
Abstract
Researchers have investigated for decades a suitable catalyst to selectively synthesize epoxides from alkenes with air. Here we show that a selective aerobic epoxidation of industrial alkyl alkenes in liquid phase occurs without any catalyst, solvent nor additive, just placing the neat alkene under 3–5 bar of O2 and heating between 100 and 200 °C. The reaction can be performed in either an autoclave, a stirring vessel with air bubbling or even a simple open flask, provided that any metal piece is not in contact with the reaction. Alkyl epoxides are directly obtained, after air venting, in yields and selectivity up to 90%. The simplicity of the set-up (just the neat liquid alkene and air) allows to engage the epoxidation reaction with either a previous alkene formation (upstreaming) or a later epoxide opening (downstreaming) reactions, to achieve a variety of industrially-relevant organic products in one-pot.
Similar content being viewed by others
Introduction
Alkyl epoxides are fundamental organic molecules in industry, with widespread application in our society on a daily basis, from surfactants to construction, electronics or adhesives, to name a few1. Indeed, technological evolution cannot be understood without alkyl epoxides. The global alkyl epoxide market has been valued in ~€70 billion in 2024 and is expected to achieve >€90 billion in 2028, with a compound annual growth rate (CAGR) between 3 and 6% (https://www.databridgemarketresearch.com/reports/global–epoxides–market), (https://www.thebusinessresearchcompany.com/report/epoxide–global–market–report), which implies that megaton amounts of these epoxides are to be manufactured.
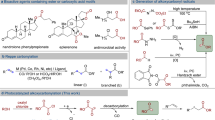
Figure 1A shows that the production of alkyl epoxides relies on the so–called epoxidation reaction of alkenes, which employs toxic, dangerous and relatively expensive oxidant agents such as water peroxide (H2O2), organic peroxides and hydroperoxides, in the presence of catalysts and with organic solvents for most of the processes except one, i.e., the epoxidation reaction of ethylene, which is carried out with molecular oxygen (O2/air) over a Ag–supported catalyst at >500 °C2. Therefore, it is not surprising that a long–sought reaction in synthetic chemistry is a general epoxidation reaction of alkenes with air3,4,5,6,7. For that, a plethora of catalysts, most of them based on metals8, have been studied, in many cases with the assistance of organic molecules able to shuttle the activated oxygen species to the alkene, such as aldehydes, ketones, or carboxylic acids9,10,11. However, none of the systems found to date is apparently suitable to achieve the efficiency of the catalysed processes with H2O2 or organic peroxides/hydroperoxides.

A State of the art for the epoxidation reaction of alkenes, with the different oxidants, industrial systems and epoxide products. B The typical reaction conditions in the literature for the aerobic epoxidation of alkenes, where the most feasible industrial conditions (diluted air, concentrated alkene) are little explored (this Figure can be considered as a cartoon of the state-of-the-art rather than a quantitative calculation of the different reports). Bottom: The step forward of the results presented here, from unsatisfactory catalysed processes under flammable conditions with organic solvents to the here reported uncatalyzed, solventless process within flammability limits.
Figure 1B depicts the reaction conditions traditionally employed during the epoxidation reaction of alkenes with air, catalyzed or not. It can be seen that, in general, O2 pressures above 5 bar (> 50 bar in many cases) are set over dissolved alkenes at high reaction temperatures12. These conditions make sense if one considers the reluctance of O2 to form the required electrophilic O atoms and react with the alkene to the target epoxide, and the expected increase in the extent of undesired alkene by–reactions at high concentrations (oligomerization, overoxidation…). Unfortunately, yields and selectivity to the desired epoxides are usually below 30%, and these precedents only highlight the uniqueness of ethylene to selectively react with O2, due to its small size and the lack of any highly reactive allylic H atom. However, after a deep analysis of the literature, it seems that reaction conditions consisting in low O2 pressures (< 5 bar) and highly concentrated alkene solutions, even with the neat alkene, have been barely explored13,14. These counterintuitive experimental reaction conditions are in turn those required by industry, where the flammability limits of the alkyl alkenes severely decrease the partial O2 pressure allowed during reaction13, and where reactor occupancy must be maximized to increase the reaction throughput. Another important factor to be considered here is the use of metal reactors, majorly used in previous studies despite metals tend to uncontrollably decompose O2 into very reactive species, which unavoidably over–oxidize the alkene and epoxide to ketones and carboxylic acids14,15,16.
Here we show that a variety of alkyl alkenes are transformed to the corresponding epoxides in high yields and selectivity when reacting in neat form with O2, at pressures below 5 bar and without metal pieces in the reactor. Regardless the auto-oxidation of alkenes, especially for aryl alkenes and cyclic alkenes, has been known for decades with peroxides17,18,19,20, a comprehensive previous study focused on the aerobic selective epoxidation reaction has not been performed as far as we know, in the absence of peroxyradicals21,22. Any catalyst, solvent nor additive is not required, just the alkene and O2, which allows to obtain the epoxide product directly after O2 venting, and to start a subsequent reaction on the epoxide if required. Complementarily, the alkene can be previously formed by conventional reactions such as isomerization23 or alkene metathesis, and then epoxidized in one-pot with just O2, and the epoxide product further elaborated, in-situ24. The epoxidation reaction can be run not only in autoclaves but also in regular glass flasks with TeflonTM–coated magnetic stirs under air–bubbling or open-flask conditions. The bottom of Fig. 1 depicts the step forward that, in our opinion, our results constitute in the field of epoxidation reactions. These results might be seen as a practical outcome of more than 75 years of research in the direct oxidation of alkenes with air. In the 50´s, several works introduced the notion of direct reaction of alkenes with O2 to give the epoxide vs. the more common allylic hydrogen removal, to give the alternative hydroperoxide25. During the next three decades, the concept was expanded with more examples26 and with new insights into the reaction mechanism, presenting alkene cation radicals and perepoxides as potential intermediate species12,27. However, the irruption of metal catalysts for the allylic hydrogen abstraction reaction to generate hydroperoxide intermediates28 and the lack of practical examples for a direct aerobic epoxidation beyond cyclooctene29 apparently diminished the interest for studying the direct epoxidation of alkenes.
Results
Optimization of the reaction conditions: from autoclaves to open flask reactors
We found by serendipity that the reaction between neat alkenes and O2 occurs in significant extent under mild reaction conditions, at low O2 pressures. Suprun and Opeida described the oxidation of styrene in the cumene solution20, and that could be the first time when it was observed, experimentally, the epoxidation without solvent and O2 as the epoxidation agent. Our attempts to reproduce some catalytic results for the epoxidation of styrene 1a under 4 bar of O2 lead us to the conclusion that neither the metal catalyst nor the solvent were required to convert the alkene, after following the reaction by gas chromatography coupled to mass spectrometry (GC–MS, see Fig. S1 in Supplementary Information). Indeed, the conversion of styrene was higher in the absence of any catalyst and solvent (compare entries 1, 5, and 7 in Table S1). The reactions were run in glass autoclaves with a TeflonTM–coated magnetic bar, which in principle discards any O2 activation by a metal. Despite the selectivity to styrene epoxide was not good in the absence of N,N–dimethylformamide, which can exert here as an O2 activator (aldehyde), we studied the possible epoxidation reaction of other alkenes under solventless reaction conditions and 4 bar of O2, and Fig. 2A shows the results with a variety of liquid alkyl and cyclic alkenes. The secondary alkyl alkene 7-tetradecene 1b (trans/cis 75:25) gives the corresponding trans–isomer enriched epoxide 2b in 74% yield after complete conversion of the starting material (18 h reaction time), tripled checked by GC–MS, 1H nuclear magnetic resonance (1H NMR) and isolated yield after purification by flash column chromatography (averaged yield). The rest of products mainly correspond to overoxidized products, such as aldehydes, carboxylic acids and alcohols. As the alkyl chains flanking the secondary alkene shorten, the selectivity and yield to the epoxide product decrease, and 3-octene 1c (pure trans) and 4-octene 1d (pure trans) give epoxides 2c and 2d in 54/40% and 16/16% selectivity/yield, respectively. The terminal alkene 1-tetradecene 1e was completely unreactive during reaction under similar reaction conditions. The cyclic alkenes cyclododecene 1 f (cis/trans 73:25), cyclooctene 1g (pure cis) and cyclohexene 1h (pure cis) followed the same trend30 than the open chain alkyl alkenes, i.e., the longer the alkyl chain the higher the yield to the epoxide product, to give epoxides 2f, 2g and 2h in 93/77%, 84/79% and 4/4% selectivity/yield, respectively. These results indicate that secondary alkenes with relatively long alkyl chains, typically more than six carbon atoms, convert to the corresponding epoxide products in good yields under 4 bar of O2 at 100 °C.

A Results for the epoxidation reaction of alkenes 1b–h under 4 bar of O2 in a glass autoclave reactor at 100 °C for 18 h. B Results for the epoxidation reaction of alkene 1b at 130 °C under the reaction conditions of (A) and in the presence or not of a metal nut. C Results for the epoxidation reaction of alkenes 1b and 1f after bubbling O2 through the neat alkene at 130 °C for 18 h. D Kinetic plot for the epoxidation reaction of alkene 1f in a round-bottomed flask with O2 bubbling at 200 °C. E Results for the epoxidation reaction of alkenes 1b, 1f, and 1g under reaction conditions in (D) (open flask) at different temperatures. F Results for the epoxidation reaction of alkenes 1b and 1f under 65 bar of diluted (5%) O2 in N2, in a TeflonTM vessel within an autoclave reactor at 130 °C for 18 h. All results refer to gas chromatography (GC) yields, tripled checked mass spectrometry (GC–MS), 1H nuclear magnetic resonance (1H NMR), and isolated yield after purification by flash column chromatography.
Figure 2B shows that a higher reaction temperature (130 °C) does not produce an increase in the yield but a decrease in selectivity for 7-tetradecene 1b, to give epoxide 2b in 64% yield. At this point, we tried a wide scope for the reaction by using a steel multi–reactor autoclave at 100 °C under 4 bar of O2, but all the alkene substrates tested, which included 1b–h, only yielded overoxidized products, mainly carboxylic acids (results not shown). It is noteworthy that the terminal alkene 1-tetradecene 1e, unreactive in the glass reactor, also decomposed to overoxidized products in the steel reactor. To check if the metal reactor was the cause of the overoxidation products, the epoxidation reaction in the glass reactor was repeated with a small metal nut inside the reaction mixture. As it can be seen in Fig. 2B, the yield of epoxide 2b decreased to just 3% (compared to 64% without the metal nut) for a complete conversion of the starting material, the rest being overoxidation products, and when the reaction was repeated again with another metal nut previously passivated with diluted HNO3, the yield of epoxide 2b was restored. These results strongly suggest that the presence of non-passivated metal parts during the aerobic epoxidation reaction leads to overoxidation products and that either glass or properly passivated metal reactors must be used to maximize the epoxide products.
The epoxidation reaction of alkenes 7-tetradecene 1b and cyclododecene 1f was then tested in a round-bottomed flask reaction after bubbling O2 through the neat alkene, at 130 °C (Fig. S2). The results in Fig. 2C shows that the epoxide products 2b and 2f were obtained in very high yields (87–91%) after 18 h reaction time and the kinetic plot in Fig. 2D shows that the yield of epoxide 2f reaches 90% after just 3.5 h reaction time. Then, we tested the epoxidation reaction in an open round-bottomed flask, just connecting a reflux condenser in order to avoid any loss of alkene. The reaction temperature was set at 200 °C in the thought that the relatively low concentration of O2 in the atmosphere will severely decrease the epoxidation reaction rate, however, this was not the case, since Fig. 2E shows that the optimal reaction temperature under open flask conditions is 150 °C, with yields between 70–80% for the different alkenes tested. A kinetic experiment shows the smooth formation of the epoxide product (Fig. S3). At lower reaction temperatures, the reactions may stop at moderate conversions (40–60%) because of the higher density of the incipient epoxide product, which hampers a suitable diffusion of O2. However, the decomposition of the epoxide at this temperature could also play a role, although we did not observe much overoxidized products (which in any case would need a good O2 mass transfer). To solve this problem, different solvents were employed, and n-dodecane and toluene showed to be compatible with the epoxidation reaction, to give epoxide 2b in high yields in concentrated solutions (7.8 M, Table S2) but not in diluted conditions (compare entries 2–5). Other solvents, such as DMF, are not suitable for the aerobic epoxidation reaction (entry 6).
In view that the epoxidation reaction proceeds in a diluted O2 atmosphere, such as ambient pressure, the reaction was carried out with diluted (5%) O2 in N2, which is a typical allowed reaction atmosphere in industry for highly flammable substances. A 65 bar total gauge pressure was set to achieve a similar O2 partial pressure (~ 3 bar) than in previous experiments. The epoxidation reactions of alkenes 1b and 1f were performed in TeflonTM vessels within metal autoclave reactors at 130 °C for 18 h, and the results in Fig. 2F show that a yield/selectivity value of 47/64% for epoxide 2b and 66/> 99% for epoxide 2f were obtained. Slight variations in the reaction temperature and O2 dilution improve the yield and selectivity for the epoxide products (Table S3). These results, together, confirm that the selective epoxidation of secondary alkenes can be performed in either autoclave reactors with 3–4 bar O2 partial pressures (including industrially feasible O2/N2 mixtures) and round-bottomed flasks after bubbling O2 or just opening the flask, with high yields of the corresponding epoxide products, and with the possibility of adding small amount of n-dodecane or toluene to better handling the reaction mixture31. Notice that all these reaction conditions are in principle, implementable under industrial environments but also practical at the laboratory scale. Indeed, since we do not have any catalyst, the productivity only depends of the scale of the reaction, and for a 10-g reaction with 90% yield (see for instance Fig. S2), the productivity after 4 h would be ≈2 g·h−1, and if scaled up to 10 Kg (which is plausible considering that any other additive or solvent is not employed, apart of the alkene substrate), the productivity after 4 h would be ≈2 Kg·h−1. These numbers might be attractive from an industrial point of view.
Scope of the reaction conditions
Figure 3 shows the results for the uncatalyzed and solventless aerobic epoxidation reaction of a variety of alkenes either in glass autoclave reactors under 4 bar of O2, or round-bottomed flasks with bubbling O2 or open reaction conditions. The results show that the three conditions can be employed with similar results (compare products 2b, 2f, 2g, 2n, and 2p). The reaction time for the glass autoclave reactors can be diminished to just a couple of hours or even to 30 min. Open and cyclic alkyl chain secondary alkenes of different sizes react well to give the epoxides in moderate to very good isolated yields (products 2b–k, 50–84%). The epoxide product is very stable under reaction conditions since purified product 2b stays unchanged for 3 h under the O2 pressure (5 bar) at 130 °C. Secondary dienes are also reactive, including bridged cycles, to give the corresponding mixture of mono- and di-epoxides in good combined yields (products 2l–n, 46–70%). Remarkably, a diene such as limonene 1o, where one of the alkenes is terminal (geminal) and the other is internal and tertiary, reacts exclusively through the internal alkene to give the industrially relevant limonene epoxide 2o in 47% yield. Trienes also selectively react by one of the alkenes, and all-trans cyclododecatriene 1p gives the monoepoxide 2p in a consistent 72–78% isolated yield with the three reaction methodologies, i.e., pressure, bubbling or open flask. It is worthy remarking here that 1p and its different cis/trans isomers constitutes the starting point to all macrocycles in the market, since the former is manufactured by trimerization of isobutene and gives access to cyclododecanone. Thus, the monoepoxide 2p here formed, with three different functional groups handles, can be used as an alternative precursor for the macrocycle market. The reaction under O2 diluted in N2 (5% in a total pressure of 65 bar) gave a 45% yield after 18 h reaction time (Fig. S4B).

Results for the epoxidation reaction of alkenes 1b–z either under glass autoclave reactors (4 bar of O2), bubbling of O2 through the neat alkene or open flask reaction conditions, under the indicated reaction conditions. Isolated yields. a 45% yield with O2 diluted in N2 (5% in 65 bar). b 18 h reaction time. c 30 min reaction time. d 3 h reaction time. e 150 °C. f 200 °C. g 100 °C. h 1H NMR combined with GC–MS yield.
Triene 1q, with two terminal alkenes and one internal secondary alkene, reacted through the two alkenes (product 2q, 22% internal + 10% terminal alkene), which may indicate that a terminal alkene becomes reactive in the presence of an internal alkene. To test this hypothesis, the epoxidation reaction of a mixture 1-tetradecene 1e and 7-tetradecene 1b was carried out, and Fig. 3 shows that the unreactive terminal alkene gives a 32% yield of the corresponding epoxide 2e. At this point, a two-pot epoxidation experiment was designed, as shown in Fig. 4. In this set-up, O2 is bubbled in a first flask containing the internal alkene 1b and the gas exit is connected to a second flask containing the terminal alkene 1e, both at 130 °C but not in contact. The kinetic plots show that the unreactive terminal alkene 1e converts to the epoxide 2e in >60% yield after the gas coming from the first flask (with the internal alkene 1b) is passed through; in other words, that the O2 gas passing through alkene 1b is active to epoxidize terminal alkene 1e. These results would be further commented in the mechanistic section. When we performed the reaction in the same pot, we obtained a similar result, i.e., a 32% yield of the epoxide 2e together with a 40% yield of 2b (Scheme 1)

A Schematic representation of the two-pot experiment where O2 is bubbled in a first flask with the internal alkene 1b and the gas exit is connected to a second flask with the terminal alkene 1e. B Kinetic plots for the epoxidation reactions of alkenes 1b and 1e in the two-pots epoxidation experiment.
Bio–based epoxides constitute a rocketing growing market, in particular from fatty acids derivatives to be employed in the adhesive and construction industry32,33. Under our aerobic reaction conditions, oleic acid 1r, methyl oleate 1s and linoleic acid 1t react to give the corresponding epoxides 2r–t in 33–66% yield after just 30 min reaction time, according to optimized results (Table S4). Other diene natural products, such as geraniol 1u and geranial 1v react under open flask conditions to give the mixture of tri- and tetra-substituted epoxides 2u and 2v in ~40% combined yield, and the latter demonstrates that aldehyde groups are significantly compatible under these reaction conditions. The tri-substituted alkene 1w gives a 74% yield of the quaternary epoxide 2w. The open flask reaction conditions also allow the synthesis of terminal epoxides when combined with internal ones, as shown above, in this case the epoxide from 1-octene 1x was obtained in 57% yield (product 2x) concomitantly with the epoxide of 2-octene 1y (>99% yield, product 2y) at just 100 °C reaction temperature (see Fig. S5 for 1H NMR spectra).
The aforementioned results demonstrate that, essentially, any sort of alkyl alkene, from terminal to highly substituted and from open to cyclic chains, can engage in the uncatalyzed solventless aerobic epoxidation reaction. In view of this, the reactivity of propylene 1z was tested. Propylene oxide 2z is, after ethylene epoxide, the highest epoxide industrially produced, despite peroxides must be employed during the catalytic fabrication process. With this in mind, we directly tested reaction conditions compatible with an industrial production of propylene oxide 2z with O2, i.e., diluted air and no additional additives nor alkenes and propylene in liquid conditions by increasing pressure. The result at the bottom of Fig. 3 shows that a 0.2% yield of neat 2z (60% selectivity) could be obtained, according to 1H NMR and GC–MS analysis under supercritical reaction conditions (70 bar and 150 °C, Fig. S6). The other 40% was the corresponding diol (glycol). Under liquid conditions (40 bar and 100 °C), any reactivity was not found. This yield for propylene oxide 2z is in line with the low reactivity of terminal alkenes under reaction conditions. We have also tried an in-flow reaction with a tubular reactor for liquid substrates, after feeding air continuously at counter-gravity to a flow of 7-tetradecene 1b (Fig. S7). However, we were not able to obtain a relevant conversion to the epoxide since it is much more difficult with this set-up to control the O2/alkene dissolution rate respect to the batch conditions, leading to some amounts of overoxidation products even at low conversion.
One-pot reactions
The absence of any additional molecule, i.e., catalyst, oxidant, solvent, or additive, in the reaction mixture beyond the starting alkene and the epoxide product, invites to telescope the synthetic procedure, either upstream (formation of the alkene)34 or downstream (transformation of the epoxide). Figure 5 shows the results obtained here. The classical alkene metathesis reaction35 can be successfully combined with the aerobic epoxidation reaction to give the rare 2,3-bisdecyloxirane (10,11-oxi-n-docosane epoxide) product 2aa in 43% yield from terminal alkene 1-dodecene 1aa (Fig. 5A). O2 cannot be present during the room temperature metathesis reaction since the 1st generation Grubbs catalyst (1st Gen Grubbs) would be decomposed, however, the catalyst and the dichloromethane solvent do not need to be removed after reaction, since O2 can be directly charged in the reactor to give the epoxide product 2aa after heating. However, a single-step protocol for the synthesis of internal epoxides from terminal alkenes would be in principle feasible since internal alkenes react with O2 much faster than terminal alkenes according to the above results. Thus, we changed the one-pot, two-step alkene metathesis-aerobic oxidation by a one-step alkene isomerization-aerobic oxidation reaction protocol, to generate an internal alkene by a simpler Ru catalyst, more tolerant to O2 than the Grubbs catalyst (Fig. 5B)36. The results with 1-tetradecene 1e as the starting alkene show that not only the internal epoxide product 2e is formed in good yield (~ 60%) but that the major product is the diastereo- and regio-isomerically defined product cis-2-methyl-3-undecyloxirane 2e–cis (47% yield, 60% selectivity, see kinetics in Fig. S8). This product comes from the rapid quenching of the so-formed cis 2-tetradecene by O2, which does not allow to generate significant amounts of neither the trans or the more internal alkenes, in an example of precise tandem reaction.

A Results for the one-pot, two-steps alkene metathesis-aerobic oxidation reaction with terminal alkene 1aa. B Results for the one-pot alkene isomerization-aerobic oxidation tandem reaction with terminal alkene 1e. C Results for different reactions of the in-situ formed epoxide 2f after uncatalyzed aerobic epoxidation reaction. The conversions obtained are given in brackets (see SI for more information about the procedures).
Regarding the downstream one-pot approach, the results for different reactions of the in-situ formed epoxide 2f (after the uncatalyzed aerobic epoxidation reaction, yield 79% and selectivity 85%), are shown in Fig. 5C. It can be seen that epoxide 2f was able to transform to different derivatives in the same reaction vessel. For instance, opening with H2O led to glycol 3f in 37% yield and complete selectivity, while reduction with LiAlH4 gave cyclododecanol 4f in 66% yield and also with complete selectivity. Less selective were the addition reactions with NH4SCN and TMSN3 (TMS is trimethylsilyl), which gave the thiirane 5f and the silyl-protected azide 6f products in 37/77% and 34/42% yield/selectivity, respectively. However, the addition of TMSCl gave the chloroalcohol 7f quantitatively. The cis diastereoisomer reacted noticeably faster than the anti epoxide during these reactions, which was checked by kinetic experiments where the rapid consumption of the cis isomer was observed. Additionally, all these reactions were repeated with a simulated epoxidation reaction mixture using the commercial alkene 1f and epoxide 2f, and the reactions took place similarly, although in 5–10% less extent in some cases. We attribute the slight improvement in the one-pot reactions, with the in-situ formed epoxide, to the generation of some amounts of carboxylic acids as by-products, which can catalyze the epoxide opening reactions.
Reaction mechanism
The rate equation for the aerobic epoxidation reaction was determined by the pseudo-stationary method, measuring the initial rates for reactions where the concentration of one of the reactants, i.e., alkene 1b and O2, was varied. The concentration of O2 was supposed to be linear with the pressure37. The kinetic results for alkene 1b (Fig. S9) and O2 (Fig. S10) show that the equation rate fits to v0 = kexp[1b][O2], where the two reactants behave with order +1, regardless if a solvent is employed (Fig. S9). These results indicate that one molecule of each reactant participates during the turnover-determining step (tds) of the reaction. Further kinetic experiments enable to calculate the activation energy (Ea) of the reaction, using an Arrhenius plot (Fig. S11), which gives Ea = 74 kJ·mol−1, within the expected value for a reaction that can proceed at 100 °C14.
The better reactivity of substituted alkyl alkenes suggests that electron-richer C = C double bonds are favored during the epoxidation reaction. Indeed, 1H and 13C NMR experiments show a shift in the signals associated to the C = C bond of 7-tetradecene 1b in concentrated solutions (Figs. S12 and S13), which indicates a gain of electron density by the alkene after aggregation38,39,40. This result is confirmed by ultraviolet–visible (UV–vis) absorption spectroscopy analyses (Fig. S14), where a shift in the maximum intensity of the band corresponding to 7-tetradecene 1b, from 240 to 320 nm, occurs, which corresponds to an energy decrease in the gap between the highest occupied and the lowest unoccupied molecular orbitals (HOMO–LUMO) of 1.2 eV. In other words, it is easier for the alkene to transfer one electron to a suitable electrophile such as O2 in concentrated, electron-rich solutions (see Fig. S15 for a schematic representation). It is noteworthy to comment that the terminal alkene 1-tetradecene 1e also aggregates in concentrated solutions and electronically enriches the C = C double bonds (Figs. S16–S18), however, which much lesser intensity than the internal counterpart 1b. Not only terminal alkenes are activated in the presence of internal alkenes (see Figs. 4 and 5), but also the reluctant to epoxidize41 cyclohexene 1h (Fig. S19). These results indicate that the more electron density the alkene shows, the better the epoxidation reaction, as it occurs with pre-activated oxidants (i.e., peroxides), and that the concentration of the alkene molecules allows more electron density to be available for O2.
The exclusive participation of O2 during the epoxidation reaction was studied with isotopically labelled and reactivity experiments. When 36O2 instead of 32O2 was used during the epoxidation reaction of 1f, the corresponding isotopically labelled epoxide product 18O–2f was obtained, according to CG–MS (Fig. S20) and Fourier–transformed infrared spectroscopy measurements (FT–IR, Fig. S21), where the expected isotopic mass and infrared shift were observed. Besides, analysis of the gas phase of the reaction by GC–MS spectroscopy revealed the progressive formation of C18O2 during reaction, as shown in Fig. 6A. This CO2 is formed after C–C bond breaking and overoxidation of the alkene to carboxylic acids.

A Mass spectrum of the gas phase during the epoxidation reaction of neat tetradecene 1b under 4 atm of 18O2 in a glass autoclave reactor at 130 °C. B Ultraviolet–visible (UV–vis) absorption spectra of 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD): in toluene (black line), with O2 (red line), with O2 and 1-tetradecene (1e, blue line), and with O2 and cyclododecene (1f, pink line) recorded at room temperature. C Proposed reaction mechanism.
Moreover, to confirm that triplet O2 is involved in the reaction mechanism, we performed the reaction using 7-tetradecene 1b in the presence of radical scavengers, such as BHT and TEMPO, obtaining 5.4% and 5.6% conversion of alkene, respectively (Fig. S22). The GC–MS analysis of the reaction performed with TEMPO shows the formation of the captured radical alkene by TEMPO. Then, we carried out the experiment using 1-tetradecene and neither the terminal epoxide, the captured radical nor TMPH formation were detected. These results indicate that a simple autooxidation of alkenes by oxygen radicals is not merely in operation, since 1-tetradecene is not reacting in any case, and an alkene radical can be trapped by TEMPO in the presence of triplet O2. Nevertheless, we cannot discard an auto-oxidation with simple oxygen radicals.
At this point, three activated O2 species were considered as potential O donors during the epoxidation reaction: ozone (O3), singlet oxygen (1O2), and superoxide (O2·−). First, a reactivity test with indigo as a probe molecule was carried out with the alkenes 1f (internal) and 1e (terminal) in toluene solution (Fig. S23), in order to seek the formation of O3 during the epoxidation reaction. In-situ UV–vis results show the disappearance of the indigo absorption band at 604 nm and the appearance of the band at 320 nm, assignable to the oxidation product with O3, only with the internal alkene 1f and not with 1e, i.e., when the epoxidation reaction occurs. These results suggest the formation of O3 during the uncatalyzed aerobic epoxidation reaction. The formation of a radical oxygen species in the gas phase42 would explain the epoxidation of the terminal alkene 1e in the two–flasks experiment of Fig. 4, since the terminal alkene can give the epoxide through the allyl mechanism. However, O3 is prone to break rather than epoxide alkenes, thus O3 could be considered here as a by-product of the actual epoxidizing species. Indeed, the selectivity to the epoxide product increases and the conversion slows down in the presence of indigo (Table S5), thus discarding O3 as the epoxidizing species.
The potential participation of singlet oxygen (1O2) was studied by reactivity experiments43. To do so, the gas phase of the epoxidation reaction was bubbled into a flask which contained anthracene (0.2 M, CDCl3), with the aiming of forming the corresponding endoperoxide44. However, neither the 1H nor the 13C NMR spectrum showed any signal different from those belonging to the starting anthracene (Fig. S24), which discards the formation of significant amounts of singlet oxygen during the epoxidation reaction, hence its possible active role.
In order to determine if the active species was the superoxide radical anion (O2·−), an aromatic derivative described in the literature for quenching this species45 was employed as a UV–vis reactive probe: 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD). As it can be seen in Fig. 6B, only the presence of the more active internal alkene 1f, and not a terminal alkene, triggered the decomposition of NBD (decrease of the band at 350 nm), thus indicating the formation of the superoxide radical anion (O2·−) during the epoxidation reaction.45 The formation of superoxide radical anions (O2·−) during reaction was further checked by employing redox metal salts such as SnO, Fe2O3, or MnO as radical generators, since they are able to react with O2 and give radical anions and ozone46,47,48. The results (Table S5) show that the presence of these metal salts significantly increased the conversion of the aerobic reaction of 1b while keeping a good selectivity for the epoxide 2b, but also forming the expected overoxidized products at high conversions. The overoxidation of the alkene 1b was also studied in the presence of different metal oxides (Fig. S25). As it can be observed, after 2–h reaction time, most of the conversions are near complete; however, the selectivity for these metal oxides is, in most cases, lower than when any catalyst is not used for the epoxidation reaction (see for instance V2O5, Fe2O3, and MgO). The other metal oxides tested did not improve the results obtained. For example, the best selectivity for the corresponding epoxide was obtained with MnO2, but it also produced a mixture of other overoxidized compounds formed from the rupture of 7-tetradecene 1b. Furthermore, after a 4-h reaction time, only MoO3 yields results comparable to those without any catalyst. To confirm whether the activity in the presence of metal oxides follows the mechanism proposed here or the well–known Mars-Van Krevelen mechanism, we performed an additional experiment in which CuO was added under a nitrogen atmosphere. After 4 h of reaction, we obtained a 23% conversion with a selectivity to the epoxide of 42%, 12% to carboxylic acid, 42% to vinyl ketone, and 4% to other compounds. With all these results in hand, we conclude that the addition of metal oxides does not improve the selectivity towards the target epoxide product, and in most cases, it leads to the formation of undesirable compounds. In addition, it is difficult to think that the generation of the superoxide is associated to the metal oxide, moreover considering the that the Mars-Van Krevelen mechanism is under operation. An organic additive such as activated carbon did not promote the epoxidation reaction. Besides, oxoneTM was used as an ozone generator during reaction in stoichiometric amounts with alkenes 1b and 1e (Fig. S26), but any epoxidation product could not be found. These results discard ozone as the epoxidating agent.
With all the above data in mind, the proposed reaction mechanism for the uncatalyzed aerobic epoxidation of alkyl alkenes is shown in Fig. 6C. In accordance with the experimental results, the first step is the one-electron transfer from the alkene to triplet O2 (3O2), to generate the corresponding alkene radical cation and the superoxide anion O2·−, respectively. This reaction constitutes the turnover-determining step (tds) of the reaction, fitting well the rate equation obtained through kinetic experiments, where one molecule of each reactant (alkene and O2) participates. The use of optimum solvents such as perflourodecaline49 to better dissolve the O2 molecules and increase the yield for epoxides, even in reluctant alkene reactants such as 1d, confirms the participation of dissolved O2 in the rds of the reaction (Fig. S27). Partial O2 solubility must be directly related to the reaction, thus, the solvent50,51, however, most of the reactions here are perfomed in neat alkene. It is known that the intermediate alkene radical cation is enough stable and does not rapidly isomerize to contiguous carbon atom positions12,44, in line with the observed epoxidation results.
The radical coupling of a second molecule of 3O2 to the intermediate alkene radical cation is in principle highly favored, to produce an intermediate zwitterionic radical species which is in equilibrium with two possible O–coupled species, the more stable peroxide intermediate (in red) and the less favorable but more reactive perepoxide (in green). All these processes, still in equilibrium, allow the intermediate cationic C–C bond to rotate, thus cis/trans isomerization may occur during the epoxidation reaction12,14. To check this, a kinetic experiment with cyclododecene 1f was performed, showing the enrichment in the thermodynamic more stable trans isomer of the epoxide product 2f during the aerobic epoxidation reaction (Fig. S28), confirming the cis/trans isomerization process during reaction.
It has been experimentally and computationally determined52,53 that the Ea for the formation of perepoxides from typical alkenes and O2 is ~80 kJ·mol−1, while the Ea for the formation of the corresponding peroxides is the half, around ~40 kJ·mol−1. These Ea values explain the easy formation of overoxidized products when alkenes are treated with O2 under inappropriate reaction conditions to generate the epoxides. However, the Ea obtained under the reaction conditions reported here [Ea = 74 kJ·mol−1 for alkene 1b, see above] strongly suggests that perepoxide formation takes over and leads to the synthesis of the epoxide by a simple coarctate reaction54. The latter occurs after reaction of the perepoxide with the previously generated radical superoxide O2·−, to finally lead to the epoxide product and ozone, the major products experimentally observed during reaction.
The difficulties on finding in the literature any method to prepare perepoxides lead us to perform computational calculations to support the proposed reaction mechanism. These calculations, based on the density functional theory (DFT), were carried out to optimize the molecules involved during the epoxidation reaction of alkene 1b (see computational details in the SI, and Table S6). The Natural Bond Orbital (NBO) analysis was performed to confirm the electronic distribution of optimized structures (see SI Fig. S29). In case of the perepoxide molecule, the Natural Population Analysis (NPA) indicates that the unpaired electron is largely localized on the terminal oxygen atom (spin density = 0.862), which is consistent with its assignment as the radical center. However, according to the NPA charges, the positive charge is delocalized between this oxygen and the two carbons that form the three-membered ring, since these atoms show partial positive charges. Both radical species shown a delocalization of the unpaired radical between the two atoms that support it, so they show the same value for NPA charge and spin density. Figure 7A shows the better energetic and symmmetric compatibility between the highest occupied molecular orbital (HOMO) of the alkene and the lowest unoccupied molecular orbital (LUMO) of O2 for the more stable β form of triplet 3O2, but not for the 3O2 α form, and also for the singlet 1O2, to generate the radical alkene cation and superoxide O2·− after the electronic transfer from the HOMO of the alkene to the LUMO of either 3O2 (β form) or 1O2. Following this molecular orbital theory approach, Fig. 7B shows that the so–generated alkene cation radical is also able to react with a second molecule of 3O2 (β form), to generate the corresponding perepoxide, which finally can react with the radical superoxide O2·− left behind, as shown in Fig. 7C. Indeed, the LUMO and HOMO orbitals of the computed structures match well after interacting each other during the different steps of the reaction (Figs. S29 and S30). These computational results are compatible with the experimental results and strongly support the mechanism proposed involving a perepoxide intermediate. It is noteworthy to comment here that this mechanism is significantly different from other proposed for the autooxidation of alkenes, since involves triplet rather than singlet O255,56, besides to occur in the liquid rather than in the gas phase57.

Calculated Gibbs energy profiles at 373.15 K for (A) highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) from both singlet (S) and triplet (T) O2 and alkene 1b, B HOMO and LUMO molecular orbitals from triplet oxygen and radical alkene 1b, and C HOMO and LUMO molecular orbitals from superoxide and perepoxide.
Implications for the ambient oxidation of alkenes
The results shown above infer that alkyl alkenes in contact with the atmosphere will generate the epoxide as the primary product, at least in small amounts. To check this, we examined through GC–MS analysis the open bottles of two representative secondary alkyl alkenes (1b and 1f) available at the laboratory and which do not contain stabilizers such as butylhydroxytoluene (BHT). Apparently, these bottles may have not been stored under inert atmosphere. The results (Fig. S31) show the formation of high amounts of the corresponding epoxides in both cases, up to a 44% of epoxides and overoxidized products. New bottles of the same alkenes were bought and analyzed, showing that the brand-new alkene materials contain some epoxides but in lower extent (Fig. S32). These results demonstrate that secondary alkyl alkenes will generate the epoxide after storage or heating under ambient conditions.
In summary, the aerobic epoxidation of alkyl alkenes can be carried out in good yields and selectivity after heating the neat alkene with O2 or air in either a pressurized reactor, under O2 bubbling or just in an open flask. The process does not require any catalyst58,59, solvent nor additive, and can be performed under industrially-feasible reaction conditions, far from flammability points. Fundamental industrial epoxides such as those derived from propylene, cyclic alkenes, limonene, fatty acids or geraniol can be obtained with this uncatalyzed procedure, which can be engaged in one-pot reactions either with different alkene formations or epoxide transformations. Mechanistic studies reveal that the turnover-limiting step of the epoxidation reaction is the one-electron transfer from the alkene to O2, and that perepoxide species are potential key intermediates to lead to the epoxide product, and circumvent undesired overoxidation products. The uncatalyzed aerobic epoxidation reaction occurs not only under synthetic conditions but also spontaneously under ambient conditions. These results open a new sustainable pathway for the synthesis of epoxides.
Methods
General
Reagents and solvents were obtained from commercial sources and were used without further purification otherwise indicated. Glassware was dried in an oven at 175 °C before use. Reactions were performed in either house-made glass reactors connected to manometers, round-bottomed flasks or 2.0 ml vials, equipped with a magnetic stirrer. Oxygen or air atmospheres were set after purging three times. 1H, 13C, and DEPT nuclear magnetic resonance (NMR) spectra were recorded at room temperature on a 400 MHz spectrometer (Bruker Ascend 400) using the appropriate solvent. Infrared spectra were recorded on an Attenuated total reflection (ATR) Fourier transform infrared (FTIR) spectroscopy, performed in a JASCO FT/IR–4700, which was employed to record the IR spectra from 400 to 4000 cm−1 of the different solid catalysts. Gas chromatographic analyses were performed in an instrument (Agilent 8860) equipped with a 30 m × 250 μm × 0.25 μm Agilent HP-50+ capillary column. N-dodecane was used as an external standard. GC–MS analyses were performed on a Agilent 8890 N spectrometer equipped with a 30 m × 250 μm × 0.25 μm Agilent HP-5MS UI capillary column and operated under the same conditions. Products were characterized by comparison with the given literature, when possible. Absorption and emission ultraviolet–visible spectroscopy measurements were performed on the same samples than that for mass determination, using an UV–vis (UV0811M209, Varian) and a LP S–220B spectrophotometer (Photon Technology International, equipped with 75 W Xe lamp), respectively. DFT electronic structure calculations were performed using the software Gaussian 09 with the M06-2× coupled with the 6–31 + G(3df,2p) basis set. Geometries of reactants, intermediates and products were optimized by calculating vibration frequencies to confirm that an energy minimum was found. A Natural Bond Orbital (NBO) analysis was performed to obtain the Natural Population Analysis (NPA) charges and NPA spin densities. Graphics were represented in the ExcelTM program and the error bars represent a particular confidence interval, in this case a 95% confidence.
Reaction procedures
Typical reaction procedure for pressurized reactors
In a 7.5 mL autoclave equipped with a magnetic bar and a manometer, 0.5 mmol of the corresponding alkene 1b–1t is added, three purges are made with oxygen and, then, the reactor is charged at 3–5 bar of oxygen pressure, heated at 130 °C for 1.5 h, and the reaction followed by GC until its complete conversion. Afterwards, if required, the mixture was purified by flash chromatography using as an eluent a hexane: ethyl acetate mixture (typically 10:1, v:v), obtaining the pure epoxide with the corresponding yield (Figs. 2 and 3).
Typical reaction procedure for O2 bubbling
In a 25 mL round-bottomed flask stoppered with a septum, 5 mmol of the corresponding alkene 1b, 1f, 1n, or 1p are introduced and heated to 130 °C, and a capillary with an oxygen flow of 1 mL·min−1 is introduced to the bottom of the flask, so that a slight bubbling is produced in the starting alkene product. An outlet orifice is let to avoid pressure inside the flask. The reaction is followed by drawing aliquots periodically and measured them on the GC, until maximum conversion. Once the reaction is finished, the mixture is purified (if required) by flash chromatography using as an eluent a hexane: ethyl acetate mixture (typically 10:1, v:v), to obtain the pure epoxide with the corresponding yield (Figs. 2 and 3).
Typical reaction procedure for open flasks
In a 10 mL round-bottomed flask equipped with a magnetic bar, 5 mmol of the corresponding alkene 1b, 1e, 1f, 1g, 1p, or 1u–1y is introduced, and a reflux condenser open to the air is adapted. The mixture is heated at 150 °C for 16–20 h, aliquots are periodically removed and measured by GC, in order to follow the reaction progress until it reaches a maximum conversion. Once finished, the mixture is purified by flash chromatography (if required) using as an eluent a hexane: ethyl acetate mixture (typically 10:1, v:v), to obtain the pure epoxide with the corresponding yield (Figs. 2 and 3).
A representative one-pot reaction
In a round-bottomed flask equipped with a magnetic bar, the corresponding terminal alkene (1 mmol) and the Grubbs´ 1st generation catalyst (5 mol%) were added and stirred at 90 °C for 18 h. Then, the flask was connected to a reflux condenser and heated at 130 °C for 1.5 h. The reaction was monitored after taking out and measuring aliquots by GC, until maximum conversion. Once the reaction is finished, the mixture is purified by flash chromatography using as an eluent a hexane: ethyl acetate mixture (10:1, v:v), to obtain the pure epoxide 2aa in 43% yield (Fig. 5A).
Computational details
DFT electronic structure calculations were performed using the software Gaussian 09 with the M06-2× coupled with the 6–31 + G(3df,2p) basis set. Geometries of reactants, radical intermediates, and products were optimized by calculating vibration frequences at 373.15 K of temperature to confirm that it is a energy minimum. A Natural Bond Orbital (NBO) analysis was performed to obtain the Natural Population Analysis (NPA) charges and NPA spin densities.
Data availability
The datasets generated during and/or analysed during the current study are included in this published article (and its supplementary information files) or available from the corresponding author on request. Datasets could be also deposited in public repositories of the UPV and CSIC. Source data are provided with this paper.
References
Meng, Y. et al. The lord of the chemical rings: catalytic synthesis of important industrial epoxide compounds. Catalysts 11, 765 (2021).
Berndt, T. & Böge, O. Gas–phase epoxidation of propylene and ethylene. Ind. Eng. Chem. Res. 44, 645–650 (2005).
Nishiyama, Y., Nakagawa, Y. & Mizuno, N. High turnover numbers for the catalytic selective epoxidation of alkenes with 1 atm of molecular oxygen. Angew. Chem. Int. Ed. 40, 3639–3641 (2001).
Aprile, C., Corma, A., Domine, M. E., Garcia, H. & Mitchell, C. A cascade aerobic epoxidation of alkenes over Au/CeO2 and Ti-mesoporous Material by “In situ” Formed Peroxides. J. Catal. 264, 44–53 (2009).
Pattisson, S. et al. Tuning graphitic oxide for initiator– and metal–free aerobic epoxidation of linear alkenes. Nat. Commun. 7, 12855 (2016).
Stamoulis, A. G., Bruns, D. L. & Stahl, S. S. Optimizing the synthetic potential of O2: implications of overpotential in homogeneous aerobic oxidation catalysis. J. Am. Chem. Soc. 145, 17515–17526 (2023).
Liu, H. et al. Modulating charges of dual sites in multivariate metal–organic frameworks for boosting selective aerobic epoxidation of alkenes. J. Am. Chem. Soc. 145, 11085–11096 (2023).
Blanckenberg, A. & Malgas-Enus, R. Olefin epoxidation with metal-based nanocatalysts. Catal. Rev. 61, 27–83 (2019).
Lu, X.-H. et al. Non-metallic ketone compounds catalyzed selective epoxidation of styrene with dry air. Catal. Lett. 132, 487–491 (2009).
Hadian-Dehkordi, L. & Hosseini-Monfared, H. Enantioselective aerobic oxidation of olefins by magnetite nanoparticles at room temperature: a chiral carboxylic acid strategy. Green. Chem. 18, 497–507 (2016).
Sanz-Navarro, S. et al. Radical α–alkylation of ketones with unactivated alkenes under catalytic and sustainable industrial conditions. Appl. Catal. A 613, 118021 (2021).
Correa, P. E., Hardy, G. & Riley, D. P. Selective autoxidation of electron-rich substrates under elevated oxygen pressures. J. Org. Chem. 53, 1695–1702 (1988).
Mahajani, S. M., Sharma, M. M. & Sridhar, T. Uncatalysed oxidation of cyclohexene. Chem. Eng. Sci. 54, 3967–3976 (1999).
Mahajan, S. S., Sharma, M. M. & Sridhar, T. Uncatalyzed liquid-phase oxidation of cyclododecene with molecular oxygen. Ind. Eng. Chem. Res. 43, 3289–3296 (2004).
Gonzalez-de-Castro, A. & Xiao, J. Green and efficient: iron-catalyzed selective oxidation of olefins to carbonyls with O2. J. Am. Chem. Soc. 137, 8206–8218 (2015).
Soler, J., Gergel, S., Klaus, C., Hammer, S. C. & Garcia-Borràs, M. Enzymatic control over reactive intermediates enables direct oxidation of alkenes to carbonyls by a P450 iron-oxo species. J. Am. Chem. Soc. 144, 15954–15968 (2022).
Mill, T. & Hendry, D. G. Chapter 1 Kinetics and mechanisms of free radical oxidation of alkanes and olefins in the liquid phase. In Liquid–Phase Oxidation (eds. Bamford, C. H.et al.) Vol. 16, 1–87 (Elsevier, 1980).
Van Sickle, D. E., Mayo, F. R., Arluck, R. M. & Syz, M. G. Oxidations of acyclic alkenes. J. Am. Chem. Soc. 89, 967–977 (1967).
Diaz, R. R., Selby, K. & Waddington, D. J. Reactions of oxygenated radicals in the gas phase. Part I. Reaction of peracetyl radicals and but-2-Ene. J. Chem. Soc. Perkin. Trans. 2, 758–763 (1975).
Suprun, W. Y. & Opeida, J. Studies on the autoxidation of aryl alkenes. Chem. Eng. Technol. 29, 1253–1261 (2006).
Metelitsa, D. I. Reaction mechanisms of the direct epoxidation of alkenes in the liquid phase. Russ. Chem. Rev. 41, 807 (1972).
Filippova, T. V. & Blyumberg, E. A. Mechanism of the epoxidation of alkenes by molecular oxygen. Russ. Chem. Rev. 51, 582 (1982).
Sanz-Navarro, S. et al. Parts–per–million of ruthenium catalyze the selective chain–walking reaction of terminal alkenes. Nat. Commun. 13, 2831 (2022).
Kumar, V. & Chimni, S. S. Metal-free ring-opening of epoxides. ChemistrySelect 8, e202301963 (2023).
Cvetanović, R. J. Molecular rearrangements in the reactions of oxygen atoms with olefins. Can. J. Chem. 36, 623–634 (1958).
Van Sickle, D. E., Mayo, F. R. & Arluck, R. M. Liquid–phase oxidations of cyclic alkenes1. J. Am. Chem. Soc. 87, 4824–4832 (1965).
Maranzana, A., Ghigo, G. & Tonachini, G. Mechanistic significance of perepoxide trapping experiments, with epoxide detection, in 1Δg dioxygen reactions with alkenes. J. Org. Chem. 68, 3125–3129 (2003).
Moriai, T., Tsukamoto, T., Tanabe, M., Kambe, T. & Yamamoto, K. Selective hydroperoxygenation of olefins realized by a coinage multimetallic 1-nanometer catalyst. Angew. Chem. Int. Ed. 59, 23051–23055 (2020).
Leach, A. G., Houk, K. N. & Foote, C. S. Theoretical prediction of a perepoxide intermediate for the reaction of singlet oxygen with trans–cyclooctene contrasts with the two–step no–intermediate ene reaction for acyclic alkenes. J. Org. Chem. 73, 8511–8519 (2008).
Alshammari, H., Miedziak, P. J., Knight, D. W., Willock, D. J. & Hutchings, G. J. The effect of ring size on the selective oxidation of cycloalkenes using supported metal catalysts. Catal. Sci. Technol. 3, 1531–1539 (2013).
Wu, Y. et al. Sustainable and practical access to epoxides: metal–free aerobic epoxidation of olefins mediated by peroxy radical generated in situ. ACS Sustain. Chem. Eng. 8, 1178–1184 (2020).
Keleş, E. & Hazer, B. Autooxidized polyunsaturated oils/oily acids: post–it applications and reactions with Fe(III) and adhesion properties. Macromol. Symp. 269, 154–160 (2008).
Westerman, C. R., McGill, B. C. & Wilker, J. J. Sustainably sourced components to generate high–strength adhesives. Nature 621, 306–311 (2023).
Garnes-Portolés, F. et al. Regioirregular and catalytic Mizoroki–Heck reactions. Nat. Catal. 4, 293–303 (2021).
Garnes-Portolés, F., Sánchez-Quesada, J., Espinós-Ferri, E. & Leyva-Pérez, A. Synthesis of dehydromuscone by an alkene metathesis macrocyclization reaction at 0.2 M concentration. Synlett 33, 1933–1937 (2022).
Garhwal, S., Kaushansky, A., Fridman, N. & de Ruiter, G. Part per million levels of an anionic iron hydride complex catalyzes selective alkene isomerization via two–state reactivity. Chem. Catal. 1, 631–647 (2021).
Sato, T., Hamada, Y., Sumikawa, M., Araki, S. & Yamamoto, H. Solubility of oxygen in organic solvents and calculation of the Hansen solubility parameters of oxygen. Ind. Eng. Chem. Res. 53, 19331–19337 (2014).
Van den Ven, L. J. M., De Haan, J. W. & Bucinska, A. Conformational equilibriums of normal alkanes, 1-alkenes, and some (E)- and (Z)-2-alkenes in neat liquids and in some selected solvents. A carbon-13 nuclear magnetic resonance study. J. Phys. Chem. 86, 2516–2522 (1982).
Li, M.-Y. et al. Solvent-free and catalyst-free direct alkylation of alkenes. Green. Chem. 25, 7073–7078 (2023).
Li, M.–Y. et al. Aggregation-enabled alkene insertion into carbon–halogen bonds. Aggregate 4, e346 (2023).
Mitsudome, T., Mizumoto, K., Mizugaki, T., Jitsukawa, K. & Kaneda, K. Wacker-type oxidation of internal olefins using a PdCl2/N,N-dimethylacetamide catalyst system under copper-free reaction conditions. Angew. Chem. Int. Ed. 49, 1238–1240 (2010).
Williams, P. J. H. et al. New approach to the detection of short-lived radical intermediates. J. Am. Chem. Soc. 144, 15969–15976 (2022).
Clennan, E. L. & Pace, A. Advances in singlet oxygen chemistry. Tetrahedron 61, 6665–6691 (2005).
Díaz-Uribe, C. E., Vallejo-Lozada, W. A. & Martínez-Ortega, F. Photooxidation of anthracene under visible light with metallocarboxyphenylporphyrins. Rev. Fac. Ing. Univ. Antioquia 73, 225–230 (2014).
Olojo, R. O., Xia, R. H. & Abramson, J. J. Spectrophotometric and fluorometric assay of superoxide ion using 4-Chloro-7-nitrobenzo-2-oxa-1,3-diazole. Anal. Biochem. 339, 338–344 (2005).
Li, M.–S. et al. Catalyst-free direct difunctionalization of alkenes with H-phosphine oxides and dioxygen: a facile and green approach to β-hydroxyphosphine oxides. Tetrahedron Lett. 57, 2642–2646 (2016).
Heisig, C., Zhang, W. & Oyama, S. T. Decomposition of ozone using carbon–supported metal oxide catalysts. Appl. Catal. B 14, 117–129 (1997).
Li, J. et al. Low pressure induced porous nanorods of ceria with high reducibility and large oxygen storage capacity: synthesis and catalytic applications. J. Mater. Chem. A 2, 16459–16466 (2014).
Riess, J. G. Understanding the fundamentals of perfluorocarbons and perfluorocarbon emulsions relevant to in vivo oxygen delivery. Artif. Cells, Blood Substit., Biotechnol. 33, 47–63 (2005).
Zhang, S., Song, P. & Li, S. Application of N-dodecane as an oxygen vector to enhance the activity of fumarase in recombinant Escherichia coli: role of intracellular microenvironment. Braz. J. Microbiol. 49, 662–667 (2018).
Wentzel, B. B., Alsters, P. L., Feiters, M. C. & Nolte, R. J. M. Mechanistic studies on the Mukaiyama epoxidation. J. Org. Chem. 69, 3453–3464 (2004).
Yamaguchi, K., Yabushita, S., Fueno, T., Kato, S. & Morokuma, K. Geometry optimization of the ring-opened oxirane diradical: mechanism of the addition reaction of the triplet oxygen atom to olefins. Chem. Phys. Lett. 70, 27–30 (1980).
Malek, B. et al. Probing the transition state-to-intermediate continuum: mechanistic distinction between a dry versus wet perepoxide in the singlet oxygen “Ene” reaction at the air-water interface. Langmuir 38, 6036–6048 (2022).
Herges, R. Coarctate and pseudocoarctate reactions: stereochemical rules. J. Org. Chem. 80, 11869–11876 (2015).
Dias, A. M. A., Freire, M., Coutinho, J. A. P. & Marrucho, I. M. Solubility of oxygen in liquid perfluorocarbons. Fluid Phase Equilib. 222–223, 325–330 (2004).
Rao, T. S. S. & Awasthi, S. Oxidation of olefins. J. Indian Chem. Soc. 84, 902–913 (2007).
Berndt, T. & Böge, O. Catalyst-free gas-phase epoxidation of alkenes. Chem. Lett. 34, 584–585 (2005).
Teržan, J., Huš, M., Likozar, B. & Djinović, P. Propylene epoxidation using molecular oxygen over copper- and silver-based catalysts: a review. ACS Catal. 10, 13415–13436 (2020).
Kiani, D. & Wachs, I. E. Practical considerations for understanding surface reaction mechanisms involved in heterogeneous catalysis. ACS Catal. 14, 16770–16784 (2024).
Acknowledgements
This work is part of the project PID2023-148441NB-I00 funded by MCIN/AEI/10.13039/501100011033MICIIN (Spain). Financial support by Severo Ochoa centre of excellence program (CEX2021–001230–S) is gratefully acknowledged. The work has also been funded by Generalitat Valenciana (Prometeo Grupos de Investigación de Excelencia PROMETEU/2021/054, and INNEST/2022/20 from Agència Valenciana de la Innovació). Funding for open acces charge: Universitat Politècnica de València. S.R.–N. thanks Agència Valenciana de la Innovació for a contract. J.O.-M. thanks the MICIIN for the concession of the Ramón y Cajal contract RYC2022-036154-I (funded by MCIN/AEI/10.13039/501100011033). S.H.–A. and F.G.–P. thanks ITQ for the concession of a contract. We thank Dr Mercedes Boronat and Miguel Ródenas Espada for unvaluable helping during the DFT calculations.
Author information
Authors and Affiliations
Contributions
S.H.-A. performed the epoxidation reactions, the one-pot isomerization-epoxidation reactions, DFT calculations, and mechanistic experiments. F.G.-P. discovered the uncatalyzed reaction, performed the epoxidation reactions and the one-pot metathesis-epoxidation reactions, and mechanistic experiments. S.R.-N. performed epoxidation reactions, the one-pot epoxidation-epoxide transformations, and mechanistic experiments. J.O.-M. epoxidation reactions and mechanistic experiments, and supervised to S.H.-A. A.L.-P. designed the experiments and supervised the whole work. The manuscript has been written with contributions from all authors, which also interpreted all the experimental part.
Corresponding authors
Ethics declarations
Competing interests
A Spanish patent (number P202430625) covering the uncatalyzed epoxidation reaction of alkenes has been filled, appearing as co-inventors S.H.-A., F.G.-P., J.O.-M., and A.L.-P. Another Spanish patent (number P202430884) covering the Ru-catalyzed one-pot isomerization-epoxidation reaction of alkenes has been filled, appearing as co-inventors S.H.-A., S.R.-N., and A.L.-P.
Peer review
Peer review information
Nature Communications thanks Landong Li, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hervàs-Arnandis, S., Garnes-Portolés, F., Rodríguez-Nuévalos, S. et al. Uncatalyzed aerobic epoxidation of liquid alkyl alkenes. Nat Commun 16, 6542 (2025). https://doi.org/10.1038/s41467-025-61711-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61711-3
