
Abstract
Predicting compound-protein interactions (CPIs) plays a crucial role in drug discovery. Traditional methods, based on the key-lock theory and rigid docking, often fail with novel compounds and proteins due to their inability to account for molecular flexibility and the high sparsity of CPI data. Here, we introduce ColdstartCPI, a framework inspired by induced-fit theory, which leverages unsupervised pre-training features and a Transformer module to learn both compound and protein characteristics. ColdstartCPI treats proteins and compounds as flexible molecules during inference, aligning with biological insights. It outperforms state-of-the-art sequence-based models, particularly for unseen compounds and proteins, and shows strong generalization capability compared to structure-based methods in virtual screening. ColdstartCPI also excels in sparse and low-similarity data conditions, demonstrating its potential in data-limited settings. Our results are validated through literature search, molecular docking, and binding free energy calculations. Overall, ColdstartCPI offers a perspective on sequence-based drug design, presenting a promising tool for drug discovery.
Similar content being viewed by others
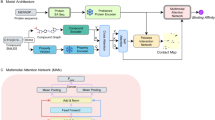

Introduction
Identifying compound-protein interactions (CPIs) plays a vital step in the process of drug discovery1. Traditional in vitro experimental biomedical measurements are reliable but costly and time-consuming and cannot reasonably be applied to large-scale data2. Due to the rapid enrichment of bioactivity databases, computational CPI prediction methods have been developed to narrow the search space for candidate compounds and proteins, thereby reducing the cost and increasing the efficiency of drug discovery3. Generally, there are three kinds of computational CPI prediction methods, structure-based methods, ligand-based methods, and proteochemometric modeling (PCM) methods4.
Structure-based methods take the precise 3D structures of proteins or compound-protein complexes as input. The classic methods include docking-based models (Glide-SP5, Vina6, Smina7, Surflex8,and DOCK6.99), machine learning scoring function (NN-score10 and RFscore11), and deep learning-based models (Pafnucy12, OnionNet13,Gnina14, BigBind15, PLANET16, ConBAP17, and DynamicBind18). While programs such as AlphaFold19 can provide structural predictions when crystal structures are not accessible, these predictions are not yet complete structural models in the classical sense of an atomic model obtained by X-ray crystallography20. And, these structure-based methods can be sensitive to errors introduced by in silico predictions21,22. The performance of ligand-based models is poor when the bioactivity data of the ligand is insufficient23. Recently, PCM methods have been considered more promising to overcome the limitations stated above and to cope with more complex scenarios with further advancements in molecular representations, machine learning techniques, and the available bioactivity data24.
The previous PCM methods were based on key-lock theory to model CPI prediction. The target protein is the lock, and the compound is the appropriate key that binds the target protein. During the feature extraction phase, the features of proteins and compounds are fixed, not changing with respect to the paired compounds, which reflects the properties of the proteins themselves, and vice versa. Machine learning-based PCM methods encode compounds and proteins into descriptors according to biochemical knowledge and are then trained and predicted using machine learning models25, e.g., Random Forest (RF)26, Support Vector Machine (SVM)27, and Multilayer Perceptron (MLP)28,29. Considering that each descriptor represents a different property, exploring the optimal combination of descriptors or building more robust descriptors is an important challenge for this class of methods4. With the successful results in computer vision, speech recognition, and bioinformatics30,31 in recent years, deep learning-based models have been developed in CPI prediction. Constructing the entire CPI prediction process as an end-to-end pipeline, the task-relevant and data-driven representations of compounds and proteins are learned, resulting in better predictive accuracy compared to high-dimensional and sparse descriptors24. This type of model typically takes SMILES strings or graphical structures of compounds and amino acid sequences of proteins as input and encodes them by utilizing various deep learning modules, such as convolutional neural network (CNN)32,33,34,35 and graph neural network (GNN)36,37,38. The above models extract the molecular features of compounds and proteins by constructing efficient modules but ignore the biological fact that only some of the amino acids of a protein or a few atoms of a compound are involved in inter-molecular interactions, not the entire molecular structure. To simulate non-covalent inter-molecular interactions between amino acids and atoms, the attention mechanism has been introduced into the DTI predictions35,39,40,41,42,43,44. These methods attempt to locate binding sites on proteins through attention mechanisms in a mode similar to rigid docking.
Among the four different realistic scenarios proposed by Pahikkala et al.45 in evaluating the generalization performance on CPI prediction, i.e., warm start and three cold-start problems (compound cold start, protein cold start, and blind start), most of the end-to-end models are mainly developed in the warm start scenario and benefited from task-relevant feature extraction. Despite the promising developments, these end-to-end models suffer from the high sparsity and biased nature of CPI data46 and cannot generalize well enough to predict CPIs among unseen compounds and unseen proteins27,28,29, i.e., the cold-start problems. Therefore, several non-end-to-end deep learning models were developed to address the cold-start problem. KGE_NFM47 is a framework, which uses a bioinformatics knowledge graph to extract compound and protein features and models the CPI prediction problem as a recommender system. KGE_NFM uses bio-network information to mitigate the cold-start problem but is limited by knowledge graph integrity and information leakage. DrugBAN_CDAN48 is a variant model of DrugBAN48, which transfers learned knowledge from the source domain to the target domain (cold-start conditions) by using a conditional domain adversarial network. DrugBAN_CDAN48 achieves promising results in the cold-start problem but suffers from the training instability of adversarial networks. Pre-training of molecular encoding in a large number of chemical libraries facilitates the mining of deep features about the biological properties of compounds/proteins49 and improves the generalization performance. AI-Bind50 is a preliminary work based on pre-training, which uses two pre-trained models to encode the global features of compounds and proteins and then leverages an MLP for CPI prediction. However, AI-Bind ignores the modeling of interactions in compound substructures and amino acid fragments, which are important features for CPI prediction by several works39,40,42,43,44,48. INGNN-DTI44 uses a cross-attention module to learn the interaction feature between the substructures of compounds and proteins and also incorporates the global features created by the unsupervised models. Many previous models are designed based on the lock-and-key theory and rigid docking, where compounds or proteins are treated as rigid and their features are fixed, thereby limiting the ability to generalize models51. But in fact, the binding of a compound to a target protein causes a change in the shape of the target protein, thus enhancing or inhibiting its activity52, i.e., induced-fit theory53. Modeling based on induced-fit theory is more realistic and might improve the prediction performance of the CPI.
To this end, we propose a two-step CPI prediction method, named ColdstartCPI, which is based on pre-trained feature encoding and the induced fit theory. To introduce semantic features of drug substructures and high-level features related to protein structure and function, we use Mol2vec54 and ProtTrans55 to extract the substructure feature matrix of the compound and the amino acid feature matrix of the protein, respectively. Then, inspired by the induced-fit theory, ColdstartCPI treats compounds and proteins as flexible molecules and uses the Transformer structure to learn compound and protein features by extracting inter- and intra-molecular interaction characteristics. In our study, for compounds, the inter- and intra-molecular interaction characteristics denote the contribution of proteins and substructures to the compound features, respectively. For protein, the inter- and intra-molecular interaction characteristics denote the contribution of compounds and amino acids to the protein features, respectively. Through comprehensive tests, we have demonstrated that ColdstartCPI can substantially improve performance over the state-of-the-art CPI prediction methods, especially under cold-start conditions. Furthermore, we analyze and validate drug candidate identification for target proteins and drug repurposing predicted by ColdstartCPI for Alzheimer’s Disease (AD), breast cancer, and COVID-19 through literature search, docking simulations, and binding free energy calculations. All these results demonstrate that induced-fit theory-inspired ColdstartCPI provides more insights into sequence-based drug design.
In summary, our contributions are as follows:
-
1.
We propose a unified CPI prediction framework based on pre-training feature engineering. This unified framework is simple to access and efficient to use pre-trained features completing the prediction of CPIs on both warm and cold starts.
-
2.
Considering the induced-fit theory, ColdstartCPI treats compounds and proteins as flexible molecules and uses the Transformer structure to learn compound and protein features by extracting inter- and intra-molecular interaction characteristics. The compound features change depending on the binding proteins and vice versa, aligning with known biological observations.
-
3.
We conduct extensive experiments under four experimental setups on three large-scale datasets and the results show that ColdstartCPI consistently outperforms existing methods. ColdstartCPI also performs better under sparse data and low similarity conditions. We also evaluate the model’s virtual screening ability and use multiple validation strategies, including literature search, molecular docking simulations, binding free energy calculations, and molecular dynamics simulations to valid the top predictions on Alzheimer’s Disease, breast cancer, and COVID-19, demonstrating the effectiveness of ColdstartCPI.
Results
Overview of ColdstartCPI
The flowchart of ColdstartCPI is illustrated in Fig. 1. The whole process consists of five parts. (1) Input. The input of ColdstartCPI is the SMILES strings of compounds and the amino acid sequences of proteins. (2) Pre-trained module. We use two pre-trained models, Mol2Vec and ProtTrans, to generate the feature matrices of compounds and proteins, respectively. These matrices include the representation of substructures (the function groups) of compounds and amino acid fragments of proteins, which represent the fine-grained properties of the molecules. Furthermore, we use the pooling function on the feature matrices to get the global representations of the compounds and the proteins. (3) Decouple module. Four different MLPs are applied here to unify the feature spaces of compounds and proteins and to decouple feature extraction with CPI prediction. (4) Transformer module. Considering the induced fit theory, we construct a joint matrix representation of the CP pair and feed it into the Transformer module to learn compound and protein features by extracting inter- and intra-molecular interaction characteristics. (5) Prediction module. The compound and protein features are concatenated and are processed by a three-layer fully connected neural network with dropout to predict the CPI probability.

The pipeline mainly consists of five parts: Input, Pre-trained module, Decouple module, Transformer module, and Prediction module.
ColdstartCPI outperforms existing methods in both warm start and cold start settings for CPI prediction
To demonstrate the advantages of ColdstartCPI, we compare it with ten SOTA methods as baselines under four realistic evaluation settings (one warm start and three cold starts) on three large-scale public datasets (BindingDB_AIBind50, BindingDB48, and BioSNAP39). The area under the receiver operating characteristic (AUC) and the area under the precision-recall curve (AUPR) are used as the major metrics. In addition, we also report accuracy, precision, recall, and F1 score. The SOTA baselines include two feature-based methods (DNN28, and DeepConv-DTI29), five end-to-end models (GraphDTA36, ML-DTI35, MolTrans39, HyperAttentionDTI42, and DrugBAN48), one domain adaptation-based method, DrugBAN_CDAN48, and two pre-training-based method, iNGNN-DTI44 and AI-Bind50. DrugBAN_CDAN is a variant model of DrugBAN48, which is evaluated under three cold start settings to enhance DrugBAN’s performance by using a conditional domain adversarial network. INFGNN-DTI44 takes contact maps of proteins as input, whereas of the three datasets, only the BioSNAP dataset contains contact map data. We removed proteins lacking contact maps from the BioSNAP dataset and constructed a dataset named BioSNAP_CM for comparing ColdstartCPI and iNFGNN-DTI. More details of datasets and evaluation settings are provided in Subsections Benchmark datasets and evaluation protocols in Section Methods, respectively.
The evaluation on the BindingDB_AIBind dataset50 is to avoid the topology of the CPI network driving the prediction task (see Supplement Note 1). The BindingDB_AIBind dataset is composed of the positives, generated from BindingDB56 and DrugBank57, and the network-derived negatives with experimentally validated non-binding CPIs to ensure sufficient positive and negative samples for each entity (i.e., compounds and proteins) in the training data. The results are shown in Table 1. We find that ColdstartCPI achieves the best results under all four scenarios. In the scenario of the warm start, ColdstartCPI achieves an average AUC of 0.970 ± 0.001 and AUPR of 0.962 ± 0.002, compared with the second-best HyperAttentionDTI’s AUC of 0.950 ± 0.003 and AUPR of 0.938 ± 0.003.
On the three cold-start settings, ColdstartCPI and AI-Bind outperform other baselines due to the incorporation of the self-supervised feature extraction trained on a larger collection of compounds and protein corpus, allowing ColdstartCPI and AI-Bind to learn a wider variety of chemical patterns. In the most challenging blind start, ColdstartCPI achieves an AUC of 0.839 ± 0.009 with a 6.8% improvement and an AUPR of 0.785 ± 0.02 with a 7.7% improvement. Performance comparisons on BindingDB_AIBind under node-centric local measures58 are provided in Supplementary Table 1, which demonstrates that ColdstartCPI outperforms the baseline models across these node-centric metrics, highlighting its effectiveness in link prediction tasks. The main reasons for the performance improvement are as follows: 1. ColdstartCPI takes pre-trained feature matrices as inputs to obtain finer features of compounds and proteins; 2. its Transformer module extracts inter- and intra-molecular interaction features of compounds and proteins to further improve their feature representation in CPI prediction. These results demonstrate that excluding annotation bias, ColdstartCPI learns positive and negative distinguishing features of compound-protein pairs from pre-trained features, thus maintaining superior and stable prediction performance.
The BioSNAP dataset39 is created from the DrugBank database57, whose compounds on average have 5-10 unique target proteins. It is often used as a benchmark for CPI prediction evaluation39,48. As shown in Fig. 2 and Supplementary Table 2, ColdstartCPI achieves the best results under all four scenarios. Specifically, in the scenario of the warm start, ColdstartCPI achieves an average AUC of 0.922 ± 0.002 and AUPR of 0.945 ± 0.003, compared with the second-best AI-Bind’s AUC of 0.927 ± 0.001 and AUPR of 0.934 ± 0.001. In the compound cold start, ColdstartCPI also outperforms the second-best AI-Bind and achieves an average AUC of 0.884 ± 0.014 and AUPR of 0.893 ± 0.025. In the scenario of the protein cold start, ColdstartCPI achieves an average AUC of 0.871 ± 0.022 and an average AUPR of 0.891 ± 0.017 with 7.3% improvements to AI-Bind. In the scenario of the blind start, ColdstartCPI achieves an AUC of 0.791 ± 0.025 with an improvement of at least 10.2% and an AUPR of 0.810 ± 0.047 with at least 7.4% improvement than the second-best models, DrugBAN_CDAN and AI-Bind. As shown in Supplementary Table 3, ColdstartCPI also outperforms iNFGNN-DTI under the four scenarios on the BioSNAP_CM dataset. ColdstartCPI achieving better and more stable performance indicates that ColdstartCPI inspired by induced fit theory obtains a more general joint representation for CPI prediction.

All results are obtained by 5-fold cross-validation (number of data points n = 5; center line, median; box limits, upper and lower quartiles; whiskers, maximum and minimum values; green triangles, mean values; dots, outliers). a The performance under warm start. b The performance under compound cold start. c The performance under protein cold start. d The performance under blind start. Source data are provided as a Source Data file.
Considering the ligand bias in the original BindingDB data59, we use a low-bias version of the binary BindingDB dataset proposed by ref. 60, with the bias-reducing preprocessing steps described in Supplementary Note 1. The results are shown in Supplementary Table 4. In the scenario of the warm start, pre-training-based methods achieve higher predictive performance than other models. Specifically, ColdstartCPI achieves the best predictive performance with an AUC of 0. 965 ± 0.002 and an AUPR of 0.953 ± 0.002. In the scenario of the compound cold start, ColdstartCPI also achieves the best performance with an AUC of 0.835 ± 0.021 and an AUPR of 0.781 ± 0.032. In the scenario of the protein cold start, ColdstartCPI achieves relative improvements in AUC (0.672 ± 0.066) with 8.7% and in AUPR (0.592 ± 0.084) with 7.2%, compared with the second-best baselines, GraphDTA (AUC = 0.585 ± 0.008) and DrugBAN (AUPR = 0.52 ± 0.073). In the scenario of the blind start, ColdstartCPI outperforms the second-best method, DrugBAN_CDAN, by 5.1% and 2.9% in AUC and AUPR, respectively. Specifically, ColdstartCPI achieves an AUC of 0.66 ± 0.041 and an AUPR of 0.584 ± 0.039. Furthermore, both ColdstartCPI and DrugBAN_CDAN have higher AUC and AUPR than those of AI-Bind, which suggests that pre-trained features alone are not enough to gain an advantage under the blind start on the BindingDB dataset. The compound and protein features containing the inter- and intra-molecular interactions extracted by the Transformer module bring a significant gain to CPI predictions for proteins and compounds. Evaluation of the BindingDB dataset can rule out the possibility that the model utilizes ligand bias rather than CPIs, further illustrating the superiority and plausibility of ColdstartCPI.
Considering that network-negative generation is a core component of AI-Bind’s pipeline, to provide a more accurate comparison with the complete AI-Bind model, we regenerated the negative samples for the BindingDB and BioSNAP datasets using AI-Bind’s Network Negative Generation method, naming the datasets BindingDB_AIBind2 and BioSNAP_AIBind, respectively. The comparison results are shown in Supplementary Table 5. The results indicate that ColdstartCPI still outperforms AI-Bind, even when network negatives are regenerated according to AI-Bind’s pipeline. This provides a fairer and more thorough comparison, highlighting ColdstartCPI’s superior performance.
Considering the superiority of AlphaFold361 for structure prediction of compound-protein complexes, we constructed an independent test set, BindingDB_AF (shown in Supplementary Table 6) for comparing AlphaFold361 and ColdstartCPI. The construction details are provided in Supplementary Note 2. Supplementary Fig. 1 and Supplementary Table 7 present detailed predictions and performance metrics for both ColdstartCPI and AlphaFold3 on the BindingDB_AF dataset. The results show that ColdstartCPI outperforms AlphaFold3 across all metrics, including Accuracy, Recall, and Sensitivity.
These results indicate that ColdstartCPI surpasses existing methods in both warm-start and cold-start scenarios for CPI prediction on large-scale public datasets (P-values tests are provided in Supplementary Tables 8-10). Despite the degradation in performance observed across all CPI models under cold start settings, especially blind start, due to significant differences in distribution between training and testing sets, ColdstartCPI consistently outperforms all state-of-the-art models.
One of the reasons for the superior performance of ColdstartCPI compared to AI-Bind is attributed to the nature of their pre-training feature vectors. ColdstartCPI’s pre-training feature matrices provide more fine-grained atom and amino acid characterizations compared to the global representations used by AI-Bind. Additionally, AI-Bind’s prediction module solely relies on a fully connected network, which overlooks the role of non-covalent inter-molecular interactions. In contrast, ColdstartCPI leverages its Transformer module to capture interaction patterns, thereby further enhancing its predictive performance.
Comparison with structure-based models
To validate the performance of ColdstartCPI in real-world virtual screening, we assess its screening power against structure-based baselines, including docking-based models (Glide-SP5, Vina6, Smina7, Surflex8, DOCK6.99), machine learning scoring function (NN-score10, RFscore11), and deep learning-based models (Pafnucy12, OnionNet13, Gnina14, BigBind15, PLANET16, ConBAP17, DynamicBind18). Screening power is defined here as a model’s ability to distinguish binders from non-binders. For benchmarking, we utilize three public virtual screening datasets, DUD-E62, LIT_PCBA63, and Antibiotics21. It is important to note that deep learning models tend to exhibit overly optimistic performance given the condition that the training set and the selected test set are homogenous in nature (e.g., splitting a common parent dataset to obtain the training and test sets)46,64,65,66. To mitigate this risk in model evaluation, ColdstartCPI is trained on the PDBbind67 dataset without any fine-tuning, promoting a more robust generalization evaluation consistent with prior deep learning baselines13,14,15,16,17.
The results of the DUD-E are presented in Table 2. One can see that the performance of all deep learning-based models originally trained on the PDBbind dataset is much less promising on the DUD-E dataset because the composition of the DUD-E dataset is distinctively different from the PDBbind dataset. In contrast, our ColdstartCPI exhibits a reasonable level of performance here. For example, ColdstartCPI (AUC = 0.765) is superior to PLANET (AUC = 0.736), Pafnucy (AUC = 0.631), and OnionNet (AUC = 0.597), given that all three of these deep learning-based models are also trained on the PDBbind dataset. Notably, Glide-SP, a conventional docking method, achieves the highest performance across AUC, BEDROC, and EF (1% and 5%), highlighting how, despite their simpler mathematical architecture, docking methods effectively capture some basic physics in compound-protein interactions. Although ColdstartCPI is second to Glide-SP on the DUD-E benchmark, it outperforms all other docking-based and deep learning-based models. Notably, ColdstartCPI achieves an enrichment of active compounds that is 21.61 times higher than random screening in the top 0.5% of ranked compounds. This result indicates that ColdstartCPI is very effective in identifying active compounds in the early screening, and can find more active molecules in fewer compounds in a concentrated manner, which is of great significance for further screening and experimental validation.
To conduct a more objective evaluation of screening power, we further test ColdstartCPI head-to-head with Glide-SP, PLANET, and ConBAP on the LIT-PCBA benchmark, where both the actives and inactives had been experimentally verified and an extreme imbalance of actives and inactives is retained to mimic the challenging real screening scenarios. Similar to the case of DUD-E, ColdstartCPI is directly evaluated on LIT-PCBA without fine-tuning. The evaluation results are illustrated in Table 2. According to the average AUC% of all the 15 targets, ColdstartCPI performs the best among all models. Here, one can see that LIT-PCBA is indeed more challenging than DUD-E, as the mean AUC scores of ColdstartCPI and Glide-SP are 0.596 and 0.536, respectively, both of which are lower than the counterparts obtained on DUD-E. To further investigate the results, we present the performance on each individual target in Supplementary Fig. 2. Compared with Glide-SP, ColdstartCPI produces higher AUC scores on 11 targets and lower scores on four targets in LIT-PCBA. We also calculate the number of targets with EF values greater than 1. Specifically, there are 10, 8, 11, and 10 targets, whose EF0.5% values are greater than 1, in the predictions of ColdstartCPI, Glide-SP, PLANET, and ConBAP, respectively. These results illustrate that ColdstartCPI performs competitively with existing structure-based methods.
Furthermore, to evaluate ColdstartCPI in a more realistic virtual screening scenario, we have conducted additional analyses using the Antibiotics benchmark21. The Antibiotics benchmark consists of 12 critical E. coli proteins and 218 active compounds, totaling 2,616 compound-protein interactions (CPIs). Wang et al.21 screened these 218 compounds for enzyme inhibition against a panel of 12 essential E. coli proteins or protein complexes, performing duplicate assays at a concentration of 100 μM. Ground truth values were derived by binarizing the relative enzyme activity data (1 if the relative enzyme activity in both biological replicates was less than 0.5, 0 otherwise), resulting in 415 positive samples in the dataset.
We also trained ColdstartCPI on the PDBbind dataset and assessed the model’s performance using the AUC metric. As shown in Supplementary Table 11, the AUC values across the 12 proteins ranged from 0.5752 (for murA) to 0.8222 (for dnaE), with an average AUC of 0.7365. ColdstartCPI outperformed several common docking methods, including Vina, as well as machine learning-based scoring methods and the state-of-the-art deep learning-based method DynamicBind18.
Additionally, Supplementary Table 12 shows that ColdstartCPI’s predicted scores have Pearson and Spearman correlation coefficients of 0.5475 and 0.5692, respectively, with the inhibition constants reported in the experimental data. ColdstartCPI achieves an enrichment of compound-protein pairs that is 4.5 times higher than random screening in the top 0.5% of ranked pairs, with 61 of the top 100 predicted compound-protein pairs identified as hits. These results further demonstrate ColdstartCPI’s potential for proteome-level virtual screening applications.
ColdstartCPI demonstrates considerable predictive power as a CPI prediction model that leverages full protein sequences, making it versatile across a wide range of protein targets beyond those with known structural information. Our virtual screening experiments on benchmark datasets DUD-E and LIT_PCBA highlight that ColdstartCPI outperforms existing deep learning-based approaches, underscoring its robust generalization capabilities.
A key advantage of ColdstartCPI lies in its basis in induced-fit theory, which allows it to capture the dynamic adaptability of protein-compound interactions. This theoretical foundation grants ColdstartCPI an edge in modeling interaction potential across diverse compound-protein complexes without requiring precise structural data for each target. Overall, ColdstartCPI provides a practical and computationally scalable alternative that maintains competitive predictive accuracy while enabling broader application across various CPI prediction tasks.
Performance evaluation with scarce data and unseen data
Based on the existing practical situation and needs, drug developers have already conducted in-depth research on some compounds and protein targets to fully explore the related CPIs. With the emergence of new challenges, only a few CPIs have been developed for relevant compounds or new targets68,69. Therefore, robustness under sparse conditions or unseen data without similarity is the focus of model development39.
To evaluate the performance of ColdstartCPI and the baselines under sparse conditions, we trained each method on 5%, 10%, 20%, and 30% of the datasets and validated and tested on the remaining 95%, 90%, 80%, and 70% of the datasets, respectively. The validation set is used for early stopping and is drawn by random sampling with a ratio of 1:9 to the test set. The model performance comparisons on the BindingDB_AIBind dataset under different sparse conditions are provided in Fig. 3a (Supplementary Table 13). AUC is used as the major metric. The missing rate indicates the percentage of non-training sets (validation and testing sets).

a Performance comparison of ColdstartCPI with baselines under different scarce conditions on the BindingDB_AIBind dataset (number of data points n = 5). The error bar indicates the standard deviation. The missing rate indicates the percentage of non-training sets (validation and testing sets). The ratio of the validation set to the testing set is 1 to 9. b The spectral performance curves of AUC and AUPR for ColdstartCPI and baselines in the BindingDB_SPECTRA dataset. Source data are provided as a Source Data file.
As shown in Fig. 3a (Supplementary Table 13), ColdstartCPI outperforms the baselines under all sparse conditions with an average of 2.5% improvement. Even with a miss rate of 95%, ColdstartCPI achieves an average AUC of 0.833 ± 0.007 with a 2.6% improvement on the BindingDB_AIBind dataset over the second-best method HyperAttentionDTI. These results show that based on the pre-trained matrices of compounds and proteins, the Transformer module mines more prevalent joint features in sparse CPIs, compared to data-driven features and pre-defined descriptors. AI-Bind does not show a significant advantage over end-to-end baselines under sparse conditions, due to the fact that it ignores substructure and subfragment features in compounds and proteins.
The model performance comparisons on the BioSNAP and BindingDB datasets under different sparse conditions are provided in Supplementary Table 13. Specifically, on the BioSNAP dataset, ColdstartCPI outperforms the baselines under all sparse conditions with an average of 3.1% improvement. Overall, ColdstartCPI is robust under sparse conditions and gives better and more stable results compared to the baselines.
Considering the impact of the similarity between the training and test sets on the assessment of the generalizability of the model, Ektefaie et al. proposed SPECTRA70, which plots model performance as a function of decreasing cross-split overlap and reports the area under this curve as a measure of generalizability. To better reduce the sequence similarity between the training set and the test set, we construct a subset of the BindingDB_AIBind dataset that has positive and negative samples containing all compounds and proteins, respectively, while minimizing the frequency of their occurrences. A dataset containing 8358 positive samples and 7684 negative samples was finally obtained, named BindingDB_SPECTRA. The data generation pseudocode is provided in Algorithm 1 in Supplementary Note 3.
Following the spectral property graph generation setting of PDBbind described in SPECTRA, we created splits with spectral parameters ranging from 0 to 1 in 0.1 increments. For each spectral parameter, a corresponding split was generated. Cross-split overlap and the data size in SPECTRA are shown in Supplementary Fig. 3 and the average protein similarity (generated by BLSTAp) between the training and test sets of the SPECTRA split are shown in Supplementary Table 14. More details are provided in Supplementary Note 3. In the SPECTRA evaluation framework, the model is trained and tested on each split to generate a plot of the model’s performance against the spectral parameter, the model’s spectral performance curve (SPC), for the molecular sequencing dataset. The area under the spectral performance curve (AUSPC) summarizes model performance across all levels of cross-split overlap and can be used to compare model generalizability to other models within and across tasks.
As shown in Fig. 3b and Supplementary Fig. 4, we provide AUSPCs for all relevant models in the BindingDB_SPECTRA dataset. Our results reveal that the performance of deep learning models generally declines as the cross-split overlap decreases and the amount of training data becomes more limited. However, ColdstartCPI achieves the highest performance across all metrics, underscoring its ability to generalize effectively to novel sequences in CPI prediction tasks.
Ablation studies on ColdstartCPI
Impact of each module of ColdstartCPI on predictive performance
Here, we conduct an ablation study to investigate the influences of the individual modules on ColdstartCPI. The details of three different variants of ColdstartCPI are illustrated as follows. WOPretrain is an end-to-end model that removes the pre-trained features and takes the SMILES sequence, amino acid sequence as input, and CNNs as feature extraction. WODecouple is a model that eliminates the Decouple module and takes the first 300 features of the protein as input, which ensures that the compound and protein features are of the same dimension. WOTransformer is a model that excludes the Transformer module, takes the global features of compounds and proteins as input, and feeds them into the fully connected network. More details of these three variants are provided in Supplementary Figs. 5-7.
The results under four scenarios on BindingDB_AIBind are shown in Fig. 4a (Supplementary Table 15). For WOPretrain, we observe a significant decrease under all four scenarios, demonstrating that the pre-trained features provide a more powerful generalization ability to unseen compounds and proteins than data-driven features. We can see that WOTransformer yields a lower performance without the Transformer module to model the inter- and intra-molecular interactions, indicating the independent embedding of compounds and proteins is less effective than the joint representation generated by the Transformer module. The potential reason is that redundant features exist in the global representation of compounds and proteins, further indicating that dynamically adjusting the local interaction features of compounds and proteins based on the induced fit theory is crucial for CPI prediction. WODecouple performs slightly worse than ColdstartCPI, indicating the necessity of the Decouple module to decouple the pre-trained feature extraction with CPI prediction.

a Radar charts illustrating the ablation study of ColdstartCPI Architecture. b Performance comparison of ColdstartCPI with MolTrans_pretrain and DrugBAN_pretrain. MolTrans_pretrain and DrugBAN_pretrain are two pre-training-based variant models based on MolTrans and DrugBAN, respectively, which use pre-trained feature matrices to replace the original end-to-end feature extraction module. c Evaluation performance under different pre-trained schemes. Source data are provided as a Source Data file.
Furthermore, we have constructed a variant model ProtTransTurning that takes protein amino acids as input and uses the end-to-end ProtTrans as an extraction module for the protein feature matrix. ProtTransTurning is loaded the pre-training weights on large-scale protein sequence databases and fine-tuning them by using the Adam optimizer with \({learning\_rate}=5\times {10}^{-4}\) on the annotated CPI dataset BindingDB_AIbind under four settings. The results are shown in Supplementary Table 16. ProtTransTurning achieved results comparable to ColdstartCPI on the warm start (S1) and compound cold start (S2) settings, but lower than ColdstartCPI on the protein cold (S1) and blind start (S4) settings. There are two reasons for this: firstly, the fine-tuning destroys the parameter distribution in the pre-trained weights, which may lose some of the generalized features; secondly, ProtTransTurning contains more trainable parameters than ColdstartCPI, which makes the model overfitted and reduces the model generalization performance.
Effectiveness of the induced-fit theory-inspired structure of ColdstartCPI
To verify the effectiveness of the model structure of ColdstartCPI designed based on the induced-fit theory, we select two classical rigid docking-inspired models, MolTrans and DrugBAN, and replace their feature extraction modules with the pre-trained feature matrices to construct two variant models, MolTrans_pretrain and DrugBAN_pretrain (Supplementary Figs. 8 and 9).
The pre-trained feature matrices of compounds and proteins are the same as ColdstartCPI. The results on the BindingDB_AIBind dataset under four scenarios are provided in Fig. 4b (Supplementary Table 17). We find that under the same feature extraction module, ColdstartCPI achieves the best results, indicating that the Decouple, Transformer, and Prediction modules of ColdstartCPI extract more effective features than the Biliner attention network and Decoder of DrugBAN, as well as the Interaction and Decoder modules of MolTrans. This result implies that CPI modeling based on induced-fit theory provides more flexible representations of compound-protein complexes compared with rigid docking.
To further explore that ColdstartCPI can capture the induced-fit phenomena, we used T-SNE for visualization and analysis on 231 proteins with more than 10 CPIs. For each protein, we paired it with the corresponding compounds and inputted them into ColdstartCPI to extract protein features.
Supplementary Fig. 10a presents a T-SNE visualization of the features, showing how protein features change according to the binding compounds in the inference process of ColdstartCPI. To provide more detail, Supplementary Fig. 10b focuses on the 10 proteins with the highest number of CPIs. It is evident from the visualization that different compounds lead to distinct feature expressions in all protein groups, except for proteins P11712 (cytochrome P450 2C9) and P33261 (cytochrome P450 2C19). These two proteins exhibit a high sequence similarity (91.72% similarity as determined by BLASTp), with 283 and 223 compounds interacting with P11712 and P33261, respectively, 157 of which overlap. This overlap leads to relatively close and even overlapping clusters for these two proteins, showing that when the same compound binds to highly similar proteins, their features are expressed in a similar manner.
C9 is the set of ligands for P11712 and C19 is the set of ligands for P33261. To further explore this, we selected 5 compounds each from three sets: the overlap of the ligands for P11712 and P33261 (C9 ∩ C19), the ligands exclusive to P11712 (C9 \ C19), and those exclusive to P33261 (C19 \ C9). We then calculated the Euclidean distance between the features of P11712 and P33261 after binding these ligands. The results, presented in Supplementary Fig. 10c, show that the features of P11712 and P33261 are more similar when both proteins bind the same ligands. This is consistent with the biological intuition that proteins within the same family tend to share similar three-dimensional structures, especially at the ligand-binding sites, and may interact with ligands in a similar manner.
We performed experiments from two perspectives, the compound and protein, respectively. We selected the protein Receptor tyrosine-protein kinase erbB-2 (ERBB2, Uniprot ID: P04626) and the drug Paclitaxel (DrugBank ID: DB01229) for our study, which are target protein and drug of breast cancer. We used ColdstartCPI to perform feature extraction on Paclitaxel paired with different proteins and ERBB2 paired with different compounds in the BindingDB_AIBind dataset. Supplementary Fig. 11 shows the feature visualization results of T-SNE, which shows the features of Paclitaxel change depending on the binding proteins and the features of ERBB2 change according to the binding compounds.
We further perform feature and structure correlation analysis on ERBB2. We downloaded the ERBB2 structure 3PP0 from the PDB database without binding to any ligand, as well as four ERBB2 resolved structures with ligands, 7JXH (bound to VOY), 3RCD (bound to 03 P), 8U8X (bound to W9N), and 7PCD (bound to 70I). Supplementary Fig. 12 illustrates their alignment structures at the binding sites, where red, blue, green, purple, and yellow bands indicate 3PP0, 7JXH, 3RCD, 8U8X, and 7PCD, respectively, and the feature visualization on TSNE. Supplementary Fig. 13 shows their specific differences from 3PP0.
As can be seen in Supplementary Fig. 12a, binding to different ligands leads to structural changes in ERBB2. T-SNE (Supplementary Fig. 12b) shows that on four resolved structures with different ligands, the features of ERBB2 generated by ColdstartCPI vary depending on the bound ligands, which captures the biological fact on ERBB2. These experiments demonstrate that ColdstartCPI treats proteins and compounds as flexible molecules in the process of inference, which captures induced-fit phenomena.
To explore the ability of models to adjust to protein features, we have chosen ML-DTI35, HyperAttentionDTI42, and iNGNN-DTI44 as representative cross-attention-based baselines for comparison. These models adjust the relative importance of amino acid residues in proteins based on compound features through cross-attention mechanisms.
To evaluate how different models adapt protein features in response to compound binding, we measure the change between apo (pre-interaction) and holo (post-interaction) protein representations using cosine similarity on the BioSNAP dataset, which includes 13,818 compound-protein interactions (CPIs). This metric provides a scale-invariant comparison that captures shifts in feature directionality rather than magnitude.
The results, shown in Supplementary Fig. 14a, illustrate the distribution of feature similarities. ML-DTI and HyperAttentionDTI have a mean similarity of 0.999. iNGNN-DTI has a mean similarity of 0.87 and a minimum similarity of 0.509. While, ColdstartCPI exhibits a mean similarity of 0.686, with a minimum similarity of 0.02. We conduct a Kruskal–Wallis H-test on the similarity distributions to statistically confirm these observations. The resulting p ≪ 0.001 confirms a highly significant difference in protein feature adaptation across the evaluated models. These findings demonstrate that ColdstartCPI more effectively differentiates between apo and holo protein representations than cross-attention-based models, supporting its stronger capacity to capture dynamic protein-compound interactions.
To further evaluate the ability of ColdstartCPI to capture dynamic protein-ligand interactions, we construct two benchmark datasets—RigidBind and FlexibleBind—based on the degree of conformational change upon ligand binding. Following descriptions from previous studies71,72,73, we identify three proteins—Apoptosis regulator Bcl-2 (Uniprot ID: P10415), Acetylcholinesterase (Uniprot ID: P22303), and HIV-1 protease (Uniprot ID: P03369)—whose ligand binding does not induce substantial conformational changes, consistent with the lock-and-key model. From the DrugBank database, we compiled 94 CPIs involving these proteins to form the RigidBind dataset (20 for Bcl-2, 65 for Acetylcholinesterase, and 9 for HIV-1 protease). In contrast, guided by works74,75,76,77, we have constructed the FlexibleBind dataset comprising 75 CPIs involving 50 proteins and 74 ligands, where binding is known to induce significant conformational rearrangements—consistent with the induced-fit model.
For each CPI in both datasets, we compute the cosine similarity between apo and holo protein features. The results are shown in Supplementary Fig. 14b. ML-DTI and HyperAttentionDTI exhibit mean cosine similarities of 0.999 across both datasets, with no significant difference. iNGNN-DTI shows moderate adaptation, with mean similarities of 0.902 (FlexibleBind) and 0.891 (RigidBind), and a Kruskal–Wallis H-test p-value of 0.523, indicating no significant difference. ColdstartCPI, however, demonstrates distinct behavior: a mean similarity of 0.653 on FlexibleBind and 0.678 on RigidBind, with a Kruskal–Wallis H-test p-value of 3.175 × 10⁻¹⁴, indicating a highly significant difference between the two settings.
These results highlight that ColdstartCPI is more responsive to binding-induced conformational changes, capturing significantly lower similarity scores (i.e., greater adaptation) for interactions governed by induced-fit mechanisms compared to lock-and-key interactions. Additionally, as shown in Supplementary Fig. 14c, ColdstartCPI demonstrates significantly greater adaptation of protein features on the FlexibleBind dataset compared to cross-attention-based models. Unlike iNGNN-DTI, which exhibits notable feature changes for only a subset of CPIs, ColdstartCPI consistently induces substantial feature changes across all CPIs in the FlexibleBind dataset—indicating a stable and systematic response to induced-fit interactions. Similarly, Supplementary Fig. 14d shows that ColdstartCPI produces more pronounced feature modifications than all baseline models across every CPI in the RigidBind dataset.
These results collectively illustrate that, in comparison to cross-attention-based models, the Transformer-based ColdstartCPI exhibits a markedly higher capacity for dynamically adjusting protein features in response to ligand context. This ability enables a broader and more expressive protein feature space during CPI prediction, contributing to its enhanced modeling flexibility and performance.
The effect of pre-trained feature combinations on ColdstartCPI performance
To assess the impact of different pre-trained feature combinations on the performance of ColdstartCPI, we incorporated two recent pre-trained models: ESM-249 for protein features and MolFormer78 for compound features. This allowed us to evaluate four different pre-trained model combinations: Mol2Vec_ESM-2, MolFormer_ESM-2, MolFormer_ProtTrans, and Mol2Vec_ProtTrans. The results of these four combinations on the BindingDB_AIBind dataset are shown in Fig. 4c (Supplementary Table 18).
Mol2Vec_ProtTrans achieves the best performance across all four scenarios. Although ESM-2 is the more recent Protein Large Language Model, ProtTrans performs much better on the CPI prediction task in our study. This may be due to ProtTrans’ strength in protein function prediction tasks. For example, Kabir et al.79 found that ProtTrans outperformed ESM-2 in remote homology prediction, while ESM-2 has an advantage in tasks requiring fine-grained information in specific sequences.
As for the compound models, Mol2Vec performed better than MolFormer, which might be explained by two factors: (1) MolFormer uses SMILES sequences as input but does not directly encode molecular topology, limiting its ability to capture certain structural features. (2) Mol2Vec directly encodes substructural features, which are crucial for capturing chemical properties that govern interactions with proteins, making it more suitable for downstream CPI prediction tasks. In addition, the combination of Mol2Vec’s compound features with ProtTrans’ protein features may have produced some synergistic effect, better capturing the complexity of compound-protein interactions and improving the predictive accuracy of the model.
Validation of ColdstartCPI predictions on Alzheimer’s Disease, breast cancer, and COVID-19
Using ColdstartCPI for drug candidate identification for target proteins and drug repurposing is promising due to its ability to recognize CPI patterns during model training, leveraging pre-trained biochemical knowledge extraction. To further confirm ColdstartCPI’s reliability, targeting diseases with high global prevalence and importance to public health, such as Alzheimer’s Disease (AD), breast cancer, and COVID-19, strengthens the significance of ColdstartCPI’s applications.
By validating predictions against experimental data, molecular docking simulations, and binding free energy calculations in these disease contexts, the reliability and effectiveness of ColdstartCPI can be better assessed, potentially leading to valuable insights for drug discovery and repurposing efforts. We selected target proteins and drugs from the Therapeutic Target Database. We chose the BindingDB_AIBind as the training set and removed the CPIs related to the target proteins and drugs. As for drug candidate identification of target proteins, we retrieved the amino acid sequences in FASTA format from the UniProt database. We tested the CPIs between the target proteins and compound candidate set, including a total of 2,743,637 small molecule compounds (see Subsection Construction of the compound candidate set and protein candidate set in Section Methods).
We ranked them by their prediction probabilities and evaluated the predicted top 100 interactions with a literature search. We also conducted docking simulations on the predicted top 100 and bottom 100 binding interactions with pocket docking (on the known binding sites) using AutoDock Vina6 and Ledock80. To cope with the challenge of unknown binding sites and missed candidates, we also performed a blind docking procedure based on AutoDock Vina6.
As for drug repurposing, we retrieved the SMILES strings from DrugBank. Drug repositioning in this section means that the target drugs are FDA-approved drugs and does not imply the presence of relevant CPIs in the training set. We tested the CPIs between the target drugs and the protein candidate set extracted from DrugBank (see Subsection Construction of the compound candidate set and protein candidate set in Section Methods). We also ranked them by their prediction probabilities and evaluate the predicted top 50 and bottom 50 interactions with literature search and docking simulations using AutoDock Vina6.
All the 3D structures of proteins are from PDB or AlphaFold DB (if not retrieved in PDB), meanwhile, all the 3D structures of drugs/ligands/compounds are from PubChem or ChemSpider (if not retrieved in the PubChem database). The database identifiers, reported experimental evidence, binding free energies, and docking affinities of candidate proteins and compounds are provided in Supplementary Tables 19–28.
Validation on Alzheimer’s Disease
As for drug candidate identification, we selected Acetylcholinesterase (UniProt ID: P22303, PDB ID: 6O4W) as our target protein for AD. We find that 49 out of the top 100 predictions from ColdstartCPI are supported by the works of literature and records (Supplementary Table 19), including three FDA-approved drugs DB04616, DB00645 (with an IC50 value of 181 nM)81, and DB00572 (with a Ki value of 0.35 nM)82.
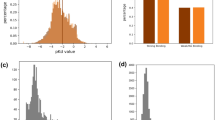
The docking results of Autoduck Vina6 for the top 100 and bottom 100 predictions made by ColdstartCPI are presented in Fig. 5. As shown in Fig. 5a, the mean pocket docking scores for all top 100 candidates, 49 experiment-validated candidates, and the remaining 51 candidates are −6.493 kcal/mol, −6.548 kcal/mol, and −6.445 kcal/mol, respectively. The mean pocket docking scores for all bottom 100 candidates is −4.341 kcal/mol. The results confirm that the top predictions of ColdstartCPI have a significantly higher propensity to bind than the bottom ones (Kruskal–Wallis H-test p-value of 6.7*10−31). To further validate the 51 non-literature-supported candidates, we employed binding free energy (BEF) calculations by Discovery Studio’s (https://www.3ds.com/products/biovia/discovery-studio) Calculate Binding Energies protocol, which is based on Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA)83,84 on the pocket docking results. As shown in Supplementary Table 20, all 51 candidates have reasonable binding free energies, 31 of which are better than the reference ligand. Furthermore, we employed Absolute Binding Affinity Free Energy Perturbation (ABFEP) for more accurate BEF calculations. Given the high computational cost of ABFEP, we focused on the top 5 candidate molecules (without experimental validation) and calculated their absolute BEFs with target proteins. For reference, we also calculated the absolute BEF for the known ligand. We used an automated ABFEP workflow (ABFE_workflow85) based on GROMACS 2022.2 to compute the absolute BEFs. The results are shown in Supplementary Table 21. For Acetylcholinesterase, the calculated ΔGFEP-ABFE values for CNP0235434, DB07618, and CHEMBL5202565 were −6.87, −7.55, and −6.75, respectively, better than the −5.24 for the known ligand E20 (dissociation constant of 8 nM).

a The distribution of pocket docking affinities of the top 100 compound candidates with Acetylcholinesterase (number of data points n = 100, 49, 51, 100 in each group; center line, median; box limits, upper and lower quartiles; whiskers, maximum and minimum values; white circles, mean values; dots, outliers). b The docking pose and non-covalent interactions of CNP0450629 with Acetylcholinesterase (UniProt ID: P22303, PDB ID: 6O4W). c The distribution of blind docking affinities of the top 100 compound candidates with Acetylcholinesterase (number of data points n = 100, 49, 51, 100 in each group). d The docking pose and non-covalent interactions of CNP0349721 with Acetylcholinesterase. e The distribution of docking affinities of the top 50 protein candidates with Donepezil (number of data points n = 50, 45, 5, 50 in each group). f The docking pose and non-covalent interactions of Donepezil (DrugBank ID: DB00843) with Alpha-2A adrenergic receptor (ADRA2A, UniProt ID: P08913, PDB ID: 6KUX). In b, d, and f, the legends show the types of protein-ligand interactions, which have been introduced in detail in Supplementary Note 4. Source data are provided as a Source Data file.
Among the 51 non-validated candidates, CNP0450629 and CNP0349721 from the COCONUT database achieve the best pocket and blind docking results with docking scores of −7.419 kcal/mol and −8.863 kcal/mol, respectively. As shown in Figs. 5b and 5d, there are 8 non-covalent interactions between CNP0450629 and Acetylcholinesterase and 5 of them are hydrogen bonds, 7 non-covalent interactions between CNP0349721 and Acetylcholinesterase, 3 of which are hydrogen bonds, indicating that CNP0450629 and CNP0349721 bind well to Acetylcholinesterase. The definition of non-covalent interactions is provided in Supplementary Note 4.
To verify the stability of the docking poses, we performed molecular dynamics simulations on them by Discovery Studio. The Root Mean Square Deviation (RMSD) imparts knowledge of the stability of the protein backbone. CNP0450629 and CNP0349721 project RMSDs of 1.43 nm and 0.98 nm (shown in Supplementary Fig. 15), respectively, demonstrating that the systems are largely stable throughout the simulation run (100 conformations), without major variations. Furthermore, compared with the reference ligand DB07701 recorded in 6O4W, CNP0450629 and CNP0349721 have shown close stability. CNP0349721 has the least RMSD average value, which illustrates better backbone stability than the reference (as shown in Supplementary Fig. 16).
As for drug repurposing, we selected Donepezil (DrugBank ID: DB00843) as our target drug, which is an acetylcholinesterase inhibitor used to treat the behavioral and cognitive effects of Alzheimer’s Disease. We find that 5 out of the top 50 predicted proteins from ColdstartCPI are indeed experiment-validated interactions (Supplementary Table 22), including two DrugBank-documented target proteins, Acetylcholinesterase (UniProt ID: P22303) and Cholinesterase (UniProt ID: P06276). Furthermore, Histamine H3 receptor (UniProt ID: Q9Y5N1), Sigma non-opioid intracellular receptor 1 (UniProt ID: Q99720) and Potassium voltage-gated channel subfamily H member 2 (UniProt ID: Q12809) get an IC50 value of 350, 14.6, 640 nM, respectively86,87,88.
The distribution of binding affinities in the docking simulations for the top 50 predicted proteins made by ColdstartCPI is presented in Fig. 5e (details are provided in Supplementary Table 22). The mean binding affinities for all candidates, 5 experiment-validated target proteins, and the remaining 45 candidate proteins are −6.805 kcal/mol, −6.357 kcal/mol, and −6.856 kcal/mol, respectively. Among the 45 non-validated candidate proteins, Alpha-2A adrenergic receptor (ADRA2A, UniProt ID: P08913, PDB ID: 6KUX) achieves the best docking pose with a binding affinity of −8.487 kcal/mol. As shown in Fig. 5f, there are 6 non-covalent interactions between Donepezil and ADRA2A and 3 of them are hydrogen bonds. The complex system consisting of Donepezil and ADRA2A has an RMSD value of 1.81 nm determining its stability.
Validation on breast cancer
We selected ERBB2 (UniProt ID: P04626, PDB ID: 3RCD) as our target proteins for breast cancer. As for the evaluation of ERBB2 in breast cancer, 75 out of the top 100 predictions derived by ColdstartCPI are reported to have a high compound-protein affinity with ERBB2 (Supplementary Table 23).
The docking results of Autoduck Vina for the top 100 and bottom 100 predictions made by ColdstartCPI are presented in Fig. 6. On the pocket docking (shown in Fig. 6a), 2 of 25 non-validation predictions cannot be reported due to a procedural error. The mean binding affinities for the top 100 candidates are −6.628 kcal/mol, which is better than −5.603 kcal/mol of the bottom 100 candidates (Kruskal–Wallis H-test p-value of 3.4*10−19). After pocket docking, we used MM-PBSA to calculate BEFs for the 23 non-validated candidates and 5 of them could not be obtained results due to a procedural error. As shown in Supplementary Table 24, 18 candidates had reasonable binding free. Among them, 6 candidates have better binding free energies than the reference ligand. As for the top five candidate molecules on which there is no experimental validation, the ΔGFEP-ABFE values generated by ABFE_workflow for CNP0443196, CNP0266780, and CNP0294175 were −7.24, −6.45, and −8.31, outperforming the known ligand 03 P’s −6.38 (IC50 of 17 nM).

a The distribution of pocket docking affinities of the top 100 compound candidates with Receptor tyrosine-protein kinase erbB-2 (number of data points n = 98, 23, 75, 98 in each group; center line, median; box limits, upper and lower quartiles; whiskers, maximum and minimum values; white circles, mean values; dots, outliers). b The docking pose and non-covalent interactions of DB00878 with ERBB2 (UniProt ID: P04626, PDB ID: 3RCD). c The distribution of blind docking affinities of the top 100 compound candidates with ERBB2 (number of data points n = 98, 23, 75, 98 in each group). d The docking pose and non-covalent interactions of CNP0266780 with ERBB2. e The distribution of docking affinities of the top 50 protein candidates with Paclitaxel (number of data points n = 50, 39, 11, 50 in each group). f The docking pose and non-covalent interactions of Paclitaxel (DrugBank ID: DB01229) with Afamin (UniProt ID: P43652, PDB ID: 5OKL). In b, d, and f, the legends show the types of protein-ligand interactions, which have been introduced in detail in Supplementary Note 4. Source data are provided as a Source Data file.
Among the 23 non-validated candidates, DB00878 from the DrugBank database yields the best docking result with a binding affinity of −7.478 kcal/mol. As shown in Fig. 6b, DB00878 has 10 non-covalent interactions with ERBB2, including 5 hydrogen bonds, which indicates that DB00878 binds well to the pocket of ERBB2.
As for the blind docking, the mean binding affinities for the top 100 candidates, 75 experiment-validated candidates, and the remaining 25 candidates are −8.575 kcal/mol, −8.835 kcal/mol, and −7.688 kcal/mol, respectively. Among the 75 experiment-validated candidates, CHEMBL1630112 and CHEMBL233325 from the ChEMBL database yield docking affinity scores below −10 kcal/mol. Among them, CHEMBL1630112 gets the best docking result with a binding affinity of −10.594 kcal/mol, which was shown to have a strong inhibitory effect on ERBB2 with an IC50 value of 4.9 nM89. Among the 25 non-validated candidates, CNP0266780 from the COCONUT database yields the best docking result with a binding affinity of −10.33 kcal/mol. As shown in Fig. 6d, there are 11 non-covalent interactions between CNP0266780 and ERBB2 and 2 of them are hydrogen bonds. Molecular dynamics simulation experiments show that DB00878 and CNP0266780 bind to ERBB2 with good backbone stability (shown in Supplementary Fig. 17).
As for drug repurposing, we selected Paclitaxel (DrugBank ID: DB01229) as our target drug, which is frequently used as the first-line treatment drug for breast cancer. We find that 11 out of the top 50 predicted proteins from ColdstartCPI are indeed experiment-validated interactions (Supplementary Table 25), including six DrugBank-documented target proteins, Nuclear receptor subfamily 1 group I member 2 (Uniprot ID: O75469), Tubulin beta-1 chain (Uniprot ID: Q9H4B7), Apoptosis regulator Bcl-2 (Uniprot ID: P10415), Microtubule-associated protein 4 (UniProt ID: P27816), Microtubule-associated protein 2 (UniProt ID: Q11137), and Microtubule-associated protein tau (UniProt ID: P10636). The distribution of docking affinities for the top 50 predicted proteins made by ColdstartCPI is presented in Fig. 6e (details are provided in Supplementary Table 25). The mean docking affinities for all candidates, 11 experiment-validated target proteins, and the remaining 39 candidate proteins are −6.954 kcal/mol, −7.109 kcal/mol, and −6.910 kcal/mol, respectively. Among the 39 non-validated candidate proteins, Afamin (UniProt ID: P43652, PDB ID: 5OKL) demonstrates the best docking with a binding affinity of −8.563 kcal/mol. As shown in Fig. 6f, there are 7 non-covalent interactions between Paclitaxel and Afamin and 3 of them are hydrogen bonds. The docking and dynamic simulation results indicate that Paclitaxel binds stably to Afamin (shown in Supplementary Fig. 17) and may help reduce insulin resistance and the risk of developing type 2 diabetes mellitus.
Validation on COVID-19
To test the model’s ability to respond quickly to public health emergencies, we also carried out a case study on COVID-19. In this regard, Replicase polyprotein 1ab (UniProt ID: P0DTD1, PDB ID: 5RMH), called R1AB here, is an attractive drug target as it plays a central role in viral replication by processing the viral polyproteins pp1a and pp1ab at several different cleavage sites90. In this case, we find that 23 out of the top 100 candidates predicted by ColdstartCPI are reported to bind with R1AB (Supplementary Table 26), including two FDA-approved drugs, Tideglusib (DrugBank ID: DB12129) and Ebselen (DrugBank ID: DB12610). Experimental evidence based on compound repurposing strategy69 reconfirms the activity of Tideglusib and Ebselen, which give IC50 values between 20 and 220 nM. The docking results of Autoduck Vina6 for the top 100 and bottom 100 predictions made by ColdstartCPI are presented in Fig. 7. As shown in Fig. 7a, the mean pocket docking scores for all top 100 candidates, 23 experiment-validated candidates, and the remaining 67 candidates are −6.385 kcal/mol, −7.064 kcal/mol, and −6.172 kcal/mol, respectively. The mean pocket docking scores for all bottom 100 candidates is −4.041 kcal/mol. The results confirm that the top predictions of ColdstartCPI have a significantly higher propensity to bind than the bottom ones (Kruskal–Wallis H-test p-value of 6.8*10−19). Among the 67 non-validated candidates, CNP0111583 from the COCONUT database achieves the best docking result with a binding affinity of −8.727 kcal/mol. As shown in Fig. 7b, the complex of R1AB with CNP0111583 has a stable backbone, which includes 8 non-covalent interactions and 4 of them are hydrogen bonds. These results indicate that CNP0111583 is a promising SARS-CoV-2 main protease targeting ligand. As for the top five candidate molecules on which there is no experimental validation, as shown in Supplementary Table 21, CNP0235766, CNP0286507, and CNP0129129 showed ΔGFEP-ABFE values of −8.98, −17.68, and −7.45, respectively, which are superior to the known ligand VX4 at −4.29.

a The distribution of pocket docking affinities of the top 100 compound candidates with R1AB (number of data points n = 100, 67, 23, 100 in each group; center line, median; box limits, upper and lower quartiles; whiskers, maximum and minimum values; white circles, mean values dots, outliers). b The docking result and non-covalent interactions of CNP0111583 with R1AB (UniProt ID: P0DTD1, PDB ID: 5RMH). c The distribution of blind docking affinities of the top 100 compound candidates with R1AB (number of data points n = 100, 67, 23, 100 in each group). d The docking pose and non-covalent interactions of CNP0290973 with R1AB. e The distribution of docking affinities of the top 50 protein candidates with Baricitinib (number of data points n = 50, 43, 7, 50 in each group). f The docking pose and non-covalent interactions of Baricitinib (DrugBank ID: DB11817) with cAMP-dependent protein kinase inhibitor alpha (PKIA, UniProt ID: P61925, PDB ID: 1CMK). In b, d, and f, the legends show the types of protein-ligand interactions, which have been introduced in detail in Supplementary Note 4. Source data are provided as a Source Data file.
As for the blind docking (Fig. 7c), the mean binding affinities for the top 100 candidates, 23 experiment-validated candidates, and the remaining 67 candidates are −6.172 kcal/mol, −6.598 kcal/mol, and −6.038 kcal/mol, respectively. Among the non-validated candidates, CNP0290973 from the COCONUT database yields the best docking result with a binding affinity of −9.259 kcal/mol. As shown in Fig. 7d, there are 11 non-covalent interactions between CNP0290973 and Replicase polyprotein 1ab and 5 of them are hydrogen bonds. Molecular dynamics simulation experiments show that CNP0111583 and CNP0290973 bind to Replicase polyprotein 1ab with good backbone stability (shown in Supplementary Fig. 18).
As for drug repurposing, we selected Baricitinib (DrugBank ID: DB11817) as our target drug, which controls SARS-CoV-2-induced Cytokine Storm in humans to reduce mortality in Critically Ill patients. We find that 7 out of the top 50 predicted proteins from ColdstartCPI are indeed experiment validated interactions (Supplementary Table 27), including four DrugBank-documented target proteins, Tyrosine-protein kinase JAK1 (Uniprot ID: P23458), Tyrosine-protein kinase JAK2 (Uniprot ID: O60674), Tyrosine-protein kinase JAK3 (Uniprot ID: P52333), and Non-receptor tyrosine-protein kinase TYK2 (Uniprot ID: P29597). The distribution of binding affinities in the docking simulations for the top 50 predicted proteins made by ColdstartCPI is presented in Fig. 7e (details are provided in Supplementary Table 27).
The mean binding affinities for all candidates, 7 experiment-validated target proteins, and the remaining 43 candidate proteins are −5.784 kcal/mol, −5.661 kcal/mol, and −5.804 kcal/mol, respectively. Of the 43 non-validated candidate proteins, 28 have resolved structures. Among them, cAMP-dependent protein kinase inhibitor alpha (PKIA, UniProt ID: P61925, PDB ID: 1CMK) achieves the best docking pose with an affinity of −6.886 kcal/mol. As shown in Fig. 7f, there are 3 non-covalent interactions between Baricitinib and PKIA, and 2 of them are hydrogen bonds. The docking and dynamic simulation results indicate that Baricitinib binds stably to PKIA (shown in Supplementary Fig. 18) and has the potential to treat tumor, cardiovascular, and metabolic diseases by modulating cAMP/PKA signaling.
As shown in Supplementary Table 28 and Supplementary Fig. 19, the docking experiments on the predicted top candidates for AD, breast cancer, and COVID-19 yield positive results compared to the bottom ones, and the non-validated candidates achieve similar docking results to the validated candidates. Molecular dynamics simulation experiments point out that predicted complexes with the best docking poses have good protein backbone stability (Supplementary Figs. 15-18, 20). These ColdstartCPI-predicted CPIs without current literature support hold promise for further study through biological experiments. Furthermore, the CPIs associated with the target proteins/drugs (e.g., P04626 and DB01229 for Breast cancer) in the BindingDB_AIBind dataset and the DrugBank database are considered as the ground truth. The numbers of hits in the top 100/50 candidates predicted by ColdstartCPI are shown in Supplementary Table 29. Specifically, for Breast cancer, there are 9 drugs recorded in BindingDB_AIBind and DrugBank that interact with the target protein P04626, 4 of which are predicted in the top 100. The details of ground truth are provided in Supplementary Table 30. In summary, the above results validate the accuracy of the predictions of ColdstartCPI and further demonstrate that it is an excellent tool to help the community accelerate drug discovery progress.
Discussion
The CPI prediction based on deep learning is a promising direction for rational drug discovery. In this study, we developed a two-step framework, named ColdstartCPI, to leverage unsupervised pre-training feature extraction for compounds and proteins to predict CPIs. ColdstartCPI extracts the feature matrices of compounds and proteins by Mol2Vec and ProtTrans, respectively, and then models the intra- and inter-molecular interactions of compounds and proteins by a Transformer-based module to yield accurate and robust prediction of CPIs. The powerful predictive ability of ColdstartCPI has been extensively validated on three benchmark datasets and compared with a total of ten state-of-the-art baseline models under four realistic evaluation settings, especially for scenarios of the cold start.
ColdstartCPI is not sensitive to hyperparameter variations, and all experiments are done with the same set of hyperparameter settings, which provides better robustness than other deep learning models. Furthermore, with the support of pre-trained feature extractors, our model can still achieve good prediction accuracy with limited training data. More importantly, unlike previous methods47,48,91,92, ColdstartCPI is a flexible framework that relies solely only on the SMILES strings of the compounds and the amino acid sequences of the proteins to complete the CPI prediction, thus simplifying data collection and processing and providing the ability to perform early-stage computer-aided drug design in the complete absence of a 3D protein structure.
Moreover, compared with AI-Bind50, which is also based on pre-trained features, we take the feature matrices as input and introduce the Transformer structure to better mine the CPI-related information hidden in the pre-training text library. ColdstartCPI is shown to be a successful pipeline for CPI prediction by literature search, docking simulations, binding free energy calculations, and molecular dynamics simulations. The case studies demonstrate that the candidates predicted by ColdstartCPI bind well to target proteins. Overall, ColdstartCPI is a stable and highly competitive CPI prediction method that promises to be a rapid drug screening method for complex diseases. The idea of introducing pre-trained models in CPI prediction has also been validated on other end-to-end models, which can be extended to other bio-interaction prediction problems, such as the prediction of drug-disease, compound-compound, and protein-protein interaction predictions to accelerate the drug discovery progress.
In ColdstartCPI, we do not incorporate any binding pocket data into model training because it is sparse. The binding pockets or exact binding sites among proteins are important for CPI prediction, which will greatly reduce the difficulty of constructing protein features and help accurately analyze the prediction results. To improve the interpretability of the model, further upgrades to the Decoupling module will be made. By collecting protein binding sites from the PDB database and incorporating them into the training process, multi-objective optimized predictive models will be constructed to improve interpretability.
Constructing a multitasking framework93 in combination with downstream tasks such as non-covalent bonding interaction prediction between amino acids and the atoms of small molecules, molecular property prediction94 can further help us reduce false positives. Furthermore, ColdstartCPI is a highly scalable framework that can further improve predictive performance by integrating pre-trained models of other modalities on compounds (i.e., the 2D graph structure95 and the 3D geometry96) and proteins (i.e., the 3D structure19,97,98). Even feature representation engineering based on contrastive learning99 and large language models49,78 can be incorporated into our framework. However, one challenge is how to effectively align and fuse multimodal data100,101,102. In addition, due to the differences between compound and protein corpora, the fitness between compound and protein pre-training features is also an issue worth investigating.
While we employed multiple validation strategies in our case study, the main limitation is the lack of in vitro or in vivo experimental validation. Computational methods such as ColdstartCPI and virtual docking tools like Vina rely on simulation to predict compound-protein interactions. However, these computational tools may not fully capture the complexity of biological interactions in a physiological setting. Wet experiments are critical to validate ColdstartCPI’s predictions in a laboratory setting, as experimental assays are the gold standard for confirming binding affinity, interaction strength, and therapeutic efficacy.
Moreover, while ColdstartCPI treats compounds and proteins as flexible entities and therefore outperforms many computational approaches, methods that resolve protein structures (e.g., X-ray crystallography or nuclear magnetic resonance) can capture details of interactions that are sometimes missed by computational predictions. This represents another limitation of ColdstartCPI. Incorporating physicochemical constraints within structural data could yield further insights, particularly for complex binding mechanisms that are ignored by existing computational models. Combining dynamic feature extraction of ColdstartCPI with interaction patterns in structures may be a valuable strategy.
In the future, we will pay more attention to pre-training-based feature extraction in our framework for further improvements in the prediction ability for CPIs. In addition, modeling of CPI prediction based on the induced-fit theory or introduction of other biological theoretical models will further facilitate the process of drug discovery and development.
Methods
Benchmark datasets
Supplementary Table 31 provides statistics for all the datasets in this study. We evaluated ColdstartCPI alongside state-of-the-art baselines using three publicly available CPI datasets: BindingDB_AIBind50, BioSNAP, and BindingDB. The BindingDB_AIBind dataset is from the BindingDB database. Given that the topology of the compound-protein interaction network drives the prediction task in the BindingDB dataset, the BindingDB_AIBind dataset is used for CPI prediction evaluation50. In the BindingDB_AIBind dataset, the compound-protein pairs, which are seven hops apart, are randomly selected as negatives to create an overall class balance between positive and negative samples in the training data, which is to restrict the models from exploiting topological shortcuts to generate CPI predictions. It contains 6788 compounds, 4472 proteins, and 50,312 CPIs, making it the largest dataset in this study.
The BioSNAP dataset, derived from the DrugBank database, maintains a balance between validated positive interactions and an equal number of negative samples sourced from unseen compound-protein pairs. It includes 4505 compounds, 2181 proteins, and 27,438 CPIs.
The BindingDB dataset is generated by the bias-reducing preprocessing steps to improve drug-wise pair class balance and reduce hidden ligand bias. It consists of 14,643 compounds, 2623 proteins, and 49,199 CPIs.
The BioSNAP_CM dataset is from the BioSNAP dataset by removing the proteins without contact maps and related CPIs.
The BindingDB_AIBind and BindingDB_AIBind2 datasets are generated by AI-Bind’s Network Negative Generation method according to positive CPIs of the BioSNAP and BindingDB datasets, respectively. The number of negative CPIs generated based on the network is equal to the number of positive CPIs.
The BindingDB_AF dataset is from the BindingDB database, of which CPIs were released between 2023 and May 2024 to ensure that there was no overlap with the training set of AlphaFold3 and the BindingDB_AIBind dataset. The compounds of BindingDB_AF belong to the 19 ligands specified in the AlphaFold3 Serve (https://alphafoldserver.com/).
BindingDB_SPECTRA is a subset of the BindingDB_AIBind dataset that has positive and negative samples containing all compounds and proteins, respectively, while reducing the number of times they appear to better reduce the similarity between the training set and the test set.
PDBbind67 is a standardized database for docking and binding affinity prediction. It consists of experimentally measured structures of compound-protein complexes and their binding affinity labels, from which true positive protein-molecule pairs with accurate structures can be extracted. We used PDBbind 2019 which contains binding affinity data for over 16,000 compound-protein complexes covering a wide range of chemical spaces and protein families. We consider all compound-protein complexes in PDBbind as positive samples and randomly disrupt compound-protein combinations to generate five times as many negative samples as positive samples to simulate the real CPI distribution.
The DUD-E62 dataset is a crucial dataset for benchmarking virtual screening protocols. DUD-E contains 102 target proteins across 8 protein families. Each target has 224 actives and over 10,000 decoys, on average. The decoys are chosen such that they are physically similar but topologically dissimilar to the actives. The final dataset contains 22,645 positive examples and 1,407,145 negative examples.
LIT-PCBA63 is claimed as an unbiased dataset designed for benchmarking ML and VS, where the bioactivities of both the active and inactive compounds are verified by experimental results based on dose-response PubChem bioassays. The full set contains a total of 15 targets, 10,030 true actives, and 2,798,737 true inactives. For each target, the ratio of actives to inactives is around 1: 1000, and this high imbalance could better mimic the challenging scenarios in real-world applications.
Antibiotics benchmark21 screened 218 compounds for enzyme inhibition against a panel of 12 essential E. coli proteins or protein complexes, performing duplicate assays at a concentration of 100 μM. Ground truth values were derived by binarizing the relative enzyme activity data (1 if the relative enzyme activity in both biological replicates was less than 0.5, 0 otherwise), resulting in 415 positive samples.
The data generation pseudocode is provided in Supplementary Note 3.
ColdstartCPI architecture
Overview
The overall framework of the ColdstartCPI is shown in Fig. 1, which is divided into four modules, namely the Pre-trained module, the Decouple module, the Transformer module, and the Predict module. Unlike the end-to-end method, ColdstartCPI is a non-end-to-end method that separates compound and protein feature extraction from CPI prediction. Taking the SMILES of the compound and the amino acid sequence of the protein as input, ColdstartCPI first extracts the compound substructure feature matrix and protein amino acid feature matrix using the pre-trained models, Mol2Vec and ProtTrans, respectively (Pre-trained module). After mapping the feature matrices to the feature space of the same dimension using the fully connected networks (Decouple module), the Transformer model is used to extract the protein global feature vector sensing compound features and the compound global feature vector sensing protein features (Transformer module). Finally, the prediction of CPI is done using a fully connected network (Prediction module).
Pre-trained module
Considering the influence of functional groups on drug-receptor binding and potency, and the fact that the one-dimensional sequence of proteins determines the three-dimensional structure and the function, ColdstartCPI uses the pre-trained Mol2Vec and ProtTrans models for feature matrix extractors to introduce semantic features of drug substructures and high-level features related to protein structures and functions. These models create 300- and 1024-dimensional embeddings for each substructure in a compound and each amino acid in a protein, respectively. Specifically, taking the SMILES of the compound as input, we used RdKit to identify the compound molecule and extract the substructure identifiers, which have a radius of 1. Given that it has \({n}_{c}\) identifiers, the atom representation matrix \({M}_{c}\in {{\mathbb{R}}}^{{n}_{c}\times 300}\) is generated by Mol2Vec. For an amino acid sequence of length \({n}_{p}\), the amino acid representation matrix \({M}_{p}\in {{\mathbb{R}}}^{{n}_{p}\times 1024}\) is generated by ProtTrans. \({M}_{c}\) and \({M}_{p}\) are fine-grained features of the compound and the protein. ColdstartCPI uses mean-pooling to transform \({M}_{c}\) and \({M}_{p}\) into the compound representation vector \({f}_{c}\in {{\mathbb{R}}}^{1\times 300}\) and the protein representation vector \({f}_{p}\in {{\mathbb{R}}}^{1\times 1024}\), respectively, which are global features of the compound and the protein.
Decouple module
The Decouple module is composed of four fully connected networks with \({M}_{c},\,{M}_{p},{f}_{c}\), and \({f}_{p}\) as inputs. Considering that different pre-trained methods yield inconsistent feature encoding dimensions, i.e., the compound-related encoding dimension is 300 and the proteins-related encoding dimension is 1024, the Decouple module serves to unify the feature dimensions of compounds and proteins, making them easy to be used by the downstream Transformer module. Furthermore, it can decouple the pre-trained feature space from the feature space of CPI prediction to improve the performance of the model. Given a uniform feature dimension \(d\), we can get the new features of compounds and proteins, \({M}_{c}\in {{\mathbb{R}}}^{{n}_{c}\times d}\), \({M}_{p}{{\mathbb{\in }}{\mathbb{R}}}^{{n}_{p}\times d}\), \({f}_{c}{{\mathbb{\in }}{\mathbb{R}}}^{1\times d}\), and \({f}_{p}{{\mathbb{\in }}{\mathbb{R}}}^{1\times d}\), through four fully connected networks. We concatenate \({f}_{c}\), \({M}_{c}\), \({f}_{p}\), and \({M}_{p}\) to form a compound-protein pair feature matrix \({M}_{{cp}}=[{f}_{c},{M}_{c},{f}_{p},{M}_{p}]\in {{\mathbb{R}}}^{(1+{n}_{c}+1+{n}_{p})\times d}\).
Transformer module
ColdstartCPI inputs the compound-protein pair feature matrix \({M}_{{cp}}\) into the Transformer module and uses the self-attention mechanism to achieve the interaction modeling of compound substructures and amino acid fragments. Given the hidden dimension \(h\) of the feed-forward layer in the Transformer module, taking \({M}_{{cp}}\) as input, the Transformer module first calculates \({M}_{{cp}\,}^{Q}\), \({M}_{{cp}\,}^{K}\), and \({M}_{{cp}\,}^{V}\in {{\mathbb{R}}}^{(1+{n}_{c}+1+{n}_{p})\times h}\) matrices as follows:
where \({W}_{Q},\,{W}_{K}\), and \({W}_{V}\in {{\mathbb{R}}}^{d\times h}\) are linear projection matrices. Then the Transformer module does a self-attention operation on \({M}_{{cp}\,}^{Q}\), \({M}_{{cp}\,}^{K}\), and \({M}_{{cp}\,}^{V}\) matrices, it is formulated as:
where \(A\in {{\mathbb{R}}}^{(1+{n}_{c}+1+{n}_{p})\times (1+{n}_{c}+1+{n}_{p})}\) is the attention matrix. \({A}^{i}\) is the vector of the i-th row, as shown in Fig. 8a, which represents the attention scores of the other items in \({M}_{{cp}\,}^{V}\) for the i-th item. As shown in Fig. 8b, the compound feature \({M}_{1}^{A}\) introduce protein and amino acid features, i.e., the inter-molecular interactions with the protein, in addition to containing their atomic features, i.e., the intra-molecular interactions of the compound. This progress allows compounds to form complexes with different proteins to produce different molecular representations, thus improving the model’s ability to express features. Such a process of changing the compound feature from \({f}_{c}\) to \({M}_{1}^{A}\) considers the influence of protein features on it. Similarly, as shown in Fig. 8c, protein features \({M}_{2+{n}_{c}}^{A}\) incorporates the effect of compound features. The extraction of \({M}_{1}^{A}\) and \({M}_{2+{n}_{c}}^{A}\) responds to the change from apo conformation to holo conformation, which is inspired by the induced-fit theory. After the self-attention operation, the finial compound-protein complex features are generated by residual connection, layer normalization, and feed-forward operations as follows:
where \({M}^{A}\in {{\mathbb{R}}}^{(1+{n}_{c}+1+{n}_{p})\times h}\). \({M}_{1}^{A}\in {{\mathbb{R}}}^{1\times h}\) and \({M}_{2+{n}_{c}}^{A}\in {{\mathbb{R}}}^{1\times h}\) are the molecular and protein global feature vectors, respectively, which are concatenated and fed into the Prediction module.

a The attention matrix. b The compound features. c The protein features.
Prediction module
The molecular global feature vector\(\,{M}_{1}^{A}\) with the protein global feature vector \({M}_{2+{n}_{c}}^{A}\) are concatenated and fed into the MLP network for CPI prediction. The last layer of MLP outputs the probability \(\hat{y}\) indicating the likelihood of the compound-protein interaction. As a binary classification task, the training objective is to minimize the binary cross-entropy loss as follows:
where N is the number of samples in the training set, \({\hat{y}}_{i}\) and \({y}_{i}\) represent the predictive score and true label of the ith sample, respectively.
Baselines
In this study, we assessed the efficacy of our ColdstartCPI method relative to state-of-the-art baselines for compound-protein interaction (CPI) prediction. We categorized these baselines into four distinct groups based on their technical approaches.
Feature-based methods: These include DNN28 and DeepConv-DTI29. These methods utilize descriptions of compounds and proteins, incorporating chemical expert knowledge as input to model interactions.
End-to-end methods: Examples are GraphDTA36, ML-DTI35, MolTrans39, HyperAttentionDTI42, and DrugBAN48. These methods process the raw representations of compounds (e.g., SMILES and graph structures) and proteins (e.g., amino acid sequences) directly to extract relevant features, driven by the underlying data.
Domain adaptation-based method: DrugBAN_CDAN48 employs conditional domain adversarial learning to adjust the interaction representations for novel compound-protein pairs from different distributions.
Pre-trained-based methods: iNGNN-DTI44 and AI-Bind50 utilize pre-training techniques to derive features for compounds and proteins.
Docking-based models: Glide-SP5, Vina6, Smina7, and Surflex8 predict ligand binding modes and affinities by simulating molecular interactions within protein binding sites. These models use scoring functions that assess fit and energetics, making them widely used tools for virtual screening in structure-based drug discovery.
ML scoring function: NN-score10 and RFscore11 apply statistical models to predict binding affinities by learning patterns from existing ligand-receptor data. NN-Score uses neural networks, while RF-Score relies on RF models, both enhancing prediction accuracy in virtual screening.
Structure-based models: Pafnucy12, OnionNet13, Gnina14, BigBind15, PLANET16, and ConBAP17 utilize 3D protein-ligand structures to predict binding interactions. These models use CNNs to detect complex spatial patterns, offering high accuracy in identifying active compounds.
For each baseline model, unless otherwise noted (e.g., structure-based models), we have re-implemented, retrained, and tested them using the recommended hyperparameter settings provided in their original publications. A comprehensive summary of these baselines is presented in Supplementary Table 32 and more details are provided in Supplementary Table 33 and Supplementary Note 5.
Evaluation protocols
Training process
The experiments all use 5-fold cross-validations. Early stopping is used in all model training. A portion of the training set is drawn from the training set to form the validation set. One round (epoch) of model training is followed by one round of validation, and when the model does not improve in 20 consecutive epochs on the performance of the validation set, the model is considered to have completed training.
Four realistic scenarios for CPI prediction
There are four different realistic scenarios proposed by Pahikkala et al.45 in evaluating the generalization performance on CPI prediction, namely, warm start, compound cold start, protein cold start, and blind start.
Warm start (S1)
Compounds and proteins present in the testing set are also present in the training set. The scenario is to find unknown CPIs in the space of known compounds and proteins, corresponding to drug repositioning103.
Compound cold start (S2)
Compounds that appear in the testing set do not appear in the training set. This scenario searches for new compound candidates for known target proteins in a wide range of unknown chemical spaces104.
Protein cold start (S3)
Proteins that appear in the testing set do not appear in the training set. This scenario corresponds to a wide range of applications, including the discovery of protein targets for complex diseases and the identification of potential side effects105.
Blind start (S4)
neither the drug nor the protein appearing in the test set appeared in the training set. This scenario is relevant to the emergence and treatment of the SARS-CoV-2 virus68.
In this study, we employed a 5-fold cross-validation (CV) approach with different strategies for warm start, compound cold start, protein cold start, and blind start scenarios:
Warm Start CV: The CPIs are randomly divided into five equal parts. For each fold, one part is selected as the testing set, and the remaining four parts are combined to form the training set.
Compound Cold Start CV: Compounds are randomly divided into five parts. For each fold, one part is designated as the testing compound set, and the remaining four parts make up the training compound set. CPIs associated with the testing compounds form the testing set, while those linked to the training compounds form the training set.
Protein Cold Start CV: Proteins are split into five random parts. Each fold involves selecting one part as the testing protein set, with the remaining four parts serving as the training protein set. CPIs related to the testing proteins comprise the testing set, whereas CPIs associated with the training proteins are used for training.
Blind Start CV: 60% of the compounds are selected as the training compound set and the remaining 40% are used as the testing compound set, 60% of the proteins are selected as the training protein set and the remaining 40% are used as the testing protein set. CPIs consisting of the training compound set and training protein set are used as the training set, CPIs consisting of the testing compound set and testing protein are used as the testing set, and the remaining CPIs are used as the validation set.
For further details on these evaluation protocols, refer to Supplementary Note 6. This structured format clarifies the methodological setup for each CV strategy and ensures easier comprehension.
Evaluation metrics
In this study, we evaluate each model’s effectiveness using both the receiver operating characteristics curve (ROC) and the precision-recall curve (PR). The ROC curve, which plots the true positive rate (TPR) against the false positive rate (FPR) at various threshold settings, is a robust visual and quantitative tool for assessing model performance. The area under the ROC curve (AUC) quantifies the probability that the classifier will rank a randomly chosen positive instance higher than a negative one, providing a comprehensive measure of the model’s binary classification accuracy.
Similarly, the precision-recall curve (PR) tracks the balance between precision and recall for different thresholds, where a higher area under the PR curve (AUPR) indicates superior precision and recall. We also report standard metrics such as accuracy, precision, recall, and the F1 score to provide a complete picture of model performance. To evaluate the performance in the link prediction task, we have included performance metrics for node-centric local measures, specifically Hits@TopK, MRR (Mean Reciprocal Rank), and VCMPR@TopK58.
In the test of virtual screening, the AUC score, BEDROC, and enrichment factor (EF) at different thresholds are computed as the measurements of screening power. BEDROC is an enhanced variant of the area under the ROC curve (AUC) and the commonly used variant is BEDROC80.5. Unlike AUC, BEDROC is particularly concerned with the top ranking of active compounds, since in practice the most critical part of virtual screening is to find the active molecule as early as possible. The enrichment factor was computed with the following formula:
where \({{NT}}^{\alpha }\) is the number of binders found among the top \(\alpha\)-ranked molecules (e.g., α = 1%) based on the predicted binding score. \({{NT}}^{{total}}\) is the total number of binders for the given target protein in the data set. EF measures the multiplicity of the number of active compounds enriched by the model in x% of the data prior to screening relative to the number of active compounds that can be found by random screening. It is generally accepted that this method can significantly enrich for active compounds when the EF value is greater than 1. Due to the limitation of testing capacity, virtual screening of large compound libraries can only test a small number of compounds for biological activity, especially those with the best scores, which makes us only concerned with the early enrichment ability of virtual screening. The EFs of 0.5%, 1%, and 2% before the highest scoring are particularly important.
Implementation
ColdstartCPI is implemented in Python 3.8 and PyTorch 1.12.0, along with functions from Scikit-learn 1.0.2, Numpy 1.18.1, Pandas 1.0.1, and RDKit 2022.9.4. The batch size is set to 64 and the Adam optimizer is used with a learning rate of 1e-5. Early stopping is applied to avoid overfitting in the training process. The model is selected at the epoch giving the best sum of AUC and AUPC scores on the validation set within 20 epochs, which is then used to evaluate the final performance on the testing set. The default hyper-parameter settings, the search scopes, and the optimized values are summarized in Supplementary Table 34. All the experiments described here adopt this set of optimal hyper-parameter settings for ColdstartCPI. ColdstartCPI was trained on one NVIDIA Tesla A100 (40 G) GPU. The details of Molecular dynamics simulation and automated absolute binding free energy calculation are provided in Supplementary Note 7.
Construction of the compound candidate set and protein candidate set
The compound candidate set is combined from five databases: DrugBank57, ChEMBL106, COCONUT107, NPASS108, and FooDB (The Food Database, http://foodb.ca/). Referring to the data preprocessing steps of Mol2Vec, we processed the compounds and composed the compound candidate set by the following cutoffs and criteria:
-
1.
Remove molecules not recognized by RDKit;
-
2.
Remove the molecules with a mass greater than 1000 Da;
-
3.
Remove the molecules that are composed of only one heave atom or ion;
-
4.
Convert the SMILES string of molecules to canonical SMILES by RDKit;
-
5.
Merge and de-duplicate the compounds from DrugBank, ChEMBL, COCONUT, NPASS, and FooDB using the canonical SMILES as identifiers to construct the compound candidate set.
Detailed statistical data are shown in Supplementary Table 35.
The protein candidate set is generated from DrugBank (version 5.1.12) including 5349 proteins.
literature-based validations
The literature-based validations were conducted manually through searches on PubMed, as well as comparisons with authoritative databases such as DrugBank and BindingDB.
File preparation for docking simulations
We perform molecular docking simulations for the compound-protein interactions predicted by ColdstartCPI. High-resolution crystallographic structure of the target protein, e.g., Acetylcholinesterase (UniProt ID: P22303, PDB ID: 6O4W), was downloaded from the RCSB PDB database. The steps to implement docking simulations in AutoDock Vina6 include:
-
1.
According to the SMILES strings, download the 3D compound structures in SDF format from PubChem and translate them into.pdb format with OpenBabel for the AutoDockTools process. If the 3D structure was not found in PubChem, we used RdKit generation to generate the initial 3D coordinates and performed minimized energy optimization to obtain the molecular 3D structure, saved as SDF format.
-
2.
Save the compound structures in.pdbqt format using AutoDockTools.
-
3.
Download the 3D protein structures in.pdb format from PDB and use AutoDockTools to remove water molecules, add all hydrogen atoms and the Kollman charge to the protein, and save the protein structures in.pdbqt format.
-
4.
Docking grids containing the entire protein structure are created to ensure a blind docking setup so that all locations on the protein are taken into account when determining binding affinity.
-
5.
Create the configuration file with the grid details and launch the docking simulation for the top 100 candidates. We consider the protein molecule to be rigid and the compound molecules to be flexible, i.e., we allow rotatable bonds for the compounds. The configurations of the grids are shown in Supplementary Table 36.
Molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) binding energy calculations
We use Discovery Studio’s Calculate Binding Energies protocol, which is based on Molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) [3,4], to estimate the binding energy between a receptor and a ligand. MM-PBSA is more accurate than most scoring functions of molecular docking and less computationally demanding than alchemical free energy methods and has been widely used in biomolecular studies such as compound–protein binding and protein–protein interactions. MM-PBSA is considered a physics-based approach because it relies on molecular mechanics force fields and continuum electrostatics to estimate binding free energy. It combines molecular mechanics (MM) calculations with implicit solvent models, such as the Poisson-Boltzmann (PB) approach, to approximate solvation effects. The binding free energy \(\Delta {G}_{{bind}}\) is calculated as follows:
where the free energy \(G\) of each component is decomposed into three terms:
\({E}_{{MM}}\): Molecular mechanics energy, including bonded (bond, angle, dihedral) and non-bonded (van der Waals and electrostatic) interactions.
\({G}_{{sol}}\): Solvation free energy, consisting of the polar solvation energy (calculated using the Poisson-Boltzmann (PB) or Generalized Born (GB) model) and the nonpolar solvation energy (typically estimated using the solvent-accessible surface area (SA) method).
\({TS}\): Entropic contribution, usually estimated via normal mode analysis (NMA) or empirical methods.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data availability is as follows. The data generated and analyzed in the study have been deposited on Zenodo [https://zenodo.org/records/15622018]. The source data of the three benchmarks (the BindingDB, BioSNAP and BindingDB_AIBind datasets) are available on the GitHub pages for DrugBAN [https://github.com/peizhenbai/DrugBAN], BioSNAP [https://github.com/kexinhuang12345/MolTrans/tree/master/dataset/BIOSNAP], and Zenodo [https://zenodo.org/record/7226641], respectively. The publicly available datasets used in this study can be found on their official websites: DrugBank [https://www.drugbank.com/], BindingDB [https://www.bindingdb.org], Drug Target Commons [http://drugtargetcommons.fimm.fi/], DUD-E [https://dude.docking.org/], LIT_PCBA [https://drugdesign.unistra.fr/LIT-PCBA/], Antibiotics [https://doi.org/10.15252/msb.202211081], PDBbind [http://pdbbind.org.cn/], Uniprot [https://www.uniprot.org/], Protein Data Bank [https://www.rcsb.org/], and PubChem [https://pubchem.ncbi.nlm.nih.gov/]. The top 100 candidates predicted by ColdstartCPI are available within Supplementary Information. Unless otherwise stated, all data supporting the results of this study can be found in the article, supplementary, and source data files. Source data are provided with this paper.
Code availability
The code of our deep learning model is provided on GitHub [https://github.com/zhaoqichang/ColdstartCPI].
References
Bai, Q. et al. Application advances of deep learning methods for de novo drug design and molecular dynamics simulation. WIREs Comput. Mol. Sci. 20, e1581 (2021).
Pan, X. Q. et al. Deep learning for drug repurposing: methods, databases, and applications. Wires Comput. Mol. Sci. 12, e1597 (2022).
Sadybekov, A. V. & Katritch, V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023).
Rifaioglu, A. S. et al. Recent applications of deep learning and machine intelligence on drug discovery: methods, tools and databases. Brief. Bioinform. 20, 1878–1912 (2019).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Koes, D. R., Baumgartner, M. P. & Camacho, C. J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model 53, 1893–1904 (2013).
Spitzer, R. & Jain, A. N. Surflex-Dock: docking benchmarks and real-world application. J. Comput Aid Mol. Des. 26, 687–699 (2012).
Allen, W. J. et al. DOCK 6: impact of new features and current docking performance. J. Comput. Chem. 36, 1132–1156 (2015).
Durrant, J. D. & McCammon, J. A. NNScore 2.0: a neural-network receptor-ligand scoring function. J. Chem. Inf. Model 51, 2897–2903 (2011).
Ballester, P. J. & Mitchell, J. B. O. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 26, 1169–1175 (2010).
Stepniewska-Dziubinska, M. M., Zielenkiewicz, P. & Siedlecki, P. Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 34, 3666–3674 (2018).
Zheng, L. Z., Fan, J. R. & Mu, Y. G. OnionNet: a multiple-layer intermolecular-contact-based convolutional neural network for protein-ligand binding affinity prediction. Acs Omega 4, 15956–15965 (2019).
McNutt, A. T. et al. GNINA 1.0: molecular docking with deep learning. J. Cheminform. 13, 43 (2021).
Brocidiacono, M. et al. BigBind: learning from nonstructural data for structure-based virtual screening. J. Chem. Inf. Model 64, 2488–2495 (2023).
Zhang, X. et al. PLANET: a multi-objective graph neural network model for protein–ligand binding affinity prediction. J. Chem. Inf. Model. 64, 2205–2220 (2024).
Luo, D., Liu, D. D., Qu, X. Y., Dong, L. A. & Wang, B. J. Enhancing generalizability in protein-ligand binding affinity prediction with multimodal contrastive learning. J. Chem. Inf. Model 64, 1892–1906 (2024).
Lu, W. et al. DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model. Nat. Commun. 15, 1071 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583 (2021).
Subramaniam, S. & Kleywegt, G. J. A paradigm shift in structural biology. Nat. Methods 19, 20–23 (2022).
Wong, F. et al. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 18, e11081 (2022).
He, X. H. et al. AlphaFold2 versus experimental structures: evaluation on G protein-coupled receptors. Acta Pharm. Sin. 44, 1–7 (2023).
Lim, S. et al. A review on compound-protein interaction prediction methods: Data, format, representation and model. Comput Struct. Biotechnol. J. 19, 1541–1556 (2021).
Zhao, Q. C., Yang, M. Y., Cheng, Z. J., Li, Y. H. & Wang, J. X. Biomedical data and deep learning computational models for predicting compound-protein relations. IEEE/ACM Trans. Comput. Biol. Bioinform. 19, 2092–2110 (2022).
Chen, X. et al. Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinform. 17, 696–712 (2016).
Wen, M. et al. Deep-learning-based drug-target interaction prediction. J. Proteome Res. 16, 1401–1409 (2017).
Jacob, L. & Vert, J. P. Protein-ligand interaction prediction: an improved chemogenomics approach. Bioinformatics 24, 2149–2156 (2008).
Tian, K., Shao, M., Wang, Y., Guan, J. & Zhou, S. Boosting compound-protein interaction prediction by deep learning. Methods 110, 64–72 (2016).
Lee, I., Keum, J., Nam, H. DeepConv-DTI: prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 15, e1007129 (2019).
Li, J. et al. DARC: deep adaptive regularized clustering for histopathological image classification. Med. Image Anal. 80, 102521 (2022).
Yue, H. et al. MLDRL: multi-loss disentangled representation learning for predicting esophageal cancer response to neoadjuvant chemoradiotherapy using longitudinal CT images. Med. Image Anal. 79, 102423 (2022).
Rifaioglu, A. S. et al. DEEPScreen: high performance drug-target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 11, 2531–2557 (2020).
Öztürk, H., Özgür, A. & Ozkirimli, E. DeepDTA: deep drug-target binding affinity prediction. Bioinformatics 34, 821–829 (2018).
Zheng, S. J., Li, Y. J., Chen, S., Xu, J. & Yang, Y. D. Predicting drug-protein interaction using quasi-visual question answering system (Feb, 10.1038/s42256-020-0152-y, 2020). Nat. Mach. Intell. 2, 551–551 (2020).
Yang, Z., Zhong, W., Zhao, L. & Chen, C. Y.-C. ML-DTI: mutual learning mechanism for interpretable drug–target interaction prediction. J. Phys. Chem. Lett. 12, 4247–4261 (2021).
Nguyen, T. et al. GraphDTA: predicting drug-target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Li, Y. Q. et al. An adaptive graph learning method for automated molecular interactions and properties predictions. Nat. Mach. Intell. 4, 645–651 (2022).
Torng, W. & Altman, R. B. Graph convolutional neural networks for predicting drug-target interactions. J. Chem. Inf. Model 59, 4131–4149 (2019).
Huang, K. X., Xiao, C., Glass, L. M. & Sun, J. M. MolTrans: Molecular Interaction Transformer for drug-target interaction prediction. Bioinformatics 37, 830–836 (2021).
Zhao, Q. C. et al. GIFDTI: prediction of drug-target interactions based on global molecular and intermolecular interaction representation learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 1943–1952 (2023).
Chen, L. F. et al. TransformerCPI: improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 36, 4406–4414 (2020).
Zhao, Q. C., Zhao, H. C., Zheng, K. & Wang, J. X. HyperAttentionDTI: improving drug-protein interaction prediction by sequence-based deep learning with attention mechanism. Bioinformatics 38, 655–662 (2022).
Cheng, Z. J., Zhao, Q. C., Li, Y. H. & Wang, J. X. IIFDTI: predicting drug-target interactions through interactive and independent features based on attention mechanism. Bioinformatics 38, 4153–4161 (2022).
Sun, Y., Li, Y. Y., Leung, C. K., Hu, P. iNGNN-DTI: prediction of drug–target interaction with interpretable nested graph neural network and pretrained molecule models. Bioinformatics 40, btae135 (2024).
Pahikkala, T. et al. Toward more realistic drug-target interaction predictions. Brief. Bioinform. 16, 325–337 (2015).
Sieg, J., Flachsenberg, F. & Rarey, M. In need of bias control: evaluating chemical data for machine learning in structure-based virtual screening. J. Chem. Inf. Model 59, 947–961 (2019).
Ye, Q. et al. A unified drug-target interaction prediction framework based on knowledge graph and recommendation system. Nat. Commun. 12, 6775 (2021).
Bai, P. Z., Miljkovic, F., John, B. & Lu, H. P. Interpretable bilinear attention network with domain adaptation improves drug-target prediction. Nat. Mach. Intell. 5, 126–136 (2023).
Lin, Z. M. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Chatterjee, A. et al. Improving the generalizability of protein-ligand binding predictions with AI-Bind. Nat. Commun. 14, 1989 (2023).
Ravindranath, P. A., Forli, S., Goodsell, D. S., Olson, A. J. & Sanner, M. F. Advances in protein-ligand docking with explicitly specified binding site flexibility. Plos Comput Biol. 11, e1004586 (2015).
Siebenmorgen, T. et al. MISATO: machine learning dataset of protein-ligand complexes for structure-based drug discovery. Nat. Comput Sci. 4, 367–378 (2024).
Koshland, D. E. Application of a theory of enzyme specificity to protein synthesis*. Proc. Natl Acad. Sci. USA 44, 98–104 (1958).
Jaeger, S., Fulle, S. & Turk, S. Mol2vec: unsupervised machine learning approach with chemical intuition. J. Chem. Inf. Model 58, 27–35 (2018).
Elnaggar, A. et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern. Anal. Mach. Intell. 44, 7112–7127 (2022).
Gilson, M. K. et al. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–D1053 (2016).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2018).
Menand, N. & Seshadhri, C. Link prediction using low-dimensional node embeddings: the measurement problem. Proc. Natl. Acad. Sci. USA 121, e2312527121 (2024).
Gao, K. Y. et al. Interpretable drug target prediction using deep neural representation. Proc. 27th International Joint Conference on Artificial Intelligence (IJCAI), 3371-3377 (2018).
Bai, P. et al. Hierarchical clustering split for low-bias evaluation of drug-target interaction prediction. 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 641-644 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594 (2012).
Tran-Nguyen, V. K., Jacquemard, C. & Rognan, D. LIT-PCBA: an unbiased data set for machine learning and virtual screening. J. Chem. Inf. Model 60, 4263–4273 (2020).
Yang, J., Shen, C. & Huang, N. Predicting or pretending: artificial intelligence for protein-ligand interactions lack of sufficiently large and unbiased datasets. Front. Pharmacol. 11, 69 (2020).
Su, M. Y., Feng, G. Q., Liu, Z. H., Li, Y. & Wang, R. X. Tapping on the black box: how is the scoring power of a machine-learning scoring function dependent on the training set?. J. Chem. Inf. Model 60, 1122–1136 (2020).
Xia, J., Tilahun, E. L., Reid, T. E., Zhang, L. G. & Wang, X. S. Benchmarking methods and data sets for ligand enrichment assessment in virtual screening. Methods 71, 146–157 (2015).
Wang, R. X., Fang, X. L., Lu, Y. P., Yang, C. Y. & Wang, S. M. The PDBbind database: methodologies and updates. J. Med. Chem. 48, 4111–4119 (2005).
Wang, G. Y. et al. Deep-learning-enabled protein-protein interaction analysis for prediction of SARS-CoV-2 infectivity and variant evolution. Nat. Med 29, 2007 (2023).
Kuzikov, M. et al. Identification of inhibitors of SARS-CoV-2 3CL-pro enzymatic activity using a small molecule in vitro repurposing screen. ACS Pharmacol. Transl. Sci. 4, 1096–1110 (2021).
Ektefaie, Y. et al. Evaluating generalizability of artificial intelligence models for molecular datasets. Nat. Mach. Intell. 6, 1512–1524 (2024).
Ramos, J. et al. Shedding light on the interaction of human anti-apoptotic Bcl-2 protein with ligands through biophysical and in silico studies. Int. J. Mol. Sci. 20, 860 (2019).
Dvir, H., Silman, I., Harel, M., Rosenberry, T. L. & Sussman, J. L. Acetylcholinesterase: from 3D structure to function. Chem.Biol. Interact. 187, 10–22 (2010).
Torbeev, V. Y. et al. Protein conformational dynamics in the mechanism of HIV-1 protease catalysis. Proc. Natl. Acad. Sci. USA 108, 20982–20987 (2011).
Stenstroem, O., Diehl, C., Modig, K., Akke, M. Ligand- induced protein transition state stabilization switches the binding pathway from conformational selection to induced fit. Proc. Natl. Acad. Sci. USA 121, (2024).
Subramanian, J., Sharma, S. & B-Rao, C. Modeling and selection of flexible proteins for structure-based drug design: backbone and side chain movements in p38 MAPK. ChemMedChem. 3, 336–344 (2008).
Seeliger, D. & de Groot, B. L. Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PloS Comput. Biol. 6, e1000634 (2010).
Meller, A. et al. Predicting locations of cryptic pockets from single protein structures using the PocketMiner graph neural network. Nat. Commun. 14, 1177 (2023).
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4, 1256–1264 (2022).
Kabir, A., Moldwin, A., Shehu, A. A comparative analysis of transformer-based protein language models for remote homology prediction. Proc. 14th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Article 97 (2023).
Liu, N. & Xu, Z. Using LeDock as a docking tool for computational drug design. IOP Conf. Ser. Earth Environ. Sci. 218, 012143 (2019).
Chen, Y. et al. Discovery of new scaffolds from approved drugs as acetylcholinesterase inhibitors. RSC Adv. 5, 90288–90294 (2015).
Sowell, J. W. et al. Synthesis and cholinergic properties of bis[[(dimethylamino)methyl]furanyl] analogs of ranitidine. J. Med. Chem. 35, 1102–1108 (1992).
Srinivasan, J., Cheatham, T. E., Cieplak, P., Kollman, P. A. & Case, D. A. Continuum solvent studies of the stability of DNA, RNA, and Phosphoramidate-DNA Helices. J. Am. Chem. Soc. 120, 9401–9409 (1998).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000).
Ries, B., Alibay, I., Anand, N. M., Biggin, P. C. & Magarkar, A. Automated absolute binding free energy calculation workflow for drug discovery. J. Chem. Inf. Model 64, 5357–5364 (2024).
Bembenek, S. D. et al. Lead identification of acetylcholinesterase inhibitors-histamine H receptor antagonists from molecular modeling. Bioorgan Med. Chem. 16, 2968–2973 (2008).
Kato, K. et al. TAK-147, an acetylcholinesterase inhibitor, increases choline acetyltransferase activity in cultured rat septal cholinergic neurons. Neurosci. Lett. 260, 5–8 (1999).
Li, X. K. et al. The novel therapeutic strategy of vilazodone-donepezil chimeras as potent triple-target ligands for the potential treatment of Alzheimer’s disease with comorbid depression. Eur. J. Med Chem. 229, 114045 (2022).
Mahboobi, S. et al. Novel chimeric histone deacetylase inhibitors: a series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J. Med. Chem. 53, 8546–8555 (2010).
Mody, V. et al. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 4, 93 (2021).
Luo, Y. A. et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8, 573 (2017).
Wang, Y. X. et al. ZeroBind: a protein-specific zero-shot predictor with subgraph matching for drug-target interactions. Nat. Commun. 14, 7861 (2023).
Li, S. et al. MONN: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10, 308–322.e311 (2020).
Shen, W. X. et al. Out-of-the-box deep learning prediction of pharmaceutical properties by broadly learned knowledge-based molecular representations. Nat. Mach. Intell. 3, 334–343 (2021).
Li, H. et al. A knowledge-guided pre-training framework for improving molecular representation learning. Nat. Commun. 14, 7568 (2023).
Fang, X. M. et al. Geometry-enhanced molecular representation learning for property prediction. Nat. Mach. Intell. 4, 127–134 (2022).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871 (2021).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Wang, Y., Wang, J., Cao, Z. & Barati Farimani, A. Molecular contrastive learning of representations via graph neural networks. Nat. Mach. Intell. 4, 279–287 (2022).
Liu, S. et al. Multi-modal molecule structure–text model for text-based retrieval and editing. Nat. Mach. Intell. 5, 1447–1457 (2023).
Wang, X. Q. et al. Multitask joint strategies of self-supervised representation learning on biomedical networks for drug discovery. Nat. Mach. Intell. 5, 445–456 (2023).
Hu, F. et al. A multimodal protein representation framework for quantifying transferability across biochemical downstream tasks. Adv. Sci. 10, e2301223 (2023).
Yang, M. Y., Wu, G. Y., Zhao, Q. C., Li, Y. H. & Wang, J. X. Computational drug repositioning based on multi-similarities bilinear matrix factorization. Brief. Bioinform 22, bbaa267 (2021).
Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 180, 688 (2020).
Lounkine, E. et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 486, 361 (2012).
Zdrazil, B. et al. The ChEMBL Database in 2023: a drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 52, D1180–D1192 (2023).
Sorokina, M., Merseburger, P., Rajan, K., Yirik, M. A., Steinbeck, C. COCONUT online: collection of open natural products database. J. Cheminform. 13, 2 (2021).
Zhao, H. et al. NPASS database update 2023: quantitative natural product activity and species source database for biomedical research. Nucleic Acids Res. 51, D621 (2022).
Shen, C. et al. Beware of the generic machine learning-based scoring functions in structure-based virtual screening. Brief Bioinform. 22, bbaa070 (2021).
Ragoza, M., Hochuli, J., Idrobo, E., Sunseri, J. & Koes, D. R. Protein-ligand scoring with convolutional neural networks. J. Chem. Inf. Model 57, 942–957 (2017).
Tsubaki, M., Tomii, K. & Sese, J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 35, 309–318 (2019).
Acknowledgements
This work has been supported by the National Natural Science Foundation of China (No. 62350004, No. U22A2041, No. 62473149), the Project of Xiangjiang Laboratory (No. 23XJ01011), Xinjiang Uygur Autonomous Region Key R & D program under Grant 2024B03039, the Science Foundation for Distinguished Young Scholars of Hunan Province (NO. 2023JJ10080), and the Academy of Finland No. 359752. This work was carried out in part using computing resources at the High-Performance Computing Center of Central South University. The authors express thanks to Hebei Medical University for the Discovery Studio software availability.
Author information
Authors and Affiliations
Contributions
J.X.W. conceived and designed this project. Q.C.Z. and J.X.W. conceived, designed, and implemented the ColdstartCPI. H.C.Z., K.Z., Y.J.L., and Q.L. analyzed the results. Q.C.Z. and J.X.W. wrote the paper. H.C.Z., J.T. and Y.H.L. contributed to the revision of the manuscript. Q.C.Z., L.Y.G. and J.T. contributed to the case study. All authors have read and approved the final version of this paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Q., Zhao, H., Guo, L. et al. ColdstartCPI: Induced-fit theory-guided DTI predictive model with improved generalization performance. Nat Commun 16, 6436 (2025). https://doi.org/10.1038/s41467-025-61745-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61745-7
This article is cited by
-
Contrastive learning in both structure and function spaces improve drug-target interaction prediction
BMC Bioinformatics (2026)
