
Abstract
Human development and physiology are fundamentally linked with the microbiome. This is particularly true during early life, a critical period for microbiome assembly and its impact on the host. Understanding microbial acquisition in early life is thus central to both our basic understanding of the human microbiome and strategies for disease prevention and treatment. Here, we review the historical approaches to categorize microbial transmission originating from the fields of infectious disease epidemiology and evolutionary biology and discuss how this lexicon has influenced our approach to studying the early-life microbiome, often leading to confusion and misinterpretation. We then present a conceptual framework to capture the multifaceted nature of human microbiome acquisition based on four key components: what, where, who, and when. We present ways these parameters may be assigned, with a particular focus on the ‘transmitted strain’ through metagenomics to capture these elements. We end with a discussion of approaches for implementing this framework toward defining each component of microbiome acquisition.
Similar content being viewed by others


Introduction
The human microbiome, the assembly of microorganisms living in and on the human body, and the genes and products of these microbes, has emerged as a pivotal determinant of host physiology and disease, influencing multiple tissues and organ systems. Early life, defined here as spanning from pregnancy to infancy, is a critical period of host-microbe interactions. The interactions range from the impact of maternally derived microbial metabolites on fetal development in utero to serving as a blueprint of current and future health1. Disturbances to the source, order of arrival, and succession of microbes during early life have been linked to infections and different physiological disorders, including cancer2.
The terminologies describing microbiome acquisition, mainly that of vertical and horizontal transmission, have roots in infectious disease epidemiology and evolutionary biology. The origin of using “transmission” to describe the transfer of microorganisms from one host to another originates from Koch’s works3,4 and studies of invertebrates causing “vector-born” infectious diseases of animals and plants5,6,7,8,9. Applied to mother-to-child transmission, the term “vertical” can be traced to the early 1950s and the transmission of microbial pathogens10,11,12,13,14,15. In the coming decades, microbial transmission between parent and offspring remained in the realm of infectious diseases, being referred to as “inheritance of infection” or “hereditary transmission” (Box 1). Although the used definitions consider single infectious agents and may give information on cross-generation microbial transfer, the microbiome field has adapted the terminology for mother-to-child microbial transmission without major modifications.
Compared to the infectious disease lens, in which the term vertical transmission focuses on generational inheritance (namely from a parent), we see an added focus from the fields of ecology and evolutionary biology. In addition to generational inheritance, these scientific fields have emphasized the mechanism and, relatedly, timing of transmission. Here, the added distinction between vertical and horizontal transmission is based on when and, by extension, where transmission occurs (Box 1). A prime example of vertical transmission is the transovarial transmission of the intracellular bacteria Wolbachia in the fruit fly Drosophila16. In comparison, horizontal transmission of microbes, in essence, is everything else, with a focus on routes such as sexual, vector-borne, and attendant-borne transmission17. Well-studied examples focus on environmental sources of microbes in open ecosystems, such as the colonization of the light organ by the marine free-living symbiont Vibrio fischeri by the Hawaiian bobtail squid Euprymna scolopes, and the acquisition of the rhizosphere microbiome from soil.
Yet, within studies of the microbiome acquisition along animal and plant life cycles, there are gray areas between vertical and horizontal transmission. For example, during trophallaxis in termites, offspring may acquire the maternal fecal microbiome after hatching. In stinkbugs of the family Plataspidae, endocellular γ-Proteobacteria are transmitted to the offspring via symbiont capsules that females produce upon oviposition18. Such scenarios have invoked the terms “social transmission, pseudo-vertical transmission, external maternal transmission, and postnatal vertical transmission” to describe such microbial acquisition routes17.
In this Perspective, we discuss how the above-described historical lexicon is often ambiguous when used in the context of human microbiome acquisition and limited in capturing the multidimensional features of microbial transmission. Given the imprecision and limitations of this status quo lexicon, we propose a conceptual framework termed 4 W to describe microbiome transmission in early life, centered on assigning four critical features: what, where, who, and when (Box 1). We then follow with a discussion of how we can capture these features in a human microbiome cohort design.
Central to an accurate description of early-life microbiome acquisition is the ability to define from ‘who’, ‘where’, and ‘when’ transmission occurs by methods enabling tracking of the ‘what’. We define the “transmitted microbial strain” based on metagenomic resolution as currently the most precise unit to determine the transmission of microbes over space and time, and discuss how it can be used to assign the parameters of microbiome acquisition. We then introduce a fifth question, ‘why’, discussing how a 4 W framework can address both mechanisms of early-life acquisition, and while not the focus of this Perspective, aspects of microbial assembly, such as succession and colonization. We end by providing recommendations for the design of studies aiming to capture the 4Ws of microbiome acquisition. The proposed framework will empower an expanded understanding of the transmission and factors shaping the human microbiome and the mechanisms governing the impact of the early-life microbiome on health and disease.
Ambiguity of existing terms for human microbiome acquisition
The term “vertical” has been widely used for human microbiome acquisition. In similar contexts, “vertical transmission” is broadly and ambiguously defined as 1) transmission from the mother or both parents, 2) from and to different body sites (in contact or not with the open environment), and 3) transmission during or also after birth. Although a commonly used term originating from infectious disease epidemiology, the description of “vertical transmission” in scientific reports often lacks important information, including simultaneous reporting of timing, source of transmission, and the microbial commodity being transmitted.
With a few exceptions, the term “horizontal transmission” is rarely used in the early-life microbiome field. Instead, other terms such as “microbial taxa dispersal from different sources” and “horizontal dispersal of microbes” have been applied. The few studies that used the term “horizontal transmission” generally indicated that it is not mother-to-infant microbial transmission; however, it is unclear whether the transmission of the microbiome is from other family members, the community, or the environment. That this term is not commonly used in the context of early-life microbiome acquisition and the absence of its definition shows that transmissions of microbes from the environment and others than the mother remain understudied.
What terms, if any, should we use to describe early-life microbiome acquisition? If based on an evolutionary biology definition focused on the timing of transmission, the use of “vertical” and “horizontal” transmission in reference to early-life microbiome transmission may similarly be imprecise and potentially inappropriate. If defined as transmission before birth, the acquisition of microbes with replicative potential is limited to pathogens in an uncomplicated pregnancy, therefore excluding “vertical transmission” from an evolutionary framework. Similarly, as we will describe below, when considering microbial transmission from a broader perspective, including microbe-derived factors such as metabolites across the placenta, indeed, “vertical transmission” from an evolutionary definition may apply, yet this situation is seldom considered in the common use of the term. For vaginal delivery, do we consider this vertical, horizontal, or rather use quasi-terms as in the evolutionary literature? As such, one of the fundamental features distinguishing vertical and horizontal transmission is whether transmission occurs in a closed (transplacental/transovarial) or open ecosystem (consider whale vaginal birth) and thus varies in ecological competition and fidelity of transmission19. We are faced with a common problem in lexicology: the misuse of commonly accepted terms or their acceptance, explicit statements of definition, and even redefinition of terms.
A conceptual framework of microbial acquisition and transmission
As reviewed above, precise terminology for different mechanisms of transmission is lacking, and the existing terms referring to the transmission of microbial strains from mother to infant fail to capture the multifaceted nature of microbial acquisition. Ultimately, what do we want to know about early-life microbiome acquisition?
Here, we provide a conceptual and operational framework of early-life microbial transmission structured around four central components (4 W): what, where, who, and when. Characterizing transmission events according to each of these components is critical to our understanding of the assembly of the human microbiome and the mechanisms by which the microbiome impacts human physiology and disease. This precise characterization should inform study design, methodology, and results interpretation when studying the early-life microbiome. Here, we lay out this conceptual framework and provide examples of how these parameters may characterize new and unanticipated transmission events.
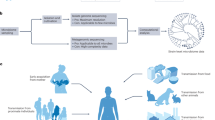
What. When considering transmission of the microbiome, we must consider “what” is the transmitted commodity. These might include cells (or, in the case of viruses, virions, viroids or even viroid-like) that have replicative potential (inclusive of microbes in dormant phases such as spores), microbially derived components, such as different structural elements of the cells (proteins/peptides, nucleic acids, mobile genetic elements, lipids and sugars), and their metabolites (Fig. 1). Classically, when thinking about the assembly of the infant microbiome, the operative “what” are microbial cells that can “seed” and subsequently replicate for “colonization”. Importantly, the workhorse of identifying the microbial “what” of the microbiome is next-generation sequencing (NGS), such as amplicon (i.e., bacterial 16S rRNA gene or fungal internal transcribed spacer region) and shotgun metagenomic sequencing, which is based on DNA present in the sample and thus not indicative of “colonization” per se. Advances in single-cell microbial genomics hold the potential for bridging this discrepancy. At present, metagenome-defined strains are the units most accessible to infer transmission and thus assess microbial acquisition in early life (Box 2; Fig. 2).

Different microbes colonize the mother at multiple body sites, although the focus is on gut bacteria here. Microbially derived metabolites (what) can translocate from the mother gut (from who) during pregnancy (when) into the intestinal lamina propria and blood circulatory system. Metabolites can also cross via the placenta and affect the developing brain of the fetus (where to). Microbially derived DNA from the parent gut can translocate into circulation, cross via the placenta, and may enter different sites in the fetus, including the gut. Sequencing-based techniques may detect the presence of microbial DNA, even when live microbes are not occurring in the system. Live organisms are not expected to translocate outside the gut or cross the placental barrier into the fetus. Pathogenic microorganisms like L. monocytogenes can translocate from the gut and infect the placenta and fetal brain tissue.

Strain boundaries should be identified on a species-by-species basis and based on a comparison of (phylo-) genetic distance distributions of strains detected in longitudinal samples from the same individual (same strain; green distribution) to those between unrelated individuals who have never been in contact (different strain; orange distribution). While some strain replacement events might occur within an individual’s microbiome even without any intervention (e.g., antibiotic treatment, diet changes), these are a limited minority in samples taken less than six months apart36. Once such thresholds are established, the origin of a strain in the infant can be inferred (maternal: pink distribution; from an unknown source: gray distribution). Sampling of more environments, individuals, and body sites thus adds to the “from where/who” and “to where/who” dimensions, while the collection of samples from multiple time points allows to establish “when” the transmission event took place.
A weak signal of the nucleic acid translocation might be detected by NGS and considered as a fetal microbiome. Relatedly, with the exception of colonization of the placenta and fetus with pathogens (such as Group B streptococci; GBS, see “when” below20), contamination with microbial DNA during post-fetal sampling has also been misinterpreted as evidence of placental or fetal colonization21. Because of the latter challenges, one must control for contamination, which is omnipresent in microbial studies and can happen at any stage of sample collection and processing, with well-to-well contamination being a major culprit in microbiome studies. Stringent negative and positive controls need to be included, especially in the case of low microbial biomass samples (e.g., human milk, skin). Several approaches are now available to prevent and detect contamination, including sterile sampling and spike-in quantitative approaches for low biomass communities20,22. Similarly, a variety of open-source tools exists, from lists of common contaminants and guidelines on how to reduce well-to-well effects to reports on technical biases in general.
In addition to using NGS approaches for defining the “what” of transmission, advances in analytical techniques such as metabolomics and metaproteomics have enabled the characterization of other essential units of the microbiome (e.g., proteins and metabolites) that may be transmitted in early life. Metaproteomics seeks to comprehensively define the proteins in a given sample. Thus, proteins or shorter peptides detected in infant samples and assigned as of microbial origin may serve both as evidence of microbiome transmission and the potential for function. At the same time, the use of metaproteomics for the study of microbial transmission in early life has been limited to a few pioneering studies so far. For example, bacterial peptides have been detected in the amniotic fluid derived from uncomplicated pregnancies, as well as within extracellular vesicles isolated from amniotic fluid23 and human milk24. The metabolome, the collection of small molecules within a given sample, can also be used to define microbiome transmission. Microbial-derived metabolites, such as short-chain fatty acids or 4-ethylphenylsulfate produced by bacteria in the maternal gut during gestation, may enter circulation and cross the placenta into fetal circulation25. Microbially-derived metabolites have been reported so far in human amniotic fluid26, fetal intestine27, and breast milk28. In addition, free nucleic acids derived from maternal microbes (most notably pathogens) may interact with the placenta, acting as agonizts of innate immunity. Effects of such microbial products can be entry into fetal blood/tissues, and subsequent activation of inflammatory programs impacting the fetus or stimulation of fetal intestinal lymphoid system (Fig. 1).
Critically, while some proteins and metabolites can be clearly defined as of microbial origin [such as distinct primary (e.g., acetate) or secondary metabolites (e.g., lantibiotics)], other proteins or metabolites can be synthesized by both host and microbes (e.g., acetate or secondary amino acid metabolites such as serotonin and polyamines). Thus, ascribing such proteins or metabolites as of microbial origin and transmitted by the microbiome requires specialized approaches to trace the source of such molecules. Two approaches, limited to animal studies, have been used to date. This first has been using 13 C or 15 N labeled substrates, such as dietary fibers or proteins, respectively, in mice with (conventionally reared) or without (germ-free) a resident microbiome. This strategy is followed by sampling of mouse tissues and detection of labeled proteins and/or metabolites, whereby comparison of labels between conventional and germ-free mice defines microbial origin. To date, such an approach has not been used to define early-life transmission. Alternatively, bacteria can be labeled isotopically in vitro, before introduction to mice, followed by sampling of mouse tissues, in essence a ‘pulse-chase’ experiment. Such an approach using auxotrophic bacteria to eliminate colonization of labeled bacteria at different timepoints has been used to define ‘when’ (gestational, postnatal) and ‘what’ (amino acid metabolite, nucleic acid, protein) of early-life transmission of microbial products in mouse models.
Where. The “where” of microbial transmission can be thought of as the route of transmission of the “what”, i.e., from where/to where, often occurring “via” conduit(s). During gestation, pathogens, classically such as Listeria monocytogenes, which originate “from” the maternal gut, translocate via the epithelial barrier to the maternal blood and the placenta, eventually disseminating “to” fetal systemic and neurovascular circulation29,30. Another example is the intracellular protozoan Toxoplasma gondii, a parasite of cats with a range of intermediate hosts (e.g., rodents) that transmits from mother to fetus at the placenta, resulting in congenital infection. In vaginal delivery, the birth canal is the main route of microbial transmission, with microbes “from” the maternal vaginal and fecal microbiomes, skin, and nearby environment, being transmitted “to” the newborn’s skin, oral cavity, and gut, the latter “via” oral ingestion (Fig. 3A). During delivery by Cesarean section, skin microbes from the mother are transferred to the infant, in addition to microbes transferred from the medical staff or operating room (Fig. 3B). Breast milk and transmission of breast milk microbiota to the infant during lactation (Fig. 4A) is another example of “where”, such as maternal bacteria transmission via the oral-entero-mammary route31,32.

A During vaginal birth (when), microbes (what) are transferred from the mother’s (who) vaginal and fecal communities (where from) to the child’s oral, gastrointestinal, and skin communities (to where). B During Cesarean section, skin microbes are transferred from the skin of the mother and health personnel to the infant.

AThe mother (who) transfers microbes and metabolites (what) to the child via breastfeeding (when) from breast milk (from where) to the mouth, gut, or skin of the infant (to where). B Transfer of microbes from the dog’s skin and mouth to the baby’s skin and mouth.
Who. Related to where is the “who”. Here, the “to who” is defined as the biological offspring. The “from who” can vary from both human (mother, father, parent, siblings, health-care providers, etc.) and non-human (pets) and sources from the abiotic environment (food, air, built environment, such as a newborn nursery). “Who” can also involve secondary actors or routes, for example, microbes transmitted from the environment via a secondary carrier. As a hypothetical example, imagine a family dog that carries goat stool microbes on its mouth and transfers them by licking an infant’s mouth or skin (Fig. 4B).
When. Finally, the time of acquisition is captured by the “when”. This can be broken down into discrete, operational periods, such as at which gestational week, at which stage of delivery (e.g., premature rupture of membranes), and post-delivery, and including important transitions in diet (suckling and weaning). This final parameter defining early-life microbial transmission emphasizes that the specific timing of when microbes and their products are transmitted is critical to ecological succession, microbial competition, and immune tolerance windows33. An example where the timing of microbial transmission/ colonization has been shown to be important is microbiome disturbance by antibiotics in infancy, which is linked to an increased disease risk later in life34. Since exposures to antibiotics might have a different effect if occurring exclusively during pregnancy or after delivery, this points to distinct time windows by which microbiome transmission impacts infant health.
The importance of microbial transmission timing can also be derived from infectious diseases, with examples such as congenital, perinatal, and postnatal acquisition of pathogens, such as cytomegalovirus, human immunodeficiency virus, and herpes simplex35. There, the timing of pathogenic microbe transmission affects the disease risk, with multiple sources having an additive effect on the rate of transmission. The “when” parameter of microbial acquisition also illustrates the multidimensional nature of the early-life microbiome since determining only “when” without any information on the microbial sources (“where” and “who”) is bound to give incomplete information. For example, GBS, which may cause severe neonatal infections such as sepsis and meningitis, is considered of maternal origin (acquired during birth) if it causes early onset infection (< 72 h after delivery). Yet for late-onset sepsis, GBS could be acquired from the mother via various routes, including breast milk or other sources. Hence, to identify the GBS source (“what”) with high certainty, samples from the infant, mother, and their environment (“where” and “who”) would have to be collected during pregnancy and the first month of life (“when”).
Everything, everywhere, from everyone, all the time
Unlike the movie, it is not feasible nor are agencies likely to fund a prospective study encompassing the multiverse of samples and methods of analysis, powered to capture the 4Ws in the scope and depth required to answer the who (from and to), what, where, and when of each microbe and microbial product of early-life transmission. Ultimately, the design of a prospective study of microbiome acquisition (although not limited to early life period) will depend on a balance of the constraints of a study (e.g., budget, sample collection infrastructure, storage, and technical/analytic capacity), the primary and secondary aims of the study, and which and how many of the 4Ws should be captured to answer specific questions. In Table 1, we highlight a selection of representative studies of early-life microbiome acquisition, presenting these studies through the lens of the 4 W framework, defining which aspects of 4 W were captured, focused on, and how these allowed for the definition of early-life microbial transmission. In Box 3, we provide case studies in which specific questions can be answered and prioritized (and ‘future-proofed’ for the potential to address additional and forthcoming questions and aspects of transmission) through weighing sample collection and analysis for specific aspects of the 4Ws. Below, we discuss theoretical approaches to capture the 4Ws in a prospective study of early-life microbial transmission. Although we focus our discussion of the 4 W framework on microbial acquisition at early life, the lens of 4 W can be applied for microbial transmission, colonization, and succession throughout a person’s lifetime36, such as defining the 4Ws of microbiome ecology during travel, hospital admissions, and microbiome repopulation through and after antibiotic use.
For the “what” component, the status quo, allowing definition and tracking of the ‘transmitted strain’, is shotgun metagenomics. As most (but not all and not all the time22) human microbiomes are dominated by bacteria, metagenomics will bias to a bacterial ‘what’. However, it is crucial to understand that microbial transmission is not restricted to bacteria, which have been the most studied to date. Viruses, fungi, and protozoa, as well as different microbial metabolites and structural components, play significant roles in early life development and deserve sufficient attention from the research community. Current and future studies need to expand the description of “what” in terms of whether targeting microorganisms or metabolites, including the resolution level of the identified organism or metabolite. This will require improved sequencing techniques, such as the use of long-read sequencing to provide better strain-specific identification, tools like Hi-C cross-linkage to map mobile elements and viruses to their host organisms, deep sequencing to allow full coverage of rare community members, and targeted enrichment of hard-to-capture organisms and clades37.
Related to the ‘what’ is ‘how much of what’. Both NGS workflows and omics-based measurements (such as metabolomics and proteomics to a certain extent) are semi-quantitative, providing the relative abundance of the ‘what’. Such readouts limit our understanding of critical aspects of microbial transmission. First, our ability to determine microbial growth and colonization versus passing through without replication is limited when we measure the relative abundance of DNA through NGS. Second, measurement of one domain of life (as above), let alone the relative abundance of this domain, curtails our ability to determine the role of interkingdom interactions in early-life transmission. Relatedly, and third, approaches have been proposed to utilize absolute quantification from meta-omics data38, such as metabolomics and proteomics, that can aid in deciphering the mechanisms of microbe-microbe and microbe-host interactions. Future studies focusing on technological and analytical quantitation of the ‘how much’ of the microbiome22,39,40,41 during early life will allow us to turn the ‘what’ of microbial transmission into the ‘how’.
The role of the “where” factor has largely been recognized as a critical influence on early microbiome development. Babies delivered vaginally vs Cesarean delivery show pronounced differences in their microbiome compositions during the first year of life42. Similarly, the effect of breastfeeding on the gut microbiome is substantial43. To address the “where” aspect comprehensively, it is essential to collect multiple potential sources such as feces, vaginal fluid, skin, saliva, and breast milk and detailed information on delivery and feeding, including specifics such as the time the amniotic sac was broken, the use of a vacuum during delivery, the use of a breast pump, intermittent formula feeding, and finally, multiple samples from the environment.
For the “who” aspect, sampling should not be restricted to babies and mothers. Samples from family members and other proximate individuals, including pets, can also carry valuable information. Specific questions around “who” could also be addressed through study designs encompassing the diversity of familial relationships: studies of microbial acquisition in children born by surrogacy or children breastfed by parents who did not give birth to them may provide insight into differences in microbial source and transmission timing. “Who” should also include health care providers, especially in the newborn nursery and neonatal intensive care units, in such critical early life periods.
Lastly, addressing the “when” aspect necessitates the longitudinal collection of samples of both the infant and other individuals; however, the frequency of sampling will depend on the life stage. Compared to a rather stable microbiome of a healthy adult, the infant microbiome experiences dynamic changes during its establishment. In contrast, the mother’s microbiome gradually changes during pregnancy and returns to a pre-pregnancy state after delivery44,45. Therefore, although challenging, samples should be collected at least once from the mother before and after birth, from all individuals involved in birth, and more frequently from the infant in the first weeks of life. Such a sampling strategy will offer the most detailed insights into the establishment of the infant microbial ecosystem. After this initial stage, monthly sampling from the infant, family members, and environment would be ideal for capturing the different sources contributing to the microbiome development. The frequency of sampling should also increase during life events that alter the microbiome composition, such as the introduction of new food and weaning in general, vaccination, or medical treatment. Finally, while some reports suggest that the infant gut microbiome starts to resemble an adult one after the age of two to three years, others show that a child’s microbiome does not reach an adult-like state until later46. These discrepancies underline the need for more frequent and longer follow-ups on the child’s microbiome development.




Practical aspects of 4 W study design
In practical terms, conducting large cohorts with extensive metadata collection, comprehensively sampling infants and their household members, plus the environment in a longitudinal design, and finally, sample characterization via multi-omics, is currently unrealistic. This issue is challenging for cohort studies in general, but is pronounced for early-life microbiome studies due to the dynamic nature of microbial communities, which are continuously adapting to the fast-developing human physiology and display a large inter-individual variability. In addition, the specifics of the country where the research takes place will have a major influence on the study design, as one must account for differences in ethical norms, jurisdiction, and geographical proximity/ remoteness that affect logistics. Because of the economic, logistic, and other practical challenges, here we present examples of how researchers have been employing strategies to describe, at least partly, the 4 W parameters (Table 1) and a theoretical approach to study design along a 4 W framework (Box 3).
Firstly, one can design nested zoom-in studies within cohorts that have a large number of participants and employ extensive longitudinal sampling47. Depending on the research question, one may apply a cross-sectional, nested case-control, or matched cohort design within a larger population study, using samples from selected timepoints (defining “when”), sources (“who” and “where”), and focusing on particular “what” (e.g., type of microorganisms or their products). From an epidemiological perspective, there will be a difference if the research aim is to study how early life microbial acquisition and transmission are affected by a rare exposure (e.g., formula contamination) or a rare outcome (e.g., necrotizing enterocolitis in premature infants). In the first case, large cohorts are necessary, yet one can opt to characterize only those with the exposure and several controls (matched cohort design). In the case of a rare outcome, one can choose to select and characterize all samples of those with the outcome, matched to one or more controls without the outcome (nested case-control design). The advantages of both approaches compared to characterizing all collected samples include lower cost and comparator groups that are more equal in size, facilitating more statistically robust bioinformatics analyses, as some analytic measures do not perform well if group sizes are unbalanced. Finally, clinical intervention studies, ideally designed as randomized control trials (RCT), can confirm associations between microbial transmission and selected early life factors, as shown in a study using maternal fecal microbiota transplantation to restore the intestinal microbiota of Cesarean section-born infants48. However, RCTs performed in the infant population require extensive ethical approvals compared to adults, which significantly extends the project planning period. Current examples of large population studies that have investigated specific questions related to early life microbial transmission include the Canadian CHILD, Swedish SweMaMi, Danish COPSAC, Dutch Lifelines-NEXT, and Finnish FinnBrain and HELMi studies. Many of these studies are still ongoing and including more 4 W aspects into their designs would help obtain novel insight into early-life microbial transmission and assembly. For empirical evidence, see Table 1 with several case studies that illustrate how the 4 W framework can guide research on human microbiome acquisition and development, when used to identify which of the components of microbial transmission have been closely investigated and which remain largely uncharacterized.
Secondly, another study design is to focus only on some of the 4 W parameters, but study them in greater depth. For instance, the BabyBiome project from Oslo sampled 12 infants daily throughout the first year of life, providing high resolution into the dynamics of gut bacterial community over time and describing a strong temporal structure and specific developmental stages of the community maturation49. More of such studies with densely sampled mother-infant pairs would give insight into the “when” of microbial transfer and the effect of the transferred microbes on the colonization dynamics of the infant microbiome. However, this is not feasible for large cohorts. Examples of other focused cohorts include the Diabimmune and DIPP studies50, designed to pinpoint specific microbial profiles in individuals predisposed to immune diseases, and the MicrobeMom study, which studies the transmission of specific bacteria from mother to infant51.
Lastly, robust findings on early life microbiome can and will be derived from joint analysis of results from various studies with differing designs52,53,54. This is indeed a good example of the way forward, which lies in collaboration, allowing researchers to connect resources (e.g., economic, know-how, personnel) and combined datasets to tackle research questions. To this end, ensuring the transparent sharing of metagenomic data and metadata is crucial55.
“Why” is this microbiome different from all other microbiomes
Defining the 4Ws of microbiome transmission is crucial to understanding microbiome acquisition in early life. Still, defining ‘what’, ‘when’, from ‘who’ and ‘where’ microbes are transmitted does not allow for full comprehension of the acquisition process and aspects of microbial ecology outside of early-life acquisition, such as primary (assembly) and secondary (after perturbation) succession and colonization over time. Indeed, a fifth question on ‘why’ is fundamental to understanding which members of the microbiome are acquired and successfully colonize specific habitats in the infant (and beyond). Ultimately, our understanding of ‘why’ requires ecological and evolutionary frameworks, and for which we can turn to the four tenets of community ecology and assembly: stochastic processes (dispersal, drift, diversification) and selection (i.e., the niche56,57). To date, most studies have focused on the latter, defining the roles of selection on microbial acquisition and colonization in early life. Many of these selective factors are intimately related to the 4Ws. For example, dietary factors such as milk-derived oligosaccharides, which may selectively feed transmitted microbes in the gut, or the developmental expression of host secreted innate (antimicrobial peptides) and adaptive (antibodies in milk or produced in the gut over time) factors specific for distinct microbes, are examples whereby ‘when’ combine with ‘why’ to determine transmission. Microbe-microbe interactions, spanning negative (e.g., antibiotics) and positive (e.g., cross-feeding), provide examples where two distinct ‘whats’ (i.e., microbial strains) act as selective factors for (co-) acquisition.
While selective (or deterministic) pressures define the microbial niche and potential for success of acquisition, stochastic (or probabilistic) processes, i.e., neutral processes, are likely to play instrumental roles in early-life microbiome transmission and assembly. At present, however, as the default or null hypothesis of transmission, dispersal (random movement of microbes across space), drift (random changes in fitness of microbial populations), and diversification (genomic changes), all defined as ‘random’, are difficult to assign a weight in early-life microbial transmission.
How can the 4 W framework offer new insights into defining stochastic versus selective aspects of microbial ecology, assembly, and succession? Indeed, combining the power of population genetic approaches with microbial source tracking will allow us to test the null hypothesis and the roles of sources and sinks by modeling neutral processes shaping the microbiota acquisition58. Operationally, a broad sampling and quantification across the domains of life of the ‘who’ and ‘where’ will allow definition of the ‘sources’ and ‘sink’. Here also lies a research potential in the use and integration of strain-tagging and controlled transmission experiments31,59.
For example, source tracking has been used to identify and categorize transmission events60. The algorithms assume a somewhat unidirectional transmission, where the infant community is the ‘sink’ and it acquires microbiomes for a set of ‘source’ reference communities. The algorithm can then either predict similarity to a source state or predict the portion of the microbial community contributed by each source61. Modeling of neutral processes during microbiota assembly can reveal the contribution of dispersal and demographic stochasticity, which has been found to explain the prevalence of a majority of infant-colonizing microbes58. With comprehensive sampling across the 4 W and population genetic modeling approaches, we can define if the transmission of a microbe obeys the features of a stochastic process. When this null hypothesis is not met, we can begin to define the contribution of selection (and then what features of microbes are selected) in the assembly and definition of the microbial niche of transmission and successful colonization. For example, by quantifying absolute abundances of a specific microbe (‘what’) in a source (e.g., vagina at delivery) and in infant feces (‘who’ and ‘where’) at birth and over time (‘when’), we could determine if such transmission was related to the microbial abundance in that particular source (stochastic) versus the microbial properties, which facilitate successful acquisition and colonization (selection).
Such a 4 W framework can be readily applied to aspects of microbial acquisition outside of the early-life period, such as repopulation of the gut microbiome after antibiotic perturbation. By capturing the 4 W of potential ‘who’s’ (the person taking the antibiotics and their contacts), ‘where’s’ (the sources of the microbes to repopulate the gut, i.e., their own oral microbiome or the gut microbiome of their household partner), ‘when’ (the timing and relation to the abundance and composition of the focal person’s gut microbiome before, during and after use of antibiotics), and ‘what’ (strain-level tracking via metagenomics or whole-genome sequencing of cultivated isolates), we can use the 4 W framework to capture the parameters needed to test the null hypothesis and by extension define role of the ecological niche in transmission. Ultimately, a combination of both deterministic and probabilistic modeling will allow a quantitative and predictive understanding of the ‘why’ of early-life microbiome transmission.
Conclusions
The current lexicon of early-life microbiome acquisition originating from the fields of infectious disease epidemiology and evolutionary biology, and namely vertical and horizontal transmission, often shows limited resolution. To achieve a deep ecological understanding of the early life microbial dynamics, we propose that efforts should be centered on deciphering the what-where-who-when aspects of microbial acquisition. By transitioning from the vertical/ horizontal transmission language toward using precise terminology of the 4 W framework, researchers can systematically examine different components of microbiome acquisition. Currently, metagenomics is the workhorse for describing microbial transmission. But also wider adoption of other methods for tracking microbial cells and their components, such as single cell-based microbial genomics, metabolomics, metaproteomics, and high-throughput culturomics, has the potential to significantly contribute to these goals, when affordable and accessible to non-specialists.
Importantly, the interpretation of transmission events carries a large degree of uncertainty and necessitates considering alternative microbial sources. A broader implementation of computational approaches, which can resolve microbial patterns and minimize degrees of freedom in interpreting transmission events, is thus a prerequisite for the study of human microbiome acquisition. Defining which aspects of the 4Ws and how we can feasibly capture these in studies of the early-life human microbiome, and human microbiome at-large, will be instrumental to future-proofing and comprehensively understanding the dynamics and significance of early-life microbial colonization and when, where, and how we should and can intervene for human health.
References
Gensollen, T., Iyer, S. S., Kasper, D. L. & Blumberg, R. S. How colonization by microbiota in early life shapes the immune system. Science 352, 539–544 (2016).
Peppas, I., Ford, A. M., Furness, C. L. & Greaves, M. F. Gut microbiome immaturity and childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 23, 565–576 (2023).
Koch, R. Vorläufige Mitteilungen über die Ergebnisse einer Forschungsreise nach Ostafrika. Dtsch Med Wochenschr 31, 1865–1869 (1905).
Berg, G. et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome 8, 103 (2020).
Shope, R. E. THE SWINE LUNGWORM AS A RESERVOIR AND INTERMEDIATE HOST FOR SWINE INFLUENZA VIRUS: II. THE TRANSMISSION OF SWINE INFLUENZA VIRUS BY THE SWINE LUNGWORM. J. Exp. Med. 74, 49–68 (1941).
Smith, T. & Kilborne, F. L. Investigations into the Nature, Causation, and Prevention of Texas or Southern Cattle Fever. (U.S. Dept. of Agriculture, Bureau of Animal Industry, Washington, D.C., 1893).
Cordy, D. R. & Gorham, J. R. The pathology and etiology of salmon disease. Am. J. Pathol. 26, 617–637 (1950).
Dutton, J. E. & Todd, J. L. The Nature of Human Tick Fever in the Eastern Part of the Congo Free State. (1905).
Montgomery, E. On a Tickborne Gastroenteritis of sheep and goats occurring in British East Africa. J. Comp. Pathol. Ther. 30, 28–57 (1917).
Gross, L. The vertical epidemic of mammary carcinoma in mice; its possible implications for the problem of cancer in general. Surg. Gynecol. Obstet. 88, 295–308 (1949).
Gross, L. The “vertical” transmission of mouse mammary carcinoma and chicken leukemia; its possible implications for human pathology. Cancer 4, 626–633 (1951).
Bevilacqua, G. The Viral Origin of Human Breast Cancer: From the Mouse Mammary Tumor Virus (MMTV) to the Human Betaretrovirus (HBRV). Viruses 14, (2022).
Burmester, B. R. The vertical and horizontal transmission of avian visceral lymphomatosis. Cold Spring Harb. Symp. Quant. Biol. 27, 471–477 (1962).
Mims, C. A. Immunofluorescence study of the carrier state and mechanism of vertical transmission in lymphocytic choriomeningitis virus infection in mice. J. Pathol. Bacteriol. 91, 395–402 (1966).
Andrewes, C. H. Factors in Virus Evolution. in Advances in Virus Research (eds. Smith, K. M. & Lauffer, M. A.) vol. 4 1–24 (Academic Press, 1957).
Hadfield, S. J. & Axton, J. M. Germ cells colonized by endosymbiotic bacteria. Nature 402, 482 (1999).
Ebert, D. The Epidemiology and Evolution of Symbionts with Mixed-Mode Transmission. Annu. Rev. Ecol. Evol. Syst. 44, 623–643 (2013).
Hosokawa, T., Kikuchi, Y., Nikoh, N., Shimada, M. & Fukatsu, T. Strict host-symbiont cospeciation and reductive genome evolution in insect gut bacteria. PLoS Biol. 4, e337 (2006).
Moran, N. A. & Sloan, D. B. The Hologenome Concept: Helpful or Hollow?. PLoS Biol. 13, e1002311 (2015).
de Goffau, M. C. et al. Human placenta has no microbiome but can contain potential pathogens. Nature 572, 329–334 (2019).
Kennedy, K. M. et al. Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 613, 639–649 (2023).
Rao, C. et al. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 591, 633–638 (2021).
Kaisanlahti, A. et al. Maternal microbiota communicates with the fetus through microbiota-derived extracellular vesicles. Microbiome 11, 249 (2023).
Kim, S. Y. & Yi, D. Y. Analysis of the human breast milk microbiome and bacterial extracellular vesicles in healthy mothers. Exp. Mol. Med. 52, 1288–1297 (2020).
Pessa-Morikawa, T. et al. Maternal microbiota-derived metabolic profile in fetal murine intestine, brain and placenta. BMC Microbiol 22, 46 (2022).
Parfieniuk, E. et al. Amniotic fluid metabolic fingerprinting indicated metabolites which may play a role in the pathogenesis of foetal Down syndrome - a preliminary report. Ginekol. Pol. 92, 188–194 (2021).
Li, Y. et al. In utero human intestine harbors unique metabolome, including bacterial metabolites. JCI Insight 5, (2020).
Stinson, L. F. et al. Human Milk From Atopic Mothers Has Lower Levels of Short Chain Fatty Acids. Front. Immunol. 11, 1427 (2020).
Wang, Z., Tao, X., Liu, S., Zhao, Y. & Yang, X. An Update Review on Listeria Infection in Pregnancy. Infect. Drug Resist. 14, 1967–1978 (2021).
Luca, C. D. et al. Listeria infection in pregnancy: A review of literature. Open Infect. Dis. J. 9, 20–25 (2015).
Zhong, Z. et al. Bifidobacterium animalis subsp. lactis Probio-M8 undergoes host adaptive evolution by glcU mutation and translocates to the infant’s gut via oral-/entero-mammary routes through lactation. Microbiome 10, 197 (2022).
Russo, M. et al. Influence of maternal oral microbiome on newborn oral microbiome in healthy pregnancies. Ital. J. Pediatr. 49, 140 (2023).
Laforest-Lapointe, I. & Arrieta, M.-C. Patterns of Early-Life Gut Microbial Colonization during Human Immune Development: An Ecological Perspective. Front. Immunol. 8, 788 (2017).
Aversa, Z. et al. Association of Infant Antibiotic Exposure With Childhood Health Outcomes. Mayo Clin. Proc. 96, 66–77 (2021).
Madrid, L., Varo, R., Sitoe, A. & Bassat, Q. Congenital and perinatally-acquired infections in resource-constrained settings. Expert Rev. Anti. Infect. Ther. 14, 845–861 (2016).
Valles-Colomer, M. et al. The person-to-person transmission landscape of the gut and oral microbiomes. Nature (2023) https://doi.org/10.1038/s41586-022-05620-1.
Pettersen, V. K., Antunes, L. C. M., Dufour, A. & Arrieta, M.-C. Inferring early-life host and microbiome functions by mass spectrometry-based metaproteomics and metabolomics. Comput. Struct. Biotechnol. J. 20, 274–286 (2022).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017).
Tkacz, A., Hortala, M. & Poole, P. S. Absolute quantitation of microbiota abundance in environmental samples. Microbiome 6, 110 (2018).
Maghini, D. G. et al. Quantifying bias introduced by sample collection in relative and absolute microbiome measurements. Nat. Biotechnol. 42, 328–338 (2024).
Roach, J. et al. Microbiome metabolite quantification methods enabling insights into human health and disease. Methods 222, 81–99 (2024).
Busi, S. B. et al. Persistence of birth mode-dependent effects on gut microbiome composition, immune system stimulation and antimicrobial resistance during the first year of life. ISME Commun. 1, 8 (2021).
van den Elsen, L. W. J., Garssen, J., Burcelin, R. & Verhasselt, V. Shaping the Gut Microbiota by Breastfeeding: The Gateway to Allergy Prevention?. Front Pediatr. 7, 47 (2019).
Koren, O. et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480 (2012).
DiGiulio, D. B. et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl Acad. Sci. Usa. 112, 11060–11065 (2015).
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765–776.e3 (2021).
Wijmenga, C. & Zhernakova, A. The importance of cohort studies in the post-GWAS era. Nat. Genet. 50, 322–328 (2018).
Korpela, K. et al. Maternal Fecal Microbiota Transplantation in Cesarean-Born Infants Rapidly Restores Normal Gut Microbial Development: A Proof-of-Concept Study. Cell 183, 324–334.e5 (2020).
de Muinck, E. J. & Trosvik, P. Individuality and convergence of the infant gut microbiota during the first year of life. Nat Commun. 9, 2233 (2018).
Lamichhane, S. et al. Dynamics of Plasma Lipidome in Progression to Islet Autoimmunity and Type 1 Diabetes - Type 1 Diabetes Prediction and Prevention Study (DIPP). Sci. Rep. 8, 10635 (2018).
Feehily, C. et al. Detailed mapping of Bifidobacterium strain transmission from mother to infant via a dual culture-based and metagenomic approach. Nat. Commun. 14, 3015 (2023).
Podlesny, D. & Fricke, W. F. Strain inheritance and neonatal gut microbiota development: A meta-analysis. Int. J. Med. Microbiol. 311, 151483 (2021).
Mancabelli, L. et al. Multi-population cohort meta-analysis of human intestinal microbiota in early life reveals the existence of infant community state types (ICSTs). Comput. Struct. Biotechnol. J. 18, 2480–2493 (2020).
Ho, N. T. et al. Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat. Commun. 9, 4169 (2018).
Mirzayi, C. et al. Reporting guidelines for human microbiome research: the STORMS checklist. Nat. Med. 27, 1885–1892 (2021).
Kuziel, G. A. & Rakoff-Nahoum, S. The gut microbiome. Curr. Biol. 32, R257–R264 (2022).
Nemergut, D. R. et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 77, 342–356 (2013).
Sprockett, D. D. et al. Microbiota assembly, structure, and dynamics among Tsimane horticulturalists of the Bolivian Amazon. Nat. Commun. 11, 3772 (2020).
Moore, R. L. et al. Ability of Bifidobacterium breve 702258 to transfer from mother to infant: the MicrobeMom randomized controlled trial. Am. J. Obstet. Gynecol. MFM 5, 100994 (2023).
Song, S. J. et al. Naturalization of the microbiota developmental trajectory of Cesarean-born neonates after vaginal seeding. Med 2, 951–964.e5 (2021).
Shenhav, L. et al. FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632 (2019).
Ferretti, P. et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 24, 133–145.e5 (2018).
Manara, S. et al. Maternal and food microbial sources shape the infant microbiome of a rural Ethiopian population. Curr. Biol. 33, 1939–1950.e4 (2023).
Mitchell, C. M. et al. Delivery Mode Affects Stability of Early Infant Gut Microbiota. Cell Rep. Med 1, 100156 (2020).
Garmaeva, S. et al. Transmission and dynamics of mother-infant gut viruses during pregnancy and early life. Nat. Commun. 15, 1945 (2024).
Dubois, L. et al. Paternal and induced gut microbiota seeding complement mother-to-infant transmission. Cell Host Microbe 32, 1011–1024.e4 (2024).
Selma-Royo, M. et al. Birthmode and environment-dependent microbiota transmission dynamics are complemented by breastfeeding during the first year. Cell Host Microbe 32, 996–1010.e4 (2024).
Schei, K. et al. Early gut mycobiota and mother-offspring transfer. Microbiome 5, 107 (2017).
Vatanen, T. et al. Mobile genetic elements from the maternal microbiome shape infant gut microbial assembly and metabolism. Cell 185, 4921–4936.e15 (2022).
Bright, M. & Bulgheresi, S. A complex journey: transmission of microbial symbionts. Nat. Rev. Microbiol. 8, 218–230 (2010).
Yan, Y., Nguyen, L. H., Franzosa, E. A. & Huttenhower, C. Strain-level epidemiology of microbial communities and the human microbiome. Genome medicine 12, (2020).
Van Rossum, T., Ferretti, P., Maistrenko, O. M. & Bork, P. Diversity within species: interpreting strains in microbiomes. Nat. Rev. Microbiol. 18, 491–506 (2020).
Ghazi, A. R., Münch, P. C., Chen, D., Jensen, J. & Huttenhower, C. Strain Identification and Quantitative Analysis in Microbial Communities. J. Mol. Biol. 434, 167582 (2022).
Altermann, E., Tegetmeyer, H. E. & Chanyi, R. M. The evolution of bacterial genome assemblies - where do we need to go next?. Microbiome Res Rep. 1, 15 (2022).
Anyansi, C., Straub, T. J., Manson, A. L., Earl, A. M. & Abeel, T. Computational Methods for Strain-Level Microbial Detection in Colony and Metagenome Sequencing Data. Front. Microbiol. 11, 1925 (2020).
Garud, N. R. & Pollard, K. S. Population Genetics in the Human Microbiome. Trends Genet 36, 53–67 (2020).
Tawk, C. et al. Infection leaves a genetic and functional mark on the gut population of a commensal bacterium. Cell Host Microbe 31, 811–826.e6 (2023).
Acknowledgements
This project was possible through a grant awarded to V.K.P. by the Center for Advanced Study (CAS) in Oslo, Norway, which funded and hosted the Young CAS research project (Infant Gut Microbiome Acquisition: Off to a Healthy Start) during the 2022/23 academic year. CAS is an independent research foundation funded by the Norwegian Ministry of Education and Research. We also acknowledge and appreciate our partners and children who enabled and enlivened our discussions during the summer of 2023 in Tromsø, Norway. S.R.-N. acknowledges support from the Mathers Foundation, a Career Award for Medical Scientists from the Burroughs Wellcome Fund, a Pew Biomedical Scholarship, and National Institutes of Health (R01DK138023, R01AI158814, R01AI171100, R01AI126915). M.V.-C. acknowledges funding from the Spanish Ministry of Science and Innovation (Knowledge Generation Project PID2022-139328OA-I00), the Spanish Ministry of Universities (Beatriz Galindo Junior fellowship BG22/00172), and from La Caixa Foundation (Junior Leader fellowship 100010434, code LCF/BQ/PI24/12040001). M.E.T. acknowledges funding from the European Union’s Horizon 2020 (MSCA-IF postdoctoral grant MicroMi-898088), and Ramón y Cajal grant RYC2023-045839-I, Ministry of Science, Innovation and Universities, the State Research Agency MCIU/AEI/10.13039/501100011033 and FSE+. The Institute of Agrochemistry and Food Technology (IATA-CSIC) is a Center of Excellence Severo Ochoa (CEX2021- 001189-S MCIN/AEI/10.13039/501100011033). A.Z. is supported by the Netherlands Organization for Scientific Research NWO-VICI grant VI.C.232.074, the NWO Gravitation grant Exposome-NL(024.004.017), the NWO KIC grant KICH1.LWV04.21.01, the ZonMW ME/CFS grant 10091012110015, EU Horizon Europe Program grant INITIALIZE (101094099), and EU Horizon Europe Program grant DarkMatter (ID-DarkMatter-NCD, 101136582).
Author information
Authors and Affiliations
Contributions
This perspective was conceived during a research sabbatical, with contributions from S.R.N., J.D., M.V.C., H.T.N., M.E.T., A.Z., N.B., and V.K.P. All of these authors contributed to the writing, editing, and critical review of the manuscript. MVC created Fig. 2, while JD designed Figs. 1, 3, and 4. J.D. finalized all figures for publication. S.R.N. used his institution’s professional BioRender account (Boston Children’s Hospital) to illustrate the 4 W study designs in Box 3. The outlines for the pregnant person and fetus used in Figs. 1, 3b were obtained from BioArt, an NIH-funded public repository accessible to J.D. The bacteria’s shapes and colors were created by J.D. V.K.P. secured the funding from the Center for Advanced Study (CAS).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rakoff-Nahoum, S., Debelius, J., Valles-Colomer, M. et al. A reconceptualized framework for human microbiome transmission in early life. Nat Commun 16, 7546 (2025). https://doi.org/10.1038/s41467-025-61998-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-61998-2
