
Abstract
RNA-binding proteins (RBP) are important for the initiation and resolution of inflammation, so better understanding of RBP-RNA interactions and their crosstalk with metabolism may provide alternate targets to controlling inflammation. Here we establish global RNA-protein interactome purification (GRPIp) to profile the RBP landscape in inflammatory primary macrophages and identify ribosomal RNA processing 1 (RRP1) as a suppressor of inflammatory innate responses. Mechanistically, RRP1 binds nuclear thymidylate synthetase (Tyms) transcript and decreases TYMS expression post-transcriptionally in inflammatory macrophages, consequently suppressing folate metabolism cycle and inhibiting one-carbon metabolism-driven inflammation. Myeloid-specific RRP1-deficient mice develop severe experimental arthritis with increased pro-inflammatory cytokines and immunologic injury. Meanwhile, in patients with rheumatoid arthritis, RRP1 expression in peripheral blood monocytes negatively correlates with TYMS expression and serum IL-1β levels. Our results thus suggest that RRP1 acts as an anti-inflammatory factor through braking one-carbon metabolism post-transcriptionally, thereby implicating potential strategies for controlling autoinflammation.
Similar content being viewed by others
Introduction
RNA-binding proteins (RBPs) play important roles in health and diseases via the universal interactions with RNA molecules in the regulation of gene expression at different steps, including RNA polyadenylation, mRNA transport, RNA decay and the translation process1. The RBP-RNA interactions vary dynamically during the diverse cellular processes to maintain the homeostasis; however, the dysregulation of RBPs and their binding to RNA targets may lead to pathogenesis2,3. Several RBP family members, such as zinc finger protein ZFP36, heterogeneous nuclear RBP hnRNP UL1, and LIN28 have been shown to mediate post-transcriptional regulation of transcripts in the immune system4,5,6. How RBPs and their RNA targets are orchestrated in the regulation of anti-inflammation or inflammation-resolution requires further investigated.
Inflammation is a multistep response to eliminate perturbation (e.g., infection or injury). However, the excessive or non-resolving undesirable inflammatory response is always to the point of being pathological7. Especially, macrophages, with great plasticity, can undergo corresponding signaling pathway activation in local inflamed tissue, and then secrete pro-inflammatory cytokines such as TNF, IL-1 and IL-6, consequently leading to the inflammatory autoimmune diseases such as rheumatoid arthritis (RA) and inflammatory bowel disease8,9,10,11. Clinically, blockade of these cytokines-mediated inflammatory pathways has been used to treating autoimmune diseases12,13. Current studies focus on elucidating molecular mechanisms for controlling the inflammatory response duration or magnitude, aiming to fish out the new anti-inflammatory signals or molecules that can alleviate the autoinflammation.
Recent studies show that endogenous anti-inflammatory factors and inflammation-resolving mediators, especially metabolites such as itaconate, mannose and sphingolipid may provide new avenues for alleviating pathological injury in autoimmune diseases14,15,16,17. Notably, folate-dependent one-carbon (1C) metabolism, has long been regarded as a critical cellular process that regulates immune response, such as controlling effector and regulatory T cell fate and function via methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), and driving inflammatory macrophages via histone methylation18,19,20. In fact, the antineoplastic and immunosuppressant agent, methotrexate (MTX) targeting the key 1C-metabolic enzyme, dihydrofolate reductase (DHFR), has been used to treat RA since 195121,22. However, these antifolate drugs have adverse side-effects due to the requirement of folate in mammalian cells, prompting a search for more selective inhibitors of immune cell metabolism or clinically useful strategies in the anti-inflammatory response.
This study develops a modified phase separation-based RNA-protein capture method (GRPIp) to profile the dynamic RNA-binding proteome in macrophages, revealing that ribosomal RNA processing protein RRP1 broadly suppresses inflammation by binding Tyms mRNA and downregulating one-carbon metabolism. The findings establish RRP1 as a key anti-inflammatory factor through both mechanistic studies and clinical correlation analyses. Collectively, the GRPIp methodology provides a robust tool for studying RNA-protein interactions in immune cells, while the discovery of RRP1’s metabolic regulation offers new therapeutic targets for autoimmune and inflammatory diseases.
Results
GRPIp screening identifies RRP1 as an inhibitor of IL-1-mediated inflammation in primary macrophages
The unbiased “Phase Separation-based RNA-protein complex-capture” approach exploits the property that crosslinked global protein-RNA complexes in total cell samples separate into the insoluble interphase during RNA extraction with acid guanidinium thiocyanate-phenol-chloroform (e.g., TRIzol)23,24. Meanwhile, our previous study delineates three distinct stages in macrophage inflammatory response: the resting state, the initiation stage, and the resolution stage25. To investigate the dynamic RBP landscape in primary macrophages and identify mechanisms that limit the IL-1β-induced inflammatory response, we treated BMDMs with or without IL-1β for 1 h or 12 h. These time points generated cells representing the three inflammatory stages, which were cross-linked by UV to preserve endogenous RNA-RBP complexes. The RNA-RBP complexes were isolated using unbiased Phase Separation23,24, digested with Trypsin/Lys-C fragmentation after DNase treatment. We optimized screening by including an ultrafiltration treatment to purify fragmented peptide samples before employing four-dimensional (4D) label-free quantification (LFQ) quantitative mass spectrometry (MS; see Methods section) (Fig. 1a).

a Schematic of the GRPIp method (See Methods). b Distribution of RNA-RBP complex extracts after proteinase K or RNase A digestion analyzed by agarose gel electrophoresis. Line 1: the purified total RNA from the BMDMs. Line 2 to Line 4: RNA-RBP extracts purified from the interphase. (QC test ① mentioned in the Schematic). c Distribution of samples after RNase digestion, trypsin/Lys-C fragmentation by HPLC analysis (QC test ② mentioned in the Schematic). GO analysis (d) and Venn diagram (e) of potential RBPs identified in BMDMs stimulated by IL-1β for 0 h, 1 h and 12 h. f, g RT-qPCR detection of Il6 and Il-1b mRNA levels in BMDMs with siRNA-mediated RRP1 silence followed by IL-1β stimulation for the indicated hours. h ELISA of IL-6 levels in the supernatant of BMDMs with siRNA-mediated RRP1 silence followed by IL-1β stimulation for the indicated hours. Data are representative of three independent experiments (b, c). Data are of technical replicates from three independent experiments (n = 3) (d, e) with hypergeometric test (one-tailed) (d). Data are presented as means ± SD of (f, g, n = 3; h, n = 4) biologically replicates from three independent experiments with student’s t test (two-tailed unpaired). Source data are provided as a Source Data file.
To validate the reliability of our modified purification methods, we performed two quality control (QC) steps to verify the composition of the RNAs or RBPs in the samples. First, RNA-RBP complex extracts were digested with proteinase K and RNase A, then separated by agarose gel electrophoresis. Taking total purified RNA from the whole cells as a reference, RNA-RBP complex extracts showed a prominent band near the loading well. In contrast, with proteinase K-treated samples showed a shift in RNA-RBP complex and the 18S/28S bands, confirming enrichment of RNA-RBP complexes in the extracts (Fig. 1b). Secondly, after RNase digestion, trypsin/Lys-C fragmentation, the peptide samples were qualitatively analyzed by high-performance liquid chromatography (HPLC) (Fig. 1c). HPLC fractionation detected peptide samples across molecular weight fractions: (1) low molecular weight (LMW) fractions containing nucleic acid fragments emerged in the first 10 min (i.e., Fraction1), and (2) high molecular weight (HMW) fraction of protein peptide segments digested by trypsin/Lys-C flowed out later (i.e., Fraction2, 3, 4) (Fig. 1c).
The two QC analysis further confirmed that cross-linked peptides in the samples were suitable for 4D-LFQ MS analysis. Based on the LFQ signal intensity, a total of 386 potential RBPs were enriched by MS detection in BMDMs from three IL-1β treated groups (Supplementary Fig. 1a). Gene Ontology (GO) analysis revealed that many identified RBPs had RNA -associated functions including RNA splicing, mRNA metabolic process and RNA binding. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that these potential RBPs were enriched in multiple basic biological processes, including ribosome and spliceosome related processes, RNA/protein transport, mRNA surveillance pathway (Fig. 1d and Supplementary Fig. 1b). Moreover, we classified these potential RBPs into three groups according to the duration of IL-1β treatment. The Venn diagram (Fig. 1e) demonstrated that 176 proteins were common to the three groups and 13, 35, and 121 proteins respectively were specific to the 0 h resting group, 1 h and 12 h IL-1β-stimulation group. These results reveal the landscape of the RBP proteome at different stages of inflammation in macrophages, providing a molecular basis for how RBPs may influence inflammation.
To further identify functional RBPs that potentially mediating the anti-inflammatory response via increasing interactions between the RBP and its target RNA molecules, we focused on proteins only enriched in the 12 h IL-1β-stimulation group. Firstly, we measured the expression of chemokines and cytokines in macrophages stimulated with IL-1β. Using small interfering RNAs (siRNAs) to silence candidate proteins in BMDMs, RT-qPCR analysis revealed significantly increased Il6、Il-1b and Ccl2 mRNA levels following ribosomal RNA processing 1 (RRP1) silencing (Fig. 1f, g; Supplementary Fig. 1c–h). Additionally, IL-6 levels in the supernatant of RRP1 silenced BMDMs followed by IL-1β stimulation were also increased (Fig.1h). These data suggest that RRP1, identified by the GRPIp method, may negatively regulate the cytokine-induced inflammation in macrophages.
RRP1 and its Nop52 domain suppress inflammatory response in macrophages upon cytokine and innate stimuli
Then we confirmed these anti-inflammatory effects in THP-1-derived human macrophages using siRNA-mediated silencing of RRP1 (Fig. 2a, b, Supplementary Fig. 2a). To determine whether RRP1 specifically inhibits IL-1β-triggered inflammatory signaling, we treated both wild-type and IL-1 receptor type 1 knockout (IL-1R1 KO) BMDMs with poly(I:C) or lipopolysaccharide (LPS). RRP1 silencing enhanced inflammatory cytokine expression in both RRP1-deficient and control BMDMs following poly(I:C) and LPS stimulation (Supplementary Fig. 2c, d). These data indicate that RRP1 broadly restrains inflammation in macrophages in response to other innate stimuli, extending beyond IL-1 signal-mediated autoinflammation.

RT-qPCR detection of IL1B (a), IL6 (b) mRNA levels in THP-1 cells upon RRP1 silencing and IL-1β stimulation for the indicated hours. RT-qPCR detection of IL-1b (c) and Il6 (d) mRNA levels in wild-type (WT) or RRP1 knock out (RRP1 KO) RAW 264.7 cells with IL-1β stimulation for the indicated hours. e RT-qPCR detection of Tnfα mRNA levels in WT or RRP1 KO RAW 264.7 cells with murine TNFα (10 ng/ml) stimulation for the indicated hours. f Cell death ratio of WT or RRP1 KO RAW 264.7 cells treated with TCZ. TCZ: Combination of murine TNFα (20 ng/ml), cycloheximide (CHX) (10 μg/ml) and Z-IETD-FMK (2 μM). See methods. Western blotting of the phosphorylation (p-)of the key inflammatory signal pathway molecules in WT and RRP1 KO RAW 264.7 cells respectively stimulated by murine IL-1β (50 ng/ml) (g), TNFα (10 ng/ml) (h), IL-6 (20 ng/ml) (i) for indicated hours. GAPDH serves as loading control. j Western blot (left) and quantitative analysis (right) of the Flag tagged full-length RRP1 (FL), Nop52 and C terminal (Cter) expression in RRP1 KO RAW 264.7 cells. Empty vector (Vector) was transfected as a control. The Flag band intensities were normalized to GAPDH. RT-qPCR detection of IL-1b (k), Il6 (l) mRNAs after over-expressing the Flag tagged full-length RRP1 (FL), Nop52 (Nop52) and C terminal (Cter) in RRP1 KO RAW 264.7 cells followed by IL-1β stimulation. WT and the RRP1 KO cells transfected with empty vector (Vector) were used as controls. Western blotting data are representative of three independent experiments. RT-qPCR and Western bloting quantitative data are presented as means ± SD of (a–e, j right, k, l, n = 3) biologically replicates from three independent experiments with student’s t test (two-tailed unpaired). Cell death data are presented as means ± SD of (n = 4) biologically replicates from three independent experiments with ANOVA-test (two way). Source data are provided as a Source Data file.
To directly confirm whether RRP1 has anti-inflammatory effects, we constructed Rrp1 gene knockout (RRP1 KO) mouse macrophages via CRISPR-Cas9-mediated editing of RAW 264.7 cells. After IL-1β treatment, RRP1 KO cells showed significantly increased mRNA levels of Il6, Il-1b, Ccl2 compared to WT control cells (Fig. 2c, d, Supplementary Fig. 2f). However, RRP1 deficiency did not affect TNFα expression in ether THP-1 or RAW 264.7 cells (Supplementary Fig. 2b, e). To investigate whether RRP1 suppresses TNFα-mediated autoinflammation independently of other pro-inflammatory cytokines, we treated RAW 264.7 cells with TNFα. Tnfα mRNA levels were significantly elevated in RRP1 KO RAW 264.7 cells compared to WT controls (Fig. 2e). Consistently, WT RAW 264.7 cells showed greater resistance to necropotosis induced by combination treatment of TNFα and cycloheximide than RRP1 KO cells, suggesting an additional contribution of RRP1 in the anti-inflammatory process (Fig. 2f). We next assessed phosphorylation of key IL-1β signaling components. ERK1/2 and P38 phosphorylation was significantly enhanced in the middle and late phase of the inflammatory response in RRP1 KO RAW 264.7 cells (Fig. 2g). Similarly, more active MAPK signaling pathway compared to WT cells after TNFα treatment (Fig. 2h). Additionally, we also detected an increased level of STAT3 phosphorylation in RRP1 KO RAW 264.7 cells stimulated with IL-6, compared that with WT cells (Fig. 2i). The broad anti-inflammatory function of RRP1 was further confirmed in the RAW 264.7 cells stimulated with poly(I:C) (Supplementary Fig. 2g, h). These observations demonstrate that RRP1 broadly suppresses the inflammatory autoimmune response in macrophages upon various cytokines and innate stimuli.
The human RRP1 is also known as NOP52 and the mouse RRP1 has two main domains: a multiple α-helical structure called the Nop52 domain and a disordered C-terminal (Cter) domain. We therefore tested which of these domains is essential for its anti-inflammatory effect26. Overexpressing full-length RRP1 or the Nop52 domain in RAW 264.7 KO cells inhibited the increased inflammatory response, whereas, overexpression of the Cter domain had only modest effects at 4 h (Fig. 2j–l and Supplementary Fig. 2i). Thus, the Nop52 domain is essential for RRP1’s anti-inflammatory function in macrophages.
RRP1 binds to the RNAs related to one-carbon metabolism in inflammatory macrophages
To explore the relationship between the RNA-binding function of RRP1 and its anti-inflammatory effects, we employed RNA immunoprecipitation followed by high-throughput sequencing (RIP-seq) to identify BMDM-expressed RNAs that differentially bind RRP1 with and without IL-1β stimulation. Both the quantity and binding intensity (fold-change) of RRP1-bound RNAs increased in BMDMs after 12 h of IL-1β stimulation compared to resting cells, supporting the original hypothesis that increasing interactions between RRP1 and its target RNAs during inflammatory macrophages (Fig. 3a). KEGG analysis of RNAs with enhanced RRP1 binding after IL-1β stimulation revealed enrichment in metabolism-related genes (Supplementary Fig. 3a), suggesting that RRP1 participates in metabolic regulation during IL-1β-induced inflammation.

a Volcano plot showing of RNA molecules that differentially bind to RRP1 in BMDMs between 0 h and 12 h of IL-1β stimulation. b Heatmap showing metabolites in BMDMs stimulated by IL-1β for 12 h or not (n = 6). c KEGG analysis of metabolic pathway-related gene sets of RIP-seq identified RNAs that increasingly binding to RRP1 following IL-1β treatment. d Schematic of 1C-metabolism, including the folate cycle and the downstream methionine cycle. Met methionine, SAM S-adenosyl methionine, SAH S-adenosyl homocysteine, Hcy homocysteine, VB12 vitamin B12, THF tetrahydrofolate, 5, 10-mTHF N5,10-methylene- tetrahydrofolate, 5-mTHF N5-methyltetrahydrofolate, DHF dihydrofolate, DHFR dihydrofolate reductase, TYMS thymidylate synthetase. e Enrichment of 1C-metabolic-related gene sets of RIP-seq identified mRNAs that differentially binding to RRP1 following IL-1β treatment. f RIP-qPCR detects RRP1 binding to the Tyms mRNA, as assessed by two pairs of primers in BMDMs stimulated by IL-1β for indicated hours. g Western blot detects the Flag tagged full-length RRP1 (FL), Nop52 and C terminal (Cter) expression in NIH/3T3 cells as long as the immunoprecipitation (IP) effect by using the anti-Flag beads. The data were quantified with Image J. h RIP-qPCR detects the Flag tagged full-length RRP1 (FL), Nop52 and C-terminal (Cter) binding to Tyms mRNA in NIH/3T3 cells followed with IL-1β treatment for indicated hours. The cells transfected with empty Vector were used as controls. The data were normalized as (2^-ΔCt RIP RNA / 2^-ΔCt Input RNA) / (integrated density IP protein / integrated density Input protein). RIP-seq data are presented with normalized FPKM values of technical replicates (n = 3) from three independent experiments (a, e). Metabolomics data are presented with normalized response peak area values (z-score) (b). KEGG enrichment are used by hypergeometric test (one-tailed) with FDR correction (Q-values) (c). Western blotting data is representative of three independent experiments. RT-qPCR data are presented as means ± SD of (f, h, n = 3) biologically replicates from three independent experiments with student’s t test (two-tailed unpaired). Source data are provided as a Source Data file.
To confirm this hypothesis, we employed an unbiased metabolomics analysis and found that IL-1β stimulation for 12 h significantly increased the level of metabolites linked to 1C-metabolism, including methionine, glycine and serine (Fig. 3b). Glycine and serine serve as 1C-units-donors, while methionine acts as a key intermediate carrier in the 1C-metabolic pathway27. We then focused on further analysis of metabolic pathway-related gene sets in the RRP1 RIP-seq data (Supplementary Fig. 3a). KEGG analysis showed that among those RNAs enriched for increased binding to RRP1, purine metabolism and carbon metabolism, including the methionine cycle and the upstream folate cycle-related enzymes ranked among the top 15 rich-factor scores, including Tyms and Mthfd1 (Fig. 3c–e). To corroborate this finding, we conducted the confirmatory RIP, and the qPCR revealed that the association of RRP1 with thymidylate synthetase (Tyms, also known as TS) mRNA was significantly enhanced after IL-1β stimulation (Fig. 3f, Supplementary Fig. 3c). To map the Tyms mRNA-interacting region within RRP1, we performed RIP assays with the Flag-tagged RRP1 truncations ectopically expressed in NIH/3T3 cells. The results demonstrated that the Nop52 domain of RRP1 is the critical region mediating its binding to Tyms mRNA (Fig. 3g, h). Furthermore, RNA fluorescence in situ hybridization (FISH) with a probes’ pool targeting Tyms mRNA combining with immunofluorescence (IF) in BMDM cells revealed their colocalization. Upon IL-1β stimulation, Tyms mRNA exhibited obvious co-localization with RRP1 in nuclear and perinuclear compartments (Supplementary Fig. 3b). The results indicated that RRP1 might modulate 1C-metabolism in inflammatory macrophages by binding mRNAs encoding key metabolic enzymes and potentially affecting their function.
RRP1 maintains a repressed one-carbon-metabolic state in inflammatory macrophages
To validate the role of RRP1 in modifying 1C-metabolism, we employed targeted metabolomic analysis to compare the metabolites present in wild-type and RRP1 KO RAW 264.7 cells upon IL-1β stimulation for 12 h. We found that a higher level of the 1C-unit donors, including serine or glycine, the 1C- unit-intermediates, such as N5-methyltetrahydrofolate (5-mTHF), and the two key components in methionine metabolic cycle, methionine and S-adenosylmethionine (SAM) in the RRP1 KO macrophages (Figs. 3d, 4a). This indicated that the whole 1C-metabolic cycle was more active in the inflammatory RRP1 KO RAW 264.7 cells than that in wild-type cells. Overexpressing either full-length RRP1 or the Nop52 domain inhibited the increased SAM, methionine and THF levels of RRP1 KO cells upon IL-1β stimulation, while the C-terminal domain had only minimal effects (Fig. 4b, Supplementary Fig. 4a, b). To determine the dynamic contributions of RRP1 to 1C-metabolic cycle, we used L-serine-[13C3] isotope tracers to assess the 1C-metabolic flux in macrophages. We observed comparable intracellular serine (M + 3) uptake and its conversion to glycine (M + 2) in wild-type RAW 264.7 cells or RRP1 KO cells after IL-1β stimulation for 4 h (Fig. 4c, h and Supplementary Fig. 4c). However, serine-derived labeling of SAMs (M + 1 to M + 5), particularly the M + 3 SAM, were significantly increased in IL-1β-stimulated RRP1 KO RAW 264.7 cells (Fig. 4e, Supplementary Fig. 4d). Accordingly, the total level of intracellular SAM also increased in the RRP1 KO RAW 264.7 cells after stimulated with IL-1β for 4 h (Fig. 4d). Interestingly, methionine labeling (M + 1 to M + 3) was obviously higher in RRP1 KO cells than that in the wild-type cells, while the M + 4 methionine accumulated only in the wild-type cells (Fig. 4f, g, Supplementary Fig. 4e). These observations indicate RRP1 directs the source of 1C-unit routing within the methionine metabolic cycle and suppresses the synthesis of SAM in inflammatory macrophages (Fig. 4h). Additionally, the 13C labeling ratio of purine, including the IMP, AMP and GMP also increased in the RRP1 KO RAW 264.7 cells after stimulated with IL-1β, indicating RRP1’s inhibition of the de novo synthesis pathway of purine nucleotides, which is one destination of 1C-units (Supplementary Fig. 4f–k).

a Heatmap of 1C-metabolites in wild-type (WT) and RRP1 KO RAW 264.7 cells upon IL-1β induction for 12 h. Color indicates normalized intensities (z-score). b ELISA detection of SAM, methionine (Met), THF levels in RRP1 KO RAW 264.7 cells after over-expressing the Flag tagged full-length RRP1 (FL), Nop52 and C-terminal (Cter) followed by IL-1β stimulation for 8 h. The RRP1 KO cells transfected with empty Vector were used as controls. c Ratio of intracellular serine (M + 3) uptake from exogenous L-serine-[13C3], out of the total serine pools in WT and RRP1 KO RAW 264.7 cells upon IL-1β induction for 4 h or not. The ratio is show as Mass Distribution Vector (MDV). d Abundance of intracellular SAM total pools in WT and RRP1 KO RAW 264.7 cells upon IL-1β induction for 4 h or not. The ratio is show as Mole Percentage Enrichment (MPE). Ratio of intracellular SAM (M + 3) (e), methionine (M + 1- M + 3) (f), methionine (M + 4) (g) derived from L-serine-[13C3], out of their respective total pools in WT and RRP1 KO RAW 264.7 cells upon IL-1β induction for 4 h or not. The ratio is show as MDV. h Schematic of L-serine-[13C3] labeling patterns. i–l RT-qPCR quantification of Il-1b, Il6 mRNAs in WT or RRP1 KO RAW 264.7 cells stimulated with IL-1β for indicated hours in medium replenishment with exogenous SAM, methionine (Met) and serine. ELISA, Isotope Tracers and RT-qPCR data are presented as means ± SD of (b, i–l, n = 3; c–g, n = 4) biologically replicates from more than two independent experiments with student’s t test (two-tailed unpaired). Source data are provided as a Source Data file.
These observations indicated that the enhanced 1C-metabolism in IL-1β- stimulated RRP1 KO macrophages may be related to their pro-inflammatory phenotype, thus raising the question of the contributions of these 1C-unit-carrying metabolites to inflammation. In fact, serine and methionine synergistically fuel SAM generation to support pro-inflammatory macrophages through H3K36me320. To determine the role of RRP1 in 1C-metabolism-mediated inflammation in IL-1β-stimulated macrophages, we added SAM, methionine, or serine respectively to the cells that pre-cultured in the medium lacking the three amino acids. The replenishment of SAM dramatically increased the expression of IL-1β and IL-6 mRNAs in IL-1β-stimulated, wild-type RAW 264.7 cells as previously reported, but not significantly when methionine was replenished (Fig. 4i, j). This result suggests that the direct supplement of exogenous SAM can promote IL-1β-mediated inflammatory response in macrophages. Conversely, in IL-1β-stimulated RRP1 KO RAW 264.7 cells, the expression of IL-1β and IL-6 mRNAs increased significantly when serine or methionine replenished, but no significant effect was observed with SAM (Fig. 4k, l). These data indicated that improved serine utilization, methionine metabolism could promote inflammation, and the endogenic SAM may be sufficient to support its function in inflammatory RRP1 KO macrophages. However, the replenishment of exogenous methionine might not increase the production of SAM in the wild-type macrophages, perhaps due to the suppressed folate cycle upstream (Fig. 4i, j). In conclusion, these differences in the effect of these 1C-metabolites between the wild-type and RRP1 KO macrophages prove that RRP1 may inhibit the inflammatory response by restricting 1C-metabolic flux.
RRP1 represses pro-inflammatory cytokine expression by inhibiting TYMS
Given that RRP1 increasingly bound to the Tyms mRNA in the inflammatory macrophages induced by IL-1β (Fig. 3e–h), we speculated that the RRP1 may inhibit the inflammatory response and 1C-metabolism through interaction with Tyms mRNA. To demonstrate this, we silenced TYMS by using siRNAs, and found that the silencing of TYMS severely reduced the mRNA levels of Il6, Il-1b in IL-1β-stimulated RRP1 KO RAW 264.7 cells (Fig. 5a–c). Additionally, the expression of IL-1β was also significantly reduced when TYMS was silenced in THP-1 cells stimulated with IL-1β, and it observably abrogated the promoting effects of RRP1 silencing (Fig. 5d, e).

RT-qPCR detection of Il-1b (a) and Il6 (b) mRNA levels in RRP1 KO RAW 264.7 cells upon TYMS silencing and IL-1β stimulation for the indicated hours. c Western blot detection of RRP1 and TYMS protein levels in RRP1 KO RAW 264.7 cells upon TYMS silencing targeted by two distinct siRNAs. GAPDH serves as loading control. d RT-qPCR detection of IL-1B mRNA levels in negative control (NC)- and RRP1, TYMS or double-silenced THP-1 cells stimulated with IL-1β for the indicated hours. e ELISA detection of IL-1β levels in the supernatant of NC- and RRP1, TYMS or double-silenced THP-1 cells stimulated with IL-1β for the indicated hours. f Heatmap of 1C-metabolites in RRP1 KO RAW 264.7 cells upon TYMS silencing and IL-1β stimulation for the indicated hours. Color indicates normalized intensities (z-score). g ELISA detection of SAM, methionine (Met), THF levels in RRP1 KO RAW 264.7 cells upon TYMS silencing and IL-1β stimulation for the indicated hours. h RT-qPCR detection of Il-1b and Il6 mRNA levels in RRP1 KO RAW 264.7 cells pre-treated with Raltitrexed in the indicated concentrations and IL-1β stimulation for the indicated hours. i Western blotting for TMYS in RRP1-silenced BMDMs stimulated by IL-1β for the indicated hours. GAPDH serves as loading control. Western blotting for TMYS in the WT and RRP1 KO RAW 264.7 cells upon poly(I:C) (j) or murine IL-6 (k) stimulation for the indicated hours. GAPDH serves as loading control. Western blotting data are representative of three independent experiments (c, i, j, k). Metabolome data are from three biologically independent samples. RT-qPCR data are presented as means ± SD (n = 3) of biologically samples from three independent experiments with student’s t test (two-tailed unpaired) (a, b, d, e, g, h). Source data are provided as a Source Data file.
TYMS is the essential enzyme responsible for sustaining the THF pools to transfer the 1C-units, also for the reductive methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), an essential precursor for DNA synthesis and repair (Fig. 3e and Supplementary Fig. 4k). To verify whether TYMS is the link between 1C-metabolism and RRP1-mediated inflammatory gene expression, we employed targeted metabolomics analyses in RAW 264.7 cells, and found that the silencing of TYMS decreased the level of 1C-metabolites in IL-1β stimulated cells in both the wild-type and RRP1 KO cells (Fig. 5f, Supplementary Fig. 5a). Meanwhile, silencing of TYMS in RRP1 KO RAW 264.7 cells led to an obvious decrease in the levels of key 1C-metabolites, including SAM, methionine, and THF, as measured by commercially available ELISA kits (Fig. 5g; Supplementary Fig. 5b, c). These results indicate that the pro-1C-metabolic effects mediated by RRP1 knockout were markedly abrogated upon TYMS silencing. Then we used three drugs, Trifluridine, Pemetrexed, or Raltitrexed to inhibit the enzymatic activity of TYMS, respectively, and found that the Il6, Il-1b expression decreased in a dose-dependent manner in RRP1 KO RAW 264.7 cells stimulated with IL-1β (Fig. 5h, Supplementary Fig. 5g, h). The three drugs-mediated inhibition of TYMS may lead to DNA damage and cell death28. Hence, we analyzed the cell death kinetics of RRP1 KO macrophages treated with the three drugs in concentration gradients, and found that low concentrations (i.e., lower than 50 μM) of TYMS inhibitors scarcely led to cell death while exhibiting significant anti-inflammatory effect (Supplementary Fig. 5d–f, Fig. 5h).
These results suggest that the functional defect of TYMS could abolish the pro-inflammatory phenotype of RRP1-deficient macrophages. Conversely, besides the increasing level of SAM, Met, and THF (Supplementary Fig. 5j), transfection-mediated TYMS overexpression in wild-type RAW 264.7 cells revealed the following phenotypes: Under the resting state, TYMS-overexpressing cells exhibited a modest increase in the baseline expression of IL-6 and IL-1β. Upon IL-1β stimulation, TYMS overexpression significantly amplified the activation of inflammatory signaling pathways, as evidenced by enhanced phosphorylation levels of ERK1/2, JNK, and P38, accompanied by further upregulation of IL-6 and IL-1β expression (Supplementary Fig. 5i, k). These results suggest that TYMS overexpression potentiates inflammatory responses. Notably, we observed a significant increase in TYMS protein levels in RRP1-silenced BMDMs as early as 1 h after IL-1β stimulation, while control cells showed no such response (Fig. 5i). Furthermore, poly(I:C) or IL-6 stimulation reduced TYMS protein levels in wild-type RAW 264.7 cells, but this reduction was abolished in RRP1 KO cells (Fig. 5j, k). These results collectively demonstrate that RRP1 functions as an endogenous suppressor of TYMS expression in macrophages upon innate immune stimulation.
RRP1 inhibits TYMS expression post-transcriptionally in inflammatory macrophages
Our findings indicate that RRP1 acts as an endogenous anti-inflammatory factor by inhibiting TYMS expression to suppress 1C-metabolism, consequently leading to the inhibition of innate stimuli-triggered inflammation. To explore how RRP1 inhibits TYMS expression, we first conducted ribosome sedimentation to analyze TYMS translation in RRP1 KO RAW 264.7 cells (Fig. 6a and Supplementary Fig. 6a). The levels of ribosome-loading Tyms mRNA increased dramatically in RRP1 KO cells stimulated with IL-1β for 4 h compared to resting cells, while no changes occurred in wild-type cells with or without IL-1β stimulation (Fig. 6b). The findings demonstrate that RRP1 exerts a suppressive effect on TYMS translation. Cytoplasmic/nuclear fractionation revealed that RRP1 predominantly localized in the nucleus of BMDMs, with no significant change in subcellular distribution upon IL-1β stimulation (Supplementary Fig. 6b). This is consistent with RRP1 primarily binding to the nuclear fraction of Tyms mRNA in RAW 264.7 cells (Supplementary Fig. 3c). We next examined Tyms mRNA subcellular localization via cytoplasmic/nuclear fractionation in RAW 264.7 cells pre-treated with actinomycin D before harvesting. Quantitative analysis revealed that Tyms mRNA transcripts spanning the exon 2-exon 3 junction accumulated significantly in the cytoplasm of RRP1 KO RAW 264.7 cells after IL-1β stimulation, while this increase was absent in wild-type cells (Fig. 6c, Supplementary Fig. 6c, g). In contrast, transcripts containing the intron region exhibited a decreased cytoplasmic/nuclear ratio in RRP1 KO cells (Supplementary Fig. 6d). These findings demonstrate that RRP1 deficiency specifically enhances cytoplasmic accumulation of mature Tyms mRNA, but not pre-mRNA.

a Spectrophotometer (A260) detection of the fractions in RAW 264.7 cells after sucrose density gradient centrifugation (see Methods section). b RT-qPCR analysis of Tyms mRNA levels residing on the ribosome (mainly in Fraction 11). c RT-qPCR analysis of the cytoplasmic/nuclear ratio of mature Tyms mRNA (Across Exon2 and Exon3) in wild-type and RRP1 KO RAW 264.7 cells stimulated by IL-1β for the indicated time. RT-qPCR detection of the mature mRNA (across exon2 and exon3, d) and the newly transcribed nascent transcription (intron3, e) of Tyms in RRP1 KO RAW 264.7 cells upon IL-1β stimulation or not. f Schematic of the reads coverage of Tyms mRNA binding to Flag-tagged RRP1 by seCLIPs performed in RAW 264.7 cells upon IL-1β stimulation or not. g Surface plasmon resonance analysis of binding between the in vitro transcribed RNAs corresponding to the first intron of Tyms mRNA identified by CLIP-seq and the recombinant RRP1 proteins at different concentrations. KD, equilibrium dissociation constant. Immunofluorescence images (h) of RRP1 in RAW 264.7 cells stimulated by IL-1β for the indicated hours and the quantitative analysis (i, >20 cells per field, see methods section). Data are presented as means ± SD of (b, n = 4; d, e, n = 3) technical replicates from three independent experiments with student’s t test (two-tailed unpaired). Data are presented as means ± SEMs of (c, n = 3) technical replicates from three independent experiments with ANOVA test (two way). Data are representative of three independent experiments (a, g, h). Source data are provided as a Source Data file.
To further investigate this, we performed nascent RNA-capture assays to examine transcript dynamics of Tyms in IL-1β-treated macrophages. RRP1 KO RAW 264.7 cells, showed significantly elevated nascent Tyms RNA transcripts spanning the exon 2-exon 3 junction compared to wild-type controls (Fig. 6d, Supplementary Fig. 6e, g), suggesting either enhanced transcriptional initiation or exon retention. Notably, intronic regions displayed comparable levels between wild-type and RRP1 KO cells under IL-1β stimulation (Fig. 6e), indicating that RRP1 regulates TYMS expression primarily through post-transcriptional suppression of pre-mRNA splicing or maturation rather than nascent transcription. Then single-end enhanced cross-linking and immunoprecipitation (seCLIPs) revealed that RRP1 binding to Tyms mRNA intron region, with several binding sites, especially the first two introns (Fig. 6f). Surface plasmon resonance (SPR) analysis further validated this interaction, showing significantly stronger binding affinity of recombinant RRP1 protein to the the first intron than the second intron (Supplementary Table. 1; Fig. 6g, Supplementary Fig. 6f). Furthermore, RNA binding motif analysis via RRP1 protein crosslinking to in vitro transcribed RNAs of Tyms first intronic region, followed by RIP-qPCR, identified amino acid residues 124-133 and 205-211 in the Nop52 domain as critical for RRP1 binding to the Tyms intronic region (Supplementary Fig. 3d–h). Collectively, these data indicate that RRP1 post-transcriptionally suppresses TYMS expression.
RRP1 primarily localizes in the nucleolus and shuttles between the nucleolus and the pre-nucleolar body, a key event that drives pre-rRNA splitting process26. Immunofluorescence imaging confirmed RRP1 predominantly located in the nucleolus in both RAW 264.7 and THP-1 cells (Fig. 6h, Supplementary Fig. 6h). Notably, increased RRP1 fluorescence signal appeared outside the nucleolus when the two cell lines were stimulated with IL-1β (Fig. 6h, i, and Supplementary Fig. 6h), suggesting that RRP1 may re-localize from the nucleolus to the nucleoplasm in macrophages in response to innate stimuli. This relocalization potentially explains altered RNA-binding dynamics of RRP1 upon IL-1β stimulation in macrophages (Fig. 3a, f, h, and Supplementary Fig. 3c). Collectively, nuclear RRP1 acts as a key metabolic checkpoint in macrophages responding to inflammation by inhibiting TYMS expression. This inhibition results in global metabolic reprogramming in the 1C-cycle, thereby inhibiting cytokine-mediated autoinflammation (Supplementary Fig. 6f).
RRP1 restrains experimental autoinflammation in vivo
To further explore the in vivo function of RRP1 in the inflammatory response and autoimmune diseases, we generated myeloid-specific RRP1-deficient mice (Supplementary Fig. 7a, b) and prepared the collagen antibody-induced arthritis (CAIA) mouse model to assess the role of RRP1 in vivo29. This experimental polyarthritis model represents the effector phase of arthritis, which is mediated by the innate immune system with IL-1 extensively involved (Supplementary Fig. 7c)30,31. Intriguingly, Rrp1fl/fl Csf1r-IRES-Cre+ mice showed more severe development of inflammatory arthritis with higher clinical scores, thicker ankle joint diameter, and severe redness and swelling of the entire paw, including digits, compared with Rrp1fl/fl Csf1r-IRES-Cre- littermates (Supplementary Fig. 7e, Fig. 7a, b). Levels of the pro-inflammatory cytokines, IL-1β and IL-6, were significantly higher in sera from Rrp1fl/fl Csf1r-IRES-Cre+ mice compared to Rrp1fl/fl Csf1r-IRES-Cre- littermates (Fig. 7c). Histological analysis of knee joints and carpal joints by H&E staining showed dramatically increased disease severity with attenuated synovial hyperplasia, greater inflammatory cell infiltration, and periosteum destruction in Rrp1fl/fl Csf1r-IRES-Cre+ mice (Fig. 7d, e). As TYMS-mediated 1C-metabolism correlates with the pro-inflammatory phenotype in vitro, we hypothesized that inhibition of TYMS alone would be sufficient to treat the autoimmune disease. And TYMS protein levels in BMDMs from Rrp1fl/fl Csf1r-IRES-Cre+ mice were increased compared to their Rrp1fl/fl Csf1r-IRES-Cre- littermates in the CAIA model (Supplementary Fig. 7d). We next evaluated the treatment efficacy of pharmacological inhibition of TYMS with Raltitrexed in established mouse models of CAIA on Day 5 and Day 7 (Fig. 7f). The results revealed that intraperitoneal injections with Raltitrexed significantly attenuated macroscopic disease severity in Rrp1fl/fl Csf1r-IRES-Cre+ mice, as evidenced by reduced joint swelling, synovial inflammation, and periosteum destruction compared to vehicle-treated controls (Fig. 7g, h). These data indicate that RRP1-deficient mice have enhanced joint inflammation and immunologic injury in an arthritis model, accompanied by increased TYMS and pro-inflammatory cytokines’ expression.

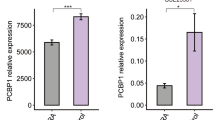
a Images of the ankle joint and the entire paw of Rrp1fl/fl Csflr-IRES-Cre+ (condition knockout, cKO) mice and the Rrp1fl/fl Csflr-IRES-Cre- (wilde-type, WT) littermates on day13 in the CAIA model. Intravenous injection with Normal saline (NS) was as control. b Clinical scores of Rrp1fl/fl Csflr-IRES-Cre+ mice and the Rrp1fl/fl Csflr-IRES-Cre- littermates of CAIA model mice (n = 10 mice per group). c ELISA of the indicated cytokines in the sera of Rrp1fl/fl Csflr-IRES-Cre+ mice and Rrp1fl/fl Csflr-IRES-Cre- littermates of CAIA model on day13 (n = 7 mice per group). H&E staining of the carpal joints (d) or knee joints (e) of Rrp1fl/fl Csflr-IRES-Cre+ (condition knockout, cKO) mice and Rrp1fl/fl Csflr-IRES-Cre- (wilde-type, WT) littermates on day13. The black arrows indicate the sites of inflammation. f Schematic for pharmacological inhibition of TYMS in established CAIA models (see methods section). g H&E staining of the carpal joints or knee joints of Rrp1fl/fl Csflr-IRES-Cre+ mice treated with Raltitrexed (right) and the vehicle-treated control littermates (left) on day13. The black arrows indicate the sites of inflammation. h Clinical scores of Rrp1fl/fl Csflr-IRES-Cre+ mice treated with Raltitrexed (10 mg/kg mouse body weight) and the vehicle-treated control littermates in CAIA model (n = 5 mice per group). Correlation analysis of the correlation between RRP1 (i) or TYMS (j) relative mRNA level in PBMCs and IL-1β serum levels in patients with RA. k Immunofluorescence analysis of RRP1 protein in PBMC nuclei from patients with RA comparing the RRP1-high-expression-group (H_1, H_2) and RRP1-low-expression-group (L). PBMC samples were classified into high and low expression based on the median value of relative RRP1 mRNA fold change. Data are presented as means ± SEMs from (b, n = 10; h, n = 5) biological mouse samples from two independent experiments with ANOVA test (two way). Data are presented as means ± SEMs from (n = 7) biological mouse samples from two independent experiments with student’s t test (two-tailed unpaired) (c). Data are representative of more than three biological samples from two independent experiments (a, d, e). Data are representative of (n = 6 per group) biological samples (k). The correlation between different variables was computed with nonparemetric Spearman correlation (two-tail) (i, j) (n = 50). Source data are provided as a Source Data file.
Low expression of RRP1 in RA patients is accompanied by increased IL-1β levels
Clinically, IL-1β is an effective target in the treatment of several diseases, including RA, as it is produced by pro-inflammatory monocytes in RA joint synovial tissues12. We speculated a correlation between RRP1 or TYMS expression and IL-1β levels in RA patients. Therefore, we isolated serum and PBMCs from blood samples of RA patients, and found that serum levels of IL-1β exhibited a significant negative correlation with RRP1 transcript and a positive correlation with TYMS mRNA levels in PBMCs (Fig. 7i, j). Furthermore, immunofluorescence analysis revealed that nuclear RRP1 was mainly dispersed in the nucleoplasm of PBMCs in the RRP1-high-expression group while RRP1 was largely nucleolar in the RRP1-low-expression group (Fig. 7k), echoing our finding that RRP1 dispersed from the nucleolus in THP-1 cells stimulated with IL-1β (Supplementary Fig. 6h). Additionally, we analyzed the GEO data (GSE55235, Woetzel, D. et al.)30 and found that TYMS mRNA levels of in synovial tissues from osteoarthritis or RA patients were significantly higher than those in normal controls (Supplementary Fig. 8a), while the RRP1 mRNA levels were lower in arthritis patients comparing to controls (Supplementary Fig. 8b). Collectively, these clinical data confirm that RRP1 functions as an anti-inflammatory factor in RA pathogenesis.
Discussion
The RNA-RBP interactions and their cross-talk with pathways such as metabolic reprogramming remain incompletely defined in the context of inflammation and autoimmunity. Specifically, key RBPs and their target RNA mediating the metabolism-autoinflammation axis await identification. In this study, we established the GRPIp method to profile RBPs in inflammatory BMDMs upon IL-1β stimulation, identifying RRP1 as a critical inhibitor of cytokine-driven autoinflammation. Mechanistically, RRP1 post-transcriptionally suppresses TYMS expression, thereby restraining one-carbon metabolism.
Profiles of dynamic RNA-RBP interactome holds potential to uncover the global landscape of RBP-RNA interactions and elucidate their remodeling under pathological conditions such as autoimmunity and autoinflammation2. However, comprehensive study of RNA-RBP interactomes have been limited because conventional RNA-specific capture methods (e.g., Oligo(dT)-RNA-MS, CHIRP-MS)32,33,34 and protein-specific approaches (e.g., CLIP-seq, ARTR-seq)34,35,36 fail to retain ~95% of mammalian cellular RNAs and numerous non-canonical RBPs. Notably, although the majority of RBPs are characterized as containing RBD(s), an emerging class of “moonlight” RBPs, including epigenetic regulators like HDAC1, DNMT3A, TET2, lacking canonical RBD or RNA-binding functional annotation but exhibit RNA-binding capacity35,36,37,38,39. Thus, an “unbiased” approach capable of isolating native protein-RNA complexes from unlabeled whole-cell samples and detecting RBD-lacking RBPs or diverse RNA classes is critically needed23,24. Moreover, primary cell studies remain scarce due to challenges in cell availability and RNA labeling, leaving RBP-RNA interactions poorly characterized in pathogenic contexts32,36. Herein, the GRPIp method, leveraging the optimized “Phase Separation” approach, addresses these limitations of preconceived functions by enabling unbiased identification of inflammation regulators in primary BMDM cells. This approach not only captures dynamic RNA-protein interactions but also facilitates the discovery of novel functional RBPs in response to cellular spatiotemporal changes23. Furthermore, the method serves as a powerful tool for constructing RBP libraries from primary cells or clinical samples, thereby providing mechanistic insights into disease pathogenesis across animal models and human disease.
Macrophages undergo dynamic metabolic shifts upon inflammatory stimulation, and that changes in metabolites selectively orchestrate the macrophage response40,41,42. Recently, attention has focused on the emerging role of 1C-metabolism, a network of metabolic cycles that supports methylation and provides 1C-bound folatesis and is linked to de novo purine and thymidine synthesis, epigenetic control of DNA or histone methylation, and redox homeostasis20,43,44. The general cognition about 1C-metabolism in inflammatory immunity centers on its role in promoting IL-1β production through increasing SAM for histone H3 lysine 36 trimethylation or serine generation for glutathione synthesis20,44. However, few studies have investigated the regulation of 1C-unit carriers, particularly the folate cycle. Within this pathway, TYMS is the key enzyme for sustaining THF pools, but its regulatory mechanisms remain poorly understood22. Thus, the impact of 1C-metabolism on inflammation appears multifaceted. Moreover, little is known about how 1C-metabolism is regulated in macrophages during inflammatory stimulation. Our study demonstrates that TYMS expression is suppressed, thereby blocking the 1C-metabolism in inflammatory macrophages induced by IL-1β. We further identify the RNA binding protein RRP1 as an endogenous inhibitor of 1C-metabolism via TYMS suppression. We also observed an increasing expression of the other key 1C-metabolic enzyme, MTHFD2L, whose mRNA was also enriched in our RIP-seq. But the further seCLIP-seq by RRP1 did not identify binding peaks on the MTHFD2L mRNAs transcript body. Whether RRP1 indeed exerts a regulatory role on MTHFD2L requires further investigation.
RRP1 has been studied for its role in rRNA processing, but its involvement in other cellular processes and pathology remains unclear. In this study, we reveal that RRP1 suppresses TYMS protein expression in macrophages through its RNA-binding activity. Although we observed increasing binding to Tyms mRNA introns in IL-1β-stimulated macrophages, the precise mechanism by which RRP1 regulates Tyms mRNA processing requires further elucidation. This process may involve additional factors such as m6A methylation or nucleolar RNA decay45,46. Furthermore, the concerted alteration-cytoplasmic enrichment of mature mRNA coupled with reduced nuclear pre-mRNA in RRP1 KO RAW 264.7 cells, indicates that RRP1 deficiency enhances cytoplasmic accumulation efficiency through dual potential mechanisms: promoting Tyms mRNA splicing maturation or facilitating nucleocytoplasmic transport, which need further validation. Finally, the kinetics by which RBPs bind to and dissociate from their RNA are critical for orchestrating cellular RNA-protein interactions. These dynamics can be determined by quantitation of specific sequence or binding affinity analysis in vitro47,48,49,50, a direction for future investigation.
One hypothesis consistent with our findings is that RRP1 re-localization from the nucleolus to the nucleoplasm may alter its RNA-binding capabilities in IL-1β-stimulated macrophages. It is also noteworthy that we observed increased fumaric acid levels in IL-1β activated BMDMs (Fig. 3b). Since fumaric acid can promote the succination of target proteins at the cysteine residues in proinflammatory macrophages51,52, we speculate that RRP1 cysteine residues might be modified by increased cellular fumaric acid upon IL-1β stimulation, thus strengthening the ability of RRP1 to bind RNAs. Recently, a time-resolved RNA-protein cross-linking method using a pulsed femtosecond ultraviolet laser, followed by immunoprecipitation and high-throughput sequencing, has allowed RBP-RNA binding and dissociation kinetics to be experimentally measured in cells50. This approach may provide a feasible method for future determination of the RNA-binding kinetics of RRP1 in macrophages during the inflammation response.
RBPs are emerging as promising therapeutic targets53. For example, conformational changes in the RBPs SF3B1 and PHF5A, which recognize pre-mRNAs, can be inhibited by anti-tumor compounds such as macrolides54. Our study identifies RRP1 as a regulator of 1C-metabolism that suppresses inflammation in autoimmune diseases, offering a novel target for RA therapy. Notably, the antifolate drug MTX has been used for the treatment of RA for about forty years as it can impair macrophage pro-inflammatory responses through DHFR enzyme inhibition55. But strategies bypassing direct inhibition of DHFR that retain the efficacy of anti-folates without the limiting toxicities are still in need56. In recent years, the ability of new-generation antifolates (e.g., Raltitrexed, PT523, and GW1843) to inhibit TYMS activity and potently suppress pro-inflammatory cytokine release from T cells of RA patients, including those clinically refractory to MTX has been demonstrated57. Consistent with these findings, our results revealed that Raltitrexed exerts a remarkable anti-inflammatory effect on macrophages and alleviates disease severity by attenuating the progression of rheumatoid arthritis in mouse CAIA models.
Encouraged by these results, RRP1-mediated suppression of TYMS-driven 1C-metabolism could, therefore, provide a specific new target for anti-rheumatic drug design. Moreover, RRP1’s subnuclear localization differs in PBMCs with high versus low RRP1 expression, suggesting its potential as a diagnostic biomarker for RA. Critically, our finding that RRP1 also constrains inflammatory response in macrophages in response to multiple pro-inflammatory cytokines including TNFα and IL-6 indicated that RRP1 may be broad applicability for treating autoimmune and inflammatory disorders.
Methods
Ethics statement
The study design and experimental procedure for peripheral blood samples collected from patients were approved by the Ethics Review Board of Peking Union Medical College Hospital and Institute of Basic Medicine, Chinese Academy of Medical Sciences with the Ethical approval number: JSS-1196 and ZS-2024006. Informed consent in writing was obtained from all participants. No sex or gender analysis was carried out.
All animal experiments conducted in this study were carried out following the guidelines established by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Laboratory Animal Science of Chinese Academy of Medical Sciences. The experimental design and procedures were reviewed and approved by the animal ethics review board (ACUC-A01-2019-006). The mice were bred in specific pathogen-free conditions under constant temperature and humidity with a 12 h light cycle. Animals were allowed food and water ad libitum. The temperature was 24 ± 2 °C and humidity was 40–70%.
PBMCs from rheumatoid arthritis patients
Peripheral blood samples were collected from patients who met the 2010 ACR/EULAR Rheumatoid Arthritis classification criteria at their first hospital admission without specific treatment in Peking Union Medical College Hospital. Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood using Lymphoprep (07861, STEMCELL).
Mice, cells and reagents
C57BL/6 mice were obtained from Joint Ventures Sipper BK Experimental Animals (Shanghai, China). IL-1 Receptor type 1 deficient mice (IL-1R1 KO) (B6.129S7-Il1rI tm1Imx/J, Stock No: 003245) were from The Jackson Laboratory. Rrp1-Flox (Rrp1fl/fl) mice generated by using CRISPR-cas9 editing (Cat. No: NM-CKO-200313) and Csf1r-IRES-Cre mice (Cat. No: NM-KI-190026) were from Shanghai Model Organisms Center, Inc. To establish myeloid-specific Rrp1-deficient mice, Rrp1fl/fl mice were crossed with Csf1r-IRES-Cre mice. Exon 4 of Rrp1 was excised by CRE recombinase in myeloid cells. The genotype primers by PCR analysis were listed in Supplementary Table 2.
THP-1 cell lines were obtained from National Collection of Authenticated Cell Cultures (NCACC). RAW 264.7, NIH/3T3 and 293 T cell lines were obtained from American Type Culture Collection (ATCC). RAW 264.7, NIH/3T3 and 293 T cells were cultured in DMEM supplemented with 10% FBS (10099141 C, Thermo Fisher) and 5% Penicillin-Streptomycin Solution. THP-1 cells were cultured in RPMI 1640 supplemented with 10% FBS and 0.05 mM β-mercaptoethanol (21985023, Thermo Fisher), and treated with 10 ng/ml PMA for 48 h to differentiate the cells into macrophages before the experiments. BMDMs culturing conditions were previously described23. Briefly, bone marrow was harvested from C57BL/6 mice and cultured in DMEM medium supplemented with 50 ng ml−1 murine M-CSF (315-02, PeproTech), 10% FBS, and 1× Penicillin-Streptomycin solution (C0222, Beyotime). The cells were cultured for 7 days with fresh media changed every other day.
The RRP1 knock out RAW 264.7 cell line was generated using CRISPR/Cas9 editing (51133, 52962, Addgene). Targeted but unedited cells were used as the wild-type (WT) control. The sequence of the sgRNA targeting the first exon of Rrp1 is 5’- GGGCAATGAACAGGTGACGC-3’ (Supplementary Table 1). RRP1-overexpressing RAW 264.7 cells were generated by infection with lentivirus containing RRP1-3×Flag-pCDH vector and then puromycin (Sigma) selection. The reagents including chemicals, peptides, and recombinant proteins used in this study are listed in Supplementary Table 7. Kits for Critical commercial assays are listed in Supplementary Table 8.
RNAi, plasmids, and lentivirus transfection
For siRNA transfection, cells were transfected with 20 nM siRNAs using Lipofectamine RNAiMAX (13778150, Thermo Fisher) according to the manufacturer’s instructions. The sequences of siRNAs are listed in Supplementary Table 3. The coding sequences of Rrp1 (gene ID: 18114) and Tyms (gene ID: 22171) with distinct tags were amplified from macrophage cDNAs. Then, the CDSs were cloned into a pCDNA3.1 (Cat# V79520, Thermo Fisher) or pCDH (BR594, Fenghui Biotechnology) eukaryotic expression vector. For transient transfection of plasmids, cells were transfected at a final concentration of 1 µg/mL plasmid using Jetprime reagents (101000046, Polyplus Transfection). For stably transfection, lentivirus containing RRP1-3×Flag-pCDH vector were produced in 293T cells by using psPAX2 and pMD2.G helper vectors (BR036, 037, Fenghui Biotechnology) and then infected cells with polybrene (GM-040901, Genomeditech) according to the manufacturer’s instructions. Cells were allowed to grow for 24–36 h before treating with 50 ng/mL IL-1β (211-11B, PeproTech), 10 ng/ml TNFα (654245, Sigma-Aldrich), 20 ng/ml IL-6 (216-16, PeproTech), 100 ng/mL LPS (L2880, Sigma-Aldrich) or 100 µg/mL Poly (I:C) (P1530, Sigma-Aldrich) transfection.
Cell nuclear and cytoplasmic separation
Separation of the cell nuclear and cytoplasmic fractions was performed using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (78833, Thermo Fisher) according to the manufacturer’s protocol. Before harvesting, the cells were treated with actinomycin D (5 µM) for 25 min in 37 °C.
RNA isolation and RT-qPCR
Total cellular RNA was extracted using an RNA extraction kit (220011, Fastagen) base on the manufacturer’s instructions. The cDNA was synthesized with HiScript III 1st Strand cDNA Synthesis Kit ( + gDNA wiper) (R312, Vazyme). ChamQ SYBR qPCR Master Mix (Q331, Vazyme) was used for RT-qPCR analysis. mRNA expression was analyzed using GAPDH as an endogenous control. Data were analyzed with the 2-ΔCt-ΔCt method. Primer sequences are listed in Supplementary Table 5.
Western blot
Whole-cell lysates were harvested and the supernatant was collected by centrifugation at 14,000 × g for 10 min. Protein concentration was determined using the BCA assay. In total, 20 µg proteins were loaded in each lane on SDS-PAGE gels and transferred to the nitrocellulose membrane after electrophoresis. 5% non-fat milk in TBST was used to block the membrane for 1 h at room temperature. The membrane was then incubated with primary antibodies overnight at 4 °C. Anti-mouse-HRP (7076, CST) and anti-rabbit-HRP (7074, CST) were used as secondary antibodies and protein bands were visualized using ECL. Western bloting bands were quantified via ImageJ-based integrated density analysis, normalized to GAPDH as the internal reference control. The antibodies used in this study are listed Supplementary Table 6.
Immunofluorescence and RNA FISH
For Immunofluorescence staining, cells were seeded on glass-bottom dishes 12 h before being treated. After stimulation, cells were fixed in 4% paraformaldehyde for 10 min and blocked in 5% BSA in PBS containing 0.1% Triton-X100 (PBST) for 1 h at room temperature. Then cells were incubated in primary antibody anti-RRP1 (GTX115107, GeneTex, 1:500) for THP-1 cells and anti-NNP1 (sc-398970, Santa Cruz, 1:200) for RAW 264.7 in PBST overnight at 4 °C. After being washed three times with PBST, the cells were further incubated with Alexa Fluor 488 conjugate anti-Rabbit IgG (4412, Cell Signaling Technology), Alexa Fluor 488 conjugate anti-mouse IgG (4408, Cell Signaling Technology) and Alexa Fluor 568 conjugate anti-mouse IgG (ab175473, Abcam) respectively in PBST for 1 h at room temperature. After being washed three times with PBST, the cells were sealed with anti-fade mounting medium containing DAPI (P0131, Beyotime). The stained cells were imaged using a TCS SP8 laser confocal microscope (Leica) with × 63 oil immersion objectives. For the quantitative analysis, the area of each channel-specific image was quantified using Image J, followed by normalization according to the following formula: (AreaRRP1-AreaFBL) / AreaRRP1 × 100%.
For RNA FISH-IF assay, Fluorescein isothiocyanate (FITC)-conjugated Tyms probes were generated by Azenta life science. Sequences are listed in Supplementary Table 4. Cells were hybridized with DNA probe sets according to the Stellaris RNA FISH Protocol from Biosearch Technologies, and then immunostained RRP1 with the antibodies and Cy5-conjugated anti-mouse IgG. Then the cells were sealed and imaged.
Cell death kinetics
Cell death kinetics were indicated using Green Nucleic acid dye (S7020, Invitrogen) and monitored using the IncuCyte S3 (Sartorius) live-cell analysis system based on the manufacturer’s instructions. Briefly, about 6 × 104 cells that well cultured in 96-well plates were changed fresh culture medium with the dye before TCZ treatment (TNFα (20 ng/ml), cycloheximide (CHX) (10 μg/ml) and Z-IETD-FMK (2 μM))58,59. Then the plate was immediately scanned for the indicated time durations, with fluorescent and phase-contrast images acquired in real-time every 2 h. Fluorescent-positive dead cells were quantified using the software package supplied with the IncuCyte imager. The cell death ratio was generated by normalization to the original total cell counts.
RNA immunoprecipitation and high-throughput sequencing
Briefly, 1 × 108 BMDMs seeded in two 150 mm cell dishes were used for IL-1β treatment at each time point. After treatment, cells were harvested using RIP lysis buffer (50 mM Tris-HCl pH=7.5, 1% NP-40, 1 mM EDTA, 0.1% SDS) and 5% of the lysate was isolated as input. A primary antibody that recognized RRP1 or control IgG was added to the supernatants overnight at 4 °C. Protein A/G Magnetic Beads (MCE, HY-K0202) were then added and gently mixed for 1 h at 4 °C. The beads were washed with NT2 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40) 5 times before digestion with Proteinase K in buffer (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM EDTA, 10 mM EGTA, 0.5 mM Triton-X100, 0.2% SDS) for 30 min at 55 °C to remove proteins. The RNA was isolated using TRIzol LS (10296028, Thermo Fisher) and further assessed by RT-qPCR or high-throughput sequencing. RNA-sequencing was performed on Illumina HiSeq 2500 with 150 bp paired-end reads. The fastq files from three biological replicates of RNA immunoprecipitation sequencing (RIP-seq) were concatenated per read pair to generate one pooled fastq file. The BAMS were used by RIP Seekerto statistically infer RIP regions. The genome version was mm10. The up-gene enrichment of the RIP samples from IL-1β treated BMDMs is list in Source Data file. The data to GEO (GSE271948) have uploaded.
seCLIP-seq
seCLIP-seq were performed as according to previous study60. Biological replicates of RAW 264.7 stably expressing Flag-tagged RRP1 that cultured with distinct initiation dates and crosslinking termination timelines were collected. seCLIP was conducted as previously reported, with the following modifications. Ultraviolet-crosslinked (400 mJ/cm2) RAW 264.7 (8 × 107) were lysed in lysis buffer (50 mM Tris-HCl pH = 7.4, 100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate and protease inhibitors) and sonicated. Lysates were treated with RNase I (Thermo Fisher Scientific) to fragment RNA and DNase I to remove DNA. Anti-Flag Beads were washed three times using lysis buffer and incubated with proteins lyastes for 6 h at 4 °C. A 3’ RNA adaptor was ligated onto the RNA with T4 RNA ligase (M0202, NEB). Protein-RNA complexes were run on a 4–12% gradient Bis-Tris Gels (Invitrogen), transferred to NC membranes, and RNA was isolated off the membrane identically to standard seCLIP. A fraction of sample was used for western blot of CLIPed Flag-tagged RRP1, indicating regions excised for seCLIP library preparation. After purification, RNAs were reverse transcribed with Superscript III reverse transcriptase (18080044, Invitrogen) with nested specific primer and a protected reverse PCR primer as previously reported, free primer was removed and a 3’ cDNA adaptor was ligated onto the cDNA product with T4 RNA ligase (M0202, NEB). Libraries were then amplified with Premix Taq PCR mix (Takara). Adapters and primers were designed according to the commercial indexing and sequencing primers. Sequencing reads were processed and mapped according to seCLIP procedure. The DNA fragments bound by proteins were then isolated and sequenced using the DNBSEQ-T7 platform. Sequencing raw reads were preprocessed by filtering out sequencing adapters, short-fragment reads and other low-quality reads. Bowtie2 (version 2.2.6) was then used to map the clean reads to the mouse mm10 reference genome. Peak detection was performed by MACS2 (version 2.2.7.1). The data to GEO (GSE292070) have uploaded.
Global RNA-protein Interactome purification (GRPIp)
After stimulation for different times, primary mouse macrophages were processed using GRPIp. Our RNA-Binding Peptide isolation and comparison method was based on “Interphase Separation” related RBP isolation methods23,24. At least 1 × 109 BMDMs per time point were seeded in several 150 mm dishes in order to obtain enough TRIzol-Chloroform interphase material. Cells were cross-linked with exposure to 200 mJ/cm2 UV at 254 nm after being washed 3 times with ice-cold PBS and dried for 3–5 min. Cells were then harvested with 8 mL TRIzol Reagent (15596018, Thermo Fisher) and placed in 15 mL centrifuge tubes. 1.6 mL chloroform was added to the tubes and mixed thoroughly. The lysates were centrifuged at 8000 g for 15 min at 4 °C. The white interphase was transferred into 2 mL tubes and gently washed once using Wash I Buffer (Tris-HCl 100 mM pH = 7.5, EDTA 1 mM, SDS 0.05%), centrifuged at 6000 g for 2 min at room temperature, and the supernatant was discarded. Immediately afterwards, 1 mL of Wash II Buffer (Tris-HCl 100 mM pH = 7.5, EDTA 1 mM, SDS 0.25%) was added to the tube to disperse the white interphase thoroughly, then centrifuged at 6000 g for 2 min at room temperature. This step was repeated once, then the supernatant was transferred to a new 2 mL tube. The solution was washed twice with 1 mL of Wash III Buffer (Tris-HCl 100 mM pH = 7.5, EDTA 1 mM, SDS 5%) and 1 mL of supernatant was harvested into a new tube after each centrifugation. After four washing steps, we obtained four tubes each containing 1 mL elutant (i.e., supernatant above). 1 mL isopropanol along with NaCl at a final concentration of 300 mM was added to each of the four 1 mL elution fractions. The mixtures were inverted 3–5 times and centrifuged for 20 min at 18,000 g at −10 °C. Supernatants were discarded and the four pellets were washed twice with 2 mL 70% ethanol. 1.8 mL RNase-free water was then used to dissolve the pellets in 2 mL tubes. 200 µL 10 × DNase Buffer (100 mM Tris-HCl, 25 mM MgCl2, 5 mM CaCl2), 10 µL RNase inhibitor (40 U/µL) and 100 µL DNase I (2 U/µL) was added and these samples were incubated at 37 °C with 800 rpm shaking for 1 h. Finally, RNA-RBP complexes were precipitated using isopropanol.
Qualitative peptide analysis by HPLC
Qualitative HPLC was performed using the Agilent Infinity 1260 LC system (Agilent) with 300SB-C18 5 µm 4.6 × 150 mM (Agilent). 20 µL 10 × dilution buffer (200 mM ammonium acetate) was added to peptides samples to a final volume of 200 µL. Samples were mixed and centrifuged at 18,000 g for 5 min. Supernatants were collected and transferred to loading bottles. Buffer A was 20 mM ammonium acetate, buffer B was 100% acetonitrile. The gradient program was: 0–2 min 100% A and 0% B, 2–60 min linear gradient to 65% B, 61–62 min linear gradient to 85% B, 62–80 min 85% B, 86–100 min 0% B.
Peptides preparation and 4D MS
At least 1000 µg RNA-RBP complexes were used to prepare peptides from RNA-associated proteins. The RNA-RBP complexes were pre-digested into RNA-peptides complexes by incubation with 10 µg trypsin/LysC at 37 °C for 1 h. 2-Chloroacetamide was added to the digestion mixture at a final concentration of 20 mM for another digestion step and alkylation for 1 h. Following this, we applied a modified method for isolating RNA-peptides complexes using a silica column (Qiagen, 163032371), as described previously23. Next, the isopropanol precipitate was used to isolate peptides cross-linked to RNAs. 50 µL 10 mM Tris-HCl was added to the pellets before RNase digestion. Finally, peptide samples were further purified with 30 kDa ultrafiltration tubes to remove RNase and any undigested proteins. Peptides were identified by 4D-LFQ MS (Bruker timsTOF Pro)61. The full MS scan range was 350–1600 m/z at 60,000 resolution, with MS/MS scans at 15,000 resolution in data-dependent acquisition (DDA) mode. Automatic gain control (AGC) was 1E5 with an intensity threshold of 230,000 ions/s. The maximum injection time was 22 ms, and dynamic exclusion was applied for 30 s to prevent redundant precursor scans. The resulting MS/MS data were processed using MaxQuant search engine (v.1.6.15.0). Search parameter setting: The database was SwissProt Mouse (17022 sequences). The enzyme digestion method was set as Trypsin/P with missing cuts number ≤2. The mass error tolerance for primary parent ions was 20 ppm and 5 ppm respectively for first search and main search, and 0.02 Da for secondary fragment ions. The alkylation of cysteine was fixed modification, and the modification was variable to the oxidation of methionine and the acetylation of the N-terminal of protein. The FDR for protein and PSM identification was 1%.
RNA binding motif-analysis in vitro
Recombinant Flag-tagged RRP1 proteins were expressed in 293 T cells purified with anti-Flag beads. In vitro transcribed RNAs (Supplementary Table 1) and purified recombinant RRP1 proteins (1 μg) were mixed into RNA-protein binding buffer (Tris-HCl, pH 7.5, 10 mM; KCl, 10 mM; MgCl2, 10 mM; DTT, 1 mM; 10% glycerin) with a molar ratios of 3:1, and incubated at 37 °C for 30 min. The reaction was terminated by immediate transfer onto ice and cross-linking with UV (254 nm, 150 mJ/cm2). The RNA-RBP complexes were subjected to reduction with 10 mM DTT and 0.1% SDS at 60 °C for 20 min, alkylated with 2-Chloroacetamide for 30 min, then pre-digested with trypsin/LysC (0.5 ng/μL) at 37 °C for 30 min (700 rpm) to generate RNA-peptide complexes. These complexes were isolated via silica column (Zymo, R1016) following the manufacturer’s instructions. Subsequent RNA digestion was performed using RNase A (20 ng/μL), RNase T1 (1 U/μL), and RNase I (10 U/μL) at 37 °C for 8 h. Peptides were further digested with trypsin/LysC (10 ng/μL) overnight, demineralized, and analyzed by LC-MS/MS. The LC-MS/MS analysis was performed on a Vanquish-Neo-UHPLC/Orbitrap Exploris 480 mass spectrometer (Thermo Fisher Scientific). The pFind software was used to identify proteins with the cutoff of peptide FDR < 1%62. The MS2 spectra were annotated using pLabel.
Surface plasmon resonance (SPR)
Biotinylated RNAs were synthesized by in vitro transcription using T7 RNA Polymerase (2050, NEB) and labeled with biotin RNA labeling mix (11685597910, Roche), followed by purification to ensure proper secondary structure formation. The biotinylated RNAs were then individually immobilized onto streptavidin (SA) sensor chips using a Biacore 1 K system (Cytiva). Recombinant RRP1 proteins (obtained from 293 T cells) diluted to the indicated concentrations were injected to associate with loaded RNA-sensors. The Response units (RU) were recorded at room temperature, and the interaction data were fitted to a 1:1 binding model with Biacore Evaluation Software (Cytiva).
ELISA
IL-6 from BMDMs culture supernatants and IL-1β from the serum of RA patients was measured using mouse IL-6 ELISA kits (ab222503, Abcam), mouse IL-1β ELISA kits (ab197742, Abcam) and human IL-1β ELISA kits (ab46052, Abcam) according to the manufacturer’s instructions. ELSIA kits (Meimian, Jiang Su, China) for SAM, Met, THF were used to detect the leves of SAM (MM-43942M1), Met (MM-45354M1), THF (MM-927831O1) in cultured RAW 264.7 cells. Briefly, RAW 264.7 (5 × 107) were suspended in 600 µL cold PBS (pH 7.2–7.4) and lysed by sonication. Centrifuged at 4 °C for 20 min (3000 rpm) and collected the supernatant for the next measurements according to the manufacturer’s instructions.
Metabolomics analysis
Sample collection and metabolomic analysis were performed as in previous reports63. In brief, 1 × 107 macrophages were cultured in 100 mm dishes and stimulated with IL-1β for indicated hours. Then, cells were collected and transferred to a 1.5 ml tube for centrifugation at 300 g for 5 min at 4 °C and then washed with cold PBS. The cell pellets were first centrifuged at 300 g for 5 min, then completely disrupted by resuspension in cold 80% methanol with vigorous vortexing, followed by ultrasonic treatment for thorough lysis. Finally, the samples were centrifuged at 14,000 g for 10 min at 4 °C and the supernatant was collected to complete the extraction process. For untargeted metabolomics profiling (n = 6 independent samples per group), the following instrumentation was used for separation, measurement and analysis: time-of-flight mass spectrometry (GC-TOF/MS) system (Pegasus HT, USA) coupled with gas chromatography (Agilent 7890B, Santa Clara, CA, USA) and a Gerstel MPS2 MultiPurpose Sampler (Gerstel, Mülheim, Germany) wer used to perform peak alignment, peak picking and quantitation for each metabolite. The iMAP platform (v1.0; Metabo-Profile, Shanghai, China) was used for further data analysis, and then significantly enriched pathways were selected using P < 0.05. For targeted metabolomics profiling (n = 5 or 3 independent samples per group), a ultraperformance liquid chromatography coupled to tandem mass spectrometry (UPLC-MS/MS) system (ACQUITY UPLC-Xevo TQ-S, Waters Corp., USA) was employed. The raw data were then processed using iMAP platform. The Z-score indicates how many standard deviations an observation is above or below the mean of the control group.
Carbon-tracing experiments
Carbon-tracing experiments were performed as in previous reports20,63. RAW 264.7 cells (6 × 106 cells) were plated in 60 mm dishes in complete medium. After overnight, the cells were carefully washed twice with DPBS and exchanged with fresh medium lacking serine, supplemented with 10 µM L-serine-[13C3] (HY-N0650S, MCE) for pre-tracing. After 2 h, cells were stimulated for 4 h with IL-1β or not. Then the cells ware harvested and flash freezing with liquid nitrogen and store at −80 °C (n = 4 independent samples per group). Samples were homogenized in 400 μL cold 80% methanol using ultrasonic disruption, followed by centrifugation at 18,000 g for 15 min at 4 °C. The supernatants were concentrated and reconstituted in 100 μL 80% methanol, with 50 μL transferred to 96-well plates for folate analysis. For amino acid quantification, 10 μL supernatant was derivatized with 70 μL borate buffer and 20 μL AQC regent, 55 °C, 10 min, and further diluted 100-fold before detection. All samples were tested by UPLC-MS/MS system (ACQUITY-I UPLC/Xevo TQS). The raw data files generated by UPLC-MS/MS were processed using the MassLynx software (v4.1, Waters, Milford, MA, USA) to perform peak integration, calibration, and quantitation for each metabolite.The Mass Distribution Vector (MDV) values of the metabolites are list in Source Data file.
Capture of the newly transcribed RNA using click chemistry
The nascent RNA was captured as previously described64. Briefly, RAW 264.7 cells were cultured in 100 mm dishes to 85% confluence (1.5 × 107) and pre-labeled with 250 µM 5-ethyluridine (EU) (HY-141140, MCE) for 5 min, and immediately stimulated with IL-1β or not for 4 h. After labeling, the cells were cross-linking with UV (254 nm, 150 mJ/cm2), fixed with cold 90% fresh ethanol and permeabilized with 0.5% Triton X-100 (in PBS). Then, the N-(3-Azidopropyl) biotinamide (HY-129832, MCE) was linked to the alkyne- labeled EU in the cells by click chemistry and the cells were collected. The biotin- EU- labeled RNA in the cells was capture with streptavidin- conjugated magnetic beads. After washing, the beads were digested with Proteinase K in buffer (20 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM EDTA, 10 mM EGTA, 0.5 mM Triton-X100, 0.1% SDS) for 1.5 h at 55 °C to remove proteins. The RNA was isolated using TRIzol LS (10296028, Thermo Fisher) and further assessed by RT-qPCR.
Ribosome sedimentation analysis
Sucrose density gradient centrifugation was used to separate ribosome fractions. 1.5 × 107 Cells were pre-treated with 100 µg/ml cycloheximide (CHX) for 10 min at 37 °C. After being washed with cold PBS containing 100 µg/ml CHX, cells were lysed in lysis buffer (10 mM Tris-HCl pH 7.5, 100 M KCl, 5 mM MgCl2, 2 mM DTT, 1% Triton X-100, 100 µg/ml CHX and 80 U/ml RNase inhibitor and 1× Protease inhibitor). After centrifugation at 13,000 g for 10 min, supernatants were collected and loaded onto a 10–50% sucrose gradient in the base buffer (lysis buffer without Triton X-100). The gradients were sedimented at 35,000 rpm for 3.5 h using a SW41 Ti rotor at 4 °C. Eleven fractions were collected (the fractions, from top to bottom, were marked as Fraction1 to Fraction 11 in turn) and used for Western blot and qPCR analysis. The most RPL35-enriched fraction was regarded to be the ribosomal fraction. Tyms mRNA levels residing on the ribosome were normalized to those in the whole cell input.
Collagen antibody-induced arthritis (CAIA) model
Sex- and age (9-week-old) -matched Rrp1fl/fl Csflr-IRES-Cre+ (condition knockout) mice and Rrp1fl/fl Csflr-IRES-Cre- (wilde type) littermates (n = 10 per group) were used to establish CAIA models according to the manufacturer’s instructions (Chondrex, Inc.). 2 mg/mouse of the mAb cocktail was injected on day 0 followed by an LPS injection (30 µg) on day 3. We evaluated the severity of redness and swelling in the wrist and paw in a scale of 0–2 from day 5 to day 9 to qualitatively score the severity of paw inflammation. Arthritis was further exacerbated using an additional LPS injection (30 µg) on day 10. Mice were euthanized by cervical dislocation following isoflurane anesthesia on day13. Knee and carpal joints were fixed in paraformaldehyde for subsequent H&E staining. Serum was collected for cytokine measurement by ELISA. The clinical score of CAIA mouse model was decided by the morphology of the indicated paws according to the manufacturer’s instructions. Score =0: Normal; Score = 1: Mild, but definite redness and swelling of the ankle or wrist, or apparent redness and swelling limited to individual digits, regardless of the number of affected digits; Score = 2: Moderate redness and swelling of ankle or wrist; Moderate redness and swelling of ankle or wrist; Score = 3: Severe redness and swelling of the entire paw including digits.
For the pharmacological inhibition of TYMS, sex-unconsidered Rrp1fl/fl Csflr-IRES-Cre+ mice were administered Raltitrexed (HY-10821, MCE) via intraperitoneal injection at 10 mg/kg on days 5 and 7. An equal volume of sterile PBS served as the vehicle control (n = 5 per group).
Statistical analysis
Results are provided as means ± the standard deviation (SD). The statistical significance of comparisons between two groups was analyzed with a two-tailed Student’s t test or one way ANOVA test the indicated biological or technical replicates from at least triplicate independent experiments or biological as described in Figure Legends. Statistical analysis was performed by GraphPad Prism v8. RIP-seq data analysis was conducted in R and statistical analyses were performed as described in Results and Figure Legends. Omicshare online tools were used for GO annotation and KEGG pathway analysis (https://www.omicshare.com/tools). Differences were considered to be statistically significant: P < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw data for RIP-seq and seCLIP seq experiments have been deposited in GEO under accession codes GSE271948 and GSE292070. Raw data of the protein mass spectrometry have been deposited in Proteome Xchange via the PRIDE partner repository with the primary accession code PXD054800. The metabolomics data were included in Excel tables in the Source Data file. All other data are included in the article and/or Supplementary Information. Source data are provided with this paper.
References
Turner, M. & Diaz-Munoz, M. D. RNA-binding proteins control gene expression and cell fate in the immune system. Nat. Immunol. 19, 120–129 (2018).
Liu, J. & Cao, X. RBP-RNA interactions in the control of autoimmunity and autoinflammation. Cell Res. 33, 97–115 (2023).
Hashimoto, S. & Kishimoto, T. Roles of RNA-binding proteins in immune diseases and cancer. Semin. Cancer Biol. 86, 310–324 (2022).
Galloway, A. et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 352, 453–459 (2016).
Ma, Z. et al. RNA-binding protein hnRNP UL1 binds κB sites to attenuate NF-κB-mediated inflammation. J. Autoimmun. 129, 102828 (2022).
Ustianenko, D. et al. LIN28 selectively modulates a subclass of Let-7 microRNAs. Mol. Cell 71, 271–283 (2018).
Medzhitov, R. The spectrum of inflammatory responses. Science 374, 1070–1075 (2021).
Na, Y. R., Stakenborg, M., Seok, S. H. & Matteoli, G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 16, 531–543 (2019).
Tang, P. M., Nikolic-Paterson, D. J. & Lan, H. Y. Macrophages: versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 15, 144–158 (2019).
Brennan, F. M. & McInnes, I. B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 118, 3537–3545 (2008).
Rose-John, S., Jenkins, B. J., Garbers, C., Moll, J. M. & Scheller, J. Targeting IL-6 trans-signalling: past, present and future prospects. Nat. Rev. Immunol. 23, 666–681 (2023).
Broderick, L. & Hoffman, H. M. IL-1 and autoinflammatory disease: biology, pathogenesis and therapeutic targeting. Nat. Rev. Rheumatol. 18, 448–463 (2022).
Burmester, G. R. & Pope, J. E. Novel treatment strategies in rheumatoid arthritis. Lancet 389, 2338–2348 (2017).
Bäck, M. et al. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Torretta, S. et al. D-mannose suppresses macrophage IL-1beta production. Nat. Commun. 11, 6343 (2020).
Faas, M. et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity 54, 2531–2546 (2021).
York, A. G. et al. IL-10 constrains sphingolipid metabolism to limit inflammation. Nature 627, 628–635 (2024).
Newman, A. C. et al. Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells. Mol. Cell 81, 2290–2302 (2021).
Sugiura, A. et al. MTHFD2 is a metabolic checkpoint controlling effector and regulatory T cell fate and function. Immunity 55, 65–81 (2022).
Yu, W. et al. One-carbon metabolism supports S-Adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell 75, 1147–1160 (2019).
Levescot, A. et al. IL-1β-driven osteoclastogenic Tregs accelerate bone erosion in arthritis. J. Clin. Investig. 131, e141008 (2021).
Lee, Y., Vousden, K. H. & Hennequart, M. Cycling back to folate metabolism in cancer. Nat. Cancer 5, 701–715 (2024).
Trendel, J. et al. The human RNA-binding proteome and its dynamics during translational arrest. Cell 176, 391–403 e319 (2019).
Urdaneta, E. C. et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 10, 990 (2019).
Zhang, Q. et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015).
Yoshikawa, H. et al. Human nucleolar protein Nop52 (RRP1/NNP-1) is involved in site 2 cleavage in internal transcribed spacer 1 of pre-rRNAs at early stages of ribosome biogenesis. Nucleic Acids Res. 43, 5524–5536 (2015).
Ducker, G. S. & Rabinowitz, J. D. One-carbon metabolism in health and disease. Cell Metab. 25, 27–42 (2017).
Dekhne, A. S., Hou, Z., Gangjee, A. & Matherly, L. H. Therapeutic targeting of mitochondrial one-carbon metabolism in cancer. Mol. Cancer Ther. 19, 2245–2255 (2020).
Khachigian, L. M. Collagen antibody-induced arthritis. Nat. Protoc. 1, 2512–2516 (2006).
Fraenkel, L. et al. American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. 73, 924–939 (2021).
Woetzel, D. et al. Identification of rheumatoid arthritis and osteoarthritis patients by transcriptome-based rule set generation. Arthritis Res. Ther. 16, R84 (2014).
Brannan, K. W. et al. SONAR discovers RNA-binding proteins from analysis of large-scale protein-protein interactomes. Mol. Cell 64, 282–293 (2016).
Castello, A. et al. Comprehensive identification of RNA-binding domains in human cells. Mol. Cell 63, 696–710 (2016).
Chu, C. & Chang, H. Y. ChIRP-MS: RNA-directed proteomic discovery. Methods Mol. Biol. 1861, 37–45 (2018).
Danan, C., Manickavel, S. & Hafner, M. PAR-CLIP: a method for transcriptome-wide identification of RNA binding protein interaction sites. Methods Mol. Biol. 2404, 167–188 (2022).
Ramanathan, M., Porter, D. F. & Khavari, P. A. Methods to study RNA-protein interactions. Nat. Methods 16, 225–234 (2019).
Xiao, Y. et al. Profiling of RNA-binding protein binding sites by in situ reverse transcription-based sequencing. Nat. Methods 21, 247–258 (2024).
Shen, Q. et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554, 123–127 (2018).
Xiao, R. et al. Pervasive chromatin-RNA binding protein interactions enable RNA-based regulation of transcription. Cell 178, 107–121 (2019).
Timblin, G. A. et al. Mitohormesis reprogrammes macrophage metabolism to enforce tolerance. Nat. Metab. 3, 618–635 (2021).
Liu, P. S. et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat. Immunol. 24, 452–462 (2023).
Wculek, S. K. et al. Oxidative phosphorylation selectively orchestrates tissue macrophage homeostasis. Immunity 56, 516–530 (2023).
Clare, C. E., Brassington, A. H., Kwong, W. Y. & Sinclair, K. D. One-carbon metabolism: linking nutritional biochemistry to epigenetic programming of long-term development. Annu. Rev. Anim. Biosci. 7, 263–287 (2019).
Rodriguez, A. E. et al. Serine metabolism supports macrophage IL-1β production. Cell Metab. 29, 1003–1011 (2019).
Liu, Y. et al. N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176 (2019).
Lee, T. A. et al. The nucleolus is the site for inflammatory RNA decay during infection. Nat. Commun. 13, 5203 (2022).
Dominguez, D. et al. Sequence, structure, and context preferences of human RNA binding proteins. Mol. Cell 70, 854–867.e9 (2018).
Kappel, K. et al. Blind tests of RNA-protein binding affinity prediction. Proc. Natl. Acad. Sci. USA 116, 8336–8341 (2019).
Qiu, C., Wine, R. N., Campbell, Z. T. & Hall, T. M. T. Bipartite interaction sites differentially modulate RNA-binding affinity of a protein complex essential for germline stem cell self-renewal. Nucleic Acids Res. 50, 536–548 (2022).
Sharma, D. et al. The kinetic landscape of an RNA-binding protein in cells. Nature 591, 152–156 (2021).
Humphries, F. et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 369, 1633–1637 (2020).
Kornberg, M. D. et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453 (2018).
Gebauer, F., Schwarzl, T., Valcarcel, J. & Hentze, M. W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 22, 185–198 (2021).
Cretu, C. et al. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 70, 265–273 (2018).
Municio, C. et al. Methotrexate limits inflammation through an A20-dependent cross-tolerance mechanism. Ann. Rheum. Dis. 77, 752–759 (2018).
Katturajan, R., Vijayalakshmi, S., Rasool, M. & Evan Prince, S. Molecular toxicity of methotrexate in rheumatoid arthritis treatment: a novel perspective and therapeutic implications. Toxicology 461, 152909 (2021).
van der Heijden, J. W. et al. Methotrexate analogues display enhanced inhibition of TNF-α production in whole blood from RA patients. Scand. J. Rheumatol. 43, 9–16 (2014).
Wang, L., Du, F. & Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Muendlein, H. I. et al. Neutrophils and macrophages drive TNF-induced lethality via TRIF/CD14-mediated responses. Sci. Immunol. 7, eadd0665 (2022).
Blue, S. M. et al. Transcriptome-wide identification of RNA-binding protein binding sites using seCLIP-seq. Nat. Protoc. 17, 1223–1265 (2022).
Meier, F. et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteom. 17, 2534–2545 (2018).
Yang, B. et al. Identification of cross-linked peptides from complex samples. Nat. Methods 9, 904–906 (2012).
Xia, J. et al. Blocking glycine utilization inhibits multiple myeloma progression by disrupting glutathione balance. Nat. Commun. 13, 4007 (2022).
Guo, X. et al. Capture of the newly transcribed RNA interactome using click chemistry. Nat. Protoc. 16, 5193–5219 (2021).
Acknowledgements
This work was supported by Grants from the National Natural Science Foundation of China (82388201, 82101868 and 32370963), National Key Research & Development Program of China (2018YFA0507403), China Postdoctoral Science Foundation (BX20200066) and Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2021-I2M-1-017 and 2022-I2M-JB-005). We thank Suzhou Municipal Key Laboratory (SZS2023005) for the helpful instrument support.
Author information
Authors and Affiliations
Contributions
X. C. designed the experimental approach and supervised the study; Y. Z. and M. L. established and applied the GRPIp method; Y. Z., M. L., K. J., and M.W. performed the RT-qPCR, Western blot, ELISA, RIP and animal experiments; M. L., and K. J. performed immunofluorescence; H.Q., Y. X., X. Z., and C.W. provided clinical samples, reagents and helpful discussion. Y. Z., M. L., and X. C. analyzed data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Li, M., Jin, K. et al. The RNA-binding protein RRP1 brakes macrophage one-carbon metabolism to suppress autoinflammation. Nat Commun 16, 6880 (2025). https://doi.org/10.1038/s41467-025-62173-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62173-3
