
Abstract
Bacterial σ factors bind RNA polymerase (E) to form holoenzyme (Eσ), conferring promoter specificity to E and playing a key role in transcription bubble formation. σN is unique among σ factors in its structure and functional mechanism, requiring activation by specialized AAA+ ATPases. EσN forms an inactive promoter complex where the N-terminal σN region I (σN-RI) threads through a small DNA bubble. On the opposite side of the DNA, the ATPase engages σN-RI within the pore of its hexameric ring. Here, we perform kinetics-guided structural analysis of de novo formed EσN initiation complexes and engineer a biochemical assay to measure ATPase-mediated σN-RI translocation during promoter melting. We show that the ATPase exerts mechanical action to translocate about 30 residues of σN-RI through the DNA bubble, disrupting inhibitory structures of σN to allow full transcription bubble formation. A local charge switch of σN-RI from positive to negative may help facilitate disengagement of the otherwise processive ATPase, allowing subsequent σN disentanglement from the DNA bubble.
Similar content being viewed by others

Introduction
Specific promoter recognition during bacterial transcription initiation requires the association of a σ factor with the RNA polymerase (RNAP) core enzyme (α2ββ‘ω; E) to form the holoenzyme (Eσ)1. Multiple alternative σ factors compete for E, enabling the cell to switch transcriptional programs. Alternative σ factors are almost all homologous to the housekeeping Escherichia coli (Eco) σ70 factor2, but nearly all bacterial clades except Actinobacteria harbor a structurally and evolutionarily unrelated σ factor, σN (also known as RpoN or σ54)3,4. σN mediates diverse transcriptional pathways, most notably the expression of virulence factors in many human bacterial pathogens, including Pseudomonas aeruginosa, Vibrio cholerae, the Lyme disease agent Borrelia burgdorferi, Chlamydia trachomatis, and Helicobacter pylori5,6,7,8,9, and the control of nitrogen metabolism in plant symbionts, such as Rhizobium sp.10.
Unlike factors of the σ70 family, transcription initiation by EσN proceeds through a stable, inactive promoter complex that requires ATP-dependent post-recruitment activation mediated by a bacterial enhancer-binding protein (bEBP)3,4. The bEBPs, which belong to the large superfamily of AAA+ proteins (ATPases associated with diverse cellular activities), activate transcription through an unknown mechanism. All known bEBPs form hexameric rings and consist of a central AAA+ domain, with variable presence of an N-terminal regulatory domain and/or a C-terminal DNA binding domain enabling recognition of promoter-distal enhancer sequence motifs11. Full DNA melting and progression to the transcription-competent ‘open promoter complex’ (RPo) are prohibited by an interaction of σN N-terminal region I (RI) with the extra-long helix (ELH) (Supplementary Fig. 1A)12,13,14,15. At promoters, EσN recognizes conserved sequence elements located 12 and 24 base pairs upstream (the −12 and −24 elements) of the transcription start site (TSS; position +1) via the σN ELH helix-turn-helix (HTH) domain and RpoN domain, respectively16,17,18, forming a stable early melted intermediate (RPem) in which two base pairs (at −12 and −11) are melted19. In RPem, an N-terminal segment of σN-RI threads through the DNA bubble20,21. The bEBP engages σN-RI on the opposite side of the DNA bubble with highly conserved GAFTGA loops in the AAA+ domain20,22.
Recent structural work revealed the interaction of σN-RI inside the pore of the hexameric ring20. The structure is trapped with ADP aluminum fluoride (ADP-AlFx) but leads to an attractive hypothesis: the ATP hydrolysis cycle of the AAA+ bEBP pulls σN-RI through its pore, mechanically unfolding σN-RI and disrupting the σN-RI:ELH interactions. The mechanical disruption of the σN-RI:ELH interaction could explain how bEBP action allows RPo formation to proceed, but this mechanistic hypothesis also raises crucial questions: How many rounds of ATP hydrolysis are required to disrupt the σN-RI:ELH interactions? Most AAA+ translocases are processive23; does the bEBP thread the entire length of the σN polypeptide (Eco σN is 477 residues in length) through the DNA bubble? Alternatively, the bEBP could disengage from σN after RPo formation, but what is the signal for disengagement?
To address these questions, we perform a kinetics-guided structural analysis of de novo formed EσN initiation complexes using cryo-electron microscopy (cryo-EM) and engineer a biochemical assay to measure bEBP-mediated σN-RI translocation in initiation complexes. We show that the bEBP translocates about 30 residues of σN-RI through the DNA bubble, converting ATP hydrolysis into mechanical force to disrupt the σN-RI:ELH inhibitory interface to allow full transcription bubble formation. Our data suggest that a switch of local net charge in σN-RI from positive to negative facilitates disengagement of the otherwise processive bEBP, permitting σN disentanglement from the DNA bubble during later steps.
Results
Kinetics-informed capture of actively initiating EσN-bEBP complexes by cryo-EM
To capture and structurally visualize intermediates during bEBP action, we first determined the kinetics of RPo formation. We employed a fluorescence assay where a Cy3 fluorophore attached to the +2 position experiences protein-induced fluorescence enhancement (PIFE)24 upon RPo formation (Fig. 1a)25. Our transcription system comprised Eco EσN, a consensus σN promoter (the Aquifex aeolicus dhsU promoter16,26), and a constitutively active variant of the A. aeolicus (Aae) bEBP NtrC1 (C1; N-terminal regulatory domain and C-terminal enhancer DNA-binding domain removed)27. To enable correct positioning of the fluorescent label, we determined the TSS for both the wild-type (+2A) and mutant (+2T) promoters (Cy3 is attached via a thymine base) under our experimental conditions (Supplementary Table 1). A Cy3-labeled dhsU promoter fragment was synthesized (dhsU+2T-Cy3; Supplementary Table 2) and used in a stopped flow assay to determine the kinetics of RPo formation. The fluorescence signal reached a plateau after about 5 min, indicating RPo formation was complete (Fig. 1b). Kinetic analysis of the data yielded a maximum apparent rate constant for RPo formation of about 0.009 s−1 (Supplementary Figs. 1B–D). Accounting for the difference in reaction temperature, these results are consistent with previously reported data28.

a Stopped-flow measurement employs a linear, Cy3-labeled (yellow) σN promoter DNA (Aae dhsU). The fluorophore is subject to protein-induced fluorescent enhancement (PIFE) when RPo forms. For the experiment, an early-melted intermediate (box 1; E + σN+dhsU+C1) is pre-assembled from core RNAP (E, gray), σN (orange), and Cy3-labeled dhsU (green) in presence of bEBP (C1; blue). ATP is prepared in buffer (box 2). Mixing the contents of boxes 1 and 2 will start the reaction and PIFE can be observed. b RPo formation-dependent fluorescence saturates at around 8–10 min reaction time and is dependent on the presence of all components (black; n = 6). Mixing ATP with complexes without C1 (red; n = 5), ATP with DNA alone (blue; n = 3), or the E + σN+dhsU+C1 with buffer (no ATP, green; n = 6) results in only background signal. Solid lines represent the average of the individual mixing events. Dotted lines indicate error bands of one standard deviation. The arrow indicates the time point used for cryo-EM sample preparation. c Cryo-EM analysis of samples prepared after 35 s reaction time yields three major populations: early-melted intermediates (RPem, 31% of particles), bEBP-bound complexes (55% of particles), and complexes with a melted promoter DNA (RPo-like, 14%). Maps from the 3D classification (see main text and methods for details) were colored according to core RNAP (gray), σN (orange), dhsU (green), and bEBP (blue). The dashed arrows indicate the sequence of the complexes along the transcription initiation pathway. Source data are provided as a Source Data file.
The population of melting intermediates should be greatest around the initial rise of the signal, which occurred around 20–50 s (Fig. 1b). Accordingly, cryo-EM samples were prepared by adding ATP to RPem + C1 and plunged into liquid ethane after 35 s reaction time (Fig. 1b). Data processing revealed a class of bEBP-bound complexes (55%), a class of RPem complexes without bEBP (31%), and a class of RPo-like complexes (no bEBP bound, DNA melted and inserted into the active site cleft of RNAP; 14%) (Fig. 1c; Supplementary Fig. 2A). Thus, our experiment captured complexes along the entire bEBP-EσN RPo formation pathway. The fraction of RPo-like complexes (14%) roughly corresponds with the fraction of maximum signal obtained during the stopped-flow assay at 35 s (25%), establishing consistency between the different experimental approaches (Fig. 1b, c; note that direct correspondence between the two assays is not expected since the fluorescence signal in the stopped-flow assay is not a linear function of RPo abundance).
Structures of EσN-bEBP initiation complexes reveal bEBP-mediated translocation and unfolding of σN-RI
The bEBP-bound class displayed a large variability of the bEBP position around the axis along the σN-bound DNA. Steps of focused classifications and local alignments (see Methods) revealed “open” and “closed” bEBP ring states defined by the presence or absence of a distinct gap in the ring, respectively (Supplementary Fig. 2A). For each ring state, maps encompassing the whole complex were obtained by merging the bEBP-focused map with a map reconstructing RNAP (i.e., without masks) and a σN-focused map. Nominal FSC resolution estimates were similar across both ring states: 3.0–3.1 Å for the bEBP and σN maps, and 2.7 Å for the RNAP maps, with homogeneous distributions of angular views (Table 1; Supplementary Figs. 3 and 4).
The structures of both ring states share several common features: Density for the dhsU promoter fragment is clearly resolved from about −35 to −2 (Fig. 2a). As in the RPem state (without bEBP)20,21, σN-RI threads between the opened DNA strands in the structures of both ring states (Fig. 2a, b). After σN-RI threads through the opened DNA strands, it engages with the pore of the bEBP hexamer on the opposite side of the DNA (Fig. 2a, c-right panel; Supplementary Fig. 5) as previously seen in a bEBP-bound RPem trapped in the presence of ADP-AlFx (RPem-bEBPADP-AlFx; PDB 7QV9; Fig. 2c, left panel)20. In RPem-bEBPADP-AlFx, σN-RI residues between 1–11 are captured in the pore20, revealing the initial capture of σN-RI by the bEBP without translocation (which is prevented by ADP-AlFx). Our EσN-bEBP open and closed ring states (in the presence of ATP) captured σN-RI residues between 3–13 – a two-residue register shift deeper into the pore (Fig. 2c; Supplementary Fig. 5B). The two-residue register shift indicates that the open and closed ring states captured in the presence of ATP underwent one translocation step analogous to the two amino acid step size of protein translocating AAA+ ATPases23. Therefore, we refer to the open and closed ring states as RPi1open and RPi1closed to indicate that they represent a translocation intermediate after one step of ATP hydrolysis.

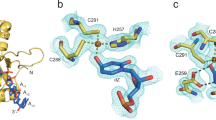
a Overview of the merged, unsharpened cryo-EM map of the “open” ring state. Map density is colored according to the location of subunits, non-template (nt) and template (t) strands. The extent of visible map density for the DNA duplex is indicated by the colored letters at the top. b Helix 1 (H1) of σN-RI exhibits three turns in RPem (left) and RPem-bEBPADP-AlFx (middle), but a full turn is unfolded in the RPi1 intermediate (right). Residue P17 (red) located at the N-terminal end of H1 in RPem serves as visual indicator of helix changes. Regulatory interactions between σN-RI and the ELH remain intact (dotted circle; residues in magenta are mutation sites for bypass variants). Bases corresponding to the −11 position in dhsU are colored in yellow. c Comparison of the σN-RI:bEBP interaction between RPem- bEBPADP-AlFx (left) and RPi1 (right), illustrating the translocation of σN-RI by two residues. Every second residue in σN-RI starting with M1 up to Q11 is highlighted in red. M1 was not modeled for the open and closed ring structures.
In both RPem (PDB 8F1K)21 and RPem-bEBPADP-AlFx (PDB 7QV9)20, Pro17 marks the N-terminal end of σN helix 1 (H1) and sits on the RNAP-side of the small DNA bubble accommodating the threaded σN-RI (Fig. 2b, left and middle panels). By contrast, the bEBP-mediated translocation of σN-RI N-terminal end in the RPi1 structures partly unravels H1, pulling Pro17 more than 9 Å through to the bEBP-side of the DNA bubble (Fig. 2b, right panel). The A-T base pair at the −11 position reforms yielding only a one nucleotide bubble consistent with the reduced spatial requirements for a partially folded H1.
All known ‘bypass’ mutants of σN—which allow RPo formation and transcription initiation without bEBP intervention12,14,15,29,30,31—participate in a network of conserved interactions that stabilize the interaction between σN-RI and the σN-ELH-HTH domain (Fig. 2b)16. Thus, this intra-protein interface is crucial for the proper regulation of σN regulons by preventing unregulated transcription. This regulatory interface includes the C-terminal end of σN-H1 but remains intact in the RPi1 structures despite the unraveling of the σN-H1 N-terminal end (Fig. 2b, right panel). Further bEBP-mediated translocation of σN-RI would presumably unravel σN-H1 completely and disrupt the regulatory interface, allowing RPo formation to proceed.
bEBP-mediated unfolding and threading of σN-RI halts at a defined position
To observe and quantify the extent of the bEBP threading and unfolding activity indicated by the structural data, we engineered a proteolytic assay to measure the extrusion of σN-RI during RPo formation. In this assay, the bEBP was engineered to interact with a compartmentalized protease, where it could translocate σN-RI into the proteolytic chamber for degradation, providing a readout of the extent of σN-RI extrusion (Fig. 3a). We designed our constructs after the proteasome from Mycobacterium tuberculosis (Mtb) and its regulator, the mycobacterial proteasome AAA+ ATPase (Mpa), which features a defined ATPase-proteasome interaction motif (GQYL) and well-characterized interaction to the “open gate” proteasome core variant32,33.

a The structures suggest that the bEBP (blue; shown with C-terminal domains) actively extrudes σN-RI (orange) from the initiation complex (left panel). The mycobacterial proteasome requires an ATPase (Mpa, purple) to feed protein substrates (green) into its proteolytic chamber (middle panel). A short C-terminal motif (GQYL) maintains interaction between proteasome and ATPase (inset). In our assay, an engineered variant of the bEBP carrying the GQYL motif (light blue) interacts with the proteasome. Proteolysis of σN-RI provides a measure for bEBP-mediated translocation during transcription initiation. b Schematic representation of the variants used for the proteolytic assay. c Distinct band shifts of 2Nt-σN, GS7-σN, and Nt-σN in presence of ATP and the bEBP variant carrying the GQYL motif (C1-GS4) demonstrate translocation of σN-RI into the proteasome by the bEBP. Reactions contained 0.5 μM RNAP, 0.5 μM σN, 0.6 μM dhsU-CT, and 0.5 μM C1 or C1-GS4 and 5 mM ATP or ATPγS as indicated. Representative gels of at least two individual experiments are shown. d Processive translocation by the bEBP occurs with σN, C1-GS4, and the proteasome. A translocation halt is only observed with the full transcription complex. Representative gels of three individual experiments are shown. e Estimated length required to reach the proteasome active site predicts bEBP position on σN-RI during translocation halt (black shading). Secondary structure elements of σN-RI present in RPem (H1 and H2) are indicated as gray dashed boxes. The experimentally determined cleavage site is indicated in red (*) on the sequence of Nt-σN and the σN-AAA variant. Conserved net charge distribution of σN-RI (solid line) changes from positive to negative around residues 28–30. Charge distribution of σN-AAA (dashed line) shown for comparison. Sequence logo is based on the alignment shown in Supplementary Fig. 9. f The surface of the inner pore of the bEBP is lined with negative charges near the hydrophobic pockets that capture residues during translocation. Solvent-excluded surface was clipped to reveal the pore interior and colored according to Coulombic electrostatic potential calculated in ChimeraX. Source data are provided as a Source Data file.
We appended the five C-terminal residues of Mpa (LGQYL; the proteasome interaction motif) to the C-terminus of C1 with a four-residue Gly-Ser (GS) linker to generate the variant C1-GS4 (Fig. 3b). C1-GS4 showed similar ATPase activity to C1 in the absence or presence of the proteasome (Supplementary Fig. 6A). Compared to C1, transcriptional activation by C1-GS4 was reduced in the absence of the proteasome but strongly increased in the presence of the proteasome (Supplementary Fig. 6B), confirming that C1-GS4 interacts with the proteasome as designed and was functional for activation of EσN. We further confirmed the formation of the complex between C1-GS4 and the proteasome by low-resolution cryo-EM analysis, demonstrating that C1-GS4 caps the proteasome as designed (Supplementary Fig. 6C).
The shortest distance of the bEBP pore pocket formed by subunits e-f to the closest active site in the proteasome measures about 90 Å, which corresponds to 26 residues assuming a fully stretched peptide chain with 3.5 Å per residue (i.e., using the Ni to Ni+1 distance in a β-sheet peptide as proxy), or roughly 30 residues accounting for structural constraints in the complex. Therefore, at least 30 residues must be translocated by the bEBP before translocation becomes detectable by proteolysis. To enhance the assay’s readout, we generated variants of σN with extended N-termini, where 14 N-terminal residues of σN were duplicated once (Nt-σN), once with a GS linker of 7 residues (GS7-σN; total additional length of 21 residues), or duplicated twice (2Nt-σN; total additional length of 28 residues; Fig. 3b). Each variant was active in a transcription assay but showed decreasing activity with increasing length of the N-terminal extension (Supplementary Fig. 6D and E). However, transcription activity of each variant was restored to near wild-type (WT) levels on a promoter template with a pre-melted two-nucleotide bubble (dhsU-CT; Supplementary Table 2; Supplementary Fig. 6D and E) previously shown to enhance RPem formation34,35. Thus, the σN variants with extended N-termini were partially defective in forming RPem but fully functional in subsequent, bEBP-dependent steps of transcription initiation.
Next, we performed the degradation assay with all σN variants in the context of the full transcription complex – i.e., reactions containing E, WT σN or σN variants, dhsU-CT, C1-GS4 and the proteasome – and analyzed the reactions by denaturing gel electrophoresis (Fig. 3c and Supplementary Fig. 7A–C). We did not detect degradation with WT σN protein, but reactions with Nt-σN, GS7-σN, and 2Nt-σN showed a small but distinct shift of the σN band to lower molecular weight. The band shift was observed in the presence of ATP but not with the non-hydrolyzable nucleotide analog ATPγS or C1, confirming that cleavage of σN-RI is dependent on ATP hydrolysis and the ATPase-proteasome interaction. Observation of a distinct band shift, but not a weaker intensity of the band, demonstrates that the bEBP halts translocation of σN into the proteolytic chamber at a defined position, possibly disengaging from the complex. The translocation/degradation halt was observed only with the full complex (for example, Nt-σN + E + dhsU-CT promoter fragment); Nt-σN alone, Nt-σN + dhsU-CT, or Nt-σN + E were all completely degraded by the C1-GS4 + proteasome combination in an ATP-dependent manner (Fig. 3d and Supplementary Fig. 8), highlighting the intrinsic processivity of C1. Degradation of Nt-σN alone in the absence of nucleotide can be explained by the elevated degradation of poorly structured/partially unstructured substrates by the open gate proteasome36,37,38. The presence of E or dhsU-CT abolishes this activity and stabilizes Nt-σN against stochastic entry into the proteasome (no loss with E or dhsU-CT in the absence of ATP; Fig. 3d and Supplementary Fig. 8).
To determine the cleavage site(s), we subjected the σN protein bands from reactions in the absence or presence of ATP with WT σN, Nt-σN, and GS7-σN to N-terminal sequencing by Edman degradation. Reactions without ATP yielded the N-terminal sequence of the full-length proteins (MKQGL; Supplementary Table 3), as expected. Sequencing the reaction with WT σN in the presence of ATP also yielded the N-terminal sequence of the full-length protein, suggesting that WT σN does not reach the active sites of the proteasome during the process of RPo formation. For reactions with Nt-σN, and GS7-σN in presence of ATP, we identified the N-terminal sequence LAMKQGL or LAGSGS, respectively (residue at the WT N-terminus marked in bold). Cleavage in both σN variants occurred at SQQ/LA, indicating that the GS linker in GS7-σN was strongly disfavored for proteasomal cleavage, which agrees with the reported substrate preference of the Mtb proteasome36,37. We can estimate the point of translocation stop of the bEBP to be roughly 30 residues away from the cleavage site in Nt-σN (Fig. 3e). Curiously, σN-RI exhibits a conserved switch in local charge environment from slightly positive to negative at this location, and the peptide chain retains an overall negative charge throughout σN-RII (Fig. 3e; Supplementary Fig. 9)39. Furthermore, while the bEBP pore pockets possess a hydrophobic character (Supplementary Fig. 5C), the inner pore surface exhibits a negative electrostatic surface charge (Fig. 3f). During translocation beyond σN-RI E32, the resulting charge repulsion between σN-RI and the environment of DNA and the bEBP inner pore may represent a crucial determinant for bEBP disengagement.
To test the hypothesis that the negative charges of the acidic patch in σN-RI promote bEBP disengagement, we substituted the first three acidic residues of σN-RI (E32, E36, E42) with charge-neutral amino acids (Q, S, L, or A; termed σN QSL, QAA or AAA). After confirming that the variants were transcriptionally active on the dhsU promoter (Supplementary Fig. 7D), we subjected them to the proteolytic assay and determined the N-terminal sequences of the products. In the absence of ATP, all variants exhibited the wild-type N-terminal sequence (Supplementary Table 4). In the presence of ATP, the N-terminal protein sequencing indicated a cleavage at SQQ/LAMTPQ (between positions +12/ + 13; shown on the example of σN AAA in Fig. 3e, Supplementary Data 1). The results demonstrate that the bEBP translocated further along σN-RI when the charge switch was located further away from the N-terminus supporting a model in which charge repulsion between σN-RI and the DNA and/or bEBP determines translocation length.
Pre- and post-catalytic states of bEBP nucleotide hydrolysis suggest a single subunit mechanism
The bEBP engages σN-RI protruding from the DNA bubble with its GAFTGA pore loops, thereby coming in close contact with the DNA. Each bEBP subunit contributes highly conserved residue K213 to form a ring of positive charges around the pore entrance. K213 of alternating bEBP subunits of both RPi1open and RPi1closed (subunits a, c, and e) are positioned to make salt bridge contacts with backbone phosphate oxygens of the DNA, possibly holding the bEBP loosely attached to the DNA during σN-RI translocation. Inside the bEBP pore, hydrophobic pockets formed by residues F216 and T217 of the GAFTGA pore loops, Y211, L263 and I203 capture every second residue of σN-RI (Fig. 4a, b; Supplementary Fig. 5B, C). Notably, σN-RI contains a loosely conserved pattern of residues such that the captured residues are most frequently L or Q (Supplementary Fig. 9).

a Conserved “GAFTGA” loops at the bEBP pore (top panel) engage with σN-RI and arrange in a spiral pattern (bottom panel; residues 209–224 of each bEBP subunit and residues 3–15 of σN-RI are shown). Map density for σN-RI (sharpened map) shown as transparent surface (orange). The small inset shows a schematic representation. b Clipped surface representation of bEBP subunits reveals captured side chains of σN-RI in hydrophobic pockets. Pocket-forming residues are shown in stick representation. Subunits a and f are hidden for visual clarity. c Open and closed ring states correspond to ATP- and ADP-bound states of subunit e. ATP colored in red, ADP colored in gray. The C-terminal domain of subunit e extends below the plane of the hexamer in the ATP-bound state (right), but swings into the plane of the ring in the ADP-bound state (left). Small insets indicate conformational change of subunit f schematically. d Active site interactions in subunit e show ATP coordination in a pre-catalytic state (left panel; open ring). Closed ring state contains ADP in the active site of subunit e, corresponding to a post-catalytic state. The arginine finger (R299, f) moved out of the active site (black arrow), and intersubunit salt bridge between E174 (e) and R253 (f) is broken. S-I = sensor I; S-II = sensor II.
In hexameric, ring-forming AAA+ proteins, six ATPase centers are located at the subunit interfaces, where one subunit provides the nucleotide-binding pocket while the neighboring subunit contributes essential residues for catalysis23. The RPi1 states are characterized by different conformations of subunit f and different nucleotide occupancies of subunit e (Fig. 4c): In RPi1open, a gap of about 10–15 Å exists between subunits a and f and subunit f is rotated out of the ring plane. In RPi1closed, the C-terminal domain of subunit f contacts subunit a, thereby bridging the gap, closing the ring and placing subunit f into the plane of the ring. ATP is bound in all active sites of both states, except for the active site of subunit e in RPi1closed, which contains ADP (Fig. 4c and d; Supplementary Fig. 10). In RPi1open, the active site of subunit e is fully formed in a pre-catalytic state poised for catalysis (Fig. 4d): The β- and γ-phosphates of ATP coordinate a Mg2+-ion together with active site residues D238 and E239 of subunit e. R299 of subunit f (the “arginine finger”) reaches into the active site contacting the γ-phosphate to provide transition state stabilization during hydrolysis40,41. By contrast, in RPi1closed, ADP is bound in the e-f pocket, and subunit f is partly dissociated from subunit e, moving the arginine finger out of the active site (Fig. 4d). In both complexes, the active site of subunit a contains an ATP-Mg2+ complex situated in a conformation compatible with catalysis. On the other hand, the binding pockets of subunits b, c and d contain ATP in a markedly different conformation (Supplementary Fig. 10): The γ-phosphate of ATP occupies the catalytic Mg2+-ion position and is far away from the arginine finger, which is incompatible with the common mechanism of catalysis for AAA+ proteins and C1 specifically23,40. The pre- and post-catalytic structures (RPi1open and RPi1closed, respectively) suggest that ATP hydrolysis only occurs in the active site of the second-to-last subunit (e) of the spiral, in agreement with the canonical substrate translocation mechanism of AAA+ proteins23.
σN-RII occupies the RNAP active site cleft
Density in the RNAP downstream channel of the bEBP-bound classes, including RPi1open and RPi1closed, exhibits features consistent with duplex DNA, presumably due to the effective high concentration of DNA ends in the sample, favoring end-binding of DNA. Although this density can be clearly interpreted as duplex DNA, it is comparably weak, suggesting heterogeneity within the particle stack. The classification of the bEBP-bound class (Fig. 1c; Supplementary Fig. 2A) using a mask around the bEBP did not reveal any correlation of the ring position with the presence or absence of DNA in the downstream channel. Hence, we performed another 3D classification of the bEBP-bound class focusing on the downstream channel of RNAP (Supplementary Fig. 2B). Approximately half of the classes (corresponding to 43% of the particles) exhibited additional density for σN-RI and σN-RII, and refinement of the best quality classes yielded map “RII” (nominal FSC resolution estimate of 2.7 Å; Supplementary Fig. 10). In the RII complex, residues 52–56 of σN-RI extend over the β’-clamp domain, loosely interacting with the β’ rudder. About 30 residues of σN-RII, including a small helical region (residues 72–75), run along the β’-side of the RNAP cleft and coil between the bridge helix and the gate loop into the vicinity of the active site (Supplementary Fig. 11A). Binding of RII in the downstream channel agrees with the prevalence of negatively charged residues in RII (Supplementary Fig. 9)39. The RII position in these states clashes with the path of the DNA template strand (t-strand) in RPo (Supplementary Fig. 11B), suggesting that RII counteracts nonspecific nucleic acid binding under physiological conditions, analogous to σ70 region 1.1 (Supplementary Fig. 11C)42. RII binding in the downstream channel was also observed in a crystal structure of EσN43, and thus appears to remain in the downstream channel through promoter binding and the early steps of initiation.
σN-RII intertwines with the t-strand DNA in RPo
To gain insight into subsequent steps of RPo formation with σN, we conducted further cryo-EM analysis of the “melted” class (Fig. 1c) by subjecting the particle stack to another 3D classification (Supplementary Fig. 2C). All classes contained a large transcription bubble with density of varying quality for the t-strand. Similar high-quality classes were pooled and refined, yielding two final classes (Fig. 5a, b; Supplementary Fig. 2C). The first class (RPo) was refined to a global FSC resolution estimate of 2.8 Å (45,631 particles; Supplementary Fig. 12). In the resulting map, the transcription bubble is opened from position −9 to −1 with the TSS forming the last base pair at the edge of the downstream fork (Fig. 5a, c), similar to a previously determined EσN RPo structure44. The bases of the small bubble present in RPem (two nucleotides; at positions −12 and −11) and RPi1 (one nucleotide; at position −12) are paired again and contained within the upstream duplex DNA (Fig. 5c). The σN-ELH is wedged into the bubble with conserved W328 located right at the edge of the upstream fork, possibly playing a role in stabilizing the fork. Density for the bEBP and σN-RI are lost entirely, suggesting that the bEBP has disengaged in the RPo state. Strikingly, σN-RII residues 90–120 loop around the DNA t-strand and exit the RNAP main channel between the β flap domain and the β protrusion (Fig. 5d), seemingly acting as an additional measure to stabilize the open transcription bubble and helping to position the t-strand.

a Unsharpened map of the RPo complex containing a transcription bubble opened from −9 to −1 (top schematic). b Unsharpened map of RPo+2 A, an initial transcribing complex containing nucleotides (red) in the active site. The transcription bubble is opened from −9 to +3 (top schematic). c Schematic comparison of melted regions in the DNA for complexes along the σN transcription initiation pathway. d In RPo (and RPo+2A), σN-RII (red) loops around the DNA t-strand (pale green; left panel) and exits the complex between β-Si2/flap domain and the β protrusion (right panel). The model of RPo is shown with β/β’ subunits in clipped surface representation to reveal the interior. Labeled arrows indicate neighboring structural elements in σN for clarification. e Active site of RPo+2A contains ADP in the i site and ATP in the i + 1 site in a pre-catalytic state. The trigger helix (blue) coordinates triphosphates of ATP (i + 1). Density of the sharpened map around DNA and key residues shown as transparent surface.
The 3D classification of the “melted” particle stack yielded an additional, distinct class (RPo+2A; Supplementary Fig. 2C), where density for the typically flexible β’-Si3 domain is well-defined and located in the cleft between β’ and β above the downstream channel (global FSC resolution of 3.1 Å; 15,649 particles; Supplementary Figs. 2C and 13). The resulting map represents an initial transcribing complex (RPitc) in which the transcription bubble is opened from position −9 to +3 (Fig. 5b, c). The t-strand is positioned in the RNAP active site and RII loops around the t-strand as seen in RPo, but the position where σN-RII crosses the template strand is shifted by one base (Supplementary Fig. 14). The active site contains two nucleotides – ADP in the “i” site (although we cannot rule out a bound ATP with a disordered γ-phosphate) and ATP in the “i + 1” site – and the trigger loop is folded, indicating that the complex is poised for catalysis (Fig. 5e). The templating bases positioned in the “i” and “i + 1” sites are +2T and +3T, placing the TSS (+1C; Supplementary Table 1) in the RNAP main channel. The conditions of complex preparation for cryo-EM included ATP as the sole source of nucleotide, presumably favoring the observed start site selection.
Discussion
Through functional and structural analysis, we show that the bEBP exerts limited ATP-powered mechanical action on σN-RI to pull it through the initial DNA bubble of RPem, thereby disrupting the σN-RI:ELH inhibitory interactions. Translocation of nucleic acids or proteins by AAA+ remodellers occurs typically with high processivity45,46,47,48,49. Although the bEBP displays processivity in the absence of the full transcription complex (degrading the entire 477 amino acid length of σN; Fig. 3d), in the presence of the full complex translocation of σN occurs only for about 15 rounds of ATP hydrolysis, corresponding to roughly 30 residues (Fig. 3e). The amino acid composition of the substrate has been shown to greatly affect translocation efficiency for ClpX50. The local negative net charge in σN-RI after residue 30 appears to create sufficient repulsion between the DNA and/or the inner pore surface of the bEBP to cause the translocation stop (Fig. 3e). To disengage, the bEBP ring may fall off as a complex or dissemble into smaller oligomers or even individual subunits. Therefore, EσN transcription activation represents the bacterial counterpart of an exclusive group of examples found in eukaryotes, where the mechanistic principle of activation by limited substrate translocation by an AAA+ ATPase is utilized: Mitochondrial ClpX activates the initiating enzyme for heme biosynthesis by partial unfolding to enable cofactor insertion51; TRIP13 remodels the cell-cycle protein MAD2 of the mitotic checkpoint complex by local unwinding at its N-terminus to trigger completion of the spindle assembly checkpoint52,53; Rubisco activase rescues the photosynthetic enzyme complex from sugar phosphate inactivation by selective engagement and local destabilization at the C-termini of the large subunits54; NSF recycles membrane fusion SNARE proteins in a single step of ATP hydrolysis, representing the lower extreme of limited translocation55,56.
Determining the kinetics of RPo formation (Fig. 1a and b) enabled timing of the cryo-EM sample preparation to obtain structures of bEBP-bound EσN initiating complexes in the presence of ATP, including intermediates of RPo formation (Figs. 1c and 2). The structures caught the bEBP in action while mechanically unfolding σN-RI, demonstrating that the bEBP works as a protein remodeller, consistent with the function of many AAA+ proteins23. Furthermore, we engineered a proteolysis assay which was used to demonstrate biochemically that the bEBP exhibits substrate translocation and unfolding activity (Fig. 3), firmly supporting the structural data. Within the bEBP-bound structures, the majority of particles were intermediate complexes where the bEBP advanced by one step (i.e., translocation of σN-RI by two residues; RPi1), which was sufficient to partially unfold H1 of σN-RI. The interaction between σN-RI and the ELH remained intact in these intermediates (Fig. 2b). No further advanced intermediates could be determined from our dataset, indicating that disrupting the σN-RI:ELH interaction represents a strong energetic barrier and a rate-limiting step for RPo formation. The lack of intermediate states between RPo and RPi1 in our data can be explained by a highly stabilized RPo state corresponding to a large free energy change28 resulting in sparsely populated intermediates following the rate-limiting step.
Determining which and how many subunits perform nucleotide hydrolysis at each step of substrate translocation in AAA+ proteins is crucial to understanding their mechanism. Our structures suggest that the bEBP works according to the canonical hand-over-hand substrate translocation mechanism of AAA+ proteins23 as follows (Fig. 6a): At each step, only subunit e is catalytically active and hydrolyzes ATP. Upon ATP cleavage, the last subunit (f) can dissociate, bind subunit a, and take the place of subunit a at the top of the spiral. The GAFTGA loop of subunit f would reach up to the protein substrate (σN-RI) and engage with the next residue of σN-RI, thereby advancing the bEBP along the substrate by one step. Highly conserved residue K213 (located right before the GAFTGA motif, i.e., …KGAFTGA…) may aid in repositioning the loop for substrate capture by interacting with the DNA phosphate backbone. Subunit e takes the position of the last subunit (f), ADP is exchanged for ATP, and the complex is ready to enter the next cycle of ATP hydrolysis and substrate translocation. Notably, in the post-catalytic state (RPi1closed), the C-terminal domain of subunit f has already established contact with subunit a, but the GAFTGA loop has yet to move to the top of the spiral. Likely, we captured this state because this intermediate has piled up at the energetic barrier of the σN-RI:ELH interaction.

a For ATP hydrolysis-coupled substrate translocation by the bEBP, the ATP-containing bEBP binds to an unstructured end of the protein substrate (σN-RI) to engage in a spiral conformation, leaving a gap between the top (blue) and bottom (yellow) subunit. ATP hydrolysis in the green subunit disrupts the intersubunit interaction between green and yellow allowing the yellow subunit to reach across the gap and contact the blue/top subunit. The pore loop of the yellow subunit engages with the substrate, and thereby, the yellow subunit assumes the position at the top of the spiral and the complex advanced one step along the substrate. Subsequently, ADP in the green subunit is exchanged for ATP and cycle can start over. b For the isomerization from RPi1 to RPo, the transcription initiation complex is viewed along the DNA axis and large subunits (β, β’, bEBP) are clipped along the axis of σN-RI (red) for visual clarity. During translocation of σN-RI by the bEBP, the ELH must insert at the minor groove near the DNA bubble to place the DNA t-strand between σN-RI and the ELH. To loop around the t-strand, σN-RI/RII passes around the ELH and inserts between β-Si2 and the β protrusion forming the arrangement observed in the RPo and RP+2A structures.
A model for the steps that must occur during the transition from RPi1 to RPo can be deduced (Fig. 6b): as the bEBP continues translocation of σN-RI, disrupting the σN-RI:ELH interaction, the ELH must insert between the DNA strands at the minor groove around position −8 to bring the t-strand between σN-RI and the ELH. Subsequently, σN-RI/RII must pass in front of the ELH in order to insert between the β-Si2/flap domain and the β protrusion to arrive at the looped arrangement observed in RPo. Experiments with slowly hydrolysable ATPγS suggest that the bEBP is required for additional steps after disruption of the σN-RI:ELH interaction57, which may be achieved early in the translocation process. It seems plausible that continued bEBP threading of σN until the translocation halt is required to pull RII out of the RNAP main channel. Thereafter, the RNAP downstream channel is free to engage with DNA, and binding free energy may drive the insertion of the downstream duplex DNA into the channel.
A remarkable feature of the RPo and RPo+2A/RPitc structures is the looping of σN-RII around the t-strand DNA (Fig. 5d). In Eσ70 transcription complexes, σ70 region 3.2 (the “σ-finger”)—which occupies a similar location in RPo as σN-RII58,59—facilitates efficient formation of the first phosphodiester bond60,61, presumably by helping to position the t-strand DNA. The RII:t-strand junction may fulfill a similar role and help to position the t-strand for initial catalysis. In recently determined RPitc structures with different lengths of nascent RNA the RII loop lies beneath the t-strand and is not topologically entangled62. However, these samples were prepared for structural analysis by assembling complexes with promoter DNA containing a pre-melted bubble, a “bypass” variant of σN, and in the absence of a bEBP and so do not reflect bona fide, bEBP-dependent EσN RPitc complexes.
The entanglement of RII with the t-strand is a topological conundrum that must be resolved before RNAP can leave the promoter. Regulated bEBP disassembly prevents the bEBP from having to unfold the entire length of the σN polypeptide (477 residues) through the DNA bubble and presumably allows σN to disentangle from the DNA t-strand (Fig. 5d) as RNAP escapes the promoter. Further work will be required to elucidate the structural details for the steps of RPo formation following RPi1, and for how the entanglement of σN with the DNA bubble is resolved for promoter escape.
Methods
Protein expression and purification
All purification steps were performed at 4 °C or on ice if not stated otherwise.
Eco core RNAP
Full-length Eco RNAP (UniProt entries P0A7Z4, P0A8V2, P0A8T7, P0A800 for α, β, β’, and ω) was expressed and purified largely following the procedure as described previously63. Specifically, a pET-based plasmid harboring full-length RNAP subunits α, β, ω as well as β’-PPX-His10 (PPX; PreScission protease site, LEVLFQGP, Cytiva) under an IPTG-inducible promoter (addgene #128940) was co-transformed with pACYC-Duet-1 encoding ω (addgene #128837) into Eco BL21(DE3). Shaking cultures of a total of 6 L LB media supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol were inoculated with an overnight preculture from freshly transformed cells and grown to an OD600 of 1 at 37 °C. Expression was induced with IPTG (0.5 mM final concentration) and cultures were incubated for 3 h at 30 °C. Cells were harvested by centrifugation (Beckman JS-4.2, 4500xg, 4 °C, 20 min) and pellets were resuspended in a total of 120 mL lysis buffer (50 mM Tris-HCl, pH 8/RT, 10 mM DTT, 1 mM ZnCl2, 5% (v/v) glycerol, 0.5x c0mplete EDTA-free protease inhibitors (Roche), 1 mM PMSF). Resuspended cells were stored at −80 °C until use. Cell suspensions were thawed, lysed by high-pressure shearing (Avestin EmulsiFlex C50), and insoluble material was removed by centrifugation (Beckman JA-20, 27,000 x g, 4 °C, 30 min). While stirring, polyethyleneimine [PEI; 10% (w/v) in water adjusted to pH 8/RT with HCl] was slowly added to a final concentration of 0.6% (w/v) to the soluble fraction, and the mixture was incubated for 25 min at 4 °C while stirring. After collecting the PEI precipitate by centrifugation (Beckman JA-20, 27,000 x g, 4 °C, 1 h), the pellets were washed three times in a total of 210 mL PEI wash buffer [50 mM Tris-HCl, pH 7.9/RT, 0.5 M NaCl, 5% (v/v) glycerol, 10 mM DTT]. For each wash step, the pellets were resuspended using a glass Dounce homogenizer (Wheaton) and the PEI precipitate was collected again by centrifugation. To elute RNAP from the PEI, the pellets from the last wash step were resuspended as above in a total of 120 mL PEI elution buffer [50 mM Tris-HCl, pH 7.9/RT, 1 M NaCl, 5% (v/v) glycerol, 10 mM DTT] and the PEI precipitate was collected again by centrifugation saving the supernatant. Elution was repeated for a total of three rounds, pooling the supernatant from each centrifugation step (final total of 360 mL). 35 g/100 mL ammonium sulfate (grounded to powder) was added slowly while stirring, and the mixture was incubated overnight at 4 °C. Ammonium sulfate precipitate was recovered by centrifugation (Beckman JA-18, 33000xg, 4 °C, 30 min) and resuspended in a total of 50 mL IMAC buffer A [20 mM Tris-HCl, pH 7.8/RT, 1 M NaCl, 5% (v/v) glycerol, 1 mM β-mercaptoethanol]. The sample was passed through 5 μm filter and applied to 2 × 5 mL HiTrap IMAC HP columns (Cytiva) connected in series, charged with Ni2+ and equilibrated in IMAC buffer A. After washing the columns with increasing imidazole concentrations in buffer A (last wash with 80 mM imidazole), protein was eluted in buffer B [20 mM Tris-HCl, pH 7.8/RT, 1 M NaCl, 250 mM imidazole, 5% (v/v) glycerol, 1 mM β-mercaptoethanol]. Fractions were pooled according to protein content and His-tagged 3 C protease was added at 1:36 molar ratio. The sample was dialyzed overnight at 4 °C in a 12–14 kDa cutoff membrane (SpectraPor) against 20 mM Tris-HCl, pH 8/4 °C, 1 M NaCl, 5% (v/v) glycerol, 0.1 mM EDTA, 1 mM β-mercaptoethanol, 0.5 mM DTT. After dialysis, the sample was passed over the IMAC columns followed by dialysis of the flow-through overnight at 4 °C in a 12–14 kDa cutoff membrane (SpectraPor) against 10 mM Tris-HCl, pH 7.8/RT, 100 mM NaCl, 0.1 mM EDTA, 5% (v/v) glycerol, 5 mM DTT. After dialysis, the sample was loaded on a 40 mL Biorex column (Biorad Biorex-70 resin #142-5842) equilibrated in Biorex A buffer [10 mM Tris-HCl, pH 7.8/RT, 0.1 mM EDTA, 5% (v/v) glycerol, 5 mM DTT] and eluted with a gradient of 20–80% Biorex B buffer [10 mM Tris-HCl, pH 7.8/RT, 1 M NaCl, 0.1 mM EDTA, 5% (v/v) glycerol, 5 mM DTT] over 15 column volumes. Fractions were pooled according to protein content. The pool was concentrated using a centrifugal filter (3500 x g, 4 °C, Amicon Ultra 100 K, EMD Millipore) and loaded on a Superdex 200 26/600 320 mL column (Cytiva) equilibrated in 20 mM HEPES-Na, pH 7.8/RT, 0.5 M NaCl, 0.1 mM EDTA, 5% (v/v) glycerol, 0.5 mM TCEP. Peak fractions were pooled, concentrated using centrifugal filters (3500 x g, 4 °C, Amicon Ultra 100 K, EMD Millipore) and mixed with buffer containing 50% (v/v) glycerol to achieve a final concentration of 20% (v/v) glycerol. Aliquots were frozen in liquid N2 and stored at −80 °C until use.
Eco σN and variants
Tagless, full-length Eco σN (RpoN, Sig54; UniProt entry P24255) was purified as described previously21. Variants of σN were obtained by cloning full-length σN into pET28a with an N-terminal His10-SUMO tag and introducing the desired mutations by PCR amplification with oligonucleotide primers carrying the desired modifications and ligation of the PCR product. Constructs were freshly transformed into Eco BL21(DE3) cells, and transformants were grown at 37 °C in 200 mL LB shaking cultures (baffled flasks) containing 50 μg/ml kanamycin. At OD600 of 0.6, the cultures were equilibrated to 16 °C for 30 min before induction with 1 mM IPTG. Expression was carried out overnight at 16 °C. Cells were harvested by centrifugation (Beckman JS-4.2, 4500 x g, 4 °C, 30 min) and resuspended in lysis buffer [20 mM Tris-HCl, pH 8/RT, 500 mM NaCl, 5 mM imidazole, 5% (v/v) glycerol, 0.5 mM DTT, 1 mM PMSF, 1x c0mplete EDTA-free protease inhibitor (Roche)]. At this point, the cell resuspensions were frozen in liquid N2 and stored at −80 °C until further processing. After thawing, cells were lysed by sonication (Branson Digital Sonifier 450) and subjected to centrifugation (Beckman JA-20, 11,000 x g, 4 °C, 1 h) to remove insoluble material. The soluble fraction was passed through 0.45 μm filter and applied to a 1 mL Ni2+-charged IMAC HiTrap (Cytiva) equilibrated in buffer A [20 mM Tris-HCl, pH 8/RT, 500 mM NaCl, 5% (v/v) glycerol, 0.5 mM DTT]. The column was washed with increasing concentrations of imidazole in buffer A (40 mM, 80 mM, 120 mM, 350 mM imidazole) while collecting fractions. Fractions containing Eco σN protein were pooled; His-tagged Ulp1 protease was added at 1:40 molar ratio and the sample was dialyzed overnight at 4 °C against buffer A in a 12–14 kDa cutoff membrane (SpectraPor). After recovering the sample from dialysis, it was passed again over the IMAC resin to remove uncleaved fusion protein and the Ulp1 protease. The flow-through was collected, concentrated using centrifugal filters (3500 x g, 4 °C, Amicon Ultra 30 K, EMD Millipore) and mixed with an equal volume of storage buffer [20 mM Tris-HCl, pH 8/RT, 500 mM NaCl, 40% (v/v) glycerol, 0.1 mM EDTA, 2 mM DTT]. Aliquots were frozen in liquid N2 and stored at −80 °C until use.
Aae C1 (bEBP) and variants
The constitutively active variant of Aae NtrC1 (UniProt entry O67198) missing the N- and C-terminal domains (C1; residues 121–387 of the full-length protein) was purified as described previously21. An expression construct for C1 carrying the proteasome interaction motif (C1-GS4) was obtained by amplification of the parent expression vector using mutagenic primers (Supplementary Table 2) to encode residues GSGSLGQYL at the C-terminus and ligation of the linearized product. Purification of C1-GS4 was carried out in analogy to the procedure described for C1 but required several modifications: Specifically, freshly transformed Eco BL21(DE3) were grown in 2 L LB medium supplemented with 100 μg/ml ampicillin as a shaking culture at 37 °C. Expression was induced with 1 mM IPTG at an OD600 of 0.6 and continued overnight at 25 °C. Cells were harvested by centrifugation (Beckman JS-4.2, 4500 x g, 4 °C, 20 min) and pellets were resuspended in lysis buffer [50 mM Tris-HCl, pH 8/RT, 500 mM KCl, 5% (v/v) glycerol, 5 mM EDTA, 1 mM TCEP] supplemented with 1x c0mplete EDTA-free (Roche) and 1 mM PMSF. After cell lysis by high-pressure shearing (Avestin EmulsiFlex C50), insoluble material was removed by centrifugation (Beckman JA-18, 30,000 x g, 4 °C, 30 min). The supernatant was recovered and placed in a water bath equilibrated at 70 °C for 30 min while stirring. Following removal of the precipitate by centrifugation (Beckman JA-18, 30,000 x g, 4 °C, 30 min), protein was precipitated with ammonium sulfate at 60% saturation at 4 °C for 1 h. Precipitate was collected by centrifugation (Beckman JA-18, 30,000 x g, 4 °C, 30 min) and resuspended in 20 mM Tris-HCl, pH 8/RT, 500 mM KCl, 1 mM EDTA, 1 mM DTT. The resuspension was dialyzed overnight at 4 °C in a 12–14 kDa cutoff membrane (SpectraPor) against 20 mM Tris-HCl, pH 8/RT, 10 mM KCl, 1 mM EDTA, 1 mM DTT. Subsequently, the sample was loaded on a 5 mL HiTrap Q HP column (Cytiva) equilibrated in low salt buffer (20 mM Tris-HCl, pH 8/RT, 50 mM KCl, 1 mM EDTA, 1 mM DTT) and eluted with a salt gradient of 50 mM to 500 mM KCl over 15 column volumes. Fractions were pooled according to protein content and dialyzed overnight at 4 °C in a 12–14 kDa cutoff membrane (SpectraPor) against 20 mM MES, pH 6.5/RT, 10 mM KCl, 1 mM DTT. The dialyzed sample was loaded on a 5 mL HiTrap SP HP column (Cytiva) and eluted with a salt gradient of 50 mM to 300 mM KCl over 20 column volumes. Fractions were pooled according to protein content and concentrated using centrifugal filters (3500 x g, 4 °C, Amicon Ultra 30 K, EMD Millipore). Aliquots were frozen in liquid N2 and stored at −80 °C until use.
Bacterial proteasome
The “open-gate” variant of the 20S core particle of the M. tuberculosis proteasome (20S-og) was expressed recombinantly in Eco BL21(DE3) from a pETDuet-1 vector encoding prcAΔN7 and prcB with a C-terminal Strep tag (WSHPQFEK). The expression construct was a gift from Eilika Weber-Ban (ETH Zurich, Switzerland). Cells were grown in ZYP-5052 autoinduction media as three 2 L shaking cultures at 25 °C overnight and harvested by centrifugation (Beckman JS-4.2, 4500 x g, 4 °C). Pellets were resuspended in buffer P [50 mM Tris-HCl, pH 7.8/RT, 150 mM NaCl, 1 mM EDTA, 10% (v/v) glycerol, 1 mM DTT) supplemented with 1 mM DTT (final 2 mM), and cells were lysed by high-pressure shearing (Avestin EmulsiFlex C50). Insoluble material was removed by centrifugation (Beckman JA-18, 33,000 x g, 4 °C, 1 h). The supernatant was recovered, passed through a 5 μm filter, and applied to a 5 mL StrepTactin XT 4Flow high-capacity resin (IBA Lifesciences) equilibrated in buffer P. After washing the resin with five column volumes of buffer P, the protein was eluted with buffer A containing 50 mM biotin. Elution fractions were pooled according to protein content and further purified by gel filtration (Superdex 200 HiLoad 26/600, 320 mL, Cytiva) in buffer P. Peak fractions were pooled, concentrated to 15–30 μM proteasome complex using centrifugal filters (3500 x g, 4 °C, Amicon Ultra 100 K, EMD Millipore) and stored at 4 °C. Aliquots of the sample were applied to a Superose 6 10/300GL (24 mL) column (Cytiva) in buffer P before use to remove large aggregates. Peak fractions were pooled, concentrated and stored at 4 °C.
TSS determination by 5’-RACE with template switching
We performed rapid amplification of cDNA ends (RACE) including reverse transcription with template switching64 to determine the 5’-end sequences of in vitro transcribed RNAs from the linear dhsU and dhsU+2 T promoter fragments ( − 60 to +30; Supplementary Table 2). Promoter DNA fragments were annealed from synthetic oligos to form dhsU (oligos dhsU_top and dhsU_bot; Supplementary Table 2) and dhsU+2 T (oligos dhsU+2T_top and dhsU+2T_bot; Supplementary Table 2) duplex DNA. Oligos were mixed at 2 μM final concentration in 10 mM HEPES-NaOH, pH 8/RT, 50 mM NaCl and subjected to the following program in a thermocycler: 95 °C for 30 s, 95 °C for 15 s for 70 cycles with −1 °C/cycle, 25 °C hold. Reactions containing 80 nM RNAP, 200 nM σN, 20 nM promoter DNA, 1 μM C1, 1 mM of each CTP/GTP/UTP, 5 mM ATP were assembled in a 120 μL reaction in buffer TXN (40 mM Tris-HCl, pH 8/RT, 200 mM KCl, 10 mM MgCl2, 5 mM DTT, 40 μg/μL BSA) as follows: RNAP and σN were mixed and incubated for 10 min at 37 °C to form holoenzyme; DNA was added and the reaction was incubated for 15 min at 37 °C to form the early-melted intermediate (RPem); C1 was added and the reaction was incubated for 5 min at 37 °C before starting the reaction with the addition of NTPs. After incubating the reaction for 30 min at 37 °C, 2 μL TURBO DNase (ThermoFisher Scientific #AM1907; final concentration 33 U/mL) was added and the reaction was incubated for 15 min at 37 °C to degrade the promoter DNA. To eliminate free Mg2+ ions, 5 μL of 0.5 M EDTA were added (final concentration 19.7 mM) and the reaction was placed on ice. RNA was purified using the Oligo Clean & Concentrator kit (Zymo #D4060), eluting in 20 μL of 20 mM Tris-HCl, pH 8/RT. RNA preparations were subjected to capping by the Vaccinia virus Capping Enzyme (VCE; NEB #M2080S) in 20 μL reactions containing 0.5 mM GTP, 1 U/μL RNase inhibitor (NEB #M0314S), 0.5 U/μL VCE in 1x capping buffer (NEB), and 12 μL purified RNA. RNA was heat-denatured at 70 °C for 5 min and cooled on ice before addition to the reaction. Capping reactions were incubated for 2 h at 37 °C and treated RNAs were purified using the Oligo Clean & Concentrator kit (Zymo #D4060), eluting in 20 μL water. 4 μL purified capped RNAs were mixed with RT primer (dhsU-RTprim; Supplementary Table 2; final concentration 1 μM) and dNTP mix (NEB #N0447S; final concentration 1 mM) in 6 μL total volume, denatured at 70 °C for 5 min and placed immediately on ice. A working solution of reverse transcriptase and template switching (RTTS) reagents (NEB #M0466S) was prepared by mixing 2.5 parts template switching RT buffer, 1 part of 10 μM RACE-TSO_fw primer (Supplementary Table 2), and 1 part of RT enzyme mix. 4 μL of RTTS mix were added to the denatured RNA-RTprimer-dNTP mix (total volume 10 μL). The reaction was incubated in a thermocycler at 42 °C for 90 min, heated to 85 °C for 5 min, and cooled to 4 °C. Reactions were diluted two-fold with water, and 2.5 μL of diluted RTTS reaction were used in 25 μL cDNA amplification reactions. PCR mixes contained 0.2 mM dNTPs, 0.5 μM RACE-PCR-TSO_fw primer (Supplementary Table 2), 0.5 μM RACE-PCR-RT_rv primer (Supplementary Table 2), 0.02 U/μL Q5 HotStart polymerase (NEB #M0493S) in Q5 reaction buffer (NEB) and were subjected to the following PCR program: initial denaturation at 98 °C for 30 s; five cycles of 98 °C for 10 s, 72 °C for 2 s; five cycles of 98 °C for 10 s, 70 °C for 2 s; 35 cycles of 98 °C for 10 s, 65 °C for 15 s, 72 °C for 15 s; final extension at 72 °C for 30 s; hold at 10 °C. Two 25 μL cDNA amplification reactions were performed for each promoter DNA. Free primers were digested by adding 0.5 μL (10 U) of Exonuclease I (NEB #M0293S) to each reaction and incubating the reactions at 37 °C for 30 min. PCR products were purified using the Oligo Clean & Concentrator kit (Zymo #D4060), eluting in 12 μL 20 mM Tris-HCl, pH 8/RT. Purified PCR products were cloned into a pTwistAmp vector (Twist Bioscience) linearized using primers RACE-pTwist_fw and RACE-pTwist_rv (Supplementary Table 2) by isothermal DNA assembly (NEB #E5520S). Assembly was performed by mixing 15 fmol linearized vector with 90 fmol cDNA PCR product in 2.5 μL total volume. An equal volume of 2x assembly enzyme mix (NEB #E5520S) was added and reactions were incubated at 45 °C for 30 min. 3 μL of the assembly reactions were transformed into 50 μL chemo-competent NEB5α cells (NEB #E5520S) according to the manufacturer’s protocol. Recovered transformants were plated on LB agar plates containing 100 μg/mL ampicillin, and plates were incubated at 37 °C overnight. Plates were subjected to direct colony sequencing (Azenta/Genewiz, New Jersey, USA) of ten random clones for each promoter DNA fragment using the M13FOR primer (Supplementary Table 2). Sequencing results were aligned using ClustalOmega and TSS were identified as being directly preceded by 4 G bases (for capped RNAs) or 3 G bases (for uncapped RNAs) at the 3’-end of the RACE-TSO_fw sequence as previously described64. Nine out of nine sequencing reactions yielded GAACAA as the 5’-end sequence for dhsU (Supplementary Table 1). Six out of seven sequencing reactions yielded GTACAA as the 5’-end sequence for dhsU+2 T. Failed sequencing reactions and transcription via end binding were eliminated from the analysis. The results established the +2 site, and that introduction of a T at this position did not change the TSS.
Stopped flow fluorescence spectroscopy for RPo formation kinetics
A dhsU promoter fragment labeled with Cy3 at the +2 position (Cy3-dhsU+2 T) was prepared by annealing 10 μM dhsU+2T-Cy3_top (Supplementary Table 2) and 11 μM dhsU+2T_bot (Supplementary Table 2) in 10 mM HEPES-Na, pH 8/RT, 50 mM NaCl in a thermocycler (program: 95 °C for 30 s, 95 °C for 15 s for 70 cycles with −1 °C/cycle, 25 °C hold). Reaction mix A was prepared in buffer S (40 mM Tris-HCl, pH 8/RT, 200 mM KCl, 10 mM MgCl2, 1 mM DTT, 10 μg/mL BSA) by first mixing RNAP and σN and incubating for 5 min at 37 °C; Cy3-dhsU+2 T was added and the reaction was incubated for 10 min at 37 °C; C1 was added and the reaction was incubated for 2 min at 37 °C. Reaction mix B was prepared by mixing ATP in buffer S. Reaction mixes A and B were loaded into a stopped flow instrument (Applied Photophysics SX20; 535 nm LED, 550 nm shortpass excitation filter, 570 longpass emission filter) equilibrated at 37 °C and incubated 1 min before starting the experiment. At least three traces were recorded and averaged. For analysis, the averaged traces were offset corrected by subtracting the value of the first data point from all data points within a series. Final concentrations after mixing in the stopped flow cell were 50 nM RNAP, 75 nM σN, 10 nM Cy3-dhsU+2 T, 250 nM C1, 2.5 mM ATP for traces shown in Fig. 1a and Supplementary Fig. 1B; 100 nM RNAP, 150 nM σN, 20 nM Cy3-dhsU+2 T, 500 nM C1, 5 mM ATP for traces shown in Supplementary Fig. 1C; 200 nM RNAP, 300 nM σN, 20 nM Cy3-dhsU+2 T, 500 nM C1, 5 mM ATP for traces shown in Supplementary Fig. 1D. Fluorescence signal F in the experiment was fit with a single exponential equation (i.e., unimolecular conversion) as follows
where a0 is the background signal, a1 is the maximum signal, and k corresponds to the apparent rate constant. Parameter estimates from the data fit are presented in Supplementary Table 5. Data fitting was performed with the GraphPad Prism v9.5.1 software.
Cryo-EM analysis of EσNdhsU-bEBP de novo complexes
dhsU promoter DNA
Commercially synthesized DNA oligos (Integrated DNA Technologies) dhsU_top and dhsU_bot (Supplementary Table 2) were resuspended in nuclease-free H2O. Equimolar amounts of each strand were mixed and annealed in 10 mM HEPES-NaOH, pH 8.0, 50 mM NaCl at a final duplex concentration of 120 μM.
Sample preparation
Purified proteins were desalted into reaction buffer (40 mM Tris-HCl, pH 8/RT, 200 mM KCl, 10 mM MgCl2, 1 mM DTT) using Zeba Spin Desalting Columns 7 K (ThermoFisher Scientific #89883) before use. For complex assembly, core RNAP (E) and σN were mixed in reaction buffer and incubated for 10 min at 37 °C to form EσN followed by addition of the dhsU promoter fragment and another incubation for 10 min at 37 °C to form the early-melted intermediate (RPem). C1 was added, and the reaction was incubated for 5 min at 37 °C. Concentrations of each component in the final mix were 4 μM E, 4.8 μM σN, 4.8 μM dhsU, and 5.6 μM C1. The EσN-dhsU-C1 complex was concentrated to 16 μM (using the concentration of E as a proxy for the complex) using Amicon Ultra 10 K centrifugal filters (Merck Millipore #UFC501096). An ATP-detergent solution containing 40 mM ATP (Sigma #A2383) and 12 mM fluorinated fos-choline-8 (FC8F; Anatrace #F300F) in reaction buffer was prepared. ATP-detergent solution and EσN-dhsU-C1 were centrifuged at 11,000 x g for 10 min at 23 °C before grid preparation.
Grid preparation
C-flat holey carbon grids (CF-1.2/1.3-4Au, EMS) were glow-discharged for 5 s at 25 mA and 0.3 mbar air atmosphere (Pelco easiGlow) before use. Sample application and vitrification was performed using a Vitrobot Mark IV (ThermoFisher Scientific) equilibrated to 37 °C and 100% relative humidity in the blotting chamber. For each grid, the following procedure was performed: A glow-discharged grid was mounted in the blotting chamber of the Vitrobot instrument. 3.5 μL EσN-dhsU-C1 were equilibrated to 37 °C on a thermoblock for at least 1 min. To start the reaction, 0.5 μL ATP-detergent solution was added. 3.5 μL of the reaction mix were immediately transferred to the grid, where reaction incubation was continued. Grids were blotted and plunged into liquid ethane with a total reaction time of 35–38 s.
Cryo-EM data acquisition and processing
Grids were imaged using a 300 kV Titan Krios (ThermoFisher Scientific) equipped with a K3 camera (Gatan), a Cs corrector and a BioQuantum imaging filter (Gatan). Images were recorded using SerialEM v4.1.0beta65 with a pixel size of 0.86 Å/px over a nominal defocus range of −0.8 to −2.2 μm and 20 eV energy filter slit width. A total of 17,199 gain-normalized movies were recorded in “super resolution” mode (K3 camera binning 0.5; image dimensions of 11,520 × 8184 px; effective image pixel size of 0.43 Å/px) with 22 e−/px/s (at the camera) in dose-fractionation mode of 0.04 s over a 1.4 s exposure (35 frames) to give a total dose of 42 e−/Å2. Dose-fractionated movies were binned by a factor of 2 (resulting image pixel size of 0.86 Å/px), drift-corrected, summed, and dose-weighted using MotionCor2 v1.1.066. The contrast transfer function (CTF) was estimated for each summed image using the Patch CTF module in cryoSPARC v4.3.167. Micrographs were curated to remove outliers in estimated CTF fit resolution ( > 10 Å discarded), astigmatism (>5000 Å discarded) and relative ice thickness (0.8 <x < 1.07 retained) resulting in a set of 16,649 images. Particles were picked using cryoSPARC Blob Picker (10,510,033 picks) retaining only picks with a normalized cross-correlation (NCC) score >0.1 and a power score between 370 and 800 (NCC and power scores were scaled based on the entire set of picks). Particles were extracted from images with a box size of 448 px, Fourier-cropped to 128 px and subjected to two rounds of cryoSPARC 2D classification (N = 200) resulting in 1,341,601 particles. Duplicate particles were removed based on a center-to-center cutoff distance of 20 Å keeping the particles with higher NCC score. Images with less than five particles were manually inspected and excluded from further analysis (64 images/161 particles removed). Initial models were generated using cryoSPARC Ab initio Reconstruction67 from a subset of 300,000 particles yielding reconstructions centered on RNAP (2 classes) or bEBP (2 classes) or “junk” (2 classes). Particles were further curated using two rounds of cryoSPARC Heterogeneous Refinement (N = 6) with ab initio classes serving as 3D references (2 RNAP classes, 2 × 2 junk classes). Particles contributing to RNAP or bEBP classes were combined and as above, duplicates and images with less than five particles were removed resulting in 1,060,259 particles (16,461 images). The particle stack was refined using cryoSPARC Non-Uniform (NU) Refinement with Defocus and Global CTF Refinement (tilt/trefoil) enabled68 and then further processed using RELION v4.0.1 Bayesian Polishing69. Polished particles were imported into cryoSPARC and subjected to NU Refinement with Defocus and Global CTF Refinement (tilt/trefoil/tetrafoil/spherical aberration) enabled, yielding a consensus reconstruction of 2.3 Å nominal resolution.
Initially, the consensus particle stack was subjected to cryoSPARC 3D classification (without masks) with various settings to explore the heterogeneity of the sample. To proceed, a 3D classification (N = 12, target resolution = 6 Å, random class initiation) using a focus mask around the bEBP and σN-RI/bubble region (mask 1) was performed, providing improved particle assignments while yielding similar results to the unmasked classification. Classes were manually inspected and pooled yielding an “early-melted” class (295,087 particles), a “bEBP-bound” class (522,509 particles), and a “melted” class (133,297 particles) (Fig. 1c). Each class was subjected to NU Refinement68.
The refined “melted” class (133,297 particles) was further divided by cryoSPARC 3D classification (N = 10, target resolution = 6 Å, no masks supplied, random class initiation). Resulting classes were manually inspected, pooled, and refined to yield maps for the “RPo” complex (45,631 particles, 2.8 Å) and “RPo+2A” complex (15,649 particles, 3.1 Å). For each complex, local refinement using a mask around σN (RPo-SigN mask) on signal-subtracted particles was carried out. Locally refined maps were merged with the parent map using phenix.combine_focus_maps v1.21.170.
Further cryoSPARC 3D classification (N = 24, target resolution = 6 Å, random class initiation) of the refined “bEBP-bound” class (522,509 particles) was carried out using the focus mask around the bEBP and σN-RI/bubble region (mask 1). Classes were manually pooled based on similarity and quality of σN-RI density yielding three groups: “full helix” (74,144 particles), “one turn” (41,788 particles), and “two turns” (378,668 particles). A mask based on all orientations of the bEBP relative to RNAP (mask 2) was generated and used for signal subtraction of the “two turns” particle stack. Subtracted particles were subjected to local refinement using mask 2 followed by another local refinement using a tight mask around the bEBP from the first local refinement (mask 3). Using the tight mask around the bEBP (mask 3), cryoSPARC 3D classification (N = 10, target resolution = 4 Å, random class initiation) was performed yielding class pools “bEBP open” (118,561 particles, 3.1 Å) and “bEBP closed” (260,106 particles, 3.0 Å). For each class, the unsubtracted particles were aligned by NU Refinement to yield “RNAP” maps. To improve map quality in σN and the DNA, a mask around most parts of σN (excluding CBDs) and the σN-bound DNA (SigN mask) was used to perform signal subtraction and local refinement yielding “SigN” maps. The bEBP and SigN maps were merged with the RNAP map using phenix.combine_focus_maps70.
In a separate approach, the “bEBP-bound” class (522,509 particles) was again subjected to cryoSPARC 3D classification (N = 12, target resolution = 6 Å, forced hard classification, random class initiation) using a focus mask around the RNAP downstream channel (ds channel mask). While seven classes exhibited presence of DNA (295,571 particles; 56.6%), the remaining five classes contained density for an extended σN-RI and σN-RII (226,938 particles; 43.4%). The quality of the density for σN-RI and σN-RII varied considerably among the classes indicating flexibility, and therefore, only the two classes with the best resolved density were pooled and refined to yield the map “RII” (95,920 particles, 2.7 Å).
Local resolution and locally filtered maps were generated using cryoSPARC67 or blocres/blocfilt (Bsoft v2.1.3)71. Most structural biology software was accessed through the SBGrid software package72.
Model building and refinement
Initial models were derived from PDBs 8F1K21, 6GH544 and 4LZZ73. The models were manually fit into the cryo-EM density maps using ChimeraX v1.674 and rigid-body refined using phenix.real_space_refine v1.21.175. Models were inspected and modified in Coot v0.9.676 or ISOLDE v1.677. Models were finalized by all-atom and B-factor refinement with Ramachandran and secondary structure restraints using phenix.real_space_refine. Maps and models were visualized in ChimeraX74.
Cryo-EM analysis of C1-GS4:proteasome complexes
400 nM 20S-og and 800 nM C1-GS4 (final concentrations) were mixed in 40 mM Tris-HCl, pH 8/RT, 200 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.5 mM ADP, 4 mM NaF, 1 mM AlCl3 and incubated for 5 min at 37 °C. Grids (Ultrathin C on lacey carbon Cu400, TedPella #01824) were glow-discharged for 10 s at 10 mA and 0.3 mbar air atmosphere (Pelco easiGlow) before use. Vitrified samples on grids were prepared using a Vitrobot Mark IV (ThermoFisher Scientific) at 10 °C and 100% relative humidity in the sample chamber. For each grid, 3.5 μL sample were applied to a continuous carbon grid and incubated for 30 s. Excess liquid was blotted away, and the grid was plunged into liquid ethane.
Data was collected on a 200 kV Talos Arctica instrument (ThermoFisher Scientific) equipped with a K2 camera (Gatan). Dose-fractionated movies were recorded in “counting” mode (image dimensions 3838 × 3710 px) using SerialEM65 with a pixel size of 1.5 Å/px, 12 s exposure, and 40 frames per movie. Dataset 1 was collected with a tilt angle of 20° over a nominal defocus range of −1 to −2.5 μm with a dose of 1.227 e−/Å2/frame and comprised 466 movies. Dataset 2 was collected with a tilt angle of 0° over a nominal defocus range of −0.8 to −3 μm with a dose of 1.12 e−/Å2/frame and comprised 503 movies. For each dataset, movies were drift-corrected, summed, and dose-weighted using MotionCor266. Micrographs were further processed in cryoSPARC v4.3.167 for each dataset separately. After CTF estimation using cryoSPARC’s PatchCTF module, particles were picked using circular blobs (BlobPicker; diameter 100–250 Å) and outliers were rejected based on NCC and power score resulting in 462,564 picks for dataset 1 and 475,471 picks for dataset 2. Particle images were extracted with a box size of 256 px and subjected to two rounds of 2D classification (120 classes, batch size 200) resulting in 291,510 particles for dataset 1 and 315,009 particles for dataset 2. For each dataset, ab initio classes were generated (3–6 classes, C7 symmetry) followed by a NU Refinement (C7 symmetry) of all particles using the ab initio volume with the best quality as input. Duplicates with less than 50 Å center-to-center distance (based on 3D alignments) were removed, and particles were re-extracted with a box size of 288 px to recenter particle images resulting in 283,067 particles for dataset 1 and 308,092 particles for dataset 2. At this point, particle stacks from both datasets were merged, and new ab initio volumes were generated (6 classes, C7 symmetry). The merged particle stack was subjected to two rounds of Heterogeneous Refinement resulting in 437,119 particles. The particles were refined (NU Refinement, C7 symmetry, per-particle defocus and global CTF refinement (tilt/trefoil) enabled) using the volume from the last Heterogenous Refinement run with the best quality as input yielding a reconstruction of 3.4 Å nominal FSC resolution. Duplicates with less than 20 Å center-to-center distance based on 3D alignments were removed and particle alignments were refined again (NU Refinement, C7 symmetry, per-particle defocus and global CTF refinement (tilt/trefoil) enabled) yielding a reconstruction of 3.3 Å nominal FSC resolution from 434,340 particles. To improve density for the bEBP, the particles were subjected to 3D classification (ten classes, filter resolution = 6 Å, O-EM epochs = 4) using a mask around the bEBP. The resulting classes showed rotational variability of the bEBP density around the central particle axis, and single classes exhibited a hexagonal outline typical for the bEBP. One class with well-defined bEBP was chosen and subjected to local refinement using a solvent mask of the entire complex to obtain a reconstruction without symmetry constraints. The final volume was reconstructed from 43,660 particles at a nominal FSC resolution of 4.6 Å.
Proteolysis assays
RPem complexes were assembled by mixing E, σN (wild-type or variant), promoter DNA (dhsU or dhsU-CT), bEBP (C1 or C1-GS4), and 20S-og in buffer T [40 mM Tris-HCl, pH 8/RT, 200 mM NaCl, 10 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol]. At each addition step, the reaction was incubated for 10 min at 37 °C to allow complex formation. Reactions were started with the addition of ATP (or alternative nucleotides or buffer as indicated) and incubated at 37 °C for the indicated time. To stop the reaction, 9 μL reaction mix were directly mixed with an appropriate volume of SDS-PAGE loading dye and heated to 95 °C for 5 min. Reaction components were separated by SDS-PAGE and stained with Coomassie for analysis. Final concentrations of the reaction components, if not stated otherwise, were 0.5 μM E, 0.5 μM σN, 0.6 μM promoter DNA, 0.5 μM bEBP, 0.6 μM 20S-og, and 5 mM ATP.
ATPase activity assays
ATPase activity was measured using the Malachite Green assay (Sigma #MAK307). Briefly, bEBP (C1 or C1-GS4) and 20S-og, if indicated, were mixed in buffer T [40 mM Tris-HCl, pH8/RT, 200 mM NaCl, 10 mM MgCl2, 1 mM DTT, 5% (v/v) glycerol] in 5 μL volume. Mixtures were equilibrated at 37 °C and 5 μL ATP was added to start reaction. After incubation at 37 °C for the given time, reactions were quenched in an ice water bath and diluted 50-fold with buffer T. 80 μL diluted sample were added to a microtiter plate containing 20 μL Malachite Green working reagent. Reactions were incubated for 30 min at room temperature before reading absorbance values at 620 nm. Standards ranging from 0 to 40 μM inorganic phosphate were used to convert the obtained sample absorbance values to phosphate concentrations. Reactions only containing ATP in buffer showed the highest free phosphate content and were used for background subtraction. Final concentrations in the reactions were 0.5 μM bEBP (C1 or C1-GS4), 0.75 μM Mtb 20S-og if indicated, and 5 mM ATP.
In vitro transcription assays
Reactions were performed in buffer R (40 mM Tris-HCl, pH 8/RT, 200 mM KCl, 10 mM MgCl2, 1 mM DTT, 5 μg/mL BSA) and assembled as follows: E and σN were mixed and incubated for 10 min at 37 °C to form EσN. Promoter fragment dhsU was added, and the reaction was incubated for 10 min at 37 °C to form RPem. bEBP (C1 or C1-GS4) was added and the reaction was incubated for up to 5 min at 37 °C. Reactions were started with addition of NTP mix (ATP, GTP, CTP, UTP, and α-32P-UTP) and incubated at 37 °C for 15 min before stopping the reaction by adding an equal volume of 2x STOP buffer [0.5x TBE, 8 M urea, 30 mM EDTA, 0.05% (w/v) bromophenol blue, 0.05% xylene cyanol]. Stopped reactions were heated at 95 °C for 5 min before separation on a 20% (1:29 acrylamide:bis-acrylamide) 1x TBE-urea gel. Bands were visualized by autoradiography. Final concentrations in the reactions were: 80 nM E, 200 nM σN, 10 nM promoter DNA, 1 μM bEBP, 1.1 μM 20S-og, 5 mM ATP, 0.5 mM GTP, 0.5 mM CTP, 0.05 mM UTP, and 0.1 μCi/μL α-32P-UTP unless noted otherwise.
Sequence alignments
Protein sequences were aligned using Clustal Omega v1.2.478. Sequence motifs were generated using WebLogo3 v3.779. The conserved net charge U at position n of the aligned sequences was calculated as follows
where X is the set of all possible amino acids, i.e., X = {Ala, Gly, Val, Leu, Met, Arg, Cys, Phe, Tyr, Trp,…}, and
Fi = frequency of residue i
Ci = net charge of residue i at physiological pH
S = total number of sequences in the alignment
Net charges at physiological pH were assumed as R/K = +1, D/E = −1 and any other = 0. A moving average over a window size of 3 residues was applied to plot the data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Model and map files generated in this study were deposited at the Protein Data Bank (PDB) and Electron Microscopy Data Bank (EMDB), respectively: RPi1open (PDB 9MSE, EMD-48586, EMD-48580, EMD-48581, EMD-48582), RPi1closed (PDB 9MSF, EMD-48587, EMD-48583, EMD-48584, EMD-48585), RPo (PDB 9MSH, EMD-48589, EMD-48576, EMD-48577), RPo+2A (PDB 9MSJ, EMD-48590, EMD-48578, EMD-48579), RII (PDB 9MSG, EMD-48588), and 20S-og+C1-GS4+ADP-AlFx (EMD-48574). All other data generated in this study are provided in the manuscript or the Supplementary Information/Source Data file. Requests for materials should be submitted to S.A.D. (darst@rockefeller.edu). Source data are provided with this paper.
References
Gruber, T. M. & Gross, C. A. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 57, 441–466 (2003).
Lonetto, M., Gribskov, M. & Gross, C. A. The sigma 70 family: sequence conservation and evolutionary relationships. J. Bacteriol. 174, 3843–3849 (1992).
Studholme, D. J. & Dixon, R. Domain architectures of σ54-dependent transcriptional activators. J. Bacteriol. 185, 1757–1767 (2003).
Buck, M., Gallegos, M.-T., Studholme, D. J., Guo, Y. & Gralla, J. D. The bacterial enhancer-dependent σ54 (σN) transcription factor. J. Bacteriol. 182, 4129–4136 (2000).
Correa, N. E., Lauriano, C. M., McGee, R. & Klose, K. E. Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Mol. Microbiol. 35, 743–755 (2000).
Feldman, M. et al. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 66, 43–51 (1998).
Hathroubi, S., Zerebinski, J. & Ottemann, K. M. Helicobacter pylori biofilm involves a multigene stress-biased response, including a structural role for flagella. mBio 9, e01973–18 (2018).
Soules, K. R., LaBrie, S. D., May, B. H. & Hefty, P. S. Sigma 54-regulated transcription is associated with membrane reorganization and type III secretion effectors during conversion to infectious forms of Chlamydia trachomatis. mBio 11, e01725–20 (2020).
Fisher, M. A. et al. Borrelia burgdorferi sigma 54 is required for mammalian infection and vector transmission but not for tick colonization. Proc. Natl. Acad. Sci. USA 102, 5162–5167 (2005).
Hauser, F. et al. Dissection of the Bradyrhizobium japonicum NifA+σ54 regulon, and identification of a ferredoxin gene (fdxN) for symbiotic nitrogen fixation. Mol. Genet. Genomics 278, 255–271 (2007).
Bush, M. & Dixon, R. The role of bacterial enhancer binding proteins as specialized activators of σ 54 -dependent transcription. Microbiol. Mol. Biol. Rev. 76, 497–529 (2012).
Wang, J. T., Syed, A., Hsieh, M. & Gralla, J. D. Converting Escherichia coli RNA polymerase into an enhancer-responsive enzyme: role of an NH2-terminal leucine patch in sigma54. Science (1979) 270, 992–994 (1995).
Syed, A. & Gralla, J. D. Isolation and properties of enhancer-bypass mutants of sigma 54. Mol. Microbiol. 23, 987–995 (1997).
Wang, J. T., Syed, A. & Gralla, J. D. Multiple pathways to bypass the enhancer requirement of sigma 54 RNA polymerase: Roles for DNA and protein determinants. Proc. Natl. Acad. Sci. USA 94, 9538–9543 (1997).
Chaney, M. & Buck, M. The sigma 54 DNA-binding domain includes a determinant of enhancer responsiveness. Mol. Microbiol. 33, 1200–1209 (1999).
Campbell, E. A., Kamath, S., Rajashankar, K. R., Wu, M. & Darst, S. A. Crystal structure of Aquifex aeolicus σN bound to promoter DNA and the structure of σN-holoenzyme. Proc. Natl. Acad. Sci. USA 114, E1805–E1814 (2017).
Merrick, M. & Chambers, S. The helix-turn-helix motif of sigma 54 is involved in recognition of the -13 promoter region. J. Bacteriol. 174, 7221–7226 (1992).
Taylor, M. et al. The RpoN-box motif of the RNA polymerase sigma factor σ N plays a role in promoter recognition. Mol. Microbiol. 22, 1045–1054 (1996).
Morris, L., Cannon, W., Claverie-Martin, F., Austin, S. & Buck, M. DNA distortion and nucleation of local DNA unwinding within sigma-54 (σN) holoenzyme closed promoter complexes. J. Biol. Chem. 269, 11563–11571 (1994).
Ye, F., Gao, F., Liu, X., Buck, M. & Zhang, X. Mechanisms of DNA opening revealed in AAA+ transcription complex structures. Sci. Adv 8, eadd3479 (2022).
Mueller, A. U. et al. A general mechanism for transcription bubble nucleation in bacteria. Proc. Natl. Acad. Sci. USA 120, e2220874120 (2023).
Chaney, M. et al. Binding of transcriptional activators to sigma 54 in the presence of the transition state analog ADP-aluminum fluoride: insights into activator mechanochemical action. Genes Dev 15, 2282–2294 (2001).
Puchades, C., Sandate, C. R. & Lander, G. C. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat. Rev. Mol. Cell Biol. 21, 43–58 (2020).
Stennett, E. M. S., Ciuba, M. A., Lin, S. & Levitus, M. Demystifying PIFE: the photophysics behind the protein-induced fluorescence enhancement phenomenon in Cy3. J. Phys. Chem. Lett 6, 1819–1823 (2015).
Ko, J. & Heyduk, T. Kinetics of promoter escape by bacterial RNA polymerase: effects of promoter contacts and transcription bubble collapse. Biochem. J. 463, 135–144 (2014).
Studholme, D. J., Wigneshwereraraj, S. R., Gallegos, M.-T. & Buck, M. Functionality of Purified ς N (ς 54) and a NifA-Like Protein from the Hyperthermophile Aquifex aeolicus. J. Bacteriol. 182, 1616–1623 (2000).
Chen, B., Sysoeva, T. A., Chowdhury, S., Guo, L. & Nixon, B. T. ADPase activity of recombinantly expressed thermotolerant ATPases may be caused by copurification of adenylate kinase of Escherichia coli. FEBS J 276, 807–815 (2009).
Friedman, L. J. & Gelles, J. Mechanism of transcription initiation at an activator-dependent promoter defined by single-molecule observation. Cell 148, 679–689 (2012).
Wang, J. T. & Gralla, J. D. The transcription initiation pathway of sigma 54 mutants that bypass the enhancer protein requirement: Implications for the mechanism of activation. J. Biol. Chem. 271, 32707–32713 (1996).
Casaz, P., Gallegos, M.-T. & Buck, M. Systematic analysis of σ 54 N-terminal sequences identifies regions involved in positive and negative regulation of transcription. J. Mol. Biol. 292, 229–239 (1999).
Wang, L. & Gralla, J. D. Roles for the C-terminal Region of Sigma 54 in Transcriptional Silencing and DNA Binding. J. Biol. Chem. 276, 8979–8986 (2001).
Kavalchuk, M., Jomaa, A., Müller, A. U. & Weber-Ban, E. Structural basis of prokaryotic ubiquitin-like protein engagement and translocation by the mycobacterial Mpa-proteasome complex. Nat. Commun. 13, 276 (2022).
Wang, T. et al. Structural Insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 17, 1377–1385 (2009).
Cannon, W., Wigneshweraraj, S. R. & Buck, M. Interactions of regulated and deregulated forms of the sigma54 holoenzyme with heteroduplex promoter DNA. Nucleic Acids Res 30, 886–893 (2002).
Gallegos, M.-T. & Buck, M. Sequences in σ 54 region I required for binding to early melted DNA and their involvement in sigma-DNA isomerisation. J. Mol. Biol. 297, 849–859 (2000).
Lin, G., Tsu, C., Dick, L., Zhou, X. K. & Nathan, C. Distinct specificities of Mycobacterium tuberculosis and mammalian proteasomes for N-Acetyl tripeptide substrates. J. Biol. Chem. 283, 34423–34431 (2008).
Lin, G. et al. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol. Microbiol 59, 1405–1416 (2006).
Delley, C. L. et al. Bacterial proteasome activator Bpa (Rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One 9, e114348 (2014).
Sasse-Dwight, S. & Gralla, J. D. Role of eukaryotic-type functional domains found in the prokaryotic enhancer receptor factor σ54. Cell 62, 945–954 (1990).
Chen, B. et al. Engagement of arginine finger to ATP triggers large conformational changes in NtrC1 AAA+ ATPase for remodeling bacterial RNA polymerase. Structure 18, 1420–1430 (2010).
Nadanaciva, S., Weber, J., Wilke-Mounts, S. & Senior, A. E. Importance of F 1 -ATPase residue α-Arg-376 for catalytic transition state stabilization. Biochemistry 38, 15493–15499 (1999).
Bae, B. et al. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ 70 domain 1.1. Proc. Natl. Acad. Sci. USA 110, 19772–19777 (2013).
Yang, Y. et al. Structures of the RNA polymerase-σ54 reveal new and conserved regulatory strategies. Science 349, 882–885 (2015).
Glyde, R. et al. Structures of bacterial RNA polymerase complexes reveal the mechanism of DNA loading and transcription initiation. Mol. Cell 70, 1111–1120.e3 (2018).
Aubin-Tam, M.-E., Olivares, A. O., Sauer, R. T., Baker, T. A. & Lang, M. J. Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 (2011).
Banwait, J. K., Islam, L. & Lucius, A. L. Single turnover transient state kinetics reveals processive protein unfolding catalyzed by Escherichia coli ClpB. Elife 13, RP99052 (2024).
Lee, C., Schwartz, M. P., Prakash, S., Iwakura, M. & Matouschek, A. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell 7, 627–637 (2001).
Kraut, D. A. et al. Sequence- and species-dependence of proteasomal processivity. ACS Chem. Biol. 7, 1444–1453 (2012).
Fernandez, A. J. & Berger, J. M. Mechanisms of hexameric helicases. Crit. Rev. Biochem Mol. Biol. 56, 621–639 (2021).
Bell, T. A., Baker, T. A. & Sauer, R. T. Interactions between a subset of substrate side chains and AAA+ motor pore loops determine grip during protein unfolding. Elife 8, e46808 (2019).
Kardon, J. R., Moroco, J. A., Engen, J. R. & Baker, T. A. Mitochondrial ClpX activates an essential biosynthetic enzyme through partial unfolding. Elife 9, e46808 (2020).
Ye, Q. et al. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. Elife 4, e07367 (2015).
Alfieri, C., Chang, L. & Barford, D. Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 559, 274–278 (2018).
Bhat, J. Y. et al. Mechanism of enzyme repair by the AAA+ chaperone rubisco activase. Mol. Cell 67, 744–756 (2017).
Ryu, J.-K. et al. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science 347, 1485–1489 (2015).
White, K. I., Zhao, M., Choi, U. B., Pfuetzner, R. A. & Brunger, A. T. Structural principles of SNARE complex recognition by the AAA+ protein NSF. Elife 7, e38888 (2018).
Burrows, P. C. et al. Coupling σ factor conformation to RNA polymerase reorganisation for DNA melting. J. Mol. Biol. 387, 306–319 (2009).
Murakami, K. S., Masuda, S. & Darst, S. A. Structural basis of transcription initiation: RNA polymerase holoenzyme at 4 Å resolution. Science 296, 1280–1284 (2002).
Zhang, Y. et al. Structural basis of transcription initiation. Science 338, 1076–1080 (2012).
Kulbachinskiy, A. & Mustaev, A. Region 3.2 of the σ subunit contributes to the binding of the 3′-initiating nucleotide in the RNA polymerase active center and facilitates promoter clearance during initiation. J. Biol. Chem. 281, 18273–18276 (2006).
Campbell, E. A. et al. Structure of the bacterial RNA polymerase promoter specificity σ Subunit. Mol. Cell 9, 527–539 (2002).
Gao, F. et al. Structural basis of σ 54 displacement and promoter escape in bacterial transcription. Proc. Natl. Acad. Sci. USA 121, e2309670120 (2024).
Chen, J. et al. E. coli TraR allosterically regulates transcription initiation by altering RNA polymerase conformation. Elife 8, e49375 (2019).
Wulf, M. G. et al. Chemical capping improves template switching and enhances sequencing of small RNAs. Nucleic Acids Res 50, e2–e2 (2022).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Cardone, G., Heymann, J. B. & Steven, A. C. One number does not fit all: Mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol. 184, 226–236 (2013).
Morin, A. et al. Collaboration gets the most out of software. Elife 2, e01456 (2013).
Sysoeva, T. A., Chowdhury, S., Guo, L. & Nixon, B. T. Nucleotide-induced asymmetry within ATPase activator ring drives σ54-RNAP interaction and ATP hydrolysis. Genes Dev 27, 2500–2511 (2013).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci 32, e4792 (2023).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D. Struct. Biol. 74, 519–530 (2018).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Crooks, G. E., Hon, G., Chandonia, J.-M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res 14, 1188–1190 (2004).
Acknowledgements
We are grateful to Ruth Saecker, Brandon Malone, Ruby Froom, James Chen, Elizabeth Campbell, and Robert Landick for constructive discussion and critical reading of the manuscript. We thank Eilika Weber-Ban (ETH Zurich, Switzerland) for the generous gift of the expression plasmid of the M. tuberculosis open-gate proteasome. We thank Johanna Sotiris, Honkit Ng, and Mark Ebrahim of the Evelyn Gruss Lipper cryo-EM Resource Center (The Rockefeller University, New York, NY 10065, USA) for support with electron microscopy instrumentation and sample handling. We thank Michael Berne of the Tufts University Core Facility (Tufts Medical School, Boston, MA 02111, USA) for the Edman degradation-based protein sequencing service. A.U.M. is an Agouron Institute Awardee of the Life Sciences Research Foundation. This work was supported by a grant from the NIH to S.A.D. (R35 GM118130).
Author information
Authors and Affiliations
Contributions
A.U.M. and S.A.D. conceived and designed the study. A.U.M., N.M., and B.T.N. prepared purified proteins and performed biochemical experiments. A.U.M. performed cryo-EM sample preparation, data collection, and processed and analyzed the data. A.U.M. and S.A.D. interpreted structural data and prepared models. A.U.M. drafted the manuscript and prepared figures. A.U.M., B.T.N., and S.A.D. wrote the full manuscript. All authors provided comments and contributed to editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kalyan Das, Dong Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mueller, A.U., Molina, N., Nixon, B.T. et al. Real-time capture of σN transcription initiation intermediates reveals mechanism of ATPase-driven activation by limited unfolding. Nat Commun 16, 7138 (2025). https://doi.org/10.1038/s41467-025-61837-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61837-4
