
Abstract
HCoV-HKU1, one of seven human coronaviruses (HCoVs) that have harmful effects on human health, accounts for a substantial portion of common cold cases and can cause severe respiratory diseases in certain populations. Currently, effective antiviral treatments against this virus are limited. Recently, TMPRSS2, a host protease long acknowledged for its role in priming the spike proteins of various CoVs and promoting viral entry, was identified as a functional receptor for HCoV-HKU1, opening an avenue for anti-HCoV-HKU1 therapy development. In this study, we elucidate the detailed molecular mechanism underlying the interaction between the HCoV-HKU1 receptor-binding domain (RBD) and TMPRSS2 via crystallography. Guided by these structural insights, we successfully develop two types of therapeutic antibodies against HCoV-HKU1. The first type neutralizes the RBD, potently disrupting its interaction with TMPRSS2 and preventing viral infection. The second type targets TMPRSS2, inhibiting its enzymatic activity and/or interfering with its binding to the RBD. The latter demonstrates broad-spectrum anti-CoV activity, as the enzymatic activity of TMPRSS2 is crucial for both HCoV-HKU1 infection and other CoV infections. Our findings provide crucial structural insights into the recognition of TMPRSS2 by HCoV-HKU1 and offer promising antibody-based strategies for combating HCoV-HKU1 and other CoV infections.
Similar content being viewed by others


Introduction
Coronaviruses (CoVs) are important etiological agents that pose great threats to global health. To date, seven human coronaviruses (HCoVs) have been identified, including four endemic seasonal CoVs (HCoV-229E, -OC43, -NL63, and -HKU1) that collectively account for 15–30% of common cold cases1 and three highly virulent CoVs (SARS-CoV, MERS-CoV, and SARS-CoV-2) that have caused devastating pandemics. Despite the availability of several treatments for SARS-CoV-2, effective therapies for other HCoVs remain limited. In antiviral therapeutic development, antibodies that block viral entry, which offer faster immune protection than vaccines, have emerged as highly effective treatment options2.
A recent study identified host transmembrane serine protease 2 (TMPRSS2) as the functional receptor for HCoV-HKU13, revealing potential possibilities for the development of therapeutics against HCoV-HKU1. TMPRSS2, a trypsin-like protease, has long been implicated in various diseases, ranging from cancer to viral infections4,5,6. Notably, previous animal studies have suggested that the function of TMPRSS2 might be redundant and potentially compensated by other proteases7, making it an attractive drug target with potentially minimal side effects. Given the role of TMPRSS2 as the receptor for the spike protein of HCoV-HKU1, there is a pressing need for structural studies to elucidate the precise molecular mechanisms underlying this interaction. Such insights would be invaluable for the development of antiviral therapeutics targeting this specific interaction, potentially offering a viable strategy against HCoV-HKU1 and possibly other TMPRSS2-associated diseases.
In this study, we determined the crystal structure of the HCoV-HKU1 spike receptor-binding domain (RBD) in complex with TMPRSS2 at a resolution of 2.75 Å. This structure demonstrated the molecular basis of their interaction, revealing two important epitopes for therapeutic antibody development: the interacting interface between the RBD and TMPRSS2 and the substrate-binding pocket of TMPRSS2. Guided by these structural insights, we successfully identified a neutralizing antibody (R7) targeting the interaction interface of the HCoV-HKU1 RBD that effectively disrupted its interaction with TMPRSS2. Furthermore, we explored two additional antibodies targeting the substrate-binding pocket of TMPRSS2. The first one (T22) abolished the catalytic activity of TMPRSS2, which is crucial for HCoV-HKU1 infection through priming its spike protein8 and plays a role in other TMPRSS2-related diseases, including various CoV infections. Notably, the second antibody (VHH77) had a distinct dual function of simultaneously targeting both the interacting interface and the substrate-binding pocket of TMPRSS2. This dual function enabled the antibody to both inhibit the enzymatic activity of TMPRSS2 and disrupt its interaction with the HCoV-HKU1 RBD, potentially offering a more comprehensive approach to blocking TMPRSS2-mediated viral entry. We further optimized this promising dual-function antibody through affinity maturation and elucidated its molecular mechanism via crystallography, providing a robust foundation for the development of antibody-based therapeutic strategies.
In conclusion, our findings provide crucial insights for the development of anti-HCoV-HKU1 antibodies and potential broad-spectrum antivirals that target TMPRSS2. These discoveries not only advance therapeutic strategies for HCoV-HKU1 but also offer promising approaches for combating other TMPRSS2-dependent infectious diseases.
Results
Molecular mechanism through which the HCoV-HKU1 RBD recognizes TMPRSS2
First, we confirmed the interaction between the HCoV-HKU1 RBD and TMPRSS2 via biolayer interferometry (BLI), and the binding affinity (KD) of TMPRSS2 to the HCoV-HKU1 RBD was determined to be 8.70 ± 0.08 nM (Supplementary Fig. 1a). Interestingly, we also discovered that the TMPRSS2 mutant harboring an S441A mutation, which abolishes its enzymatic activity, displays a comparable binding affinity to the HCoV-HKU1 RBD, with a measured binding affinity of 8.36 ± 0.08 nM (Supplementary Fig. 1b). This finding suggests that the interaction between the HCoV-HKU1 RBD and TMPRSS2 is independent of the catalytic activity of TMPRSS2. To further reveal the precise mechanism by which the HCoV-HKU1 RBD recognizes TMPRSS2, we solved the high-resolution crystal structure of the HCoV-HKU1 RBD in complex with the TMPRSS2 S441A mutant.
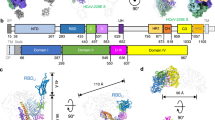
As illustrated in Fig. 1a, the HCoV-HKU1 RBD features a “U”-shaped receptor-binding motif (RBM, spanning residues 504–530) that interacts with the serine protease (SP) domain of TMPRSS2. Nearly all the RBD residues involved in the intermolecular interaction with TMPRSS2 are located within this RBM. Notably, the RBM does not engage the catalytic center located within the SP domain, which is consistent with our previous BLI experiments. The interface between the RBM and TMPRSS2 has a buried surface area of approximately 770 Å2, which can be further subdivided into a central hydrophilic region flanked by two hydrophobic regions (Fig. 1b, c).

a Crystal structure of the HCoV-HKU1 RBD in complex with the TMPRSS2 S441A mutant. The RBD (residues 323–609) is shown in wheat, with its RBM (residues 504–530) highlighted in salmon. TMPRSS2 is depicted with its SP domain (residues 256–492) in mint, while its low-density lipoprotein receptor type-A (LDLRA) and scavenger receptor cysteine-rich (SRCR) domains (residues 112–249) are colored slate. The Cα atom of residue 441 in TMPRSS2 is represented by a hot pink sphere. b, c Open-book view of the interaction interface between the HCoV-HKU1 RBD and TMPRSS2. The surfaces are colored according to hydrophobicity, with a spectrum ranging from dark cyan (most hydrophilic) through white to dark goldenrod (most lipophilic). d–f Detailed views of the three interaction regions between the HCoV-HKU1 RBD and TMPRSS2. Key residues involved in the RBD:TMPRSS2 interaction are depicted as sticks, with hydrogen bonds illustrated by dashed lines.
The central hydrophilic region is formed by hydrogen bonds and ionic interactions (Fig. 1d). This interface region features two central arginine residues, R470TMPRSS2 and R517RBD, which are oriented antiparallel to each other. R470TMPRSS2 forms ionic interactions with the side chain carboxyl group of E505RBD, whereas R517RBD forms hydrogen bonds with the main chain atoms of Y414TMPRSS2 and V415TMPRSS2. Furthermore, the hydrogen bond formed between S463TMPRSS2 and T508RBD, as well as the hydrogen bond between D417TMPRSS2 and H488RBD, also contributes to the stability of this region.
In hydrophobic region I (Fig. 1e), residues Y414TMPRSS2, L430TMPRSS2, and Y469TMPRSS2 form a hydrophobic socket on TMPRSS2 that perfectly accommodates the hydrophobic residues L521RBD, P522RBD, and Y528RBD on the RBM. In addition, this region is further stabilized by hydrogen bonds formed between the phenolic hydroxyl group of Y469TMPRSS2 and the main chains of L521RBD. Similarly, in hydrophobic region II, the aliphatic or aromatic residues V509RBD, L510RBD, H512RBD, and W515RBD neatly reside within a hydrophobic pocket on TMPRSS2 formed by Y416TMPRSS2, L419TMPRSS2, W461TMPRSS2, and the alkyl portions of T341TMPRSS2 and K342TMPRSS2 (Fig. 1f).
In addition, during the preparation of this manuscript, several papers reporting similar structures were published9,10,11. When these recently published structures were superimposed with our high-resolution structure, the root mean square deviation (RMSD) values were consistently less than 1.0 Å. This close alignment not only validates the accuracy of our structural determination but also corroborates the findings of these other studies.
The R7 antibody targeting the HCoV-HKU1 RBD blocks the binding of the HCoV-HKU1 RBD to TMPRSS2
Since HCoV-HKU1 binds to host TMPRSS2 through its RBD domain, which enabling viral attachment and entry, neutralizing antibodies that disrupt the RBD:TMPRSS2 interaction could block viral entry, offering a strategy for the prevention or treatment of HCoV-HKU1 infection. With this rationale, we focused our efforts on developing antibodies targeting the RBD:TMPRSS2 interface to interrupt this critical interaction.
By screening a human naïve single-chain variable fragment (scFv) phage display antibody library using the HCoV-HKU1 RBD and excluding redundant sequences, 17 antibodies were identified and successfully converted into the scFv-Fc format by fusing the human Fc to the C-terminus of the scFv (Fig. 2a). We then characterized the ability of these 17 scFv-Fcs to block the RBD:TMPRSS2 interaction via BLI-based competition analysis (Supplementary Fig. 2a, b). Through this analysis, we successfully identified several antibodies capable of effectively disrupting the RBD:TMPRSS2 interaction.

a Flowcharts of the human naïve scFv phage library panning and antibody selection process. The number within parentheses indicates the quantity of phages and scFv-Fc antibodies that were tested. b Binding of R7 to the HCoV-HKU1 RBD, as measured via BLI. Biotinylated HCoV-HKU1 RBDs were immobilized on SA biosensors and then exposed to twofold dilutions of R7. The thick red curves represent the raw data, and the thin black curves are the corresponding fitted curves. c Effect of R7 on HCoV-HKU1-mediated cell‒cell fusion. HEK293T GFP-split cells were transfected with HCoV-HKU1 spike and TMPRSS2 for 20 h. Fusion was quantified 24 h post-coculture in the presence of different concentrations of R7. The data were normalized to the nontreated conditions. The data points represent the means ± SDs from biological triplicates (n = 3). d Overall structure of the HCoV-HKU1 RBD:R7 Fab complex. The RBD is depicted in wheat, with its RBM highlighted in salmon. The R7 heavy chain is shown in dark gray, with its CDRs in orange. The R7 light chain is displayed in light gray, with its CDRs in marine blue. e–h Detailed views of the interactions between R7 and the RBD. Panel (e) highlights the central interacting region, whereas panels (f–h) focus on additional interface areas. Panel (a) was created in BioRender. Duan, Y. (2025) https://BioRender.com/ie8n6b3.
Among these, R7 demonstrated exceptional blocking efficacy and strong binding affinity to the RBD (KD = 0.65 ± 0.01 nM) (Fig. 2b and Supplementary Fig. 2c, e). A subsequent cell-based assay further validated the ability of R7 to abrogate the RBD:TMPRSS2 interaction (Fig. 2c). To elucidate the detailed molecular mechanism underlying this inhibition, we determined the crystal structure of R7 in complex with the HCoV-HKU1 RBD at a resolution of 1.80 Å.
As shown in Fig. 2d, the R7 epitope is located primarily on the RBM of the HCoV-HKU1 RBD, preventing the binding of host TMPRSS2 to this “U”-shaped motif in the RBD. The interface between R7 and the RBD has a buried surface area of approximately 1100 Å2 and encompasses complementarity-determining region (CDR) H2 and CDRH3 on the heavy chain, as well as CDRL1 and CDRL3 on the light chain. Apart from CDRs, heavy chain framework region (FR) H2 and FRH3 also contribute significantly to the intermolecular interactions, covering approximately 550 Å2 of the buried surface area.
The interface between R7 and the RBM on the RBD is characterized by a central interacting region, where a distinctive groove formed between the heavy and light chains of the antibody is highly complementary to the protruding Y528RBD. This structural arrangement allows for precise accommodation of the Y528RBD residue, forming a key anchor point in the antibody‒antigen interaction. This structural arrangement facilitates extensive interactions between R7 and the RBD, collectively contributing to the high affinity and specificity of R7 for the HCoV-HKU1 RBD (Fig. 2e). Specifically, Y528RBD engages in a network of interactions: its benzyl ring forms hydrophobic or π–π stacking interactions with W34, H59, and Y109 on the antibody’s heavy chain while simultaneously participating in a cation‒π interaction with R96 on the light chain. In addition, the side chain hydroxyl group of Y528RBD forms hydrogen bonds with S51 and E99 on the heavy chain, and its main chain carboxyl group forms hydrogen bonds with R96 on the light chain.
Beyond this central interacting region, RBD:R7 binding is further reinforced by multiple hydrogen bonds, which form between the RBM and the CDRH2, CDRH3, and FRH3 regions of the antibody (Fig. 2e, f), as well as between residues adjacent to the RBM and the CDRL1, CDRL3, and CDRH3 regions of the antibody (Fig. 2g, h). This extensive network of interactions significantly enhances the binding affinity of R7 to the HCoV-HKU1 RBD. Importantly, R7 directly occupies the RBM, including key residues such as R517RBD, Y528RBD, and D529RBD, which were shown to be critical for the RBD:TMPRSS2 interaction. By engaging these specific residues within the RBM—a region essential for viral attachment to TMPRSS2—R7 has strong potential to interfere with HCoV-HKU1 infection.
The T22 antibody is a potent inhibitor of TMPRSS2
In addition to its role as the direct receptor for HCoV-HKU1, TMPRSS2 has also been found to be a crucial cofactor facilitating the entry of a wide range of CoVs, including HCoV-HKU1, through its enzymatic activity. Consequently, the substrate-binding pocket of TMPRSS2 presents a promising target for developing not only anti-HCoV-HKU1 antibodies but also potentially broad-spectrum therapeutic antibodies against multiple CoVs.
Therefore, we focused on the development of antibodies that target the substrate-binding pocket of TMPRSS2. By screening a human naïve scFv phage display antibody library against TMPRSS2, we successfully identified and converted 62 antibodies into the scFv-Fc format (Fig. 3a). We then characterized the inhibition efficiency of these 62 scFv-Fcs against TMPRSS2 via continuous kinetic measurements based on fluorescence resonance energy transfer (FRET). In this assay, we employed Boc-Gln-Ala-Arg-AMC as the fluorogenic substrate. Three of the scFv-Fcs antibodies demonstrated > 90% inhibition of TMPRSS2 at a concentration of 1 µM. These inhibitors were selected for further analysis of their half-maximal inhibitory concentrations (IC50) (Supplementary Fig. 3a, b).

a Flowcharts of the scFv phage library panning and antibody selection process. The number within parentheses indicates the quantity of phage and scFv-Fc antibodies that were tested. b Dose‒response curve illustrating T22-mediated inhibition of TMPRSS2 catalytic activity. The data points represent the means ± standard deviations (SDs) from biological quadruplicates (n = 4). Error bars may not be visible because of their small magnitude relative to the data points. c Binding of T22 to TMPRSS2 measured by BLI. Biotinylated TMPRSS2 was immobilized on SA biosensors and then exposed to twofold dilutions of T22. d Effect of T22 on HCoV-HKU1-mediated cell‒cell fusion. The data points represent the means ± SDs from biological triplicates (n = 3). e Vero E6-TMPRSS2 cells were pretreated with different concentrations of T22 for 1 h and then infected with wild-type SARS-CoV-2 or its delta variant (MOI = 0.01). The cytopathic effect (CPE) rates were determined with a Celigo image cytometer at 72 h post-infection. The data points represent the means ± SDs from biological triplicates (n = 3). f Crystal structure of the T22 Fab in complex with TMPRSS2. TMPRSS2 is depicted with its SP domain in mint, whereas its LDLRA and SRCR domains are colored slate. The Cα atom of residue 441 in TMPRSS2 is represented by a hot pink sphere. The T22 heavy chain is shown in dark gray, with its CDRs in orange. The T22 light chain is displayed in light gray, with its CDRs in marine blue. g Structural comparison between the RBD:TMPRSS2 and T22:TMPRSS2 complexes. TMPRSS2 from both structures are aligned, with only TMPRSS2 from the T22:TMPRSS2 complex shown owing to negligible conformational changes between the two structures. TMPRSS2 is displayed as a surface representation, whereas RBD and T22 are shown as cartoons. h, i Zoomed-in views of the interactions between T22 and TMPRSS2. j Illustration of CDRH3 binding in the TMPRSS2 substrate-binding pocket. The structure of the peptidomimetic inhibitor Ac-KQLR-cmk in complex with TMPRSS1 (homologous to TMPRSS2, PDB ID: 1Z8G) is superimposed. The inhibitor and catalytic serine residue S353 are shown as deep blue sticks. Panel (a) was created in BioRender. Duan, Y. (2025) https://BioRender.com/ul5s7vf.
Among these antibodies, T22 emerged as the most potent TMPRSS2 inhibitor in subsequent enzymatic assays, with an IC50 of 29.3 ± 0.4 nM (Fig. 3b and Supplementary Fig. 3). Furthermore, T22 exhibited exceptional binding affinity to TMPRSS2, with a KD of 0.20 ± 0.02 nM (Fig. 3c). The efficacy of T22 was further validated through multiple cell-based assays. A cell‒cell fusion assay demonstrated the capacity of T22 to successfully inhibit HCoV-HKU1-mediated cell‒cell fusion (Fig. 3d). An antiviral assay confirmed the ability of T22 to inhibit the infection of both SARS-CoV-2 and its delta variant (Fig. 3e). In addition, pseudovirus infection experiments revealed that T22 interferes with HCoV-HKU1 pseudovirus infection (Supplementary Fig. 6). These comprehensive results strongly suggest that T22-mediated inhibition of TMPRSS2 catalytic activity can effectively prevent HCoV-HKU1 virus entry and has the potential to serve as a broad-spectrum antiviral agent targeting TMPRSS2-dependent viral infections.
To elucidate the structural basis of the ability of T22 to inhibit TMPRSS2 activity, we determined its crystal structure in complex with the TMPRSS2 ectodomain at 1.90 Å resolution (Fig. 3f). The crystal structure of T22 in complex with the TMPRSS2 ectodomain revealed an interaction interface of approximately 850 Å2. CDRH3 composes the majority of the contact area, accounting for approximately 500 Å2, whereas the three CDRs on the light chain collectively contribute approximately 350 Å2. As illustrated in Fig. 3g, the T22 epitope is located on the serine protease (SP) domain of TMPRSS2, and the binding position of T22 is near the RBD binding site without exhibiting any notable steric clash.
As depicted in Fig. 3h, i, the interaction between T22 and TMPRSS2 is predominantly mediated by polar interactions. The CDRH3 of T22 accounts for the majority of T22:TMPRSS2 interface. In addition, residues from the light chain also contribute to binding. Specifically, S31 on CDRL1 and S96 on CDRL3 form hydrogen bonds with TMPRSS2, whereas D52 on CDRL2 establishes a salt bridge with K392TMPRSS2.
As shown in Fig. 3h, CDRH3 forms an extensive network of hydrogen bonds with TMPRSS2. Notably, as shown in Fig. 3h, j, residue R107 in CDRH3 mimics the P1 residue of a substrate and is inserted into the deep S1 substrate-binding subsite of TMPRSS2, which is highly selective for arginine at this position5. The guanidine group of R107 forms key interactions with residues in the S1 subsite, including hydrogen bonds with S436TMPRSS2 and G464TMPRSS2. Beyond the S1 subsite, although the binding pose of CDRH3 does not closely resemble a peptide substrate or occupy corresponding substrate-binding subsites, it still clashes with the substrates from the P1 to the P3 subsites. This steric interference could potently disrupt substrate binding to TMPRSS2, thereby inhibiting its enzymatic activity. Importantly, while CDRH3 occupies the substrate-binding pocket, it does not reach the catalytic center where S441 can catalyze peptide bond hydrolysis, suggesting that T22 is not cleaved by TMPRSS2 and could maintain its potent inhibitory efficacy.
Nanobody VHH77, which targets TMPRSS2, exerts dual functions during viral infection
The structural basis of TMPRSS2 inhibition by T22, which involves the insertion of its CDRH3 into the enzymatic pocket of TMPRSS2, inspired us to explore nanobodies as potential enzyme inhibitors. As the smallest fully functional antibody fragments, nanobodies do not have the problem of heavy chain and light chain mispairing, which makes them ideal for drug target development. Moreover, nanobodies are known for their ability to insert their CDR3 loops into cryptic, concave epitopes, such as the substrate-binding pocket of enzymes, thereby inhibiting their catalytic activity12,13. This distinctive property stems from their prolate shape, featuring a convex paratope as well as a long, flexible CDR3 loop, which allows them to preferentially recognize and interact with cleft regions on target proteins14,15.
To leverage these advantageous properties, we created a synthetic nanobody phage display library with mutant CDR3 regions consisting of 16, 17, and 19 amino acids. The library size exceeded 1010, and next-generation sequencing (NGS) analysis confirmed its high level of diversity, indicating that the library was successfully constructed (Supplementary Fig. 4).
Through screening the synthetic nanobody library with TMPRSS2, we identified 30 nanobodies that were subsequently converted into the VHH-Fc format (Fig. 4a). Furthermore, we found that 17 of the identified VHH-Fc antibodies demonstrated > 90% inhibition against TMPRSS2 at a concentration of 1 µM (Supplementary Fig. 5), which demonstrated the effectiveness of our strategy. Among these, VHH77 emerged as a standout candidate, demonstrating remarkable binding affinity to and inhibitory potency against TMPRSS2 (Supplementary Fig. 5). Specifically, VHH77 exhibited a binding affinity (KD) of 2.64 ± 0.03 nM and a half-maximal inhibitory concentration (IC50) of 54.7 ± 0.6 nM (Fig. 4b, c). The efficacy of VHH77 was further validated through cell-based assays and HCoV-HKU1 pseudovirus experiments, which confirmed its ability to block HCoV-HKU1 infection (Fig. 4d and Supplementary Fig. 6). Notably, the performance of VHH77 was superior to that of T22 in the pseudovirus assay. As a potent TMPRSS2 inhibitor, VHH77 also shows potential as a broad-spectrum therapeutic candidate for other TMPRSS2-dependent diseases. This broader applicability is evidenced by its potent anti-SARS-CoV-2 effect (Fig. 4e), which again surpassed that of T22. To gain further insights into the structural basis of the blocking mechanism of VHH77, we determined the crystal structure of VHH77 in complex with the TMPRSS2 ectodomain at a resolution of 2.00 Å via X-ray crystallography.

a Flowcharts of the synthetic nanobody phage library panning and antibody selection process. The number within parentheses indicates the quantity of phage and VHH-Fc antibodies that were tested. b Binding of VHH77-Fc antibodies to TMPRSS2 measured by BLI. Biotinylated TMPRSS2 was immobilized on SA biosensors and then exposed to twofold dilutions of VHH77-Fc antibodies. c Dose‒response curve showing the inhibition of TMPRSS2 catalytic activity by VHH77-Fc antibodies. The data points represent the means ± SDs from biological quadruplicates (n = 4). Error bars may not be visible because of their small magnitude relative to the data points. d Effects of VHH77-Fc antibodies on HCoV-HKU1-mediated cell‒cell fusion. The data points represent the means ± SDs from biological triplicates (n = 3). e Vero E6-TMPRSS2 cells were pretreated with different concentrations of VHH77-Fc antibodies for 1 h and then infected with wild-type SARS-CoV-2 or its delta variant (MOI = 0.01). The CPE rates were determined with a Celigo image cytometer at 72 h post-infection. The data points represent the means ± SDs from biological triplicates (n = 3). f Crystal structure of VHH77 in complex with TMPRSS2. TMPRSS2 is depicted with its SP domain in mint, while its LDLRA and SRCR domains are colored slate. The Cα atom of residue 441 in TMPRSS2 is represented by a hot pink sphere. VHH77 is shown in gray, with its CDRs in orange. g Structural comparison of the RBD:TMPRSS2 and VHH77:TMPRSS2 complexes. TMPRSS2 from both structures are aligned, with only TMPRSS2 from the VHH77:TMPRSS2 complex shown owing to negligible conformational changes between the two structures. TMPRSS2 is displayed as a surface representation, whereas RBD and VHH77 are shown as cartoons. The clash region between the RBD and VHH77 is highlighted and shown in the inset. h, i Zoomed-in views of the interactions between VHH77 and TMPRSS2. j Illustration of CDR3 binding in the TMPRSS2 substrate-binding pocket. The structure of the peptidomimetic inhibitor Ac-KQLR-cmk in complex with TMPRSS1 (homologous to TMPRSS2, PDB ID: 1Z8G) is superimposed. The inhibitor and catalytic serine residue S353 are shown as deep blue sticks. Panel (a) was created in BioRender. Duan, Y. (2025) https://BioRender.com/dae9m8h.
Similar to T22, as illustrated in Fig. 4f, the VHH77 epitope is located on the SP domain of TMPRSS2, adjacent to the binding region of the HCoV-HKU1 RBD. Surprisingly, when the complex structures of TMPRSS2:VHH77 and TMPRSS2:RBD were superimposed, residue Q108 from VHH77 sterically clashed with the β-turn on the RBM, which was composed of residues V509RBD, L510RBD, D511RBD, and H512RBD (Fig. 4g). These RBM residues were identified as crucial components of hydrophobic region II in the TMPRSS2:RBD interaction (Fig. 1f). Consequently, this steric hindrance imposed by VHH77 could abolish the interaction between TMPRSS2 and the RBD by disrupting their interactions in hydrophobic region II. These structural observations are further corroborated with BLI-based competition analysis (Supplementary Fig. 2d, e).
The interface between VHH77 and TMPRSS2 had a buried surface area of approximately 1000 Å2, which is predominantly mediated by the three CDRs of VHH77. As illustrated in Fig. 4h, a hydrophobic interface is formed by several VHH77 residues, including L33 on CDR1, L57 on CDR2, and F47 and Y59 on the framework regions. This hydrophobic surface aligns with a complementary hydrophobic region on TMPRSS2, comprising P301TMPRSS2, W306TMPRSS2, H307TMPRSS2, and the alkyl portion of K300TMPRSS2. This interaction is further stabilized by a network of hydrogen bonds, increasing the overall binding affinity.
In addition to the contributions from CDR1 and CDR2, the CDR3 of VHH77 also plays an important role in the interaction with TMPRSS2. As depicted in Fig. 4i, CDR3 forms an extended, flexible loop that creates a substantial interface with TMPRSS2. At one side, P99, W102, and V103 on CDR3 form hydrophobic interactions with V275TMPRSS2, V280TMPRSS2, H296TMPRSS2, C297TMPRSS2, and L302TMPRSS2. On the other side, Q100 and V103-D106 establish an extensive hydrogen bond network with TMPRSS2.
Notably, similar to T22, the CDR3 of VHH77 is also accommodated within the substrate-binding pocket of TMPRSS2. As shown in Fig. 4j, the CDR3 resides in the substrate-binding pocket of TMPRSS2. Residue R105 on CDR3 mimics the P1 residue of the substrate, positioning its guanidine group within the S1 pocket. There, it establishes hydrogen bonds and salt bridges with S436TMPRSS2, G464TMPRSS2, and D435TMPRSS2. Adjacent residues on CDR3 also occupy the substrate-binding pocket, creating steric interference with substrate binding.
Overall, VHH77 has dual functions: it not only disrupts the binding between the HCoV-HKU1 RBD and host TMPRSS2 but also inhibits the enzymatic activity of TMPRSS2, which explains the enhanced antiviral effects of VHH77 compared with those of T22 (Supplementary Fig. 6). This dual action effectively neutralizes the role of TMPRSS2 in HCoV-HKU1 infection by preventing it from serving as the viral receptor and blocking its ability to prime the HCoV-HKU1 spike protein. Moreover, we evaluated the efficacy of VHH77 in preventing SARS-CoV-2 infection. As illustrated in Fig. 4e, our results demonstrate that VHH77 indeed exhibits broad-spectrum antiviral activity against both wild-type SARS-CoV-2 and its delta variant. These findings underscore the potential of VHH77 as a versatile therapeutic agent against multiple CoVs.
Structure guided maturation of nanobody VHH77
Given VHH77’s promising dual-mechanism antiviral activity, we sought to further optimize this antibody to enhance its efficacy. Guided by the crystallographic structural insights, we engineered affinity maturation of VHH77 through rational site-directed mutagenesis concentrated on the CDR3 loop architecture to maximize VHH77’s dual functionality. Two VHH77 mutation libraries with two or three random mutations within CDR3 were generated. Following three rounds of affinity-driven phage display selection against recombinant TMPRSS2 extracellular domain, the sequence of enriched phages was analyzed. The highly enriched variants identified mutation hotspots at positions 107–109 of CDR3, with Val at position 107 commonly substituted by Arg (V107R) and residues Q108-A109 frequently replaced by Ser or Asp (Supplementary Fig. 7a, b).
Based on this mutation profile, we engineered five VHH77 variants (VHH77-9 through VHH77-13) and systematically evaluated their efficacy in both binding and functional assays (Fig. 5a and Supplementary Fig. 7c, d). The V107R single mutation variant (VHH77-9) demonstrated significantly enhanced binding affinity to TMPRSS2 and increased inhibition of catalytic activity (Fig. 5a). Additional mutations at positions 108 and 109 further improved both binding affinity and inhibitory potency. Notably, variants with aspartate substitutions at position 108 (VHH77-10/11) exhibited superior performance compared to those with serine mutations (VHH77-12/13).

a Binding affinities (KD) and half-maximal inhibitory concentrations (IC50) of VHH77 variants for TMPRSS2 catalytic activity. b BLI-based competition analysis of VHH77 variants with HCoV-HKU1 RBD to TMPRSS2. c, d Vero E6-TMPRSS2 cells were pretreated with different concentrations of VHH77-10-Fc (c) or VHH77-11-Fc (d) antibodies for 1 h and then infected with wild-type SARS-CoV-2 or its delta variant (MOI = 0.01). The CPE rates were determined with a Celigo image cytometer at 72 h post-infection. The data points represent the means ± SDs from biological triplicates (n = 3). e Effects of VHH77-Fc, VHH77-10-Fc, and VHH77-11-Fc antibodies on HCoV-HKU1-mediated cell‒cell fusion. The data points represent the means ± SDs from biological triplicates (n = 3). f–h Structural comparisons highlight the differences between the VHH77-10:TMPRSS2 and VHH77:TMPRSS2 interactions. The structures of the VHH77-10:TMPRSS2 and VHH77:TMPRSS2 complexes were overlaid, with CDR3 of VHH77-10 colored orange and CDR3 of VHH77 colored wheat. The remainder of VHH77-10 is shown in gray, and the remainder of VHH77 is in white. TMPRSS2 in the VHH77-10:TMPRSS2 complex is colored mint, while TMPRSS2 in the VHH77:TMPRSS2 complex is colored light mint. Hydrogen bonds and ionic interactions in the VHH77-10:TMPRSS2 complex are represented by black dashed lines. In panel (g), the structure of the HCoV-HKU1 RBD in the RBD:TMPRSS2 complex is overlaid, with the RBM colored salmon.
In RBD competition assays, VHH77-9,VHH77-10 and VHH77-11 demonstrated markedly improved efficacy in blocking RBD binding to TMPRSS2. However, VHH77-12/13 variants containing the Q108S substitution showed slightly reduced RBD blocking capability (Fig. 5b). To further validate the efficacy of the most promising variants, VHH77-10 and VHH77-11, we performed cell-based assays. Consistent with our biochemical data, both variants showed enhanced inhibition of SARS-CoV-2 infection and better blockade of HCoV-HKU1-mediated cell-cell fusion compared to wild-type VHH77 (Fig. 5c–e), confirming successful structure-guided optimization of this dual-function therapeutic candidate.
To elucidate the molecular mechanism underlying the enhanced efficacy of these mutations, we solved the crystal structure of TMPRSS2:VHH77-10 complex at a resolution of 2.0 Å. As shown in Fig. 5f, the substitution of Val with Arg at position 107 enables R107 to form additional interactions with TMPRSS2, including a hydrogen bond with S460TMPRSS2, a salt bridge with D345TMPRSS2, and a π-cation interaction with H296TMPRSS2. These additional interactions explain the increased binding affinity of the V107R mutant.
Furthermore, R107 occupies the position previously inhabited by K342TMPRSS2, causing the latter to swing its sidechain outward. This conformational change is further stabilized by an ionic interaction between K342TMPRSS2 and the substituted D108 (Q108D mutation). Interestingly, when superimposed with TMPRSS2:RBD complex (Fig. 5g), it becomes evident that the outward-pointing K324TMPRSS2 clashes with L510RBD, therefore blocking RBD binding. This structural feature explains why the R107 mutation enhances VHH77’s ability to block RBD binding to TMPRSS2. In addition, since Ser is shorter and uncharged compared to Asp, the Q108S substitution in VHH77-12/13 variants fails to form polar contacts with K342TMPRSS2 and cannot stabilize its outward conformation. This likely accounts for the weaker RBD blocking capability observed in these variants containing the Q108S mutation.
As for the third mutation at residue 109, we observed a notable conformational change in the subsequent residues, particularly W111(Fig. 5h), which flips to the opposite side and forms additional van der Waals interactions with TMPRSS2. The substituted D109 forms a hydrogen bond with the main chain of W111, likely contributing to the stabilization of this altered conformation and consequently enhancing binding to TMPRSS2. In conclusion, our structure-guided maturation of VHH77 successfully yielded several variants with both improved affinity to TMPRSS2 and enhanced blocking of RBD binding. Meanwhile, the crystal structure revealed a distinct mechanism whereby the V107R mutation on VHH77 not only strengthens intermolecular interactions but also induces a conformational change in TMPRSS2 that contributes to more effective blockade of RBD binding.
Discussion
TMPRSS2, a type II transmembrane serine protease, has long been recognized for its notable role in various human diseases. Initially, TMPRSS2 was strongly implicated in prostate cancer, which is the second most common cancer in men16 and the second leading cause of cancer-related mortality among American males17. In this context, TMPRSS2 promotes cancer cell invasion, tumor growth, and metastasis through its protease activity, primarily by proteolytically activating matriptase and hepatocyte growth factor (HGF) and degrading the extracellular matrix5,6. Furthermore, TMPRSS2 has been implicated in inducing cancer-related pain through its proteolytic activation of protease-activated receptor 2 (PAR2)18.
In addition to its role in cancer, TMPRSS2 has emerged as a crucial factor in facilitating viral invasion. Analogous to its function in cancer, the protease activity of TMPRSS2 is also essential for the entry of various respiratory viruses, including several CoVs, and, moreover, influenza viruses. Notable influenza strains that are dependent on TMPRSS2 include H1N119,20, which led to the 2009 swine flu pandemic, the Russian flu in 1977, and the devastating Spanish flu pandemic of 1918–1920. TMPRSS2 is essential for the entry of SARS-CoV21, MERS-CoV4, and SARS-CoV-222, which have caused serious epidemics or pandemics in the past two decades. In addition, it facilitates the entry of seasonal HCoVs, including HCoV-229E23, -NL6324, -OC43, and -HKU18, which collectively account for a considerable proportion of common cold cases annually. Together, all these viruses have claimed not thousands but millions of lives globally. In all these cases, TMPRSS2 performs its crucial function through a common mechanism in which it cleaves viral surface proteins—hemagglutinin on influenza viruses and spike proteins on CoVs—triggering membrane fusion between the viral envelope and the host cell membrane, thereby enabling viral entry.
Given that the physiological role of TMPRSS2 appears to be redundant and potentially compensated by other similar proteases25, it has emerged as an attractive target for the development of both cancer therapeutics and antiviral drugs. Current interventions targeting TMPRSS2 include the administration of antiandrogens that downregulate its expression25 and the use of TMPRSS2 inhibitors such as nafamostat mesilate, which have demonstrated promising anti-SARS-CoV-2 activity with favorable safety profiles in clinical trials26. Surprisingly, despite TMPRSS2 being a membrane protein exposed on the cell surface and thus highly accessible, research on antibody development targeting TMPRSS2 has been relatively limited.
Recently, a paradigm-shifting discovery has further elevated the importance of TMPRSS2 as a druggable target in human diseases. While its function as an enzyme that cleaves various substrates to promote prostate cancer and processes viral surface proteins to facilitate viral infection is well established, TMPRSS2 has been found to play an unexpected role. Notably, it has been identified as the direct receptor for the spike protein of HCoV-HKU13. This finding not only expands our understanding of the multifaceted nature of TMPRSS2 in human diseases but also opens an avenue for developing antibody-based antiviral treatments that target the TMPRSS2:HCoV-HKU1 RBD interface, potentially revolutionizing our approach to combating this viral infection.
In our study, we not only elucidated the detailed molecular mechanism by which the HCoV-HKU1 RBD recognizes and attaches to TMPRSS2, precisely identifying the TMPRSS2:RBD interface, but also identified antibodies targeting either TMPRSS2 or the HCoV-HKU1 RBD that potently abrogate their interaction. Notably, we discovered that two TMPRSS2-targeting antibodies, T22 and VHH77, exhibit distinct mechanisms of action. These antibodies insert their CDRH3 or CDR3 into the substrate-binding pocket of TMPRSS2, specifically occupying the S1 substrate-binding subsite with an arginine residue. This mechanism enables T22 and VHH77 to potently inhibit TMPRSS2 activity, suggesting their potential as versatile therapeutic candidates. Furthermore, through structure-guided maturation of VHH77, we identified several mutations in the CDR3 region that substantially enhanced its performance. The resulting variants, VHH77-10 and VHH77-11, demonstrated not only improved efficacy in blocking SARS-CoV-2 infection but also enhanced blockade of HCoV-HKU1-mediated cell-cell fusion. Structural analysis revealed the distinct molecular mechanism by which these mutations simultaneously strengthen TMPRSS2 binding and induce conformational changes that more effectively prevent RBD:TMPRSS2 interaction. In conclusion, these antibodies could serve not only as treatments for HCoV-HKU1 infection but also more broadly as candidates for cancer therapy or antiviral treatments against influenza viruses and various CoVs that rely on TMPRSS2 for cell entry.
Methods
Expression and purification of the HCoV-HKU1 RBD and TMPRSS2
The genes encoding the HCoV-HKU1 RBD (residues 323–609) and human TMPRSS2 (wild type and S441A mutant, residues 109–492) were subsequently cloned and inserted into pFastBac1 vectors. These vectors included an N-terminal GP64 signal peptide (MVSAIVLYVLLAAAAHSAFA) and a C-terminal 10 × His-tag27,28. For the TMPRSS2 construct, the autocleavage sequence (250SSRQSR255) was replaced with an enteropeptidase cleavage sequence (DDDDK).
The plasmid was transformed into E. coli (DH10Bac strain) competent cells to produce recombinant bacmid DNA. Sf9 cells cultured in Sf-900 II SFM (Thermo Fisher Scientific) were transfected with bacmid DNA via the FuGENE 6 Transfection Reagent (Promega). The initial P0 viral stock was collected 72 h post-infection and amplified to produce P1–P3 stocks with high titers. For protein expression, cells at a density of 2.5 × 106 cells/mL were infected with P3 virus and harvested after 4 days.
Proteins were then purified from the culture supernatant by incubation with Ni Sepharose Excel resin (Cytiva) at 4 °C for 2 h. After washing with TBS 1 buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl) with 20 mM imidazole, the proteins were eluted with TBS 1 containing 300 mM imidazole. TMPRSS2 WT and S441A were further digested with enterokinase (New England Biolabs) overnight at 20 °C. All proteins were purified via gel filtration chromatography using a Superdex 200 10/300 column (Cytiva) with TBS 2 buffer (25 mM Tris-HCl, pH 8.0; 75 mM NaCl; 2 mM CaCl2). The proteins were successfully purified as shown in Supplementary Fig. 8.
Construction of a nanobody phage display library
We constructed a synthetic phage display library of humanized Lama single-domain antibodies on the basis of a humanized synthetic single-domain antibody (hs2dAb) scaffold optimized for intracellular stability29. The diversity of CDRs was introduced by randomizing the oligonucleotide DNA using the degenerate codon NNK (N denotes a 25% mixture of adenine, thymine, cytosine, and guanine nucleotides each; and K denotes a 50% mixture of thymine and guanine nucleotides each). A constant length of 7 amino acids was chosen for CDR1 and CDR2. A large spectrum of Camelidae VHH CDR3 lengths is naturally observed. Thus, 16, 17, or 19 random amino acids (excluding cysteine) were introduced into CDR3. Large-scale PCR was then carried out to ensure that many diverse nanobodies were generated, and the fragments were cut and inserted into the pCGMT3 phagemid. The phagemid was transformed into electrocompetent E. coli (XL1-Blue strain) cells. A 1 × 1010 diversity library was generated. The transformed bacteria were plated on 2×YT-Amp agar plates (150 mm), grown overnight at 37 °C, harvested, and stored at − 80 °C in 30% glycerol.
Quality control was carried out using next-generation sequencing (NGS). We first merge the paired-end NGS sequencing reads, then extract the CDR1, CDR2, and CDR3 regions using the constant regions of the antibody scaffold, and translate them into amino acid sequences by the software CLC Genomics Workbench (V11.0.1, https://digitalinsights.qiagen.com/). We retain CDR sequences without stop codons that can be correctly translated into 20 amino acids. Finally, the proportion of each of the 20 amino acids at every position in the CDR sequences is calculated.
About the maturation of nanobody VHH77, we constructed two maturation libraries. Using the nucleic acid sequence of VHH77 as a template, one library (M1 library) introduced random amino acid mutations (degenerate codon NNK) at 2 random positions at CDR3, and the other library (M2 library) introduced random amino acid mutations at 3 consecutive amino acid positions at CDR3. The mutated nanobody VHH77 library DNA fragment was inserted into pCGMT3 phagemid. The follow-up method is the same as above.
Phage display selection
Two phage display antibody libraries were used for panning, including a human naïve scFv library constructed as previously described30 and a synthetic nanobody library constructed as described above. The phage libraries were amplified in E. coli (XL1-Blue strain). The HCoV-HKU1 RBD and TMPRSS2 WT proteins were biotinylated with EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific) and purified using Zeba Spin Desalting Columns (Thermo Fisher Scientific).
For selection, phage libraries were incubated with biotinylated HCoV-HKU1 RBD or TMPRSS2 WT protein for 2 h at room temperature. Phage‒antigen complexes were captured via Dynabeads M-280 (Thermo Fisher Scientific). After 8 washes with PBS, the bound phages were eluted with 0.2 M glycine-HCl (pH 2.2) for 10 min at room temperature and then neutralized with 1 M Tris-HCl (pH 8.0) to pH 7.4. Eluted phages were added to XL1-blue for infection following the addition of the M13 helper phage. The infected bacteria were cultured under selection with kanamycin and carbenicillin to amplify the phage for the next round of panning. After three rounds of panning, the phage eluates were used to infect XL1-Blue to grow single colonies for phage preparation. Individual phage clones were also amplified, and the phage-containing supernatant was harvested by centrifugation for subsequent phage ELISA.
For affinity maturation of VHH77, the amount of antigen used in screening was reduced by 10 times round by round, and the screening method was the same as above.
Phage ELISA
Phage ELISA was performed according to standard protocols. First, 96-well half-area high-binding plates (Corning) were coated with HCoV-HKU1 RBD or TMPRSS2 WT (5 µg/mL) overnight at 4 °C. The plates were then blocked with 6% (w/v) skim milk in PBST buffer (M-PBST) for 1 h at room temperature. Phage supernatant was added to the wells, followed by eight washes with PBST buffer. Bound phages were detected via incubation with an HRP-conjugated M13 bacteriophage antibody (Sino Biological) diluted 1:5000 in 6% M-PBST for 1 h. The reaction was developed by the addition of 50 µL of ABTS solution (Thermo Fisher Scientific). The absorbance at 405 nm was monitored using an EnSpire multimode plate reader (Perkin Elmer). The positive phages were defined by the ratio of the absorbance of the target binding to that of BSA binding being greater than two. The positive phages were added to bacteria to amplify phagemids for Sanger sequencing to identify the antibody sequences.
Expression and purification of scFv-Fc and VHH-Fc antibodies for screening
Phagemids (returned by the sequencing company) and the pFuse-Fc vector were digested with the SfiI restriction endonuclease (New England Biolabs). The VHH and scFv genes were ligated into the pFuse-Fc vector using T4 DNA ligase (New England Biolabs). The pFuse-Fc vector included an N-terminal IL-2 signal peptide (MYRMQLLSCIALSLALVTNS) and a C-terminal human IgG1 Fc fragment.
The plasmids were transiently transfected into FreeStyle 293-F cells cultured in SMM 293-TII expression medium (Sino Biological) using PEI MAX (Polysciences). Cells at a density of 2.5 × 106 cells/mL were transfected with a mixture of 100 μg of plasmid and 300 μg of PEI MAX in 2.5 mL of medium and incubated for 20 min before being added to 100 mL of cell culture. The culture supernatant was collected after 72 h.
The cell culture supernatant was incubated with Protein A resin (GenScript) at 4 °C for 2 h. The resin was then washed with PBS and eluted with 0.1 M glycine-HCl (pH 3.0). The eluate was neutralized with 1 M Tris-HCl (pH 8.5) to pH 7.4. The buffer of the antibody was further replaced with TBS 1. scFv-Fc and VHH-Fc antibodies were used for screening for competition analysis and inhibition analysis.
BLI-based competition analysis
BLI-based competition analysis was performed on an Octet RED96 machine (ForteBio). The HCoV-HKU1 RBD and TMPRSS2 were biotinylated and purified as previously described. All the assays were conducted in kinetics buffer (PBS with 0.02% v/v Tween-20 and 1 mg/mL BSA) at 1000 rpm shaking speed and 30 °C.
To screen for antibodies with blocking ability, the biotinylated RBD or TMPRSS2 was immobilized on SA biosensors to 2 nm. Loaded biosensors were exposed to antibody (scFv-Fc or VHH-Fc) solutions (400 nM) for 200 s, followed by 200 s of dissociation in kinetics buffer. The sensors were then exposed to TMPRSS2 or RBD solutions (100 nM) for 200 s to assess competition binding, and the TMPRSS2 or RBD response values were normalized to their respective starting points. The human isotype IgG1 (Selleck) served as a control.
To compare the blocking efficacy of antibodies (R7 IgG and VH77-Fc) blocking the RBD:TMPRSS2 interaction, a modified BLI method was employed. The procedure was similar to that of the screening method, but loaded biosensors were exposed to threefold dilutions of antibody solutions (900–1.23 nM). Blocking percentages were calculated using the following formula: Blocking% = [(normalized response of isotype antibody - normalized response of test antibody)/normalized response of isotype antibody] × 100%. The analysis results are shown in Supplementary Fig. 2.
Enzymatic activity inhibition analysis
Inhibitory antibodies against human TMPRSS2 were screened using an in vitro enzymatic inhibition assay. TMPRSS2 enzymatic activity was measured via a continuous kinetic assay based on fluorescence resonance energy transfer (FRET), employing the fluorogenic substrate Boc-QAR-AMC (GL Biochem). The excitation and emission wavelengths of the fluorogenic substrate were 340 nm and 460 nm, respectively. The fluorescence intensity was monitored using an EnVision multimode plate reader (Perkin Elmer).
Screening assays were performed in 384-well black microplates (PerkinElmer) with a total volume of 50 µL. A Bravo automated liquid handling platform (Agilent) was utilized to add antibodies to the enzymatic reaction mixture. The final reaction system comprised the following: enzymatic reaction buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.1 mg/mL BSA, 0.01% v/v Triton X-100), 1 nM TMPRSS2, 10 μM substrate, and 1 μM antibodies (scFv-Fc/VHH-Fc). TBS 1 buffer treatment served as a negative control.
The inhibitory effect of the antibodies was evaluated by comparing the initial velocities of the reactions containing antibodies to those of the negative control. Antibodies demonstrating over 90% inhibition were classified as hits and selected for further testing. All the statistical analyses were performed using GraphPad Prism 9.5.
IgG (R7 and T22) and VHH77-Fc expression and purification
Using scFv-phagemids as templates, the VH domain was subcloned and inserted into the pFuse IgG heavy chain vector. The VL domain was subcloned and inserted into the pFuse light chain vector. The plasmids were transiently transfected into FreeStyle 293-F cells. Cells at a density of 2.5 × 106 cells/mL were transfected with a mixture of 1 mg of plasmid (the molar ratio of plasmid expressing heavy chain to light chain was 1:1) and 2.5 mg of PEI MAX in 40 mL of medium and incubated for 20 min before being added to 1 L of cell culture. The culture supernatant was collected after 72 h. The purification method used for VHH77-Fc and IgG was consistent with that used for scFv-Fc. These antibodies were further purified by gel filtration chromatography using a Superdex 200 Increase 10/300 column with PBS buffer. The antibodies were successfully purified as shown in Supplementary Fig. 8.
Binding affinity assays
Kinetic analyses were performed on an Octet RED96 machine (ForteBio). To measure the binding affinity of the HCoV-HKU1 RBD for TMPRSS2 and its S441A mutant, biotinylated HCoV-HKU1 RBDs were immobilized on SA biosensors (Sartorius) to 2 nm and incubated with twofold dilutions of TMPRSS2 and the S441A mutant. To measure antibody binding affinity to its target protein, biotinylated target proteins were immobilized on SA biosensors (Sartorius) to 2 nm and incubated with twofold dilutions of antibodies. Curves were fitted via a 1:1 binding model. The data were exported using ForteBio Data Analysis 11.1 software, and the binding profile was drawn via GraphPad Prism 9.5.
IC50 analysis
The IC50 values of the TMPRSS2 inhibitory antibodies were measured following a previously published protocol28. The total volume of each enzyme activity reaction was set at 50 μL. TMPRSS2 was diluted in enzymatic reaction buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.1 mg/mL BSA, 0.01% v/v Triton X-100). Assays were conducted using 1 nM TMPRSS2 and the 10 μM fluorogenic substrate Boc-QAR-AMC (GL Biochem) in the presence of varying concentrations of antibodies (IgG/VHH-Fc). Prior to substrate addition, the antibodies were incubated with TMPRSS2 at room temperature for 1 h. The fluorescence intensity was monitored using an EnVision multimode plate reader (Perkin Elmer), with excitation and emission wavelengths set at 340 nm and 460 nm, respectively. The initial velocities were determined by fitting the linear portion of the curves to a straight line. IC50 values were calculated by fitting the data to a nonlinear regression curve via GraphPad Prism 9.5.
VHH and Fab expression and purification
For VHH expression, the VHH domain was subcloned and inserted into the pFuse VHH vector, which included a C-terminal 6 × His-tag. For Fab expression, using scFv-phagemids as templates, the VH domain was subcloned and inserted into the pFuse Fab heavy chain vector. The pFuse Fab heavy chain vector included a C-terminal 6 × His-tag. The plasmids were transiently transfected into FreeStyle 293-F cells as previously described. The cell culture supernatant was incubated with Ni Sepharose Excel resin at 4 °C for 2 h. After washing with TBS 1 buffer containing 20 mM imidazole, the proteins were eluted with TBS 1 buffer containing 250 mM imidazole. Final purification was performed by gel filtration chromatography using a Superdex 75 Increase 10/300 column (Cytiva) with TBS 2 buffer. The proteins were successfully purified as shown in Supplementary Fig. 8.
Crystallization, data collection and structure determination
Protein complexes for crystallization were prepared by mixing HCoV-HKU1 RBD and the TMPRSS2 S441A mutant or R7 Fab at a 1:1 molar ratio and TMPRSS2 WT with T22 Fab, VHH77, or VHH77-10 at a 1:1.2 molar ratio. The mixtures were incubated at room temperature for 2 h and purified with a Superdex 200 Increase 10/300 column in TBS 2 buffer. These complexes were successfully purified as shown in Supplementary Fig. 8. Protein samples were concentrated (RBD:TMPRSS2 S441A mutant and RBD:R7 Fab complexes to 8 mg/mL, TMPRSS2:T22 Fab to 17 mg/mL, and TMPRSS2:VHH77 and TMPRSS2:VHH77-10 complexes to 11 mg/mL), crystallized by vapor diffusion at 20 °C, and mixed with well buffer at a 1:1 (v/v) ratio.
The optimal crystallization conditions were as follows. For the RBD:TMPRSS2 S441A mutant complex: 0.2 M Tris pH 7.0, 0.2 M MgCl2, 10% (w/v) Polyethylene glycol 8000; for the RBD:R7 Fab complex: 0.2% (w/v) Ala-Ala, 0.2% (w/v) Ala-Gln, 0.2% (w/v) Gly-Glu, 0.2% (w/v) Gly-L-Ala, 0.2% (w/v) Gly-L-Asp, 0.2% (w/v) Gly-Sar, 0.2% (w/v) L-Carnosine, 0.2% (w/v) Leu-Ala hydrate, 0.1 M Buffer System 3 [Tris (base); BICINE] pH 8.5, 20% (v/v) Polyethylene glycol 500* MME, 10% w/v Polyethylene glycol 20,000; for the TMPRSS2:T22 Fab complex, 50 mM MES pH 5.6, 8.6% (w/v) Polyethylene glycol 4000, 17.1% (v/v) Polyethylene glycol 600; for the TMPRSS2:VHH77 complex, 0.2 M sodium malonate pH 6.0, 10% (w/v) Polyethylene glycol 3350; and for the TMPRSS2:VHH77-10 complex, 100 mM HEPES/Sodium hydroxide pH 7.0, 10% (w/v) Polyethylene glycol 6000. Crystals were cryoprotected in well buffer containing 20% (v/v) glycerol before flash-freezing in liquid nitrogen.
X-ray diffraction data were collected at 100 K on the BL02U1, BL10U2, and BL19U1 beamlines at the Shanghai Synchrotron Radiation Facility (SSRF), China. Data integration and scaling were performed using XDS31. Structures were determined via molecular replacement with PHASER32, using the following search templates: the structure of the HCoV-HKU1 RBD (PDB ID: 5KWB), the structure of TMPRSS2 in complex with camostat (PDB ID: 7Y0E), and the predicted structures of the antibodies generated using SWISS-MODEL33. The models were iteratively adjusted manually with Coot34 and refined with Phenix REFINE35. The representative electron density map for each crystal structure are illustrated in Supplementary Fig. 9. The phasing and refinement statistics are summarized in Supplementary Tables 1 and 2.
Cell culture
Vero E6-TMPRSS2 (JCRB, 1819) and HEK293T (ATCC, CRL-3216) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin. All the cells were cultured at 37 °C in a fully humidified atmosphere containing 5% CO2 and tested negative for mycoplasma infection.
GFP-split fusion assay
HEK293T GFP-split cells (GFP1-10 and GFP11) were transfected separately with 1000 ng of spike or TMPRSS2 plasmid for 20 h. The cells were washed twice, and acceptor and donor cells were mixed and seeded at 6 × 104 cells/well. At 24 h of coculture, images were acquired on an Operetta CLS high-content screening system (PerkinElmer). The GFP area was quantified via ImageJ analysis software.
Generation of HCoV-HKU1 pseudoparticles
Lentivirus-based pseudotypes bearing the HCoV-HKU1 spike protein were generated in HEK293T cells via cotransfection with plasmids encoding lentiviral proteins, a luciferase reporter, and the HCoV-HKU1 spike plasmid as previously reported3. Briefly, HEK293T cells were seeded into 6-cm-diameter plates (1 × 106 cells per well) 1 day before transfection with 0.75 μg psPAX2, 0.75 μg pWPI-Fluc, and 1.5 μg HCoV-HKU1 spike expression construct pcDNA3.1-S using PEI Max as a transfection reagent. Threefold volumes of a PEI working solution were mixed with the indicated DNA diluted in Opti-MEM prior to transfection of the HEK293T cells. The supernatant was harvested after 48 h by centrifugation at 4000 rpm for 10 min at 4 °C. Vero E6-TMPRSS2 cells were infected and assayed for luciferase activity at 48 h post-infection using a One-Lite luciferase assay system kit (DD1203-03, Vazyme) to evaluate the efficiency of pseudotyped virus infection.
Pseudotyped HCoV-HKU1 virus inhibition experiments
A total of 1.5 × 104 HEK293T-TMPRSS2 cells were seeded in a 96-well plate 1 day before pretreatment with 5 μM antibody (T22 IgG/VHH77-Fc) for 2 h. 100 μL of pseudotyped HCoV-HKU1 virus was added to the monolayer of HEK293T-TMPRSS2 cells. Luciferase activity was detected 48 h after the incubation of the cells with the virus at 37 °C with 5% CO2. The inhibition rate was calculated from the luciferase activity of the pseudotyped HCoV-HKU1 virus. Two independent experiments were performed with quintuplicate infections, and data from one representative experiment are shown.
SARS-CoV-2 preparation and titrations
The SARS-CoV-2 wild-type strain (WT-IQTC02-16#, WT) and Delta variant (IQTC-IM2175251) were propagated in Vero E6 cells and stored at − 80 °C. Virus titers were determined via 10-fold serial dilutions in confluent Vero E6 cells in 96-well microtiter plates. Three days after inoculation, the CPE was scored, and the Reed‒Muench formula was used to calculate the TCID50. All of the infection experiments were performed at BSL-3 in the Guangzhou Customs Inspection and Quarantine Technology Center (IQTC).
In vitro anti-SARS-CoV-2 assay
SARS-CoV-2 experiments were performed as previously described28,36. Briefly, 2 × 104 Vero E6-TMPRSS2 cells were seeded in a 96-well plate for 24 h. The antibodies at different dilutions were incubated with the cells for 1 h at 37 °C, and then, monolayers of Vero E6-TMPRSS2 cells were inoculated with SARS-CoV-2 (MOI = 0.01). Seventy-two hours after inoculation, the CPE was scored with a Celigo image cytometer. The inhibition of antibodies and the EC50 value were calculated from the SARS-CoV-2 CPE rates. Two independent experiments were performed with eight concentration gradients, each with triplicate wells, and one representative is shown.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All experimental data are provided in the manuscript. The structures determined in this study have been deposited in the Protein Data Bank (PDB) under accession codes: 9JCX (HCoV-HKU1 RBD in complex with the TMPRSS2 S441A mutant), 9JCY (HCoV-HKU1 RBD in complex with the R7 Fab), 9JD1 (TMPRSS2 in complex with the T22 Fab), 9JD0 (TMPRSS2 in complex with VHH77), and 9U8G (TMPRSS2 in complex with VHH77-10). The structure of the peptidomimetic inhibitor Ac-KQLR-cmk in complex with TMPRSS1 was downloaded from PDB under the accession code 1Z8G. Materials used in this study will be made available under an appropriate Materials Transfer Agreement. Source data are provided in this paper.
References
Liu, D. X., Liang, J. Q. & Fung, T. S. In Encyclopedia of Virology. (Fourth Edition) (eds. Bamford, D. H. & Zuckerman, M.) 428–440 (Academic Press, Oxford, 2021).
Nathan, R. et al. A narrative review of the clinical practicalities of bamlanivimab and etesevimab antibody therapies for SARS-CoV-2. Infect. Dis. Ther. 10, 1933–1947 (2021).
Saunders, N. et al. TMPRSS2 is a functional receptor for human coronavirus HKU1. Nature 624, 207–214 (2023).
Shen, L. W., Mao, H. J., Wu, Y. L., Tanaka, Y. & Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 142, 1–10 (2017).
Lucas, J. M. et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 4, 1310–1325 (2014).
Ko, C. J. et al. Androgen-induced TMPRSS2 activates matriptase and promotes extracellular matrix degradation, prostate cancer cell invasion, tumor growth, and metastasis. Cancer Res. 75, 2949–2960 (2015).
Kim, T. S., Heinlein, C., Hackman, R. C. & Nelson, P. S. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell Biol. 26, 965–975 (2006).
Shirato, K., Kawase, M. & Matsuyama, S. Wild-type human coronaviruses prefer cell-surface TMPRSS2 to endosomal cathepsins for cell entry. Virology 517, 9–15 (2018).
McCallum, M. et al. Human coronavirus HKU1 recognition of the TMPRSS2 host receptor. Cell 187, 4231–4245 (2024).
Wang, H. et al. TMPRSS2 and glycan receptors synergistically facilitate coronavirus entry. Cell 187, 4261–4271 (2024).
Fernández, I. et al. Structural basis of TMPRSS2 zymogen activation and recognition by the HKU1 seasonal coronavirus. Cell 187, 4246–4260 (2024).
Desmyter, A. et al. Crystal structure of a camel single-domain VH antibody fragment in complex with lysozyme. Nat. Struct. Biol. 9, 803–811 (1996).
Zavrtanik, U., Lukan, J., Loris, R., Lah, J. & Hadži, S. Structural basis of epitope recognition by heavy-chain camelid antibodies. J. Mol. Biol. 430, 4369–4386 (2018).
Lauwereys, M. et al. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 17, 3512–3520 (1998).
De Genst, E. et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 103, 4586–4591 (2006).
Rebello, R. J. et al. Prostate cancer. Nat. Rev. Dis. Prim. 7, 9 (2021).
Siegel, D. A., O’Neil, M. E., Richards, T. B., Dowling, N. F. & Weir, H. K. Prostate cancer incidence and survival, by stage and race/ethnicity - United States, 2001-2017. MMWR Morb. Mortal. Wkly Rep. 69, 1473–1480 (2020).
Lam, D. K., Dang, D., Flynn, A. N., Hardt, M. & Schmidt, B. L. TMPRSS2, a novel membrane-anchored mediator in cancer pain. Pain 156, 923–930 (2015).
Limburg, H. et al. TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J. Virol. 93, e00649-19 (2019).
Hatesuer, B. et al. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 9, e1003774 (2013).
Zhou, Y. et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 116, 76–84 (2015).
Hoffmann, M. et al. SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280 (2020).
Bertram, S. et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 87, 6150–6160 (2013).
Kawase, M., Shirato, K., van der Hoek, L., Taguchi, F. & Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86, 6537–6545 (2012).
Stopsack, K. H., Mucci, L. A., Antonarakis, E. S., Nelson, P. S. & Kantoff, P. W. TMPRSS2 and COVID-19: Serendipity or opportunity for intervention?. Cancer Discov. 10, 779–782 (2020).
Okugawa, S. et al. Antiviral effect and safety of nafamostat mesilate in patients with mild early-onset COVID-19: An exploratory multicentre randomized controlled clinical trial. Int. J. Antimicrob. Agents 62, 106922 (2023).
Fraser, B. J. et al. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat. Chem. Biol. 18, 963–971 (2022).
Wang, H. et al. Structure-based discovery of dual pathway inhibitors for SARS-CoV-2 entry. Nat. Commun. 14, 7574 (2023).
Moutel, S. et al. NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies. ELife 5, e16228 (2016).
Wang, Y. et al. High-throughput functional screening for next-generation cancer immunotherapy using droplet-based microfluidics. Sci. Adv. 7, eabe3839 (2021).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr. 68, 352–367 (2012).
Guo, Y. et al. Discovery and characterization of potent pan-variant SARS-CoV-2 neutralizing antibodies from individuals with Omicron breakthrough infection. Nat. Commun. 14, 3537 (2023).
Acknowledgements
We would like to acknowledge the Discovery Technology Platform of the Shanghai Institute for Advanced Immunochemical Studies (SIAIS) and the Molecular and Cell Biology Core Facility of the School of Life Science and Technology (SLST), ShanghaiTech University, for their instrumentation and technical assistance. We would also like to thank the Shanghai Synchrotron Radiation Facility of BL02U1, BL10U2, and BL19U1 (https://cstr.cn/31124.02.SSRF) for the assistance on data collection. This work was supported by grants from the National Natural Science Foundation of China (grant number 92469202 to H.Y.); the Shanghai Municipal Science and Technology Major Project (grant number ZD2021CY001 to H.Y.); Guangzhou Laboratory (grant numbers SRPG22-003); the Postdoctoral Fellowship Program of CPSF (grant number BX20250142 to Y.D.); and the Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine of ShanghaiTech University.
Author information
Authors and Affiliations
Contributions
H.Y. and H.Z. conceived the project; Z.Z., Y.H., H.L. and M.D. designed and performed the phage display screening assay; M.D., H.L. and W.W. constructed a synthetic nanobody phage library; Z.Z., X.L., M.L. and Y.H. cloned, expressed, purified, and crystallized proteins; Z.Z., X.L., M.L., Y.D. and H.W. collected diffraction data and solved the crystal structures; Z.Z. and Y.L. performed biolayer interferometry binding assays; Z.Z., X.L. and X.Z. performed the enzymatic activity inhibition assays; Q.Y. and A.Z. performed the cell-based antiviral assays; Z.Z., Q.Y., X.L., M.L., Y.D., H.W., X.Y., H.Z. and H.Y. analyzed and discussed the data; Z.Z., Y.D., H.W., X.Y., H.Z., X.C., Z.R. and H.Y. wrote the manuscript with inputs from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Levon Halabelian, Andrey Kovalevsky, and the other anonymous reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Z., Yang, Q., Liu, X. et al. The crystal structure of coronavirus RBD-TMPRSS2 complex provides basis for the discovery of therapeutic antibodies. Nat Commun 16, 6636 (2025). https://doi.org/10.1038/s41467-025-62023-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62023-2
