
Abstract
ATP is an important allosteric regulator of many enzymes, but these enzymes typically utilize ATP or other nucleotides as substrates. Acyl-CoA oxidase (ACOX) enzymes are central players in peroxisomal fatty acid metabolism, as well as in secondary metabolism in nematodes and plants. These dimeric enzymes have been shown to bind to ATP at unusual sites that are buried at the dimer interface. Here, we show that ATP stimulates the activity of an ACOX enzyme by increasing the binding affinity of the enzyme for its FAD cofactor. The effect of ATP is highly specific as other nucleotides do not stimulate the activity of the enzyme and mutation of the ATP binding site blocks the effect. We use X-ray crystallography and molecular dynamics simulations of the apo dimeric enzyme to identify an unprecedented mechanism whereby ATP can reach its binding site through a shift in an α-helix bundle. An allosteric network connects the ATP and FAD binding sites, enabling ATP to enhance FAD binding affinity and thus enzymatic activity. In summary, the binding of ACOX enzymes to FAD is allosterically controlled by ATP, potentially providing a link between ATP levels and primary and secondary metabolism.
Similar content being viewed by others
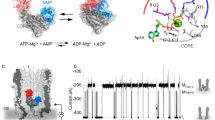

Introduction
Acyl-CoA oxidase (ACOX) enzymes play a central role in primary metabolism, catalyzing the first and rate-limiting step in the peroxisomal β-oxidation cycle that shortens long-chain fatty acids, branched-chain fatty acids, and bile acid intermediates. In humans, there are three ACOX enzymes (ACOX1, 2, and 3) with each specializing in different types of substrates1,2,3,4. The crystal structures of human ACOX1 and homologs from a variety of organisms reveal that these enzymes have a homodimeric structure with FAD cofactors noncovalently bound to each subunit at the dimeric interface. These enzymes use a two-step mechanism to install α,β-unsaturation in their acyl-CoA substrates. First, a catalytic glutamate abstracts the α-proton of the CoA-thioester, leading to hydride transfer from the β-position to FAD. Then, FADH2 is reoxidized through the reduction of molecular oxygen to form hydrogen peroxide5,6. In contrast, the mitochondrial counterpart to the peroxisomal ACOX enzymes, the acyl-CoA dehydrogenases, transfer the electrons to FAD and ultimately the electron transport chain7,8.
In addition to primary metabolism, ACOX enzymes participate in secondary metabolism in many organisms, including nematodes and plants9,10,11. In C. elegans, ACOX enzymes control the production of the ascaroside pheromones, a broad family of pheromones that mediate chemical communication in the worm, enabling it to coordinate its development and behavior12. There are seven C. elegans ACOX enzymes, with five of these directly implicated in ascaroside biosynthesis (ACOX-1.1, −1.2, −1.3, −1.4, and −3, referred to as ACOX-1, −2, −3, −4, and −6 in earlier literature)13,14,15,16,17. These enzymes act as gatekeepers in the β-oxidation cycle that shortens the side chains of the ascarosides. Different ACOX enzymes have different side-chain length preferences, leading to a diverse pool of ascarosides with different side-chain lengths.
Remarkably, structural analysis of the ACOX enzymes from C. elegans revealed that these enzymes have an ATP-binding site that is critical for enzymatic activity15. The crystal structures of ACOX-1.1 and ACOX-1.2 containing mutations in the catalytic glutamate (E434A and E432A, respectively) revealed that, in addition to FAD, these enzymes bind to molecules of ATP at the dimer interface at sites adjacent to the FAD-binding sites (Fig. 1a). This result is surprising, as ATP is not involved in the catalytic mechanism of ACOX enzymes and has also not been shown to regulate the activity of any ACOX enzyme or other type of oxidase6. The ATP-binding sites appear to be conserved in several ACOX enzymes in C. elegans (including ACOX-1.1, -1.2, -1.3, and -1.4) and in other nematode species15.

a The crystal structure of the ACOX-1.1(E434A) homodimer (PDB: 5K3I) shown as both a cartoon and surface representation with subunit A in orange and subunit B in cyan. The two ATP molecules are boxed and labeled, while the two FAD molecules are only labeled at their corresponding binding sites. Both of the FAD and ATP-binding sites are at the interface between the two subunits in the dimer. However, for clarity in this manuscript, we refer to one of the FAD/ATP as being predominantly associated with subunit A (given that more of the binding site residues are associated with subunit A) and one of the FAD/ATP as being predominantly associated with subunit B. b A cartoon representation of ACOX-1.1(E434A) (PDB: 5K3I), with bound FAD and ATP, overlaid with that of ACOX-1.1 ApoForm I (PDB: 5K3G) in white. The FAD/ATP-binding sites that are shown are predominantly associated with subunit A. Residues, which when mutated result in a lack of retention of both FAD and ATP to purified ACOX-1.1, are highlighted, and they include W189, Q340, K391, N437, H396, and R53615. The subunit that they are part of is indicated with a superscript. In the holo ACOX-1.1(E434A) structure, K391 hydrogen bonds to main-chain carbonyls in the catalytic loop (which forms a hydrophobic interaction surface for FAD) and to N437 (which in turn hydrogen bonds to ATP). In the apo ACOX-1.1 structure, the catalytic loop, K391, and N437 are disordered, and H396 is in a different position than it is in holo ACOX-1.1(E434A). The catalytic residue, E434, is not depicted in the catalytic loop in this figure because it is mutated to Ala in the holo ACOX-1.1(E434A) structure, and it is disordered in the apo ACOX-1.1 structure.
In the ACOX-1.1(E434A) structure, we identified a hydrogen bond network between the FAD and ATP-binding sites involving two critical residues, K391 and N437, that likely stabilize the catalytic loop that connects two α-helices (αK and αL) and contains the catalytic residue, E434 (Fig. 1b)15. On the other hand, in the wild-type ACOX-1.1 apo-structure, which is not bound to either FAD or ATP, there is no electron density for K391, N437, or the catalytic loop. Single residue mutations to K391, N437, or distal residues in either the ATP or FAD-binding sites resulted in a lack of retention of both ATP and FAD during purification, suggesting that ATP and FAD binding are correlated15. Furthermore, an ATP-binding mutant (H396G) of ACOX-1.1 exhibited impaired in vitro enzymatic activity, and a mutant worm strain expressing this mutant ACOX-1.1 displayed defects in ascaroside biosynthesis in vivo15,16. The ATP-binding site, however, is buried at the dimer interface. Thus, it was speculated that the ATP might play a structural role in stabilizing the dimer, rather than an allosteric role in regulating enzymatic activity.
Here, we show that binding of the enzyme to ATP dramatically enhances the binding affinity of the enzyme for FAD and thus stimulates enzymatic activity. This effect is specific to ATP as it is not seen with other nucleotides or with ACOX-1.1 ATP-binding site mutants. We propose a model whereby the enzyme binds to ATP, thereby shifting the EC50 of the enzyme for FAD from the mid-micromolar range to the mid-nanomolar range. ATP binding is thus essential for ACOX-1.1 to bind FAD at concentrations that are found in the cell. Although the ATP-binding sites are buried at the dimer interface, molecular dynamics (MD) analysis of the apo enzyme suggests a mechanism whereby ATP can reach its binding site through a shift in an α-helix bundle. An allosteric network connects the ATP and FAD-binding sites, demonstrating how ATP enhances FAD-binding affinity and thus enzymatic activity.
Results
A fraction of the enzyme is purified in an inactive form
Human ACOX1a reportedly is purified in an apo form (that is, lacking FAD), unless FAD is included in the purification buffers, in which case it can be purified in a holo form4. Other ACOX1 homologs are purified as a mixture of apo- and holo-enzyme, regardless of whether FAD is included in the purification buffers4,15,18,19,20. When we purified C. elegans ACOX-1.1 using our standard method with FAD in all purification buffers except those used in the final step of purification (size exclusion), approximately half of the enzyme contained bound FAD when averaged across a large number of purification trials (Supplementary Fig. 1a). Previously, we showed by HPLC that the amount of FAD and ATP bound to ACOX-1.1 is correlated15. Specifically, for wild-type enzyme, using a similar purification method as is described here, roughly 50% of the enzyme is bound to FAD and slightly less is bound to ATP, for the E434A catalytic mutant, roughly 100% of enzyme is bound to FAD and ATP, and for a variety of mutants in either the FAD or ATP-binding sites, none of the enzyme is bound to either FAD or ATP15. We attempted to enhance the amount of holo-enzyme by including 10 μM FAD plus 500 μM ATP in all purification buffers, including those used in the final size-exclusion step. However, the enzyme with added FAD and ATP had only slightly enhanced enzymatic activity in comparison to enzyme that was purified using our standard method (Supplementary Fig. 1b). We considered two possibilities: (1) a certain fraction of the enzyme is purified with cofactors and is active while a certain fraction is purified without cofactors and is either inactive or has a very low affinity for FAD, or (2) only one subunit in the dimer is bound to cofactors and is active while the other is not.
To monitor the enzymatic activity of ACOX-1.1, we utilized an enzyme-coupled kinetic assay in which production of hydrogen peroxide leads to generation of a UV-active product. For all assays we utilized a commercially available C12 fatty acyl-CoA substrate, lauroyl-CoA, which had previously been demonstrated to be a substrate for ACOX-1.116. Addition of 10 μM FAD and 350 μM ATP to the enzymatic reaction did not lead to an immediate enhancement of the activity of ACOX-1.1 (Supplementary Fig. 1b). This experiment further confirmed that a portion of ACOX-1.1 is purified in an inactive state and cannot be converted to the active state through the addition of 10 μM FAD and/or 350 μM ATP. Our subsequent work suggests that this portion of the enzyme does not respond to ATP and thus has a very low affinity for FAD (see data presented in later sections below).
Given that FAD and ATP bind at the dimer interface, we considered the possibility that they might affect enzymatic activity by enhancing dimer formation. Indeed, it has been shown for human ACOX1 that succinylation by SIRT5 or a gain-of-function mutation (N237S) can stimulate formation of the active dimer in vivo, although the molecular basis for this increased dimer formation is not well understood18,19. However, we were not able to detect a monomer form of the enzyme during purification (Supplementary Fig. 2). Furthermore, we could not promote monomer formation through dilution of the enzyme. The addition of FAD and ATP to the enzyme did not result in more dimer (Supplementary Fig. 3). Indeed, the crystal structures of ACOX1 homologs across organisms are dimeric, and the dimeric state is known to be the enzymatically active state4,15,18,19,20. Two ACOX-1.1 ATP-binding site mutants (H396G and R536E) purify without bound FAD or ATP, as previously shown15. However, the mutants are still dimers based on size-exclusion chromatography and display a similar overall fold to wild type, as judged by circular dichroism (Supplementary Fig. 4)15. Furthermore, we confirmed that the ACOX-1.1(H396G) mutant, like the holo wild-type enzyme, is a dimer using dynamic light scattering (DLS) (Supplementary Fig. 5). Thus, FAD and ATP are unlikely to affect the dimerization state of ACOX-1.1.
ATP modulates the affinity of ACOX-1.1 for FAD
Incubation of ACOX-1.1 in the absence of added FAD or ATP results in a time-dependent decrease in enzymatic activity (Fig. 2a, b). Interestingly, this decrease can be prevented completely by including FAD and ATP in the incubation buffer, but can be prevented only partially by FAD alone (Fig. 2a). Importantly, the effects of FAD and ATP on enzyme activity are immediate. That is, if the enzyme is incubated for a period of time without FAD and ATP, such that its activity declines, and then FAD and ATP are added and enzyme activity immediately assayed, the FAD and ATP can immediately rescue the time-dependent decrease in activity that is otherwise seen (Fig. 2b). Thus, the time-dependent loss of activity is not due to degradation or unfolding of the enzyme. We confirmed the stability of the enzyme during the incubation by analyzing it by SDS-PAGE and circular dichroism before and after incubation (Supplementary Fig. 6).

a, b The value of kcat as a function of incubation time. a ACOX-1.1 was incubated either alone or with FAD or with FAD/ATP, and then the reaction was initiated after a certain amount of time. Due to constraints of the method, the enzyme at the initial time point had been treated for about 30 s with either nothing, FAD, or FAD/ATP, resulting in some difference in the kcat value at this time point between samples. b ACOX-1.1 was incubated alone for a certain amount of time until just before assay, at which point either no FAD/ATP, FAD, or FAD/ATP was added, and then the reaction was initiated. The colored dashed lines are for visualization purposes only. c, d Determination of kcat after 1 h incubation as the chosen time point in the presence of either no FAD/ATP, FAD, FAD plus ATP, or FAD plus other nucleotides. e The rate normalized to that of ACOX-1.1 alone as a function of increasing FAD only (green bars) or holding FAD constant at 10 μM and increasing ATP (red bars). f The fit of the data for increasing ATP concentration in (e) using nonlinear regression. g Measurement of the fluorescence anisotropy of FAD for wild-type ACOX-1.1 or ACOX-1.1(E434A) over time to determine the FAD koff. a–d The concentration of FAD and ATP were 10 μM and 350 μM, respectively. d Other nucleotides were tested at >350 μM (except AMP was tested at 310 μM). a–g The measured amount of FAD bound per dimer was 50% (a) 36% (b) 56% (c, d) 47% (e, f—replicate 1), 50% (e, f—replicate 2), 46% (g—wild type), and 66% (g—E434A). a, b Data represent the mean of n = 3 technical replicates, and error bars correspond to the 95% confidence interval for the kcat parameter. c, d Data represent the mean of n = 3 technical replicates ± standard error. e, f Data represent the mean of n = 2 biological replicates. Source data are provided as a Source Data file.
The kcat almost doubles utilizing enzyme that was incubated with FAD and ATP relative to enzyme that was not incubated with FAD and ATP (Fig. 2c). The Hill coefficient averaged across conditions is equal to 1.75 ± 0.12, suggesting that the two subunits of the enzyme cooperatively bind substrate (Fig. 2c). To test the specificity of the effect of ATP on the rate of the enzyme, the incubation was repeated with FAD plus other nucleotides, including deoxy-ATP, ADP, AMP, and GTP. None of these nucleotides are able to rescue the time-dependent decrease in enzymatic activity as well as ATP (Fig. 2d). When utilizing 10 μM FAD, the concentration of ATP needed for half-maximal rescue is 248.7 μM (95% CI: 151.6–484.4 μM), and it plateaus at mid to high-μM ATP (Fig. 2e, f and Supplementary Fig. 7). To obtain a similar amount of rescue with FAD alone (that is, with no ATP), at least 100 μM FAD is needed, suggesting that ATP increases the affinity of the enzyme for FAD (Fig. 2e). Although the Khalf is not a direct measurement of substrate affinity, its minimal change in the presence of FAD and ATP suggests that FAD and ATP help to maintain the kcat during incubation but do not significantly affect the affinity of the enzyme for its substrate (Supplementary Fig. 8).
The enzyme-coupled kinetic assay is an indirect measure of enzymatic activity. To confirm that the effect of ATP and FAD on rate is correlated to substrate turnover, a more direct assay for product formation based on HPLC was developed. These data confirmed that indeed ATP increases the rate of product formation (Supplementary Fig. 9a). It was also confirmed by HPLC that ATP is not hydrolyzed by ACOX-1.1, since the rate of ATP hydrolysis when ACOX-1.1 was incubated with ATP did not surpass that when ATP was incubated with FAD in the absence of enzyme (Supplementary Fig. 9b). Thus, ATP can increase the rate of substrate turnover in the presence of FAD cofactor without the hydrolysis of ATP.
To verify that the loss of enzyme activity during incubation was due to loss of FAD, we developed an assay to measure the koff based on the fluorescence anisotropy of FAD. Incubation of wild-type holo ACOX-1.1 led to a gradual decrease in anisotropy with a koff of 1.93 h−1, indicating that roughly half of the bound FAD is lost in 20 minutes (Fig. 2g). This rate is consistent with the rate of the loss of enzyme activity upon incubation (Fig. 2a, b). Meanwhile, the catalytic mutant ACOX-1.1(E434A) loses FAD much more slowly with a koff of 0.72 h−1, indicating that about half of the bound FAD is lost in an hour (Fig. 2g). Consistent with this result, wild-type ACOX-1.1 is purified with roughly 50% FAD and ATP bound, while the catalytic mutant is isolated with 100% FAD and ATP bound (that is, with one FAD and ATP bound per subunit). FAD may be more tightly bound by the catalytic mutant because it is less flexible or possibly because this mutant cannot process substrate.
FAD and ATP synergistically increase the activity of the apo enzyme
To directly characterize the enzyme without any bound FAD or ATP, we generated the apo enzyme by purifying the enzyme using our standard method but without FPLC purification, dialyzing the enzyme overnight, and then purifying it through FPLC. We confirmed that we had obtained apo enzyme by measuring the amount of bound FAD and ATP using UV spectroscopy and HPLC (Supplementary Fig. 10). Analysis of the apo enzyme by SDS-PAGE confirmed that the enzyme did not degrade during dialysis (Supplementary Fig. 11a). Furthermore, CD spectroscopy confirmed that the apo enzyme remained well-folded (Supplementary Fig. 11b). In addition, we also used DLS to confirm that the apo enzyme, like holo-enzyme, was a dimer (Supplementary Fig. 11c).
In the absence of added FAD, the apo enzyme had virtually no activity. A dramatic enhancement in kcat was seen only by adding both FAD and ATP, not either alone (Fig. 3a and Supplementary Fig. 12). The effect of FAD and ATP on rate was immediate; that is, it did not require any pre-incubation with FAD and ATP before initiating the reaction with substrate. The effect of ATP was specific, as no other nucleotide could enhance the binding of the enzyme for FAD as much as ATP (Fig. 3a). We also confirmed by DLS and size-exclusion chromatography-multiangle light scattering (SEC-MALS) that the enzyme remained a dimer even when diluted to concentrations used in the enzymatic assay (Supplementary Figs. 13 and 14). Thus, FAD and ATP synergistically enhance the activity of the dimeric enzyme.

a Determination of kcat of wild-type apo ACOX-1.1 immediately after adding FAD, ATP, or FAD plus ATP (or other nucleotides). The concentrations of FAD and ATP (or other nucleotides) were 10 μM and 350 μM, respectively. b Rate of wild-type apo ACOX-1.1 immediately after adding 350 μM ATP and various concentrations of FAD. c Rate of wild-type apo ACOX-1.1 immediately after adding various concentrations of FAD (but no ATP). d, e Rate of wild-type apo ACOX-1.1 immediately after adding 10 μM FAD (d) or 200 μM FAD (e) and various concentrations of ATP. f Measurement of FAD anisotropy for 100 nM FAD in the presence of different apo ACOX-1.1 concentrations (0–10 μM) with either no ATP or 350 μM ATP. Specifically, each individual curve represents the FAD anisotropy across different enzyme concentrations in the presence of a constant ATP concentration. The enzyme concentration in the figure has been normalized based on the activity of the apo ACOX-1.1, relative to holo ACOX-1.1. g Measurement of FAD anisotropy for 4 μM apo ACOX-1.1 and 100 nM FAD in the presence of varying concentrations of ATP. h, i Rate of ACOX-1.1(H396G) (h) or ACOX-1.1(R536E) (i) immediately after adding 350 μM ATP with various concentrations of FAD or after adding 10 μM FAD with various concentrations of ATP. a, d Data represent the mean of n = 3 biological replicates ± standard deviation. b, c, e, h, i Data represent the mean of n = 3 technical replicates ± standard deviation. a Statistical analysis was done using one-way ANOVA with Dunnett’s multiple comparison test. Source data are provided as a Source Data file.
When including 350 μM ATP in the enzymatic reaction, a careful titration of FAD revealed that there were two plateaus to the curve, with one EC50 calculated at 385 nM FAD (95% CI: 358–476 nM) and one EC50 estimated at 177 μM FAD (95% CI: 74–203 μM) (Fig. 3b). Thus, the enzyme appears to have a high-affinity site for FAD and a low-affinity site for FAD. When the experiment was repeated in the absence of ATP, addition of FAD still gave two plateaus, but the lower EC50 was increased from 385 nM FAD to 13.0 μM (95% CI: 6.1–27.5 μM) (Fig. 3c). Thus, the effect of ATP appears to be to increase the affinity of the high-affinity FAD-binding site, as well as increase the overall activity level of the enzyme, while the low-affinity FAD-binding site does not appear to be significantly affected by ATP. The Hill coefficient of the high-affinity FAD EC50 is at least 2, suggesting that the ATP-bound enzyme likely binds to two molecules of FAD in a cooperative manner. One possible interpretation of our data is that there are two populations of the enzyme: one population, which can bind to ATP and thus binds to FAD with high affinity, and another population, which cannot bind to ATP and thus only binds to FAD with low affinity.
To characterize the enzyme’s binding of ATP, we titrated the concentration of ATP in the assay while including an FAD concentration (10 μM) that was greater than the high-affinity FAD EC50 (Fig. 3d). The EC50 for ATP was 69.4 μM (95% CI: 54.8–87.1 μM) with 10 μM FAD. The Hill coefficient for the ATP EC50 was 1.65, indicating that the enzyme may bind ATP cooperatively. We also titrated the concentration of ATP in the assay while including an FAD concentration (200 μM) that was greater than the low-affinity FAD EC50. The EC50 for ATP with 200 μM FAD was 110.9 μM (95% CI: 40.5–328.8 μM), which is not significantly different from the EC50 for ATP with 10 μM FAD (Fig. 3e). However, it was difficult to measure activity at 200 μM FAD due to interference of the UV signal for FAD with the signal of the enzyme-coupled kinetic assay, and thus there was a greater amount of error in this assay. Our data would suggest that there is a population of enzyme that binds to ATP and has a high-affinity EC50 for FAD, and there is a population of enzyme that does not bind to ATP and has a low-affinity EC50 for FAD. However, it is also possible that this second population of enzyme also binds to ATP but with a lower affinity that is difficult to measure.
To further probe the nature of the FAD and ATP-binding sites, the KD of ACOX-1.1 for FAD and for ATP were determined using the fluorescence anisotropy of FAD (Fig. 3f, g). In the presence of ATP, the KD of ACOX-1.1 for FAD is about 450 ± 60 nM, which is consistent with the high-affinity FAD EC50, while in the absence of ATP, the KD of ACOX-1.1 for FAD could not be measured because it is too high (Fig. 3f). Thus, ATP increases the binding affinity of the enzyme for FAD. Meanwhile, the KD of ACOX-1.1 for ATP is 20.7 μM (95% CI: 6.1–27.5 μM), which is roughly consistent with the ATP EC50 (Fig. 3g).
ATP-binding site mutations block the effect of ATP on enzyme activity
To confirm whether the effects of ATP on ACOX-1.1 are mediated through the enzyme’s ATP-binding sites, two ATP-binding site mutants were used, ACOX-1.1(H396G) and ACOX-1.1(R536E). The ACOX-1.1(H396G) mutant lacks the histidine that π-stacks with the adenine of ATP, while the ACOX-1.1(R536E) mutant lacks an arginine, which hydrogen bonds to the α and β phosphates of ATP in the crystal structure (Fig. 1b). The purification of the ACOX-1.1(H396G) mutant or the ACOX-1.1(R536E) mutant results in an enzyme preparation that is completely lacking in bound FAD or ATP15. It was also previously shown that the ACOX-1.1(H396G) mutant is completely inactive unless FAD is included in the enzymatic reaction buffer, in which case it has only a low level of activity15. Not surprisingly, both the ACOX-1.1(H396G) mutant and the ACOX-1.1(R536E) mutant do not respond to ATP (Fig. 3h, i). Thus, the effects of ATP on ACOX-1.1’s affinity for FAD are likely due to it binding to the ATP-binding sites on ACOX-1.1.
Identification of an entry/exit mechanism for FAD and ATP
In the ACOX-1.1(E434A) holo-crystal structure, the FAD-binding sites are located at the dimer interface near the surface of the enzyme, while the ATP-binding sites are located at the dimer interface but are buried from the outer surface of the enzyme (Fig. 1a). Thus, it is difficult to envision how ATP could enter or exit these binding sites from the static structure. However, our data have demonstrated that exogenously provided ATP can immediately enhance the activity of the dimeric enzyme in the presence of FAD and that this effect requires ATP binding specifically to the ATP-binding sites. To investigate how FAD and ATP might be exchanged from the enzyme, we performed MD simulations of apo ACOX-1.1. Our starting structure was the wild-type apo ACOX-1.1 crystal structure, in which we modeled disordered residues using AlphaFold21. The MD simulations revealed a potential entry/exit mechanism for both FAD and ATP (Fig. 4 and Supplementary Table 1).

a Representative structures from the MD simulations of the apo enzyme with an open versus a closed FAD-binding site. For the apo enzyme with the FAD site in the open state, subunit A is in orange and subunit B is in cyan. For the apo enzyme with the FAD site in the closed state, both subunits are in white. b Comparison of a surface view of the representative structures of the apo enzyme from (a) showing an open and closed FAD-binding site. c Representative structures from the MD simulations of the apo enzyme with an open versus a closed ATP-binding site. For the apo enzyme with the ATP site in the open state, subunit A is in orange and subunit B is in cyan. For the apo enzyme with the ATP site in the closed state, both subunits are in white. d Comparison of a surface view of the representative structures of the apo enzyme from (c) showing an open and closed ATP-binding site. a–d The representative structures shown were obtained from the most populous clusters from clustering analysis of the MD simulations performed as described in “Methods”. a–d ATP and FAD have been modeled into the structures based on their positions in the holo ACOX-1.1(E434A) crystal structure.
To visualize the conformational changes that enable FAD and ATP exchange, we overlaid representative structures from the MD simulations of apo ACOX-1.1 in which either the FAD-binding site (Fig. 4a, b) or ATP-binding site (Fig. 4c, d) was either closed or open. Comparing the closed and open states of the FAD binding site, the loop region between β-strands β1 and β2, as well as the entire β-sheet in which β1 and β2 reside, shifts away from FAD (Fig. 4a). In particular, the loop between β-strands β1 and β2 closes off the FAD-binding site in the closed state but shifts away from FAD in the open state. The loop region between β1 and β2 and the loop between α-helices αG and αH contain many residues strongly correlated in the allosteric network linking FAD and ATP (see next section). The binding site for ATP converts between its closed and open states through the outward movement of an α-helix bundle: αM and αN, which lie anti-parallel to each other, and αL, which lies perpendicular to αM and αN and connects to the catalytic loop that contains Glu434 (Fig. 4c). The openings for FAD and ATP are more apparent in the surface representations (Fig. 4b, d).
To quantify the opening of the FAD and ATP-binding sites in wild-type apo ACOX-1.1, we ran ten independent simulations of 2 μs each. For these simulations, we then calculated the shortest distance between any atom in the residues surrounding the ligand-binding sites. Specifically, for the FAD-binding site, we used residues in the loop between β1 and β2 and in the catalytic loop (see Fig. 4a and Supplementary Table 2), and for the ATP-binding site, we used specific residues in the α-helices that bracket the opening to the ATP-binding site (αM, αN, αH, αO, and αQ; see Fig. 4c and Supplementary Table 2). To illustrate the overall motion of the apo enzyme, we plotted the size of the ATP opening on subunit A versus the ATP opening on subunit B, and the size of the FAD opening on subunit A versus the FAD opening on subunit B (10 simulations in Fig. 5a(i), b(i) and Supplementary Figs. 15–17). These data quantitatively show that the FAD-binding sites open to some degree in most simulations, while the ATP-binding sites open in a much more stochastic manner (Supplementary Figs. 15 and 16).

a, b The size of the FAD and ATP-binding site openings for each system, plotting (a) the size of the ATP opening on subunit A versus the size of the ATP opening on subunit B and (b) the size of the FAD opening on subunit A versus the size of the FAD opening on subunit B. The systems included apo ACOX-1.1, holo ACOX-1.1, apo ACOX-1.1 replicating the waters from subunit A for both subunits, and apo ACOX-1.1 replicating the waters from subunit B for both subunits. A plot was done for each system for 10 MD simulations—2 μs each (first row), 20 MD simulations—2 μs each (second row) or 20 MD simulations—4 μs each (third row).
We next performed MD simulations of holo ACOX-1.1 bound to FAD and ATP. Our starting structure was the holo ACOX-1.1(E434A) crystal structure, in which A434 had been changed in silico to the catalytic glutamate residue found in the wild-type enzyme (Supplementary Table 1)21. These data show that the holo-enzyme is less flexible and more closed for the ATP sites than the apo enzyme (10 simulations in Fig. 5a(ii), b(ii) and Supplementary Figs. 15 and 16). This result is consistent with our experimental result that the holo-enzyme has a slow off-rate for ATP and FAD, while the apo enzyme responds immediately to the addition of ATP and FAD.
While the starting apo-structure was largely symmetrical, some asymmetry is seen in the MD simulations. In particular, the ATP-binding site was shown to primarily open on subunit A (Fig. 5a(i), b(i)). We anticipated that if sufficient simulations were run with sufficient sampling time, ATP-binding sites on subunits A and B of the apo enzyme would show a similar degree of opening. Thus, we increased the number of simulations of the apo and holo structures to 20, and then extended these simulations from 2 μs to 4 μs (20 simulations—2 μs each, 20 simulations—4 μs each in Fig. 5a(i–ii), b(i–ii)). Increasing and extending the simulations increased the symmetry of the simulations. To eliminate the asymmetry of the apo simulations, we increased the symmetry of the starting structure, by using the same position of the waters in both subunits (that is, replicating the waters from subunit A to subunit B and vice versa) (Fig. 5a(iii–iv), b(iii–iv), Supplementary Figs. 15–17, and Supplementary Table 1). By starting with more symmetrical starting structures, we were able to achieve symmetrical opening of the ATP sites on both subunits, confirming the mechanism of ATP binding to the apo enzyme.
To characterize the nature of the movement between the closed and open conformations of the apo and holo enzymes, clustering was performed on the MD simulations to select representative structures corresponding to the closed and open states. In this manner, we obtained eight subsets in total for the apo and holo systems in either the closed or open states of the ATP and FAD sites. Each subset was clustered using the pairwise RMSD of the relevant ligand-binding sites. In each subset of frames, the centroid of the cluster with the highest population was selected as the representative structure. The representative structures for closed and open apo states of the FAD and ATP sites are the ones shown in Fig. 4a–d. For a comparative analysis of the ligand-binding site flexibility between apo and holo systems, we calculated the ligand-binding site opening distances of the centroids for the top ten clusters (Supplementary Fig. 18). Overall, these data confirm that the FAD and ATP sites are more closed in the holo-enzyme than in the apo enzyme (Supplementary Fig. 19).
Identification of an allosteric network that couples the FAD and ATP-binding sites
To analyze the allosteric network mediating interactions between FAD and ATP in holo ACOX-1.1, we used SenseNet, a framework for extracting allosteric networks by analyzing interactions that occur during MD simulations22. A time series of atomic interactions in apo and holo ACOX-1.1 was built with a 3.5 Å distance cutoff for hydrogen bond interactions and a 5 Å distance cutoff for the heavy atom interaction network. We performed Difference Node Correlation Factor (DNCF) analysis to identify the allosteric network and allosteric correlation values in the holo system using the interaction network of the apo system as the reference. The DNCF analysis estimates the impact of an interacting residue pair on the immediate neighboring interactions within a system, using another system as a reference. In our study, this analysis reveals the effect of ATP and FAD binding on the ACOX-1.1 enzyme. Each residue in the enzyme was represented as a node, and the interactions between residues were represented as edges. For clarity, we show only the residues that belong to the top 10% of the allosteric values, and node color is normalized from white-to-red based on the allosteric correlation value (Fig. 6a, b and Supplementary Fig. 20). Overall, the allosteric network is strongest between residues surrounding the FAD and ATP associated with each subunit, but there are also interactions across subunits (Fig. 6a).

a Interaction network for residues with the top 10% of DNCF scores for the holo ACOX-1.1 network with apo ACOX-1.1 as a reference. Individual nodes represent FAD, ATP, or the top 10% of coupled residues, with the color of each node corresponding to the strength of the allosteric connection to other nodes and visualized with a gradient of white-to-red. FAD and ATP had very high correlation values and were not included in the normalization of the entire network. Solid edges indicate heavy atom contacts, and dashed lines indicate hydrogen bond interactions. The edge width signifies the frequency of a particular interaction (or contacts) in the interaction timeline built from the MD trajectories. b The ACOX-1.1 starting structure with residues colored according to the z-score of their DNCF value for the holo ACOX-1.1 network with apo ACOX-1.1 as a reference. Residues with negative z-scores are colored white, and those with positive z-scores are colored white-to-red. Residues with z-scores >2 have been explicitly shown. c, d The allosteric network between the FAD and ATP that are more closely associated with subunit A. The subunit B network is similar. Residues with z-scores >2 have been explicitly shown. Color scheme is the same as in (b). Residues that are part of subunit A are indicated with an A superscript, and residues that are part of subunit B are indicated with a B superscript. The catalytic loop containing E434 is also indicated. d Closeup of the allosteric network shown in (c) with hydrogen bonds indicated. In addition, K391, which mediates interactions between N437 and the catalytic loop, and the catalytic loop containing E434 have been shown. e–h Rates of apo ACOX-1.1(R343Q) and ACOX-1.1(R343A) (e), ACOX-1.1(N437A) (f), ACOX-1.1(K391N) (g), and ACOX-1.1(R295Q) and ACOX-1.1(R295A) (h) after adding 350 μM ATP and various concentrations of FAD. As the rates were extremely low, they were monitored over the course of several hours. e–h Data represent the mean of n = 3 technical replicates ± standard deviation. Source data are provided as a Source Data file.
The most correlated residues in the allosteric network are clustered around the ATP and FAD-binding sites, as well as the catalytic loop (Fig. 6c). The strongest single residue path between FAD and ATP is through R343, which forms hydrogen bonds with the 2′-hydroxyl of the ATP ribose ring, the N3 of the ATP adenine base, and the 2′-hydroxyl of the FAD ribose ring (Fig. 6d). To test the importance of this residue, we generated two mutants, ACOX-1.1(R343Q), and ACOX-1.1(R343A) (Supplementary Fig. 21). Although both of these mutant enzymes appeared well-folded, they were purified in the absence of any bound FAD and had extremely low activities, regardless of the amount of FAD or ATP added (Supplementary Fig. 22). Testing the activity of the enzyme in the presence of 350 μM and varying amounts of FAD showed that the EC50 for FAD is extremely high ( > 1 mM) (Fig. 6e). Varying the amount of ATP indicated that ATP has no influence on the activity of the enzyme (Supplementary Fig. 23). Thus, the allosteric network had been disrupted.
In addition to R343, additional residues appeared important in the allosteric network. The strongest two-residue paths include N437, which hydrogen bonds to N1 of ATP, and E436, a neighboring residue which forms a hydrophobic surface for binding to FAD, as well as Q404 and R343, which form a hydrogen bond network between ATP and FAD. N437 also forms a hydrogen bond bridge between the N1 of the ATP adenine base and K391, which forms hydrogen bonds with two main-chain carbonyls in the catalytic loop containing E434. This catalytic loop also forms a hydrophobic surface that stacks favorably with the isoalloxazine ring of FAD. To test the importance of N437 and K391, we generated the N437A and K391N mutants (Supplementary Fig. 21). Despite being well-folded, both of these mutant enzymes were purified in the absence of any bound FAD, had extremely low activities, had a high EC50 for FAD, and did not respond to ATP (Fig. 6f, g and Supplementary Figs. 22 and 23).
Interestingly, the allosteric network also implicated R295, which is adjacent to the catalytic loop and hydrogen bonds to the main-chain carbonyl of the catalytic residue E434 and likely also interacts with substrate. The two mutants that were generated in this residue, R295Q and R295A, have extremely low activities, but have EC50’s for FAD that are similar to that of the wild-type enzyme in the absence of ATP and these mutants also do not respond to ATP (Fig. 6h and Supplementary Figs. 21–23). On the FAD side, R320 and Q322, which are in the loop between α-helices αG and αH, hydrogen bond to the phosphates in FAD (Fig. 6c). Y337 π-stacks with the adenine ring of FAD. H156 is in the loop between β1 and β2 that forms a lid across the FAD-binding site. Meanwhile, on the ATP side, multiple residues (e.g., Y581) in αΜ and αΝ on either side of the ATP-binding site, are potentially allosterically regulated (Fig. 6a, b).
Discussion
Through X-ray crystallography, enzymology, and MD simulations, we have uncovered a unique role for ATP in the allosteric regulation of an oxidase enzyme. Previously, the structure of the ACOX-1.1(E434A) catalytic mutant homodimer suggested that the enzyme is bound to both its cofactor FAD and ATP at two symmetric sites at the interface between the two subunits, with one FAD/ATP primarily associated with each subunit15. The ATP-binding sites in ACOX-1.1 do not resemble any previously identified conserved ATP-binding domain based on a search of two different databases, one of nucleotide binding domains and another of allosteric modulators23,24. Typically, ATP is bound by loop motifs that recognize the phosphates and/or adenine ring of ATP, but the ATP-binding domain in ACOX-1.1 has a purely helical fold made up of α-helices contributed by both subunits of the dimer25,26,27. Because these sites are buried at the dimer interface, our initial analysis of the crystal structure did not reveal how ATP might enter/exit its binding site, enabling it to allosterically regulate the enzyme. However, our MD simulations of wild-type apo ACOX-1.1 have revealed a potential mechanism whereby ATP could enter/exit the enzyme through the movement of an α-helix bundle. Furthermore, they have revealed a potential mechanism whereby FAD could enter/exit the enzyme through the movement of a β-sheet and associated loops, as well as an allosteric network through which ATP could allosterically regulate the binding of FAD.
To characterize the role of ATP and FAD in the enzymatic activity of the enzyme, we took two complementary approaches. In the first approach, we showed that incubation of the enzyme at room temperature for an extended period resulted in the gradual loss of activity of the enzyme due to the gradual loss of bound FAD and ATP. This loss could be rescued fully at any point in the incubation by adding both FAD and ATP (but not either alone) to the incubation buffer and immediately assaying the activity of the enzyme. These results might suggest a slow off-rate. In the second approach, we generated an apo enzyme through overnight dialysis, followed by FPLC purification. For the apo enzyme, which we verified is dimeric, we could show an immediate, synergistic increase in activity of the enzyme upon addition of both FAD and ATP. This effect was dependent on the ATP-binding site, since ATP-binding mutants did not respond to ATP. When assaying the enzyme’s activity in the absence of ATP with varying concentrations of FAD, we detected an EC50 in the low-micromolar range and an EC50 in the mid-micromolar range for FAD. However, when assaying the enzyme’s activity in the presence of ATP, the low-micromolar EC50 shifted to the low-nanomolar range, indicating the formation of a high-affinity FAD-binding site in the presence of ATP. Furthermore, the KD of the enzyme for FAD is much lower in the presence of ATP. The overall level of the activity of the enzyme in the presence of ATP is also higher. Because of the biphasic response of ACOX-1.1 for FAD, we suggest a model in which there are two populations of the ACOX-1.1 enzyme, one which does not bind ATP and has a low affinity for FAD and one which binds ATP and cooperatively binds FAD with high affinity. We only detected one EC50 for ATP, possibly indicating that ATP only affects the activity of one of the two populations of the enzyme. These two populations of enzyme may explain why we typically isolate wild-type “holo” ACOX-1.1 with roughly 50% of the protein bound to FAD, and why we cannot force the other 50% of the enzyme to bind to significantly more FAD or enhance its activity using low-micromolar concentrations of FAD.
The fact that ATP can enhance the affinity of the enzyme for FAD is consistent with our MD simulations of the enzyme. Based on these simulations, the apo enzyme has sufficient flexibility to open the ATP-binding site through the movement of an α-helix bundle including αM, αL, and αN and to open the FAD-binding site through the movement of the β-sheet in which β1 and β2 reside. Given that an allosteric network links ATP and FAD, ATP binding to the enzyme likely enhances the binding affinity of the enzyme for FAD, specifically through interactions between ATP and FAD mediated by R343 and other residues. In contrast to the apo enzyme, which is potentially flexible enough to allow ATP binding, the holo-enzyme is almost completely closed at the ATP-binding sites. This result agrees with the slow off-rate of the holo enzyme for ATP and FAD.
The EC50’s of ACOX-1.1 for FAD and ATP are potentially consistent with a model in which cellular concentrations of FAD and ATP regulate the activity of the enzyme in vivo. In yeast and mammalian cells, ATP concentration is reported to be relatively stable and to be in the low-millimolar range28,29. In contrast, in bacteria, ATP concentration has been found to significantly fluctuate and to be less than 500 μM in about 10% of cells30. Furthermore, in certain organelles and tissues of plants, ATP concentration is reported in the low-to-mid-micromolar range31,32. In C. elegans, to our knowledge, ATP concentration has only been reported as a relative number; for example, Hibshman et al. showed that ATP levels drop about 75% during starvation for several days33. Thus, it is unknown what the concentration of ATP is in different organelles and tissues in C. elegans. The KD of ATP-regulated allosteric sites in enzymes ranges from the low-micromolar to low-millimolar range34,35,36. For example, the KD of the high-affinity ATP site of the ATP-activated aspartate transcarbamoylase is in the 100–200 μM range37,38. The concentration of free (non-protein-bound) FAD has also been demonstrated to fluctuate, but is typically reported to be in the low-micromolar range39,40,41. Our data would suggest that if the ATP concentration is below the EC50 that we detected for ACOX-1.1 (about 69.4 μM), FAD concentrations would have to be in the hundred micromolar range in order to activate the enzyme, which is a much higher concentration than is typically reported for the cell. Thus, sufficient ATP in C. elegans would potentially enable ACOX-1.1 to utilize its high-affinity, nanomolar FAD-binding site. However, it is not known whether ATP concentrations dip low enough in C. elegans for ATP to enable this type of regulatory mechanism. Furthermore, although we have shown that ATP can rapidly bind to and regulate the activity of apo ACOX-1.1, the slow off-rate of holo ACOX-1.1 for ATP suggests that additional factors would be necessary in vivo for the enzyme to rapidly lose ATP at low ATP concentrations.
In summary, we uncovered the molecular mechanism whereby ATP binds to an oxidase enzyme, ACOX-1.1, and increases its affinity for its FAD cofactor. This mechanism likely regulates the activity of other ACOX enzymes with an ATP-binding site, including those in C. elegans and in other nematodes. Furthermore, we identified a potential entry/exit mechanism for ATP involving movement of helices that is quite different from mechanisms in other ATP-regulated enzymes involving movement of flexible loops. Overall, our work potentially reveals how ATP regulates ACOX enzymes, thereby linking ATP levels in the cell to fatty acid β-oxidation and pheromone biosynthesis. The central role of ACOX enzymes in primary and secondary metabolism requires that their function is tightly regulated, and here, we have potentially uncovered a regulatory mechanism that can be targeted to modulate their activity.
Methods
Expression and purification of ACOX-1.1
ACOX-1.1a (the longest splice variant of ACOX-1.1 in C. elegans) and mutants were recombinantly expressed in BL21(DE3) cells. The construction of the H396G and R536E mutants was previously published, and the R343Q, R343A, N437A, K391N, R295Q, and R295A mutants were generated using the Q5 kit (New England Biolabs) using the primers in Supplementary Table 315. Protein expression was induced with 0.6 mM isopropyl β-D-1-thiogalactopyranoside at 25 °C for 20 h15. Cells were lysed in lysis buffer (100 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM MgCl2, 10 μM FAD) with a microfluidizer and centrifuged. The proteins were purified using Ni-NTA resin (Thermo Fisher), washing the resin with lysis buffer and binding buffer (100 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM MgCl2, 50 mM imidazole, 10 μM FAD) and then eluting with elution buffer (100 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM MgCl2, 250 mM imidazole, 10 μM FAD). The elution fraction from the Ni-NTA resin was concentrated with either a 10 kDa- or 30 kDa-cutoff Amicon centrifugal filter (SigmaMillipore), as indicated, and then loaded onto a HiLoad 16/600 Superdex 200 column (GE Healthcare) on an FPLC using running buffer (50 mM Tris-HCl, pH 7.5, and 150 mM NaCl, 10 mM MgCl2) and a flow rate of 0.5 mL/min. The protein was concentrated with either a 10 kDa- or 30 kDa-cutoff Amicon centrifugal filter, as indicated, to 3–4 mg/mL.
Quantifying FAD bound to ACOX-1.1 by UV spectroscopy
The concentration of the enzyme was obtained using the molar absorption coefficient calculated by the ExPASy server using the amino acid sequence of the full-length protein42. Next, the molar absorption coefficient of FAD in size-exclusion running buffer was calculated using a calibration curve generated by serial dilution of a known concentration of an FAD standard. The coefficient for ACOX-1.1 at 280 nm is 79,190 M−1 cm−1 and for FAD it is 20,230 M−1 cm−1, and the coefficient for FAD at 450 nm is 11,420 M−1 cm−1. A previously published method was used to quantify the concentration of FAD bound to ACOX-1.143. In brief, the concentration of holo-enzyme is expressed as the total UV absorption at 280 nm from which the contribution by FAD is subtracted to yield the concentration of apo enzyme. Following size-exclusion separation of each sample and collection of the dimeric peak, the concentration of apo enzyme and FAD of each sample was used to calculate the % of FAD bound to ACOX-1.1. For the purifications that included FAD and ATP in the size-exclusion buffer, the UV absorption of the buffer without ACOX-1.1 was subtracted from the UV absorption of the sample containing ACOX-1.1.
Enzyme-coupled kinetic assay
The assay was adapted from a previously published protocol with modifications made to accommodate a 96-well plate setup2. In brief, the assay couples substrate-dependent hydrogen peroxide production by the enzyme to the formation of a UV-active product. A standard curve of the concentration of hydrogen peroxide versus the measured signal at 500 nm yielded a molar absorption coefficient of 6900 M−1 cm−1 that is consistent with previous literature1. The assay buffer was the same as the size-exclusion running buffer, and the substrate used was lauroyl-CoA (C12-CoA). The substrate concentration was either held constant at 25 µM for initial rate measurements or varied from 3 or 5 µM to 50 µM for the determination of kinetic parameters. A minimum of seven substrate concentrations were used for experiments with a 1-h incubation, and a minimum of four substrate concentrations were used for other experiments. ACOX-1.1 was at a concentration of 335 nM during incubation in a total volume of 112 µL, and the final concentration was 250 nM at the final assay volume of 150 µL. ACOX-1.1 was mixed with all components except substrate in a tube and then aliquoted to each well of a 96-well plate, and aliquoting after mixing the enzyme with the components was staggered in such a way that every replicate for each condition tested was incubated for an equivalent time. A CLARIOstar microplate reader (BMG Labtech) operating the MARS data analysis software (4.20) was used with the double injection system, where one syringe delivered substrate and the second delivered high-purity water. Two syringes always delivered the same total volume of 38 µL (one syringe contained substrate dissolved in ultrapure water and the other ultrapure water only) to the remaining reaction components in 112 µL with 1.10 mM 4-aminoantipyrine (0.82 mM final concentration), 14.2 mM phenol (10.6 mM final concentration), 5 IU horseradish peroxidase, 0.335 µM ACOX (0.250 µM final concentration), and +/− ATP/FAD or buffer (ACOX only control) in each well to give a total volume of 150 µL14. The blank wells used to subtract any background signal contained all components except for substrate, and each condition tested contained its own set of blank measurements. The start time for measurement was at the end of injection from both syringes, which takes less than 5 s, and a 30 s measurement window was used since it allowed the inclusion of more samples given the incubation period and was sufficient to obtain the initial rate. Other nucleotides tested were at the following concentrations: 310 μM for AMP, 470 μM for ADP, 420 μM for deoxy-ATP, and 470 μM for GTP. The incubation was done at 21–23 °C, and the assay was done at 25–26 °C.
Determining kinetic parameters
All analysis was done using models contained within the GraphPad Prism 9.4.1. For Fig. 2c, where seven different substrate concentrations were tested for each of the conditions, the average Hill coefficient was 1.75 ± 0.12. Because it was not practical to test this number of substrate concentrations in all cases, the Hill coefficient was constrained to 2 for Figs. 2a–d and 3a, as the data could not always be fitted without setting some constraints. For Fig. 3b, c, where the FAD concentration was varied, the Hill coefficient for the first EC50 was 2.6 and 2.8, respectively. Because it was difficult to measure the second EC50 at high FAD concentrations, the Hill coefficient was set to 2 for Fig. 3b, c to enable fitting of the data. For Fig. 3d, where ATP concentration was varied, the Hill coefficient was 1.65. Thus, in both Fig. 3d, e, the Hill coefficient was set to 2 to enable fitting of the data in Fig. 3e, in which a high FAD concentration was used, resulting in large error bars.
Dilution of dimer and molecular weight determination of the unknown peak
To determine whether dilution of the dimer enabled the formation of monomer, the ACOX-1.1 dimer peak was collected following size-exclusion chromatography. This dimer peak was diluted from a protein concentration of 2.0 mg/mL to 0.2 mg/mL (10× dilution) using the size-exclusion running buffer. After the dilution, the purified enzyme was once again injected onto the size-exclusion column for separation.
Analyzing the ACOX-1.1 dimer incubated with and without FAD and ATP
ACOX-1.1 (45 μM) was incubated at room temperature for 1 h with either water or FAD (50 µM) plus ATP (500 μM). The samples were analyzed by FPLC on a 10/300 GL Superdex 200 column (GE Healthcare) with a flow rate of 0.25 mL/min. An additional control with ACOX-1.1 (10 μM) incubated on ice was done to monitor for temperature-dependent oligomerization.
Circular dichroism spectroscopy of ACOX-1.1
In total, 0.1 mg/mL ACOX-1.1 samples were incubated in gel filtration buffer (50 mM Tris-HCl pH 7.65, 150 mM NaCl, 10 mM MgCl2) at room temperature for either 0 h or 1 h. Then, the circular dichroism spectra of the far UV range (200–280 nm) were measured by Chirascan V100 (Applied Photophysics, Inc.). Before each measurement, the instrument was blanked with gel filtration buffer.
Analysis of enzyme activity and ATP hydrolysis by HPLC
The assay conditions were the same as those for the coupled enzyme assay (total volume of 150 μL), except that the coupled enzyme components (4-aminoantipyrone, phenol, horseradish peroxidase) were absent. In brief, 38 μL of substrate dissolved in ultrapure water was added to a volume of 112 μL with 0.33 µM ACOX-1.1 (0.25 μM final concentration) and +/− ATP/FAD or buffer (ACOX-1.1 only control) to give a total volume of 150 μL. At 30 s, 60 s, and 120 s after the reaction start, a 50 µL aliquot was taken from the reaction, heated in a 90 °C heat block for 10 min, and then centrifuged at 14,674×g. In total, 40 μL of sample was loaded into a 100 µL injection loop on an Agilent 1200 series HPLC connected to a ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm × 5 μm). The solvent system was adapted from the literature44, with mobile phase A (100 mM ammonium dihydrogen phosphate (NH4H2PO4), pH 6.0, 1% methanol, filtered) and mobile phase B (1% methanol). The gradient consisted of holding for 18 min with 100% A, then ramping to 90% B over 10 min, holding at 90% B for 3 min, followed by column re-equilibration to 100% A for 6 min. The flow rate was 0.8 mL/min. The elution times for different compounds were as follows: ATP (3.7 min), ADP (4.3 min), FAD (24.7 min), substrate C12-CoA (30.2 min), and product ΔC12-CoA (30.0 min). The identities of all but the product were confirmed with synthetic standards, while the identity of the product was confirmed by LC-MS. The substrate and product were quantified using their extinction coefficients of 16,400 cm−1 M−1 and 22,400 cm−1 M−1, respectively at 260 nm45. The initial rate was approximated using a linear regression of the product formed at 0, 30, and 60 s and the assumption that the initial (0 s) product concentration was zero. Nonlinear regression spanning the 30, 60, and 120 s measurements was best fit to a Hill equation with a Hill coefficient of 1.5. In determining the rate of ATP hydrolysis, the sum of the concentration of ADP and AMP (hydrolysis products of ATP) was divided by the total time and enzyme concentration. Conditions used for the assay to determine hydrolysis of ATP are as follows: 30 µM FAD, 50 µM lauroyl-CoA, 50 µM ATP (or 200 µM ATP), and 0.50 µM enzyme.
Generation of apo ACOX-1.1 through dialysis
ACOX-1.1 was purified by using Ni-NTA resin (Thermo Fisher) as described above. The eluted protein (11 mL) was concentrated by 10-kDa-cutoff Amicon centrifugal filter (MilliporeSigma) to 1 mL, diluted to 11 mL with gel filtration buffer, and then concentrated to 1 mL again. This buffer-exchange process was repeated two more times. The sample was then dialyzed using 10-kDa-cutoff SnakeSkin dialysis tubing (Fisher) in 1 L gel filtration buffer at room temperature for 16 h. The dialyzed protein was filtered through a 0.2 mm syringe filter (Sigma-Aldrich). The filtered sample was then further purified on a HiLoad 16/600 Superdex 200 column (GE Healthcare) attached to an FPLC (GE AKTA).
Dynamic light scattering (DLS)
The specified concentration of freshly purified protein was used to confirm the molecular weights of apo and holo ACOX-1.1 by DLS. 1 mL of protein was added to a disposable semi-micro cuvette, and the protein size was measured by a Malvern Zetasizer Nano ZS operating Zetasizer software (7.13).
Size-exclusion chromatography-multiangle light scattering (SEC-MALS)
The SEC-MALS instrument consisted of an Agilent 1260 Infinity II pump for flow, a Cytiva Superdex 200 Increase 10/300 size-exclusion column for separation, a Wyatt Eclipse DualTech separation system to control flowpaths, a Wyatt Optilab T-rEX interferometer to measure concentration, and a Wyatt DAWN HELIOS II multiangle light scatterer for size determination. Flow and run control are handled by Wyatt Voyager CDS v2, and all processing is done in Wyatt Astra 7. A 42 μM stock of apo ACOX-1.1 and a 35 μM stock of holo ACOX-1.1 were analyzed at a flow rate of 4 mL/min in gel filtration buffer.
Measurement of the FAD k off rate using fluorescence polarization
In this assay, 150 μL of 5 μM wild-type ACOX-1.1 or ACOX-1.1(E434A) with FAD bound (not dialyzed) was used to measure the koff rate of FAD. The triplicate samples were added to a black 96-well plate (Thermo Scientific). The fluorescence anisotropy of FAD was measured using a CLARIOstar microplate reader (BMG Labtech) in fluorescence polarization mode over 1 h, and 5 μM FAD alone was used as the control sample for focus and gain adjustment. The koff rate was calculated by fitting the curve into the dissociation kinetics equation in GraphPad.
Measurement of FAD and ATP dissociation constants using fluorescence polarization
A series of triplicate samples with 100 nM FAD, varying amounts of apo ACOX-1.1, and a constant amount of ATP were set up 0.5× gel filtration buffer in a black 96-well plate (Thermo Scientific). Each sample had a total volume of 150 μL. The fluorescence anisotropy of FAD in each well was measured using a CLARIOstar microplate reader (BMG Labtech) in fluorescence polarization mode, and the control sample without ACOX-1.1 but with everything else was used for focus and gain adjustment. The concentrations of apo ACOX-1.1 were normalized by comparing the enzymatic activity of the apo enzyme to that of the holo-enzyme46. The KD of FAD was calculated by fitting a single titration curve into the one-site specific binding equation in GraphPad. The KD of ATP was calculated by fitting the titration curve into the allosteric modulator titration equation in GraphPad.
Enzyme-coupled kinetic assay of ACOX-1.1 ATP-binding mutants
The activity of the ACOX-1.1 ATP-binding mutants with or without FAD/ATP incubation was analyzed with the coupled enzyme assay using a setup analogous to the one of the microplate reader, except that the incubation of enzyme and FAD/ATP was done in a 20 µL total volume and then this sample was manually mixed with the remaining reaction components already in a cuvette in a UV–Vis spectrophotometer (Agilent 8453). The concentration of substrate was 50 µM, and the concentration of enzyme was 500 nM. The concentrations of FAD and ATP are stated in the relevant figures. The experiments where FAD and ATP concentration were varied used the same assay setup as previously described for the wild type, using the microplate reader.
MD simulations
To generate starting structures for MD simulations, the wild-type apo ACOX-1.1 and holo ACOX-1.1(E434A) crystal structures with Protein Data Bank (PDB) codes 5K3H and 5K3I, respectively, were downloaded from the RCSB PDB database47. These structures contain four homodimers within the unit cell, but only one homodimer consisting of subunits A and B was used in this study. For the holo ACOX-1.1(E434A), the structure includes FAD and ATP molecules bound to both subunits A and B. For the wild-type holo starting structure, the A434 residue in the holo ACOX-1.1(E434A) crystal structure was mutated in silico back to glutamate to make the structure wild-type in sequence. Because some residues are missing in both the apo and holo-crystal structures, these residues were built into the starting structures using AlphaFold2 and SWISS-MODEL, respectively48,49. For example, for the wild-type apo ACOX-1.1 starting structure, the catalytic loop including residues 431 to 436 was missing and thus was built into the apo starting structure using AlphaFold2 with apo ACOX-1.1 structure as a template. The ProteinPrepare tool from PlayMolecule package was used to estimate the protonation states for the histidines, especially those near ATP50. The respective crystal structures water molecules were transferred to the AlphaFold-modeled structure of apo and the SWISS-MODEL-modeled structure of holo (i.e., waters from 5K3H were transferred to the modeled apo and waters from 5K3I were transferred to the modeled holo). For the apo 2A and 2B starting structures, the crystal waters from either subunit A or subunit B were duplicated. The characteristics of each system are summarized in Supplementary Table 1.
The AMBER20 software suite was used for setting up and running the MD simulations51. FAD was parametrized with GAFF using the antechamber tool from AMBER52. The parameters for ATP were taken from the AMBER parameter database53. The ff19SB force field was used for the enzyme with the OPC water model54,55. Minimization was carried out in six stages (4000 steps in each of them using the steepest descent algorithm for the first 2000 steps and using the conjugate gradient algorithm for the last 2000 steps while slowly decreasing the heavy atom restraints from 30 kcal/mol to 056,57. Then the system was gradually heated to 298.15 K for 1 ns with a timestep of 1 fs under a constant volume condition. The heated system was then equilibrated with constant pressure (Berendsen thermostat58) and temperature (Langevin thermostat59), first for 1.1 ns with 5.0 kcal/mol restraints on the heavy atoms (where first 0.1 ns were run on CPU and then moved to GPU for the next 1 ns), then for 5 ns without any restraints. For equilibration, we used timesteps of 1 fs, which is the same as we used in the heating stage. Afterward, all the equilibrated systems were simulated for production for 2 µs and then extended to 4 µs at 298.15 K with a constant pressure of 1 atm (Monte Carlo barostat) and constant temperature conditions (Langevin thermostat)60. Skin-permit parameter was set to 0.75 for better efficiency at the production stage without limiting the accuracy (https://ambermd.org/GPULogistics.php), similar to as described61. At the production stage, we used hydrogen mass repartition and SHAKE constraints, allowing us to use a timestep of 4 fs62,63. A cutoff of 8 Å was used to approximate the long-range electrostatic interaction using the Particle–Mesh Ewald (PME) method64. Throughout the process, the collision frequency was set to 1 ps−1. The production stage was run in 20 copies with unique “random numbers” for different copies to ensure the uniqueness of different copies. The reliability and reproducibility checklist for MD simulations is given in Supplementary Table 4.
To calculate the sizes of the openings to the FAD and ATP-binding sites, we calculated the shortest distances between any atom surrounding the ligand-binding sites. We used different sets of residues for calculating the closest distance, and they are summarized in Supplementary Table 2. The distances in each frame in the production trajectory (4 μs) were calculated using the MDTraj package (1.9.9)65.
Clustering analysis
For the ATP and FAD sites in the apo and holo MD simulations, we extracted the top 5% of frames with the largest opening distance and top 5% of frames with the smallest opening distance using the MDTraj package. Each subset of frames was clustered independently to identify a representative structure in each of those eight categories. We clustered each subset using the pairwise RMSD of the relevant ligand-binding sites as the similarity metric with the hierarchical agglomerative clustering algorithm implemented in cpptraj from the AMBER20 suite. In all cases, average linkage was used to calculate the distance between clusters with an RMSD cutoff of 1.5 Å. The ligand-binding site opening distances of the centroids for the top ten clusters in each category were calculated using the MDTraj package.
Allosteric network analysis
SenseNet (1.1.1) was used to calculate the allosteric network as a plugin of Cytoscape (3.10.1)22,66. For this purpose, two different time series were built using cpptraj tool from the AMBER20 suite, one for the hydrogen bond interaction network (with a 3.5 Å distance cutoff) and one for the heavy atom interaction network (with a 5 Å distance cutoff)67. For memory efficiency, we only used 500 equally spread-out frames from the first 10 (2 µs) MD simulations of the apo and holo enzymes for this analysis. After generating the time series, we used a JAVA (1.8.0_441) environment-dependent tool AIFgen (1.0.9) to convert the cpptraj output to the SenseNet-supported format AIF. The AIF file was loaded into SenseNet, ignoring the interactions that occur less than 50% of the time. Each residue in the enzyme was represented as a node, and the interactions between residues were represented as edges. First, the Edge Correlation Factor (ECF) was calculated using the “correlation” function using mutual information mode from the SenseNet GUI. This calculation resulted in correlation values for all residue pairs. The correlation values for each residue were summed up using the “Degree” function. These summed up correlation values for each residue were saved as allosteric correlation values. SenseNet allows us to estimate allosteric correlation in several different ways, such as Node Correlation Factor (NCF) and Difference Node Correlation Factor (DNCF). NCF is calculated for a single interaction system resulting from one system. Whereas the DNCF uses two interaction networks: one as a target network and another as a reference network. In this work, we focused on the DNCF network as our goal is to understand the role of the cofactors (ATP and FAD in this case) in the system. Therefore, we used the interaction network from the holo system as the target and the interaction network of the apo system as the reference network. In Fig. 6a, residues with allosteric values in the top 10% were shown in the network for better visualization and the color was normalized from white-to-red for these residues based on their raw allosteric correlation values. In Fig. 6b–d, allosteric values were normalized with z-score values, and graphically the network was visualized as white for nodes with negative z-score values and as a white-to-red gradient for positive z-score values. The most important path between two selected nodes was calculated by summing up the number of edges between pairs of residues.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available without restriction and will be provided upon request. The initial models used in the MD simulations, as well as selected frames used for analysis, are deposited in the GitHub repository: https://github.com/arupmondal835/ACOX-simulations68. Source data are provided with this paper.
Code availability
The setup and analysis scripts for the MD simulations, the custom parameters for ATP and FAD, the clustering scripts, and the SenseNet network building scripts are deposited in the GitHub repository: https://github.com/arupmondal835/ACOX-simulations68.
References
Oaxaca-Castillo, D. et al. Biochemical characterization of two functional human liver acyl-CoA oxidase isoforms 1a and 1b encoded by a single gene. Biochem. Biophys. Res. Commun. 360, 314–319 (2007).
Chu, R. et al. Overexpression and characterization of the human peroxisomal acyl-CoA oxidase in insect cells. J. Biol. Chem. 270, 4908–4915 (1995).
Ferdinandusse, S. et al. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 952–958 (2018).
Sonani, R. R., Blat, A. & Dubin, G. Crystal structures of apo- and FAD-bound human peroxisomal acyl-CoA oxidase provide mechanistic basis explaining clinical observations. Int. J. Biol. Macromol. 205, 203–210 (2022).
Tokuoka, K. et al. Three-dimensional structure of rat-liver acyl-CoA oxidase in complex with a fatty acid: insights into substrate-recognition and reactivity toward molecular oxygen. J. Biochem. 139, 789–795 (2006).
Romero, E., Gomez Castellanos, J. R., Gadda, G., Fraaije, M. W. & Mattevi, A. Same substrate, many reactions: oxygen activation in flavoenzymes. Chem. Rev. 118, 1742–1769 (2018).
Burgener, S., Schwander, T., Romero, E., Fraaije, M. W. & Erb, T. J. Molecular basis for converting (2S)-methylsuccinyl-CoA dehydrogenase into an oxidase. Molecules https://doi.org/10.3390/molecules23010068 (2017).
Kim, J. J., Wang, M. & Paschke, R. Crystal structures of medium-chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. USA 90, 7523–7527 (1993).
Arent, S., Pye, V. E. & Henriksen, A. Structure and function of plant acyl-CoA oxidases. Plant Physiol. Biochem. 46, 292–301 (2008).
Pedersen, L. & Henriksen, A. Acyl-CoA oxidase 1 from Arabidopsis thaliana. Structure of a key enzyme in plant lipid metabolism. J. Mol. Biol. 345, 487–500 (2005).
De Bellis, L. et al. Purification and characterization of a novel pumpkin short-chain acyl-coenzyme A oxidase with structural similarity to acyl-coenzyme A dehydrogenases. Plant Physiol. 123, 327–334 (2000).
Bhar, S., Perez, D. H. & Butcher, R. A. In Comprehensive Natural Products III: Chemistry and Biology Vol. 3 (ed. Tadhg, P. Begley Hung-Wen (Ben) Liu) 3–30 (Elsevier, 2020).
von Reuss, S. H. et al. Comparative metabolomics reveals biogenesis of ascarosides, a modular library of small-molecule signals in C. elegans. J. Am. Chem. Soc. 134, 1817–1824 (2012).
Zhang, X. et al. Acyl-CoA oxidase complexes control the chemical message produced by Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 112, 3955–3960 (2015).
Zhang, X., Li, K., Jones, R. A., Bruner, S. D. & Butcher, R. A. Structural characterization of acyl-CoA oxidases reveals a direct link between pheromone biosynthesis and metabolic state in C. elegans. Proc. Natl. Acad. Sci. USA 113, 10055–10060 (2016).
Zhang, X., Wang, Y., Perez, D. H., Jones Lipinski, R. A. & Butcher, R. A. Acyl-CoA oxidases fine-tune the production of ascaroside pheromones with specific side chain lengths. ACS Chem. Biol. 13, 1048–1056 (2018).
Zhou, Y. et al. Biosynthetic tailoring of existing ascaroside pheromones alters their biological function in C. elegans. eLife. https://doi.org/10.7554/eLife.33286 (2018).
Chen, X. F. et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. https://doi.org/10.15252/embr.201745124 (2018).
Chung, H. L. et al. Loss- or gain-of-function mutations in ACOX1 cause axonal loss via different mechanisms. Neuron 106, 589–606.e586 (2020).
Freitas, M. O. et al. The peroxisomal protein import machinery displays a preference for monomeric substrates. Open Biol. 5, 140236 (2015).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Schneider, M. & Antes, I. SenseNet, a tool for analysis of protein structure networks obtained from molecular dynamics simulations. PLoS ONE 17, e0265194 (2022).
Huang, Z. et al. ASD v2.0: updated content and novel features focusing on allosteric regulation. Nucleic Acids Res. 42, D510–D516 (2014).
Zheng, Z., Goncearenco, A. & Berezovsky, I. N. Nucleotide binding database NBDB-a collection of sequence motifs with specific protein-ligand interactions. Nucleic Acids Res. 44, D301–D307 (2016).
Lu, S. Y. et al. The structural basis of ATP as an allosteric modulator. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1003831 (2014).
Narunsky, A. et al. On the evolution of protein-adenine binding. Proc. Natl. Acad. Sci. USA 117, 4701–4709 (2020).
Longo, L. M., Petrovic, D., Kamerlin, S. C. L. & Tawfik, D. S. Short and simple sequences favored the emergence of N-helix phospho-ligand binding sites in the first enzymes. Proc. Natl. Acad. Sci. USA 117, 5310–5318 (2020).
Takaine, M., Ueno, M., Kitamura, K., Imamura, H. & Yoshida, S. Reliable imaging of ATP in living budding and fission yeast. J. Cell Sci. https://doi.org/10.1242/jcs.230649 (2019).
Imamura, H. et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 106, 15651–15656 (2009).
Yaginuma, H. et al. Diversity in ATP concentrations in a single bacterial cell population revealed by quantitative single-cell imaging. Sci. Rep. 4, 6522 (2014).
Voon, C. P. et al. ATP compartmentation in plastids and cytosol of Arabidopsis thaliana revealed by fluorescent protein sensing. Proc. Natl. Acad. Sci. USA 115, E10778–E10787 (2018).
Gout, E., Rebeille, F., Douce, R. & Bligny, R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. USA 111, E4560–E4567 (2014).
Hibshman, J. D. et al. Nonselective autophagy reduces mitochondrial content during starvation in Caenorhabditis elegans. Am. J. Physiol. Cell Physiol. 315, C781–C792 (2018).
Clarke, R. J., Apell, H. J. & Kong, B. Y. Allosteric effect of ATP on Na(+),K(+)-ATPase conformational kinetics. Biochemistry 46, 7034–7044 (2007).
Rozman Grinberg, I. et al. Novel ATP-cone-driven allosteric regulation of ribonucleotide reductase via the radical-generating subunit. eLife https://doi.org/10.7554/eLife.31529 (2018).
Klyuyeva, A., Tuganova, A. & Popov, K. M. Allosteric coupling in pyruvate dehydrogenase kinase 2. Biochemistry 47, 8358–8366 (2008).
Allewell, N. M., Friedland, J. & Niekamp, K. Calorimetric analysis of aspartate transcarbamylase from Escherichia coli: binding of cytosine 5’-triphosphate and adenosine 5’-triphosphate. Biochemistry 14, 224–230 (1975).
Gray, C. W., Chamberlin, M. J. & Gray, D. M. Interaction of aspartate transcarbamylase with regulatory nucleotides. J. Biol. Chem. 248, 6071–6079 (1973).
Bennett, B. D. et al. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5, 593–599 (2009).
Yamada, Y., Merrill, A. H. Jr. & McCormick, D. B. Probable reaction mechanisms of flavokinase and FAD synthetase from rat liver. Arch. Biochem. Biophys. 278, 125–130 (1990).
Huhner, J., Ingles-Prieto, A., Neususs, C., Lammerhofer, M. & Janovjak, H. Quantification of riboflavin, flavin mononucleotide, and flavin adenine dinucleotide in mammalian model cells by CE with LED-induced fluorescence detection. Electrophoresis 36, 518–525 (2015).
Duvaud, S. et al. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic Acids Res. 49, W216–W227 (2021).
Kutta, R. J., Archipowa, N., Johannissen, L. O., Jones, A. R. & Scrutton, N. S. Vertebrate cryptochromes are vestigial flavoproteins. Sci. Rep. 7, 44906 (2017).
Menegollo, M., Tessari, I., Bubacco, L. & Szabadkai, G. Determination of ATP, ADP, and AMP levels by reversed-phase high-performance liquid chromatography in cultured cells. Methods Mol. Biol. 1925, 223–232 (2019).
Peter, D. M., Vogeli, B., Cortina, N. S. & Erb, T. J. A chemo-enzymatic road map to the synthesis of CoA Esters. Molecules 21, 517 (2016).
Jarmoskaite, I., AlSadhan, I., Vaidyanathan, P. P. & Herschlag, D. How to measure and evaluate binding affinities. eLife https://doi.org/10.7554/eLife.57264 (2020).
Berman, H. M. et al. The Protein Data Bank and the challenge of structural genomics. Nat. Struct. Biol. 7, 957–959 (2000).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Martinez-Rosell, G., Giorgino, T. & De Fabritiis, G. PlayMolecule ProteinPrepare: a web application for protein preparation for molecular dynamics simulations. J. Chem. Inf. Model 57, 1511–1516 (2017).
Case, D. A. et al. The Amber biomolecular simulation programs. J. Comput Chem. 26, 1668–1688 (2005).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput Chem. 25, 1157–1174 (2004).
Meagher, K. L., Redman, L. T. & Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput Chem. 24, 1016–1025 (2003).
Tian, C. et al. ff19SB: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput 16, 528–552 (2020).
Izadi, S., Anandakrishnan, R. & Onufriev, A. V. Building water models: a different approach. J. Phys. Chem. Lett. 5, 3863–3871 (2014).
Meza, J. C. Steepest descent. WIREs Comput. Stat. 2, 719–722 (2010).
Shewchuk, J. R. An introduction to the conjugate gradient method without the agonizing pain. Carnegie Melon University (Pittsburgh, PA, 1994). http://www.cs.cmu.edu/~quake-papers/painless-conjugate-gradient.pdf.
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Davidchack, R. L., Handel, R. & Tretyakov, M. V. Langevin thermostat for rigid body dynamics. J. Chem. Phys. 130, 234101 (2009).
Faller, R. & de Pablo, J. J. Constant pressure hybrid molecular dynamics-Monte Carlo simulations. J. Chem. Phys. 116, 55–59 (2002).
Evans, R. T. et al. Optimizing GPU-Enhanced HPC System and Cloud Procurements for Scientific Workloads, Vol. 12728 (Springer, 2021).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. Numerical-integration of cartesian equations of motion of a system with constraints—molecular-dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Darden, T., York, D. & Pedersen, L. Particle Mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Schneider, M. & Antes, I. Comparison of allosteric signaling in DnaK and BiP using mutual information between simulated residue conformations. Proteins 91, 237–255 (2023).
Roe, D. R. & Cheatham, T. E. 3rd. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 (2013).
Perez, D. H. et al. ATP allosterically regulates an acyl-CoA oxidase. Zenodo https://doi.org/10.5281/zenodo.15702808 (2025).
Acknowledgements
The authors would like to thank Prof. Michael Harris (University of Florida) for many helpful discussions and the use of a CLARIOstar microplate reader and the Chirascan V100 CD spectrometer. The authors would also like to thank Dr. Peter Randolph (Florida State University) for performing and analyzing the SEC-MALS experiments. This study was supported by funding from the National Institutes of Health (R35 GM144076 to R.A.B.) and the National Science Foundation (CHE-1555050 to R.A.B.).
Author information
Authors and Affiliations
Contributions
D.H.P., W.X., V.B., and H.E.C. performed all biochemical experiments. A.M. performed all MD simulations. D.H.P., A.M., W.X., A.P., and R.A.B. designed the approach and interpreted data. D.H.P., A.M., and R.A.B. wrote a first draft of the manuscript, which was edited and approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jianping Ding and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Perez, D.H., Mondal, A., Xu, W. et al. ATP allosterically regulates an acyl-CoA oxidase. Nat Commun 16, 7318 (2025). https://doi.org/10.1038/s41467-025-61905-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61905-9
