
Abstract
The development of efficient and general strategies for constructing alkyl germanes is crucial due to their significant roles in various areas that impact human quality of life. Herein, we report a robust electrochemically driven method for the construction of alkyl germanes, utilizing a variety of functionalized alkyl nitriles and commercially available chlorogermanes. The developed decyanative germylation approach allows for the efficient and modular preparation of a wide range of structurally diverse alkyl germanes under mild reaction conditions. Notably, primary, secondary, and tertiary alkyl nitriles are all compatible. Furthermore, a series of natural product and drug-derived substrates undergo smooth late-stage functionalization, yielding the corresponding complex alkyl germanes.
Similar content being viewed by others
Introduction
Transforming functional groups plays a vital role in organic synthesis1,2. Beyond simply adding or converting functional groups, the strategic removal of these groups has also been essential in the construction of structurally complex organic compounds3,4,5,6. Given the extensive use of functional group removal, the beneficial features of the cyano group7 make the transformation of organic cyanides into parent molecules particularly interesting8. However, traditional strategies often require alkali metals and harsh reaction conditions, significantly limiting the application of decyanation reactions9. Over the past few decades, the cleavage of the C(sp2)-CN bond has been extensively studied using photochemical10,11,12,13, electrochemical14,15,16, and thermal17,18 synthetic approaches (Fig. 1a). Nevertheless, the high bond dissociation energies (BDEs, >100 kcal/mol) associated with aliphatic C-CN bonds and selectivity issues19 have limited the exploration of C(sp3)-CN activation in cross-coupling reactions20,21,22, with no electrochemical methods reported to date (Fig. 1b). However, developing catalytic methodologies to cleave carbon-cyano bonds electrochemically and introducing functional groups, is highly desirable in organic and pharmaceutical chemistry23,24. Benzylic C(sp3)-H coupling methods are recognized as step-economical and cost-effective strategies for forging valuable chemical bonds and elaborating complex structures25,26,27,28,29,30. Numerous elegant reports have disclosed the utility of this chemistry, including transition metal catalysis31,32,33,34, organocatalysis35, radical chemical oxidation36, photochemistry37, and electrochemistry38,39,40, creating new opportunities for the modification of complex reagents via late-stage C-H functionalization. Among these, the C-H functionalization of benzyl cyanide groups presents a significant opportunity to expand this field31,34,41,42,43,44. Existing research has primarily focused on reactions with metal catalysts, oxidative reagents, or HAT reagents. Developing new, efficient methods for synthesizing cyano-containing molecules without metal catalysts or additional oxidative reagents has garnered significant attention yet remains largely unexplored.

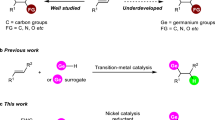
a The cleavage of the C(sp3)-CN bond. b The cleavage of the C(sp2)-CN bond. c Recent development of C(sp3)-germylation with chlorogermane. d Challenge. e Our work: Switchable Electrochemical Pathways for the Selective C(sp3)-Ge Bond Formation. XEC cross-electrophile coupling.
Organogermanium compounds have numerous applications in pharmaceuticals, material science, and organic transformations as effective catalysts or coupling partners45,46,47,48,49,50,51. Therefore, developing diverse methods to introduce germane motifs into organic molecules is a prominent topic in synthetic chemistry52,53,54,55,56. Reductive catalytic strategies for building C-Ge bonds have only recently been explored (Fig. 1c)57,58,59,60,61,62,63,64,65. For instance, our group reported an electro-catalyzed XEC cross-coupling to form C-Ge bonds60,61,66. Oestreich and co-workers disclosed the formation of C-Ge via nickel-catalyzed cross-coupling67, and Shu showcased an efficient C-Ge coupling63. However, the complexity and diversity of C(sp3)-Ge compounds, particularly those bearing tertiary and quaternary stereocenters, remain underdeveloped in organic chemistry62. Thus, exploring new strategies to transfer versatile and accessible functionalities into corresponding C-Ge bonds could lead to significant advances in germanium chemistry, which remains largely unexplored (Fig. 1d).
Owing to its adjustable redox potentials68,69,70,71,72, organic electrosynthesis73, particularly radical-involved reductive electrocatalysis74,75 and metalloelectrocatalysis76,77, has emerged as an innovative platform for constructing diverse and complex molecules efficiently and in an environmentally friendly manner78,79,80,81,82,83,84,85,86,87. A series of elegant electrochemically catalyzed cross-electrophile coupling (eXEC) reactions have been realized by researchers such as Lin65,88,89, Sevov90,91,92,93, Gosmini94,95,96,97 and others98,99,100,101,102. Despite these pioneering studies, electrochemical reductive germylation remains challenging due to the inherent difficulties in avoiding reduction and homocoupling (Fig. 1d). Therefore, developing reliable catalytic procedures would provide access to previously inaccessible reaction pathways, offering rational and predictable routes to form C(sp3)-Ge bonds from common precursors under electrolytic conditions. To our knowledge, electrocatalyzed decyanation or direct C-H activation to form C(sp3)-Ge bonds has not been established, which is more challenging than cleavage C-X or C-O bonds. Toward this end, we demonstrate the electrochemically driven formation of C(sp3)-Ge bonds (Fig. 1e). Our protocol achieves the synthesis of organogermanes through C-CN cleavage or α-H activation of the cyan group, representing a streamlined approach to organogermane synthesis and expanding the existing repertoire of methodologies.
Results and discussion
Reaction optimization
We began optimizing the electrochemically cobalt-catalyzed decyanation reductive cross-coupling using phenylacetonitrile (1a) and trimethylgermanium chloride (2a) as standard coupling partners (Table 1). Applying CoBr2 as the catalyst, L1 as the ligand, Me4NPF6 as the electrolyte, and SS//SS electrodes in DMF with 4 mA current, the desired product was obtained in 81% yield (entry 1). Using RVC//RVC electrodes and TMEDA as the electrolyte inhibited the reaction (entry 2). Other electrode combinations were tested but did not yield good results (entries 3–7). Screening other metal catalysts also showed no improvement (entries 8–10). Lower yields were observed with different ligands (entries 11–14). Other reaction parameters, including different electrolytes and solvents, also succeeded in the electrochemical process, albeit with lower efficiency (entries 15–22). Applying currents of 2 and 6 mA resulted in lower yields (entries 23 and 24). Control experiments indicated that all reaction parameters, including the cobalt catalyst, ligand, electrolyte, and electricity, were essential for achieving high reaction efficiency (entries 25–28). We also tried to perform the reaction using metal reductant (Mn/Zn) in place of electricity; however, no products were detected, which further illustrating the advantage of our protocol.
Substrate scope
With the optimized reaction conditions in hand, we extensively evaluated the generality of primary phenylacetonitrile in this decyanation cross-coupling reaction. As shown in Fig. 2, phenylacetonitrile bearing para, meta, and ortho methoxyl groups on the phenyl ring all served as effective coupling partners, yielding the corresponding products with reasonable efficiency (3a, 3b, and 3c). This indicates that steric hindrance from the substituents has little effect on the reaction activity. A wide range of electron-donating groups substituted on phenylacetonitrile, including phenyl, trifluoromethoxyl, tert-butyl, diphenylamino, methanesulfonyl, methyl sulfide, and ester, were tolerated, furnishing the corresponding products (3d-3n) in moderate to excellent yields. Methoxy-substituted naphthalene acetonitrile also performed well (3o). Electron-neutral phenylacetonitriles, including those involving phenyl, naphthyl, phenanthrene, and pyrene, proceeded smoothly under this protocol, yielding the corresponding products (3p-3s) in moderate yields. Additionally, phenylacetonitriles bearing electron-withdrawing groups such as ester, trifluoromethyl, fluoro, amide, and mesylate were all compatible, providing the corresponding products (3t-3x) in moderate to excellent yields. Notably, reactive groups such as fluoro, amide, mesylate, and methyl sulfide remained intact, allowing for further late-stage modification, and demonstrating the excellent chemoselectivity of this protocol. Di-substituted and tri-substituted substrates also worked well, giving the corresponding products in moderate yields (3y-3ab). Significantly, several heterocyclic substrates bearing quinolone, benzofuran, dibenzothiophene, pyridine, thiophene, and indole motifs also participated, albeit with moderate yields (3ac-3ah).

Reactions were performed with 0.2 mmol of phenylacetonitrile, and 0.6 mmol of trimethylgermanium chloride. Yields are of the isolated products.
Various secondary and tertiary nitrile coupling partners were also investigated and proceeded smoothly (Fig. 3). A wide range of symmetric and unsymmetric nitriles served as efficient coupling partners in this electrochemical decyanation protocol. Secondary nitriles, regardless of their electronic properties, were employed efficiently. The compatibility of this decyanation protocol was well demonstrated by the tolerance of a wide variety of synthetically useful functional groups, including phenyl, naphthyl, ester, fluoro, methoxyl, trifluoromethyl, amide, furan, thiophene, and pyridine (5a-5n). The scope of tertiary nitriles was also attractive. A wide range of cyclic tertiary nitriles, including three- (5o), four- (5p), and six-membered rings (5q), served as efficient coupling partners, producing the desired products in moderate yields. Remarkably, methyl-substituted substrates (5r) provided the product in acceptable yields. The triarylmethane motif is a privileged structure with versatile applications. Despite steric hindrance, our protocol effectively introduced triarylmethane derivatives, delivering germanium products in moderate to good yields (5s-5w). Additionally, heterocyclic motifs, including N-Boc indole (5x) and thiophene (5y), coupled smoothly in this decyanation protocol. It is worth noting that entirely unsymmetrical triarylacetonitriles provided the desired coupling products in moderate yields (5aa-5ac).

Reactions were performed with 0.2 mmol of phenylacetonitrile and 0.6 mmol of trimethylgermanium chloride. Yields are of the isolated products.
Inspired by the successful substrate scope of this electrochemical protocol, we then investigated its robustness and potential synthetic utility, as illustrated by the modification of drug-like molecules (Fig. 4). Various primary and secondary nitriles derived from Sesamol, Clofibrate, Tocopherol, Estrone, Loratadine, Amoxapine, Pyrazole, Fenofibrate, Isoxepac, and precursors for Cytenamide and Ketoprofen proceeded smoothly in this electrochemical decyanation reaction, furnishing decorated alkylgermanes (6-16) in moderate to good yields. This further highlighted the potential utility in the diversification of complex molecules. Notably, aryl chlorides were tolerated, allowing for further cross-coupling (12 and 13).

Reactions were performed with 0.2 mmol of phenylacetonitrile, and 0.6 mmol of trimethylgermanium chloride. Yields are of the isolated products.
To further demonstrate the utility of this transformation, we evaluated the effect of various R3Ge−Cl compounds. Similarly, Me3GeCl with different chain lengths and functionalities proceeded well under this protocol, yielding desired germanium products in good yields (2b-2d). Phenyl-substituted chlorogermanes also reacted well, providing alkylgermanes in moderate to good yields (2e-2h). Electron-donating (OMe) and electron-withdrawing (CF3) group-substituted aryl germanes were tolerated and afforded products in moderate yields (2i-2j). Interestingly, thiophene-substituted chlorogermane also provided the desired product (2k), albeit in low yield.
During the optimization of the decyanation reaction, we detected a trace amount of direct C-H activation to form the C-Ge bond product in the absence of the cobalt catalyst. Considering the importance and difficulty of synthesizing such compounds, we explored whether the desired product could be obtained exclusively through a mild electrochemical method without the catalyst (Table S1). Initial screening of current settings resulted in a 22% yield (entries 1–3). The solvent evaluation revealed that MeCN was the most effective, offering a 33% yield (entries 4–6). Other electrode combinations suppressed the reaction (entries 7–13). Base optimization yielded inferior results (entries 14–17). Substituting Me4NPF6 with other electrolytes resulted in lower or no yields of the desired product (entries 18–23). Further voltage change showed that using 2.5 V led to a significantly improved yield of 48% (entries 24–26). Monitoring the reaction revealed that extending the reaction time increased the yield (entries 27 and 28). Control experiments confirmed that electricity was essential for achieving high reaction efficiency (entries 29 and 30).
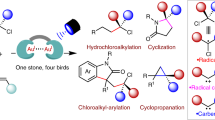
Under optimized conditions, the generality of the aliphatic C-H germylation reaction of benzylacetonitriles with trimethylgermanium chloride (2a) was investigated (Fig. 5). An array of benzylacetonitriles bearing different substituents on the aromatic ring were all compatible, producing the corresponding products in moderate-to-good yields. Para-, meta-, and ortho-methoxy-substituted benzylacetonitriles effectively yielded the corresponding molecules, indicating that steric hindrance from the substituents has minimal effect on the reaction activity (17a-c). A series of functional groups, including electron-rich BzO, Ph, t-Bu, Me, MeS, and Ph2N, were tolerated, resulting in the desired germylation products in moderate yields (17d-i). 7-Methoxy-substituted 1-naphthylacetonitrile also furnished the product 17j with a 62% yield. Notably, electron-neutral benzylacetonitrile and 2-naphthylacetonitrile were also compatible, producing 17k and 17l in 61% and 53% yields, respectively. However, F-substituted benzylacetonitrile gave a lower yield of product 17m. Replacing F with other strong electron-withdrawing groups, including CF3 and CO2Me, resulted in dramatically lower yields (<20%). Additionally, di- and tri-methoxy-substituted substrates yielded the corresponding products in moderate yields (17n, o). Moreover, heteroaryl acetonitrile provided the germylation products 17p, 17q, and 17r in 63%, 71% and 46% yields, respectively. Next, we assessed the scope of chlorogermanes. Chlorotriethylgermane yielded 17r in a 69% yield. Substituting the methyl group with phenyl, thienyl, and vinyl groups also furnished products 17s-17u in moderate yields.

Reactions were performed with 0.2 mmol of phenylacetonitrile, and 0.6 mmol of trimethylgermanium chloride. Yields are of the isolated products.
Mechanistic studies
To gain further insights into the reaction mechanism, a series of experiments were conducted. Initially, a radical clock experiment using 2-cyclopropyl-2-(4-methoxyphenyl)acetonitrile as the substrate produced the alkene-tethered germanium-containing product 19a in 33% yield (Fig. 6a). Furthermore, the addition of 3.0 equivalents of TEMPO, BHT, and 1,1-diphenylethene inhibited the formation of alkylgermanes (Fig. 6b). These observations suggest the involvement of an alkyl radical intermediate in the decyanation process. In the reaction with cyclopropanyl chlorogermane 20, the coupling product 21 was obtained in 44% yield without detecting any ring-opening product (Fig. 6c), indicating that the activation of chlorogermane proceeds through a non-radical pathway. When substrate 22 was reacted with 2a, only the uncyclized product 23 was obtained in 43% yield, with no cyclization product 23a detected (Fig. 6d). Additionally, product 17a was obtained in 44% yield under standard reaction conditions with the addition of 3.0 equivalents of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO) (Fig. 6e). These results indicate that C-H activation does not proceed via a radical pathway. Subsequently, a germanium radical clock reaction was performed to determine whether a germanium radical was generated in this process. The coupling of cyclopropanyl chlorogermane 20 produced product 24 in 51% yield without any ring-opening product being detected (Fig. 6f). This observation confirmed that no germanium radical was generated under the standard reaction conditions. Reaction monitoring showed that decyanation was completed within 5 h, whereas C-H germylation required a longer reaction time (24 h, Figs. S1 and S2). Moreover, several cyclic voltammetry (CV) experiments were conducted to determine the redox potential of the substrates (Fig. 7a). For the decyanation reaction, the cobalt complex (CoBr2·bpy) undergoes reduction at potentials of −1.09 V, −1.52 V, and −2.36 V vs. SCE, corresponding to the CoII/CoI, CoI/Co0, and Co0/Co−I reduction peaks, respectively. The reduction peaks for 2a and 1a were found to be −2.73 V and <−3.0 V vs. SCE, respectively, indicating they are more difficult to be reduced than [bpy-CoII]Br2. Additionally, the addition of varying equivalents of 2a increased the currents of the CoII/CoI, CoI/Co0, and Co0/Co−I reduction peaks, suggesting that 2a reacts with the reduced forms of the cobalt complexes. In contrast, only the current of the Co0/Co−I reduction peak increased when 1a was introduced. These results imply that the low-valent cobalt intermediate may initially undergo oxidative addition with 2a. For the C-H germylation reaction, the oxidation potential of 1a was determined to be +1.83 V. Thus, 1a appears to undergo anodic oxidation easily under the given conditions, whereas 2a does not undergo oxidation by the anode.

a Alkyl radical clock experiment for decyanation. b Radical scavengers experiment for decyanation. c Germanium radical clock experiment for decyanation. d Radical cyclization experiment for C-H germylation. e Radical scavengers experiment for C-H germylation. f Germanium radical clock experiment for C-H germylation. TEMPO 2,2,6,6-tetramethylpiperidine-1-oxyl, BHT 2,6-di-tert-butyl-4-methylphenol.

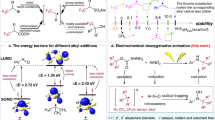
a Cyclic voltammetry (CV). b DFT calculated catalytic cycle for decyanative germylation.
To further elucidate the reaction mechanism of the cobalt-catalyzed decyanation reaction, we performed DFT calculations of the catalytic cycle. Building on the CV experimental results, we examined the oxidative addition of electrophiles 1a and 2a with CoI (singlet and triplet states), Co0, and Co-I (singlet and triplet states) intermediates (Figs. 7b and S6–S9). In the CoI-triplet catalytic cycle, trimethylgermanium chloride (2a) undergoes concerted oxidative addition with the triplet CoI complex T-A1 (ground state) via transition state T-TSA1 in triplet state, with a lower energy barrier of 13.5 kcal/mol. In contrast, the oxidative addition of phenylacetonitrile (1a) exhibits a higher energy barrier of 27.8 kcal/mol in triplet state. In the favored catalytic pathway, the generated CoIII complex T-B1 is reduced by the cathode to form the triplet CoI intermediate T-C1 (ground state). Phenylacetonitrile (1a) then coordinates to T-C1, yielding intermediate T-D. This species undergoes a barrierless CN abstraction, affording a benzylic radical and the Co(II) intermediate D-E. Next, the benzylic radical recombines with the intermediate D-E with an energy barrier of 5.6 kcal/mol. The reductive elimination from the CoIII intermediate T-F has an energy barrier of 0.2 kcal/mol, leading to the formation of the final product and the CoI-CN intermediate T-A2. For the next catalytic cycle, we also calculated the oxidative addition of trimethylgermanium chloride (2a) and phenylacetonitrile (1a) with the intermediate T-A2. The results also showed that 2a oxidative addition has a lower energy barrier (Fig. S9). Next, the CoI-singlet catalytic cycle was also explored. Here, the overall intrinsic chemical energy barrier is 19.9 kcal/mol, which is higher than that of the triplet-state cycle, indicating a less favorable pathway (Fig. S5). For the Co0 catalytic cycle, the oxidative addition of 1a has an energy barrier of 25.9 kcal/mol. In the case of 2a, the energy barrier was found to be prohibitively high, preventing precise determination of the transition state. These results suggest that this pathway is an alternative, albeit less favorable, route (Fig. S7). In the Co-I-triplet catalytic cycle, the cathodic reduction from CoI to Co-I is thermodynamically challenging (+68.8 kcal/mol). Additionally, the oxidative addition of 1a in this pathway presents an energy barrier of 25.8 kcal/mol, which is higher than that observed in the CoI catalytic cycle, indicating a less favorable alternative pathway (Fig. S8).
Mechanistic proposal
Based on our findings and previous insights, we propose plausible mechanisms for the electrochemical germylation reaction, as illustrated in Fig. 8. The decyanation reaction begins with the reduction of the pre-catalyst, LnCoII-Br2, at the cathode to form a CoI species A. Under the applied electrochemical conditions, the activation of germyl chloride occurs via a non-radical pathway, involving a concerted oxidative addition with the cathodically reduced CoI species. This step generates the Ge-CoIII-ClX intermediate B. Subsequent reduction of intermediate B at the cathode produces the Ge-CoI intermediate C. Next, benzyl cyanide undergoes CN abstraction by intermediate C, resulting in the formation of a benzylic radical and a CoII intermediate D. The benzylic radical then recombines with intermediate D, leading to the formation of a CoIII species E. Finally, reductive elimination of the CoIII intermediate yields the germylation product and regenerates the CoI species A, completing the catalytic cycle. In the C-H germylation reaction, benzylacetonitrile is oxidized at the anode to form the benzyl cationic radical, which is further oxidized to produce the benzylacetonitrile cation. Concurrently, chlorogermane is reduced at the cathode via a 2-electron process, yielding the germanium anion, which subsequently reacts to form the desired product.

a Proposed mechanism for electrochemical cobalt-catalyzed decyanation germylation reaction. b Proposed mechanism for electrochemical metal-free direct C-H germylation reaction.
Discussion
In conclusion, we have developed an efficient electrochemical germylation process using readily available benzylacetonitriles and trimethylgermane chloride as coupling partners under mild reaction conditions. Notably, direct C-H germylation was achieved in the absence of a transition-metal catalyst by optimizing the solvent and voltage combination. The key to success lies in the combination of electricity and a cobalt catalyst to control the selective cleavage of C-CN and C-H bonds. This approach enabled the synthesis of a wide array of primary, secondary, and tertiary organogermanes, including compounds with natural product or drug motifs, with good efficiency and regioselectivity, showcasing the broad applicability of this sustainable electrochemical process. Furthermore, the mechanism of these transformations was elucidated through a series of control experiments and DFT calculations. We believe this versatile electrochemical platform will advance both academic and industrial research on organogermanes and inspire chemists to develop new methodologies using electrochemical strategies.
Methods
General Procedure for the electrochemical cobalt-catalyzed decyano-germylation (GP-A)
A dry 5-mL vial equipped with a Teflon-coated magnetic stir bar (10 × 3 mm) was charged with phenylacetonitrile (0.2 mmol, 1 equiv., if solid), CoBr2 (4.4 mg, 0.02 mmol, 10 mol %), 2, 2′-bipyridine (bpy) (3.7 mg, 0.024 mmol, 12 mol%) and Me4NPF6 (0.8 mmol, 4 equiv.) in glovebox. Anhydrous and degassed DMF (4.0 mL), phenylacetonitrile (0.2 mmol, 1 equiv., if liquid), trimethylgermanium chloride (0.6 mmol, 3.0 equiv.) were added via syringe. Then, it was capped with a Teflon lid equipped with stainless steel (4 × 50 mm) as the anode and stainless steel (4 × 50 mm) as the cathode. The reaction mixture was stirred and electrolyzed at a constant current of 4 mA under room temperature for overnight. After the reaction is completed, the mixture was transferred to a 50 mL round bottom flask, electrodes were washed with ethyl acetate. Then H2O (20 mL) was added and the mixture was extracted with EtOAc (20 mL) for three times. The combined organic layer was washed with H2O (20 mL) and brine (20 mL). The organic layer was dried with anhydrous Na2SO4, then concentrated under vacuum. The product was purified by flash column chromatography on silica gel using hexane/EtOAc as eluent.
General Procedure for the electrochemically driven C-H germylation (GP-B)
A dry 5-mL vial equipped with a Teflon-coated magnetic stir bar (10 × 3 mm) was charged with phenylacetonitrile (0.2 mmol, 1 equiv., if solid), and Me4NPF6 (0.8 mmol, 4 equiv) in a glovebox. Anhydrous and degassed MeCN (4.0 mL), phenylacetonitrile (0.2 mmol, 1 equiv., if liquid), trimethylgermanium chloride (0.6 mmol, 3.0 equiv) were added via syringe. Then, it was capped with a Teflon lid equipped with stainless steel (4 × 50 mm) as the anode and stainless steel (4 × 50 mm) as the cathode. The reaction mixture was stirred and electrolyzed at a constant voltage of 2.5 V under room temperature for 24 h. After the reaction is completed, the mixture was purified by flash column chromatography on silica gel using hexane/EtOAc as eluent under N2 atmosphere. Note: The desired product was not stable in air, keep in nitrogen and fridge.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information Files (including experimental details, NMR data, and NMR spectra). Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Smith, M. B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure 8th edn, (John Wiley & Sons, 2020).
Yu, X. Y., Chen, J. R. & Xiao, W. J. Visible light-driven radical-mediated C-C bond cleavage/functionalization in organic synthesis. Chem. Rev. 121, 506–561 (2021).
Tobisu, M., Nakamura, R., Kita, Y. & Chatani, N. Rhodium-catalyzed reductive decyanation of nitriles using hydrosilane as a reducing agent: scope, mechanism and synthetic application. Bull. Korean Chem. Soc. 31, 582–587 (2010).
Liu, J., Ye, Y., Sessler, J. L. & Gong, H. Cross-electrophile couplings of activated and sterically hindered halides and alcohol derivatives. Acc. Chem. Res. 53, 1833–1845 (2020).
Xue, W. & Oestreich, M. Beyond carbon: enantioselective and enantiospecific reactions with catalytically generated boryl- and silylcopper intermediates. ACS Cent. Sci. 6, 1070–1081 (2020).
Feng, J. J., Mao, W., Zhang, L. & Oestreich, M. Activation of the Si-B interelement bond related to catalysis. Chem. Soc. Rev. 50, 2010–2073 (2021).
Murahahi, S.-I. In Science of Synthesis 19 (ed. Murahashi, S.I.) 345–402 (Thieme: Stuttgart, 2004).
Tobisu, M. & Chatani, N. Catalytic reactions involving the cleavage of carbon-cyano and carbon-carbon triple bonds. Chem. Soc. Rev. 37, 300–307 (2008).
Mattalia, J. M., Marchi-Delapierre, C., Hazimeh, H. & Chanon, M. The reductive decyanation reaction: chemical methods and synthetic applications. Arkivoc. 2006, 90–118 (2006).
McNally, A., Prier, C. K. & MacMillan, D. W. Discovery of an alpha-amino C-H arylation reaction using the strategy of accelerated serendipity. Science 334, 1114–1117 (2011).
Chen, M., Zhao, X., Yang, C. & Xia, W. Visible-light-triggered directly reductive arylation of carbonyl/iminyl derivatives through photocatalytic PCET. Org. Lett. 19, 3807–3810 (2017).
Zhu, S., Qin, J., Wang, F., Li, H. & Chu, L. Photoredox-catalyzed branch-selective pyridylation of alkenes for the expedient synthesis of Triprolidine. Nat. Commun. 10, 749 (2019).
Huang, J. & Chen, Z. Radical decyanations of unactivated carbon-CN bonds: recent achievements and mechanistic studies. Adv. Synth. Catal. 365, 2058–2091 (2023).
Mo, Y. et al. Microfluidic electrochemistry for single-electron transfer redox-neutral reactions. Science 368, 1352–1357 (2020).
Huang, B., Guo, L. & Xia, W. A facile and versatile electro-reductive system for hydrodefunctionalization under ambient conditions. Green. Chem. 23, 2095–2103 (2021).
Zhang, X. et al. Reductive arylation of aliphatic and aromatic aldehydes with cyanoarenes by electrolysis for the synthesis of alcohols. Org. Lett. 23, 3472–3476 (2021).
Dorval, C., Tricoire, M., Begouin, J.-M., Gandon, V. & Gosmini, C. Cobalt-catalyzed C(sp2)–CN bond activation: cross-electrophile coupling for biaryl formation and mechanistic insight. ACS Catal. 10, 12819–12827 (2020).
Wu, K. et al. Nickel-catalyzed amination of aryl nitriles for accessing diarylamines through C−CN bond activation. Adv. Synth. Catal. 363, 4708–4713 (2021).
Nakao, Y. Metal-mediated C-CN bond activation in organic synthesis. Chem. Rev. 121, 327–344 (2021).
Tobisu, M., Kita, Y., Ano, Y. & Chatani, N. Rhodium-catalyzed silylation and intramolecular arylation of nitriles via the silicon-assisted cleavage of carbon-cyano bonds. J. Am. Chem. Soc. 130, 15982–15989 (2008).
Chatani, N., Tobisu, M., Kinuta, H., Kita, Y. & Rémond, E. Novel synthetic approach to arylboronates via rhodium-catalyzed carbon–cyano bond cleavage of nitriles. Synthesis 44, 2999–3002 (2012).
Mills, L. R., Edjoc, R. K. & Rousseaux, S. A. L. Design of an electron-withdrawing benzonitrile ligand for ni-catalyzed cross-coupling involving tertiary nucleophiles. J. Am. Chem. Soc. 143, 10422–10428 (2021).
Paul, N., Patra, T. & Maiti, D. Recent developments in hydrodecyanation and decyanative functionalization reactions. Asian J. Org. Chem. 11, 128–140(2021).
Chen, Z. H. et al. From quaternary carbon to tertiary C(sp(3))-Si and C(sp(3))-Ge bonds: decyanative coupling of malononitriles with chlorosilanes and chlorogermanes enabled by Ni/Ti dual catalysis. J. Am. Chem. Soc. 146, 14445–14452 (2024).
Yi, H. et al. Recent advances in radical C-H activation/radical cross-coupling. Chem. Rev. 117, 9016–9085 (2017).
Yazaki, R. & Ohshima, T. Recent strategic advances for the activation of benzylic C–H bonds for the formation of C–C bonds. Tetrahedron Lett. 60 151225 (2019).
Wang, F. & Stahl, S. S. Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron-proton transfer mediators. Acc. Chem. Res. 53, 561–574 (2020).
Oliva, M., Coppola, G. A., Van der Eycken, E. V. & Sharma, U. K. Photochemical and electrochemical strategies towards benzylic C−H functionalization: a recent update. Adv. Synth. Catal. 363, 1810–1834 (2021).
Golden, D. L., Suh, S. E. & Stahl, S. S. Radical C(sp3)-H functionalization and cross-coupling reactions. Nat. Rev. Chem. 6, 405–427 (2022).
Zhang, Y., Zhang, T. & Das, S. Selective functionalization of benzylic C(sp3)–H bonds to synthesize complex molecules. Chem 8, 3175–3201 (2022).
Jiao, Z., Chee, K. W. & Zhou, J. S. Palladium-catalyzed asymmetric alpha-arylation of alkylnitriles. J. Am. Chem. Soc. 138, 16240–16243 (2016).
Fan, W. et al. Electrophotocatalytic decoupled radical relay enables highly efficient and enantioselective benzylic C-H functionalization. J. Am. Chem. Soc. 144, 21674–21682 (2022).
Chen, S.-J. et al. Accessing three-dimensional molecular diversity through benzylic C–H cross-coupling. Nat. Synth. 2, 998–1008 (2023).
Zhang, R., Zhou, Q., Wang, X., Xu, L. & Ma, D. Copper-catalyzed asymmetric arylation of alpha-substituted cyanoacetates enabled by chiral amide ligands. Angew. Chem. Int. Ed. 62, e202312383 (2023).
Neil, B., Saadi, L., Fensterbank, L. & Chauvier, C. Organopotassium-catalyzed silylation of benzylic C(sp(3))-H bonds. Angew. Chem. Int. Ed. 62, e202306115 (2023).
Ye, Z. et al. Photochemical diversification of strong C(sp3)–H bonds enabled by allyl bromide and sodium fluoride. Nat. Synth. 2, 766–777 (2023).
Meng, Q. Y., Lezius, L. & Studer, A. Benzylic C-H acylation by cooperative NHC and photoredox catalysis. Nat. Commun. 12, 2068 (2021).
Zhang, L. & Hu, X. Nickel catalysis enables convergent paired electrolysis for direct arylation of benzylic C-H bonds. Chem. Sci. 11, 10786–10791 (2020).
Chu, Q., Feng, Z., Zhang, S., Liu, P. & Sun, P. Three-component reaction for the synthesis of imides enabled by electrochemical C(sp3)–H functionalization. Green. Chem. 25, 6728–6732 (2023).
Li, A., Li, X., Ma, F., Gao, H. & Li, H. Cyclization of azobenzenes via electrochemical oxidation induced benzylic radical generation. Org. Lett. 25, 5978–5983 (2023).
Nerush, A. et al. Template catalysis by metal-ligand cooperation. C-C bond formation via conjugate addition of non-activated nitriles under mild, base-free conditions catalyzed by a manganese pincer complex. J. Am. Chem. Soc. 138, 6985–6997 (2016).
Bihani, M. et al. Scalable α-arylation of nitriles in aqueous micelles using ultrasmall Pd nanoparticles: surprising formation of carbanions in water. ACS Catal. 10, 6816–6821 (2020).
Liu, X., Zhao, C., Zhu, R. & Liu, L. Construction of vicinal quaternary carbon stereocenters through diastereo- and enantioselective oxidative 1,6-conjugate addition. Angew. Chem. Int. Ed. 60, 18499–18503 (2021).
Sheng, C. et al. Enantio- and diastereoselective synthesis of chiral tetrasubstituted alpha-amino allenoates bearing a vicinal all-carbon quaternary stereocenter with dual-copper-catalysis. Angew. Chem. Int. Ed. 62, e202305680 (2023).
Buriak, J. M. Organometallic chemistry on silicon and germanium surfaces. Chem. Rev. 102, 1271–1308 (2002).
Kachian, J. S., Wong, K. T. & Bent, S. F. Periodic trends in organic functionalization of group IV semiconductor surfaces. Acc. Chem. Res. 43, 346–355 (2010).
Allard, N. et al. Germafluorenes: new heterocycles for plastic electronics. Macromolecules 43, 2328–2333 (2010).
Fujii, S., Miyajima, Y., Masuno, H. & Kagechika, H. Increased hydrophobicity and estrogenic activity of simple phenols with silicon and germanium-containing substituents. J. Med. Chem. 56, 160–166 (2013).
Su, T. A. et al. Silane and germane molecular electronics. Acc. Chem. Res. 50, 1088–1095 (2017).
Fricke, C. & Schoenebeck, F. Organogermanes as orthogonal coupling partners in synthesis and catalysis. Acc. Chem. Res. 53, 2715–2725 (2020).
Xi, Y. et al. Application of trimethylgermanyl-substituted bisphosphine ligands with enhanced dispersion interactions to copper-catalyzed hydroboration of disubstituted alkenes. J. Am. Chem. Soc. 142, 18213–18222 (2020).
Song, H.-J., Jiang, W.-T., Zhou, Q.-L., Xu, M.-Y. & Xiao, B. Structure-modified germatranes for Pd-catalyzed biaryl synthesis. ACS Catal. 8, 9287–9291 (2018).
Xu, M. Y. & Xiao, B. Germatranes and carbagermatranes: (hetero)aryl and alkyl coupling partners in Pd-catalyzed cross-coupling reactions. Chem. Commun. 57, 11764–11775 (2021).
Marciniec, B., Pietraszuk, C., Pawluc, P. & Maciejewski, H. Inorganometallics (transition metal-metalloid complexes) and catalysis. Chem. Rev. 122, 3996–4090 (2022).
Pang, X., Su, P. F. & Shu, X. Z. Reductive cross-coupling of unreactive electrophiles. Acc. Chem. Res. 55, 2491–2509 (2022).
Rogova, T., Ahrweiler, E., Schoetz, M. D. & Schoenebeck, F. Recent developments with organogermanes: their preparation and application in synthesis and catalysis. Angew. Chem. Int. Ed. 63, e202314709 (2024).
Su, P. F. et al. Nickel-catalyzed reductive C-Ge coupling of aryl/alkenyl electrophiles with chlorogermanes. Angew. Chem. Int. Ed. 60, 26571–26576 (2021).
Gu, R., Feng, X., Bao, M. & Zhang, X. Modular access to alkylgermanes via reductive germylative alkylation of activated olefins under nickel catalysis. Nat. Commun. 14, 7669 (2023).
Qiao, Y.-W., Zhou, B.-Q. & Huang, Y. Ni-catalyzed reductive alkylgermylation of activated alkenes: Modular access to polyfunctionalized alkyl germanes. Chem. Catal. 3 100819 (2023).
Chen, H., Zhu, C., Yue, H. & Rueping, M. Carbon–germanium bond formation via low-valent cobalt-catalyzed cross-electrophile coupling. ACS Catal. 13, 6773–6780 (2023).
Chen, H., Zhu, C., Yue, H. & Rueping, M. Group 14 elements hetero-difunctionalizations via nickel-catalyzed electroreductive cross-coupling. Angew. Chem. Int. Ed. 62, e202306498 (2023).
You, M.-X., Su, P.-F., She, Z.-H. & Shu, X.-Z. Nickel-catalyzed cross-electrophile C-Ge coupling of benzyl pivalates and chlorogermanes. Sci. China Chem. 66, 3562–3566 (2023).
Han, G.-Y., Su, P.-F., Pan, Q.-Q., Liu, X.-Y. & Shu, X.-Z. Enantioconvergent and regioselective reductive coupling of propargylic esters with chlorogermanes by nickel catalysis. Nat. Catal. 7, 12–20 (2023).
Lu, L., Siu, J. C., Lai, Y. & Lin, S. An electroreductive approach to radical silylation via the activation of strong Si-Cl bond. J. Am. Chem. Soc. 142, 21272–21278 (2020).
Zhang, W. et al. Electrochemically driven cross-electrophile coupling of alkyl halides. Nature 604, 292–297 (2022).
Chen, H., Zhai, C., Zhu, C. & Rueping, M. Allylgermane synthesis via facile and general nickela-electrocatalyzed electrophile coupling. Chem. Catal. 5, 101257 (2025).
Xue, W., Mao, W., Zhang, L. & Oestreich, M. Mechanistic dichotomy of magnesium- and zinc-based germanium nucleophiles in the C(sp(3))-Ge cross-coupling with alkyl electrophiles. Angew. Chem. Int. Ed. 58, 6440–6443 (2019).
Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300–2347 (2008).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Mohle, S. et al. Modern electrochemical aspects for the synthesis of value-added organic products. Angew. Chem. Int. Ed. 57, 6018–6041 (2018).
Rein, J., Zacate, S. B., Mao, K. & Lin, S. A tutorial on asymmetric electrocatalysis. Chem. Soc. Rev. 52, 8106–8125 (2023).
Li, N. et al. A review of recent advances in electrochemical and photoelectrochemical late-stage functionalization classified by anodic oxidation, cathodic reduction, and paired electrolysis. Beilstein J. Org. Chem. 20, 2500–2566 (2024).
Liu, J., Lu, L., Wood, D. & Lin, S. New redox strategies in organic synthesis by means of electrochemistry and photochemistry. ACS Cent. Sci. 6, 1317–1340 (2020).
Sauer, G. S. & Lin, S. An electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal. 8, 5175–5187 (2018).
Chen, H. & Rueping, M. Facile, general allylation of unactivated alkyl halides via electrochemically enabled radical-polar crossover. Chem. Sci. 16, 6317–6324 (2025).
Meyer, T. H., Finger, L. H., Gandeepan, P. & Ackermann, L. Resource economy by metallaelectrocatalysis: merging electrochemistry and C H activation. Trends Chem. 1, 63–76 (2019).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3D metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254–4272 (2020).
Yoshida, J., Kataoka, K., Horcajada, R. & Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299 (2008).
Waldvogel, S. R. & Janza, B. Renaissance of electrosynthetic methods for the construction of complex molecules. Angew. Chem. Int. Ed. 53, 7122–7123 (2014).
Jutand, A. In Organic Electrochemistry 1393–1432 (CRC Press, 2015).
Horn, E. J., Rosen, B. R. & Baran, P. S. Synthetic organic electrochemistry: an enabling and innately sustainable method. ACS Cent. Sci. 2, 302–308 (2016).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Ma, C., Fang, P. & Mei, T.-S. Recent advances in C–H functionalization using electrochemical transition metal catalysis. ACS Catal. 8, 7179–7189 (2018).
Tang, S., Liu, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution: a green and sustainable way for bond formation. Chem 4, 27–45 (2018).
Zhu, C., Ang, N. W. J., Meyer, T. H., Qiu, Y. & Ackermann, L. Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 7, 415–431 (2021).
Malapit, C. A. et al. Advances on the merger of electrochemistry and transition metal catalysis for organic synthesis. Chem. Rev. 122, 3180–3218 (2022).
Zhang, W., Guan, W., Martinez Alvarado, J. I., Novaes, L. F. T. & Lin, S. Deep electroreductive chemistry: harnessing carbon- and silicon-based reactive intermediates in organic synthesis. ACS Catal. 13, 8038–8048 (2023).
Lu, Z. et al. Regioselective aliphatic C-H functionalization using frustrated radical pairs. Nature 619, 514–520 (2023).
Liu, J. et al. Co-catalyzed hydrofluorination of alkenes: photocatalytic method development and electroanalytical mechanistic investigation. J. Am. Chem. Soc. 146, 4380–4392 (2024).
Truesdell, B. L., Hamby, T. B. & Sevov, C. S. General C(sp(2))-C(sp(3)) cross-electrophile coupling reactions enabled by overcharge protection of homogeneous electrocatalysts. J. Am. Chem. Soc. 142, 5884–5893 (2020).
Walker, B. R., Manabe, S., Brusoe, A. T. & Sevov, C. S. Mediator-enabled electrocatalysis with ligandless copper for anaerobic Chan-Lam coupling reactions. J. Am. Chem. Soc. 143, 6257–6265 (2021).
Hamby, T. B., LaLama, M. J. & Sevov, C. S. Controlling Ni redox states by dynamic ligand exchange for electroreductive Csp3-Csp2 coupling. Science 376, 410–416 (2022).
Al Zubaydi, S., Onuigbo, I. O., Truesdell, B. L. & Sevov, C. S. Cobalt-catalyzed electroreductive alkylation of unactivated alkyl chlorides with conjugated olefins. Angew. Chem. Int. Ed. 63, e202313830 (2024).
Gomes, P., Fillon, H., Gosmini, C., Labbe, E. & Perichon, J. Synthesis of unsymmetrical biaryls by electroreductive cobalt-catalyzed cross-coupling of aryl halides. Tetrahedron 58, 8417–8424 (2002).
Gomes, P., Gosmini, C. & Périchon, J. Cobalt-catalyzed electrochemical vinylation of aryl halides using vinylic acetates. Tetrahedron 59, 2999–3002 (2003).
Gomes, P., Gosmini, C. & Perichon, J. Cobalt-catalyzed direct electrochemical cross-coupling between aryl or heteroaryl halides and allylic acetates or carbonates. J. Org. Chem. 68, 1142–1145 (2003).
Schiltz, P. & Gosmini, C. In Science of Synthesis: Electrochemistry in Organic Synthesis 1, 73–107 (Theime, 2021).
Kumar, G. S. et al. Nickel-catalyzed chain-walking cross-electrophile coupling of alkyl and aryl halides and olefin hydroarylation enabled by electrochemical reduction. Angew. Chem. Int. Ed. 59, 6513–6519 (2020).
Zhu, C., Yue, H., Jia, J. & Rueping, M. Nickel-catalyzed C-heteroatom cross-coupling reactions under mild conditions via facilitated reductive elimination. Angew. Chem. Int. Ed. 60, 17810–17831 (2021).
Liu, Y., Li, P., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling (eXEC) reactions. Angew. Chem. Int. Ed. 62, e202306679 (2023).
Zhu, C., Chen, H., Yue, H. & Rueping, M. Electrochemical chemo- and regioselective arylalkylation, dialkylation and hydro(deutero)alkylation of 1,3-enynes. Nat. Synth. 2, 1068–1081 (2023).
Shen, Z. J. et al. Organoboron Reagent-controlled selective (deutero)hydrodefluorination. Angew. Chem. Int. Ed. 62, e202217244 (2023).
Acknowledgements
This work was financially supported by the King Abdullah University of Science and Technology (KAUST), Saudi Arabia, Office of Sponsored Research (URF/1/4405). C. Zhai and C. Zhu thank the KAUST Supercomputing Laboratory (k1284) and High Performance Computing Centers at Ningbo Institute of Digital Twin for providing the computational resources.
Author information
Authors and Affiliations
Contributions
H.C. and M.R. conceived the idea of this work. H.C. carried out the reaction optimization and substrate scope. C. Zhai and C. Zhu performed the DFT calculation. H.C., C. Zhu, H.Z. and M.R. co-wrote the manuscript. M.R. directed the whole research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, H., Zhai, C., Zhang, H. et al. Switchable electrochemical pathways for the selective C(sp3)-Ge bond formation. Nat Commun 16, 7247 (2025). https://doi.org/10.1038/s41467-025-62141-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62141-x
