
Abstract
Electrochromic technology has been considered as an energy-efficient approach to reduce the energy consumption in buildings and vehicles. Studies of electrochromic devices (ECDs) have so far focused mainly on control of cations (for example, H+, Li+, Na+, K+, and Zn2+, etc), while anions were rarely considered. Here, X-ray photoelectron spectroscopy (XPS) provides direct evidence that the transformation of Prussian blue (PB) to Prussian green (PG) occurs due to an anion intercalation process, in addition to the cation intercalation-induced switching between PB and Prussian white (PW). Co-intercalation of cations and anions is found in an ECD combining Nb18W16O93 and PB as complementary electrochromic layers: cations (for example, K+) insertion into Nb18W16O93 leads to its colored state and anions (for example, Cl−) insertion into PB forms PG. Benefiting from the co-intercalation of both cations and anions, the Nb18W16O93/PB based ECD can achieve diverse color and spectral modulations while maintaining excellent performance retention, thanks to the charge balance design. The concept of co-intercalation of cations and anions in an ECD provides a new approach to the development of next-generation high-performance ECDs.
Similar content being viewed by others


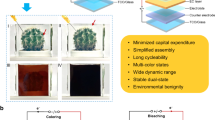
Introduction
Energy consumption in buildings accounts for 39% of the primary energy, and this ratio is more than those in industry (33%) and transportation (28%)1. Traditional windows fail to modulate the flow of solar light (either visible or near-infrared) passing through2,3; thus lighting, heating, ventilation, and air-conditioning (HVAC) systems are employed to maintain the indoor comfortable scenarios4,5,6. Electrochromic smart windows can dynamically control the transmittance of the solar spectra through a small external bias, are considered as a promising solution for energy-efficient buildings7,8,9,10,11. In addition to the two transparent electrodes (mostly indium tin oxide: ITO or fluoride doped tin oxide: FTO), the core configuration of an electrochromic device (ECD) is stacking electrochromic layer, electrolyte, and ion storage layer in series12,13,14. The ion storage layer can also be electrochromic-active to match the diverse color appearance of the full device through color superposition of the two electrodes. For example, lamination of WO3 and NiO layers by a Li+-containing electrolyte is the classical combination, which results in the appearance of transparent at the initial state, and gray at the colored state by overlaying the bluish WO313,14,15,16,17,18,19 and brownish NiO20,21 under a bias22,23,24,25. In ECD with Li+ based electrolytes, the optical modulation is achieved through cations (Li+ ions) shuttling between the electrochromic layer and the ion storage layer while the anions are rarely considered11,26,27,28. For a full ECD possessing excellent performance retention, one of the main concerns is that the two electrodes should have similar charge storage capacity, which is called charge balance of the two electrodes, i.e., the extracted ions from one electrode can be fully stored by the other electrode29,30,31,32,33. The mismatch of cation storage capacity leads to poor optical modulation, as well as complex parasitic side reactions on electrodes with weak storage capacity34,35,36, for electrochromic energy storage displays and fast response14,37,38,39,40,41,42. This is also one of the main reasons for the limited lifetime of WO3/NiO ECDs39.
Therefore, in order to maximize the merits of the two electrochromic layers in a full device to achieve dynamic spectra variation and superior cycling performance, co-intercalation of cation and anion could be promising. NiO exhibits outstanding electrochromism in alkaline electrolyte solution (such as KOH) due to anion (OH⁻) intercalation; however, no suitable counter electrode has been identified for assembling a full ECD. Fe4[Fe(CN)6]3, commonly known as Prussian blue (PB), can transform into Prussian white (PW), Prussian green (PG), and Prussian yellow (PY); however, its origin is not well understood.
In this article, we first present the direct experimental evidence from X-ray photoelectron spectroscopy (XPS) spectra that the electrochromism in the transition between PB and PG is attributed to anion (e.g., Cl−) intercalation/deintercalation, in addition to the well-documented cation intercalation/deintercalation involved in the switching between PB and PW. We further demonstrate that by intercalation of both cations and anions, the complementary ECDs can achieve diverse color and spectra modulations while maintaining excellent performance retention due to the charge balance design. Specifically, Nb18W16O93 and PB were employed as the complementary electrodes, and KCl in polyacrylamide (PAM) hydrogel as electrolyte. The device can be dynamically switched among four states by adjusting the bias, including transparent for both visible and near-infrared light, bright blue, dark blue, and green for low transmittance of visible and near-infrared light. We found that K+ ions can be inserted into Nb18W16O93 with high-rate diffusion kinetics, leading to switching between transparent and blue. More importantly, the large charge storage capacity of Nb18W16O93 is complementary to PB in a matched potential window, thereby maximizing the electrochromic performance of the full device, including large optical modulation, fast response, and long-term cycling stability. The co-intercalation of cations and anions for multi-band regulations in complementary electrodes may serve as a promising approach to achieve electrochromic smart windows with superior performance.
Results
Electrochromic device with multi-color and spectra modulations based on co-intercalation of anions and cations
The selection of KCl as the electrolyte is due to the very close ionic conductivities (73.5 and 76.4 S cm−2 mol−1 for K+ and Cl−, respectively) and mobilities (7.6 × 10−4 and 7.9 × 10−4 cm2 V−1 s−1 for K+ and Cl−, respectively) for K+ and Cl− ions43,44,45. PAM hydrogel has very high transparency and ionic conductivity similar to that of aqueous solutions46, as well as good interfacial adhesion, making it ideal as an electrolyte for ECD. The bright blue color of the assembled device is due to the combination of transparent Nb18W16O93 and intrinsic blue color of PB films (Fig. 1a). In the initial state, the open circuit potential (OCP) is 0.025 V, and both the visible and near-infrared transmittance is relatively low. Under a positive bias (1.5 V for 30 s), PB turns into PW (transparent) under K+ ions (1.33 Å) insertion, while the Cl− ions (1.81 Å) are accumulated on the Nb18W16O93 electrode instead of diffused into Nb18W16O93 lattice due to the larger ionic radius. At this stage, the device is transparent and possesses high transmittance at both visible and near-infrared light (Fig. 1b, and red line in Fig. 1e). Once the bias is switched back to OCP, the device returns to its initial state because of the transition from PW to PB through the K+ ions releasing process47. Under a negative bias (−1.8 V for 30 s), the Nb18W16O93 film changes to blue upon K+ ions insertion, while PB changes to PG upon Cl− ions insertion. Due to the overlay of blue (KxNb18W16O93) and green (PG), the device displays a dark blue and lower transmittance of both visible and near-infrared light (Fig. 1c, and blue line in Fig. 1e). As the negative bias increases (−2.4 V for 30 s), PG further transitions into PY because of the increased number of inserted Cl− ions, and the device appears to be green (Fig. 1d, and green line in Fig. 1e) from the superposition of blue (KxNb18W16O93) and yellow (PY). At this stage, both the transmittance of the visible and near-infrared light is also low. The device can return to its initial state once the bias returns to OCP. Overall, it shows that under the action of bias, the color and spectra modulations of the device can be manipulated by intercalation of both cations (K+) and anions (Cl−). This co-intercalated cation and anion in achieving dynamic spectra modulations is also found to be effective in other K⁺-based electrolyte systems (Figs. S1 and S2).

a Bright blue, as assembled. b Transparent, under a positive bias. c Dark blue, under a negative bias. d Green, under a negative bias. The insets are digital images of the device and the diagrams for different states of Nb18W16O93 and PB films. e Transmittance spectra of the device in the wavelength range of 400–2200 nm. The bias for the marked modes is OCP (bright blue), 1.5 V for 30 s (transparent), −1.8 V for 30 s (dark blue), and −2.4 V for 30 s (green), respectively. f Solar irradiance of the device in different modes in the wavelength range of 400–2200 nm. g Commission Internationale de l´Eclairage (CIE) chrominance coordinates L*, a* and b* in the CIELAB color space of the device at marked modes. h In-situ transmittance variation of the device under square-wave (−2.5 V for 20 s and 1.5 V for 20 s) cycling. i Changes in the optical density at 633 nm with respect to the intercalated charge density. j Cycling performance of the device under the square-wave (−2.3 V for 15 s and 1.3 V for 15 s) cycling for 6000 cycles. Chronoamperometry curves for (h) and (j) are shown in Fig. S4. The inset shows the transmittance at the 10th, 2000th, 4000th and 6000th cycles. All the transmittance data were recorded at 633 nm.
It is worth noting that when the device switches between transparent and dark blue mode, the average optical modulation in the solar spectrum range of 400–1400 nm is calculated to be 50.8% with a maximum optical modulation of 70.1% at 633 nm (Table S1), which is quite considerable, especially when taking into account the fact that light has to pass through the electrolyte and two sheets of FTO/glass substrates (Fig. S3). The solar irradiance transmittances of the device in different states are also calculated (Fig. 1f) and summarized in Table S2. It shows that the device at dark blue and green states possesses similar solar irradiation blocking effect (87.7%) in the visible light region (400–780 nm), and is higher than the one for bright blue state (70.1%). In short range of visible light (400–550 nm), the green state has the maximum solar radiation blocking capability (79.2%), whereas the dark blue state reveals the maximum solar radiation blocking (90.4%) in the near-infrared region (780–1400 nm). The above optical modulation data have demonstrated the multi-band electrochromism potential of this device. The corresponding chrominance coordinates L*, a*, and b* in the CIELAB color space are shown in Fig. 1g, and the distribution of color coordinates of the device in the chrominance space is intuitively seen, where the labels correspond to the modes presented in Fig. 1a–d. The corresponding chrominance coordinates in the 1931 color space (CIExy) are also shown in Table S3. Under negative potential bias (−2.4 V for 20 s), the switching time of the device from transparent to dark blue mode is only 1.0 s, and takes 3.9 s to return to transparent mode under positive bias (1.5 V for 20 s), as shown in Fig. 1h and Fig. S3, which demonstrates the potential of this device for fast switching between dark blue and green states (Fig. S5). At the wavelength of 633 nm, the coloration efficiency of the first step is 152.2 cm2 C−1, and the second step is −71.5 cm2 C−1 (Fig. 1i). The change of coloration efficiency from positive to negative switching reflects the shift of the central absorption wavelength during the color change from transparent to blue and green. Under the square wave potentials of −2.3 V for 15 s and 1.3 V for 15 s, the device switches between transparent and dark blue modes, and possesses excellent performance retention as subjecting to long-term cycling, i.e., 95.2% after 6000 cycles (Fig. 1j). The slight attenuation of bleaching transmittance may be associated with the capture of K+ in PB25,48. The slowdown of coloration speed after 4000 cycles may be related to the captures of K+ in Nb18W16O93 and Cl− in PB. We assembled more than 10 devices and the results are consistent. Compared with rocking-chair ECDs that only use cations (Table S4), the as-prepared ECDs employing co-intercalation of anions and cations demonstrate significant advantages in terms of multi-band control, response time and cycling stability.
Electrochromism for single layer of Fe4[Fe(CN)6]3 and Nb18W16O93
In order to provide a deep insight of the co-intercalation of anions and cations in the devices, the electrochromism in PB and Nb18W16O93 films was comprehensively investigated. A three-electrode configuration was employed where Ag/AgCl and Pt are used as reference electrode and counter electrode. Figure 2a shows the crystal structure of PB, which has a cubic stacking contains high spin Fe3+ ions and low spin Fe2+ ions as marked by the orange and green arrows, respectively. X-ray diffraction (XRD) patterns confirm the fcc structure (PDF# 97-002-3102) of the water-insoluble PB (Fig. 2b, blue line), and is free of any other impurities. As PB is subjecting to electrochemical cycling (100 mV s−1) in the range of −0.6–1.4 V vs Ag/AgCl in 1.0 mol L−1 of KCl hydrogel, two pairs of peaks can be noted, as denoted as pair i/ii, and iii/iv (Fig. 2c), which correspond to the insertion and extraction of K+ and Cl− ions, respectively, as validated next. The in-situ transmittance spectrum in a single cycle, i.e., PW to PY to PW, has been recorded in the two-dimensional contour plot (Fig. 2d, the associated variation of CIE and CIELAB chrominance coordinates are provided in Fig. S6 and Table S5). A negative bias is applied to yield the color change from bright blue to transparent (PW), XPS spectra in Fig. 2e–h show that K element is detected at the transparent state, in addition to other elements (Fe, C, N) from initial PB (i.e., there is no K or Cl in the as-deposited PB, as shown in Fig. S7a–c). XRD pattern reveals a shift of (200) peak to lower angles (Fig. 2b, gray line), suggesting an enlarged lattice due to the insertion of K+ ions into PB. The ratio of high-spin Fe3+ and low-spin Fe3+ in PW is found to be lower than that in PB (Fig. 2e and Table S6), which indicates that Fe3+ ions are reduced to Fe2+ ions. Meanwhile, binding energy of Fe-N in PW is slightly reduced due to the bonding of K to N (Fig. 2e–g). This result is also supported by the significant weakening of the C≡N bond stretching vibration in the PW sample, as shown in Raman spectra (Figs. S8 and S9a). Depth profile of the K element in the XPS measurement confirms that K+ ions insertion in PW film sample is vertically uniform (Figs. 2h and S7d). Therefore, we conclude that the pair peak of i/ii is due to K+ ions insertion/extraction, and the optical modulation is over 60% in the wavelength range of 550–1100 nm (Figs. 2d and S10a), the associated K+ ions intercalation process can be written as49:

a Crystal structure of PB. b XRD patterns of PB in different states (PW, PB, PG, and PY), the right is a magnified view of the (200) crystal plane. c Cyclic voltammetry (CV) curve of PB at a scan rate of 100 mV s−1 between −0.6 and 1.4 V and its in-situ transmittance curve. d Two-dimensional contour plot of in-situ transmittance spectrum change with voltage. XPS spectra of (e) Fe 2p, f C 1s and g N 1s of PW, PB, PG and PY. XPS spectra of (h) K 2p of PW, Cl 2p of PG. i Crystal structure schematic diagram of possible ion intercalation situations of PW, PB, PG, and PY. j Lattice constants “a” and their volume of PB in different states (PW, PB, PG, and PY). Note: Prussian blue (PB), Prussian white (PW), Prussian green (PG) and Prussian yellow (PY).
As the bias polarized to 1.4 V, a current density peak of iii at 1.1 V is observed and the film changes from bright blue to green (PG). XPS results reveal the presence of Cl at this stage, with no detection of K element (Figs. 2h and S7e), and the ratio of low spin Fe3+ in PG is high than that in PB. The (200) peak also shifts to lower angles, indicating an enlarged lattice (Fig. 2b, green line). This lattice expansion becomes more obvious as the increase of inserted Cl− ions (Fig. 2b, yellow line) and the associated films change from green to yellow (PY). This is also due to the slow oxidation of Fe2+ to Fe3+ (Fig. 2e)50, as an increased intensity of Cl has been detected in PY sample (Figs. 2h and S11). The Cl− ions are bonded to the low spin Fe3+ ions, and the stretching vibration of the C≡N bond is not affected, but only slightly shifted to the high wavenumber, which may be due to the weakening of coordination between the low spin Fe3+ and C≡N (Figs. S8 and S9b). The binding energy of Fe-N in PG and PY are low than that in PB (Fig. 2g). The depth profile of the Cl element in the XPS measurement confirms that Cl− ions insertion in PG and PY film samples are vertically uniform (Fig. S12). Therefore, the peak iii corresponds to Cl− ions insertion into PB while the peak iv belongs to Cl− releasing. The decline of transmittance is over 35% in the wavelength range of 400–550 nm, the increase of transmittance is over 50% in the wavelength range of 550–1100 nm (Figs. 2d and S10b). The associated intercalation of Cl− ions can be written as:
Figure 2i provides the schematic diagram of possible ion diffusion tunnel for K+ (1.33 Å) and Cl− ions (1.81 Å). It can be noted that the open tunnel with a diameter of 5.44 Å, which allows both K+ and Cl− ions to diffuse. According to Fig. 2c, the amounts of charges for insertion of K+ and Cl− ions are calculated to be 8.6 mC cm−2 and 6.2 mC cm−2, respectively (Fig. S13). The amounts of charges satisfy the change of PB to PW (ΔT = 54.5%) and PB to PY (ΔT = 51.3%). The lattice expansion and volume change of PB after K+ and Cl− intercalation are calculated and revealed in Fig. 2j. The maximum lattice expansion is 9.5%, which is induced by the intercalation of K+ ions and the increment of the radius for Fe3+ ions (Fe3+ radius is 0.79 Å and Fe2+ radius is 1.03 Å)51,52,53. Only 3.3% lattice expansion occurs after Cl− intercalation (PB to PY), confirming the unprecedented performance retention upon cycling of the device (Fig. 1j and Supplementary Note 1). To more clearly understand the influence of ion intercalation, we use an ex-situ measurement to show the structure evolution process. The results show that the crystal structure of PB does not change significantly (Supplementary Note 2 and Fig. S14), and its maximum volume change is only 11.0%, which is due to the larger ionic radius of Fe2+ than that of Fe3+. The results presented above provide clear evidence for the intercalation of K⁺ and Cl⁻ in PB, while also uncovers the origin of the observed diverse color variations.
Figure 3a shows the view along the c-axis for a 1 × 3 × 1 superstructure, where trilateral, quadrilateral, and pentagonal tunnels can be clearly seen. XRD measurements confirm that the tetragonal tungsten bronze of the pristine (bleached) Nb18W16O93 (Fig. 3b, red line). The size of the inscribed circle for trilateral, quadrilateral, and pentagonal tunnels are calculated as 0.78, 1.49, and 1.62 Å, respectively. It is found that only quadrilateral and pentagonal tunnels allow for K+ (1.33 Å) ions intercalation, while none of the three polygon tunnels could allow the insertion of Cl− (1.81 Å) ions. This clarifies our earlier statement that when a positive bias is applied on the device, K+ ions are inserted into PB to form PW while Cl− ions are accumulated on the surface of (rather than inserted into) Nb18W16O93.

a Crystal structure of Nb18W16O93, viewing along c-axis. b XRD patterns of Nb18W16O93 in bleached and colored states. c CV curve of Nb18W16O93 at a scan rate of 100 mV s−1 vs Ag/AgCl between −1.6 and 0.4 V and d the two-dimensional contour plot of in-situ transmittance spectrum change with potential. The distribution of CIE and CIELAB color coordinates for the color of film under different bias is provided in Fig. S15 and Table S7. e XPS spectra of Nb 3 d, W 4 f of Nb18W16O93 in bleached/colored states and the K 2p of Nb18W16O93 in colored state.
Figure 3c, d show the CV curve and in-situ transmittance spectrum of the Nb18W16O93 film record in 1.0 mol L−1 of KCl hydrogel. It can be seen that Nb18W16O93 only begins to change color when the applied potential is lower than −0.6 V, which possesses a higher reduction potential matching to the high oxidation potential of PB, and exhibits significant dual-band modulation properties in the visible and near-infrared regions (Fig. S16). XPS spectra of the bleached and colored states of the Nb18W16O93 film show that coloration is mainly from the reduction of W6+ to W5+ and W4+ (i.e., the valence of Nb rarely varies), as revealed in Fig. 3e. Meanwhile, only K element is detected at this state, in addition to native elements (Nb, W, O) from pristine Nb18W16O93. XRD pattern shows a slight shift to low angels of (001) plane at the colored state (Fig. 3b, blue line), suggesting an enlarged lattice due to K+ ions insertion. The depth profile of K element confirming the uniform distribution of K+ ions in Nb18W16O93 (Fig. S17a). In the entire cycling process, no Cl signal is detected (Figs. S17b and S18). Therefore, it is concluded that the observed peaks in Fig. 3c is due to the intercalation of K+ ions and can be written as:
As noted from the above XPS results (Fig. 3e), K+ ions insertion mainly leads to reduction of W6+ to W5+ and W4+, which results in a blue color of the film. For the Nb18W16O93 electrode, the amount of charge for insertion is calculated to be 14.5 mC cm−2 (Figs. 3c and S19). At the wavelength of 633 nm, the charge quantities involved in coloration and bleaching are sufficient to enable an optical modulation exceeding 70%. The lattice constant of the Nb18W16O93 electrode at its colored state is calculated according to the XRD measurements where the lattices contract along the a-axis and expand in the b-axis and c-axis upon K+ ions insertion, resulting in a maximum expansion of only 2.9% of the unit cell volume (Fig. 3b, Supplementary Note 3 and Table S8). Similarly, the crystal structure variation of the Nb18W16O93 electrode is insignificant, i.e., maximum volume change is only 6.5%, despite the injection of more charge (Fig. S20). The above results indicate that Nb18W16O93 is also an ideal electrode material for the intercalation of K+ ions.
Charge balance design of the electrochromic device with co-intercalation of anions and cations
Charge balance between the two electrodes is the key prerequisite to maximize the electrochromic performance in a full device54. The thicknesses of the PB and Nb18W16O93 films used in this work are 330 nm and 220 nm, respectively (Fig. S21). By applying a positive potential to the PW film (Fig. 4a, b), it can be calculated that the amount of charge required to change from PW to PB is Q1 (7.04 mC cm−2) under K+ ion releasing, and that from PB to PY is Q2 + Q3 (5.11 mC cm−2) under Cl− ions insertion. The ratio of Q1/(Q2 + Q3) is 1.37, which is close to the ratio (1.33) of Fe3+/Fe2+ in PB molecule. This evidence further confirms that the PB film used in this work is water-insoluble PB50,55. As shown in Fig. 4c, d, the charge for complete coloration of the Nb18W16O93 film is Q4 (13.74 mC cm−2) under K+ ions insertion, which is larger than Q2 + Q3 for Cl− ions insertion into PB. This indicates that the number of Cl− ions required for the oxidation of the PB electrode to PG or PY can fully match the number of K+ ions required for the coloration process of the Nb18W16O93 film in the assembled device. Based on the electrochromic and crystallography results of PB and Nb18W16O93 electrodes, this further indicates that the co-intercalation of anions and cations in the two electrodes can lead to a stable realization of deep blue and green states of the device.

a Direction of ion movement during the different color switching of the PB film. b In-situ transmittance curve (blue line) of the PB film under applied square-wave potentials (−0.6 V for 15 s; 1.2 V for 15 s) and its current density-time curve (red line, Fig. S22). The color of the shadow corresponds to the color of the film state, blue PB), green (PG), and yellow (PY). c Direction of ion movement during the color switching of the Nb18W16O93 film in hydrogel electrolyte. d In-situ transmittance curve (blue line) of the Nb18W16O93 film under applied square-wave potentials (−1.4 V for 15 s; 0.4 V for 15 s) and its current density-time curve (red line, Fig. S23). The color of the shadow corresponds to the color of the film state, blue (Colored Nb18W16O93). e CV curve (red line) of the ECD at a sweep rate of 100 mV s−1 between 1.6 and −2.5 V and its in-situ transmittance curve (blue line). The gray arrows indicate the sweep direction, and the sweep starts from OCP (−0.025 V). f Transmittance change of the device in its transparent, dark blue and green states over a 12-h period without bias. Single-wavelength transmittance data at 633 nm were taken. g Schematic diagram of temperature control of the building model (Fig. S24). h Temperature change rate and the temperature of the device used in the building model in four states after heating for 15 min and 40 min, respectively (The red and blue histograms represent the heating rates for the first 15 min and the next 25 min, respectively; the red and blue dotted lines represent the temperatures in the building model after heating for 15 min and 40 min, respectively). Note: Prussian blue (PB), Prussian white (PW), Prussian green (PG) and Prussian yellow (PY).
Figure 4e shows the CV curve of the assembled device based on the two electrodes as well as the in-situ optical modulation at 633 nm. The potential window is −2.4–1.6 V and the sweep rate is 100 mV s−1. It can be found that two pairs of peaks from the PB film (i.e., i/iv, ii/iii) and one pair peak from the Nb18W16O93 film (v/vi) are clearly visible, suggesting a good match of the two electrodes. Indeed, in a single cycle, the device displays almost the same amounts of charges for insertion (19.34 mC cm−2) and extraction (19.68 mC cm−2), confirming an excellent match of the two electrodes. Therefore, the co-intercalation of K+ and Cl− ions in the two electrodes results in a charge balance in the device, and the color appearance and spectra modulation appear as a superposition of the two electrodes in different states.
Finally, the “optical memory” of the device was examined. As shown in Fig. 4f, we cut off the biases when the device reached to transparent, dark blue, and green states, respectively. The states of dark blue and green are quite stable due to the fact of intercalations of Cl− ions in the PB film and K+ ions in the Nb18W16O93 film (the associated variation of OCP was recorded in Fig. S25). However, the optical memory effect of transparent state is relatively poor. It should be pointed out that, the achievement of transparent state is due to the insertion of K+ ions into the PB film and the accumulation of Cl− ions on the surface of the Nb18W16O93 film. The poor optical memory of the transparent state is due to the weak interaction between the Nb18W16O93 film and Cl− ions. It is worth noting that the dark blue and green states result from different degrees for co-intercalation of anions and cations. The heating tests of the building model have shown that the device in the green state provides the slowest heating rate (0.84 °C min−1) for the first 15 min, and the dark blue state provides the slowest heating rate (0.27 °C min−1) for the next 25 min, as shown in Fig. 4g, h. It is clear that the transparent state can keep the building model at a high temperature, which is beneficial for the use of smart windows in winter. After heating for 40 min, the building model with green state maintains a lowest temperature. The different heating rates of the four states broaden the range of options available to users for desired temperature. In order to evaluate the thermal stability, a square-wave potential is applied to the device at 45 °C for 60,000 s, and the corresponding spectra of the device in its four states before and after cycling have been recorded. The results show that the electrochemical capacitance of the device is well maintained (Fig. S26), and only the transmittance in the transparent state is slightly decreased compared to the pristine state (Fig. S27).
Though PB was previously reported to transform into PW, PG and PY, the origin of the transitions was not well understood. We first provide clear evidence that the transitions between PB and PG, PG and PY are attributed to anions intercalation/deintercalation, in addition to the well-known cation intercalation/deintercalation involved in the switching between PB and PW. ECDs were designed by using Nb18W16O93 and PB films as two electrochromic layers, and KCl hydrogel as the electrolyte. Through systematic XPS characterizations, it is found that cations (K+) insertion into Nb18W16O93 leads to its colored state and anions (Cl−) insertion into PB forms PG, which is co-intercalation of cations and anions in one device. Benefiting from the co-intercalation dynamics in the Nb18W16O93/PB system, ECDs are able to possess four modes: transparent for both visible and near-infrared light, bright blue, dark blue, and green for low transmittance of visible and near-infrared light, exhibiting diverse spectra modulations. Ion intercalation in either the Nb18W16O93 and PB films leads to small volume change, supporting the excellent cycling stability of the two electrochromic layers. The redox potential of the Nb18W16O93 and PB films are in a good match, yielding an excellent electrochromic performance of the ECD with multiple color and spectra modulations, fast response. We found that the charge for achieving various states in the PB film can be well stored in the Nb18W16O93 film, as confirmed by un-noticed degradation after 6000 cycles. We believe that the co-intercalation of cations and anions could provide an efficient approach to develop superior ECDs in the future.
Methods
Materials
Ammonium niobate oxalate (C4H4NNbO9, 99.99%), ammonium paratungstate ((NH4)10H2(W2O7)6, >99%), oxalate dihydrate (C2H2O4·2H2O, ≥99%), anhydrous ethanol (C2H5OH, 99.8%), potassium ferricyanide (K3[Fe(CN)6], 99%), ferric chloride (FeCl3, 98%), acrylamide (AM, 99%), potassium persulfate (KPS, 99.5%), N, N-methylenebisacrylamide (MBAA, 99%), potassium chloride (KCl, 99.5%), potassium bromide (KBr, 99%), potassium sulfate (K2SO4, 99%) and potassium bis(fluorosulfonyl) imide (K[(FSO₂)₂N]; KFSI, 99%). All chemicals were purchased from Shanghai Titan Scientific Co., Ltd (China), and used without further purification. Fluorine-doped tin oxide (FTO) transparent conductive glasses (square resistance <15 Ω sq−1, optical transmittance >83%) were purchased from Zhuhai Kaivo Optoelectronic Technology Co., Ltd (China).
Preparation of Nb18W16O93 films
Nb18W16O93 films were in situ grown on the surface of FTO-coated glasses by the hydrothermal method56. First, the hydrothermal precursor solution can be obtained by following process. C4H4NNbO9 and (NH4)10H2(W2O7)6 were weighed according to an atomic ratio of niobium to tungsten as 9:8 and dissolved in 35 mL of 40% C2H5OH aqueous solution containing 5 g of C2H2O4⋅2H2O at room temperature. Second, the FTO glasses should be diagonally inserted into a polytetrafluoroethylene (PTFE) liner containing 40 mL of precursor solution. After being kept at 180 °C for 4 h and cooled to room temperature, the films were taken out and cleaned with deionized water, and then dried at room temperature in air. The area of FTO glass is 2.5 × 5 cm2, and the area infiltrated by the hydrothermal precursor solution is 1.5 × 3 cm2.
Preparation of Prussian blue films
For the electrodeposition of PB films11,57, the electrolyte solution was composed of 0.05 mol L−1 of KCl, 0.01 mol L−1 of FeCl3 and 0.01 mol L−1 of K3[Fe(CN)6]. The electrodeposition was conducted under a constant current density of −50 μA cm−2 for 300 s. The area of FTO glass is 2.5 × 5 cm2, and the area infiltrated by the electrodeposition solution is 1.5 × 3 cm2.
Assembly of the device
The assembly of the device adopts a traditional sandwich structure. Two electrochromic electrodes were bonded with double-sided tape, and then a gel electrolyte was constructed by in-situ polymerization of PAM hydrogels. The preparation of PAM hydrogels is described below. First, 1.0 g of AM monomer and 0.7455 g of KCl were dissolved in 10.0 mL of deionized water. Then, 1 mg of MBAA cross-linker and 5 mg of KPS initiator were added to the above AM solution. After stirring for 30 min, the solution was sonicated for 10 min to remove the dissolved air bubbles, and injected the gap of two electrodes separated by double-sided tape. The cross-linking polymerization were finished at 60 °C for 2 h to obtain PAM hydrogels.
Materials characterization
X-ray diffraction (XRD) for crystal phase analysis was measured using a Rigaku Smartlab with a Cu Kα X-ray source operating at 40 kV and 15 mA. Multiple scans were collected between 2θ = 10° and 70° with a step size of 0.2°, a grazing incidence angle of 0.4° and a scan rate of 10° min−1. For the X-ray photoelectron spectroscopy (XPS) measurements, samples in the electrolytic cell were washed in acetone in a glove box to remove the residual electrolyte on the surface, and transferred to the XPS chamber through a vacuum transfer box. XPS spectra of the Nb18W16O93 and PB films were conducted with Escalab Xi+ from Thermo Fisher Scientific with Al Kα radiation, hυ = 1486.7 eV. The XPS depth profiles of the Nb18W16O93 and PB films in their charge/discharge states were measured. All binding energies of the Nb18W16O93 film were calibrated to the surface-contaminated C1s peak (284.8 eV). Due to the presence of CN, all binding energies of the PB film were not calibrated and the XPS peak was fitted by the Avantage software curve. The surface morphology and cross-section thickness of the samples were assessed by field emission scanning electron microscopy (FESEM, ZEISS Gemini SEM 300, Germany). Raman spectra were recorded using an inVia Qontor (Renishaw, UK) system at the wavelength of 532 nm.
Measurements for electrochemical and electrochromic properties
The electrochemical behavior of the Nb18W16O93 and PB films were investigated by using an electrochemical workstation (CHI 760E, China) in a three-electrode system, where the FTO coated glasses grown Nb18W16O93 and PB films were used as the working electrodes, Pt electrode as the counter electrode, PAM hydrogel containing 1 mol L−1 of KCl as the electrolyte. Ag/AgCl electrode was used as the reference electrode. Charge capacity (in unit of mC cm−2) was determined from cyclic voltammetry data by:\(C=\int {jdV}/{\mbox{s}}\), where j is current density (in mA cm−2), s is the sweep rate (in V s−1), and V is the voltage. Electrochromic properties of the Nb18W16O93 and PB films were measured by using a UV–Vis spectrophotometer (SHIMADZU UV-1900i, Japan) coupled to the electrochemical workstation. The raw data of the films were directly measured including the transmittances of the film and FTO glass, and the raw data of the ECDs were measured including the transmittances of the Nb18W16O93 film, PB film, PAM hydrogels and two layers of FTO glass. The optical modulation is defined as ΔT = Tb – Tc, where Tb is the transmittance of the bleached state of the film, \(T{\mbox{c}}\) is the transmittance of the colored state of the film. The response times (coloration time tc and bleaching time tb) are defined as the times taken for the optical transmittance of the film to reach 90% of the maximum optical modulation at the specified voltages.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available from the corresponding authors upon request. Source data are provided with this paper.
References
Aneke, M. & Wang, M. H. Energy storage technologies and real-life applications—a state of the art review. Appl. Energy 179, 350–377 (2016).
Sengupta, M. et al. The national solar radiation data base (NSRDB). Renew. Sust. Energy Rev. 89, 51–60 (2018).
Zhang, N. et al. Refining defect states in W18O49 by Mo doping: a strategy for tuning N2 activation towards solar-driven nitrogen fixation. J. Am. Chem. Soc. 140, 9434–9443 (2018).
Nguyen, T. A. & Aiello, M. Energy intelligent buildings based on user activity: a survey. Energy Build. 56, 244–257 (2013).
Oldewurtel, F., Sturzenegger, D. & Morani, M. Importance of occupancy information for building climate control. Appl. Energy 101, 521–532 (2013).
Waqas, A. & Din, Z. U. Phase change material (PCM) storage for free cooling of buildings-a review. Renew. Sust. Energy Rev. 18, 607–625 (2013).
Zhou, Q. et al. Photo-electrochemical synergistically induced ion detrapping for electrochromic device rejuvenation. Matter 8, 1–10 (2024).
Zhang, R. et al. Polaron hopping induced dual-band absorption in all amorphous cathodic electrochromic oxides. Appl. Phys. Rev. 12, 011404 (2025).
Wang, Y., Runnerstrom, E. L. & Milliron, D. J. Switchable materials for smart windows. Annu. Rev. Chem. Biomol. Eng. 7, 283–304 (2016).
Cai, G. F., Wang, J. X. & Lee, P. S. Next-generation multifunctional electrochromic devices. Acc. Chem. Res. 49, 1469–1476 (2016).
Wang, J. et al. A bi-functional device for self-powered electrochromic window and self-rechargeable transparent battery applications. Nat. Commun. 5, 4921 (2014).
Wen, R.-T., Granqvist, C. G. & Niklasson, G. A. Anodic electrochromism for energy-efficient windows: cation/anion-based surface processes and effects of crystal facets in nickel oxide thin films. Adv. Funct. Mater. 25, 3359–3370 (2015).
Hu, J. et al. Combining Bragg reflector and Fabry–Pérot cavity into a solid-state electrochromic device for dynamic modulation. J. Opt. Microsyst. 4, 031205 (2024).
Wang, J., Khoo, E., Lee, P. S. & Ma, J. Synthesis, assembly, and electrochromic properties of uniform crystalline WO3 nanorods. J. Phys. Chem. C 112, 14306–14312 (2008).
Shao, Z. et al. All-solid-state proton-based tandem structures for fast-switching electrochromic devices. Nat. Electron. 5, 45–52 (2022).
Deb, S. K. Opportunities and challenges of electrochromic phenomena in transition metal oxides. Sol. Energy Mater. Sol. Cells 25, 327–338 (1992).
Granqvist, C. G. Oxide electrochromics: an introduction to devices and materials. Sol. Energy Mater. Sol. Cells 99, 1–13 (2012).
Zhang, R., Zhou, Q., Huang, S., Zhang, Y. & Wen, R.-T. Capturing ion trapping and detrapping dynamics in electrochromic thin films. Nat. Commun. 15, 2294 (2024).
Ma, D., Wang, H., Zhang, Q. & Li, Y. Self-weaving WO3 nanoflake films with greatly enhanced electrochromic performance. J. Mater. Chem. 22, 16633–16639 (2012).
Wen, R.-T., Niklasson, G. A. & Granqvist, C. G. Electrochromic nickel oxide films and their compatibility with potassium hydroxide and lithium perchlorate in propylene carbonate: optical, electrochemical and stress-related properties. Thin Solid Films 565, 128–135 (2014).
Passerini, S. & Scrosati, B. Electrochromism of thin-film nickel-oxide electrodes. Solid State Ion. 53, 520–524 (1992).
Cai, G. F. et al. Electrochromo-supercapacitor based on direct growth of NiO nanoparticles. Nano Energy 12, 258–267 (2015).
Huang, H. et al. Electrochromic properties of porous NiO thin film as a counter electrode for NiO/WO3 complementary electrochromic window. Electrochim. Acta 56, 4281–4286 (2011).
Niklasson, G. A. & Granqvist, C. G. Electrochromics for smart windows: thin films of tungsten oxide and nickel oxide, and devices based on these. J. Mater. Chem. 17, 127–156 (2007).
Wang, K. et al. Long-term-stable WO3-PB complementary electrochromic devices. J. Alloy. Compd. 861, 158534 (2021).
Huang, B. et al. Prolonging lifespan of Prussian blue electrochromic films by an acid-free bulky-anion potassium organic electrolyte. Chem. Eng. J. 449, 137850 (2022).
Li, H. Z., Zhang, W. & Elezzabi, A. Y. Transparent zinc-mesh electrodes for solar-charging electrochromic windows. Adv. Mater. 32, 2003574 (2020).
Wen, R.-T., Granqvist, C. G. & Niklasson, G. A. Eliminating degradation and uncovering ion-trapping dynamics in electrochromic WO3 thin films. Nat. Mater. 14, 996–1001 (2015).
Wang, X. et al. Unlocking anionic redox by breaking metal–oxygen bonds in aqueous zinc batteries. ACS Energy Lett. 8, 4547–4554 (2023).
Lu, N. et al. Electric-field control of tri-state phase transformation with a selective dual-ion switch. Nature 546, 124–128 (2017).
Huang, Z. et al. Anion chemistry in energy storage devices. Nat. Rev. Chem. 7, 616–631 (2023).
Kim, Y. et al. Energy saving electrochromic polymer windows with a highly transparent charge-balancing layer. Adv. Funct. Mater. 27, 1701192 (2017).
Zhao, F. F. et al. Counterbalancing the interplay between electrochromism and energy storage for efficient electrochromic devices. Mater. Today 66, 431–447 (2023).
Choi, D. et al. Investigation of dry-deposited ion storage layers using various oxide particles to enhance electrochromic performance. Sol. Energy Mater. Sol. Cells 174, 599–606 (2018).
Zhao, Q. et al. Electrochromic-induced rechargeable aqueous batteries: an integrated multifunctional system for cross-domain applications. Nano Micro Lett. 15, 87 (2023).
Guo, Q. et al. High performance multicolor intelligent supercapacitor and its quantitative monitoring of energy storage level by electrochromic parameters. ACS Appl. Energy Mater. 3, 2727–2736 (2020).
Guo, Q., Zhao, X., Li, Z., Wang, D. & Nie, G. A novel solid-state electrochromic supercapacitor with high energy storage capacity and cycle stability based on poly(5-formylindole)/WO3 honeycombed porous nanocomposites. Chem. Eng. J. 384, 123370 (2020).
Ma, D., Yang, B. & Wang, J. Boosting the self-recharging of Polypyrrole/Prussian blue electrochromic device by potential difference-driven alternative redox. ACS Appl. Mater. Interfaces 15, 56041–56048 (2023).
Wang, K. et al. Boosting charge-transfer kinetics and cyclic stability of complementary WO3-NiO electrochromic devices via SnOx interfacial layer. J. Sci. Adv. Mater. Dev. 6, 494–500 (2021).
Pehlivan, E., Granqvist, C. G. & Niklasson, G. A. Electrochromic nickel-oxide-based thin films in KOH electrolyte: Ionic and electronic effects elucidated by impedance spectroscopy. Sol. Energy Mater. Sol. Cells 269, 112795 (2024).
Krishna, V. S. G. et al. Ultrathin sputtered NiO films for enhanced electrochromic performance in smart windows. J. Mater. Chem. A 12, 19378–19391 (2024).
Usha, K. S., Lee, S. Y., Sivakumar, R. & Sanjeeviraja, C. Ultra-fast switching of energy efficient electrochromic nickel oxide thin films for smart window applications. Ceram. Int. 50, 36651–36665 (2024).
Cassone, G., Creazzo, F., Giaquinta, P. V., Sponer, J. & Saija, F. Ionic diffusion and proton transfer in aqueous solutions of alkali metal salts. Phys. Chem. Chem. Phys. 19, 20420–20429 (2017).
Morgan, B. & Madden, P. A. Ion mobilities and microscopic dynamics in liquid (Li, K). Cl. J. Chem. Phys. 120, 1402–1413 (2004).
Gee, M. B. et al. A Kirkwood-Buff derived force field for aqueous alkali halides. J. Chem. Theory Comput. 7, 1369–1380 (2011).
Cai, H., Chen, Z., Guo, S., Ma, D. & Wang, J. Polyacrylamide gel electrolyte for high-performance quasi-solid-state electrochromic devices. Sol. Energy Mater. Sol. Cells 256, 112310 (2023).
Wu, C. et al. High-performance complementary electrochromic batteries using Nb18W16O93 by the synergistic effects of aqueous Al3+/K+ dual-ion. Angew. Chem. Int. Ed. 64, e202415050 (2024).
Liu, Q. R. et al. Electrolytes-relevant cyclic durability of nickel oxide thin films as an ion-storage layer in an all-solid-state complementary electrochromic device. Sol. Energy Mater. Sol. Cells 157, 844–852 (2016).
Itaya, K., Uchida, I. & Toshima, S. Mediated electron transfer reactions between redox centers in Prussian blue and reactants in solution. J. Phys. Chem. 87, 105–112 (1983).
DeLongchamp, D. M. & Hammond, P. T. High-contrast electrochromism and controllable dissolution of assembled Prussian blue/polymer nanocomposites. Adv. Funct. Mater. 14, 224–232 (2004).
Ling, C., Chen, J. & Mizuno, F. First-principles study of alkali and alkaline earth ion intercalation in iron hexacyanoferrate: the important role of ionic radius. J. Phys. Chem. C. 117, 21158–21165 (2013).
Li, K. et al. Lattice-contraction triggered synchronous electrochromic actuator. Nat. Commun. 9, 4798 (2018).
Gu, G. et al. V2O5 nanofibre sheet actuators. Nat. Mater. 2, 316–319 (2003).
Kim, S. Y., Yun, T. Y., Yu, K. S. & Moon, H. C. Reliable, high-performance electrochromic supercapacitors based on metal-doped nickel oxide. ACS Appl. Mater. Interfaces 12, 51978–51986 (2020).
Itaya, K., Ataka, T. & Toshima, S. Spectroelectrochemistry and electrochemical preparation method of Prussian blue modified electrodes. J. Am. Chem. Soc. 104, 4767–4772 (1982).
Sun, J., Wan, X., Yang, T., Ma, D. & Wang, J. High-performance tungsten-niobium bimetallic oxide films with designable electrochromic properties. Sol. Energy Mater. Sol. Cells 256, 112318 (2023).
Ma, D., Eh, A. L. S., Cao, S., Lee, P. S. & Wang, J. Wide-spectrum modulated electrochromic smart windows based on MnO2/PB films. ACS Appl. Mater. Interfaces 14, 1443–1451 (2022).
Acknowledgements
This work is financially supported by National Natural Science Foundation of China (No. 62275154, 52172294, 61775131), National Natural Science Foundation of Guangdong (No. 4685326), the “Shenzhen Science and Technology Innovation Commission” (Grant Nos. JCYJ20210324105402007 and 2022081509567001), Guangdong Provincial Innovation and Entrepreneurship Project (No. 2017ZT07C071), and Guangdong Provincial Key Laboratory Program (2021B1212040001) from the Department of Science and Technology of Guangdong Province. The characterization work used the resources from SUSTech Core Research Facilities that receives support from the Shenzhen Municipality. We also acknowledge the support from the Major Science and Technology Infrastructure Project of Material Genome Big-science Facilities Platform, supported by the Municipal Development and Reform Commission of Shenzhen. We thank Mr. Wenqi Wang for his help in supplementary experiments and Dr. Yilin Ding for her help in CIELAB result analysis.
Author information
Authors and Affiliations
Contributions
J.W.S., J.M.W., R.T.W. and D.Y.M. conceived the idea and designed the experiments. J.M.W., D.Y.M. and R.T.W. supervised the research. J.W.S. prepared all the films, devices and carried out all the electrochemical and electrochromic testing for them. Z.C. carried out the synthesis experiment of PAM hydrogels electrolyte and assisted in the assembly of the devices. R.F.Z. and M.H.Y. helped the analysis and testing of XPS data, Y.Z. and J.C.H. helped the testing of SEM, Q.Q.Z. helped the testing of XRD data, P.P.S. helped the calculation of charge amount for device. J.W.S. and M.H.Y. designed and drew the schematic diagrams for device structure. Q.J.H. helped the drawing of crystal structure. J.W.S., J.M.W., R.T.W. and D.Y.M. analyzed the data and co-wrote the manuscript. All authors analyzed and discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Guangming Nie and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, J., Chen, Z., Zhang, R. et al. Electrochromic smart windows with co-intercalation of cations and anions for multi-band regulations. Nat Commun 16, 6993 (2025). https://doi.org/10.1038/s41467-025-61854-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61854-3
