
Abstract
Targeted protein degradation is a powerful tool for biological research, cell therapy, and synthetic biology. However, conventional methods often depend on pre-fused degrons or chemical degraders, limiting their wider applications. Here we develop a guided protein labeling and degradation system (GPlad) in Escherichia coli, using de novo designed guide proteins and arginine kinase (McsB) for precise degradation of various proteins, including fluorescent proteins, metabolic enzymes, and human proteins. We expand GPlad into versatile tools such as antiGPlad, OptoGPlad, and GPTAC, enabling reversible inhibition, optogenetic regulation, and biological chimerization. The combination of GPlad and antiGPlad allows for programmable circuit construction, including ON/OFF switches, signal amplifiers, and oscillators. OptoGPlad-mediated degradation of MutH accelerates E. coli evolution under protocatechuic acid stress, reducing the required generations from 220 to 100. GPTAC-mediated degradation of AroE enhanced the titer of 3-dehydroshikimic acid to 92.6 g/L, a 23.8% improvement over the conventional CRISPR interference method. We provide a tunable, plug-and-play strategy for straightforward protein degradation without the need for pre-fusion, with substantial implications for synthetic biology and metabolic engineering.
Similar content being viewed by others


Introduction
In synthetic biology, precise manipulation of cellular activities and functions typically requires the regulation of intracellular protein abundance at the DNA, RNA, and protein levels. Compared with DNA/RNA level regulation, directly regulating the protein level by degradation technology does not require genome modification and provides benefits such as rapid response1, high tunability2,3, and broad applicability4. Additionally, it can also be used to study the functions of lethal or essential genes, eliminate long-lived proteins, and regulate protein levels in specific organelles. In recent years, protein degradation approaches have achieved rapid advancements in eukaryotic systems. For instance, chemically induced strategies such as PROTACs5, molecular glues6, and LYTACs7, as well as degron-based fusion systems like degronLOCKR8 and SD409, have emerged as powerful tools. These methods have significantly advanced both therapeutic development and fundamental research, enabling precise control over disease-related proteins10 and cellular processes11. However, the protein degradation methodologies in bacterial systems remain limited and underdeveloped. This scarcity of versatile tools has hindered the application and advancement of protein-level regulation in bacterial synthetic biology and related fields.
Bacterial protein degradation methods can be categorized into two types: degrader addition and degron fusion. BacPROTAC (Bacterial-based PROTAC), currently the only reported degrader in bacteria, is a typical PROTAC-like chemical molecule composed of a phosphorylated arginine analog (pArg) linked to a target protein ligand (biotin) via a chemical linker. In Bacillus subtilis, the pArg is responsible for binding the protease ClpC, while biotin recruits the target protein, enabling the ClpCP protease complex to proximity-induce degradation12. According to this design, recent works successfully achieved specific degradation of multiple target proteins in B. subtilis and Mycobacteria, and highlight the potential of BacPROTAC for treating bacterial infections and antibiotic tolerance12,13,14. However, its high cost and reliance on membrane permeability limit its scalability for microbial manufacturing and industrial applications. In contrast, degron fusion—characterized by simple design, editability, and low cost—has been widely adopted in bacterial systems, particularly in synthetic biology and industrial biotechnology. Representative bacterial degron fusion strategies fall into two categories: (i) Orthogonal degron degradation systems and (ii) Degron masking/exposure systems. The first systems fuse a degron sequence to the terminal of target proteins, requiring co-expression of a cognate heterologous protease15,16,17. For example, GFP fused to the Mesoplasma florum degron tag (mf-ssrA) is stable in E. coli but rapidly degraded upon mf-Lon protease induction, forming an orthogonal degradation system15. For degron masking/exposure systems, a masking sequence (e.g., a TEV protease cleavage site) is fused alongside the degron to shield it during protein folding, ensuring stable target expression. Inducing TEV protease expression removes the masking sequence, exposing the degron and triggering degradation18. This approach has been implemented in genetic circuit construction19, cellular function regulation20, and metabolic flux control21. Despite these advances, degron fusion methods require the pre-fusion of degron sequences to the target protein, which is time-consuming and may disrupt the protein’s normal function. To address these challenges, there is an urgent need to develop degradation strategies that: (i) operate without exogenous degraders; (ii) eliminate the need for pre-fused degrons; (iii) directly recognize and degrade target proteins.
In this study, we develop a protein-targeted degradation technology termed the Guided Protein Labeling and Degradation (GPlad) system. The GPlad system comprises three key components: the marking protein (MP), the guide protein (GP), and the protease. Its mechanism operates as follows: first, a de novo-designed guide protein positions the marking protein close to the target protein; next, the marking protein introduces labels to the target protein; finally, the labeled target protein is degraded by a specific protease (Fig. 1a). With this design, directly target protein degradation can be achieved without the need for additional degraders or prior degron fusion. Only a plasmid expressing the GPlad system needs to be constructed. This method has the advantages of simple operation, low cost, and plug-and-play, making it particularly suitable for application in synthetic biology and related fields. As proof of concept, various types of proteins, including fluorescent proteins, metabolic enzymes, and human proteins, are precisely degraded by the GPlad system. Additionally, we develop regulatory tools such as antiGPlad, OptoGPlad, and GPTAC to enable reversible inhibition, optogenetic regulation, and biological chimerization, respectively. Furthermore, these tools are applied to design programmable protein switches, accelerate adaptive laboratory evolution, and rewire metabolic flux in microbial cell factories.

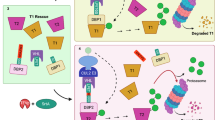
a Schematic representation of GPlad. First, a guide protein (GP) is fused with a marking protein (MP) via a linker, forming a GP-MP complex. GP binds the target protein and brings MP close to the target protein. The MP labels the target protein, and then the labeled target protein is subsequently recognized and degraded by the corresponding proteases. b Left: Western blot (WB) analysis showing McsBE121A and McsB-mediated arginine phosphorylation of mKate2 guided by 1/1′ heterodimer. Right: WB analysis showing PafAH123A and PafA-mediated ubiquitin-like modification of mKate2 guided by 1/1′ heterodimer. c Flow cytometry analysis of intracellular mKate2 fluorescence intensity at 0, 3, and 6 h after induction of McsB or PafA expression. P-value was determined by a two-tailed unpaired Student’s t-test. ****p < 0.0001. d WB analysis of intracellular mKate2 abundance after 6 h of induction using the McsB or PafA system. e Visualization of the selected binding interface of the GP on the mKate2 surface. Red indicates hydrophobic amino acids (Ala, Val, Thr, Ile, Pro, Leu, Met, Phe, Tyr, Trp) on the mKate2 surface, yellow represents PatchDock residues. f Left: Design model of GPmKate2 in complex with mKate2. Right: Confocal imaging of the co-location of GPmKate2-McsB-eGFP (Green) with mKate2-PopZ (Red) at cell poles. g Plasmid map of the GPlad degradation system, with GP-McsB placed under the Ptet promoter and ClpCP under the PLtetO-1 promoter. h GP-guided arginine phosphorylation of McsBE121A and McsB for mKate2. i Fluorescence changes at 0, 3, and 6 h after GPlad induction, assessed by flow cytometry. j Intracellular mKate2 changes at 0, 3, and 6 h after GPlad induction, using laser scanning confocal microscopy. k Intracellular mKate2 abundance at 0 and 6 h after GPlad induction, detected by WB. All microscopy experiments were performed in triplicate with similar results. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file. (a) is created in BioRender https://BioRender.com/3ihz25i.
Results
Design and construction of the GPlad system
To select a marking protein (MP) to label the target protein for degradation upon spatial proximity, we selected two proteins known for their marking and degradation functions in prokaryotic cells: McsB, an arginine kinase from B. subtilis22, and PafA, a ubiquitin-like protein ligase from Mycobacterium tuberculosis23. To test the marking ability of McsB and PafA to neighboring proteins in E. coli, mKate2 was selected as the reporter protein, and a pair of high-affinity orthogonal heterodimers, 1/1′ (37ABXB_A and 37ABXB_B), were used as proximity scaffolds24. Monomer 1 was fused with McsB or PafA, and monomer 1′ was fused with the mKate2 (Supplementary Fig. 1a). To observe the spatial proximity, an in vivo co-localization observation experiment based on the cell pole localization protein PopZ was designed25 (Fig. 1b). Confocal microscopy showed that cells using 1/1′ as the fusion scaffold exhibited red and green fluorescent foci at the poles, indicating successful spatial proximity of McsB/PafA and mKate2 (Supplementary Fig. 2). Following spatial proximity, both McsB and PafA successfully marked mKate2 in E. coli, while their inactive mutants, McsBE121A and PafAH123A, failed to label mKate2 (Fig. 1b). Subsequently, we introduced the protease ClpCP and 20S, corresponding to McsB and PafA, respectively, to degrade the marked mKate2. In the McsB test strain, the fluorescence and the abundance of mKate2 decreased by 49.5% and 56.7%, respectively, after 6 h. In contrast, the PafA test strain exhibited no significant changes in mKate2 fluorescence or abundance compared to the control (Fig. 1c, d, Supplementary Fig. 3). Collectively, the PafA-20S system failed to degrade mKate2 likely due to incomplete assembly of the 20S proteasome in E. coli. Given the simplicity and good performance of the McsB system, which was chosen as the marking protein in the GPlad system.
Building on the recent development of de novo protein-binding proteins based solely on target protein structures26, we attempted to design a guide protein specifically tailored to bind the targeted protein (mKate2) to replace the 1/1′ heterodimers. The design procedure mainly included binding interface selection, guide protein library design, and candidate screening for suitable proteins. When selecting binding interfaces on the target protein surface, two design principles were followed: (i) avoiding arginine in the binding interface of the target protein to prevent interference from McsB, and (ii) exclusively selecting hydrophobic amino acids (Ala, Val, Thr, Ile, Pro, Leu, Met, Phe, Tyr, Trp) on the target protein surface as the PatchDock residues to enhance the design success rate of guide proteins. According to the design program referred to from Cao et al.26 (see “Methods” section), we designed the guide protein of mKate2. By analyzing the amino acid distribution on the surface of mKate2, a hydrophobic region on the top side of its barrel structure was selected as the binding interface, and A143, T145, Y195, V217, V219, Y222, and L225 were selected as PatchDock27 residues (Fig. 1e). A candidate pool of 60,000 binders was generated using RifGen28,29, Rifdock28,29, and two rounds of Interface Design26. This binder pool was then narrowed down based on six recommended parameters and further filtered according to four critical metrics (see “Methods” section). To efficiently obtain a guide protein that can bind to mKate2, we developed a two-round screening procedure based on an improved bacterial two-hybrid system30,31 and PopZ co-localization experiments25, and the top 30 candidates from the pool were selected for testing (Supplementary Note 1). Five guide proteins (GP01458, GP15750, GP01684, GP05757, GP12959) that significantly activated the BFP expression levels were selected based on the bacterial two-hybrid system (Supplementary Fig. 4). Confocal microscopy further revealed that the GP12959-eGFP-expressing strain formed most bright green fluorescent clusters at cell poles, indicating that GP12959 exhibited good solubility and successfully bound mKate2 in vivo (Fig. 1f). However, other guide proteins either failed to form distinct green fluorescent clusters at the cell poles or exhibited weaker fluorescence (Supplementary Fig. 5). Consequently, GP12959 (referred to as GPmkate2 hereafter) was selected as the guide protein targeting mKate2.
Next, the GPlad system was constructed in E. coli with three components: the guide and marking protein complex GPmkate2-McsB, the degradation machinery ClpCP, and the reporter protein mKate2. In this system, GPmkate2-McsB and ClpCP were expressed in a previously constructed low-copy plasmid backbone (p15A-Ptet-ParaBAD)32 to develop the plasmid pGPlad. Specifically, the expression of GPmkate2-McsB was controlled by the Ptet promoter, while ClpCP was regulated by the anhydrotetracycline (aTc) induced PLtetO-1 promoter. The LacI-induced mKate2 expression cassette was integrated into the genome to mitigate plasmid instability, where the PlacIq promoter was used to increase lacI expression to strictly control mKate2 expression (Fig. 1g). In the GPmKate2-McsB complex, GPmKate2 binds to a hydrophobic interface on mKate2, bringing McsB into proximity. McsB phosphorylates surface-exposed arginine residues on mKate2, which are subsequently recognized by ClpC. ClpC unfolds the phosphorylated target and translocates it to ClpP for proteolytic degradation. As shown in Fig. 1h, the phosphorylated mKate2 was detected after inducing GPmkate2-McsB expression for 2 h, while no arginine-phosphorylated mKate2 was observed in the GPmkate2-McsBE121A expressing strain. Compared to the control group, the GPmkate2-McsB expression led to (i) a 43.0% reduction in mKate2 fluorescence (Fig. 1i); (ii) a gradual decrease in mKate2 fluorescence over time (Fig. 1j); and (iii) a 40.8% reduction in mKate2 abundance (Fig. 1k). To confirm that the degradation was specifically induced by McsB in the GPlad system, we designed a control group co-expressing GPmkate2-McsB and the arginine dephosphorylase YwlE33 (Supplementary Fig. 6a). Co-expression did not significantly decrease mKate2 fluorescence, indicating that YwlE inhibited the specific marking of mKate2 by McsB (Supplementary Fig. 6b).
Optimization and characterization of the GPlad system
The degradation efficiency of the GPlad system was enhanced through three key approaches: (i) co-expression of the activator protein; (ii) reduction of ClpCP expression; and (iii) optimization of the linker design. First, McsA34,35, the activator protein of McsB, was introduced into the p15A plasmid, resulting in an 89% decrease in mKate2 fluorescence within 6 h, a 51.7% improvement over the absence of McsA (Fig. 2a). However, the expression of McsA resulted in cell growth inhibition (Supplementary Fig. 7), likely due to the nonspecific degradation from McsBA’s excessive marking capability. Reducing the aTc concentration had no significant effect in mitigating cell growth inhibition. Therefore, we optimized the expression of McsB and McsA by using weaker ribosome binding sites (RBS) (RBS29, RBS32, RBS31). Among them, the RBS32 for both McsB and McsA led to the least growth inhibition, while maintaining an 84.8% reduction in mKate2 fluorescence (Supplementary Fig. 8a), making it a candidate for further optimization. Next, the expression level of ClpCP was reduced to eliminate cell growth inhibition. Five weaker promoters (Pj23109, Pj23114, Pj23115, Pj23105, Pj23107) were used to replace the strong promoter PLtetO-1. The results indicated that utilizing Pj23105 to drive ClpCP expression led to an 80.8% decrease in mKate2 fluorescence within 6 h, with no observable growth inhibition (Fig. 2b, Supplementary Fig. 8b). To further investigate the cytotoxicity of individual components in GPlad, we separately expressed the four proteins McsB, McsA, ClpC, and ClpP. Growth assays revealed that expression of McsB and ClpC caused significant growth inhibition, reducing the OD600 by 45.8% and 26.4%, respectively. Furthermore, the inactive McsBE121A mutant and the N-terminal-deleted ClpC (ClpCΔNTD, Δ2-148, responsible for recognizing and binding arginine phosphorylation sites) abolished the growth inhibition (Supplementary Fig. 9). These results indicate that both McsB and ClpC disrupt normal cellular functions in E. coli. McsB likely interferes by phosphorylating specific arginine residues on critical proteins, impairing their function. More importantly, E. coli may inherently possess proteins with arginine phosphorylation-like modifications, enabling the N-terminal domain of ClpC to recognize these modifications and induce protein unfolding, leading to functional disruption. Finally, the impact of the linker length between GPmkate2 and McsB on the efficiency of GPlad was examined. The degradation of mKate2 fluorescence was approximately 80% when the GS linker length was 10, 15, or 20 amino acids; however, when the linker length increased to 30 amino acids, the mKate2 fluorescence only decreased by 56.3%. Therefore, a GS linker length of 15 amino acids was chosen for GPlad construction (Fig. 2c). To rule out the possibility that the intrinsic disorder of the His-tag contributes to GPlad-mediated degradation, we evaluated the degradation efficiencies of mKate2 and mKate2-6His. The results showed no significant difference in degradation rates between the two proteins (Supplementary Fig. 10), demonstrating that GPlad is indeed capable of degrading unmodified, structured proteins in vivo.

a Changes in fluorescence with the introduction of McsA in the GPlad system. Corresponding cells were induced with 20 ng/mL aTc for 6 h. b Evaluation of GPlad degradation with ClpCP expression controlled by Pj23105. Corresponding cells were treated with 20 ng/mL aTc for 6 h. c Optimization of the linker length between GP and McsB. Corresponding cells were treated with 20 ng/mL aTc for 6 h, and the mKate2 fluorescence was measured using a microplate reader. d, e. Binding interface selection (Left) and design models (Right) for GP complexes. Left: Target protein surfaces showing hydrophobic residues (red) and Patchdock-selected binding sites (yellow). Right: Designed complexes of GPErmB-ErmB and GPAroK-AroK, with binding sites (yellow) and targets (gray). f WB analysis of ErmB abundance in genomic expression after 2 h of GPErmB-McsB induction. g Spot plating assays of E. coli harboring GPlad for ErmB under 150 μg/mL erythromycin stress for 12 h on plates. h WB analysis of AroK abundance in genomic expression after 2 h of GPAroK-McsB induction. i Evaluation of changes in the kinase activity of AroK degradation following GPlad induction after 8 h. j Degradation assay of FGFR2 and VirB8. Corresponding cells were induced with 20 ng/mL aTc for 6 h, and protein abundance was measured by WB analysis. k Volcano plot showing fold changes in protein abundance from global proteomics analysis of E. coli cells induced with GPlad targeting mKate2 for 6 h. A two-sided unpaired t-test was performed on the relative quantitative value of each protein from the two sample groups. p < 0.01 was usually considered the threshold for significance. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file.
The GPlad system was characterized by four aspects: the fusion format of GP and McsB, the universality of target proteins, spatially dependent degradation capacity, and protein degradation specificity. First, we found that fusing the guide proteins to the C-terminus of McsB did not affect the spatial proximity but prevented the degradation of mKate2 (Supplementary Fig. 11). This fusion likely interfered with dimerization at the C-terminus of McsB, which was critical for its phosphorylation capability. Second, we evaluated the universality of target proteins using two endogenous proteins and two heterologous proteins. ErmB confers resistance to macrolide antibiotics, and its degradation renders E. coli sensitive to erythromycin36. AroK catalyzes the conversion of shikimate to shikimate-3-phosphate, and its degradation blocks the biosynthesis of aromatic amino acids and inhibits cell growth. Guide proteins GPErmB (Fig. 2d, Supplementary Fig. 12) and GPAroK (Fig. 2e, Supplementary Fig. 13) were designed to construct two GPlad systems targeting endogenous ErmB and AroK from their native genomic loci. Consequently, the ErmB abundance was decreased by 93.5% (Fig. 2f), and spot plating assays exhibited significant cell growth inhibition under 150 μg/mL erythromycin compared to the control cells (Fig. 2g). Additionally, the AroK abundance was decreased by 84.5% (Fig. 2h), resulting in a 94.2% reduction in shikimate kinase activity compared to the control strain after 8 h (Fig. 2i). We also investigated whether GPlad could degrade heterologous proteins in E. coli. The binders for human FGFR2 ectodomains (residues 22–366) and Rickettsia typhi protein VirB8 from Cao et al.26 were utilized as GPFGFR2 and GPVirB8 to construct GPlad. An MBP solubility-enhancing tag was added to FGFR2 (residues 22–366) to facilitate heterologous protein expression in E. coli. Western blot analysis indicated that after inducing GPlad, the protein abundances of MBP-FGFR2 and VirB8 decreased by 76.1% and 84.7%, respectively (Fig. 2j). Third, we evaluated the ability of GPlad to degrade mKate2-PopZ localized in cell poles, to test the spatially dependent degradation capacity of GPlad. As shown in Supplementary Fig. 14, GPlad reduced the fluorescence of mKate2-PopZ by 43.0% within 6 h. Although this reduction rate is lower than the degradation rate of mKate2 in the cytoplasm (86.0%), it still demonstrates that GPlad can target proteins in specific compartments. Lastly, the protein degradation specificity was studied from the cell growth lag time and quantitative proteomics. No lag time was observed when GPmKate2-McsB was induced to degrade mKate2, in contrast to the strain lacking GP guidance, which exhibited a lag time of 3.5 h. This finding suggested that the GPlad system has minimal off-target effects (Supplementary Fig. 15). To further evaluate the global impact of GPlad on the E. coli proteome, we conducted 4D-FastDIA quantitative proteomics. Among the 2758 proteins identified and quantified, mKate2 was the only protein with significantly decreased abundance after GPlad induction. This indicated that GPlad could induce the degradation of target proteins with high specificity while minimizing significant off-target degradation (Fig. 2k).
Extending the toolbox of GPlad
To extend the biological applications of GPlad, we developed three regulatory tools—antiGPlad, OptoGPlad, and GPTAC—to achieve reversible inhibition, optogenetic regulation, and biological chimerization of GPlad, respectively.
Our results in Supplementary Fig. 6 have demonstrated that the introduction of YwlE can effectively facilitate reversible control of GPlad degradation. However, to avoid introducing another heterologous protein, we sought to design a binder that can bind to the active site of McsB and inhibit its marking function (Fig. 3a). To achieve this, amino acids surrounding the key active residue E212 of the McsB protein (V187, I192, I196, P197, V206, A216, F221) were selected as PatchDock residues for binder design (Fig. 3b). The resulting binders were ranked following filtering, and the top 30 candidates were further evaluated through two rounds of bacterial two-hybrid tests and a PopZ-based co-localization experiment. Among these, Binder-05592 and Binder-03404 demonstrated targeted binding capabilities (Supplementary Fig. 16). Western blot analysis further revealed that Binder-03404 exhibited the most significant inhibitory effect on the arginine phosphorylation of McsB (Fig. 3c). Therefore, Binder-03404 was selected as the anti-protein for GPlad (hereafter referred to as antiMcsB). Subsequently, the antiMcsB gene was inserted into the pGPlad plasmid, with its expression regulated by the arabinose-inducible ParaBAD promoter and a strong RBS30, to evaluate its reversible control of GPlad (Supplementary Fig. 17a). When 1–2% arabinose was applied, the expression of antiMcsB did not result in significant changes in mKate2 abundance, while the control group exhibited a 91% decrease in mKate2 abundance (Fig. 3d, Supplementary Fig. 17b). These results suggested that an antiGPlad was constructed by introducing antiMcsB, effectively inhibiting the degradation process of GPlad.

a Schematic representation of antiGPlad. A binder was designed to specifically bind to the activation site of McsB, thereby inhibiting its marking ability. b Binding interface selection (Left) and design models (Right) for antiMcsB complexes. Left: McsB surface showing hydrophobic residues (red) and Patchdock-selected binding sites (yellow). Right: Designed complexes of antiMcsB-McsB, with binding sites (yellow) and targets (gray). c Inhibition of McsB phosphorylation by antiMcsB expression. Corresponding cells were treated with 1% arabinose to induce antiMcsB, leading to the inhibition of arginine phosphorylation, which was detected by western blotting. d Inhibition of protein degradation by antiMcsB induction. Corresponding cells were treated with 0.5% arabinose to induce antiMcsB, inhibiting protein degradation. e Schematic representation of OptoGPlad. Expression of OptoGPlad was inhibited by EL222. Under blue light (450 nm) and arabinose induction, the OptoGPlad system was activated, forming a complex with GPmKate2-pMagFast1 and nMagHigh1-McsB, which induced mKate2 degradation. In the darkness, the complex dissociates, halting OptoGPlad expression and thus stopping degradation. f The plasmid used in OptoGPlad; g OptoGPlad-mediated protein degradation. Corresponding cells were treated with 1% arabinose to induce OptoGPlad. h Schematic representation of GPTAC. In GPTAC, one GP was bound to McsB while the other was bound to the target protein, positioning McsB close to the target protein. i Binding interface selection (Left) and design models (Right) for GP complexes. Left: McsB surface showing hydrophobic residues (red) and Patchdock-selected binding sites (yellow). Right: Designed complexes of GPMcsB-McsB, with binding sites (yellow) and targets (gray). j GPTAC-mediated protein degradation. Corresponding cells were treated with 1% arabinose to induce GPTAC. k Schematic representation of GPTAC-mediated mKate2 and eGFP degradation. The GPTAC for mKate2 and eGFP was under the control of ParaBAD. l Design model of GPeGFP in complex with eGFP, with binding sites (yellow) and targets (gray). m GPTAC-induced mKate2 and eGFP degradation. Corresponding cells were treated with 2% arabinose to induce GPTAC. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file. (a, e, h, k) are created in BioRender https://BioRender.com/8h6634z.
To achieve optogenetic control of GPlad, we designed an OptoGPlad system based on a blue light-inducible Magnet system. The Magnet system, composed of pMagFast1 and nMagHigh1, demonstrated strong dimerization under blue light compared to the original VVD, with a switch-off kinetics half-life (t1/2) of only 10 min37. By leveraging its high affinity and rapid dissociation, we fused nMagHigh1 with McsB (nMagHigh1-McsB) and pMagFast1 with GPmkate2 (GPmkate2-pMagFast1). Under blue light (450 nm), the strong interaction between pMagFast1 and nMagHigh1 brought McsB in proximity to mKate2, while in the darkness, their rapid dissociation prevented McsB from marking mKate2 (Fig. 3e). When expressing GPmkate2-pMagFast1 under blue light, green fluorescence clustered at the poles of the cells, whereas the control group displayed a uniform distribution of green fluorescence (Supplementary Fig. 18a). This observation indicated that the binding affinity of the Magnet system remained intact and that spatial proximity was successfully achieved. Subsequently, we constructed the OptoGPlad system for mKate2 degradation by co-expressing GPmkate2-pMagFast1 and nMagHigh1-McsB in the pGPlad plasmid. To prevent non-targeted degradation due to prolonged expression of nMagHigh1-McsB following dissociation induced by aTc, we utilized the blue light-induced EL222 protein and the PBlind-V1 promoter to replace aTc induction. This modification allows for the shutdown of GPlad component expression under dark conditions38,39 (Fig. 3f). When 1% arabinose was added, the fluorescence of mKate2 decreased by 77.4% after 6 h compared to the control group under blue light (450 nm) (Supplementary Fig. 18b, Fig. 3g), demonstrating that OptoGPlad could precisely and remotely degrade intracellular proteins.
Based on the conventional PROTAC design, we also constructed a biological guide protein targeting proteolysis chimera termed GPTAC. GPTAC consisted of two guide proteins connected by a linker (GPtarget-GPMcsB), where GPtarget recognized and bound to the target protein such as mKate2, and GPMcsB recognized and bound to McsB, thereby bringing McsB and mKate2 into spatial proximity (Fig. 3h). The GPMcsB was designed by avoiding the active site and the dimerization site of McsB, ensuring that its phosphorylation and dimerization functions remain unaffected (Fig. 3i, Supplementary Fig. 19). After inducing the expression of GPmKate2-GPMcsB, green fluorescence was concentrated at cell poles, accompanied by red fluorescence, indicating that GPTAC can bring McsB and mKate2 into spatial proximity (Supplementary Fig. 20a). Subsequently, we constructed GPTAC for mKate2 degradation in the pGPlad plasmid (Supplementary Fig. 20b). When 1% arabinose was used, the fluorescence of mKate2 decreased significantly by 79% (Supplementary Fig. 20c, Fig. 3j). These results indicated that GPTAC effectively facilitated the degradation of the target protein. Furthermore, the multiple targeted degradation ability of GPTAC was assessed by constructing a GPTAC for degrading fluorescent proteins mKate2 and eGFP (Fig. 3k). We designed a guide protein specific for eGFP (Fig. 3l; Supplementary Fig. 21) and incorporated the expression cassette of GPeGFP-GPMcsB, along with RBS34, following GPmKate2-GPMcsB. When 2% arabinose was used, the fluorescence of eGFP and mKate2 decreased by 68.3% and 77.1%, respectively (Supplementary Fig. 22, Fig. 3m). These results demonstrated that GPTAC could simultaneously target and degrade multiple proteins.
Building programmable protein switches using GPlad and antiGPlad
Based on the functionalities of GPlad and antiGPlad, we developed three types of protein circuits: a reversible switch, an amplifier, and an oscillator. The first reversible switch was built by an ON- and OFF-switch unit to facilitate protein degradation, followed by accumulation. As illustrated in Fig. 4a, the ON-switch unit consists of a GP-McsB, which marks and degrades mKate2, while the OFF-switch unit contains an antiMcsB, which binds to McsB to protect mKate2 from degradation. The kinetic analysis showed that mKate2 fluorescence initially decreased during the first three hours following GPlad induction, then increased between three to six hours after antiGPlad induction (Fig. 4b). Specifically, mKate2 abundance decreased by 82.9% within three hours of GP-McsB (ON-switch), and when 1% arabinose was added to induce antiMcsB expression, mKate2 levels subsequently increased by 973.8% within three hours (OFF-switch) (Fig. 4c).

a Schematic representation of the reversible switch. b Degradation and accumulation of mKate2 controlled by the reversible switch. Initially, mKate2 expression was induced for 2 h by adding 0.1 mM IPTG (t = 0 h time point). Subsequently, 20 ng/mL aTc was added to induce GPlad, leading to the degradation of mKate2. Finally, 1% arabinose was added to induce antiMcsB, thereby halting mKate2 degradation and allowing for its gradual accumulation. c Flow cytometry was used to continuously monitor the degradation and accumulation of mKate2 under the control of a reversible protein switch. d Designed complexes of GPLacI-LacI, with binding sites (yellow) and targets (gray). e Schematic representation of an amplifier. The LacI and mKate2 were expressed from the genome, and the GP-McsB was expressed from plasmid pGPlad. f Evaluation of the minimum IPTG induction concentration threshold with the assistance of an amplifier in E. coli. Corresponding cells harboring the amplifier were treated with different concentrations of aTc to detect mKate2 fluorescence by a microplate reader. g Schematic representation of an oscillator. The LacI and mKate2 were expressed from the genome, while the GP-McsB and antiMcsB were expressed from plasmid pAntiGPlad. h Optimization of the antiMcsB promoter to achieve oscillatory changes in intracellular mKate2 abundance. Data represent mean ± s.d. from three independent experiments. i Oscillation curve of mKate2. The addition of 20 ng/mL aTc at 6 h extended the oscillation period to 14 h. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file. (a, e, g) are created in BioRender https://BioRender.com/912hekk.
The second protein circuit developed was an amplifier (Fig. 4d, Supplementary Fig. 23). In this design, IPTG relieved LacI repression on both mKate2 and GP-McsB expression, while the expression of GP-McsB subsequently triggered LacI degradation, further alleviating LacI repression of mKate2. This interaction created a feedback loop that amplified mKate2 expression (Fig. 4e). We first evaluated whether this amplifier could lower the minimum IPTG induction threshold. Flow cytometry analysis revealed that 0.1 μM IPTG was sufficient to induce mKate2 expression and accumulation, whereas cells lacking the amplifier required 5 μM IPTG (Fig. 4f). Moreover, the amplifier significantly enhanced mKate2 fluorescence by approximately 7.6-fold at 10 μM IPTG (Fig. 4f).
Finally, we constructed a programmable oscillator to periodically regulate target protein abundance. In this oscillator design, GP-McsB targets and degrades LacI, leading to a reduction in LacI levels that permits the expression of antiMcsB. Subsequently, antiMcsB inhibits GP-McsB through targeted binding, resulting in a negative feedback loop among the three components that controlled mKate2 abundance oscillations (Fig. 4g). Next, 20 ng/mL aTc was added to initiate oscillation and monitor the changes in mKate2 fluorescence. However, initial tests showed that oscillation was not observed, and the mKate2 fluorescence was very low, likely due to excessive antiMcsB expression disrupting the loop. To address this issue, we replaced the ParaBAD promoter with four weak promoters: Pj23118, Pj23107, Pj23116, and Pj23117. Oscillations in mKate2 were observed with the Pj23107 and Pj23116 promoters, with the Pj23116 promoter maintaining the oscillation period for six hours, compared to four hours for Pj23107 (Fig. 4h). The decline in mKate2 fluorescence after six hours suggested that mKate expression was inhibited by LacI. To address this, 20 ng/mL of aTc was added at six hours to increase GP-McsB expression and promote LacI degradation. Degradation kinetics showed that mKate2 continued to exhibit periodic fluctuations for up to 14 h (Fig. 4i). These results suggested that balancing the levels of the three mutually inhibitory components was crucial for the success of the oscillator.
Accelerating adaptive laboratory evolution using OptoGPlad
To verify the application of protein circuits in synthetic biology, the OptoGPlad system was employed to degrade a key protein in the DNA mismatch repair system under blue light, thereby accelerating the adaptive laboratory evolution (ALE) of E. coli40,41. This degradation could be halted in darkness, enabling strains with optimal performance to cease mutations and achieve enrichment in culture—a capability that cannot be achieved through direct knockout of the DNA mismatch repair genes (Fig. 5a).

a Schematic representation of OptoGPlad-assisted ALE. Under light exposure, continuous intracellular degradation of MutH increased the mutation rate and accelerated strain evolution. In the darkness, degradation of MutH ceased, allowing beneficial mutant strains to accumulate. b Binding interface selection (Above) and design models (Down) for GP complexes. Above: MutH surface showing hydrophobic residues (red) and Patchdock-selected binding sites (yellow). Right: Designed complexes of GPMutH-MutH, with binding sites (yellow) and targets (gray). c Left: WB analysis of intracellular MutH abundance after two hours of light-induced OptoGPlad expression. Right: WB analysis showing MutH accumulation four hours after transitioning to dark conditions. d Changes in the mutation rate of E. coli under the influence of OptoGPlad and PCA. Data represent mean ± s.d. from six biological replicates, each consisting of 20 parallel cultures. e Schematic representation of the evolution process and strain screening procedure. E. coli PCA06 was initially illuminated under blue light for 8 h and subsequently incubated for 16 h, representing round n. Following every n + 19 generation, cells with the highest fluorescence intensity were sorted by flow cytometry and used as chassis strains for the next round of evolution. f Propidium iodide (PI) staining was performed to assess the survival rate of strains at different stages of evolution in a medium containing 40 g/L PCA. The percentage of fluorescence intensity corresponded to the survival rate, where “−“ represented the live cell population and “+“ denoted the dead cell population. E. coli PCA05 strain cultured in fermentation medium without PCA was used as the control group. g PCA accumulation curves of E. coli PCA05 and E. coli PCA07 in the 5-L fermenter. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file. (a, e) are created in BioRender https://BioRender.com/ansn29h.
Protocatechuic acid (PCA) is a natural phenolic acid with antioxidant properties and is widely utilized in pharmaceuticals, health foods, cosmetics, and biomaterials42. However, PCA is highly toxic to E. coli, significantly hindering its biosynthetic efficiency43. Therefore, screening for PCA-tolerant strains is critical for developing efficient PCA cell factories. To achieve this goal, the mismatch repair endonuclease MutH was recruited in the OptoGPlad system. Inactivation of the MutH could increase the mutation rate of E. coli from 0.0022 to 0.32 mutations/h44. We designed the guide protein GPMutH (Fig. 5b, Supplementary Fig. 24) and fused it with pMagFast1 to construct the plasmid pOptoGPlad for targeting MutH. Subsequently, the plasmid pOptoGPlad was transformed into a previously constructed PCA-producing strain E. coli PCA0343 (termed E. coli PCA06), to accelerate its ALE under protocatechuic acid (PCA) stress. Under blue light, two hours after the addition of 1% arabinose, the intracellular abundance of MutH decreased to only 13.5% of that in the control group. However, after being transferred to darkness for four hours, MutH levels recovered, increasing 11.2-fold compared to those under light conditions (Fig. 5c). Additionally, the induction of OptoGPlad led to a 215-fold increase in the mutation rate under 10 g/L PCA, representing a 1613-fold increase in comparison to the mutation rate without PCA (Fig. 5d).
Then, we initiated a new cycle of ALE of E. coli PCA06 under PCA stress. Each generation comprised two phases: the mutation phase (0–8 h), during which blue light activated OptoGPlad to degrade MutH and increase the mutation rate, and the enrichment phase (8–24 h), during which OptoGPlad was turned off in the dark condition, allowing MutH accumulation and the enrichment of beneficial mutants. We introduced the PCA-responsive PyhcN-mKate sensor to perform high-throughput screening via flow cytometry every 20 generations, selecting the strains with the best performance for the next round of evolution43 (Fig. 5e). Through 100 generations, an evolved strain, E. coli PCA07, was screened, whose survival rate was increased from 45% to 88% under 40 g/L PCA. However, the strain E. coli PCA0543, which underwent traditional ALE (without OptoGPlad), only reached a 57% survival rate at 100 generations and ultimately achieved a survival rate of 80% after 220 generations (Fig. 5f).
Finally, we conducted fermentation of E. coli PCA07 in a 5 L fermentor. The fermentation time was reduced from 32 h to 28 h, resulting in a productivity of 1.73 g/L/h, which represents a 27.2% increase compared to E. coli PCA05 (Fig. 5g). Additionally, at the end of the fermentation, E. coli PCA07 achieved a survival rate of 76.2%, whereas E. coli PCA05 only had a survival rate of 41.2% (Supplementary Fig. 25). Genome re-sequencing identified numerous mutants in E. coli PCA07 genome, including several transcriptional regulators (such as RpoS and AcrR) and transporters (OmpF, TnaA, and Puup) (Supplementary Data 2), providing insights into the PCA tolerance mechanism of E. coli. These results demonstrated that OptoGPlad could be utilized to accelerate adaptive laboratory evolution.
Engineering dynamic pathway regulation using GPTAC
We also evaluated GPTAC for targeted protein degradation, which facilitated the rapid degradation of both newly synthesized and pre-existing proteins. This feature was particularly advantageous in metabolic engineering, as the rapid dynamic regulation of enzyme levels can prevent carbon flux loss caused by pre-existing enzymes or slow transcriptional regulation21,45,46.
3-Dehydroshikimic acid (DHS) is a key precursor for various valuable polymers and pharmaceuticals. The direct knockout of the aroE gene (encoding shikimate dehydrogenase) in E. coli enables the accumulation of DHS; however, this disruption also impairs the biosynthesis of aromatic amino acids, requiring the use of expensive supplements such as aromatic amino acids in the medium47,48. To address this issue, GPTAC was employed to dynamically regulate AroE abundance for DHS production. Specifically, aroE was normally expressed during the early stages of fermentation to facilitate cell growth. Once the cell density reached the desired level, GPTAC was activated to degrade AroE, thereby redirecting carbon flux from cell growth to DHS synthesis (Fig. 6a). To achieve this goal, we designed the guide protein GPAroE and fused it with GPMcsB to develop GPMcsB-GPAroE (Fig. 6b, Supplementary Fig. 26). To prevent glucose inhibition of the arabinose-induced GPMcsB-GPAroE, we replaced the system with an IPTG-inducible promoter in the pGPTAC plasmid (Supplementary Fig. 27). Additionally, to enable the shikimate-producing E. coli SA05 to synthesize aromatic amino acids47, the genes aroK (encoding shikimate kinase I) and aroL (encoding shikimate kinase II) were reintroduced into its genome, resulting in the strain termed E. coli DHS01. After implementing GPTAC in E. coli DHS01, the AroE level decreased by 82.6% within one hour upon the addition of 0.2 mM IPTG (Fig. 6c), and cell growth was severely inhibited in the M9 medium (Fig. 6d), demonstrating that GPTAC achieved rapid degradation of AroE in E. coli DHS01.

a Schematic representation of GPTAC-controlled metabolic flux regulation. In the DHS production phase, GPTAC expression not only continuously degraded AroE expressed from the genome but also rapidly eliminated previously expressed AroE, enabling a swift blockage of metabolic flux and facilitating DHS accumulation. b Binding interface selection (Left) and design models (Right) for GP complexes. Left: AroE surface showing hydrophobic residues (red) and Patchdock-selected binding sites (yellow). Right: Designed complexes of GPAroE-AroE, with binding sites (yellow) and targets (gray). c A dose-escalation experiment demonstrated GPTAC-mediated AroE degradation after one hour of treatment with different concentrations of IPTG (0.05 mM, 0.1 mM, 0.2 mM). d Detection of AroE degradation during the growth of E. coli DHS01 in M9 medium without essential amino acids. Data are reported as the average of three independent repeats. e Effect of continued addition of aTc or IPTG at 40 h on DHS production by E. coli DHS01. The first addition of the inducer occurred at 24 h. Furthermore, a decrease in DHS accumulation rate was observed at 40 h, prompting a second addition of the inducer at that time. All experiments are presented as mean ± s.d. from three biologically independent replicates. Source data are provided as a Source Data file. (a) is created in BioRender https://BioRender.com/a1rvfbu.
Next, we conducted fermentation of E. coli DHS01 in a 5-L fermentor. By optimizing the induction time and concentration, the addition of 1 mM IPTG and 50 ng/mL aTc after 24 h induced GPTAC, resulting in a DHS titer of 68.5 g/L after 60 h in NBS minimal medium (Supplementary Fig. 28). However, we observed that from hours 40 to 60, DHS accumulation slowed while cell growth continued, reaching an OD600 of 70. This indicated insufficient degradation of AroE after 40 h. This issue may be attributed to the decay of aTc (with an average half-life of 15–20 h)49 or insufficient IPTG levels. Consequently, an additional 50 ng/mL aTc or 1 mM IPTG was added at 40 h. As a result, the DHS titer reached 92.6 g/L with minimal changes in OD600 when adding 50 ng/mL aTc, whereas the addition of IPTG had no significant effect (Fig. 6e).
To evaluate the advantages of GPTAC over conventional DNA-level regulation, we employed CRISPR interference (CRISPRi), which also does not require pre-modification of the aroE sequence in the genome to inhibit aroE expression. Five sgRNAs were designed, and the most effective one, sgRNA-D, was identified by fusing eGFP with aroE and detecting the resulting changes in fluorescence (Supplementary Fig. 29a). Then, the CRISPRi system targeting aroE was introduced into E. coli SA05 for fermentation. However, only 74.8 g/L of DHS was produced, and the OD600 increased to 62 (Supplementary Fig. 29b). These results demonstrated the advantages of GPTAC in rapidly switching metabolic flux.
Discussion
In this study, a tunable, plug-and-play, and pre-editing-free method termed GPlad was proposed for targeted protein degradation based on de novo protein design and post-translational modification. Leveraging de novo protein design, we further developed antiGPlad, OptoGPlad, and GPTAC, enabling reversible inhibition, optogenetic regulation, and biological chimerization, thereby expanding their potential applications and versatility. Furthermore, these tools were utilized to design programmable protein switches, accelerate adaptive laboratory evolution, and rewire metabolic flux in microbial cell factories, demonstrating the significant utility of GPlad in synthetic biology, cellular activity, and metabolic engineering.
To the best of our knowledge, GPlad represents the first application of a de novo-designed guide protein for targeted protein degradation in vivo. Compared with current bacterial protein degradation technologies, GPlad exhibits both differences as well as unique advantages. GPlad shows significant differences from existing bacterial protein degradation technologies in terms of degradation mechanisms, target protein scope, and specificity. (i) Degradation mechanism: The representative bacterial degrader BACPROTAC recruited BsClpC or MtClpC1 close to target proteins for proteasomal recognition and degradation12, while the degron-fused based target protein degradation was controlled by mf-Lon15 or TEVP50. In contrast, GPlad utilized an in vivo-expressed guide protein and a fusion complex to arginine-phosphorylate the target protein, enabling its recognition and degradation by the corresponding protease. (ii) Range of targeted proteins: While degron fusion could theoretically be attached to the terminal of any proteins, its effectiveness might be constrained by the efficiency of degron integration. BacPROTAC specifically recognizes proteins that contain pArg analog. In contrast, GPlad-targeted proteins with hydrophilic arginine residues, which constituted an average of 5.7% of most proteins51. This suggested that GPlad exhibited the potential to target a broader spectrum of proteins for degradation. Besides, previous studies suggest that target proteins with low folding propensity or containing intrinsically disordered domains (IDDs) are more susceptible to ClpCP-mediated degradation12,52. To validate this, we calculated the folding propensities of GPlad-targeted proteins (mKate2, ErmB, AroK, MBP-FGFR2, VirB8, and LacI) and predicted their IDDs (see Supplementary Note 2). Our results demonstrate that GPlad degrades targets meeting either criterion: folding propensity <0 (indicating inherent structural instability) or presence of IDDs. For instance, mKate2 lacked predicted IDDs but exhibited a folding propensity of −0.002114, reflecting its susceptibility to unfolding, and achieved an 80.8% degradation rate. However, degradation efficiency is not only determined by folding propensity or IDDs. For example, larger proteins (e.g., LacI and MBP-FGFR2), showed lower degradation rates (74.2% and 76.1%, respectively) compared to smaller targets like ErmB (27 kDa, 93.5%) and VirB8 (25 kDa, 84.7%), suggesting a positive correlation between protein size and degradation efficiency. This implies that the degradation of the target integrates multiple factors—structural flexibility (governed by folding propensity/IDDs), protease accessibility, and possibly substrate size. (iii) Specificity: Unlike degron systems, where specificity is determined by protease recognition of a fixed degron tag, GPlad employs a dual-component specificity mechanism: the guide protein binds the target protein, and the protease recognizes the arginine phosphorylation residues. This dual specificity acts as a biological logic gate, requiring both components to act in concert for degradation, thereby enhancing target specificity. In contrast to BacPROTAC, which relies on small ligand-protein interactions, GPlad utilizes protein-protein interactions between the guide protein and the target protein. This distinction provides flexibility in selecting specificity mechanisms tailored to different applications. More importantly, GPlad offers unique advantages in operational simplicity and cost efficiency. (i) Operational simplicity: Degron fusion requires the pre-insertion or fusion of bacterial degron sequences at one end of the target gene via CRISPR or homologous recombination53. This pre-editing process was time-consuming and might lead to genome instability and disruption of target protein function. In contrast, GPlad featured a permanently designed guide protein that can be widely employed through a plug-and-play plasmid. (ii) Cost control: BacPROTAC faced challenges such as complex ligand screening and high synthesis costs. In contrast, GPlad did not necessitate in vitro synthesis and could be produced on a large scale through fermentation, thus lowering production expenses.
Various GPlad-derived regulatory tools—antiGPlad, OptoGPlad, and GPTAC—could be developed to provide unique features and broader application potential. Current strategies for inhibiting bacterial protein degradation primarily involve two approaches. The first approach uses growth-dependent promoters PrrnC to control GFP-ssrA expression. Degradation occurs during the exponential growth phase, but stops in the stationary phase54. Another method inserts a TEV protease cleavage site (TEVpS) between GFP and LAA to form GFP-TEVpS-LAA, where TEVp cleaves TEVpS to prevent GFP degradation18. However, the first approach suffers from poor tunability and slow response, while the second requires target protein modification and is labor-intensive. In contrast, antiMcsB offered a straightforward method to rapidly halt protein degradation in vivo through plasmid expression, featuring high controllability and operational simplicity. Light-controlled regulation of bacterial protein degradation employs two main strategies: light-induced degron degradation and dimerization. The former developed a blue-light-inducible LOVdeg tag, enabling efficient degradation of LacI, CRISPRa, and AcrB in E. coli55. The latter uses blue-light-inducible VVD dimers to induce spatial proximity between split TEV protease fragments (nTEVp and cTEVp), triggering degradation. OptoGPlad adopts an optogenetic dimerization approach, eliminating the need for pre-modification of the target. More importantly, the Magnet system56—featuring stronger binding affinity and faster dissociation kinetics—outperforms the VVD system by reducing homotypic interference while enhancing interaction efficiency and dissociation dynamics37. Recently, cost-effective biodegraders EndoTag have achieved remarkable advancement in eukaryotic cells57. However, biodegraders for bacteria remain unexplored. Here, GPTAC, the protein degrader developed for bacteria, features a simple design (consisting of only two guide proteins), a compact size (approximately 130 amino acids), and excellent orthogonality (due to its de novo design). Beyond these advancements, GPlad could be further enhanced by developing high-affinity competitive binding proteins against the guide proteins to create more intricate and sophisticated protein circuits58,59, as well as improving the marking capability of McsB through protein engineering34,60. GPlad demonstrated considerable potential in designing programmable protein circuits, accelerating adaptive laboratory evolution, and dynamically rewiring metabolic flux in microbial cell factories. First, our GPlad/antiGPlad design provided an alternative tool for constructing protein circuits in bacteria. The use of protein degradation systems to construct biological circuits has achieved progress in bacteria. For instance, Gao et al. engineered different viral proteases and their corresponding recognition sequences as modular components to create dynamic regulation circuits, inverters, and oscillators3. Besides, based on mf-ssrA and repressor LacI and TetR, a transcription-based toggle switch was created to control core bacterial processes and antibacterial targets11. Herein, three regulatory tools, including reversible switches, amplifiers, and oscillators, were developed based on GPlad/antiGPlad. Notably, the amplifier reduces the minimum activation threshold of IPTG by 50-fold while amplifying the signal by 7.6-fold. The oscillator could sustain oscillations for 14 h, extending the duration by 8 h compared to our previously constructed protease-based oscillator21. Second, OptoGPlad could be used to regulate cell activities such as mutation rates. Traditional methods for altering cell mutation rate included deleting genes involved in the DNA mismatch repair system61, using mutated DNA polymerases62, and employing DNA deaminases63. However, these methods lacked a system for halting mutations and enriching high-performance strains. OptoGPlad-controlled degradation of MutH divided the evolutionary process into mutation and enrichment phases, reducing the evolution time of E. coli under PCA stress by 120 generations. Third, GPTAC exhibited a significant role in metabolic engineering. Traditionally, dynamic regulation of carbon flux in metabolic pathways relied on transcriptional regulation methods, such as biosensor64, CRISPRi65, and gene circuits66,67 were adopted to control the gene expression of key enzymes. However, these methods often suffered from delayed response and inevitable noise68. In contrast, GPTAC could directly target key enzymes, achieving rapid redirection of carbon flux while minimizing losses compared to the CRISPRi method.
In the future, we believe that GPlad can be further improved in the following aspects to endow it with better performance and broader universality. First, the degradation efficiency of GPlad can be further optimized. Although GPlad requires an additional mark step compared with the mf-ssrA system, it achieves similar degradation efficiency to the mf-ssrA system in E. coli (Supplementary Table 7). This result indicates that the marking process of GPlad is very rapid and has less role in degradation efficiency, with efficiency primarily limited by the ATP-dependent unfolding and hydrolysis activity of the ClpCP protease. Therefore, engineering the ClpCP activity represents critical optimization targets for further enhancing the degradation efficiency of GPlad. Second, the current GPlad system requires four heterologous proteins. Screening for the enzymes inherent in E. coli that are responsible for degradation marking could significantly optimize the GPlad system. Since expression of ClpCΔNTD alone does not inhibit growth in E. coli, this suggests the presence of endogenous proteins with arginine phosphorylation-like modifications in E. coli that allow ClpC NTD to recognize and bind these modifications, triggering unfolding. This finding implies that investigating which proteins are degraded upon ClpC overexpression, combined with analyzing their PTMs and degradation pathways, could provide a more effective strategy for uncovering analogous arginine phosphorylation-mediated degradation mechanisms in E. coli. Besides, the current development of large-scale identification of enzymes similar to McsB in E. coli and other bacteria through omics mining provides a powerful technology69. Third, exploring the transferability of GPlad. In theory, both the GPlad system and its underlying design principle possess excellent transferability to other bacteria. Since GPlad operates as a complete, self-contained labeling and degradation system that does not rely on host-specific components, it should theoretically be transferable to most bacterial species. The key requirements for successful implementation include: (i) compatibility of the expression system, (ii) maintenance of component activity, and (iii) proper system assembly. More importantly, the core concept of using de novo-designed guide proteins to drive targeted protein degradation offers even broader expansion potential. By leveraging endogenous degradation-tagging systems native to host organisms, we can avoid introducing heterologous proteins. For instance, the arginine phosphorylation system comes from B. subtilis, which means that only expressing GP-McsB or GPTAC would enable targeted protein degradation, potentially offering better performance than in E. coli. In Corynebacterium glutamicum, an important industrial strain, there exists a Pup-like ubiquitination system component (CaPafA)70. Introducing guide proteins to create a GP-guided CaPafA system could enable endogenous targeted protein degradation. Finally, affinity measurements demonstrate that GPAroE remains efficiently degraded AroE at a KD of 710 nM, indicating that the binding affinities between the guide protein and target protein around 1 μM are sufficient for marking and degradation, highlighting the robustness of the GPlad system. However, the design of guide proteins remains time-consuming, and the binding affinity can be further increased to avoid non-target degradation. Recently, many de novo design methods for binders, such as RFdiffusion71, BindCraft, which can reduce the range of candidate proteins and design time.
Methods
Strains and plasmids
All bacterial strains and plasmids used in this study are listed in Supplementary Data 4 and 5. The construction maps of key plasmids are supplied in Supplementary Fig. 30. Escherichia coli JM109 and E. coli DH5α served as the hosts for vector construction. For the degradation assays, E. coli MG1655pro* (ΔlacI, ΔaraC, ΔtetR) was used. E. coli PCA05 and E. coli SA05 were employed for MutH and AroE degradation assays, respectively. E. coli BTH101 was utilized for bacterial two-hybrid assays. The synthetic low-copy plasmid p15A-Ptet-ParaBAD, previously constructed, was used as a backbone for developing GPlad, antiGPlad, OptoGPlad, GPTAC, protein circuits, bacterial two-hybrid assays, and PopZ-based spatial proximity experiments. The synthetic plasmid pEtrcQ, a medium-low-copy vector with enhanced lacI expression driven by the PlacIq promoter to minimize target protein leakage, was constructed in this study and used as the backbone for reporter plasmids. Genome editing was conducted using pCas9 and pTargetF, while dCas9 and pCDF-CriRNA were employed to suppress aroE expression. The sequences of promoters and ribosome binding sites (RBS) utilized in this study are detailed in Supplementary Table 1.
Genetic manipulations
CRISPR-Cas9 technology was employed for the genetic manipulation of the E. coli genome, enabling chromosomal edits such as deletions and replacements of genetic sequences. All plasmids were constructed using standard ligation cloning techniques from Takara Bio (Dalian, China) or assembled using the 2× MultiF Seamless Assembly Mix from ABclonal (Wuhan, China). The coding sequences for mcsB, mcsA, clpC, clpP, and ywlE were PCR-amplified from B. subtilis strain 168. Additionally, sequences for 1/1′ (37ABXB_A and 37ABXB_B), PafA, Pup(E), 20S (PrcA(Δ2–8) and PrcB(Δ2–57)), FGFR2(22–366), VirB8, T25, T18, pMagFast1, and nMagHigh1 were synthesized by Suzhou Genewiz Biotechnology with codon optimization for E. coli. All protein sequences used in this study are listed in Supplementary Data 3. To facilitate the detection of target protein degradation via Western blotting, a 6 × His-tag was added to the C-terminus of all target proteins through either gene cloning or CRISPR-based genome integration. For the repression of aroE, suitable sgRNAs were designed using the CHOPCHOP web tool (http://chopchop.cbu.uib.no/), and the selected sgRNAs are listed in Supplementary Table 4. Besides, the key primers used in this study were listed in Supplementary Table 3.
Culture conditions
Luria-Bertani (LB) medium was used for cell growth, and plasmids and strains construction. M9 minimal medium containing 1% glucose and 0.05% casamino acids (M9GC) was used for the degradation assay15. For the ALE and PCA production of E. coli PCA05, the medium contains 10 g/L glucose, 5 g/L yeast tract, 3 g/L KH2PO4, 2 g/L MgSO4·7H2O, 5 g/L (NH4)2SO4, 2 g/L citric acid, 0.4 g/L L-phenylalanine, 0.4 g/L L-tyrosine, 0.4 g/L L-tryptophan, 10 mg/L biotin, 1 mL metal ion solution. For DHS production, the NBS minimal medium contains 3.5 g/L KH2PO4, 5.0 g/L K2HPO4, 3.5 g/L (NH4)2HPO4, 0.25 g/L MgSO4·7H2O, 15 mg/L CaCl2·2H2O, 0.5 mg/L vitamin B1, 1 ml/L trace element solution (1.6 g/L FeCl3, 0.2 g/L CoCl2·6H2O, 0.1 g/L CuCl2, 0.2 g/L ZnCl2·4H2O, 0.2 g/L NaMnO4, 0.05 g/L H3BO3, dissolve in 0.1 M HCl), and 5% glucose was used21. Kanamycin (50 mg/L), spectinomycin (30 mg/L), ampicillin (100 mg/L), and chloramphenicol (30 mg/L) were added appropriately.
Bacterial two-hybrid assay execution
The GP candidates were first synthesized by Suzhou Genewiz Biotechnology (with codon optimization for E. coli) and subsequently cloned into the bacterial two-hybrid assay plasmid pBTH. Both pBTH and the reporter plasmid pBTHR were co-transformed into E. coli BTH101, which was cultured overnight in an LB medium supplemented with chloramphenicol and ampicillin. The following morning, the culture was inoculated into 30 mL of fresh LB medium containing antibiotics at an initial OD600 of 0.01 and grown at 37 °C for 2 h until the OD600 reached 0.1. Then, 100 ng/mL of aTc, 1% arabinose, and 0.1 mM IPTG were simultaneously added to three replicates of the cultures in shake flasks. The flasks were incubated at 37 °C for 6 h. After incubation, samples were diluted 1:10 in 200 μL of fresh LB containing antibiotics and inducers for measurement. The BFP signal was measured using the Synergy H1 Multi-Mode Microplate Reader (BioTek Instruments, USA) with an excitation wavelength (EX) of 402 ± 5 nm and an emission wavelength (EM) of 457 ± 10 nm. OD600 was also measured using the Synergy H1 Microplate Reader.
Confocal imaging of cells
The GPs selected from the bacterial two-hybrid assay were cloned into the PopZ co-localization plasmid, pCoGP, and transformed into E. coli MG1655pro*. After 8 h of induction with 50 ng/mL aTc and 1% arabinose, 200 μL of the culture was collected and centrifuged at 8000 × g for 5 min. The cell pellets were washed twice with phosphate-buffered saline (PBS) and resuspended to an OD600 of approximately 0.2. The cells were then fixed with 4% paraformaldehyde and held on ice. Following fixation, the pellets were washed with PBS, resuspended in PBS to an OD600 of approximately 0.2, and 3–6 μL of the cell suspension was transferred onto glass slides. The samples were sealed under a coverslip using polyvinylpyrrolidone mounting medium (PVP) (Sangon Biotech, Shanghai) to prevent evaporation and were immediately imaged. Confocal microscopy of the E. coli cells was performed using a Leica TCS SP8 microscope with a 63× oil-immersion objective (Leica Microsystems). Specific filters were used to excite the fluorophores: eGFP was excited with a 488-nm laser, and mKate2 with a 561-nm laser.
Arginine phosphorylation and ubiquitination assay
Specifically, mKate2 expression was induced in LB medium with 0.1 mM IPTG for 6 h, after which the cells were collected and washed with sterile PBS to remove IPTG. The cells were then transferred to a fresh M9GC medium, where the expression of either 1-McsB or GP-McsB was induced.
The detection of arginine phosphorylation referred to previously developed methods34. Specifically, mKate2 expression was induced in LB medium with 0.1 mM IPTG for 6 h, after which the cells were collected and washed with sterile PBS to remove IPTG. The cells were then transferred to a fresh M9GC medium, where the expression of either 1-McsB or GP-McsB was induced. After 1 h, the cells were harvested and centrifuged at 8000 × g for 5 min. The pellets were washed twice with Tris-buffered saline (TBS) and lysed using TieChui E. coli Lysis Buffer (ACE Biotechnology, Changzhou, China) along with a protease inhibitor cocktail. The cell lysates were centrifuged at 12,000 × g for 10 min at 4 °C, and the supernatant was mixed with 4× LDS loading buffer and incubated at room temperature for 10 min without boiling. Proteins were separated via SDS-PAGE electrophoresis at 120 V on ice and transferred to a PVDF membrane in a transfer buffer for 90 min at 0 °C. After blocking the membrane with 5% BSA in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.1% (v/v) Tween-20) for 60 min, the membrane was incubated with anti-pArg antibody34,72 (Rabbit polyclonal antibody, 1:15,000, Xiamen University, Xiamen, China) diluted in TBST with 1% BSA for 45 min at room temperature. After washing, the membrane was incubated with HRP-conjugated goat anti-rabbit IgG (dilution: 1:40,000, Proteintech, Wuhan, China). The signal was visualized using the ECL Basic Kit (ABclonal, Wuhan, China) for 2 min, and the results were captured using the iBright FL1500 Imaging System.
For Pup-mediated ubiquitination detection, the protocol followed was similar to that of arginine phosphorylation detection with several modifications: (i) a 6× His-tag was added to Pup(E) to assess whether it can be bound to mKate2 by PafA; (ii) simultaneous induction of 1-PafA and Pup(E) expression was required; (iii) the supernatant mixed with 4× LDS loading buffer was boiled for 10 min; and (iv) the primary antibody used was an HRP-conjugated mouse anti-His-tag monoclonal antibody (dilution: 1:40,000, Proteintech, Wuhan, China).
In vivo degradation assays
The in vivo protein degradation assay was followed by previously established methods15,21. E. coli cells carrying the relevant plasmids were cultured overnight in 30 mL of LB medium containing antibiotics at 37 °C. The next morning, the culture was diluted to an OD600 of 0.1 and transferred to 30 mL of fresh LB medium supplemented with 0.1 mM IPTG to induce the expression of the target protein, mKate2. For proteins expressed from the genome, such as ErmB, AroK, MutH, and AroE, IPTG induction was not required. When the OD600 reached 0.5 (after approximately 3 h), 1 mL of the culture was collected, centrifuged at 5000 × g for 5 min, and resuspended in PBS (37 °C) twice. The samples were then inoculated into 1 mL of fresh M9GC medium (0.5% glucose, 0.05% casamino acids, 20 ng/mL aTc) in a 24-deep-well plate and incubated at 37 °C. Time-course measurements were initiated at t = 0 h. To maintain the cells in the exponential growth phase, the culture was diluted 5-fold every 2 h, and the aTc concentration was under 20 ng/mL in the medium. Samples were collected as needed for analysis. Three methods were employed to assess protein degradation: fluorescence intensity measurement, flow cytometry, and Western blotting. Fluorescence intensity and flow cytometry were used to evaluate the degradation of the reporter protein mKate2, while Western blotting was applied to assess the degradation of other target proteins.
For fluorescence intensity measurement, samples were collected as needed and diluted 1:10 in 200 μL of fresh M9 medium containing antibiotics and inducers. Measurements were performed using a Synergy H1 Multi-Mode Microplate Reader (BioTek Instruments, USA). The abundance of mKate2 was quantified at an excitation wavelength of 588 ± 10 nm and an emission wavelength of 644 ± 10 nm, while eGFP abundance was quantified at an excitation wavelength of 488 ± 10 nm and an emission wavelength of 511 ± 10 nm.
For flow cytometry measurement, the samples were washed twice with PBS and then resuspended to an OD600 of 0.2. Then the cell was fixed with 1% paraformaldehyde and stored at 4 °C. The assays were performed using a cell analyzer (FACSAria III, BD Biosciences, USA) using fluorescein isothiocyanate (FITC-A) (GFP) and PE-Texas Red-A (mKate2) channels. The voltage gains for each detector were set to FITC-A, 407 V, and PE-Texas Red-A, 650 V. For each sample, at least 20,000 counts were recorded using a 0.5 mL s−1 flow rate. All data were exported in FCS3 format and processed using FlowJo software (FlowJo V10).
For western blotting, cells were washed twice with TBS and subsequently lysed by adding TieChui E. coli Lysis Buffer (ACE Biotechnology, Changzhou, China) and a protease inhibitor kit. The cell lysate was centrifuged at 12,000 × g for 10 min at 4 °C, and the supernatant was mixed with 4× LDS loading buffer and boiled for 10 min. Proteins were separated on SDS-PAGE in a cold room at 120 V and electro-transferred to a PVDF membrane in a transfer buffer for 90 min at 4 °C. The membrane was blocked with 5% non-fat milk in TBST for 60 min, followed by three washes (10 min each). It was then incubated with HRP-conjugated Mouse anti-His-tag monoclonal antibody (1:50,000, Proteintech, Wuhan, China) and HRP-conjugated Mouse anti-GAPDH Tag monoclonal antibody (1:2000, HUABIO, Hangzhou, China) in TBST for 90 min at room temperature. After washing, signals were visualized by incubating the membrane with an ECL Basic Kit (ABclonal, Wuhan, China) for 1 min and documented using the iBright FL1500 Imaging System. The full uncropped scans of the blot can be found in Supplementary Fig. 34.
De novo designs of guide protein
The guide proteins were de novo designed using a Rosetta-based binder design method26. First, a suitable region was selected as the binding interface, ideally characterized by three features: a relatively flat surface, a high content of hydrophobic residues, and the absence of steric hindrance around the arginine residues. The target protein was then relaxed using the Rosetta energy function, and then trimmed of the residues that were away from the selected interface in the target protein (Retention of too many residues may result in calculation failure of Rifgen). Next, Patchdock residues (3–9 hydrophobic residues) and Rifgen residues (which must include Patchdock residues) were selected to run RifGen using RIF docking software. A miniprotein scaffold library was rigidly docked to the selected target residues using Patchdock. Subsequently, the file rifdock.flag was prepared (with settings for hydrophobic_target_res, require_hydrophobic_residue_contacts, and large_sampling_10deg_by1.1_2A_by0.35.x) to execute a local search optimizing sidechain-sidechain interactions using Rifdock. The sequences and structures of the initial docks were optimized with RosettaFastdesign, followed by one round of diversification through RosettaMotifgraft, and further optimization with RosettaFastdesign. The output designs from this process were filtered based on 6 recommended parameters: ddG, contact_molecular_surface, score_per_res, mismatch_probability, sap_score, and binder_delta_sap. 4 metrics (ddG, contact_patch, target_delta_sap, and contact_molec_sq5_apap_target) were then optimized to achieve results in the ≥90th percentile. The remaining designs were further filtered based on ddG < −45 and contact_molecular_surface >450, as these two metrics have been reported to significantly influence the success rate of binders. The designs were then ranked by contact_patch. Finally, the top 30 binders were selected for initial screening in a bacterial two-hybrid assay, and the top 10 binders exhibiting the best activation of BFP expression were subjected to further screening. The binders demonstrating the highest activation levels in both rounds of screening underwent cell co-localization testing. The sequences of the selected guide proteins are listed in Supplementary Table 2, and their PDB structures are provided in Supplementary Data 1.
Biolayer interferometry
Biolayer interferometry (BLI) was performed using Octet 96e (ForteBio). All target proteins (5–10 μg/mL) were biotinylated and immobilized onto streptavidin-coated biosensors (SA Sartorius) in PBS (pH 7.4) with 0.02% TWEEN (PBST). The biosensors were equilibrated in binding buffer for 60 s and then dipped into binder solution for 120 s (association step), and then dipped into binding buffer for 280 s (dissociation step). The experiment was carried out at 30 °C. In the affinity analysis experiment of eGFP with GPeGFP, three concentrations of 1 μM, 0.5 μM, and 0.25 μM were set up in parallel with a PBST buffer control group (baseline). In all other tests, three concentrations of 2 μM, 1 μM, and 0.50 μM were set up in parallel with a PBST buffer control group (baseline). After subtracting the baseline signal, the binding and dissociation signal curves for each concentration were obtained, and a 1:1 global fitting model was used to fit the binding and dissociation signal curves. The related figures are shown in Supplementary Fig. 33.
Measurement of cell growth
For analyzing the spot assay of E. coli under erythromycin treatment, cells were suspended in the LB medium and grown overnight. The following day, the cells were diluted in fresh medium to an OD600 of 0.1. Then, a 1 mL aliquot of the bacterial solution was washed three times and resuspended in 1 mL of sterile PBS. The bacterial suspension was then diluted to 10⁻¹, 10⁻², 10⁻³, 10⁻⁴, and 10⁻⁵ according to a gradient. Subsequently, 2 μL of the bacterial solution was pipetted onto a solid medium containing chloramphenicol (30 mg/L), aTc (20 ng/mL), and erythromycin (150 μg/mL) and incubated at 37 °C for 12 h for observation. Additionally, all OD600 values were measured using a Synergy H1 Microplate Reader. For analyzing the cell growth of E. coli under GPlad induction, overnight cultures of the relevant cells were inoculated into fresh LB medium with an initial OD600 of 0.1. After incubating at 37 °C for OD600 reached 0.4, 20 ng/mL aTc and 0.1 mM IPTG were added to induce the expression of GPlad and mKate2, respectively. This time point was designated as 0 h for the time-course experiment. Changes in mKate2 abundance and OD600 were monitored using a Synergy H1 Microplate Reader.
Shikimate kinase activity assay
After 8 h of inducing the GPAroK, the cells were harvested, centrifuged at 12,000 × g for 10 min, and washed with cold PBS. The cell pellets were then resuspended in 0.05 M barbital buffer (pH 7.0). Crude extracts were obtained by sonication and subsequently centrifuged at 12,000 × g for 10 min. Protein concentrations were determined using the Bradford assay. SHK kinase activity was assessed21 in a 1 mL reaction mixture containing 4 μM ATP, 1 μM shikimate, 10 μM NaF, 5 μM MgCl₂, 25 μM barbital buffer (pH 9.0), and 0.1–1.0 mg of protein from the cell extract. One unit of shikimate kinase activity was defined as the consumption of 1 μM shikimate per minute.
4D-FastDIA–based quantitative proteome analysis
Quantitative proteomic analysis of E. coli cells was performed using a 4D-FastDIA approach by Jingjie PTM Biolabs (Hangzhou, China). Following 8 h of GPlad-induced degradation of mKate2, samples were subjected to three rounds of sonication on ice with an ultrasonic processor in a lysis buffer consisting of 8 M urea and 1% protease inhibitor cocktail. The lysate was then centrifuged at 12,000 × g for 10 min at 4 °C, and the supernatant was collected for protein concentration measurement using a BCA kit. Subsequently, the supernatant was treated with 5 mM dithiothreitol for 30 min at 56 °C to reduce disulfide bonds, followed by alkylation with 11 mM iodoacetamide for 15 min at room temperature in the dark. To reduce the urea concentration to below 2 M, the sample was diluted with 100 mM TEAB. For overnight digestion, trypsin was added at a ratio of 1:50, followed by a second digestion for 4 h at a 1:100 ratio. Peptides were desalted using a C18 SPE column. The resulting tryptic peptides were dissolved in solvent A (0.1% formic acid, 2% acetonitrile in water) and separated on a nanoElute UHPLC system (Bruker Daltonics) using a gradient from 6% to 80% solvent B (0.1% formic acid in acetonitrile) over 90 min, at a consistent flow rate of 450 nL/min. The peptides were introduced into a capillary source and analyzed with a timsTOF Pro mass spectrometer (Bruker Daltonics), applying an electrospray voltage of 1.60 kV. Precursors and fragments were detected by the TOF detector, with the MS/MS scan range set between 100 and 1700 m/z. Data processing was performed using the MaxQuant search engine (v.1.6.15.0). Differential proteins were identified after sample qualification, with differences in relative quantification between the two groups compared using a t-test, and the corresponding p-values were calculated. The DIA data were processed using the DIA-NN search engine (v.1.8). Tandem mass spectra were searched against XB02168DA.fasta (5499 entries) concatenated with a reverse decoy database. Trypsin/P was specified as a cleavage enzyme allowing up to 1 missing cleavage. Excision on N-term Met and carbamidomethyl on Cys were specified as fixed modifications. FDR was adjusted to <1%. For the Quantitative Analysis, the raw LC–MS datasets were first searched against the database and converted into matrices containing normalized intensity (the raw intensity after correcting the sample/batch effect) of proteins. For proteins with unique peptides ≥2, the Normalized intensity (I) was transformed to the relative quantitative value (R) after centralization. The formula is listed as follows, where i represents the sample and j represents the protein:
For the differential analysis, firstly, the samples to be compared were selected in pairwise groups, and the fold change (FC) was then calculated by the ratio of the mean intensity for each protein in the two sample groups. For example, to calculate the fold change between sample A and sample B, the formula is shown as follows: R denotes the relative quantitative value of the protein, i denotes the sample, and k denotes the protein.
To calculate the statistical significance of the difference between groups, the two-sided unpaired t-test was performed on the relative quantitative value of each protein from the two sample groups. P-value < 0.01 was usually considered the threshold for significance. Therefore, the relative quantitative value of proteins was applied with log2 transformation. The formula is shown as follows:
The protein with P-value < 0.01, the fold change >2 was regarded as a significantly up-regulated protein, while the protein with P-value < 0.01, the fold change <0.5 was regarded as a significantly down-regulated protein. The “MS_identified_information” and raw data were uploaded to the ProteomeXchange Consortium (Project accession: PXD063956). The software and databases used in 4D-FastDIA quantitative proteomics are listed in Supplementary Table 5.
Fluctuation assay
The mutant rate assessment under stress was conducted as previously research73,74. E. coli containing the pOptoGPlad plasmid was grown overnight at 37 °C in the dark to reach the stationary phase. The cells were then transferred into 30 mL of fresh LB medium (containing either 0 or 5 g/L PCA), with an initial OD600 of 0.1. Simultaneously, 1% arabinose was added to induce the expression of GPMutH-pMagFast1, and 1 mL of the culture was exposed to blue light at 37 °C in transparent 24-deep-well plates with clear and sterile self-adhesive sealing film (Axygen, USA) for 12 h. For each strain, at least 20 parallel replicate cultures, divided into 6 biological repeats, were used to determine the number of spontaneous mutants. Sixteen cultures were then plated on LB plates (containing 100 µg/mL rifampicin) for mutant selection and incubated at 37 °C for 24 h in the dark, after which colonies were counted. Four cultures were diluted and plated on LB to determine total cell counts. Next, FALCOR was used to estimate mutation rates, employing the Ma–Sandri–Sarkar maximum likelihood method for analysis75.
Optogenetic adaptive laboratory evolution
Adaptive laboratory evolution was performed with slight modifications to the previous E. coli PCA05 evolution protocol43. First, the OptoGPlad plasmid, which targets the genome-expressed MutH, along with the PyhcN-mKate PCA sensor plasmid, was transformed into E. coli SA05, resulting in the creation of E. coli PCA06. An overnight culture of E. coli PCA06 was then inoculated into three 100 mL shake flasks containing 30 mL of medium with an initial OD600 of 0.1 and 7.5 g/L PCA. Subsequently, 1% arabinose was added to induce the expression of GPMutH-pMagFast1, and the cultures were exposed to blue light at 37 °C with shaking at 200 × g for 8 h. Afterward, the cells were harvested, washed twice, and then re-inoculated into fresh medium, switching to the dark for a 16-h incubation. The culture exhibiting the highest OD600 was selected for the subsequent round of evolution. If the OD600 exceeded 2.0, the PCA concentration was increased by 0.5 g/L for the next selection round. Every 20 generations, 80,000 cells displaying high fluorescence intensity were sorted using a flow cytometer and subsequently cultivated for the next evolutionary cycle. After 100 generations, cells within the top 10% for fluorescence intensity were sorted, plated on agar containing 15 g/L PCA, and transferred to a 96-deep-well plate with 40 g/L PCA for survival testing via flow cytometry. The two plasmids in the evolved strains were then eliminated in a non-antibiotic evolution medium.
For the optogenetic LED setup, the same conditions and equipment used in our previous work were employed39. Cells were illuminated using a custom-built LED blue light panel measuring 300 mm × 300 mm × 50 mm, equipped with adjustable ON/OFF pulsing and intensity settings. Two 450 nm blue light panels (characteristics: 22 W, 24 V, 1.7 A; MODEL: HF-FX160, square light source) were activated to illuminate the shake flasks for gene expression activation. These devices were sourced from KOMA Vision Technology Company and LEMONS Co., Ltd, China. A switching power supply (AC/DC ADAPTER, MODEL: SPF-1210) powered the lighting apparatus simultaneously (AC 100–240 V, 50/60 Hz; OUTPUT: DC 12 V, 10 A).
PI staining
For the propidium iodide (PI) staining assay, E. coli cells were incubated overnight in LB at 37 °C. The following morning, cultures were inoculated into 30 mL of fresh LB and grown until an OD600 of 0.6 was achieved. The cells were then transferred to fresh LB media containing 40 g/L PCA and subjected to treatment at 37 °C with shaking at 200 × g for 6 h. After treatment, the cells from the shake flasks were centrifuged, washed twice with PBS, and resuspended to an OD600 of 0.2. Next, 3 μL of PI (Sangon Bio, Shanghai, China) was added to the samples, which were incubated at 4 °C for 20 min in the dark. PI, a nuclear fluorescent dye, is unable to penetrate intact cell membranes and thus does not bind to intracellular DNA. However, when the cell membrane is compromised, PI can enter the cell, bind to DNA, and emit red fluorescence. The number of dead cells was then quantified using the PE-Cy5-A Red channel (detection filter 695/40 nm) of a flow cytometer. A minimum of 20,000 events were recorded for each sample, and all data were analyzed using FlowJo software (FlowJo V10).
Fermentation condition
For PCA fermentation in a 5 L bioreactor (T&J Bio-engineering, Shanghai, China), 100 ml of seed culture was inoculated into 1.9 L of fermentation medium. An initial air supply of 2 vvm and a stirring speed starting at 300 × g was automatically controlled to maintain dissolved oxygen (DO) levels above 10% air saturation, while the fermentation temperature was kept at 37 °C. The pH of the medium was adjusted and maintained at 7.0 using 25% (v/v) NH4OH. Glucose feeding (800 g/L) was initiated after the depletion of the initial glucose supply.
For DHS fermentation in a 5 L bioreactor (T&J Bio-engineering, Shanghai, China), the working volume was set at 40%. The seed culture (OD600 = 3) was inoculated at a volume ratio of 12.5%. During fermentation, dissolved oxygen levels were kept between 20 and 30% saturation by adjusting agitation speed, while pH was automatically regulated to 7.0 using 25% (v/v) NH4OH, and the temperature was maintained at 35 °C. Glucose feeding (800 g/L) was implemented to sustain a glucose concentration of 0.1–5 g/L once the initial glucose was exhausted. If necessary, an antifoaming agent (DF-103) was added to control foam formation.
DNA re-sequencing and data analysis
E. coli strain PCA07 was inoculated into 200 mL of LB and cultured at 37 °C for 12 h at 200 × g. E. coli cells were harvested after 10 min centrifugation at 12,000 × g. Genomic DNA was extracted using the FastPure Gel DNA Extraction Mini Kit (Vazyme). 100 ng genomic DNA was randomly fragmented to <500 bp by sonication (Covaris S220). Whole genome sequencing was performed on the Illumina PE150 platform and the PacBio Sequel system. For the PacBio sequencing library, 5–10 μg genomic DNA was sheared into 10–15 Kb fragments using a g-TUBE device. The library was constructed using the SMRTbell® Express Template Preparation Kit 2.0. The library was sequenced using the PacBio Sequel platform. PacBio reads were assembled using Hifiasm/Canu. And then we recorrect the genome with the software Pilon using previous Illumina data. The Prodigal/Augustus gene-finding software has been used for finding coding genes. The coding genes were annotated with the National Center for Biotechnology Information (NCBI) nr database by Diamond. Then the functions of genes were annotated by the GO (Gene Ontology) database, and the pathways were annotated using the KEGG (Kyoto Encyclopedia of Genes and Genomes) database. The proteins encoded by genes were classified on a phylogenetic classification by the database of COG (Clusters of Orthologous Groups).
Statistical and reproducibility
The significance of data was determined by a two-tailed Student’s t-test by using GraphPad Prism 10. Significant differences were marked as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. All data were presented as mean ± s.d. All experiments in this study were performed in three biological repeats, and randomized sampling was employed for the analysis of all microbial samples.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data needed to evaluate the conclusions are present in the paper and supplementary information, and supplementary data. The mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository under the code of PXD063956. The genome re-sequencing data reported in this paper have been deposited to the Genome Sequence Archive and are available under accession code CRA027564. Source data are provided with this paper and can also be found in Figshare https://doi.org/10.6084/m9.figshare.29510372.v176. Source data are provided with this paper.
References
Clift, D. et al. A method for the acute and rapid degradation of endogenous proteins. Cell 171, 1692–1706.e1618 (2017).
Schapira, M., Calabrese, M. F., Bullock, A. N. & Crews, C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. 18, 949–963 (2019).
Gao, X. J., Chong, L. S., Kim, M. S. & Elowitz, M. B. Programmable protein circuits in living cells. Science 361, 1252–1258 (2018).
Shen, F. & Dassama, L. M. K. Opportunities and challenges of protein-based targeted protein degradation. Chem. Sci. 14, 8433–8447 (2023).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Hsia, O. et al. Targeted protein degradation via intramolecular bivalent glues. Nature 627, 204–211 (2024).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Langan, R. A. et al. De novo design of bioactive protein switches. Nature 572, 205–210 (2019).
Mercer, J. A. et al. Continuous evolution of compact protein degradation tags regulated by selective molecular glues. Science 383, eadk4422 (2024).
Tsai, J. M., Nowak, R. P., Ebert, B. L. & Fischer, E. S. Targeted protein degradation: from mechanisms to clinic. Nat. Rev. Mol. Cell Biol. 25, 740–757 (2024).
Ng, A. H. et al. Modular and tunable biological feedback control using a de novo protein switch. Nature 572, 265–269 (2019).
Morreale, F. E. et al. BacPROTACs mediate targeted protein degradation in bacteria. Cell 185, 2338–2353. e2318 (2022).
Junk, L. et al. Homo-BacPROTAC-induced degradation of ClpC1 as a strategy against drug-resistant mycobacteria. Nat. Commun. 15, 2005 (2024).
Won, H. I. et al. Targeted protein degradation in mycobacteria uncovers antibacterial effects and potentiates antibiotic efficacy. Nat. Commun. 15, 4065 (2024).
Cameron, D. E. & Collins, J. J. Tunable protein degradation in bacteria. Nat. Biotechnol. 32, 1276–1281 (2014).
Lei, Y. et al. Engineering an SspB-mediated degron for novel controllable protein degradation. Metab. Eng. 74, 150–159 (2022).
Chan, C. T. Y., Lee, J. W., Cameron, D. E., Bashor, C. J. & Collins, J. J. Deadman’ and ‘Passcode’ microbial kill switches for bacterial containment. Nat. Chem. Biol. 12, 82–86 (2016).
Fernandez-Rodriguez, J. & Voigt, C. A. Post-translational control of genetic circuits using Potyvirus proteases. Nucleic Acids Res. 44, 6493–6502 (2016).
Zheng, Y. et al. A tight cold-inducible switch built by coupling thermosensitive transcriptional and proteolytic regulatory parts. Nucleic Acids Res. 47, e137–e137 (2019).
Diao, W. et al. Reprogramming microbial populations using a programmed lysis system to improve chemical production. Nat. Commun. 12, 6886 (2021).
Gao, C. et al. Programmable biomolecular switches for rewiring flux in Escherichia coli. Nat. Commun. 10, 3751 (2019).
Trentini, D. B. et al. Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 539, 48–53 (2016).
Pearce, M. J., Mintseris, J., Ferreyra, J., Gygi, S. P. & Darwin, K. H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322, 1104–1107 (2008).
Chen, Z. et al. Programmable design of orthogonal protein heterodimers. Nature 565, 106–111 (2019).
Holmes, J. A. et al. Caulobacter PopZ forms an intrinsically disordered hub in organizing bacterial cell poles. Proc. Natl. Acad. Sci. USA 113, 12490–12495 (2016).
Cao, L. et al. Design of protein-binding proteins from the target structure alone. Nature 605, 551–560 (2022).
Schneidman-Duhovny, D., Inbar, Y., Nussinov, R. & Wolfson, H. J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 33, W363–W367 (2005).
Dou, J. et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018).
Cao, L. et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 370, 426–431 (2020).
Boldridge, W. C. et al. A multiplexed bacterial two-hybrid for rapid characterization of protein–protein interactions and iterative protein design. Nat. Commun. 14, 4636 (2023).
Karimova, G., Pidoux, J., Ullmann, A. & Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 95, 5752–5756 (1998).
Guo, L. et al. Engineering Escherichia coli lifespan for enhancing chemical production. Nat. Catal. 3, 307–318 (2020).
Zhou, B. et al. Arginine dephosphorylation propels spore germination in bacteria. Proc. Natl. Acad. Sci. USA 116, 14228–14237 (2019).
Lu, K. et al. Complex structure and activation mechanism of arginine kinase McsB by McsA. Nat. Chem. Biol. 21, 402–411 (2025).
Arifuzzaman, M., Kwon, E. & Kim, D. Y. Structural insights into the regulation of protein-arginine kinase McsB by McsA. Proc. Natl. Acad. Sci. USA 121, e2320312121 (2024).
Arenz, S. et al. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 5, 3501 (2014).
Kawano, F., Suzuki, H., Furuya, A. & Sato, M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun. 6, 6256 (2015).
Jayaraman, P. et al. Blue light-mediated transcriptional activation and repression of gene expression in bacteria. Nucleic Acids Res. 44, 6994–7005 (2016).
Ding, Q. et al. Light-powered Escherichia coli cell division for chemical production. Nat. Commun. 11, 2262 (2020).
Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 85–98 (2008).
Supek, F. & Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 521, 81–84 (2015).
Song, J. et al. New progress in the pharmacology of protocatechuic acid: a compound ingested in daily foods and herbs frequently and heavily. Pharm. Res. 161, 105109 (2020).
Wang, M. et al. Efficient production of protocatechuic acid using systems engineering of Escherichia coli. Metab. Eng. 82, 134–146 (2024).
Robert, L. et al. Mutation dynamics and fitness effects followed in single cells. Science 359, 1283–1286 (2018).
Xu, P. Production of chemicals using dynamic control of metabolic fluxes. Curr. Opin. Biotech. 53, 12–19 (2018).
Tang, A. Y., Jung, S., Carrasco-López, C. & Avalos, J. L. Light-Induced nanobody-mediated targeted protein degradation for metabolic flux control. ACS Synth. Biol. 13, 4110–4118 (2024).
Li, Z. et al. Systems engineering of Escherichia coli for high-level shikimate production. Metab. Eng. 75, 1–11 (2023).
Sheng, Q. et al. Shikimic acid biosynthesis in microorganisms: current status and future direction. Biotechnol. Adv. 62, 108073 (2023).
Politi, N. et al. Half-life measurements of chemical inducers for recombinant gene expression. J. Biol. Eng. 8, 5 (2014).
Chung, H. K. & Lin, M. Z. On the cutting edge: protease-based methods for sensing and controlling cell biology. Nat. Methods 17, 885–896 (2020).
Tekaia, F., Yeramian, E. & Dujon, B. Amino acid composition of genomes, lifestyles of organisms, and evolutionary trends: a global picture with correspondence analysis. Gene 297, 51–60 (2002).
Lunge, A., Gupta, R., Choudhary, E. & Agarwal, N. The unfoldase ClpC1 of Mycobacterium tuberculosis regulates the expression of a distinct subset of proteins having intrinsically disordered termini. J. Biol. Chem. 295, 9455–9473 (2020).
Izert, M. A., Klimecka, M. M. & Górna, M. W. Applications of bacterial degrons and degraders—Toward targeted protein degradation in bacteria. Front. Mol. Biosci. 8, 669762 (2021).
Hou, J. et al. Rewiring carbon flux in Escherichia coli using a bifunctional molecular switch. Metab. Eng. 61, 47–57 (2020).
Tague, N., Coriano-Ortiz, C., Sheets, M. B. & Dunlop, M. J. Light-inducible protein degradation in E. coli with the LOVdeg tag. eLife 12, RP87303 (2024).
Sun, Y. et al. Design and engineering of light-induced base editors facilitating genome editing with enhanced fidelity. Adv. Sci. 11, 2305311 (2024).
Huang, B. et al. Designed endocytosis-inducing proteins degrade targets and amplify signals. Nature 638, 796–804 (2025).
Chen, Z. & Elowitz, M. B. Programmable protein circuit design. Cell 184, 2284–2301 (2021).
Chen, Z. et al. De novo design of protein logic gates. Science 368, 78–84 (2020).
Fu, S. et al. Widespread arginine phosphorylation in human cells—a novel protein PTM revealed by mass spectrometry. Sci. China Chem. 63, 341–346 (2020).
Wang, S., Hou, Y., Chen, X. & Liu, L. Kick-starting evolution efficiency with an autonomous evolution mutation system. Metab. Eng. 54, 127–136 (2019).
Eom, G. -e, Lee, H. & Kim, S. Development of a genome-targeting mutator for the adaptive evolution of microbial cells. Nucleic Acids Res. 50, e38–e38 (2022).
Wang, Q. et al. Dual genetic level modification engineering accelerate genome evolution of Corynebacterium glutamicum. Nucleic Acids Res. 52, 8609–8627 (2024).
Ream, M. & Prather, K. L. Engineered autonomous dynamic regulation of metabolic flux. Nat. Biomed. Eng. 2, 233–243 (2024).
Wu, Y. et al. CRISPR–dCas12a-mediated genetic circuit cascades for multiplexed pathway optimization. Nat. Chem. Biol. 19, 367–377 (2023).
Xu, X. et al. Pyruvate-responsive genetic circuits for dynamic control of central metabolism. Nat. Chem. Biol. 16, 1261–1268 (2020).
Gupta, A., Reizman, I. M. B., Reisch, C. R. & Prather, K. L. J. Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat. Biotechnol. 35, 273–279 (2017).
Ni, C., Dinh, C. V. & Prather, K. L. Dynamic control of metabolism. Annu. Rev. Chem. Biomol. Eng. 12, 519–541 (2021).
Poirson, J. et al. Proteome-scale discovery of protein degradation and stabilization effectors. Nature 628, 878–886 (2024).
Özcelik, D. et al. Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat. Commun. 3, 1014 (2012).
Watson, J. L. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023).
Ouyang, H. et al. Development of a stable phosphoarginine analog for producing phosphoarginine antibodies. Org. Biomol. Chem. 14, 1925–1929 (2016).
Voordeckers, K. et al. Ethanol exposure increases mutation rate through error-prone polymerases. Nat. Commun. 11, 3664 (2020).
Swings, T. et al. Adaptive tuning of mutation rates allows fast response to lethal stress in Escherichia coli. eLife 6, e22939 (2017).
Hall, B. M., Ma, C.-X., Liang, P. & Singh, K. K. Fluctuation anaLysis calculatOR: a web tool for the determination of mutation rate using Luria–Delbrück fluctuation analysis. Bioinformatics 25, 1564–1565 (2009).
Li, Z. et al. Source data for: De novo designed protein guiding targeted protein degradation. Dataset. Figshare. https://doi.org/10.6084/m9.figshare.29510372.v1 (2025).
Acknowledgements
We thank Prof. Dr. Chuan Fu and Yan Liu (Xiamen University, China) for providing the pArg antibody. We also thank Prof. Dr. Longxin Cao and Prof. Dr. Zibo Chen (West Lake University, China) for their suggestions on protein design. Additionally, we appreciate Dr. Erez Zerbib (Ben-Gurion University of the Negev, Israel) for providing the plasmid pETDuet-1(OG). This work was financially supported by the Science Fund for Creative Research Groups of the National Natural Science Foundation of China (32021005) to L.L.; the Major Project of Natural Science Foundation of Jiangsu Province (BK20212013 to L.L., BK20220022 to J.W.); the Basic Research Center for Synthetic Biology of Jiangsu Province (BK20233003 to L.L.); the General Program of National Natural Science Foundation of China (22378164 to L.L.).
Author information
Authors and Affiliations
Contributions
Z.L., C.G., and L.L. conceived the project and wrote the manuscript. Z.L. and G.Q. designed the protein. Z.L., X.W., M.W., J.C., X.L., J.L., G.H., and J.W. designed and performed all the experiments. Z.L., C.G., and L.L. analyzed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Qiao, G., Wang, X. et al. De novo designed protein guiding targeted protein degradation. Nat Commun 16, 6598 (2025). https://doi.org/10.1038/s41467-025-62050-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62050-z
This article is cited by
-
Recent advances in microbial production of nutraceuticals: a sustainable approach
3 Biotech (2026)
-
Recent advances in targeting protein degradation for tumor immunotherapy
Journal of Hematology & Oncology (2025)
