
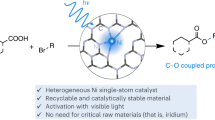
Abstract
Combining transition-metal catalysis with photocatalysis has emerged as a valuable, complementary approach for achieving carbon–heteroatom cross-coupling reactions. However, the need to use blue or high-energy near-UV light leads to problems with scalability, chemoselectivity, and catalyst deactivation, which have limited the synthetic applications of this combination. Herein, we report a method for red-light-driven nickel-catalyzed cross-coupling reactions of aryl halides with 11 different types of nucleophiles using a polymeric carbon nitride (CN-OA-m) as a photocatalyst. This semiheterogeneous catalyst system enabled the formation of four different types of carbon–heteroatom bonds (C–N, C–O, C–S, and C–Se) with a wide range of substrates (more than 200 examples) with yields up to 94%. Moreover, the photocatalyst is easily recovered and recycled, which makes it a promising new tool for the development of other reactions involving red-light metallaphotoredox catalysis.
Similar content being viewed by others

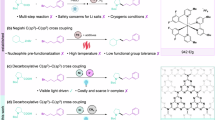
Introduction
Carbon–heteroatom cross-coupling reactions catalyzed by Pd1,2, Cu3, Ni4, and other metals5,6,7,8,9,10 offer an efficient method for the synthesis of structurally complex pharmaceuticals, pesticides, functional materials, and fine chemicals11,12,13,14,15. However, the success of these reactions heavily depends on the selection of appropriate parameters, including the ligand (e.g., phosphines, carbenes, or oxamides), precatalyst, base, additive, solvent, and reaction temperature, as well as the electronic and structural properties of the substrates16,17,18,19,20,21,22,23,24. Thus, case-by-case optimization of the reaction conditions is often required. Combining transition-metal catalysts with well-established photocatalysts such as metal complexes, organic dyes, and organic semiconductors has emerged as a valuable approach to overcoming some of the above-mentioned challenges (Fig. 1A)25,26,27,28,29,30,31,32,33. However, the light-promoted reactions reported to date require blue or high-energy near-UV light, which lead to problems with scalability, chemoselectivity, and catalyst deactivation33 that result from competitive absorption of light by the substrates and intermediates. Therefore, the synthetic utility of the combination of transition-metal catalysis and photocatalysis is currently limited34,35,36,37,38. Recently, the Rovis group pioneered red-light-driven C–N cross-coupling reactions mediated by the combination of a metal catalyst and an osmium photocatalyst (Fig. 1B)39,40,41. These investigators found that a dual Ni/Os catalyst system has good substrate compatibility and is easy to scale up, showcasing the potential utility of red-light-driven coupling reactions42. This study shows that red light can enhance the desired reactivity by inhibiting unwanted photodegradation pathways (which are usually caused by the use of blue or high-energy near-UV light), which may provide new ideas for breaking through bottlenecks in the development of cross-coupling reactions. Despite some successes in the field42,43,44,45,46,47,48, the lack of inexpensive, readily available, versatile, recyclable red-light catalyst systems has hindered the development of red-light metallaphotoredox catalysis.

A The homogeneous photocatalysts for C(sp2)–heteroatom cross-couplings formation between aryl halides with different nucleophiles. B The Red-light metallaphotocatalytic C-N cross-coupling reactions. C This work. General C-heteroatom cross-coupling reactions with semiheterogeneous red light metallaphotocatalysis.
Heterogeneous semiconductors such as carbon nitride (C3N4) are promising alternatives to other photocatalysts because the former are easy to prepare and can be recovered by filtration or centrifugation49,50,51,52,53,54. Although these semiconductors have been applied to transition-metal-catalyzed coupling reactions29,30,31,33,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74, they have rarely been used for red-light-catalyzed cross-coupling reactions. In this context, we report a method for red-light-driven Ni-catalyzed cross-coupling of aryl halides with 11 different nucleophiles using the polymeric carbon nitride CN-OA-m as a photocatalyst under the same reaction conditions (same pre-catalyst, temperature, solvent, and base, Fig. 1C). This versatile, semiheterogeneous catalyst system enables the formation of four different types of carbon–heteroatom bonds (C–N, C–O, C–S, C–Se) and exhibits a broad substrate scope (more than 200 examples, and with yields up to 94%).
Results and discussion
Investigation of the reaction conditions
In 2017, Zhang et al. reported the synthesis of CN-OA-m from urea and oxamide in a molten salt75. The conduction band potential of this polymer is at −1.65 V vs Ag/AgCl, and the valence band potential is at 0.88 V vs Ag/AgCl33, and an obvious absorption band in the 460–700 nm region75,76. These investigators also demonstrated that CN-OA-m can absorb visible light at wavelengths of >500 nm and exhibits high hydrogen-production activity. For our initial experiments, we selected 3,5-dimethylbromobenzene (1) and pyrrolidine (2) as model substrates to optimize the conditions for the desired coupling reaction with CN-OA-m as a photocatalyst (Table 1)77. Target amination product 3 was obtained in 91% isolated yield upon red-light irradiation (660–670 nm) of the substrates under argon at 85 °C for 24 h in dimethylacetamide (DMAc) containing NiBr2·glyme and 1,4,5,6-tetrahydro-1,2-dimethylpyrimidine (mDBU) (entry 1). Other photocatalyst—carbon nitride (C3N4, entry 2)78, mesoporous graphitic carbon nitride (mpg-C3N4, entry 3)79, P-doped carbon nitride prepared from diammonium hydrogen phosphate and urea (p-C3N4, entry 4)80, graphitic carbon nitride (g-C3N4, entry 5)81, RP-C3N4 (a red polymeric carbon nitride containing Na and K atoms; entry 6)82, and MC-C3N4 (a reddish-brown polymeric carbon nitride containing Na, K, and Li atoms; entry 7)83—gave lower yields (43–83%) than CN-OA-m (see also Table S1). Our findings revealed that these organic semiconductors are efficient red-light photocatalysts. The solvent also had a significant impact on the reaction yield, with DMAc being the most effective (Table S3). Of the various tested Ni catalysts, NiBr2·glyme exhibited the highest activity (Table S4). The wavelength of the light also played an important role, with 660–670 nm red light exhibiting the highest yield (compare entry 1 with entries 8–10, and see Table S6). The reaction was also sensitive to temperature; no C–N coupling product was obtained below 45 °C (entry 11, See SI, section 6). As the temperature increases, the yield increases significantly; however, once the threshold of 90 °C is surpassed, further increasing the temperature leads to negligible changes in yield (See SI section 6). These findings further underscore the importance of temperature control in this transformation. The choice of base also played a crucial role (entry 12); only organic bases were effective, with the mild, soluble base mDBU giving the best results (Table S7). Organic bases may serve two functions: one is to act as a base for deprotonation, and the other may involve serving as an electron donor to complete the photocatalytic cycle. By testing the oxidation potential of organic bases, it was revealed that the oxidation potential of mDBU is Ep/2 = + 1.39 V (Ag/AgCl in MeCN), which has a better matching oxidation potential (See SI section 13). Moreover, the reaction did not occur in the absence of light (entry 13), even at high temperatures (entry 14). Control experiments revealed that the reaction failed to proceed in the absence of a Ni catalyst (entry 15) 77.
Substrate Scope of C–Heteroatom Couplings
With the optimized conditions in hand, we next investigated the scope with respect to the nucleophiles and aryl halides (Figs. 2, 3, 4). We found that 11 different types of nucleophiles efficiently coupled with 3,5-dimethylbromobenzene (1) or 4-bromobenzonitrile to afford the corresponding products in high yields. The reaction conditions were compatible with primary amines bearing a wide range of functional groups, including straight chains (4 and 5), a ketal (6 and 8), hydroxyl groups (7 and 12), a vinyl group (9), and a carbamate ester (10). Notably, no C–O coupling was observed for the amino alcohol substrates under the standard conditions (7 and 12). Cyclic primary amines also coupled efficiently (10–14), as did secondary amines (16–19). Notably, reactions of secondary aliphatic amines did not require an external organic base, perhaps because they can act as both a substrate and a base. Primary aliphatic (20–26), aryl (27–29), heteroaryl (30 and 31), and secondary aliphatic (32–34) amides also coupled successfully, providing N-aryl amides in 53–78% yields. In addition, sulfonamides with various substituents delivered the desired products (35–45) in 43–69% yields. Pyrazole also gave corresponding C–N coupling product 46 in 63% yield. Aniline, as an important substrate, undergoes a C–N cross-coupling reaction with 4,4-dimethyl-2,2’-bipyridine (d-Mebpy) as the ligand to produce the desired diarylamine product (47–55); however, without the ligand, the reaction hardly occurs.

a C-N cross-coupling reaction conditions, unless otherwise noted: aryl bromide (0.2 mmol), amine (0.4 mmol), CN-OA-m (10 mg mL−1), NiBr2·glyme (10.0 mol%), DMAc (1 mL), mDBU (1.5 equiv.), red light, 85 °C, Ar, 24 h. b 48 h. c with d-Mebpy as a ligand. Isolated yields are provided. For details, see Supplementary Information.
Next, we also explored the reaction scope with respect to the aryl halides (Figs. 2, 3). Aryl halides containing various functional groups efficiently underwent C–N coupling with n-butylamine, delivering the desired aryl C–N coupling products in generally high yields. Electron-rich aryl halides with para substituents such as Me (56), Bpin (57), SMe (58), OMe (59), and OCHF2 (60) exhibited high reactivity. Less reactive ortho-substituted aryl bromides also afforded the target products (74 and 75). Electron-deficient aryl halides were also suitable substrates for this red-light-driven C–N coupling reaction: products bearing a sulfoxide group (63), an unprotected sulfonamide (64), a ketone (65 and 66), a cyano group (67), a trifluoromethyl group (68), a halogen atom (69) could be obtained. Satisfactory yields were also obtained with multisubstituted aryl halides (72–75). Aryl halides with a series of highly π-conjugated and photosensitive substrates such as fluorene (76), 9-fluorenone (77), benzophenone (78–79), naphthalene (80), anthracene (81), phenanthrene (82–83), triphenylene (84), perylene (85) or pyrene (86) also afforded the desired products in satisfactory yields. Aryl halides containing a heterocycle—such as pyridine (88–94), pyrimidine (95), pyrazine (96), quinoline (97–98), isoquinoline (99), quinoxaline (100), benzothiophene (101), thiophene (102), or carbazole (103)—were transformed into the corresponding heteroarylamines. Such structures are important in pharmaceutical chemistry.
Further, we explored C-O/S/Se coupling reactions with nucleophiles other than nitrogen (Figs. 3 and 4). It should be emphasized that the coupling reactions of oxygen-containing, sulfur-containing, and selenium-containing substrates as nucleophiles are not observable under standard conditions without a ligand. However, by introducing bipyridine ligands, and adjusting the wavelength of red light to 620–630 nm (Table S13), the efficiency of the reaction is significantly improved, facilitating the smooth progression of the coupling process. Aliphatic alcohols underwent C–O coupling with 4,4-di-tert-butylbipyridine (d-tbbpy) as a ligand to afford the corresponding ether products (104–133), showcasing the practicality of this method. Other oxygen-containing nucleophiles—phenols (134–139), carboxylic acids (140–145), H2O and H218O (146–154)—were also good coupling partners, yielding the desired C–O coupling products in 50–91% yields. Notably, 2-bromopyridine and 3-bromoisoquinoline afforded the lactam products (153–154). Thiol coupling products 155–160 and selenium coupling product 161 were also obtained in satisfactory yields. Simultaneously, we explored the C-O coupling reaction of aryl halides with n-butyl alcohol (162–196), delivering the desired aryl C–O coupling ether products in generally high yields.

a C-N/O cross-coupling reaction conditions, unless otherwise noted: aryl bromide (0.2 mmol), amine (0.4 mmol), CN-OA-m (10 mg mL−1), NiBr2·glyme (10.0 mol%), DMAc (1 mL), mDBU (1.5 equiv.), red light, 85 °C, Ar, 24 h. b 48 h. c with d-Mebpy as a ligand. eC-O/S/Se cross-coupling reaction conditions, aryl bromide (0.2 mmol), alcohol (0.6 mmol), CN-OA-m (10 mg mL−1), d-tbbpy (10.0 mol%), NiBr2·glyme (10.0 mol %), DMAc (1 mL), mDBU (1.5 equiv.), red light (620-630 nm), 85 °C, Ar, 24 h. Isolated yields are provided. For details, see Supplementary Information.

a unless otherwise noted: aryl bromide (0.2 mmol), alcohol (0.6 mmol), CN-OA-m (10 mg mL−1), d-tbbpy (10.0 mol%), NiBr2·glyme (10.0 mol %), DMAc (1 mL), mDBU (1.5 equiv.), red light (620-630 nm), 85 °C, Ar, 24 h. Isolated yields are provided. For details, see Supplementary Information.
To explore further applications of our methodology, we attempted to expand the reaction scope to more affordable yet more challenging aryl chlorides. Given the stronger C-Cl bond, it was not unexpected that reactions yielded products in low yields under the conditions used for aryl bromides. However, after adding TBAI as an additive and extending the reaction time, satisfactory yields could be attained (Fig. 5). The reaction provided the cross-coupling products in good yields with aryl chlorides (197–228), containing electron-rich (Me, OMe) and electron-deficient (ester, ketone, nitrile) groups, as well as disubstituted aryl chlorides (3, 208–209). Notably, it also showed excellent compatibility with the aldehyde group (207). Notably, more complex, biologically relevant substrates were also tolerated. For example, fenofibrate (212) and clofibrate (213) gave excellent yields. Meanwhile, a series of nucleophilic groups—including aliphatic amines (214–216), aromatic amines (217–218), amides (219–220), sulfonamides (221–222), alcohols (223–224), phenols (225–226), thiols (227), and selenol (228)—all provided the corresponding coupling products in excellent yields.

a Reaction conditions, unless otherwise noted: aryl chloride (0.2 mmol), NuH (0.4 mmol), CN-OA-m (10 mg mL−1), NiBr2·glyme (10.0 mol %), DMAc (1 mL), mDBU (1.5 equiv.), red light (660–670 nm), 85 °C, Ar, 36 h. b 48 h. c with d-tbbpy (10.0 mol%). d 620–630 nm, NuH (0.6 mmol). Isolated yields are provided. For details, see Supplementary Information.
Then, we also investigated the C–heteroatom bond coupling of various bioactive molecules (Fig. 6). Specifically, the following bioactive molecules containing C(sp2)–Br bonds were efficiently converted to the desired C–X coupling products: gemfibrozil (229), diacetonefructose (230 and 237), epiandrosterone (231), diacetone-d-glucose (232), tocopherol (233 and 234), celecoxib (235), l-menthol (236), stigmasterol (238) and epicholesterol (239). Notably, celecoxib bromide was compatible with the reaction conditions and could be used to obtain excellent yields of coupling products 240–262, making our method highly attractive for the modification of bioactive molecules. These examples further demonstrate the broad applicability of this red-light-driven Ni-catalyzed cross-coupling reaction, highlighting its significant potential utility for the synthesis of drug-like molecules.

a Brominated-bioactive molecule (0.2 mmol), amine (0.4 mmol), CN-OA-m (10 mg mL−1), NiBr2·glyme (10.0 mol%), DMAc (1 mL), mDBU (1.5 equiv.), red light (660-670 nm), 85 °C, Ar, 24 h. b CN-OA-m (10 mg mL−1), NiBr2·glyme (10.0 mol%), alcohol (0.6 mmol), d-tbbpy (10.0 mol%), red light (660-670 nm). c 48 h. d with d-Mebpy as a ligand. Isolated yields are provided. For details, see Supporting Information.
Structural characterization
In order to clarify the relationship between the structures of those catalysts and their activity in the reactions, a series of catalysts (C3N4, p-C3N4, g-C3N4, mpg-C3N4, RP-C3N4, MC-C3N4, CN-OA-m) were systematically investigated in terms of their physicochemical properties and structural features, including X-ray photoelectron spectroscopy (XPS), specific surface area, photoluminescence (PL), photocurrent responses, electrochemical impedance spectroscopy (EIS), time-resolved photoluminescence (TRPL) spectra, and band structures analyses. XPS analysis of CN-OA-m confirms the presence of C, N, and K elements on the surface (Figs. 7A–C, S18). The analysis of the N 1 s spectrum for CN-OA-m reveals three peaks at 399.2, 398.4, and 397.7 eV, which can be attributed to C–NHx, N–C3, and C–N = C, respectively. Additionally, a weak peak at 396.7 eV is observed, which is attributed to a negatively charged C = N–C moiety, potentially balancing the positive charge of K⁺ ions. Furthermore, the C 1 s spectrum of CN-OA-m shows peaks at 288.0, 286.0, and 284.7 eV, which are attributed to N–C = N, C–NHx, C–C bonds, respectively (Fig. 7A–C). These findings further suggest that defect energy levels can be generated between the conduction band and the valence band. Such defect levels can effectively trap photogenerated electrons, thereby suppressing the recombination of photogenerated electron–hole pairs in CN-OA-m, which in turn promotes enhanced photoredox activity. The N2 adsorption isotherm of all semiheterogeneous photocatalysts reveals that CN-OA-m signifying a highly developed porous network (Fig. S20). The Brunauer–Emmett–Teller (BET) surface area of CN-OA-m are 43.8 cm2g−1, with an average pore diameter of 13.80 nm. The BET pore size distribution of CN-OA-m exhibits the micro-meso-macropores characteristics (Figs. 7D, S21), with particularly significant proportion of macropores compared to other semiheterogeneous photocatalysts. The plentiful pores in CN-OA-m generate a high specific surface area, fully exposing catalytic sites while shortening diffusion paths for photo-induced electron–hole pairs, which collectively boosts its photocatalytic efficiency. By comparison, p-C₃N₄, g-C₃N₄, and mpg-C₃N₄ display much stronger photoluminescence signals, pointing to pronounced recombination of the photogenerated charge carriers. In contrast, the weakest photoluminescence (PL) intensity observed in CN-OA-m indicates suppressed carrier recombination (Figs. 7E, S22). This trend facilitates efficient electron transfer from the emissive state to Ni(II), potentially leading to the formation of Ni(I) species. The transient photocurrent response of different C3N4 electrodes were measured to gain in-depth insight into charge separation efficiency (Figs. 7F, S23). In contrast to the other C₃N₄, the CN-OA-m photoelectrode delivers a markedly higher photocurrent, underscoring its superior electron–hole separation and transport. Electrochemical impedance spectroscopy (EIS) supports this observation, the CN-OA-m (Figs. 7G and S24) displays a significantly smaller semicircle radius compared to the other C₃N₄ samples, signifying lower interfacial charge-transfer resistance and faster electron mobility. These findings further corroborate the efficient electron transfer from the emissive state to Ni(II), facilitating the generation of Ni(I) species. Time-resolved photoluminescence (TRPL) spectra monitored at the corresponding emission peaks give the average radiative lifetimes (τ) of the recombining charge carriers. The radiative lifetimes of p-C3N4, g-C3N4, mpg-C3N4, C3N4, PR-C3N4, MC-C3N4, CN-OA-m were 1.52, 2.04, 2.48, 2.78, 3.62, 3.73, and 3.88 ns, respectively (Figs. 7H, S25). Among these catalysts, CN-OA-m exhibits the longest fluorescence lifetime, suggesting a reduced recombination rate of photogenerated electron-hole pairs. This extended lifetime implies that photogenerated electrons are more effectively sustained during the reaction, thereby enhancing the overall photocatalytic efficiency. The electrochemical potential diagrams (Fig. 7I) of several catalysts29,33,55,84,85,86,87 further show that CN-OA-m possesses the highest reduction potential, indicating the possibility of a photoredox reaction. Combined with the above analysis, porous CN-OA-m possess the efficient separation efficiency of photogenerated electronhole pairs, exhibits relatively superior properties and may be the best photocatalyst for the reactions examined, benefiting the photocatalytic carbon–heteroatom cross-coupling reaction.

A–C XPS spectra of CN-OA-m of C 1 s, N 1 s, K 2p; (D) The pore size distribution; (E) The photoluminescence (PL) spectra; (F) Photocurrent responses; (G) The electrochemical impedance spectra (EIS); (H) Time-resolved photoluminescence (TRPL) spectra; (I) Electrochemical potential diagrams.
Synthetic application and mechanistic considerations
Red light penetrates approximately 23 times further into reaction solutions than blue light, reaching a depth of 12 cm before 90% of its power is absorbed by the solution42. To highlight the utility of red light-promoted cross-coupling reactions, we performed a gram-scale reaction of aryl bromide and n-butylamine in a round-bottom flask with a diameter of 10 cm, which yielded tetracaine (263) in 85% yield (Fig. 8A), further demonstrating the utility of red light’s penetration ability in this semiheterogeneous catalysis. Impressively, even when simulated solar light was used as the light source, tetracaine was obtained in 86% yield (Fig. 8A, See SI sections 4.5 and 4.7). Additionally, the yield of tetracaine was 59% when the reaction was carried out under solar light for 5 h (Fig. 8A, See SI section 4.4). Furthermore, a significant advantage of this method is that the photocatalyst CN-OA-m could easily be recycled and reused (Fig. 8B; and see section 5 in SI). After the reaction was complete, the CN-OA-m powder was recovered by centrifugation and then washed. Fourier transform IR spectroscopy (Fig. S29), UV–vis spectroscopy (Fig. S30), scanning electron microscopy (Fig. S31 and S32), transmission electron microscopy (Fig. S33 and S34), and solid-state NMR spectroscopy (Fig. S35) confirmed that the recovered CN-OA-m retained its original structural characteristics. It could be reused five times under identical conditions with only a slight decrease in the yield of the coupling product77. Taken together, the post-characterization data for the recovered CN-OA-m clearly demonstrated the robustness, stability, and durability of this semiheterogeneous photocatalyst.

A Synthesis of tetracaine via coupling reactions under various light sources; (B) Recycling of CN-OA-m after coupling reactions under standard conditions for 24 h; (C) Electron paramagnetic spectroscopic detection of a nitrogen vacancy (298 K); (D) Experiments involving electron and hole scavengers; (E) Detection of Ni(I) species (100 K); (F) Ni(I) initiated catalytic coupling reaction. For details, see Supporting Information.
Next, we carried out mechanistic studies. The UV–vis spectrum of the prepared CN-OA-m showed an absorption band in the red-light region (Fig. S16-17). Additionally, after a solution of CN-OA-m in DMAc was irradiated with red light for 10 min, a signal with a g value of 2.003 was observed in the electron paramagnetic resonance spectrum (Fig. 8C; and See SI section 11.2), a finding that is consistent with literature reports61,65 and that suggests that red-light irradiation produced electrons. Moreover, the progress of the red-light-promoted reaction slowed dramatically after the addition of an electron (e−) (AgNO3 or KI) or a hole (h+) scavenger (Na2S/Na2SO3 or EDTA) (Fig. 8D; and See SI section 7)66,88. These results confirm that red-light excitation of photogenerated charges played a major role in this photocatalytic coupling reaction. Furthermore, when CN-OA-m (2 mg) was used as the photocatalyst and a Ni(II)(ligand)Br2 complex was the metal catalyst under red-light irradiation for 90 min, electron paramagnetic resonance spectroscopy revealed a distinct signal at gavg = 2.204 (Fig. 8E; and See SI section 11.1)89,90,91, indicating the formation of a Ni(I) species. Therefore, we speculate that upon red-light excitation, CN-OA-m served as an electron donor to reduce the Ni(II)(ligand)Br2 complex to a Ni(I) species via a single-electron transfer. In addition, in the absence of substrates, the Ni(II)(ligand)Br2 complex was irradiated with red light for 90 minutes in the presence of CN-OA-m, aiming to in situ produce the presumed Ni(I) species; and then an aryl bromine and an amine were introduced and allowed to react in the dark for 24 h. This experiment delivered the desired product in 29% yield by NMR spectroscopy (Fig. 8F; and see SI section 10.7). These results indicate that under red-light irradiation, a catalytically active Ni(I) species was generated.
Our previous work89,92,93,94,95,96,97 and the above-mentioned observations support the view that CN-OA-m absorbs photons of enough energy under red light irradiation, excitation of electrons from the valence band potential (VB, EVB = 0.88 V) to the conduction band potential (CB, ECB = −1.65 V) occurs with consequent charge separation (formation of a hole-electron pair), the as-formed electrons (e-) from the emissive state to reduce Ni(II) (Ei [NiII/NiI] = −1.43 V vs SCE)98,99 to generate a Ni(I) species via SET, suggesting that this cross-coupling reaction likely proceeds via a Ni(I)/Ni(III) cycle (Fig. 9). Specifically, the absorption of red light by CN-OA-m triggers a charge-separation process that produces redox centers in the form of electron–hole pairs. The electrons in the conduction band in the photogenerated hole reduce Ni(II) via a single-electron transfer to form the active Ni(I) species, and the electron of organic base (mDBU, Ep/2 = +1.39 V vs Ag/AgCl in MeCN) transfer to the holes58,59,62,66 to complete the photocatalytic cycle. Concurrently, the Ni(I) species undergoes an oxidative addition reaction with the aryl halide to form a Ni(III) complex, which undergoes ligand exchange with the nucleophile. The resulting species is deprotonated by the organic base, yielding an Ar-Ni(III)-Nu species, which then undergoes a reductive elimination reaction to release the ArNu coupling product and a Ni(I) species to start the next catalytic cycle. When the illumination is stopped, the formation of a catalytically inactive Ni(II) species by comproportionation of Ni(III) and Ni(I) species89,92,93,94,95,96,98,99 or reoxidation60 of Ni(I) by the organic base radical cation interrupts the reaction. Continuous illumination is crucial to converting these inactive, off-cycle Ni(II) species back to the active Ni(I) species, thereby maintaining the catalytic cycles.

Proposed catalytic cycle for the red-light-driven Ni-catalyzed cross-coupling reaction.
In summary, we have developed a method for red-light-driven Ni-catalyzed C–heteroatom cross-coupling reactions using CN-OA-m as a photocatalyst. The versatile Ni/red-light semiheterogeneous catalyst system enables the formation of four different types of C–heteroatom bonds and has a broad substrate scope. Notably, the use of a low-energy red light results in broad functional group tolerance, demonstrating the method’s excellent potential utility for pharmaceutical chemistry. Catalyst-recycling experiments demonstrated that the CN-OA-m could be recovered and reused at least five times. This method provides a new platform for the development of other reactions involving red-light metallaphotoredox catalysis.
Methods
Standard procedure for the C-N couplingh
To an oven-dried 10 mL of storage tube were added NiBr2·glyme (10.0 mol%), 1 mL of DMAc and a magnetic stir bar under argon atmosphere. The mixture was evacuated and backfilled with argon for 3 times. Then the aryl bromide (0.2 mmol), amine (0.4 mmol), CN-OA-m (10 mg/mL) and mDBU (1,4,5,6-tetrahydro-1,2-dimethylpyrimidine) (1.5 eq., 0.3 mmol) were added. The tube was sealed with a Teflon screw valve. The reaction mixture was then irradiated with 10 W red LEDs (0.5 cm away from the tube, 660–670 nm) at 85 °C. After the reaction was completed, the mixture was diluted with ethyl acetate after cooling to room temperature. The organic phases were washed with saturated ammonium chloride (3 × 10 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by flash column chromatography using petroleum ether and ethyl acetate as eluent to afford aryl amines.
Standard procedure for the C-O coupling
To an oven-dried 10 mL of storage tube were added d-tbbpy (4,4’-Di-tert-butyl-2,2’-dipyridyl) (10.0 mol%), NiBr2·glyme (10.0 mol%), 1 mL of DMF and a magnetic stir bar under argon atmosphere. The mixture was evacuated and backfilled with argon for 3 times. Then aryl bromide (0.2 mmol), O-nucleophilic reagent (0.6 mmol), CN-OA-m (10 mg/mL) and mDBU (1,4,5,6-tetrahydro-1,2-dimethylpyrimidine) (1.5 eq., 0.3 mmol) were added. The tube was sealed with a Teflon screw valve. The reaction mixture was then irradiated with 10 W red LEDs (0.5 cm away from the tube, 620–630 nm) at 85 °C. After the reaction was completed, the mixture was diluted with ethyl acetate after cooling to room temperature. The organic phases were washed with saturated ammonium chloride (3 × 10 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by flash column chromatography using petroleum ether and ethyl acetate as eluent to afford aryl ethers.
Standard procedure for the C-S/Se coupling
To an oven-dried 10 mL of storage tube were added d-tbbpy (4,4’-Di-tert-butyl-2,2’-dipyridyl) (10.0 mol%), NiBr2·glyme (10.0 mol%), 1 mL of DMAc and a magnetic stir bar under argon atmosphere. The mixture was evacuated and backfilled with argon for 3 times. Then aryl bromide (0.2 mmol), S-nucleophilic reagent (0.4 mmol), CN-OA-m (10 mg/mL) and mDBU (1,4,5,6-tetrahydro-1,2-dimethylpyrimidine) (1.5 eq., 0.3 mmol) were added. The tube was sealed with a Teflon screw valve. The reaction mixture was then irradiated with 10 W red LEDs (0.5 cm away from the tube, 620–630 nm) at 85 °C. After the reaction was completed, the mixture was diluted with ethyl acetate after cooling to room temperature. The organic phases were washed with saturated ammonium chloride (3 × 10 mL), dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by flash column chromatography using petroleum ether and ethyl acetate as eluent to afford aryl sulfides.
Data availability
Data are available in this study are provided in the Supplementary Information. All other data are available from the authors upon request.
References
Dorel, R., Grugel, C. P. & Haydl, A. M. The Buchwald–Hartwig amination after 25 years. Angew. Chem. Int. Ed. 58, 17118 (2019).
Ingoglia, B. T., Wagen, C. C. & Buchwald, S. L. Biaryl monophosphine ligands in palladium-catalyzed C–N coupling: An updated User’s guide. Tetrahedron 75, 4199 (2019).
Bhunia, S., Pawar, G. G., Kumar, S. V., Jiang, Y. & Ma, D. Selected copper-based reactions for C−N, C−O, C−S, and C−C bond formation. Angew. Chem. Int. Ed. 56, 16136 (2017).
Marín, M., Rama, R. J. & Nicasio, M. C. Ni-catalyzed amination reactions: an overview. Chem. Rec. 16, 1819 (2017).
Correa, A. & Bolm, C. Iron-catalyzed N-arylation of nitrogen nucleophiles. Angew. Chem. Int. Ed. 46, 8862 (2007).
Zhang, J., Wang, S., Zhang, Y. & Feng, Z. Iron-catalyzed cross-coupling reactions for the construction of carbon-heteroatom bonds. Asian J. Org. Chem. 9, 1519 (2020).
Cahiez, G. & Moyeux, A. Cobalt-catalyzed cross-coupling reactions. Chem. Rev. 110, 1435 (2010).
Han, J.-F., Guo, P., Zhang, X.-G., Liao, J.-B. & Ye, K.-Y. Recent advances in cobalt-catalyzed allylic functionalization. Org. Biomol. Chem. 18, 7740 (2020).
Carney, J. R., Dillon, B. R. & Thomas, S. P. Recent advances of manganese catalysis for organic synthesis. Eur. J. Org. Chem. 2016, 3912 (2016).
Sortais. J.-B. Manganese Catalysis in Organic Synthesis, WILEY-VCH GmbH, 2022.
Levin, M. D., Kim, S. & Toste, F. D. Photoredox catalysis unlocks single-electron elementary steps in transition metal catalyzed cross-coupling. ACS Cent. Sci. 2, 293 (2016).
Shaw, M. H., Twilton, J. & MacMillan, D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 81, 6898 (2016).
Twilton, J. et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 1, 0052 (2017).
Marzo, L., Pagire, S. K., Reiser, O. & König, B. Visible-light photocatalysis: Does it make a difference in organic synthesis?. Angew. Chem. Int. Ed. 57, 10034 (2018).
Cavedon, C., Seeberger, P. H. & Pieber, B. Photochemical strategies for carbon–heteroatom bond formation. Eur. J. Org. Chem. 2020, 1379 (2020).
Hartwig, J. F. Palladium-catalyzed amination of aryl halides and sulfonates. John Wiley & Sons, Ltd. 853 (2020).
Marín, M., Rama, R. J. & Nicasio, M. C. Ni-catalyzed amination reactions: An overview. Chem. Rec. 16, 1819 (2016).
Baumgartner, L. M., Dennis, J. M., White, N. A., Buchwald, S. L. & Jensen, K. F. Use of a droplet platform to optimize Pd-catalyzed C–N coupling reactions promoted by organic bases. Org. Process Res. Dev. 23, 1594 (2019).
Voloshkin, V. A., Zorba, L. P. & Nolan, S. P. The influential IPr: 25 years after its discovery. Chem. Sci. 16, 2062 (2025).
Forero-Cortés, P. A. & Haydl, A. M. The 25th anniversary of the Buchwald–Hartwig amination: development, applications, and outlook. Org. Process Res. Dev. 23, 1478 (2019).
Reichert, E. C., Feng, K., Sather, A. C. & Buchwald, S. L. Pd-catalyzed amination of base-sensitive five-membered heteroaryl halides with aliphatic amines. J. Am. Chem. Soc. 145, 3323 (2023).
Kim, S.-T., Strauss, M. J., Cabré, A. & Buchwald, S. L. Room-temperature Cu-catalyzed amination of aryl bromides enabled by DFT-guided ligand design. J. Am. Chem. Soc. 145, 6966 (2023).
Strauss, M. J. et al. Cu-catalyzed amination of base-sensitive aryl bromides and the chemoselective N- and O- arylation of amino alcohols. J. Am. Chem. Soc. 146, 18616 (2024).
Choi, K. et al. Palladium-catalyzed amination of aryl halides with aqueous ammonia and hydroxide base enabled by ligand development. J. Am. Chem. Soc. 146, 19414 (2024).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322 (2013).
Skubi, K. L., Blum, T. R. & Yoon, T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 116, 100359 (2016).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075 (2016).
Candish, L. et al. Photocatalysis in the Life Science Industry. Chem. Rev. 122, 2907 (2022).
Markushyna, Y. C., Smith, A. & Savateev, A. Organic photocatalysis: carbon nitride semiconductors vs. molecular catalysts. Eur. J. Org. Chem. 2019, 1294 (2019).
Ruban, S. M. et al. Organocatalysis with carbon nitrides. Sci. Technol. Adv. Mater. 24, 2188879 (2023).
Zhou, P., Yang, Q., Tang, Y. & Cai, Y. Recent trends in metal-doped carbon nitride-catalyzed heterogeneous light-driven organic transformations. Nano Trends 4, 100019 (2023).
Ghosh, I. et al. General cross-coupling reactions with adaptive dynamic homogeneous catalysis. Nature 619, 87 (2023).
Gisbertz, S., Reischauer, S. & Pieber, B. Overcoming limitations in dual photoredox/nickel-catalysed C–N cross-couplings due to catalyst deactivation. Nat. Catal. 3, 611 (2020).
Shen, Z. et al. Red-light-mediated photoredox catalysis enables self-reporting nitric oxide release for rfficient antibacterial treatment. Angew. Chem. Int. Ed. 60, 20452 (2021).
Zeng, L., Huang, L., Lin, W., Jiang, L.-H. & Han, G. Red light-driven electron sacrificial agents-free photoreduction of inert aryl halides via triplet-triplet annihilation. Nat. Commun. 14, 1102 (2023).
Ravetz, B. D. et al. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 565, 343 (2019).
Glaser, F. & Wenger, O. S. Sensitizer-controlled photochemical reactivity via upconversion of red light. Chem. Sci. 14, 149 (2023).
Mato, M. et al. Oxidative addition of aryl electrophiles into a red-light-active bismuthinidene. J. Am. Chem. Soc. 145, 18742 (2023).
Goldschmid, S. L. et al. Overcoming photochemical limitations in metallaphotoredox catalysis: Red-light-driven C–N cross-coupling. J. Am. Chem. Soc. 144, 22409 (2022).
Cabanero, D. C., Nguyen, J. A., Cazin, C. S. J., Nolan, S. P. & Rovis, T. Deep red to near-infrared light-controlled ruthenium-catalyzed olefin metathesis. ACS Catal. 13, 4384 (2023).
Tay, N. E. S. et al. Targeted activation in localized protein environments via deep red photoredox catalysis. Nat. Chem. 15, 101 (2023).
Ravetz, B. D. et al. Development of a platform for near-infrared photoredox catalysis. ACS Cent. Sci. 6, 2053 (2020).
Sellet, N., Cormier, M. & Goddard, J.-P. The dark side of photocatalysis: near-infrared photoredox catalysis for organic synthesis. Org. Chem. Front. 8, 6783 (2021).
Schade, A. H. & Mei, L. Applications of red light photoredox catalysis in organic synthesis. Org. Biomol. Chem. 21, 2472 (2023).
Bols, M. L. et al. In situ UV–Vis–NIR absorption spectroscopy and catalysis. Chem. Rev. 124, 2352 (2024).
Mei, L., Veleta, J. M. & Gianetti, T. L. Helical carbenium ion: a versatile organic photoredox catalyst for red-light mediated reactions. J. Am. Chem. Soc. 142, 12056 (2020).
Mei, L., Moutet, J., Stull, S. M. & Gianetti, T. L. Synthesis of CF3-containing spirocyclic indolines via a red-light mediated trifluoromethylation/dearomatization cascade. J. Org. Chem. 86, 10640 (2021).
Hossain, M. M., Shaikh, A. C., Kaur, R. & Gianetti, T. L. Red light–blue light chromoselective C(sp2)–X bond activation by organic helicenium-based photocatalysis. J. Am. Chem. Soc. 146, 7922 (2024).
Lang, X., Chen, X. & Zhao, J. Heterogeneous visible light photocatalysis for selective organic transformations. Chem. Soc. Rev. 43, 473 (2014).
Chen, J., Cen, J., Xu, X. & Li, X. The application of heterogeneous visible light photocatalysts in organic synthesis. Catal. Sci. Technol. 6, 349 (2016).
Ong, W.-J., Tan, L.-L., Ng, Y., Yong, H. & Chai, S.-T. S.-P. Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: Are we a step closer to achieving sustainability?. Chem. Rev. 116, 7159 (2016).
Savateev, A., Ghosh, I., König, B. & Antonietti, M. Photoredox catalytic organic transformations using heterogeneous carbon nitrides. Angew. Chem. Int. Ed. 57, 15936 (2018).
Gisbertz, S. & Pieber, B. Heterogeneous photocatalysis in organic synthesis. ChemPhotoChem 4, 456 (2020).
Mazzanti, S. & Savateev, A. Emerging concepts in carbon nitride organic photocatalysis. ChemPlusChem 85, 2499 (2020).
Ghosh, I. et al. Organic semiconductor photocatalyst can bifunctionalize arenes and heteroarenes. Science 365, 360 (2019).
Zhao, X. et al. Nickel-coordinated carbon nitride as a metallaphotoredox platform for the cross-coupling of aryl halides with alcohols. ACS Catal. 10, 15178 (2020).
Vijeta, A., Casadevall, C. & Reisner, E. An integrated carbon nitride-nickel photocatalyst for the amination of aryl halides using sodium azide. Angew. Chem. Int. Ed. 61, e202203176 (2022).
Qin, Y., Martindale, B. C. M., Sun, R., Rieth, A. J. & Nocera, D. G. Solar-driven tandem photoredox nickel-catalysed cross-coupling using modified carbon nitride. Chem. Sci. 11, 7456 (2020).
Liu, Q. et al. Vacancy engineered polymeric carbon nitride nanosheets for enhanced photoredox catalytic efficiency. Cell Rep. Phy. Sci. 2, 100491 (2021).
Li, Z. et al. Integrated nickel/polymer dual catalytic system for visible-light-driven sulfonamidation between aryl halides and aryl sulfonamides. Chem. Catal. 2, 3546 (2022).
Li, Z. et al. Engineering single–atom active sites anchored covalent organic frameworks for efficient metallaphotoredox single bond C-N cross–coupling reactions. Sci. Bull. 67, 1971 (2022).
Kwak, M. et al. Ni single atoms on carbon nitride for visible-light promoted full heterogeneous dual catalysis. Chem. Sci. 13, 8536 (2022).
Xing, L. et al. Poly(heptazine imide) ligand exchange enables remarkable low catalyst loadings in heterogeneous metallaphotocatalysis. Nat. Commun. 14, 1501 (2023).
Pieber, B. et al. Semiheterogeneous dual nickel/photocatalysis using carbon nitrides: esterification of carboxylic acids with aryl halides. Angew. Chem. Int. Ed. 58, 9575 (2019).
Cavedon, C., Madani, A., Seeberger, P. H. & Pieber, B. Semiheterogeneous dual nickel/photocatalytic (thio)etherification using carbon nitrides. Org. Lett. 21, 5331 (2019).
Wang, K. et al. Visible light-catalytic hydroxylation of aryl halides with water to phenols by carbon nitride and nickel complex cooperative catalysis. Green. Chem. 22, 7417 (2020).
Vijeta, A., Casadevall, C., Roy, S. & Reisner, E. Visible-light promoted C–O bond formation with an integrated carbon nitride–nickel heterogeneous photocatalyst. Angew. Chem. Int. Ed. 60, 8494 (2021).
Nikitin, M. et al. C(sp2)–S cross-coupling reactions with nickel, visible light, and mesoporous graphitic carbon nitride. Green. Chem. 26, 5845 (2024).
Khamrai, J. et al. Photo-Ni-dual-catalytic C(sp2)–C(sp3) cross-coupling reactions with mesoporous graphitic carbon nitride as a heterogeneous organic semiconductor photocatalyst. ACS Catal. 10, 3526 (2020).
Filippini, G. et al. Light-driven, heterogeneous organocatalysts for C-C bond formation toward valuable perfluoroalkylated intermediates. Sci. Adv. 6, eabc9923 (2020).
Das, S. et al. Photocatalytic (Het)arylation of C(sp3)–H bonds with carbon Nnitride. ACS Catal. 11, 1593 (2021).
Schirmer, T. E. et al. Mesoporous graphitic carbon nitride as a heterogeneous organic photocatalyst in the dual catalytic arylation of alkyl bis(catecholato)silicates. Org. Lett. 24, 2483 (2022).
Dan, X. et al. Heterogeneous metallaphotocatalytic C(sp2)–C(sp3) cross-coupling reactions with integrated bipyridyl-Ni(II)-carbon nitride. Org. Lett. 25, 4124 (2023).
Song, H.-Y. et al. Semi-heterogeneous g-C3N4/NaI dual catalytic C–C bond formation under visible light. Green. Chem. 25, 3292 (2023).
Zhang, G. et al. Optimizing optical absorption, exciton dissociation, and charge transfer of a polymeric carbon nitride with ultrahigh solar hydrogen production activity. Angew. Chem. Int. Ed. 56, 13445 (2017).
Schmermund, L. et al. Chromoselective photocatalysis enables stereocomplementary biocatalytic pathways. Angew. Chem. Int. Ed. 60, 6965 (2021).
For details, see Supplementary Information.
Teter, D. M. & Hemley, R. J. Low-compressibility carbon nitrides. Science 271, 53 (1996).
Goettmann, F., Fischer, A., Antonietti, M. & Thomas, A. Chemical synthesis of mesoporous carbon nitrides using hard templates and their use as a metal-free catalyst for friedel–crafts reaction of benzene. Angew. Chem. Int. Ed. 45, 4467 (2006).
Liu, S., Zhu, H., Yao, W., Chen, K. & Chen, D. One step synthesis of P-doped g-C3N4 with the enhanced visible light photocatalytic activity. Appl. Surf. Sci. 430, 309 (2018).
Molaei, M. Graphitic carbon nitride (g-C3N4) synthesis and heterostructures, principles, mechanisms, and recent advances: a critical review. Int. J. Hydrog. Energ. 48, 32708 (2023).
Yang, Y. et al. An unusual red carbon nitride to boost the photoelectrochemical performance of wide bandgap photoanodes. Adv. Funct. Mater. 28, 1805698 (2018).
Zhai, B. et al. A crystalline carbon nitride based near-infrared active photocatalyst. Adv. Funct. Mater. 32, 2207375 (2022).
Hong, J., Xia, X., Wang, Y. & Xu, R. Mesoporous carbon nitride with in situ sulfur doping for enhanced photocatalytic hydrogen evolution from water under visible light. J. Mater. Chem. 22, 15006 (2012).
Zeng, W. et al. Crystalline carbon nitride with in-plane built-in electric field accelerates carrier separation for excellent photocatalytic hydrogen evolution. Chin. Chem. Lett. 35, 109252 (2024).
Ran, J., Ma, T. Y., Gao, G., Duc, X.-W. & Qiao, S. Z. Porous P-doped graphitic carbon nitride nanosheets for synergistically enhanced visible-light photocatalytic H2 production. Energy Environ. Sci. 8, 3708 (2015).
Luo, Q.-Z. et al. Steering K/C dual sites to orient lewis acid/oxygen species. Rare Met. https://doi.org/10.1007/s12598-024-03176-9 (2025).
Deshmukh, S. A. & Bhagat, P. R. Efficient and reusable benzimidazole based sulphonic acid functionalized porphyrin photocatalyst for C–N bond formation under visible light irradiation. Catal. Lett. 153, 3230 (2023).
Song, G. et al. Werner salt as nickel and ammonia source for photochemical synthesis of primary aryl amines. Angew. Chem. Int. Ed. 63, e202314355 (2024).
Johnson Humphrey, E. L. B., Kennedy, A. R., Sproules, S. & Nelson, D. J. Evaluating a dispersion of sodium in sodium chloride for the synthesis of low-valent nickel complexes. Eur. J. Inorg. Chem. 2022, e202101006 (2022).
Ting, S. I., Williams, W. L. & Doyle, A. G. Oxidative addition of aryl halides to a Ni(I)-bipyridine complex. J. Am. Chem. Soc. 144, 5575 (2022).
Li, G. et al. Light-promoted C–N coupling of aryl halides with nitroarenes. Angew. Chem. Int. Ed. 60, 5230 (2021).
Song, G. et al. Chiral arylated amines via C−N coupling of chiral amines with aryl bromides promoted by light. Angew. Chem. Int. Ed. 60, 21536 (2021).
Song, G. et al. Photochemical synthesis of anilines via Ni-catalyzed coupling of aryl halides with ammonium salts. ACS Catal. 12, 15590 (2022).
Song, G. et al. Light-promoted efficient generation of Fe(I) to initiate amination. ACS Catal. 14, 4968 (2024).
Song, G. et al. Adaptive photochemical amination via Co(II) catalysis. J. Am. Chem. Soc. 146, 26936 (2024).
West, T. C–N coupling with cobalt. Nat. Synth. 3, 1186 (2024).
Till, N. A., Tian, L., Dong, Z., Scholes, G. D. & MacMillan, D. W. C. Mechanistic analysis of metallaphotoredox C–N coupling: photocatalysis initiates and perpetuates Ni(I)/Ni(III) coupling activity. J. Am. Chem. Soc. 142, 15830 (2020).
Lin, Q., Dawson, G. & Diao, T. Experimental electrochemical potentials of nickel complexes. Synlett 32, 1606 (2021).
Acknowledgements
This research is supported by the National Natural Science Foundation of China (Grant No. 22171174, 22471150 to D.X.; Grant No. 22402113 to G.S., Grant No. 21872089 to Q.G.), the Fundamental Research Funds for the Central Universities (Grant No. GK202406026 to T.K.), the Innovation Capability Support Program of Shaanxi (Grant No. 2023-CX-TD-28 to D.X.), the Fundamental Science Research Project of Shaanxi for Chemistry, Biology (Grant No. 22JHZ002 to D.X.), the Natural Science Foundation of Shaanxi Province (Grant No. 2019JM-043 to D.X., Grant No. 2024JC-YBQN-0075 to G.S., Grant No. 2025JC-YBQN-144 to T.K., Grant No. 2025JC-YBMS-148 to Q.G.), and the State Key Laboratory of Natural and Biomimetic Drugs (Grant No. K202437 to to G.S.).
Author information
Authors and Affiliations
Contributions
D.X. conceived and directed the project. G.S. synthesized catalysts, designed and conducted experiments, analyzed data. W.Z. synthesized various C3N4 catalysts, conducted their characterization and discussed their catalytic activity. J.S. and Q.L. participated the substrate scope expansion. Y.F., J.F., H.L. helped with catalyst synthesis and characterization. T.K., J.D., G.L., X.P.Z., C.W. contributed to project discussion. Q.G. participated in the guidance of synthetic catalysts and revised the manuscript. G.S. prepared the manuscript. D.X. finally wrote the manuscript. All authors discussed the experimental results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qiong Liu, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, G., Zhang, W., Song, J. et al. General method for carbon–heteroatom cross-coupling reactions via semiheterogeneous red-light metallaphotocatalysis. Nat Commun 16, 7045 (2025). https://doi.org/10.1038/s41467-025-61812-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-61812-z
