
Abstract
Oral facial cleft (OFC) comprises cleft lip with or without cleft palate (CL/P) or cleft palate only. Genome wide association studies (GWAS) of isolated OFC have identified common single nucleotide polymorphisms (SNPs) in many genomic loci where the presumed effector gene (for example, IRF6 in the 1q32 locus) is expressed in embryonic oral epithelium. To identify candidates for functional SNPs at eight such loci we conduct a massively parallel reporter assay in a fetal oral epithelial cell line, revealing SNPs with allele-specific effects on enhancer activity. We filter these SNPs against chromatin-mark evidence of enhancers and test a subset in traditional reporter assays, which support the candidacy of SNPs at loci containing FOXE1, IRF6, MAFB, TFAP2A, and TP63. For two SNPs near IRF6 and one near FOXE1, we engineer the genome of induced pluripotent stem cells, differentiate the cells into embryonic oral epithelium, and discover allele-specific effects on the levels of effector gene expression, and, in two cases, the binding affinity of transcription factors FOXE1 or ETS2. Conditional analyses of GWAS data suggest the two functional SNPs near IRF6 account for the majority of risk for CL/P at this locus. This study connects genetic variation associated with OFC to mechanisms of pathogenesis.
Similar content being viewed by others


Introduction
Oral facial cleft (OFC) is a multifactorial disorder that can present as a cleft lip with or without cleft palate (CL/P) or a cleft palate only (CP) and has a genetic predisposition. More than one hundred syndromes include OFC as a phenotype, and overall, about 30% of CL/P cases are syndromic1. Such syndromes are generally caused by mutations in single genes and follow a Mendelian inheritance pattern. The remaining cases are non-syndromic or isolated. The etiology of non-syndromic OFC is partially genetic, as the concordance of non-syndromic OFC is 50% in identical twins but just 3–5% in other first-degree relatives2,3,4. Multiple independent genome-wide association studies (GWAS), and meta-analyses thereof, have identified more than 40 loci where alleles of common single-nucleotide polymorphisms (SNPs) are over-represented in cases versus in controls with the same ancestry (reviewed in ref. 5). Importantly, however, most of the heritable risk for isolated OFC has not been assigned to any gene or locus. Moreover, GWAS results alone do not illuminate the mechanisms of pathogenesis attributable to genetic variation at each locus. Understanding these mechanisms may point to additional genes in which mutations will contribute risk for non-syndromic OFC and may guide the design of preventative therapies.
Identifying functional (causal) SNPs among those in linkage disequilibrium with them is challenging. Most SNPs lie in non-coding DNA, and the functional subset of them presumably disrupt cis-regulatory sequences (i.e., enhancers and promoters). However, the sequence constraints of cis-regulatory sequences remain poorly understood. We and others have used machine learning to mine sets of tissue-specific enhancers for sequence patterns6,7,8, and there are in silico tools for inferring variants that affect enhancer function9,10, but currently there is no tool that can robustly identify non-coding variants that alter enhancer activity. An alternative approach is the massively parallel reporter assay (MPRA). MPRAs have been widely used to detect elements with enhancer activity, and in some cases to detect the effect of variants on enhancer activity11,12,13,14,15,16,17,18,19,20. A challenge in deploying MPRA to identify functional SNPs is that, because the results are dependent on the cellular context15, it may be necessary to use a cell line that models the embryonic cell type in which the SNPs affect disease risk.
The 1q32/IRF6 locus is associated with OFC in multiple ethnic groups21,22,23,24,25,26,27,28,29,30,31,32. IRF6, encoding the transcription factor Interferon Regulatory Factor 6, is strong candidate for the risk-relevant gene (i.e., the effector gene) in this locus because mutations in IRF6 are found in about 70% of patients with Van der Woude syndrome, the most common syndromic form of OFC (VWS1, OMIM # 119300). Finding the functional SNPs at this locus would yield insight into the regulation of IRF6 gene expression during morphogenesis of the face and, if the transcription factors whose binding is affected by those SNPs are identified, would identify new candidate genes for OFC. However, a meta-analysis of several GWAS identified more than 600 SNPs at this locus with P values indicating at least a suggestive association with OFC23. One of these SNPs, rs642961, resides in an enhancer of IRF6 and alters the binding of the transcription factor AP2-α (TFAP2A) in an electrophoretic mobility shift assay, suggesting it is functional33. However, this conclusion is uncertain because this SNP did not have allele-specific effects in a standard reporter assay33 or in an MPRA34. The large number of OFC-associated SNPs at this locus, and others, makes it difficult to determine which are functional.
Here, we deployed an MPRA in a fetal oral epithelium cell line to nominate candidate functional SNPs among those associated with OFC and within loci where the presumed effector gene is expressed in oral epithelium. We validated a subset of the MPRA results using traditional luciferase reporter assays in the cell line and in primary keratinocytes. For two promising SNPs near IRF6 and one near FOXE1, we engineered the genotype of the SNPs in induced pluripotent stem cells, differentiated the cells into embryonic oral epithelium, and then assessed allele-specific effects on gene expression and transcription factor binding. These studies support six SNPs as being functional, with varying levels of support, and two of these SNPs as accounting for most of the heritable risk for CL/P phenotype attributed to the IRF6 locus in the cohort analyzed here.
Results
A massively parallel reporter assay reveals candidate functional SNPs at eight OFC-associated loci
For analysis by MPRA, we picked 887 SNPs from eight loci associated with OFC and in which the currently presumed effector gene is expressed in oral epithelium, although not necessarily only there, and which regulates differentiation of an epithelial tissue35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60 (1q32/IRF6, 2p21/THADA, 3q28/TP63, 6p24.3/TFAP2A, 9q22.2/GADD45G, 9q22.33/FOXE1, 12q13.13/KRT18, and 20q12/MAFB) (Table 1 and Supplementary Data 1, 2). At seven of the loci, we picked SNPs based on their significance in a genome-wide meta-analysis of two prior OFC studies24,30 (Table 1 and Supplementary Data 2). We additionally included SNPs at the TFAP2A locus identified in an independent GWAS of CL/P in Han Chinese26. At an eighth locus (9q22.33/FOXE1), we picked SNPs in strong linkage disequilibrium with the GWAS lead SNP (rs12347191) and annotated as being within regulatory elements23 (Table 1 and Supplementary Data 2). As outlined in Fig. 1a, we synthesized a library of reporter plasmids containing 161 base-pairs (bp) genomic test elements each centered on an OFC-associated SNP; OFC risk and non-risk alleles of each SNP were represented in four replicate constructs with distinct barcodes (Supplementary Data 3, 4). We performed the MPRA in GMSM-K cells (Supplementary Data 5), a cell line derived from human fetal oral mucosa61. The results were strongly correlated (R2 ≥ 0.876) across four replicate experiments (Supplementary Fig. 1a).

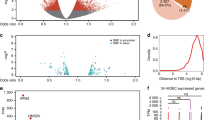
a Schematic of MPRA library construction and execution. BC, barcode. Yellow and red asterisks: non-risk and risk allele, respectively. b Scatter dot plot showing (black dots) 65 SNPs with significant allele-specific effects on reporter activity in the MPRA and (gray dots) 822 SNPs without them. Arrows indicate the functional SNPs identified in this study (two near IRF6, rs11119348 and rs661849, and one near FOXE1, rs10984103). Dashed lines indicate the 95th percentile of the reporter activity of scrambled elements; on both axes, zero, i.e., log2(1) represents the reporter activity of the empty vector.
In the MPRA results we assessed the enhancer activity of each element and whether the SNP allele affected it. Of the 887 elements evaluated, one or both variants of 480 (54.1%) had activity significantly different from that of the basal promoter (false discovery rate (FDR) <0.01); 180 (20.3%) of them exhibited higher activity than that of the basal promoter (Supplementary Data 6 and Supplementary Fig. 1b). As expected, the average activity of 84 scrambled genomic test elements was very close to that to the basal promoter (Supplementary Fig. 1c and Supplementary Data 7). Unexpectedly, the enhancer activities of the test elements were only weakly correlated with levels of histone H3 lysine 27 acetylation (H3K27Ac), a mark of active enhancers, in primary normal human epidermal keratinocytes (NHEK) (Pearson correlation coefficient r = 0.1126) (Supplementary Fig. 1d). Further, the enhancer activities of four elements from within a known oral epithelium enhancer (IRF6 MCS9.76,33,62) were not higher than that of the basal promoter (Supplementary Fig. 1c). These unexpected findings may reflect the semi-epithelial phenotype of this cell line, discussed below. Nonetheless, 65 (7%) of the SNPs tested had allele-specific enhancer activity in the MPRA (Fig. 1b and Supplementary Data 6) (FDR <0.05), nominating these SNPs as candidates to be functional. These SNPs were enriched among elements whose activity was different from that of the basal promoter (P < 0.0001, one-sided Fisher’s exact test). Each of the eight loci had at least one of the 65 MPRA-nominated SNPs; notably, at the 1q32/IRF6 locus, there were 46 such SNPs (Figs. 1b, 2a, Table 1, and Supplementary Data 6).
Filtering candidate SNPs for those present in annotated enhancers
We filtered MPRA-nominated SNPs for those lying within enhancers active in embryonic oral epithelium. In one approach to identify such enhancers, we used NHEKs as a model for embryonic oral epithelium and the activity-by-contact (ABC) method to identify enhancers for the presumed effector genes63. This method incorporates cell type-specific RNA-seq, DNAse hypersensitivity, and H3K27Ac ChIP-seq datasets, all of which are available for NHEKs, and averaged HiC chromatin-contact data from ten ENCODE cell types63,64. Two apparent enhancers for IRF6 and one for FOXE1 contained MPRA-nominated SNPs (i.e., rs11119348 and rs661849 near IRF6, rs10984103 near FOXE1) (Fig. 2a–c). The first candidate SNP near IRF6, rs11119348, lies within IRF6 MCS9.7. This element is the site of a rare single-base-pair duplication that appears to cause Van der Woude syndrome65. It also contains three other common SNPs (i.e., in addition to rs11119348) that are associated with non-syndromic OFC, including rs642961, the focus of an earlier study33. We tested all four of these common SNPs (separately) in the MPRA but only rs11119348 had allele-specific effects in it (Supplementary Fig. 1c). We will refer to this as the “IRF6 −10 kb SNP” (Fig. 2b).

a–c UCSC browser views of the human genome, GRCh37/hg19, focused on the OFC-associated SNPs at a, b IRF6 and c FOXE1. OFC SNPs, SNPs identified by GWAS and evaluated by MPRA at each locus. One SNP at the FOXE1 locus, which was tested in the MPRA but lacked significant effects in it, rs1877431, is outside of the range presented here. MPRA-sig SNPs, SNPs with allele-specific effects in the MPRA. IRF6 enhancers, FOXE1 enhancers, and enhancers for the indicated gene identified using the activity-by-contact (ABC) method and datasets from NHEK (see Methods). Gray arrows, GWAS-meta-analysis P values of the indicated SNPs. Two-sided P values are calculated with an inverse-variance weighted fixed-effects meta-analysis of a transmission disequilibrium test without adjustment for multiple testing and a logistic regression (that had 18 principal components of ancestry as covariates). GM12878 B-cell derived cell line, H1-ESC embryonic stem cells, K562 myelogenous leukemia cell line, HepG2 liver cancer cell line, HUVEC human umbilical vein endothelial cells, HMEC human mammary epithelial cells, HSMM human skeletal muscle myoblasts, NHEK normal human epidermal keratinocytes, NHLF normal human lung fibroblasts. CS 13-20, Carnegie stages for human embryonic face explants70. Colored bars, inferred chromatin state from combinatorial analysis of multiple chromatin mark datasets118. Orange and yellow, active and weak enhancer element, respectively; bright red, active promoter; light red, weak promoter; purple, inactive/poised promoter; blue, insulator; light green, weakly transcribed; gray, Polycomb repressed; light gray, heterochromatin/repetitive. Additional color bars in the facial explants dataset, green, transcription; green-yellow, transcription weak enhancer; purple, bivalent promoter; light purple, poised promoter.
The second candidate SNP in this locus, rs661849, resides within an evolutionarily conserved sequence 21.7 kb upstream of the IRF6 promoter and within the UTP25 promoter (Fig. 2b). There are no other OFC-associated SNPs within this element. We will refer to rs661849 as the “IRF6 −22 kb SNP”. This SNP is an expression QTL (eQTL) in GTEx for several genes and tissues in the region, including IRF6 in cerebellum (P value = 1.5e-8), and is a splicing QTL (sQTL) for IRF6 in sun-exposed skin (P value = 8.3e-9) and for neighboring gene TRAF3IP3 in whole blood66,67. Of note, rs642961, mentioned above, is in linkage disequilibrium with the IRF6 −22 kb SNP (R2 = 0.769) but not with the IRF6 −10 kb SNP (R2 = 0.033) (based on data from the 1000 Genomes project) (Supplementary Fig. 2 and Supplementary Data 8). The candidate SNP within a FOXE1 enhancer, rs10984103, lies within a predicted enhancer 23.8 kb downstream of the FOXE1 transcription start site (Fig. 2c). This SNP is an eQTL for FOXE1 in multiple tissues, including the testis, esophageal mucosa, nerve, and cerebellum66. We will refer to this SNP as the “FOXE1 24 kb SNP”.
In a second, broader approach to identify potential embryonic oral epithelium enhancers that is agnostic to the identity of the effector gene we used chromatin-mark evidence of enhancers active (1) in NHEK (ENCODE), (2) in human immortalized oral epithelial cells (HIOEC), which are potentially a better model of embryonic oral epithelium than NHEK68,69, or (3) in human embryonic faces70. Within elements marked as enhancers in one or more of these contexts there were fourteen MPRA-nominated SNPs, including the three discussed above (Fig. 2b, c); the additional SNPs were detected in the 1q32/IRF6, 6p24.3/TFAP2A, 3q28/TP63, and 20q12/MAFB loci (Supplementary Data 9 and Supplementary Figs. 3–9). In summary, intersecting MPRA-nominated SNPs with predicted enhancers strengthened the candidacy of 14 SNPs as being functional (Table 1 and Supplementary Data 9).
MPRA results tested with luciferase reporter assays
We next tested a subset of the MPRA results in traditional luciferase reporter assays in GMSM-K cells, using reporter elements arbitrarily chosen to be 701 bp long and centered on the SNP (Fig. 3a and Supplementary Data 10, 11). For these tests, we picked six SNPs with allele-specific effects in the MPRA and seven SNPs without them. The former were all among the fourteen candidates discussed above and included IRF6 −10 kb, IRF6 −22 kb, FOXE1 24 kb, rs4812449 (near MAFB), rs201265 (near TFAP2A), and rs75436877 (near TP63); the latter included rs642961 within IRF6 MCS9.7. All six SNPs with allele-specific effects in the MPRA also had them in the luciferase reporter assays, although in two cases, FOXE1 24 kb and rs201265 (near TFAP2A), the direction of effect was reversed (Fig. 3a and Supplementary Data 11, 12). Discordance in the direction of effects of SNP alleles between traditional reporter assays and MPRA has been reported in other studies12,71 and presumably reflects that the short elements used in MPRAs are not full-sized enhancers (addressed below). Among the seven SNPs that did not have allele-specific effects in the MPRA, five also did not have them in the luciferase reporter assays. These five included rs642961 within IRF6 MCS9.7, in agreement with a previous report33. In summary, for eleven of thirteen SNPs tested, luciferase reporter assays agreed with MPRA regarding whether enhancer activity was affected by a SNP allele; this rate of concordance between luciferase and MPRA results is significant (P = 0.0163, one-sided Fisher’s exact test) and matches or exceeds that of other studies using MPRAs to detect functional non-coding SNPs12,13.

a Bar chart of MPRA and luciferase reporter activities for the indicated SNPs at various loci in GMSM-K using elements of the indicated length centered on the SNP. MPRA data are represented as mean ± standard error of the mean (SEM) from the indicated number of replicates. Statistical significance (P value, two-tailed) of the difference between major and minor allele’s reporter activity is determined by Welch’s t-test, followed by Benjamini–Hochberg false discovery rate correction. P (MPRA) = 0.0452 (rs11110348), 0.0057 (rs661849), 0.9097 (rs642961), 0.6942 (rs4844939), 0.4487 (rs12104876), 0.0238 (rs75436877), 0.7100 (rs79482068), 0.0734 (rs79792381), 0.0125 (rs201265), 0.1206 (rs10124184), 0.0067 (rs10984103), 0.0458 (rs4812449), 0.2945 (rs57369620). For luciferase reporter activities, data are represented as mean ± standard deviation (SD) from four independent experiments. Statistical significance (P value, two-tailed) is determined by Student’s t-test. P (luciferase) = 0.0006 (rs11110348), <0.0001 (rs661849), 0.3822 (rs642961), <0.0001 (rs4844939), 0.14 (rs12104876), <0.0001 (rs75436877), 0.0693 (rs79482068), 0.5115 (rs79792381), 0.002 (rs201265), <0.0001 (rs10124184), <0.0001 (rs10984103), <0.0001 (rs4812449), 0.1421 (rs57369620). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, NS non-significant. b–d Scattered dot plots of relative luciferase activity (using the longer elements described in Results) for non-risk and risk alleles of rs11119348, rs661849 and rs10984103 in primary neonatal keratinocytes (HEKn). Value of 1 is that of the empty pGL3 promoter vector. Data are represented as mean ± standard deviation (SD) from four independent experiments. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. e–g Scattered dot plot of relative levels of e, f IRF6 or g FOXE1 mRNA in edited induced oral epithelium (iOE) cells homozygous for the non-risk or risk alleles of each SNP, as indicated, assessed by qRT-PCR. Expression levels are normalized against those of ACTB, GAPDH, HPRT1, UBC and CDH1. Data were represented as mean ± SD from e, f six replicates or g nine replicates of cells harboring each genotype, as indicated in the plot. Each dot represents three technical qPCR replicates. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. Source data are provided as a Source Data file.
We found that the length of the element tested, and whether it matched the extent of open chromatin of an endogenous enhancer, affected a SNP’s direction of effect in reporter assays. First, for the two SNPs with opposite directions of effect in MPRA and luciferase reporter assays (FOXE1 24 kb and rs201265), we repeated the latter using shorter elements that matched the lengths used in the MPRA (i.e., 161 bp). In these luciferase reporter assays, the directions of effect concorded with those from the MPRA (Supplementary Fig. 10a and Supplementary Data 12). Second, for three promising SNPs (IRF6 −10 kb, IRF6 −22 kb and FOXE1 24 kb) with allele-specific effects in the MPRA and in the luciferase reporter assays, we repeated the latter using slightly longer elements (936 bp to 1.1 kb) that aligned with open chromatin (i.e., ATAC-seq peaks) in HIOEC and in NHEK (Supplementary Fig. 10b–d and Supplementary Data 10). Remarkably, for all three SNPs the directions of effect were reversed relative to those using 701 bp elements: in the assays with the longer elements, the risk alleles of IRF6 −10 kb and IRF6 −22 kb SNPs reduced the activity, and the risk allele of FOXE1 24 kb elevated the activity, of the enhancers in which they reside (Supplementary Fig. 10b–d). To summarize, in reporter assays, the length of the element tested affected a SNP’s direction of effect but not whether there was an effect.
At this point in the study, we conducted RNA-seq on our stock of GMSM-K cells and compared expression levels of epithelial and mesenchymal markers to those in primary adult keratinocytes (NHEK) and in human embryonic palatal mesenchymal (HEPM) cells (Supplementary Data 13 and Supplementary Fig. 11a). As expected, GMSM-K expressed higher levels of oral epithelium markers PITX1 and FOXE1 transcripts than either NHEK or HEPM, consistent with their origin from fetal oral keratinocytes61. In addition, they expressed high levels of epithelial markers KRT8 and KRT18 and low levels of mesenchymal markers VIM and CDH2, similar to NHEK and unlike HEPM. However, unexpectedly, they expressed much lower levels of the epithelial marker CDH1 and keratinocyte markers KRT14, TP63, and IRF6 than NHEK did; we confirmed three of these markers with immunostaining (Supplementary Fig. 11a, b). We concluded that our stock of GMSM-K had lost robust epithelial features without fully converting to a mesenchymal phenotype. Therefore, we repeated the luciferase reporter assays in primary neonatal keratinocytes (HEKn). We found that, again, the results concorded with those from the MPRA at 11 of 13 SNPs regarding whether the SNP had allele-specific effects on activity (Fig. 3b–d and Supplementary Fig. 12). In summary, although our stock of GMSM-K lacks robust epithelial features, the results from an MPRA conducted in GMSM-K and those from luciferase reporter studies in HEKn largely concorded.
In chromatin of induced oral epithelial cells the IRF6 −10 kb and IRF6 −22 kb SNPs affect IRF6 expression and the FOXE1 24 kb SNP affects FOXE1 expression
To test whether the IRF6 −10 kb and IRF6 −22 kb SNPs affect enhancer activity in their native genomic contexts we engineered the genotype of WTC-11 induced pluripotent stem cells (iPSCs), which are heterozygous for both SNPs. For each SNP, we generated two clones each that were homozygous for the risk allele or for the non-risk allele (Supplementary Figs. 13a, b, 14a, b). Next, we subjected three replicates of each clone to a 10-day protocol that induces iPSCs to differentiate into embryonic oral epithelial cells (induced oral epithelial cells, iOE)72 (Supplementary Fig. 13a, b). Quantitative RT-PCR (qRT-PCR) revealed that the average level of IRF6 expression in cells homozygous for the risk allele of either SNP (and heterozygous at the other) was lower than that in clones homozygous for the non-risk allele of that SNP (Fig. 3e, f), consistent with the luciferase reporter assays using the longest elements. We also engineered the genotype of these SNPs in GMSM-K cells. Despite the much lower expression of IRF6 in GMSM-K compared to in iOE cells, again, cells homozygous for risk alleles had lower IRF6 expression than cells homozygous for non-risk alleles (Supplementary Fig. 15a–f). These results indicate that the IRF6 −10 kb SNP and the IRF6 −22 kb SNP both directly affect IRF6 expression.
Similarly, we engineered the genotype of the FOXE1 24 kb SNP. The parental iPSCs were heterozygous, and we engineered three clones each to be homozygous for the risk allele or the non-risk allele and differentiated them in triplicate into iOE cells (Supplementary Fig. 16a, b). qRT-PCR revealed that the average level of FOXE1 in cells homozygous for the risk allele was higher than that of the homozygous for the non-risk allele (Fig. 3g), consistent with the luciferase reporter assays using the longest elements, indicating that the FOXE1 24 kb SNP directly affects FOXE1 expression.
The IRF6 −10 kb SNP risk allele promotes binding of the OFC-associated transcriptional repressor FOXE1
Using an online tool73 we predicted that the risk allele of the IRF6 −10 kb SNP diminishes the affinity for two transcription factors (i.e., AR and SOX10) and elevated it for POU5F1B plus sixteen members of the FOX family, including FOXE1 (JASPAR score for FOXE1 site MA1487.1: risk, 14.1; non-risk, 8.6) (Fig. 4a and Supplementary Data 14, 15). Members of the FOX family can function as transcriptional activators or repressors74,75. Mutations in FOXE1 cause Bamforth–Lazarus syndrome76 and SNPs near it are associated with risk for non-syndromic OFC23,77,78. Using an antibody against FOXE1, we conducted ChIP in iOE cells generated from the parental iPSCs. qPCR revealed more chromatin precipitated at the IRF6 −10 kb SNP locus by anti-FOXE1, and by anti-H3K27Ac, than at an intergenic region, consistent with the SNP being in an enhancer bound by FOXE1 (Fig. 4b, c). While the iOE cells were heterozygous at this SNP, like the parental iPSCs from which they were derived (Fig. 4d), PCR and sequencing revealed that chromatin precipitated by the anti-FOXE1 antibody was enriched for the risk allele (Fig. 4d, two additional replicates in Supplementary Fig. 17a). By contrast, the anti-H3K27Ac antibody pulled down more of the non-risk allele (Fig. 4d and Supplementary Fig. 17a), consistent with higher activity of the enhancer and higher expression of IRF6 in cells with the non-risk genotype. Similarly, anti-FOXE1 ChIP in GMSM-K homozygous for the risk or non-risk allele of IRF6 −10 kb SNP revealed stronger binding of FOXE1 to the risk allele (Supplementary Fig. 18a). These results indicate that the IRF6 −10 kb SNP risk allele favors the binding of FOXE1.

a Consensus FOXE1 binding motif from the JASPAR database of transcription factor DNA-binding preferences (Matrix ID: MA1847.1) and alignment of the variant site in several mammals. The risk allele (A) is the reference allele and has a higher frequency than the non-risk allele (C) in most populations. b, c Percent input identified by ChIP-qPCR for anti-FOXE1 and anti-H3K27Ac, respectively, in iOE cells heterozygous for rs11119348 using primers specific to the IRF6 -10 kb enhancer site or, as a negative control, to a region 103.7 kb upstream of the IRF6 transcription start site that lacked ATAC-Seq and H3K27Ac ChIP-Seq signals in HIOEC or NHEK. Error bars refer to three ChIP replicates and expressed as mean ± SD. Each dot represents three technical qPCR replicates. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. NS non-significant. d Sequencing of anti-FOXE1 and anti-H3K27Ac ChIP-PCR products from cells heterozygous for rs11119348 using the indicated antibody. e Scattered dot plot of relative luciferase activity of the IRF6 -10 kb reporter construct (longest version) harboring the risk allele of rs11119348, in wildtype (WT) or a clone of FOXE1 homozygous knockout (KO) GMSM-K cells. Data are represented as mean ± SD from three independent experiments. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. f Scattered dot plot of relative levels of IRF6 mRNA in WT and FOXE1-KO GMSM-K cells assessed by qRT-PCR. Expression levels of IRF6 are normalized against ACTB. Data are represented as mean ± SD from three replicates. Each dot represents three technical qPCR replicates. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. g Model showing binding of FOXE1 to the IRF6 -10 kb enhancer, which is favored by the risk allele and which reduces IRF6 expression. Source data are provided as a Source Data file.
To test the importance of FOXE1 in regulating the IRF6 −10 kb enhancer we repeated the luciferase reporter assays using the IRF6 −10 kb enhancer (longest version) harboring the risk allele of the IRF6 −10 kb SNP, and compared the results between parental GMSM-K and those in which we had deleted the FOXE1 using CRISPR/Cas9 reagents (Supplementary Figs. 18b, 19). The FOXE1 knockout cells were clonal isolates and therefore entirely or mostly pure for the loss of FOXE1. We chose GMSM-K over HEKn because the expression of FOXE1 is higher in GMSM-K than in HEKn (Supplementary Fig. 11c). GMSM-K are homozygous risk for the IRF6 −10 kb SNP, where FOXE1 binds more avidly. Interestingly, in cells lacking FOXE1 the activity of the IRF6 −10 kb reporter was higher than in the control cells (Fig. 4e). We saw analogous results in cells transfected with siRNA targeting FOXE1 or a non-targeting siRNA (Supplementary Fig. 18c, d). Moreover, levels of IRF6 expression were higher in FOXE1-knockout or FOXE1 knockdown GMSM-K than in the respective controls (Fig. 4f and Supplementary Fig. 18e). While the binding of other transcription factors may be affected by the allele of the IRF6 −10 kb SNP, these results suggest its effect on binding of FOXE1 is sufficient to alter the activity of this enhancer (Fig. 4g).
The IRF6 -22 kb SNP risk allele promotes binding of the transcriptional repressor ETS2
The risk allele of the IRF6 -22 kb SNP diminished the predicted affinity for two transcription factors (GATA2 and HSF4) and elevated it for NFKB2, ZNF454, and ten members of the ETS family, including ETS2 (JASPAR score for ETS2 site MA1484.1, risk: 4.9; non-risk, -4.1) (Fig. 5a and Supplementary Data 16, 17). ETS family members, particularly ETS1, regulate differentiation of keratinocytes and other epithelial cell types79,80. ETS2 can function as a transcriptional repressor81,82. Anti-ETS2 and anti-H3K27Ac antibodies yielded higher ChIP-qPCR signals in iOE cells at the IRF6 -22 kb SNP than at an intergenic region (Fig. 5b, c), indicating ETS2 binds at this SNP and the SNP is within an active regulatory element. PCR and sequencing of chromatin precipitated by anti-ETS2 from iOE cells heterozygous for this SNP revealed an enrichment of ETS2 binding to the risk allele, while anti-H3K27Ac pulled down more of the non-risk allele (Fig. 5d, two additional replicates in Supplementary Fig. 17b). These findings are consistent with higher activity of the enhancer and higher expression of IRF6 in cells with the non-risk genotype. Similarly, anti-ETS2 ChIP-qPCR in GMSM-K homozygous for either the risk or non-risk allele of IRF6 -22 kb SNP revealed stronger binding of ETS2 to the risk allele (Supplementary Fig. 18f). We found that reporter activity of the IRF6 -22 kb enhancer harboring the risk allele of the IRF6 -22 kb SNP was higher in ETS2-depleted HEKn relative to those transfected with a control siRNA (Fig. 5e and Supplementary Fig. 18g); we observed analogous results in GMSM-K (Supplementary Fig. 18h, i). IRF6 expression was also higher in ETS2-depleted HEKn or GMSM-K than in control transfected cells (Fig. 5f and Supplementary Fig. 18j). Together these results support the model that the risk allele of the IRF6 -22 kb SNP results in lower expression of IRF6 by promoting the binding of ETS2, potentially among effects on the binding of other transcription factors (Fig. 5g and Supplementary Data 16, 17).

a Consensus ETS2 binding motif from the JASPAR database of transcription factor DNA-binding preferences (Matrix ID: MA1484.1) and alignment of the variant site in several mammals. The risk allele (C) is the reference allele but has a lower frequency than the non-risk allele (T) in most populations. The risk allele improves the match to the ETS2 binding site which remains partial. b, c Percent input identified by ChIP-qPCR for anti-ETS2 and anti-H3K27Ac respectively in iOE cells heterozygous for rs661849 using primers specific to the IRF6 −22 kb enhancer site or, as a negative control, to a region 103.7 kb upstream IRF6 transcription start site lacking ATAC-Seq and H3K27Ac ChIP-Seq signals in HIOEC or NHEK. Error bars refer to three ChIP replicates and expressed as mean ± SD. Each dot represents three technical qPCR replicates. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. NS non-significant. d Sequencing of anti-ETS2 and anti-H3K27Ac ChIP-PCR product of cells heterozygous for rs661849 using the indicated antibody. e Scattered dot plot of relative luciferase activity of the IRF6 −22 kb reporter construct (longest version) harboring the risk allele of rs661849 in control versus ETS2-depleted primary neonatal keratinocytes (HEKn). Data are represented as mean ± SD from three independent experiments. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. f Scattered dot plot of relative levels of IRF6 mRNA in control versus ETS2-depleted HEKn assessed by qRT-PCR. Expression levels of IRF6 are normalized against ACTB. Data are represented as mean ± SD from three replicates. Each dot represents three technical qPCR replicates. Statistical significance (P value, two-tailed) is determined by Student’s t-test and P value is indicated on the plot. g Model showing binding of ETS2 to the IRF6 −22 kb enhancer, which is favored by the risk allele and which reduces IRF6 expression. Source data are provided as a Source Data file.
Similarly, we observed that the risk allele of the FOXE1 24 kb SNP diminished the predicted affinity for four transcription factors (GATA2, FOXH1, GCM2, and NRL) and elevated it for three transcription factors (TFAP2A, SP2, and KLF12) (Supplementary Data 18, 19). While we did not attempt chromatin immunoprecipitation (ChIP) with antibodies to transcription factors at this locus, ChIP with an anti-H3K27Ac antibody precipitated more of the risk allele of the FOXE1 24 kb SNP from heterozygous iOE cells (Supplementary Fig. 20a–c), consistent with higher activity of the enhancer and higher expression of FOXE1 in cells with the risk genotype.
Conditional analysis of haplotypes containing IRF6 −10 kb and IRF6 -22 kb SNP
Finally, to assess whether the IRF6 −10 kb (rs11119348) and IRF6 -22 kb (rs661849) SNPs can explain the heritable risk for CL/P associated with 1q32/IRF6 we conducted conditional analyses. We previously found evidence for multiple independent signals at IRF6 by LD-clumping, and different patterns of association with OFC subtypes have been reported by several studies33,83,84,85. We therefore performed conditional analyses separately for cleft lip only (CL) and cleft lip and palate (CLP) cases to explore the contributions of each SNP to each phenotype (Fig. 6a–h and Supplementary Data 20, 21). As expected, the IRF6 locus was associated with CL (lead SNP rs12403599, P value = 3.29 × 10−9) (Fig. 6a) and CLP (lead SNP rs2076149, P value = 5.24 × 10−10) (Fig. 6e and Supplementary Data 22). Conditioning on either the IRF6 -22 kb or IRF6 −10 kb SNPs explained some of the signal at the IRF6 locus for both CL and CLP but neither SNP alone reduced the association peak fully, as evidenced by P values and odds ratios indicating residual evidence of association (Supplementary Data 22). Importantly, the impact of conditioning on either SNP alone was different for CL than for CLP due to differences in linkage disequilibrium between the functional SNPs and other SNPs at the locus associated with each phenotype. Neither the IRF6 −10 kb SNP nor the IRF6 -22 kb SNP, separately or in combination, completely accounted for the signal tagged by lead SNP rs2076149 in CL, which saw some increases in P value but only minor changes in effect size in each analysis. For CLP, the IRF6 −10 kb SNP accounted for more of the signal than the IRF6 -22 kb SNP and conditioning on both SNPs showed no remaining evidence of association at the 1q32/IRF6 locus by both P value and effect size. Together, we conclude that both SNPs account for the majority of the association signal at 1q32/IRF6 observed in this dataset for CL and CLP.

a–h Locus Zoom plots of a–d cleft lip only and e–h cleft lip and palate; a, e unconditioned; b, f conditioned on rs661849 (IRF6 −22 kb); c, g conditioned on rs1119348 (IRF6 −10 kb); d, h conditioned on both SNPs simultaneously. Two-sided P values are calculated with a logistic regression with sex without adjustment for multiple testing and ten principal components of ancestry as covariates (and the genotype of the SNP being conditioned on). Points are color-coded based on linkage disequilibrium (r2) in Europeans for each SNP with rs661849.
Discussion
To understand how common variants predispose individuals to orofacial cleft (OFC) here we tested 887 OFC-associated SNPs in a massively parallel reporter assay (MPRA) as has been done in other studies seeking to identify SNPs that affect enhancer activity12,13,86. Because enhancers are often cell type specific, we limited the study to SNPs from loci where the presumed effector gene is expressed in oral epithelium (as opposed to oral mesenchyme or brain) and used a cell line model of this tissue for the MPRA. Filtering SNPs with allele-specific effects in the MPRA for those lying in chromatin with evidence of enhancer activity in relevant cell types, or in explants of human embryonic faces, promoted the candidacy of MPRA-nominated SNPs near FOXE1, IRF6, MAFB, TFAP2A, and TP63. We then used luciferase reporter assays to confirm the findings of the MPRA at six SNPs. At five of these six SNPs, either or both variants had activity different from that of the basal promoter (FDR <0.01), the exception being a SNP at TP63. Finally, we engineered the genome of iPSCs to be homozygous for the risk or non-risk allele of the three top-candidate SNPs (separately) and differentiated the cells into embryonic oral epithelium. All three SNPs had allele-specific effects on expression levels of the respective effector gene. The risk alleles of both functional SNPs at IRF6 decreased the expression level of IRF6, which is consistent with loss-of-function mutations in IRF6 causing Van der Woude syndrome87. The risk allele of the functional SNP at FOXE1 increased the expression level of FOXE1. Both gain- and loss-of-function mutations in FOXE1 are detected in individuals with Bamforth–Lazarus Syndrome (which includes OFC)41 and, similarly, both a loss-of-function mutation in Foxe1 and over-expression of Foxe1 can cause cleft palate in mice46,47. We identified transcription factors whose binding is affected by the SNP allele for both SNPs at IRF6, and showed that they regulate the expression of the enhancers harboring the SNPs, and of IRF6 expression levels, in the predicted direction. Finally, we returned to the GWAS data and found that the haplotypes containing the two SNPs at IRF6 explain most of the CLP association signal at this locus in the cohorts evaluated. In summary, we found strong evidence for three functional SNPs and suggestive evidence for three more.
An objective in identifying functional SNPs is to determine the transcription factors whose binding is impacted by the SNPs allele, as the genes encoding such TFs may be associated with OFC risk. Supporting this possibility, earlier we found that a functional SNP in the enhancer IRF6 MCS9.7 affects binding of IRF6 itself 88, and here we found that the IRF6 −10 kb SNP affects the binding affinity of FOXE1, encoded by a gene implicated in risk for syndromic89 and nonsyndromic23,26,78,90 OFC. We infer that FOX family members expressed in oral epithelium are candidates to be OFC risk genes. In this light, it is intriguing that chromatin immunoprecipitation with sequencing (ChIP-seq) revealed binding of FOXA1 and FOXA2 in bronchial epithelial cells at the IRF6 −10 kb enhancer, and knocking down expression of either transcription factor led to upregulation of IRF691. Another implication is that there may be a greater-than-additive effect on OFC risk in individuals harboring risk-associated haplotypes at both IRF6 and FOXE1. Unfortunately, current GWAS data are underpowered to reveal such gene-by-gene interactions. It would be interesting to test for genetic interactions between IRF6 and FOXE1 in model organisms or iPSCs. The risk allele of the IRF6 −22 kb SNP creates a low-affinity ETS family binding site, and ChIP-qPCR indicated that the risk allele promotes binding of ETS2. Low-affinity ETS binding sites are important for the regulation of SHH in the limb92. Mice embryos homozygous for a strong loss-of-function mutation in ETS2 die by E8.593; heterozygous mutants exhibit craniosynostosis, probably reflecting a role for ETS2 in osteogenesis within the fibroblast growth factor (FGF) signaling pathway94. Mutations in ETS2 have been detected in patients with craniosynostosis94, and with a developmental disorder that includes cleft palate in some individuals95. It is expressed in differentiated keratinocytes and glandular epithelium96,97 and regulates the differentiation of trophoblast stem cells98. We infer that ETS2 and is paralogs, in particular ETS1, which inhibits differentiation of keratinocytes99,100, are potential contributors to isolated OFC, and that genetic interaction between IRF6 and ETS2 in OFC patients and animal models are worth exploring.
The IRF6 -10 kb SNP is the third example of a SNP with relatively strong evidence of directly influencing risk for cleft lip, cleft palate, or both within the same evolutionarily conserved enhancer of IRF6, MCS9.76,33,50,62,65,88. Of note, this enhancer was identified through investigation of a non-syndromic CL/P-associated SNP, rs642961, also with some evidence of being functional33. In stable transgenic mice, MCS9.7 (from the human genome) drove reporter expression in oral epithelium33,62; in stable transgenic zebrafish, it drove reporter expression in periderm in the entire embryo, including in the oral cavity6. While the risk-associated allele of rs642961 disrupts binding of the transcription factor AP2-α (TFAP2A) in an electrophoretic mobility shift assay33, current and previous MPRA and luciferase results33,34 all did not detect allele-specific effects of rs642961 on enhancer activity. It is possible that the IRF6 −22 kb SNP, which is in linkage disequilibrium with rs642961, is the functional SNP underlying the association of rs642961 with OFC; supporting this notion both IRF6 −22kb SNP and rs642961 are more strongly associated with CL than with CLP33. By contrast, a rare, single-nucleotide insertion in MCS9.7 segregating with cleft palate and other characteristic symptoms in a Brazilian family with Van der Woude syndrome strongly affected the enhancer activity of MCS9.7 65. In addition, we recently found that a SNP in MCS9.7, rs570516915, is associated with CP only in populations from Finland and Estonia and that the risk allele reduces IRF6 expression level in engineered induced oral epithelial cells88. The current study shows that IRF6 -10 kb (rs11119348), associated with CL and CLP and in the same enhancer, affects IRF6 expression potentially by altering the binding of FOXE1. Taken together, these studies emphasize the extraordinary importance of a single enhancer in genetic predisposition for orofacial cleft, and raise the question of how different variants in this enhancer elevate risk for cleft lip with or without cleft palate or for cleft palate only.
There remains a need for high-throughput methods to screen functional SNPs in OFC and other common disorders. The MPRA we performed required the synthesis of a library of oligos. This step was avoided in an MPRA that used reporter constructs from sheared chromatin from individuals of distinct genotypes; this study managed to score more than 50 million SNPs in two cell types15. However, the success of MPRAs at identifying functional SNPs at many loci is likely to rely on the cell line deployed being a reasonable model of the embryonic tissue where the SNP affects disease risk in vivo. While results from an MPRA helped us identify six OFC-associated SNPs that alter enhancer activity in luciferase reporter assays, we only pursued three of them with the in-depth experiments, confirming that they can alter IRF6 or FOXE1 expression in induced oral epithelium cells. It will be interesting to assess whether additional MPRA-nominated SNPs within active enhancers near IRF6 are indeed functional, and if they affect IRF6 expression in combination. However, the step of engineering the genome in iPSCs and creating induced oral epithelium or oral mesenchyme remains laborious and expensive. One promising approach to prioritize non-coding SNPs is chromatin-accessibility QTL analyses101 but these, too, require appropriate cell line models. It will also be interesting to assess the effect of functional SNPs on the expression of genes downstream of the directly affected transcription factor, and to establish OFC-prone animal models where the phenotypic effects of single SNPs might be evident. In many GWAS loci, the effector gene is unclear, which increases the challenge of identifying functional SNPs; in addition, validated functional SNPs may contribute to disease etiology by directly affecting the expression of genes other than the apparent effector gene. Despite these challenges, identifying functional SNPs remains an essential step in translating the findings of GWAS into an understanding of how common genetic variants contribute to the pathogenesis of OFC and other congenital anomalies.
Methods
Our research complies with all relevant ethical regulations. All GWAS data used in this study have been previously published24,30 and were anonymized/deidentified to all authors on this manuscript. The WTC-11 cell line (UCSFi001-A (RRID:CVCL_Y803)) was ethically derived (https://hpscreg.eu/cell-line/UCSFi001-A#ethics). The details of the donor are provided at the human pluripotent stem cell registry, and the derivation of the cells underwent ethical review by a committee at the University of California, San Francisco. The IRB protocol and approval numbers are both 10-02521.
Meta-analysis
We performed a genome-wide meta-analysis of CL/P, CP, and all OFCs, from European and Asian ancestries from two consortia24,30. The details of the sample collections, genotyping, and genotype quality control (QC) procedures have been described previously24,30,83,102. Briefly, the original studies recruited individuals with OFCs, their unaffected relatives, and unrelated controls (individuals with no known family history of OFCs or other craniofacial anomalies; N = 1626) from 18 sites across 13 countries from North America, Central or South America, Asia, Europe and Africa. Samples from the GENEVA consortium were genotyped for 589,945 SNPs on the Illumina Human610-Quadv.1_B BeadChip and imputed to the 1000 Genomes Phase 1 release (June 2011) reference panel. Samples from the POFC consortium were genotyped for approximately 580,000 SNPs from the Illumina HumanCore+Exome array, of which ~539,000 SNPs passed quality control filters recommended by the Center for Inherited Disease Research (CIDR) and the Genetics Coordinating Center (GCC) at the University of Washington24. These data were then phased with SHAPEIT2103 and imputed with IMPUTE2104 to the 1000 Genomes Project Phase 3 release (September 2014) reference panel. The most likely imputed genotypes were selected for statistical analysis if the highest probability (r2) > 0.9. SNP markers showing deviation from Hardy–Weinberg equilibrium in European controls, a minor allele frequency or MAF <5%, or imputation INFO scores <0.5 were filtered out of all subsequent analyses. The information for the genotyped markers was retained after imputation, and the imputed values for these variants were only used to assess concordance. Our methods for meta-analysis were previously published23. Case-parent trios were analyzed with the allelic transmission disequilibrium test (TDT) implemented in PLINK. Unrelated cases and controls, matched for population of origin, were tested for association using a logistic regression under an additive genetic model while including 18 principal components of ancestry to adjust for population structure. Within each OFC subtype, the resulting effect estimates for the TDT and logistic regression were combined in an inverse-variance weighted fixed-effects meta-analysis.
Selection of SNPs for analysis by MPRA
We selected 887 SNPs from eight loci that include a gene known to be expressed in oral epithelium and in most cases, regulates differentiation of an epithelial tissue (1q32/IRF6, 2p21/THADA, 6p24.3/TFAP2A, 9q22.2/GADD45G, 12q13.13/KRT18, 20q12/MAFB and 9q22.33/FOXE1) (Table 1 and Supplementary Data 1, 2). We selected SNPs with P values suggestive of association from seven loci (IRF6, THADA, TFAP2A, GADD45G, KRT18, and MAFB) from the meta-analysis (Table 1 and Supplementary Data 2). We additionally included 59 SNPs at the TFAP2A locus identified in an independent GWAS of CL/P in Han Chinese26. Because of the financial constraints on the library size, for one locus (FOXE1) we instead picked nine SNPs in strong linkage disequilibrium with the lead SNP (rs12347191) in a European population and annotated by Haploreg as being within regulatory elements23 (Supplementary Data 2).
Reporter library construction
We designed an oligonucleotide library in which each oligo contained the sequence of the SNP of interest, a unique barcode, and priming and restriction sites necessary for amplification and cloning into a pGL-hsp68 reporter plasmid105. The sequence of human genomic DNA corresponding to 161 bp windows, centered on each of 887 GWAS-identified SNPs, was downloaded from the UCSC Table browser tool (genome build: hg19). Two additional SNPs, absent from all tables, supplementary data and schematics, were included in the MPRA as part of a separate preliminary study. We generated two versions of each cis-regulatory element (CRE) that were identical except for harboring the major or minor allele of the SNP, respectively (1774 elements) (Fig. 1a and Supplementary Data 3). We ordered a pool of 7500 unique 230-mer oligonucleotides (elements) through a limited licensing agreement with Agilent Technologies (Santa Clara, CA). To establish an empirical null distribution of activity in the assay, we included 90 sequences that were scrambled versions of the sequences of other elements (Supplementary Data 4). To account for noise in the assay, each sequence was assigned four unique barcodes. Finally, to create constructs with only the basal promoter and no upstream regulatory sequence, we included an additional random 28 filler elements, which were excised in the second step of cloning, yielding 28 basal promoter-only controls with one barcode each. We synthesized 7484 elements corresponding to the 887 SNPs (7096 + 360 + 28) (Supplementary Data 4 and Fig. 1a).
Oligos were designed with the following structure (Supplementary Fig. 21): a forward priming sequence (PS1) with NheI restriction site, (primer for this site is KC_1F), 161 base cis-regulatory element (Supplementary Data 3), AgeI restriction site, NruI restriction site, MluI restriction site, 9 base barcode, and a reverse priming sequence (PS2) with XhoI site (primer for this site is KC_1R) (Fig. 1a, Supplementary Fig. 21, and Supplementary Data 4).
We amplified the oligo pool using five cycles of PCR with Phusion High-Fidelity polymerase (New England Biolabs) and primer set KC_1 (Supplementary Data 23). We cloned the pool of amplicons into the pGL-hsp68 backbone using NheI and XhoI, which excludes the hsp68 promoter fused to DsRed. We prepared DNA from 41,000 colonies to generate library KC_L1. We then amplified a DNA fragment containing the hsp68 promoter and the DsRed cDNA using primers KC_2F and KC_2R (Supplementary Data 23) containing AgeI and MluI restriction sites, respectively, and cloned this fragment into the KC_L1 library such that the CREs lie upstream of the hsp68 promoter driving DsRed and the barcodes; this procedure created library KC_L2.
To assess the completeness of the cloned library, we sequenced the barcodes in the final plasmid pool in library KC_L2. It was achieved by amplifying the barcode fragments using primer set KC_BC containing with NheI and MfeI restriction sites, followed by specific Illumina adapters ligation (P1 and PE2 adapters), and Illumina enrichment PCR using KC_3 set of primers, and sequencing (Supplementary Data 23).
Cell lines
The human fetal oral epithelial cell line (GMSM-K)61 (a kind gift from Dr. Daniel Grenier, Université Laval) was maintained in keratinocyte serum-free medium (KSFM) (Gibco, Catalog no. 17005-042) supplemented with human recombinant epidermal growth factor (EGF 30 µg/ml) and bovine pituitary extract (BPE 0.2 ng/ml). Human Epidermal Keratinocytes from neonatal foreskin (HEKn; primary neonatal keratinocytes) purchased from ATCC (ATCC Catalog no. PCS-200-010) were maintained and cultured as per the manufacturer’s instructions in dermal cell basal medium (ATCC Catalog no. PCS-200-030) supplemented with keratinocyte growth kit (ATCC, Catalog no. PCS-200-040). The Normal Human Skin Keratinocyte (N-HSK-1)106 cell line was grown in KSFM or on an irradiated NIH3T3 feeder layer in Dulbecco’s Modified Eagle’s Medium (DMEM) and HamF12 (3:1 ratio) according to Rheinwald and Green107. WTC-11 human induced pluripotent stem cells (iPSCs) (Coriell Institute for Medical Research, Catalog no. GM25256), were maintained in mTeSR Plus media (StemCell Technologies, Catalog no. 100-0276) on Matrigel (Corning, Catalog no. 356231)-coated plates and passaged with StemPro Accutase (Gibco, Catalog no. A11105-01). Cells were incubated at 37 °C in 5% CO2. Details of cell lines/primary cells/induced pluripotent cells used in the study are provided in Supplementary Data 5. Sex was not considered in tests of allele-specific effects of SNPs on enhancer activity.
Transfection of CRE-library (plasmid pool)
A pool of plasmids (from library KC_L2; as described in Methods, Reporter library construction) in four replicates, 2.5 µg each, was transfected into 1 million GMSM-K with Amaxa Cell Line Nucleofector Kit V (Lonza, Catalog no. VCA-1003) using Nucleofector II (Lonza) (program T-020). Forty-eight hours post-transfection, RNA and genomic DNA was extracted from each set using Quick-DNA/RNA Miniprep kit (Zymo Research, Catalog no. D7001) as per the manufacturer’s instructions.
Sample preparation for RNA-Seq
Isolated RNA was treated with Turbo DNase I (Thermo Fisher, Catalog no. AM1907) as per the manufacturer’s instructions. The first strand of cDNA was synthesized with SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Catalog no. 18080051). The barcodes including the flanking sequence were amplified from both cDNA, genomic DNA (transfected plasmid DNA) and plasmid DNA samples (that was used for transfection) with Phusion High-Fidelity PCR Master Mix (NEB, Catalog no. M0531S) using primer set KC_BC (Supplementary Data 23) and that yielded barcode sequences that were flanked with NheI and MfeI restriction enzyme sites. The products were digested with NheI and MfeI and then ligated with Illumina adapter sequences P1 and PE2 (Supplementary Data 23). This product was further amplified using primer set KC_3 (Supplementary Data 23), followed by sequencing.
To measure expression of the barcoded library, electroporation replicates were multiplexed and run on two lanes of an Illumina HiSeq machine, which generated 48.2 million sequence reads corresponding to cDNA and 48.8 million reads corresponding to DNA (from plasmids). Sequencing reads that matched the first 20 nucleotides of the designed sequence were counted, regardless of quality score.
Statistical analysis to determine the regulatory effects of SNPs by MPRA
We filtered out barcodes that were low abundance in the plasmid pool (<50 reads), then set any cDNA barcodes that were detection-limited in at least one sample (<50 reads for replicates run on the first lane, <100 reads for the replicates run on the second lane) to zero across all cDNA samples. We normalized each sample to the total counts per million (CPM). RNA barcode levels were highly correlated among four replicates (Supplementary Fig. 22). There was a strong correlation in the barcode counts in the plasmid library before transfection and after harvesting plasmid from transfected cells (Supplementary Fig. 23). Therefore, we used plasmid barcode counts (pre-transfection) for further analyses. We further normalized RNA barcode counts by the plasmid barcode counts (RNA/DNA). We checked the reproducibility based on log2RNA/DNA counts (Supplementary Fig. 24). To calculate the reporter activity for each sequence we averaged the log2RNA/DNA counts within a replicate, normalized to the within-replicate mean basal barcode activity and then averaged across all the replicates (Supplementary Fig. 1a). The standard error of the mean (SEM) was calculated using the basal-normalized activities across replicates. Variant significance was determined by performing Welch’s t-test between major and minor alleles, followed by Benjamini–Hochberg false discovery rate (FDR) correction (Supplementary Data 6). We used our previously published108 Python code for processing and analyzing MPRA reads, which is available on Github at [https://github.com/barakcohenlab/CRX-Information-Content].
Activity-by-contact to assign SNP-containing-enhancers to promoters
The activity-by-contact method (ABC) utilizes cell type-specific chromatin accessibility (ATAC-seq or Dnase hypersensitivity) and chromatin activity (H3K27ac) and HiC data that is either cell-type specific or averaged over ten cell types—the performance of this tool is similar with either type of HiC data63. To calculate the ABC scores for keratinocyte enhancers we used publicly available datasets from primary Normal Human Epidermal Keratinocytes (NHEK) cells for H3K27ac (NCBI GEO Identifier GSM733674), Dnase hypersensitivity (NCBI GEO Identifier GSM736545), RNA-seq (NCBI GEO Identifier GSM958736) and averaged HiC data from 10 cell types as reported in ref. 63. All elements with an ABC score >0.01 were included in the analysis.
Electroporation and dual luciferase reporter assay
For dual luciferase reporter assays, each reporter construct (Supplementary Data 10) was cloned into the pGL3 promoter vector, and constructs were co-transfected with Renilla luciferase plasmid. Briefly, GMSM-K or HEKn cells were electroporated with plasmid using the Amaxa Cell Line Nucleofector Kit V (Lonza, Catalog no. VCA-1003) and the Nucleofector II instrument (Lonza) program T-020. We used a dual luciferase reporter assay system (Promega, Catalog no. E1980) and FB12 Luminometer (Berthold Detection Systems) to evaluate the luciferase activity following the manufacturer’s instructions. Relative luciferase activity was the ratio of Firefly and Renilla activities. At least three independent measurements were performed for each transfection group. All results were presented as mean ± standard deviation (SD). Statistical significance was determined using a two-tailed Student’s t-test.
Immunofluorescence detection and imaging
GMSM-K and normal human skin keratinocytes (N-HSK-1) were grown on glass coverslips, fixed in 4% paraformaldehyde for 10 min, permeabilized in 0.2% Triton X-100 for 2 min, then rinsed in PBS. Cells were incubated in 3% goat serum (Vector Laboratories, Catalog no. S-1000-20) in 2% PBS-bovine serum albumin for 30 min to block non-specific binding, followed by incubation in primary then secondary fluorescent antibodies 1 h each at room temperature with PBS washes in between the two incubations. Primary antibodies that were used are: mouse monoclonal anti-E-cadherin (BD Bioscience, Catalog no. 610181, 1/100), mouse monoclonal anti P63 (clone 4A4, Santa Cruz, Catalog no. SC-8431, 1/250), mouse monoclonal anti human Keratin 14 (clone LL002, Novocastra, Catalog no. NCL-LL002, 1/200). Goat anti-mouse Alexa Fluor 488 (Invitrogen, Catalog no. A-11001) was used as a secondary antibody. Following PBS washes, coverslips were dried and mounted on a microscope slide using ProLong Diamond antifade mounting medium containing DAPI (ThermoFisher Scientific, Catalog no. P36962) and cured for 24 h at room temperature in the dark. Images were collected with Zeiss 880 or 980 confocal microscopes using the ZEN software (Zeiss). Confocal images were processed using Fiji software.
CRISPR-Cas9-mediated genome editing
Genome engineering for each SNP (rs11119348, rs661849, and rs10984103) in iPSCs or GMSM-K was achieved using a previously described protocol88. Sequences of CRISPR RNAs (crRNA) and repair templates specific to each SNPs are provided in (Supplementary Data 24). Briefly, equimolar concentrations of specific crRNA and trans-activating crRNA (tracrRNA, IDT, Catalog no. 1072532) were annealed at 95 °C for 5 min, followed by cooling at room temperature (RT) to form functional gRNA duplexes. Further, the ribonucleoprotein (RNP) complex was prepared 15 min before transfection by mixing gRNAs (1.5 µM) with Cas9 protein (0.45 µM) (Sigma, Catalog no. CAS9PROT for iPSCs and IDT, Catalog no. 1081058 for GMSM-K) at RT. RNP complexes together with the repair template were transfected into 1 million iPSCs with Amaxa nucleofector (Human Stem Cell kit 2, Lonza, Catalog no. VPH-5022, program: A-023 for iPSCs) in the presence of ROCK inhibitor (Y-27632 – 10 mM stock, Stemgent, Catalog no. 04-0012-10). Lonza Kit V, program: T-020 was used for transfecting GMSM-K. Individual colonies were hand-picked and plated into 96-well plates. DNA was extracted using Quick Extract DNA extraction solution (Epicentre, Catalog no. QE09050). Colonies were screened by PCR and sequenced using primers flanking each SNP (Primer sequences are provided in Supplementary Data 25). From iPSCs transfection experiment, two independent clones each of homozygous risk (AA for rs11119348; CC for rs661849) and homozygous non-risk (CC for rs11119348; TT for rs661849) for each SNP near IRF6, and three independent clones of each of homozygous risk (CC) and non-risk (AA) genotype for rs10984103 near FOXE1 were isolated (Supplementary Figs. 13a, b, 16a). From the GMSM-K transfection experiment, multiple independent clones, seven clones of homozygous risk (AA for rs11119348), six clones of homozygous non-risk (CC for rs11119348) and four independent clones each of the homozygous risk (CC for rs661849), and homozygous non-risk (TT for rs661849) were isolated. Deletion of FOXE1 gene in GMSM-K was achieved using a pair of gRNA (Supplementary Fig. 19). A set of three primers was used for colony screening, and three independent clones were isolated (Supplementary Data 26). Sequencing of three independent clones revealed that all but 30 bp at the 3’ end of the single coding exon gene was deleted (Supplementary Fig. 19).
In vitro differentiation of iPSCs into embryonic oral epithelial cells
Two independent samples of each genotype (two clones of the homozygous risk genotype, and two clones of the homozygous non-risk genotype) for both SNPs at IRF6 locus and three independent samples of each genotype (three clones of the homozygous risk genotype, and three clones of the homozygous non-risk genotype) for rs10984103 at FOXE1 locus were seeded in triplicate in 12-well plates (50,000 cells per well). Additionally, three independent samples of heterozygous genotype for each SNP individually were seeded (Supplementary Figs. 13a, b, 16a). Further, the cells were induced to differentiate into embryonic oral epithelial (iOE) cells using a 10-day differentiation protocol described previously72,88 (Supplementary Figs. 13a, b, 16a). Briefly, on the first day of differentiation stem cell media is replaced with media consisted of EpiCult-C media (StemCell Technologies, Catalog no. 05630) mixed with EpiLife (Thermo, Catalog no. MEPI500CA) at 1:1 ratio, supplemented with 0.1x supplement S7 (Thermo, Catalog no. S0175), 0.1 μM β-mercaptoethanol (Sigma, Catalog no. M7522) and 400 μM Smoothened agonist (Selleckchem, Catalog no. S7779). 150pM of Bone Morphogenic Protein-4 (R&D Systems, Catalog no. 314-BP-010) is continuously added daily starting from day 3 to day 7 of differentiation. At day 8, the media is supplemented with 1 uM of BMP-I inhibitor (LDN-193189) (Tocris, Catalog no. 6053), 5 uM of GSK3-Inhibitor (CHIR99021) (Selleckchem, Catalog no. 4423), 500 pM Epidermal Growth Factor (R&D systems, Catalog no. 236-EG) and 3.5 μM of Neurotrophin-4 (R&D systems, Catalog no. 268-N4). The cultures were then harvested at day 10 at an oral epithelium stage.
Quantitative RT-PCR on induced embryonic oral epithelial (iOE) cells or GMSM-K
RNA was extracted from multiple samples of each genotype (homozygous risk and homozygous non-risk) for rs11119348, rs661849 and rs10984103 (iOE cells—six each genotype for both SNPs near IRF6 and nine each genotype for rs10984103 near FOXE1; GMSM-K—six non-risk and seven risk genotype for rs11119348 and four each genotype for rs661849) using Quick-DNA/RNA Miniprep kit (Zymo Research, Catalog no. D7001) followed by Dnase treatment using manufacturer’s instructions (Thermo Fisher Scientific, Catalog no. AM1907). Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Catalog no. 4368814). Real-time PCR was performed using SYBR Green qPCR mix (Bio-Rad) in the Bio-Rad CFX Connect Real-Time System. Quantitative RT-PCR reaction for each sample was performed in triplicate. Data analysis was performed using previously described methods109,110. Briefly, expression levels of IRF6 were normalized against ACTB109 for GMSM-K and expression levels of IRF6 or FOXE1 were normalized against ACTB, GAPDH, HPRT1, UBC and CDH1110 for iOE cells (mentioned in the respective figure legends). Data were presented as mean ± SD. Statistical significance was determined using a two-tailed Student’s t-test. Primer sequences used in the study for qRT-PCR are provided in Supplementary Data 27.
Transcription factor binding site analysis using JASPAR 2022
Elements of 21 nucleotides, including ten nucleotides on each side of the IRF6 -10 kb, IRF6 −22 kb, and FOXE1 24 kb SNPs were used for transcription factor binding site (TFBS) analysis using JASPAR 202273. An 80% threshold was used to detect transcription factor binding sites, and all those with a score below 4.0 were discarded, to focus on binding sites of at least medium affinity. For TFBS that, at an 80% threshold, were specific for the risk or non-risk allele, we dropped the threshold to 50% to learn the affinity of such TFBSs with the other allele.
Chromatin immunoprecipitation qPCR
Induced oral epithelial cells (iOE cells) derived from iPSC that are heterozygous for risk and non-risk alleles of rs11119348, rs661849, and rs10984103 or GMSM-K homozygous for risk or non-risk alleles for rs11119348 and rs661849 were harvested (3.5 million cells) and fixed in 1% formaldehyde for 15 min at RT. Fixation was stopped with 125 mM glycine for 5 min at RT. Chromatin immunoprecipitation (ChIP) on iOE cells was performed using a semi-automated protocol described previously111. Briefly, cells were sheared using PIXUL or VirSonic 600 (5 min with 30/40 s on/off cycles for GMSM-K for IRF6 −22 kb SNP and iOE cells for FOXE1 24 kb SNP) to obtain genomic DNA fragments averaging 300 to 600 bp. The sonicated cell lysates were subjected to chromatin immunoprecipitation with specific antibody (1 μg of anti-H3K27Ac from Millipore-Sigma, Catalog no. 07-360 and 1 μg of anti-ETS2 from GeneTex, Catalog no. GTX104527 and 1 μg of anti-FOXE1, a kind gift from Pilar Santisteban, Universidad Autónoma de Madrid, as previously described112). Normal rabbit IgG (Millipore-Sigma, Catalog no. 12-370) was used as a negative antibody control. Quantitative PCR was performed with an equal volume of DNA in triplicate from three ChIP replicates of each antibody, and percent input DNA was calculated using the method described previously113. Primer sequences are provided in Supplementary Data 28. Negative control PCR primers were designed to target a sequence 103.7 kb upstream of the IRF6 transcription start site or 28.2 kb downstream of the FOXE1 gene that did not harbor active regulatory elements identified from ATAC-Seq and H3K27Ac ChIP-seq in human immortalized oral epithelial cells (HIOEC) or NHEK. Data were presented as mean ± SD. Statistical significance was determined using a two-tailed Student’s t-test.
ETS2 and FOXE1 knockdown
siRNAs targeting FOXE1 (Santa Cruz, Catalog no. sc-44175) or ETS2 (Santa Cruz, Catalog no. sc-37855) and siRNA controls (Santa Cruz, Catalog no.sc-37007) were transfected into one million HEKn (heterozygous for IRF6 −22 kb SNP) or GMSM-K (homozygous risk for IRF6 -10 kb or −22 kb SNPs). RNA extraction and qRT-PCR were performed on three replicates. Data analysis was performed using the previously described method109. Briefly, expression levels of genes of interest (FOXE1, ETS2, or IRF6) were normalized against ACTB109 for GMSM-K or HEKn. Data were presented as mean ± SD. Statistical significance was determined using a two-tailed Student’s t-test. Primer sequences used in the study for qRT-PCR are provided in Supplementary Data 27. RT-PCR primer pair B from Santa Cruz (Catalog no. sc-44175-PR) for FOXE1 was used.
Further, siRNA targeting FOXE1 or ETS2 and control siRNA were co-transfected with respective risk harboring luciferase constructs along with Renilla luciferase plasmid. Three independent transfections were performed. Cells were harvested 48 h post-transfection, and dual luciferase reporter assays were performed as described earlier in the “Electroporation and dual luciferase reporter assay” section. The RNA expression and luciferase reporter assays were carried out on FOXE1 KO GMSM-K as well.
Conditional analysis methods
Conditional analyses were conducted using genome-wide genotyping data from the Pittsburgh Orofacial Cleft (POFC) Study on the case-control subset of the dataset (described earlier). A total of 394 cleft lip only cases and 1995 cleft lip and palate cases were used in the analysis, with 1626 unrelated controls. Common variants (MAF >5%) near the IRF6 locus were included in this analysis, and the association between each variant and OFCs was calculated using a logistic regression in PLINK (v1.9)114 controlled for sex and ten principal components of ancestry. Conditional analyses also controlled for the genotype of either rs661849 or rs11119348 alone, or the genotypes of both rs661849 and rs11119348. Since the data came from a genome-wide analysis, SNPs with association P values less than 5 × 10−8 were considered genome-wide significant. Regional association plots were made with LocusZoom, where the linkage disequilibrium (LD) blocks and recombination rates were estimated from European populations115. LD between pairs of SNPs was calculated in the data from the 1000 Genomes Project using LDLink116,117.
Statistics and reproducibility
Details of statistical analyses used in GWAS, MPRA, conditional analyses, qRT-PCR, and ChiP-PCR, including numbers of replicates, are presented above. GraphPad Prism 10 was used for the statistical data analyses. A priori power analyses were not performed to pick the number of replicates used in each assay. No data were excluded. We did all the calculations of MPRA based on major versus minor allele, and later lifted the blind regarding which allele was the risk-associated allele. Investigators were not blinded to genotypes in measurements of luciferase levels, mRNA levels and ChIP-qPCR levels.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw and processed sequencing data generated in this study have been deposited at the NCBI Gene Expression Omnibus under accession numbers GSE297145 (MPRA: ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE297145) and GSE297146 (RNA-Seq, GMSM-K: ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE297146). Processed MPRA and RNA-Seq data of GMSM-K are provided in Supplementary Data 6 and 13, respectively. Data used for conditional analyses are publicly available at the database of Genotypes and Phenotypes (dbGaP) (https://www.ncbi.nlm.nih.gov/gap/), dbGaP Study Accession: phs000774.v2.p1. Source data are provided with this paper. Biological materials are available for non-commercial purposes upon request with an appropriate material transfer agreement. Source data are provided with this paper.
References
Stanier, P. & Moore, G. E. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 13, R73–R81 (2004).
Klotz, C. M. et al. Revisiting the recurrence risk of nonsyndromic cleft lip with or without cleft palate. Am. J. Med. Genet. Part A 152, 2697–2702 (2010).
Sivertsen, Å. et al. Familial risk of oral clefts by morphological type and severity: population based cohort study of first degree relatives. BMJ 336, 432–434 (2008).
Grosen, D. et al. A cohort study of recurrence patterns among more than 54 000 relatives of oral cleft cases in Denmark: support for the multifactorial threshold model of inheritance. J. Med. Genet. 47, 162–168 (2010).
Nasreddine, G., El Hajj, J. & Ghassibe-Sabbagh, M. Orofacial clefts embryology, classification, epidemiology, and genetics. Mutat. Res. Rev. Mutat. Res. 787, 108373 (2021).
Liu, H. et al. Analysis of zebrafish periderm enhancers facilitates identification of a regulatory variant near human KRT8/18. Elife 9, e51325 (2020).
Ghandi, M., Lee, D., Mohammad-Noori, M. & Beer, M. A. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput. Biol. 10, e1003711 (2014).
Kelley, D. R., Snoek, J. & Rinn, J. L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990–999 (2016).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 (2012).
Ward, L. D. & Kellis, M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016).
Inoue, F. et al. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 27, 38–52 (2017).
Klein, J. C. et al. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434 (2019).
Abell, N. S. et al. Multiple causal variants underlie genetic associations in humans. Science 375, 1247–1254 (2022).
Kheradpour, P. et al. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 23, 800–811 (2013).
van Arensbergen, J. et al. High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 51, 1160–1169 (2019).
Vockley, C. M. et al. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 25, 1206–1214 (2015).
Tewhey, R. et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165, 1519–1529 (2016).
Ulirsch, J. C. et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 165, 1530–1545 (2016).
Liu, S. et al. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 18, 1–14 (2017).
Zhang, P. et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 9, 2022 (2018).
Butali, A. et al. Replication of genome wide association identified candidate genes confirm the role of common and rare variants in PAX 7 and VAX 1 in the etiology of nonsyndromic CL (P). Am. J. Med. Genet. Part A 161, 965–972 (2013).
Camargo, M. et al. GWAS reveals new recessive loci associated with non-syndromic facial clefting. Eur. J. Med. Genet. 55, 510–514 (2012).
Leslie, E. J. et al. Genome-wide meta-analyses of nonsyndromic orofacial clefts identify novel associations between FOXE1 and all orofacial clefts, and TP63 and cleft lip with or without cleft palate. Hum. Genet. 136, 275–286 (2017).
Leslie, E. J. et al. A multi-ethnic genome-wide association study identifies novel loci for non-syndromic cleft lip with or without cleft palate on 2p24. 2, 17q23 and 19q13. Hum. Mol. Genet. 25, 2862–2872 (2016).
Sun, Y. et al. Genome-wide association study identifies a new susceptibility locus for cleft lip with or without a cleft palate. Nat. Commun. 6, 1–7 (2015).
Yu, Y. et al. Genome-wide analyses of non-syndromic cleft lip with palate identify 14 novel loci and genetic heterogeneity. Nat. Commun. 8, 1–11 (2017).
Birnbaum, S. et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat. Genet. 41, 473–477 (2009).
Rojas-Martinez, A. et al. Genetic risk factors for nonsyndromic cleft lip with or without cleft palate in a Mesoamerican population: evidence for IRF6 and variants at 8q24 and 10q25. Birth Defects Res. Part A Clin. Mol. Teratol. 88, 535–537 (2010).
Ludwig, K. U. et al. Genome-wide meta-analyses of nonsyndromic cleft lip with or without cleft palate identify six new risk loci. Nat. Genet. 44, 968–971 (2012).
Beaty, T. H. et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42, 525–529 (2010).
Lee, T.-H. et al. Association between IRF6 rs642961 polymorphism and non-syndromic cleft lip with or without a cleft palate: a systematic review and meta-analysis. Int. J. Clin. Exp. Med. 3033–3042 (2018).
Butali, A. et al. Rare functional variants in genome–wide association identified candidate genes for nonsyndromic clefts in the African population. Am. J. Med. Genet. Part A 164, 2567–2571 (2014).
Rahimov, F. et al. Disruption of an AP-2α binding site in an IRF6 enhancer is associated with cleft lip. Nat. Genet. 40, 1341–1347 (2008).
Kircher, M. et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 10, 3583 (2019).
Siewert, A., Hoeland, S., Mangold, E. & Ludwig, K. U. Combining genetic and single-cell expression data reveals cell types and novel candidate genes for orofacial clefting. Sci. Rep. 14, 26492 (2024).
Yankee, T. N. et al. Integrative analysis of transcriptome dynamics during human craniofacial development identifies candidate disease genes. Nat. Commun. 14, 4623 (2023).
Kondo, S. et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 32, 285–289 (2002).
Celli, J. et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 99, 143–153 (1999).
Tekin, M., Sırmacı, A., Yüksel-Konuk, B., Fitoz, S. & Sennaroğlu, L. A complex TFAP2A allele is associated with branchio-oculo-facial syndrome and inner ear malformation in a deaf child. Am. J. Med. Genet. Part A 149, 427–430 (2009).
Milunsky, J. M. et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am. J. Hum. Genet. 82, 1171–1177 (2008).
Carré, A. et al. A novel FOXE1 mutation (R73S) in Bamforth–Lazarus syndrome causing increased thyroidal gene expression. Thyroid 24, 649–654 (2014).
Ingraham, C. R. et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 38, 1335–1340 (2006).
Thomason, H. A., Dixon, M. J. & Dixon, J. Facial clefting in Tp63 deficient mice results from altered Bmp4, Fgf8 and Shh signaling. Dev. Biol. 321, 273–282 (2008).
Brewer, S., Feng, W., Huang, J., Sullivan, S. & Williams, T. Wnt1-Cre-mediated deletion of AP-2α causes multiple neural crest-related defects. Dev. Biol. 267, 135–152 (2004).
Lu, Y. et al. Mutations of GADD45G in rabbits cause cleft lip by the disorder of proliferation, apoptosis and epithelial-mesenchymal transition (EMT). Biochim. Biophys. Acta Mol. Basis Dis. 1865, 2356–2367 (2019).
Meng, T. et al. Overexpression of mouse TTF-2 gene causes cleft palate. J. Cell. Mol. Med. 16, 2362–2368 (2012).
De Felice, M. et al. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat. Genet. 19, 395–398 (1998).
Knight, A. S., Schutte, B. C., Jiang, R. & Dixon, M. J. Developmental expression analysis of the mouse and chick orthologues of IRF6: the gene mutated in Van der Woude syndrome. Dev. Dyn. 235, 1441–1447 (2006).
Laurikkala, J. et al. p63 regulates multiple signalling pathways required for ectodermal organogenesis and differentiation. Development 133, 1553–1563 (2006).
Kousa, Y. A., Fuller, E. & Schutte, B. C. IRF6 and AP2A interaction regulates epidermal development. J. Invest. Dermatol. 138, 2578–2588 (2018).
Schreurs, O. et al. Expression of keratins 8, 18, and 19 in epithelia of atrophic oral lichen planus. Eur. J. Oral. Sci. 128, 7–17 (2020).
Paul, B. J. et al. The Mafb cleft-associated variant H131Q is not required for palatogenesis in the mouse. Dev. Dyn. 250, 1463–1476 (2021).
Lidral, A. C. et al. A single nucleotide polymorphism associated with isolated cleft lip and palate, thyroid cancer and hypothyroidism alters the activity of an oral epithelium and thyroid enhancer near FOXE1. Hum. Mol. Genet. 24, 3895–3907 (2015).
Biggs, L. C., Rhea, L., Schutte, B. C. & Dunnwald, M. Interferon regulatory factor 6 is necessary, but not sufficient, for keratinocyte differentiation. J. Invest. Dermatol. 132, 50–58 (2012).
Kloth, L. et al. Decrease in thyroid adenoma associated (THADA) expression is a marker of dedifferentiation of thyroid tissue. BMC Clin. Pathol. 11, 1–9 (2011).
Truong, A. B., Kretz, M., Ridky, T. W., Kimmel, R. & Khavari, P. A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 20, 3185–3197 (2006).
Ishida, K. et al. Gadd45g regulates dental epithelial cell proliferation through p38 MAPK-mediated p21 expression. Genes Cells 18, 660–671 (2013).
Liang, X. et al. KRT18 regulates trophoblast cell migration and invasion which are essential for embryo implantation. Reprod. Biol. Endocrinol. 21, 78 (2023).
Miyai, M. et al. Transcription factor MafB coordinates epidermal keratinocyte differentiation. J. Invest. Dermatol. 136, 1848–1857 (2016).
Liu, M. et al. FOXE1 contributes to the development of psoriasis by regulating WNT5A. J. Invest. Dermatol. 143, 2366–2377.e7 (2023).
Gilchrist, E. P., Moyer, M. P., Shillitoe, E. J., Clare, N. & Murrah, V. A. Establishment of a human polyclonal oral epithelial cell line. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod.90, 340–347 (2000).
Fakhouri, W. D. et al. MCS9. 7 enhancer activity is highly, but not completely, associated with expression of Irf6 and p63. Dev. Dyn. 241, 340–349 (2012).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 (2012).
Fakhouri, W. D. et al. An etiologic regulatory mutation in IRF6 with loss-and gain-of-function effects. Hum. Mol. Genet. 23, 2711–2720 (2014).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Garrido-Martin, D., Borsari, B., Calvo, M., Reverter, F. & Guigo, R. Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 12, 727 (2021).
Xiao, Y. & Liu, H. Intrinsic differences between the open chromatin regions of oral and epidermal keratinocytes. Chin. J. Dent. Res 23, 119–130 (2020).
Botti, E. et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc. Natl Acad. Sci. USA 108, 13710–13715 (2011).
Wilderman, A., VanOudenhove, J., Kron, J., Noonan, J. P. & Cotney, J. High-resolution epigenomic atlas of human embryonic craniofacial development. Cell Rep. 23, 1581–1597 (2018).
Madan, N. et al. Functionalization of CD36 cardiovascular disease and expression associated variants by interdisciplinary high throughput analysis. PLoS Genet. 15, e1008287 (2019).
Alghadeer, A. et al. Single-cell census of human tooth development enables generation of human enamel. Dev. Cell 58, 2163–2180. e9 (2023).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Katoh, M., Igarashi, M., Fukuda, H., Nakagama, H. & Katoh, M. Cancer genetics and genomics of human FOX family genes. Cancer Lett. 328, 198–206 (2013).
Golson, M. L. & Kaestner, K. H. Fox transcription factors: from development to disease. Development 143, 4558–4570 (2016).
Bamforth, J., Hughes, I., Lazarus, J., Weaver, C. & Harper, P. Congenital hypothyroidism, spiky hair, and cleft palate. J. Med. Genet. 26, 49 (1989).
Ludwig, K. et al. Strong association of variants around FOXE1 and orofacial clefting. J. Dent. Res. 93, 376–381 (2014).
Moreno, L. M. et al. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum. Mol. Genet. 18, 4879–4896 (2009).
Guan, Y., Yang, Y. J., Nagarajan, P. & Ge, Y. Transcriptional and signalling regulation of skin epithelial stem cells in homeostasis, wounds and cancer. Exp. Dermatol 30, 529–545 (2021).
Oliver, J. R., Kushwah, R. & Hu, J. Multiple roles of the epithelium-specific ETS transcription factor, ESE-1, in development and disease. Lab Invest. 92, 320–330 (2012).
Panagoulias, I. et al. ets-2 acts as a Transcriptional repressor of the human immunodeficiency virus type 1 through binding to a repressor–activator target sequence of 5′-lTr. Front. Immunol. 8, 1924 (2018).
Wei, G., Schaffner, A. E., Baker, K. M., Mansky, K. C. & Ostrowski, M. C. Ets-2 interacts with co-repressor BS69 to repress target gene expression. Anticancer Res. 23, 2173–2178 (2003).
Carlson, J. C. et al. A systematic genetic analysis and visualization of phenotypic heterogeneity among orofacial cleft GWAS signals. Genet. Epidemiol. 43, 704–716 (2019).
Huang, L. et al. Genetic factors define CPO and CLO subtypes of nonsyndromicorofacial cleft. PLoS Genet. 15, e1008357 (2019).
Yang, Y., Suzuki, A., Iwata, J. & Jun, G. Secondary genome-wide association study using novel analytical strategies disentangle genetic components of cleft lip and/or cleft palate in 1q32.2. Genes 11, 1280 (2020).
Ajore, R. et al. Functional dissection of inherited non-coding variation influencing multiple myeloma risk. Nat. Commun. 13, 1–15 (2022).
de Lima, R. L. et al. Prevalence and nonrandom distribution of exonic mutations in interferon regulatory factor 6 in 307 families with Van der Woude syndrome and 37 families with popliteal pterygium syndrome. Genet. Med. 11, 241–247 (2009).
Rahimov, F. et al. High incidence and geographic distribution of cleft palate in Finland are associated with the IRF6 gene. Nat. Commun. 15, 9568 (2024).
Clifton-Bligh, R. J. et al. Mutation of the gene encoding human TTF-2 associated with thyroid agenesis, cleft palate and choanal atresia. Nat. Genet. 19, 399–401 (1998).
Marazita, M. L. et al. Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage and association results. Hum. Hered. 68, 151–170 (2009).
Paranjapye, A., Mutolo, M. J., Ebron, J. S., Leir, S.-H. & Harris, A. The FOXA1 transcriptional network coordinates key functions of primary human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 319, L126–L136 (2020).
Lim, F. et al. Affinity-optimizing enhancer variants disrupt development. Nature 626, 151–159 (2024).
Yamamoto, H. et al. Defective trophoblast function in mice with a targeted mutation of Ets2. Genes Dev. 12, 1315–1326 (1998).
Twigg, S. R. et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat. Genet. 45, 308–313 (2013).
Micale, L. et al. Heterozygous variants disrupting the interaction of ERF with activated ERK1/2 cause microcephaly, developmental delay, and skeletal anomalies. Eur. J. Hum. Genet. 33, 718–726 (2024).
Oettgen, P. et al. Characterization of ESE-2, a novel ESE-1-related Ets transcription factor that is restricted to glandular epithelium and differentiated keratinocytes. J. Biol. Chem. 274, 29439–29452 (1999).
Andreoli, J. M. et al. The expression of a novel, epithelium-specific ets transcription factor is restricted to the most differentiated layers in the epidermis. Nucleic Acids Res. 25, 4287–4295 (1997).
Lackner, A. et al. The Fgf/Erf/NCoR1/2 repressive axis controls trophoblast cell fate. Nat. Commun. 14, 2559 (2023).
Chin, S. S., Romano, R. A., Nagarajan, P., Sinha, S. & Garrett-Sinha, L. A. Aberrant epidermal differentiation and disrupted DeltaNp63/Notch regulatory axis in Ets1 transgenic mice. Biol. Open 2, 1336–1345 (2013).
Nagarajan, P. et al. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 123, 3566–3575 (2010).
Tehranchi, A. et al. Fine-mapping cis-regulatory variants in diverse human populations. Elife 8, e39595 (2019).
Leslie, E. J. et al. A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am. J. Hum. Genet. 98, 744–754 (2016).
Delaneau, O., Zagury, J.-F. & Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Kwasnieski, J. C., Fiore, C., Chaudhari, H. G. & Cohen, B. A. High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 24, 1595–1602 (2014).
Gourronc, F. A. et al. Proliferative defects in dyskeratosis congenita skin keratinocytes are corrected by expression of the telomerase reverse transcriptase, TERT, or by activation of endogenous telomerase through expression of papillomavirus E6/E7 or the telomerase RNA component, TERC. Exp. Dermatol. 19, 279–288 (2010).
Rheinwald, J. G. & Green, H. Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265, 421–424 (1977).
Friedman, R. Z. et al. Information content differentiates enhancers from silencers in mouse photoreceptors. Elife 10, e67403 (2021).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408 (2001).
Taylor, S. C. et al. The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends Biotechnol. 37, 761–774 (2019).
Bomsztyk, K. et al. PIXUL-ChIP: integrated high-throughput sample preparation and analytical platform for epigenetic studies. Nucleic Acids Res. 47, e69 (2019).
Morillo-Bernal, J., Fernández, L. P. & Santisteban, P. FOXE1 regulates migration and invasion in thyroid cancer cells and targets ZEB1. Endocr. Relat. cancer 27, 137–151 (2020).
Solomon, E. R., Caldwell, K. K. & Allan, A. M. A novel method for the normalization of ChIP-qPCR data. MethodsX 8, 101504 (2021).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, s13742-015-0047-8 (2015).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Alexander, T. A. & Machiela, M. J. LDpop: an interactive online tool to calculate and visualize geographic LD patterns. BMC Bioinformatics 21, 1–4 (2020).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 (2010).
Acknowledgements
The authors thank Pilar Santisteban, Universidad Autónoma de Madrid, for the FOXE1 antibody, and Greg Bonde (University of Iowa) and Josh Rosswork (University of Washington) for technical assistance throughout the project. We thank Jessica Hoisington-Lopez and MariaLynn Crosby in the DNA Sequencing Innovation Lab (Washington University) for assistance with high-throughput sequencing. We thank the Ellison Stem Cell Core at ISCRM and Jennifer Hesson for technical assistance in screening and genotyping of iPSCs. We are grateful for many discussions with members of the Craniofacial Interest Group weekly meeting hosted at the University of Iowa. This project was funded by grants from the US National Institutes of Health, DE027362 (R.A.C.), R01DE028599 (A.V.), R01DE033016 (R.A.C., H.R.-B., and J.M.), R01AR067739 (M.D.), HG010855 (K.B.), CA246503 (K.B.), and NHGRI F31 HG011431 (R.Z.F.). Research conducted at the E.O. Lawrence Berkeley National Laboratory was performed under Department of Energy Contract DE-AC02-05CH11231, University of California.
Author information
Authors and Affiliations
Contributions
P.K. contributed to the design, data acquisition, analysis, interpretation, drafting and editing the manuscript; R.Z.F.—data analysis, interpretation and editing the manuscript, S.W.C.—data analysis, interpretation, drafting and editing the manuscript; L.P.—data analysis; K.P. and A.V.—generation and preliminary analyses of murine in vivo reporter assays for IRF6 -10 kb and −22 kb SNPs; L.R.—data acquisition and analysis; M.D.—data acquisition, analysis, and interpretation; A.P.P.—data acquisition; D.M. and K.B.— design and data acquisition; J.M.—data acquisition and analysis; H.R.-B.—design and data interpretation; E.J.L.-C.—design and data interpretation, drafting, and editing the manuscript; M.A.W.—design, data acquisition, analysis, interpretation, and editing the manuscript, B.A.C.— design and data interpretation, R.A.C.—conception, design, data acquisition, analysis, interpretation, drafting and editing the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
L.P. is an employee of Cencora-PharmaLex. K.B. is co-founder of Matchstick Technologies, Inc., and co-inventor of PIXUL, used here for chromatin immunoprecipitation experiments. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kumari, P., Friedman, R.Z., Curtis, S.W. et al. Identification of functional non-coding variants associated with orofacial cleft. Nat Commun 16, 6545 (2025). https://doi.org/10.1038/s41467-025-61734-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61734-w
