was aspirated from each well, and the cells were fixed with 4% paraformaldehyde solution at room temperature for 30 min. After fixation, the cells were permeabilized with Perm/Wash buffer (BD Biosciences) containing 0.1% Triton X-100 and incubated with HRP-conjugated anti-MPXV polyclonal antibodies (Invitrogen) overnight at 4 °C. The reactions were developed with KPL TrueBlue Peroxidase substrates (Seracare Life Sciences). The number of viral foci was counted using an EliSpot reader (Cellular Technology, Ltd), and the number of FFUs per mL was calculated using the following formula: average spots × virus dilution times × 10 (FFU/mL).
scRNA-seq by chromium 10× genomics
Spleens were randomly isolated from groups of BALB/c mice (n = 3) treated with either AALI or LNPs. Following isolation, the spleens were thoroughly rinsed in cold PBS and then transferred onto ice in an RNase-free culture dish containing calcium-free and magnesium-free PBS. The samples were subsequently finely cut into pieces measuring ~0.5 mm2. The tissues were then dissociated into single cells using a dissociation solution composed of 0.35% collagenase IV, 2 mg/mL papain, and 120 units/mL DNase I and incubated for 20 min at 37 °C. The reaction was subsequently terminated by adding PBS containing 10% FBS. The cell suspensions were filtered with a cell strainer (aperture width of 30–70 μm) and centrifuged at 250 × g for 5 min at 4 °C. After the lysis of red blood cells and the removal of dead cells, the overall cell viability was confirmed to be greater than 90%. Single-cell suspensions were then counted and adjusted to a concentration of 800–1200 cells/mL.
Single-cell suspensions were loaded onto 10× chromium to capture 8000 single cells, following the manufacturer’s instructions outlined in the 10× genomics chromium Next GEM single cell 5’ kit (v2). The following cDNA amplification and library construction steps were performed according to the standard protocol. Libraries were sequenced on an Illumina NovaSeq 6000 sequencing system (Illumina Inc., USA) by LC-Bio Technology (Hangzhou, China) at a minimum depth of 20,000 reads per cell (double-end sequencing, 150 bp). The sequencing data were demultiplexed and converted to FASTQ format using Illumina bcl-convert software (version 3.9.3). The scRNA-seq data were aligned to the Ensembl genome GRCm38 reference genome using CellRanger software (version 7.2.0), and cellular and individual 5’ end transcripts were identified and counted in the sequenced samples. A total of 44333 single cells captured from 3 AALI- and 3 LNPs-treated mice were processed using 10× genomics chromium single cell solution. The output expression profile matrix generated by CellRanger was imported into Seurat (version 4.1.1) to filter out low-quality cells from the scRNA-seq data. The filtered dataset was subjected to downsampling and clustering. The criteria for filtering low-quality cells included the following: all genes expressed in less than 1 cell were removed, the number of genes expressed per cell >500 was set as low, <Inf was set as the high cutoff, UMI counts were less than Inf, and the percentage of mitochondrial DNA-derived gene expression <25%. The dimensionality of all the cells was projected into 2D space using UMAP. The marker genes for each cluster were identified with FindAllMarker, and the marker genes were selected on the basis of the following criteria: expression in more than 10% of the cells in each cluster, p value ≤ 0.01, and gene expression ploidy logFC ≥0.26. Hypergeometric testing was used to conduct GO and KEGG enrichment analyses on the differentially expressed identified within each cluster, as obtained from the FindAllMarkers analysis, in comparison with the genes expressed in other clusters.
TCR and BCR V(D)J immune repertoire sequencing and analysis
From a single aliquot of 5ʹ gene expression libraries, full-length TCR/BCR V(D)J segments were enriched from transcriptome cDNA via PCR amplification using the Chromium Single-Cell V(D)J Enrichment Kit (10× Genomics, PN-1000254 for TCR, PN-1000255 for BCR) following the manufacturer’s protocol. Immune repertoire preprocessing was performed using the CellRanger (version 7.2.0) vdj command with vGRCm38 as a reference, involving demultiplexing by index and barcodes, TCR/BCR V(D)J sequence discovery, and clonotype assignment to each cell. Productive chains were identified and linked with the cell’s barcode information. TCR clonotypes are defined by unique TCR α-chain (TRA) and TCR β-chain (TRB) pairs, and BCR clonotypes are defined by unique IGH-IGK/IGL pairs. Clonotypes present in at least two cells were considered clonal, with the number of such cells indicating clonality. With barcode information, T cells and B cells with prevalent clonotypes were mapped onto UMAP plots.
Statistical analysis
The data are expressed as the means ± SDs, with dots indicating individual values. The sample sizes are indicated in the figure legends. Statistical analyses were performed using the SPSS (version 26.0) and GraphPad Prism (version 8.0) software. Comparisons of two groups were performed with Student’s t-test. Comparisons of more than two groups were performed with one or two-way ANOVA with Tukey’s post hoc test when one or more independent factors were involved, respectively. *p < 0.05, ** p < 0.01, and *** p < 0.001; ns, not significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw data of scRNA-seq and scV(D)J-seq that support the findings of this study have been deposited in Sequence Read Archive (SRA) with the accession code PRJNA1156919, and the processed data of scRNA-seq and scV(D)J-seq have been deposited in Gene Expression Omnibus (GEO) with the accession code GSE276667. Source data are provided with this paper.
References
Faye, O. et al. Genomic characterisation of human monkeypox virus in Nigeria. Lancet Infect. Dis. 18, 246 (2018).
Shchelkunov, S. N. An increasing danger of zoonotic Orthopoxvirus infections. PLoS Pathog. 9, e1003756 (2013).
Henderson, D. A. The eradication of smallpox–an overview of the past, present, and future. Vaccine 29, D7–D9 (2011).
Shchelkunov, S. N. et al. Human monkeypox and smallpox viruses: genomic comparison. FEBS Lett. 509, 66–70 (2001).
Nalca, A. & Zumbrun, E. E. ACAM2000: the new smallpox vaccine for United States Strategic National Stockpile. Drug Des. Dev. Ther. 4, 71–79 (2010).
Frey, S. E. et al. Phase II randomized, double-blinded comparison of a single high dose (5×10(8) TCID50) of modified vaccinia Ankara compared to a standard dose (1×10(8) TCID50) in healthy vaccinia-naïve individuals. Vaccine 32, 2732–2739 (2014).
Jacob-Dolan, C. et al. Comparison of the immunogenicity and protective efficacy of ACAM2000, MVA, and vectored subunit vaccines for Mpox in rhesus macaques. Sci. Transl. Med. 16, eadl4317 (2024).
Petersen, B. W. et al. Clinical guidance for smallpox vaccine use in a postevent vaccination program. MMWR Recomm. Rep. 64, 1–26 (2015).
Berhanu, A. et al. Treatment with the smallpox antiviral tecovirimat (ST-246) alone or in combination with ACAM2000 vaccination is effective as a postsymptomatic therapy for monkeypox virus infection. Antimicrob. Agents Chemother. 59, 4296–4300 (2015).
Frey, S. E. et al. Safety and immunogenicity of IMVAMUNE® smallpox vaccine using different strategies for a post event scenario. Vaccine 31, 3025–3033 (2013).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines - a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Hou, F. et al. mRNA vaccines encoding fusion proteins of monkeypox virus antigens protect mice from vaccinia virus challenge. Nat. Commun. 14, 5925 (2023).
Sang, Y. et al. Monkeypox virus quadrivalent mRNA vaccine induces immune response and protects against vaccinia virus. Signal Transduct. Target Ther. 8, 172 (2023).
Pütz, M. M., Midgley, C. M., Law, M. & Smith, G. L. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat. Med. 12, 1310–1315 (2006).
Smith, G. L. & Law, M. The exit of vaccinia virus from infected cells. Virus Res. 106, 189–197 (2004).
Pauli, G. et al. Orthopox viruses: infections in humans. Transfus. Med. Hemother. 37, 351–364 (2010).
Couñago, R. M. et al. Structures of Orf virus chemokine binding protein in complex with host chemokines reveal clues to broad binding specificity. Structure 23, 1199–1213 (2015).
Roper, R. L., Wolffe, E. J., Weisberg, A. & Moss, B. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Virol. 72, 4192–4204 (1998).
Chung, C. S., Hsiao, J. C., Chang, Y. S. & Chang, W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72, 1577–1585 (1998).
Ho, Y. et al. The oligomeric structure of vaccinia viral envelope protein A27L is essential for binding to heparin and heparan sulfates on cell surfaces: a structural and functional approach using site-specific mutagenesis. J. Mol. Biol. 349, 1060–1071 (2005).
Kaufman, D. R. et al. Differential antigen requirements for protection against systemic and intranasal vaccinia virus challenges in mice. J. Virol. 82, 6829–6837 (2008).
Chiu, W. L., Szajner, P., Moss, B. & Chang, W. Effects of a temperature sensitivity mutation in the J1R protein component of a complex required for vaccinia virus assembly. J. Virol. 79, 8046–8056 (2005).
Chiu, W. L. & Chang, W. Vaccinia virus J1R protein: a viral membrane protein that is essential for virion morphogenesis. J. Virol. 76, 9575–9587 (2002).
Dahlgren, M. W. et al. Type I interferons promote germinal centers through B cell intrinsic signaling and dendritic cell dependent Th1 and Tfh cell lineages. Front. Immunol. 13, 932388 (2022).
Lindahl, P., Gresser, I., Leary, P. & Tovey, M. Enhanced expression of histocompatibility antigens of lymphoid cells in mice treated with interferon. J. Infect. Dis. 133, Suppl, A66–68 (1976).
Lindahl, P., Gresser, I., Leary, P. & Tovey, M. Interferon treatment of mice: enhanced expression of histocompatibility antigens on lymphoid cells. Proc. Natl. Acad. Sci. USA 73, 1284–1287 (1976).
Curtsinger, J. M., Valenzuela, J. O., Agarwal, P., Lins, D. & Mescher, M. F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174, 4465–4469 (2005).
Nolan, L. S. & Baldridge, M. T. Advances in understanding interferon-mediated immune responses to enteric viruses in intestinal organoids. Front. Immunol. 13, 943334 (2022).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005).
Lukhele, S., Boukhaled, G. M. & Brooks, D. G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 43, 101277 (2019).
Kolumam, G. A., Thomas, S., Thompson, L. J., Sprent, J. & Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650 (2005).
de Avila Botton, S. et al. Immunopotentiation of a foot-and-mouth disease virus subunit vaccine by interferon alpha. Vaccine 24, 3446–3456 (2006).
Jiang, W., Ren, L. & Jin, N. HIV-1 DNA vaccine efficacy is enhanced by coadministration with plasmid encoding IFN-alpha. J. Virol. Methods 146, 266–273 (2007).
O’Brien, L. et al. Alpha interferon as an adenovirus-vectored vaccine adjuvant and antiviral in Venezuelan equine encephalitis virus infection. J. Gen. Virol. 90, 874–882 (2009).
Tang, C. K. et al. Mannan-mediated gene delivery for cancer immunotherapy. Immunology 120, 325–335 (2007).
Hattori, Y., Kawakami, S., Suzuki, S., Yamashita, F. & Hashida, M. Enhancement of immune responses by DNA vaccination through targeted gene delivery using mannosylated cationic liposome formulations following intravenous administration in mice. Biochem. Biophys. Res. Commun. 317, 992–999 (2004).
Hattori, Y., Kawakami, S., Nakamura, K., Yamashita, F. & Hashida, M. Efficient gene transfer into macrophages and dendritic cells by in vivo gene delivery with mannosylated lipoplex via the intraperitoneal route. J. Pharmacol. Exp. Ther. 318, 828–834 (2006).
Zuiani, A. et al. A multivalent mRNA monkeypox virus vaccine (BNT166) protects mice and macaques from Orthopoxvirus disease. Cell 187, 1363–1373.e1312 (2024).
Boehm, O. et al. Clinical chemistry reference database for Wistar rats and C57/BL6 mice. Biol. Chem. 388, 547–554 (2007).
Fernandes, D. P. et al. Hematological and biochemical profile of BALB/c nude and C57BL/6 SCID female mice after ovarian xenograft. An. da Acad. Bras. de Cienc. 90, 3941–3948 (2018).
Serfilippi, L. M., Pallman, D. R. & Russell, B. Serum clinical chemistry and hematology reference values in outbred stocks of albino mice from three commonly used vendors and two inbred strains of albino mice. Contemp. Top. Lab. Anim. Sci. 42, 46–52 (2003).
Théry, C. & Amigorena, S. The cell biology of antigen presentation in dendritic cells. Curr. Opin. Immunol. 13, 45–51 (2001).
Quan, F. S., Huang, C., Compans, R. W. & Kang, S. M. Virus-like particle vaccine induces protective immunity against homologous and heterologous strains of influenza virus. J. Virol. 81, 3514–3524 (2007).
Slifka, M. K. & Ahmed, R. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr. Opin. Immunol. 10, 252–258 (1998).
Cheng, L. et al. Isolation and characterization of mpox virus from the first mpox case in Shenzhen, China. Virol. Sin. 39, 335–337 (2024).
Xu, J. L. & Davis, M. M. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 13, 37–45 (2000).
Valkenburg, S. A. et al. IL-15 adjuvanted multivalent vaccinia-based universal influenza vaccine requires CD4+ T cells for heterosubtypic protection. Proc. Natl. Acad. Sci. USA 111, 5676–5681 (2014).
Zhang, Y. et al. Microbiota-mediated shaping of mouse spleen structure and immune function characterized by scRNA-seq and Stereo-seq. J. Genet. Genom.50, 688–701 (2023).
Wang, Y. et al. Influenza vaccination features revealed by a single-cell transcriptome atlas. J. Med Virol. 95, e28174 (2023).
Voskoboinik, I., Smyth, M. J. & Trapani, J. A. Perforin-mediated target-cell death and immune homeostasis. Nat. Rev. Immunol. 6, 940–952 (2006).
Pieper, K., Grimbacher, B. & Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 131, 959–971 (2013).
Briney, B., Inderbitzin, A., Joyce, C. & Burton, D. R. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566, 393–397 (2019).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 (2009).
Robins, H. S. et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 114, 4099–4107 (2009).
Cheng, L. et al. Pathogenic BALB/c mice infection model for evaluation of mpox countermeasures. Cell Discov. 10, 105 (2024).
Zhang, G. et al. Up-regulation of granzyme B and perforin by staphylococcal enterotoxin C2 mutant induces enhanced cytotoxicity in Hepa1-6 cells. Toxicol. Appl. Pharmacol. 313, 1–9 (2016).
Zhou, J. et al. Circular RNA vaccines against monkeypox virus provide potent protection against vaccinia virus infection in mice. Mol. Ther. J. Am. Soc. Gene Ther. 32, 1779–1789 (2024).
Wang, S. et al. Aberrant METTL1-mediated tRNA m(7)G modification alters B-cell responses in systemic autoimmunity in humans and mice. Nat. Commun. 15, 10599 (2024).
Kang, H. Sample size determination and power analysis using the G*Power software. J. Educ. Eval. Health Prof. 18, 17 (2021).
Iparraguirre Nuñovero, M. F. et al. The effect of intracanal cryotherapy with and without foraminal enlargement on pain prevention after endodontic treatment: a randomized clinical trial. Sci. Rep. 14, 19905 (2024).
Meng, W. et al. Rapid generation of human-like neutralizing monoclonal antibodies in urgent preparedness for influenza pandemics and virulent infectious diseases. PLoS ONE 8, e66276 (2013).
Li, Y. et al. A recombinant protein containing influenza viral conserved epitopes and superantigen induces broad-spectrum protection. eLife 10, e71725 (2021).
Li, Y., Olson, V. A., Laue, T., Laker, M. T. & Damon, I. K. Detection of monkeypox virus with real-time PCR assays. J. Clin. Virol. 36, 194–203 (2006).
Fu, X. et al. Staphylococcal enterotoxin C2 mutant-directed fatty acid and mitochondrial energy metabolic programs regulate CD8(+) T cell activation. J. Immunol. 205, 2066–2076 (2020).
Acknowledgements
The authors acknowledge the grants from Shenzhen Key Laboratory of Microbiology in Genomic Modification & Editing and Application (Shenzhen Science and Technology Program, ZDSYS20230626090759006 to S.C.), Shenzhen Medical Research Funds (B2302052 to Z.Z., E24010010 to Z.Z., E24010012 to Z.Z., E24010013 to Z.Z.), the Guangdong Basic and Applied Basic Research Foundation (2022A1515110536 to Y.S.L.), Guangdong Science and Technology Plan Project, Construction of high-level biosafety laboratories (2021B1212030010 to Z.Z.), Shenzhen Science and Technology Program (ZDSYS20210623091810030 to Z.Z., JCYJ20220530150404009 to B.C.X.), and Shenzhen Portion of Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone (HTHZQSWS-KCCYB-2023060 to S.C.).
Author information
Authors and Affiliations
Contributions
Conceptualization: S.C., Z.Z., L.W. Methodology: Y.S.L., L.C., F.Y.Z., T.W., J.R., B.C.X., Y.H. Investigation: Y.S.L., L.X.J., L.C. Visualization: Y.S.L., L.X.J., J.R., Z.H.L. Funding acquisition: S.C., Z.Z., L.W. Project administration: S.C., L.W., Y.S.L. Writing—original draft: Y.S.L., S.C., L.X.J. Writing— review and editing: Y.S.L., S.C., Z.Z., L.W. Supervision: L.W., Z.Z., S.C.
Corresponding authors
Ethics declarations
Competing interests
The authors S.C., S.Y.L., and L.W. have filed a Chinese patent application (number: 202410069336.7) for the MPXV mRNA vaccines and their construction method and application. The other authors do not claim competing interests.
Peer review
Peer review information
Nature Communications thanks Milad Zandi and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Cheng, L., Jiang, L. et al. A multivalent mRNA vaccine elicits robust immune responses and confers protection in a murine model of monkeypox virus infection. Nat Commun 16, 7373 (2025). https://doi.org/10.1038/s41467-025-61699-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61699-w
