
Abstract
Catalytic reduction of carboxylic acids to valuable chemicals is highly desirable yet challenging for both biomass conversion and organic synthesis. Here we describe an efficient and sustainable electrocatalytic hydrogenation of carboxylic acids with amines utilizing protons as the hydrogen source. The application of an earth-abundant cobalt complex enables electrochemical generation of a cobalt-hydride intermediate, which serves as the key catalytically active species for this reductive process. Obviating the need for flammable H2 gas or sensitive hydrides, this general hydrogenative coupling of carboxylic acids with amines and nitroarenes allows producing a wide range of structurally diverse complex alkylamines under mild electrocatalytic conditions. Furthermore, the practicality and versatility of this protocol are demonstrated through its application in valuable isotope labeling using readily available deuterium sources.
Similar content being viewed by others
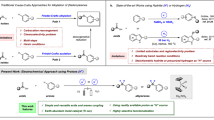

Introduction
Direct N-alkylation of amines has long-standing interest to synthetic chemists, as the resulting products are highly valuable bulk and fine chemicals such as pharmaceutical agents, agrochemicals, dyes, and natural products1. Conventional nucleophilic substitution approach to N-alkylated amines mainly relies on the use of alkyl halides as alkylating reagents (Fig. 1a)2. However, it suffers from the limitations of stoichiometric amounts of halide waste generation and poor chemoselectivity due to the undesired overalkylations. Although carbonyl reductive amination with aldehydes or ketones has been the benchmark method3,4, in some cases, their availability, sensitivity, and unwanted side reactions such as aldol condensations still restrict their applications. Carboxylic acids represent an important class of carbonyl compounds that are naturally abundant and structurally diverse from biomass feedstock5. The N-alkylation of amines using carboxylic acids, a higher-order variant of classical reductive aminations, provides an attractive alternative for the streamlined synthesis of complex alkylamines (Fig. 1b)6,7. Pioneered by Gribble and Marchini, the plausibility of reductive alkylation of amines with carboxylic acids was first demonstrated in the 1970s8,9. However, the requirement for relatively harsh reaction conditions and superstoichiometric metal hydrides (H−) significantly limited the scope of these early methods. Subsequently, a surge of synthetically useful methods have been developed with a diversity of catalytic systems, ranging from the noble metals10,11,12,13 to base metals14,15,16,17 even main group elements18,19,20 in combination with hydrosilanes as terminal reductants (Fig. 1c). More recently, Cole-Hamilton21, Beller22,23, and Sundararaju24 demonstrated the feasibility of this transformation using molecular hydrogen (H2) in the presence of Ru or Co/triphos catalysts. Despite these elegant advances, the utilization of sensitive hydrides or pressurized H2 gas as hydrogen sources might lead to potential security risk, as well as elaborate autoclave manipulation, which restricts their specific utility in synthetic chemistry.

a Classical methods for N-alkylation of amines. b Opportunities and challenges for carboxylic acid reduction. c Reductive amination of carboxylic acids using different hydrogen sources.
A rising trend in catalytic hydrogenation is the harness electricity as a renewable energy source for green and sustainable synthesis25,26,27,28,29,30,31,32,33. Complementary to classical thermal hydrogenations, electrohydrogenation directly employs protons (H+) and electrons (e−) as the hydrogen source and redox equivalent to substitute for traditional molecular hydrogen (H2) or hydride (H−) donors. Related to this strategy, the electrohydrogenation of various unsaturated bonds, such as carbonyl compounds, has received widespread attention34,35,36,37. The early examples of electrocatalytic hydrogenation of carbonyl compounds mainly focused on the reduction of relatively high reactive aldehydes and ketones38,39,40,41. We recently reported selective and efficient electrohydrogenation of less electrophilic nitriles to secondary and tertiary amines catalyzed by a cobalt bipyridine complex42. However, to the best of our knowledge, the electroreduction of more challenging carboxylic acids has seriously lagged behind related reductions of other carbonyl compounds (Fig. 1b). This is due to the low electrophilicity and thermodynamic stability of the carboxyl group, which renders it less reactive and difficult to reduce, therefore a high negative reductive potential is required43,44. Additionally, the Kolbe decarboxylation as a side reaction also competes with the desired carbonyl reduction process45,46,47. Moreover, the interaction of carboxylic acids with the metal catalyst, such as carboxylate overcoordination, may result in detrimental catalyst deactivation48,49,50. Consequently, it is highly desirable to develop a general and robust electrocatalyst system that can reduce carboxylic acids to value-added chemicals, considering their conceivable advantages of natural availability and structurally diversity.
In light of these challenges and opportunities, we herein report a mild and efficient electroreductive amination with carboxylic acids catalyzed by an earth-abundant cobalt complex (Fig. 1c). By utilizing protons as the hydrogen source, this method enables direct N-alkylations, including trifluoroethylation and methylation, to access a broad range of complex amines derived from carboxylic acids. Particularly, a key advantage of this electrocatalytic transformation is the feasible and valuable divergent incorporation of deuterium (D) into the resulting amines from readily available deuterated acids.
Results
Optimization reaction conditions
At the beginning of this study, a bench-stable, inexpensive, and bulk trifluoroacetic acid (TFA) was selected as the model substrate, as the resulting trifluoroethylation products are highly attractive in medicinally relevant fluorinated building blocks51. Compared to volatile trifluoroacetaldehyde or expensive trifluoroethylation reagents, TFA offers a cost-effective and practical alternative for synthesizing valuable β-fluoroalkylamines. Furthermore, as the simplest and ultrashort-chain perfluoroalkyl substance, the efficient and sustainable conversion of TFA into value-added chemicals has garnered significant attention52. To explore this potential, we investigated the reaction parameters for the electrocatalytic hydrogenative coupling of TFA 1 with 4-phenylaniline 2 (Table 1). Based on previous studies53,54,55, a key intermediate cobalt-hydride can be generated by cathodic reduction of a cobalt (II) precatalyst and followed by protonation in an acidic medium. Therefore, we evaluated the electrocatalytic performance of cobalt complexes with various ligands. We were pleased to obtain the desired β-fluorinated amine 3 in 93% yield using the commercially available diphosphine dppf (L1). Notably, the transformation exhibited good chemoselectivity, as neither reductive defluorination byproducts nor dialkylated species were observed. Other bidentate phosphines such as dppe (L2) and binap (L3) exhibited moderate reactivity in the model reaction, with the formation of a corresponding amide as a side product. The monodentate as well as tridentate phosphine ligands such as PPh3 (L4) and CH3C(CH2PPh2)3 (L5) also resulted in lower yields. Additionally, bipyridine (L6) and other ligands (Supplementary Table 1) did not improve the yield of the N-trifluoroethylation product. With the optimal ligand L1 identified (Table 1, entry 1), we further optimized other reaction parameters. While Co(OTf)2 provided the best results, other cobalt salts yielded slightly lower efficiencies, and non-noble metal salts such as Fe or Ni failed to produce the desired product (entries 2 and 3). The choice of Lewis acid additive proved critical for effective carbonyl group activation. For instance, ZnCl2 or BF3·Et2O instead of Ti(OnBu)4 did not give better results in this reaction (entry 4). When the reactions were performed at higher or lower temperatures, decreasing yields were observed (entry 5). As to solvent, the use of MeOH or THF suppressed the reaction completely (entry 6). Other sacrificial anodes, such as Mg, Al, or Fe, were inactive under these electrochemically conditions (entry 7). As comparison, stoichiometric reductants like Zn, Mn, Mg dust, PhSiH356, or atmospheric H2 gas, used in place of electrolysis, showed very low or no catalytic activity in the model reaction (entries 8–10). Control experiments confirmed that the cobalt catalyst was essential, and Ti(OnBu)4 significantly promoted the reaction (entries 11 and 12).
Substrate scope
With the optimal reaction conditions established, we explored the substrate scope of this cobalt-electrocatalytic hydrogenative transformation (Fig. 2). Initially, reductive N-trifluoroethylations using TFA were performed to access a variety of structurally diverse β-fluoroalkylamines (Fig. 2A). In addition to the model substrate 4-phenylaniline, 2-naphthylamine and anilines bearing alkyl, alkoxy, and aryloxy substituents all reacted efficiently, yielding fluorinated amines 4–12 in moderate to good yields (52–83%). A critical issue in hydrogenative reaction is the challenging but highly desirable chemoselectivity of diverse functional groups. Remarkably, anilines containing chlorine, trifluoromethyl, thioether, and sulfonamide functionalities underwent the transformation smoothly, providing synthetically useful yields of products 13–20. Notably, substrates with free hydroxyl and amino groups were well-tolerated without the need for pre-protection, yielding the corresponding products 21 and 22. A 2 mmol-scale reaction using 4-(2-aminoethyl)aniline as the substrate afforded product 22 in 63% yield. Under these electrohydrogenative conditions, sensitive and reducible functional groups such as nitrile, ester, and amide were also compatible, and their successful conversion to β-fluoroalkylamines 23–26 further expanded the reaction scope. Additionally, heteroaromatic amines, including quinoline, indazole, benzothiazole, and dibenzofuran, proved to be effective coupling partners, delivering the corresponding products 27–30 in 40–77% yields. Lenalidomide, an immunomodulatory drug used to treat multiple myeloma and anemia, furnished the desired product 31 in 48% yield. Furthermore, diamines reacted smoothly, providing the corresponding dialkylated amines 32 and 33 in 47% and 53% yields, respectively. While alkylamines such as benzylamine or morpholine failed to undergo the desired N-alkylations, this method offered unique advantages in isotopic labeling. By substituting CF3COOD instead of CF3COOH57, we achieved direct deuterium incorporation into β-fluorinated amine 3-d. This transformation enables the direct introduction of two important functional groups, CF3 and D, to an amine in catalytically one-pot process, highlighting the synthetic utility of this method.

a Reaction condition (A) as shown in Table 1. b Reaction condition (B): HCOONa or carboxylic acid (2.0 mmol), amine (0.2 mmol), Co(OTf)2 (10 mol%), L1 (10 mol%), BF3·Et2O (1.0 mmol), HCl (2.4 mmol, for methylation) or HFIP (0.2 mL, for alkylations), MeCN and toluene in an undivided cell with zinc cathode and anode, 80 mA, 110 °C, 1 h, N2.
Next, we turned our attention to the scope in terms of other less electrophilic carboxylic acids, including formic acid and aliphatic carboxylic acids (Fig. 2B). Installing a “magic methyl” group into molecules is highly desirable in modern drug discovery and chemical synthesis58,59,60,61,62. Among various methylation reagents, formic acid (HCOOH) stands out as one of the most convenient, non-toxic, easily manipulated, and readily available C1 sources, derived from biomass fermentation or CO2 reduction63,64. In many cases, the activation and reduction of less electrophilic carboxylic acids often require strong Brønsted or Lewis acid additives65. We were pleased to discover that BF3·OEt2, rather than Ti(OnBu)4, served as the optimal Lewis acid additive for the electroreduction of aliphatic carboxylic acids. With this additive, the molecular cobalt-electrocatalyst system enabled efficient N-methylation of diverse amines 34–38 using HCOOH or HCOONa as the methyl source. To further demonstrate the synthetic utility of this methylation method, we applied it to the direct functionalization of several natural products and pharmaceutical molecules. For instance, Cinacalcet 39, Meclizine 40, Norquetiapine 41, and Desloratadine 42 underwent this methylating transformation efficiently, highlighting the compatibility of the electroreductive protocol and its potential utility in late-stage functionalization. In addition to TFA and HCOOH, we also explored electrohydrogenative alkylations with less electrophilic aliphatic carboxylic acids. A representative set of chain and cyclic aliphatic carboxylic acids bearing olefinic and perfluoroalkyl functionalities proved compatible with this reductive coupling, yielding the corresponding alkylated anilines 43–52 in moderate yields, whereas substituted quinolines as the side products were observed through the Doebner von Miller pathway (Supplementary Fig. 8). Notably, aromatic acids showed poor conversion under these electroreductive conditions. Despite this limitation, the protocol offers a sustainable alternative to classical Eschweiler-Clarke reaction and reductive amination method, eliminating the need for toxic formaldehyde or unstable aldehydes.
Synthetic applications
Subsequently, we explored the electroreductive N-alkylation of readily available and inexpensive nitroarenes, given that most anilines are derived from their corresponding nitroarene precursors (Fig. 3). This hydrogenative coupling strategy with nitroarenes not only eliminates at least one synthetic step but also leverages bulk and cost-effective starting materials. Using TFA as the carboxylic acid substrate, the cobalt-electrocatalytic trifluoroethylation of various substituted nitroarenes proceeded smoothly, yielding the desired products 53–58 in 41–69% yields. Notably, pharmaceutical molecules such as Niclosamide 59 and Nimesulide 60 also performed comparably well under these conditions. A variety of reducible or sensitive functional groups in these molecules, including hydroxyl, ether, chlorine, and amide functionalities, were tolerated. Furthermore, aliphatic carboxylic acids, such as propionic acid and cyclobutanecarboxylic acid, were successfully converted into their corresponding N-alkylated products 61 and 62. By eliminating the need for pre-reducing nitroarenes to anilines, this electrochemical approach demonstrates remarkable versatility and practicality, enabling a convenient and economical one-pot synthesis of N-alkylamines directly from commercially accessible nitroarenes.

a 5 equiv. BF3·Et2O instead of Ti(OnBu)4, HFIP (0.2 mL), 40 mA, 100 °C, 3 h.
Organic molecules labelled with hydrogen isotopes have garnered considerable attention due to their potential applications in pharmaceutical discovery and radiation chemistry66,67,68,69. Traditional reductive deuteration processes often rely on expensive and non-recoverable D2 gas or stoichiometric deuteride reagents. Consequently, the development of cost-effective and readily accessible deuterium sources, such as deuterated protons (D+), would be highly desirable and demanding. In this work, we present an electrocatalytic approach for the incorporation of deuterium (D) at the α-positions of amines (Fig. 4). Remarkably, we achieved a practical and chemodivergent deuteration of N-methylation by strategically tuning the isotopic formates (HCOONa or DCOONa) and protic acids (HCl or DCl) as hydrogen (H) or deuterium (D) sources. Using this method, three distinct deuterium-labeled amines (–CH2D, –CHD2, –CD3) were obtained in a selective manner. For instance, the d1-methylated amines 63–66 were efficiently synthesized with high deuterium incorporation by employing DCOONa as the C1 source and aqueous HCl as the proton donor. When commercially available DCl was used, the valuable d2-methylation of amines with HCOONa was achieved, yielding products 67–70 with over 80% deuterium incorporation. Furthermore, the combination of DCOONa with DCl enabled full d3-methylation of N-methyl-4-phenylaniline 71, tetrahydroquinoline 72, meclizine 73, and dibenzylamine 74. This divergent incorporation of deuterated magic methyl groups represents a powerful tool for isotope labeling of small molecules, offering substantial potential for drug optimization and development.

a Installation of N-CH2D groups using DCOONa and HCl. b Installation of N-CHD2 groups using HCOONa and DCl. c Installation of N-CD3 groups using DCOONa and DCl.
Mechanistic investigations
To gain some insights into this electroreduction of carboxylic acids, a series mechanistic experiments were conducted. As shown in Fig. 5a, two plausible pathways were proposed for the hydrogenative coupling of carboxylic acids with amines. The carboxylic acid can be reduced to an aldehyde, which subsequently undergoes reductive amination to form the desired alkylamine. In addition, the amide formation followed by deoxygenative reduction might also exist as an alternative pathway. To elucidate the mechanism, we investigated the potential intermediacy of amide or aldehyde in this electroreduction. When the reaction was conducted with amides under electrocatalytic conditions, the expected corresponding amines were not formed. In contrast, treatment of trifluoroacetaldehyde hydrate as the alkylating reagent in place of TFA, the desired N-trifluoroethylative product was obtained in 35% yield. These findings suggest that amide formation prior to reduction is not involved in this transformation.

a Possible pathways and intermediate experiments. b Kinetic isotope effect experiments. c Reaction kinetics. d Cyclic voltammetry experiments. e 1H NMR experiments for CoH species. f DFT calculations of reaction profile.
Next, we conducted further kinetic studies to elucidate the rate-determining step of the carboxylic acid electrohydrogenation process. Initially, kinetic isotope effect (KIE) experiments were performed using deuterated and non-deuterated TFA as substrates (Fig. 5b). The rate profiles from the KIE experiments revealed a KH/KD value of 1.84, indicating a primary KIE and suggesting that hydrogen transfer is a key step in the reaction mechanism. Subsequently, we investigated the reaction kinetics by varying the concentrations of the cobalt catalyst, TFA, 4-tert-butylaniline, and Ti(OnBu)4 within the first 50 min of the reaction (Fig. 5c). The kinetic analysis demonstrated a first-order dependence on the concentrations of both the cobalt catalyst and TFA, while the reaction exhibited zero-order dependence on the concentrations of 4-tert-butylaniline and Ti(OnBu)4. Although Ti(OnBu)4 is not involved in the rate-determining step, its role as a Lewis acid in activating the carboxylic acid carbonyl group was confirmed by 13C NMR spectroscopy (Supplementary Fig. 30). These findings suggest that the reduction of the carboxylic acid by the cobalt catalyst is involved in the turnover-limiting step.
Moreover, cyclic voltammetry (CV) experiments were investigated to acquire further understanding for this transformation. As shown in Fig. 5d, measurement of CV curve for the ligand-free cobalt catalyst showed weak redox peaks in a wide range scan from +1.5 to −2.2 V vs. Ag/AgCl (red line). Notably, the application of a dppf-ligated cobalt complex displayed three distinct cathodic peaks at +0.3, −0.7, and −2.0 V vs. Ag/AgCl, corresponding to the Co(III/II), Co(II/I), and Co(I/0) redox processes, respectively (black line). To further probe the interaction between the cobalt complex and TFA, CV measurements were performed with the addition of 2–6 mM TFA to the cobalt complex solution. This resulted in the disappearance of the Co(I/0) peak and the emergence of a new potential (P1) at −1.5 V vs. Ag/AgCl, accompanied by an enhanced current response. This observation suggests the coordination of trifluoroacetate to the cobalt complex. Additionally, a more negative peak (P2) at −2.1 V vs. Ag/AgCl was detected, indicating the possible formation of a more reductive cobalt species (Co-H) following acid addition70. To further characterize this key intermediate, NMR experiments were performed. The reactions were carried out by direct electrolysis of carboxylic acids (TFA or propionic acid) with the cobalt complex in CD3CN for 1.5 h. As shown in Fig. 5e, the 1H NMR spectra exhibited similar chemical shifts at approximately −13.1 ppm, which can be attributed to the hydride signal of a Co(I)-H species71,72. Furthermore, the corresponding aldehyde and alcohol products resulting from acid reduction were also detected (Supplementary Fig. 29). These findings provide evidence for the generation of an active cobalt(I) hydride intermediate under electrocatalytic conditions, which plays a pivotal role in the hydrogenation of carboxylic acids.
Furthermore, we employed density functional theory (DFT) calculations to investigate mechanistic details of the key step of hydride transfer to carboxylic acid (Fig. 5f and Supplementary Data 1). Based on the experimental results, the active species is identified as a cobalt(I) hydride complex. Previous studies have shown that cobalt(I) hydride complexes typically form dimeric structures73,74, which are considered the resting state and thus serve as the starting point for the energy profile in our DFT calculations. Initially, the Co(I) dimer transforms into the active Co(I)-H species (Int-1) with the assistance of solvents, which is endergonic by 9.7 kcal/mol. Next, the Co(I) hydride undergoes migratory insertion into TFA, proceeding through the transition state TS-1 to form a hemiacetal complex, Int-2. This hydride transfer step exhibits an overall activation barrier of 18.9 kcal/mol, representing the rate-determining step of the reaction75. Following this, the protonation process occurs. The proton of TFA initially approaches Int-2, forming a hydrogen bond with the hemiacetal oxygen atom, resulting in Int-3, with a decrease in energy by 3.6 kcal/mol. Subsequently, the proton transfer proceeds readily via the transition state TS-2, leading to the formation of 2,2,2-trifluoroethane-1,1-diol and a carbonate complex Int-4. This protonation process is energetically favorable, with a decrease of 13.3 kcal/mol from Int-2 to Int-4. The transition state TS-2, while identifiable in terms of electronic energy in the gas phase, disappears after corrections for solvation effects and thermal contributions, indicating that the proton transfer can proceed smoothly. The resulting 2,2,2-trifluoroethane-1,1-diol is unstable and readily converts to the more stable trifluoroacetaldehyde and H2O, which is energetically downhill by 4.4 kcal/mol. The subsequent reaction processes and competitive reaction pathways have also been calculated and are presented in the Supplementary Figs. 31–36.
Based on these results, we propose a plausible mechanism in Fig. 6. Firstly, the monoligated cobalt complex is reduced to Co(I) A and then to Co(0) B species through a stepwise electron reduction process on cathode. In the presence of an acid, this complex B is further converted to the corresponding Co(I) hydride complex C, which is the key active species to initiate the following catalytic cycles. In cycle I, which involves the reduction of the carboxylic acid, the electrogenerated Co(I)−H species C transfers a hydride to the carboxylic group, forming a hemiacetal intermediate D. This hydride transfer step has been identified as the rate-determining step of the reaction. Following this, intermediate D undergoes rapid protonation by an acid, releasing the aldehyde E and regenerating the Co(I) species A, thereby completing cycle I. In cycle II, the Co(I)−H species C reacts with the imine F, which is generated through the rapid condensation of the aldehyde E with the added amine, to form the cobalt complex G. Finally, protonation of complex G yields the desired amine product and regenerates the Co(I) species A, thus completing cycle II.

a Acid reduction (Cycle I) b Imine reduction (Cycle II).
Discussion
In summary, we have developed a selective and efficient cobalt-electrocatalytic reductive amination method using stable and versatile carboxylic acids as substrates. Mechanistic studies reveal that an electrochemically generated Co(I)−H species serves as the key intermediate for carboxylic acid reduction, with hydride transfer to the carboxylic acid identified as the rate-determining step. By utilizing readily available protons as the hydrogen source, this electrochemical approach offers a mild, safe, and easily manipulated method for synthesizing complex alkylamines with a broad range of functionally and structurally diverse substituents. Furthermore, this protocol is applicable for the practical and cost-effective incorporation of deuterium isotopes into various amines, including pharmaceutical molecules. We anticipate that this transformation will enable robust and sustainable access to value-added alkylamines, providing a viable alternative to current benchmark methods.
Methods
Procedure for the synthesis of compound 3
A 10 mL glass tube equipped with a magnetic stir bar was charged with 4-phenylaniline 2 (0.2 mmol), Co(OTf)2 (0.02 mmol), and dppf (0.02 mmol). Toluene (2.0 mL), acetonitrile (2.0 mL), Ti(OnBu)4 (0.2 mmol), and TFA 1 (4.0 mmol) were then added sequentially. The reactor was equipped with Zn plates as both the cathode and anode (size 2.5 cm × 1.0 cm × 0.05 cm). The reaction was purged with N2 for three minutes, and the tube was wrapped with tape. The reaction was then electrolyzed under a constant current of 20 mA for 3 h at 70 °C. Upon completion of the reaction, the mixture was quenched with saturated NaHCO3 solution and diluted with ethyl acetate. The organic layer was washed with saturated NaHCO3, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The resulting residue was purified by silica gel flash chromatography to afford the product 3 in 91% isolated yield.
Data availability
The data reported in this paper are available within the article and its Supplementary Information files. All data is also available from the corresponding author upon request.
References
Trowbridge, A., Walton, S. M. & Gaunt, M. J. New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev. 120, 2613–2692 (2020).
Ricci, A. & Bernardi, L. Methodologies in Amine Synthesis: Challenges and Applications (Wiley-VCH, 2021).
Murugesan, K. et al. Catalytic reductive aminations using molecular hydrogen for synthesis of different kinds of amines. Chem. Soc. Rev. 49, 6273–6328 (2020).
Irrgang, T. & Kempe, R. Transition-metal-catalyzed reductive amination employing hydrogen. Chem. Rev. 120, 9583–9674 (2020).
Li, J.-B., Huang, C.-Y. & Li, C.-J. Deoxygenative functionalizations of aldehydes, ketones and carboxylic acids. Angew. Chem. Int. Ed. 61, e202112770 (2022).
Dong, J. & Yang, Z. Catalytic one-pot reductive amination of carboxylic acids. ChemCatChem 16, e202301427 (2024).
Cabrero-Antonino, J. R., Adam, R. & Beller, M. Catalytic reductive N-alkylations using CO₂ and carboxylic acid derivatives: recent progress and developments. Angew. Chem. Int. Ed. 58, 12820–12838 (2019).
Gribble, G. W. et al. Reactions of sodium borohydride in acidic media. I. Reduction of indoles and alkylation of aromatic amines with carboxylic acids. J. Am. Chem. Soc. 96, 7812–7814 (1974).
Marchini, P. et al. Sodium borohydride-carboxylic acid systems. Useful reagents for the alkylation of amines. J. Org. Chem. 40, 3453–3456 (1975).
Sorribes, I., Junge, K. & Beller, M. Direct catalytic N-alkylation of amines with carboxylic acids. J. Am. Chem. Soc. 136, 14314–14319 (2014).
Andrews, K. G., Summers, D. M., Donnelly, L. J. & Denton, R. M. Catalytic reductive N-alkylation of amines using carboxylic acids. Chem. Commun. 52, 1855–1858 (2016).
Nguyen, T. V. Q., Yoo, W. J. & Kobayashi, S. Chelating bis(1,2,3-triazol-5-ylidene) rhodium complexes: versatile catalysts for hydrosilylation reactions. Adv. Synth. Catal. 358, 452–458 (2016).
Ouyang, L., Miao, R., Yang, Z. & Luo, R. Iridium-catalyzed reductive amination of carboxylic acids. J. Catal. 418, 283–289 (2023).
Wu, J., Tongdee, S., Ammaiyappan, Y. & Darcel, C. A concise route to cyclic amines from nitroarenes and ketoacids under iron-catalyzed hydrosilylation conditions. Adv. Synth. Catal. 363, 3859–3865 (2021).
Stoll, E. L. et al. A practical catalytic reductive amination of carboxylic acids. Chem. Sci. 11, 9494–9500 (2020).
Trillo, P. & Adolfsson, H. Direct catalytic reductive N-alkylation of amines with carboxylic acids: chemoselective enamine formation and further functionalizations. ACS Catal. 9, 7588–7595 (2019).
Qiao, C., Liu, X.-F., Liu, X. & He, L.-N. Copper(II)-catalyzed selective reductive methylation of amines with formic acid: an option for indirect utilization of CO₂. Org. Lett. 19, 1490–1493 (2017).
Fu, M.-C., Shang, R., Cheng, W.-M. & Fu, Y. Boron-catalyzed N-alkylation of amines using carboxylic acids. Angew. Chem. Int. Ed. 54, 9042–9046 (2015).
Ogiwara, Y., Uchiyama, T. & Sakai, N. Reductive amination/cyclization of keto acids using a hydrosilane for selective production of lactams versus cyclic amines by switching of the indium catalyst. Angew. Chem. Int. Ed. 55, 1864–1867 (2016).
Wei, Y., Xuan, Q., Zhou, Y. & Song, Q. Reductive N-alkylation of primary and secondary amines using carboxylic acids and borazane under mild conditions. Org. Chem. Front. 5, 3510–3514 (2018).
Shi, Y., Kamer, P. C. J. & Cole-Hamilton, D. J. A new route to α,ω-diamines from hydrogenation of dicarboxylic acids and their derivatives in the presence of amines. Green. Chem. 19, 5460–5466 (2017).
Liu, W. et al. Tailored cobalt-catalysts for reductive alkylation of anilines with carboxylic acids under mild conditions. Angew. Chem. Int. Ed. 57, 11673–11677 (2018).
Sorribes, I. et al. Catalytic N-alkylation of amines using carboxylic acids and molecular hydrogen. J. Am. Chem. Soc. 137, 13580–13587 (2015).
Emayavaramban, B., Chakraborty, P. & Sundararaju, B. Cobalt-catalyzed reductive alkylation of amines with carboxylic acids. ChemSusChem 12, 3089–3093 (2019).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Novaes, L. F. T. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941–8002 (2021).
Zhu, C., Ang, N. W. J., Meyer, T. H., Qiu, Y. & Ackermann, L. Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 7, 415–431 (2021).
Malapit, C. A. et al. Advances on the merger of electrochemistry and transition metal catalysis for organic synthesis. Chem. Rev. 122, 3180–3218 (2022).
Kaefer, N. & Leitner, W. Electrocatalysis with molecular transition-metal complexes for reductive organic synthesis. JACS Au 2, 1266–1289 (2022).
Cheng, X. et al. Recent applications of homogeneous catalysis in electrochemical organic synthesis. CCS Chem. 4, 1120–1152 (2022).
Li, Y., Wen, L. & Guo, W. A guide to organic electroreduction using sacrificial anodes. Chem. Soc. Rev. 52, 1168–1188 (2023).
Liu, Y., Li, P., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling (eXEC) reactions. Angew. Chem. Int. Ed. 62, e202306679 (2023).
Zeng, L., Wang, J., Wang, D., Yi, H. & Lei, A. Comprehensive comparisons between directing and alternating current electrolysis in organic synthesis. Angew. Chem. Int. Ed. 62, e202309620 (2023).
Yang, J. et al. Advances in electrochemical hydrogenation since 2010. Adv. Synth. Catal. 363, 5407–5416 (2021).
Shi, Z. et al. Recent advances in the electrochemical hydrogenation of unsaturated hydrocarbons. Curr. Opin. Electrochem. 28, 100713 (2021).
Durin, G., Kaeffer, N. & Leitner, W. Electrocatalytic hydrogenation of unsaturated organic compounds with molecular complexes: mechanistic views. Curr. Opin. Electrochem. 41, 101371 (2023).
Hua, Y., Bi, H. & Liu, J. Electrohydrogenation of unsaturated bonds catalyzed by earth-abundant metal complexes. ChemElectroChem 11, e202400462 (2024).
Chen, Z., Glasson, C. R. K., Holland, P. L. & Meyer, T. J. Electrogenerated polypyridyl ruthenium hydride and ligand activation for water reduction to hydrogen and acetone to iso-propanol. Phys. Chem. Chem. Phys. 15, 9503–9507 (2013).
Armstrong, K. C. & Waymouth, R. M. Electroreduction of benzaldehyde with a metal–ligand bifunctional hydroxycyclopentadienyl molybdenum(II) hydride. Organometallics 39, 4415–4419 (2020).
Fokin, I. & Siewert, I. Chemoselective electrochemical hydrogenation of ketones and aldehydes with a well-defined base-metal catalyst. Chem. Eur. J. 26, 14137–14143 (2020).
Fokin, I., Kuessner, K.-T. & Siewert, I. Electroreduction of carbonyl compounds catalyzed by a manganese complex. ACS Catal. 12, 8632–8640 (2022).
Wang, T., He, F., Jiang, W. & Liu, J. Electrohydrogenation of nitriles with amines by cobalt catalysis. Angew. Chem. Int. Ed. 63, e202316140 (2024).
Qu, R., Junge, K. & Beller, M. Hydrogenation of carboxylic acids, esters, and related compounds over heterogeneous catalysts: a step toward sustainable and carbon-neutral processes. Chem. Rev. 123, 1103–1165 (2023).
Korstanje, T. J., van der Vlugt, J. I., Elsevier, C. J. & de Bruin, B. Hydrogenation of carboxylic acids with a homogeneous cobalt catalyst. Science 350, 298–302 (2015).
Li, L., Yao, Y. & Fu, N. Free carboxylic acids: the trend of radical decarboxylative functionalization. Eur. J. Org. Chem. 26, e202300166 (2023).
Ramadoss, V. et al. Advances in electrochemical decarboxylative transformation reactions. Chem. Eur. J. 27, 3213–3228 (2021).
Chen, N., Ye, Z.-H. & Zhang, F.-Z. Recent progress on electrochemical synthesis involving carboxylic acids. Org. Biomol. Chem. 19, 5501–5520 (2021).
Yoshioka, S. & Saito, S. Catalytic hydrogenation of carboxylic acids using low-valent and high-valent metal complexes. Chem. Commun. 54, 13319–13330 (2018).
Pritchard, J. et al. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: history, advances and future directions. Chem. Soc. Rev. 44, 3808–3833 (2015).
Dub, P. A. & Ikariya, T. Catalytic reductive transformations of carboxylic and carbonic acid derivatives using molecular hydrogen. ACS Catal. 2, 1718–1741 (2012).
Zhou, M.-X. et al. Recent advances in trifluoroethylation reaction. Org. Chem. Front. 10, 5986–6009 (2023).
Arp, H. P. H. et al. The global threat from the irreversible accumulation of trifluoroacetic acid (TFA). Environ. Sci. Technol. 58, 19925–19935 (2024).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953 (2019).
Jana, S., Mayerhofer, V. J. & Teskey, C. J. Photo- and electrochemical cobalt catalysed hydrogen atom transfer for the hydrofunctionalisation of alkenes. Angew. Chem. Int. Ed. 62, e202304882 (2023).
Li, Y. et al. Recent advances in cobalt-catalyzed regio- or stereoselective hydrofunctionalization of alkenes and alkynes. CCS Chem. 6, 1130–1156 (2024).
Andrews, K. G., Faizova, R. & Denton, R. M. A practical and catalyst-free trifluoroethylation reaction of amines using trifluoroacetic acid. Nat. Commun. 8, 15913 (2017).
Wang, B., Huang, X., Bi, H. & Liu, J. Electroreductive alkylations of (hetero)arenes with carboxylic acids. Nat. Commun. 15, 4970 (2024).
Chen, Y. Recent advances in methylation: a guide for selecting methylation reagents. Chem. Eur. J. 25, 3405–3439 (2019).
Steverlynck, J., Sitdikov, R. & Rueping, M. The deuterated “magic methyl” group: a guide to site-selective trideuteromethyl incorporation and labeling by using CD₃ reagents. Chem. Eur. J. 27, 11751–11772 (2021).
Aynetdinova, D. et al. Installing the “magic methyl”-C-H methylation in synthesis. Chem. Soc. Rev. 50, 5517–5563 (2021).
Huang, J., Chen, Z. & Wu, J. Recent progress in methyl-radical-mediated methylation or demethylation reactions. ACS Catal. 11, 10713–10732 (2021).
Goyal, V. et al. Recent advances in the catalytic N-methylation and N-trideuteromethylation reactions using methanol and deuterated methanol. Coord. Chem. Rev. 474, 214827 (2023).
Sorribes, I., Junge, K. & Beller, M. General catalytic methylation of amines with formic acid under mild reaction conditions. Chem. Eur. J. 20, 7878–7883 (2014).
Savourey, S., Lefèvre, G., Bertheta, J.-C. & Cantat, T. Catalytic methylation of aromatic amines with formic acid as the unique carbon and hydrogen source. Chem. Commun. 50, 14033–14036 (2014).
Lu, G.-S. et al. Catalytic reductive amination and tandem amination-alkylation of esters enabled by a cationic iridium complex. Angew. Chem. Int. Ed. 63, e202422742 (2024).
Li, H., Shabbir, M., Li, W. & Lei, A. Recent advances in deuteration reactions. Chin. J. Chem. 42, 1145–1156 (2024).
Ou, W., Qiu, C. & Su, C. Photo- and electro-catalytic deuteration of feedstock chemicals and pharmaceuticals: a review. Chin. J. Catal. 43, 956–970 (2022).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Zhang, Z. et al. Semiconductor photocatalysis to engineering deuterated N-alkyl pharmaceuticals enabled by synergistic activation of water and alkanols. Nat. Commun. 11, 4722 (2020).
Ciancanelli, R., Noll, B. C., DuBois, D. L. & DuBois, M. R. Comprehensive thermodynamic characterization of the metal-hydrogen bond in a series of cobalt-hydride complexes. J. Am. Chem. Soc. 124, 2984–2992 (2002).
Liu, B., Li, Y. & Liu, Q. Cobalt/Lewis acid cooperative catalysis for reductive etherification of ketones and aldehydes with alcohols. Chem. Catal. 2, 883–897 (2022).
Kong, D. et al. Cobalt-catalyzed (E)-selective hydrosilylation of 1,3-enynes for the synthesis of 1,3-dienylsilanes. Organometallics 40, 2070–2080 (2021).
Li, Y. et al. Ligand-controlled cobalt-catalyzed regiodivergent alkyne hydroalkylation. J. Am. Chem. Soc. 144, 13961–13972 (2022).
Zhang, Z.-L. et al. Cobalt-catalyzed facial-selective hydroalkylation of cyclopropenes. Angew. Chem. Int. Ed. 62, e202306381 (2023).
Wang, J., Wu, K. & Qi, X. Theoretical study of the ligand effect on NHC cobalt-catalyzed hydrogenation of ketones. Catal. Sci. Technol. 9, 5315–5321 (2019).
Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (22301073, J.L.), the Science and Technology Innovation Program of Hunan Province (2021RC3053, S.Q. and 2021RC3056, J.L.), and the Fundamental Research Funds for the Central Universities. The authors also thank the Analytical Instrumentation Center of Hunan University for mass spectrometry analysis.
Author information
Authors and Affiliations
Contributions
H.B., S.Q., and J.L. conceived and designed the project. H.B. and C.B. conducted the experimental part. Z.C. conducted the theoretical calculations. S.Q. and J.L. wrote the manuscript. All authors contributed to analyzing the data and editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yixin Luo, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bi, H., Chen, Z., Bi, C. et al. Electroreductive amination of carboxylic acids by cobalt catalysis. Nat Commun 16, 7167 (2025). https://doi.org/10.1038/s41467-025-62396-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62396-4
