
Abstract
The rapid emergence of difficult-to-treat multidrug-resistant pathogens, combined with the scarcity of antibiotics possessing novel mechanisms, poses a significant threat to global public health. Here, we integrate the synthetic-bioinformatic natural product approach with peptide optimization to unveil the antibiotic-producing potential of Paenibacillaceae bacteria. Our culture-independent approach led to the discovery of paenimicin, a novel 11-mer depsi-lipopeptide featuring an unprecedented dual-binding mechanism. By sequestering the phosphate and hydroxyl groups of lipid A in Gram-negative bacteria, as well as the phosphate groups of teichoic acids in Gram-positive bacteria, paenimicin exhibits potent and broad-spectrum efficacy against MDR pathogens in vitro and in vivo models. Paenimicin demonstrates no detectable resistance, favorable pharmacokinetics and low nephrotoxicity, positioning it as a promising candidate for treating severe and urgent MDR infections.
Similar content being viewed by others


Introduction
Over the past century, antibiotics have revolutionized the treatment of infectious diseases and saved billions of lives1. However, the rapid emergence and spread of multidrug-resistant (MDR) bacteria posed a significant threat to global public health and resulted in ~1.14 million deaths worldwide in 20212,3. There is an urgent need to develop novel antibiotics featuring distinct mode of action to treat MDR bacteria. The Paenibacillaceae family of bacteria, renowned as the source of pivotal antibiotics such as colistin, cilagicin, tyrocidine, and gramicidin, represents a treasure trove of invaluable antimicrobial natural products. To date, over 39 types of antimicrobial compound have been isolated and characterized from this family using traditional culture-dependent approach (Fig. 1a and Supplementary Fig. 1). Recent advancements in genomic sequencing and bioinformatic analysis have revealed that most biosynthetic gene clusters (BGCs) within Paenibacillaceae remain uncharacterized. The silence and extremely low yields of these BGCs under laboratory cultivation conditions (culture-dependent approach) hindered the isolation of new antibiotics from this family4.

a Representative antibiotics identified within Paenibacillaceae family. b Overview of the culture-independent antibiotic discovery approach utilized in the present present study. Note: SPPS, Solid phase peptide synthesis. c Sequence similarity network analysis of all NRP biosynthetic gene clusters (BGCs) of Paenibacillaceae family. Known antibiotic BGCs are highlighted in red, unclassified BGCs in blue, and BNP37 BGC is indicated in yellow.
In recent years, advances in understanding the biosynthetic machinery of microbial-derived secondary metabolites have made it increasingly feasible to predict their structures directly from BGC information with high accuracy5,6,7. This allows structure prediction without requiring the expression of BGC or reliance on spectroscopy-based structure elucidation. Currently, the biosynthesis of two major families of peptide secondary metabolites—non-ribosomal peptides (NRPs) and ribosomally synthesized and post-translationally modified peptides (RiPPs)—is particularly well understood8,9. Their overall structures can be predicted based on adenylation domain specificity for NRPs and precursor peptide sequences for RiPPs and synthesized directly using chemical reaction (i.e., synthetic bioinformatic natural product (synBNP))10. This approach integrates predictive algorithms with the feasibility of peptide synthesis, bypassing the need for in vivo expression and addressing the challenges posed by silent BGCs11,12,13,14 (Fig. 1b).
In this work, we integrated the synBNP approach with peptide structure optimization to systematically investigate the antimicrobial capabilities of NRPs encoded by bacteria in the family of Paenibacillaceae. This strategy led to the discovery of a novel broad-spectrum antibiotic, paenimicin, which exhibited potent activity against both Gram-positive bacteria and Gram-negative bacteria in vitro and in vivo. Mechanistic studies showed that paenimicin uniquely targets two sites: lipid A in Gram-negative bacteria and teichoic acids in Gram-positive bacteria. Unlike colistin, which targets phosphate group of lipid A15,16, paenimicin binds to a different site on lipid A, enabling it to maintain potent efficacy against colistin-resistant pathogens in both in vitro and in vivo models. Notably, in contrast to colistin, which is associated with well-documented nephrotoxicity17,18, paenimicin exhibits significantly reduced in vivo nephrotoxicity. Given its potent and broad-spectrum antibacterial activity, unique dual mode of action and low nephrotoxicity, paenimicin emerges as a promising candidate for the development of new antimicrobial therapies to treat complex infections caused by MDR pathogens.
Results
Discovery of paenimicin
To systematically explore the antimicrobial secondary metabolites encoded by the bacteria in Paenibacillaceae family, we retrieved 1245 genomes from public databases along with 11 genomes from our in-house collection. Our study focuses on NRPs, the predominant class of antimicrobial peptides produced by the Paenibacillaceae family. AntiSMASH analysis of these genomes identified 17,037 BGCs, which were subsequently deduplicated based on type of BGC, adenylation domain specificity and further refined to include only complete BGCs with 5−15 adenylation domain. This process led to the identification of 901 NRP BGCs, which were grouped into 45 clusters and 27 single dots in a sequence similarity network (SSN) analysis using a similarity cutoff of 0.4 (Fig. 1c). We then prioritized 48 representative BGCs from the clusters and singletons, focusing exclusively on those not associated with any known NRP BGCs, thereby ensuring their novelty. To obtain the products of the prioritized NRP BGCs, we employed the synBNP approach involving peptide sequence prediction and solid-phase peptide synthesis. This culture-independent method overcomes the low expression or silence of BGCs typically encountered in culture-dependent antibiotic discovery. The NRP structure prediction was based on the following criteria: (i) the amino acids activated by each adenylation domain in NRPS modules are predicted in silico using 10 signature codes19,20; (ii) the presence of an epimerization (E) domain indicates the incorporation of D-amino acids21; (iii) the order of NRPS modules corresponds to the amino acid sequence in the peptide22; (iv) the presence of a condensation starter (Cs) domain suggests a lipid chain forms an acyl group with the N -terminus of the peptide core23. However, genomic analysis cannot predict the type of acyl group or the topology of NRPs. Since myristic acid is one of the most common lipids found in bacterial NRPs, all predicted lipopeptides were chemically synthesized with their N-terminal acylated by myristic acid13. NRPs can exist in either linear or cyclized forms. In the cyclized forms, the C-terminal carboxyl group may form an amide bond with either the terminal amino group (head-tail cyclization) or with the side-chain amino group of basic residues (e.g., lysine, 2,4-diaminobutyric acid) (branch-to-tail cyclization). Alternatively, it may form an ester bond with hydroxylated residues (e.g., threonine or serine) in a branch-to-tail topology. To explore all potential structural possibilities, we synthesized the three major topological patterns—linear, head-to-tail cyclization, and branch-to-tail cyclization—for preliminary activity screening. The predicted peptides were chemically synthesized using the gold-standard solid-phase method with HBTU/PyBOP as the coupling agent. This approach yielded a collection of 74 purified peptides predicted from 48 representative NRP BGCs (Supplementary Table 2).
We next screen the antimicrobial activity of 74 synthetic peptide against clinically significant and challenging MDR ESKAPE pathogens (i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter cloacae), which account for a large proportion of the resistant infections encountered in clinical medicine (Supplementary Fig. 2). Notably, the BNP37 peptide family, comprising five variants (i.e. BNP37L, BNP37C1, BNP37C2, BNP37C3, BNP37C4) with identical sequences but different topologies, exhibited inhibitory activity against ESKAPE pathogens. Their minimum inhibitory concentrations (MICs) ranged from 2 to 64 μg/mL, depending on the specific peptide and targeted strain (Fig. 2a). The structures of the BNP37 peptides were predicted based on the adenylation domain specificity of the pae cluster, which is exclusively present in the genome of Paenibacillus caseinilyticus GW78 (Fig. 2b). The pae cluster comprises three NRPS genes (pae-B, pae-C, pae-D), two transporter genes (pae-E and pae-F), and one regulator gene (pae-A). The three NRPS genes together encode an 11 mer peptide predicted to consist of five proteinogenic amino acids (L-Thr, 2× L-Leu, L-Val, L-Asp) and six non-proteinogenic amino acids (3× L-Dab, D-Dab, D-Phe, D-Tyr) with a confidence level of at least 80%. Furthermore, the presence of Cs domain suggests the pae cluster encodes lipopeptides. Attempts to isolate the natural peptides encoded by the pae cluster from P. caseinilyticus GW78 using various media were unsuccessful, likely due to the cluster being silent under these growth conditions. Consequently, we chemically synthesized five BNP37 lipopeptides, one linear form (BNP37L) and four cyclized forms (BNP37C1, BNP37C2, BNP37C3, BNP37C4), and all N-acylated with myristic acid. Among the five variants, the linear peptide BNP37L exhibited the most potent antibacterial activity (MIC 2-4 μg/mL) but showed strong serum binding affinity (Fig. 2a), which may limit its in vivo efficacy. In contrast, the cyclized peptide BNP37C2 maintained comparable activity against most ESKAPE strains (MIC 2-8 μg/mL) while showing reduced serum binding ability (Fig. 2a). To enhance druggability, we synthesized 37 BNP37C2 analogues with diverse lipid chains differing in chain length, degree of branching, and substitutions (Supplementary Fig. 3). Antimicrobial and cytotoxicity evaluations revealed that none of these analogues outperformed BNP37C2, which features myristic acid as the acyl substituent (Supplementary Fig. 4a). BNP37C2 demonstrated an optimal balance between antimicrobial activity, serum binding affinity, and cytotoxicity, making it the most promising candidate for further optimization. To assess the contribution of individual amino acid residues to the antibacterial activity of BNP37C2, we synthesized ten peptide variants, each with a single amino acid replaced by alanine. Thr-2, critical for cyclization, was excluded from this analysis (Supplementary Fig. 3 and 4b). Substituting the Dab residues at positions 1, 3, 5, and 8 with alanine resulted in a 2- to 16-fold increase in MIC values. These results highlight the pivotal role of the positively charged Dab residues in maintaining the antibacterial activity of BNP37C2. Considering that BNP37C2 inherently contains four positively charged residues, we investigated the effect of adding extra cationic Dab residues on its activity. Remarkably, substituting the nonpolar D-Tyr residue at position 9 with a D-Dab residue reduced the MIC against carbapenem-resistant Acinetobacter baumannii (CRAB) and methicillin resistant Staphylococcus aureus (MRSA) by 2-fold and eliminated serum binding (Fig. 2c). This optimized structure, derived from targeted modifications of BNP37C2, has been named paenimicin. Paenimicin is a depsi-lipopeptide consisting of 11 amino acids, including five positively-charged Dab residues and five proteinogenic amino acids (Fig. 2d). The peptide is cyclized via an ester bond between the hydroxyl group of threonine at position 2 and the α-carboxyl group of the terminal aspartic acid, with an N-terminal myristic acid anchoring the structure.

a Diagrams of five peptide topologies predicted to be arisen from BNP37 BGC, along with their antimicrobial activity and serum binding efficacy against ESKAPE pathogens of all the peptides. (E. faecium BNCC 186301, S. aureus BNCC 186335, K. pneumoniae BNCC 186113, A. baumannii BNCC 194496, P. aeruginosa BNCC 186070, E. cloacae BNCC 185028). b The BGC of BNP37. 10-signature code sequences were used to predict each adenylation (A) domain, with the prediction accuracy was deemed reliable when it reached 80%. c Fold changes in MIC following 2,4-Diaminobutyric Acid (Dab) screening. Specifically, all non-Dab positions were replaced with Dab respectively, MIC values against methicillin-resistant S. aureus (MRSA, S. aureus ATCC BAA44) and carbapenems-resistant A. baumannii (CRAB, A. baumannii ATCC BAA1605) with 10% serum were evaluated. N/A: not applicable. d Structure of paenimicin.
Antimicrobial spectrum
To comprehensively investigate the antimicrobial spectrum of paenimicin, we conducted microbroth susceptibility tests against a range of clinically prevalent MDR Gram-positive and Gram-negative pathogens, including the common hospitalized ESKAPE pathogens and WHO priority pathogens. Paenimicin exhibited broad-spectrum activity against all tested strains, with MIC values ranging from 2−4 μg/mL (Table 1). Notably, paenimicin demonstrated exceptional efficacy against Gram-negative bacteria, particularly those classified as critical and high priority by the WHO, such as carbapenem-resistant Acinetobacter baumannii (CRAB), third-generation cephalosporin-resistant or carbapenem-resistant Enterobacterales. While colistin is regarded as the last resort for treating MDR Gram-negative infections, its effectiveness is increasingly compromised by the global spread of the plasmid-borne mobilized colistin-resistance gene mcr-124. To date, 34 genetic variants and 10 alleles of mcr-1 have been identified worldwide, indicating its ongoing evolution and dissemination25,26. Paenimicin maintains potent activity (MIC = 2 μg/mL) against colistin-resistant bacterial pathogens, including both acquired and intrinsic colistin-resistant strains. Furthermore, unlike colistin, paenimicin also shows comparable inhibitory activities against MDR Gram-positive bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA) and erythromycin resistant Streptococcus pyogenes (ERSP), with a MIC value of 2 μg/mL. Importantly, paenimicin exhibits no activity against fungal pathogens at concentrations up to 64 μg/mL, indicating a low potential for cross-toxicity with mammalian cells.
Antimicrobial mode of action
Given the notable activity of paenimicin against both MDR Gram-negative and Gram-positive bacteria, we next investigated its bactericidal mechanism. Through bacterial killing assays and scanning electron microscopic (SEM) analysis (Supplementary Fig. 5a–c), we confirmed that paenimicin is a bactericidal antibiotic. At 1×MIC, it completely eradicated bacterial populations within 4 h of incubation, causing cell collapse. The bacterial killing rate of paenimicin is faster than that of vancomycin while slower than that of colistin. Paenimicin did not induce cell depolarization but disrupted cell membrane integrity, leading to a dose-dependent release of potassium ions (Supplementary Fig. 5d–f). Given its broad-spectrum activity, these findings suggest that paenimicin likely targets the cell wall or cell membrane but employs a killing mechanism distinct from those of the narrow-spectrum antibiotics vancomycin and colistin. To identify the molecular target of paenimicin, we attempted to raise resistant mutants of E. coli ATCC 25922 and S. aureus BNCC 186335. Bacteria were exposed to serially increasing concentrations of paenimicin or maintained at sub-MIC levels of paenimicin for a continuous 30 day period (Supplementary Fig. 6). Despite testing under a variety of extreme conditions, we did not find any mutations resulting in an MIC increase >2-fold. Furthermore, to determine the resistance rate of paenimicin, we exposed bacterial cultures with concentration as high as 109 CFU/mL, to 2×, 4×, and 16×MIC of paenimicin. However, no resistant clones were observed. is most likely not proteins, as such targets are typically prone to mutations that can confer resistance. To further investigate the target of paenimicin, we evaluated its antibacterial activity in the presence of key bacterial cell components, including total genomic DNAs, total proteins, and cell wall components such as peptidoglycan and lipopolysaccharide (LPS) (Fig. 3a). The addition of DNA or proteins did not significantly alter the MIC values of paenimicin, although the possibility remains that some DNA or protein complexes are dismantled or partially denatured during extraction, preventing interaction with paenimicin. In contrast, LPS inhibited the antimicrobial activity of paenimicin in a dose-dependent manner (Fig. 3b), suggesting that LPS might be the primary target of paenimicin, consistent with the results from mutation assays. Furthermore, isothermal titration calorimetry (ITC) revealed a strong binding affinity between paneimycin and LPS, with an apparent dissociation constant (Kd) of 2.18 ± 0.82 μM (Fig. 3c), suggesting that LPS is the primary target of paenimicin.

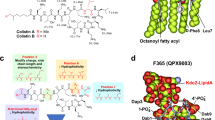
a, b Fold change in MIC of paenimicin against E. coli ATCC 25922 in the presence of genomic DNA (0.05 mg/mL) and total protein (0.38 mg/mL) extracted from the bacteria, alongside fixed concentration (0.5 mg/mL) and varying concentrations (0.0078-0.5 mg/mL) of peptidoglycan (PGN) and lipopolysaccharide (LPS). The experiments were repeated three independent times. c Isothermal titration calorimetry (ITC) assay determining the binding of paenimicin and LPS. d Schematic chemical structures and MIC value changes of three different LPS molecules in E. coli MG1655 WT and gene knockout strains: E. coli MG1655 Δ waaG and E. coli MG1655 Δ waaC. The experiments were repeated three independent times. e Fold change in MIC of paenimicin against E. coli ATCC 25922 in the presence of different concentrations of lipid A (0.0078-0.5 mg/mL). Data are presented as means ± SD. f Displacement of BODIPY TR fluorescence dye from lipid A binding upon addition of colistin, paenimicin, and kanamycin, with dye concentrations ranging from 0.0 to 10.0 mg/mL The experiments were conducted in trilicates. Data are presented as means ± SD. g Binding snapshots of paenimicin (left) and colistin (right) to lipid A from molecular dynamics (MD) simulations. Paenimicin and colistin are depicted as sticks, while lipid A is represented as a line; carbon atoms are colored cyan for paenimicin and green for colistin. Atomic interactions are shown as dashed purple lines. h, i Fold changes in MIC of paenimicin against S. aureus BNCC 186335 in the presence of fixed concentration (0.5 mg/mL) and varying concentrations (0.0078-0.5 mg/mL) of PGN of Gram-positive bacteria and lipoteichoic acid (LTA). The experiments were conducted in trilicates. Data are presented as means ± SD. j Changes in the content of the remaining paenimicin and vancomycin in the solution after addition of wall teichoic acid (WTA) at different ratios.
LPS is a vital glycolipid and a key component of the outer membrane bilayer in Gram-negative bacteria. It consists of three structural domains: lipid A (a disaccharide backbone linked to multiple fatty acid chains anchoring LPS to the outer membrane), an core region (a branched oligosaccharide domain containing nine or ten sugars composed of an inner core and an outer core), an O-antigen (a repetitive carbohydrate polymer covalently attached to the core region)27,28 (Fig. 3d). To identify the specific LPS region that interacts with paenimicin, we knocked out the waaC and waaG genes29. These genes, which are non-essential for bacterial survival, encode enzymes responsible for synthesizing the inner and outer cores of the LPS structure, respectively (Supplementary Fig. 7). Paenimicin maintained strong antimicrobial activity against the strains deficient in either the outer core or those lacking both the inner and outer cores (Fig. 3d), indicating that the inner and outer core regions of LPS are not binding sites for paenimicin. Next, to determine whether paenimicin binds lipid A, a crucial outer membrane component and primary target of colistin16, we performed MIC assays of paenimicin against E. coli in the presence of varying concentrations of lipid A. Lipid A inhibited the antibacterial activity of paenimicin in a dose-dependent manner (Fig. 3e). Furthermore, paenimicin competitively displaced the lipid A-specific fluorescent dye BODIPY-TR, confirming its strong binding affinity for lipid A30 (Fig. 3f). Although both paenimicin and colistin share lipid A as a cellular target, paenimicin retains potent antibacterial activity against both intrinsic (e.g. EptA, PmrA/B and PhoP/Q genes)31 and acquired (e.g. mcr-1 gene)24 colistin resistance strains, with a MIC value of 2 μg/mL (Table 1). Consistently, we observed that adding phosphoethanolamine (pEtN)-modified lipid A (31, 32), which is catalyzed by EptA and contributes to colistin resistance, reduced the activity of paenimicin in antibacterial assays, while it had no effect on colistin (Supplementary Fig. 8). This suggests that paenimicin employs a novel mechanism of action, targeting a distinct site on lipid A to kill Gram-negative bacteria. To predict the specific binding model, we performed molecular dynamics (MD) simulations (Fig. 3g and Supplementary Fig. 9). The MD results revealed that both paenimicin and colistin exhibited noncovalent binding with lipid A, primarily through electrostatic interactions between their positively charged Dab residues and the negatively charged phosphate groups of lipid A. Notably, the fifth amino acid of paenimicin also formed a hydrogen bond with the C-6 hydroxyl group of the glucosamine moieties, an interaction not observed with colistin. This additional binding with the hydroxyl group likely account for paenimicin’s sustained efficacy against colistin-resistant strains. It should be noted that our current MD simulation captures only the initial stage of the interaction between paenimicin and lipid A. The outward orientation of paenimicin’s myristic acid tail likely reflects the limited simulation timescale and the use of a hydrophilic water box, both of which may have hindered insertion of the lipid tail into the DPPC membrane, a process that typically occurs over microsecond to millisecond timescales32,33. A more advanced simulation model could be developed to capture the full binding process, including initial phosphate group interactions followed by hydrophobic tail insertion.
Unlike colistin, paenimicin is a broad-spectrum antibiotic with potent activity against both Gram-negative bacteria and Gram-positive bacteria. Since lipid A, the target in Gram-negative bacteria, is absent in Gram-positive bacteria, this suggests that paenimicin may interact with an alternative target in Gram-positive bacteria. To investigate this issue, systematic feeding studies were conducted, revealing that peptidoglycan extracted from Gram-positive bacteria reduces the antibacterial activity of paenimicin against Gram-positive bacteria, whereas peptidoglycan from Gram-negative bacteria does not exhibit the same effect (Fig. 3h). The key difference between the peptidoglycan of Gram-positive bacteria and Gram-negative bacteria peptidoglycan is the presence of teichoic acids (TAs), including lipoteichoic acid (LTA) and wall teichoic acid (WTA)34,35. This led us to hypothesize that LTA or WTA might serve as the molecular target of paenimicin. To test this hypothesis, we conducted MIC assays of paenimicin with the addition of LTA. Results showed that LTA exhibited a dose-dependent inhibition of paenimicin’s activity, with 0.5 mg/mL completely abolishing its antibacterial effect (Fig. 3i). ITC analysis further confirmed a strong binding affinity between paenimicin and LTA, with an apparent Kd value of 5.61 ± 3.53 μM (Supplementary Fig. 10a). Moreover, WTA also formed a complex with paenimicin, resulting in a measurable concentration decrease in the solution (Fig. 3j). Structurally, both LTA and WTA contain multiple phosphate groups, forming highly anionic substructures that may interact with the cationic regions of paenimicin. Molecular dynamics simulations further confirmed that the phosphate groups in LTA and WTA serve as the primary binding site of paenimicin (Supplementary Fig. 10b).
Our findings indicate that paenimicin binds to the phosphate and hydroxyl groups of lipid A of LPS in Gram-negative bacteria, as well as to the phosphate groups in LTA and WTA within the peptidoglycan of Gram-positive bacteria. This dual binding mechanism explains paenimicin’s broad-spectrum activity against MDR pathogens without detectable resistance (Fig. 4a). Given the broad-spectrum antimicrobial activity and attractive dual binding targets, we subsequently investigated the potential of paenimicin as an antimicrobial therapeutic agent. Paenimicin exhibited low toxicity against mammalian cells, with HC50 > 100 μg/mL and IC50 > 30 μg/mL against HepG2 cells, and LD50 > 500 mg/kg (Fig. 4b, Supplementary Fig. 11a–c), suggesting paneimicin harbors a wide therapeutic window. Additionally, paenimicin showed favorable pharmacokinetic parameters using subcutaneous injection (s.c.), with the half-life time of 20.2 ± 1.9 h and bioavailability of 102 ± 13% (Fig. 4c and Supplementary Table6), which is 10 times higher than that of colistin. Notably, unlike the pharmacokinetic pattern of colistin, the half-life time of paenimicin administrated using s.c. is 2.4-fold longer than that administrated using intravenous (i.v.) injection (Supplementary Fig. 11d and Supplementary Table 6). Based on these findings, s.c. administration was selected as the primary route for in vivo studies of paenimicin. Given that nephrotoxicity is a primary concern associated with the use of antibiotics (i.e., colistin17,18, amphotericin B36), next generation of colistin analogues with decreased nephrotoxicity have been developed, including SPR-206 (phase I)37, QPX-9003 (phase II)38, and MRX-8 (phase I)39. To evaluate the nephrotoxicity of paenimicin, mice were administrated with high concentration of paenimicin (i.e., 40 mg/kg) for a continuous 7 days. The kidney tissues were then analyzed using kidney injury biomarkers (including KIM-1, LCN2, SPP1 and TIMP-1) as well as histological evaluation, with colistin serving as a control antibiotic. Even at high concentration (20-fold of MIC) and prolonged exposure, paenimicin did not induce any detectable kidney injury. In contrast, colistin caused a severe nephrotoxicity, as indicated by both biomarkers and histological changes (Fig. 4d and Supplementary Fig. 11e).

a Schematic models illustrating the proposed mode of action of paenimicin for Gram-negative (above) and Gram-positive (below) bacteria. b Cytotoxicity assay of paenimicin against human hepatocellular carcinomas (HepG2) cell line, The experiments were repeated three independent times. Amphotericin B was used as a toxic control. Data are presented as means ± SD. c The pharmacokinetic curves of paenimicin and colistin over time after administration of 10 mg/kg via subcutaneous injection, (n = 3 rats per group). Data are presented as means ± SD. d In vivo nephrotoxicity assays of paenimicin and colistin. The concentrations of biomarkers (SPP1, TIMP-1, LCN2 and KIM-1) in mice serum were measured at 1 day and 7 days post administration of 40 mg/kg of paenimicin and colistin, respectively (s.c., qd, n = 3 mice per group). Significant differences were analyzed by one-way analysis of variance (ANOVA) (ns, no significance). e Neutropenic thigh infection model using drug resistant strains (n = 3 mice/ 6 thighs per group). All therapeutic compounds and vehicles were administrated via subcutaneous injection once daily and the bacteria burden were counted after 24 h post drug treatment. Significant differences were analyzed by one-way ANOVA, (ns, no significance). Data are presented as means ± SD. f Vaginitis infection model using N.gonorrehea ATCC 31426. All therapeutic compounds and vehicles were administrated via subcutaneous injection once daily for 3 consecutive days. On the day after the last administration, vaginal lavage fluid were collected and diluted for bacteria counting. Significant differences between each group with placebo were analyzed by one-way ANOVA. Data are presented as means ± SD. g Skin infection model using MRSA ATCC BAA44. Paenimicin and vancomycin were administrated via subcutaneous injection once daily for 5 consecutive days and the bacteria burden of wound tissues were counted 15 days after infection (n = 5 mice per group). Photographic records were taken. Significant differences were analyzed by one-way ANOVA. Data are presented as means ± SD.
In vivo efficacy in mice
Given the potent antimicrobial activity and favorable pharmacokinetic parameters, we next investigated the in vivo efficacy of paenimicin against MDR bacteria using three different types of infection models, including neutropenic thigh infection model, skin infection model, and vaginal infection model. Our initial evaluation focused on paenimicin’s efficacy against CRAB using neutropenic thigh infection mouse model. Paenimicin exhibited a dose-dependent reduction in bacterial burden. At a dosage of 20 mg/kg administered once daily, the bacterial burden decreased by 2.40 log10 compared to the vehicle control, which is comparable with that of colistin, demonstrating potent efficacy in vivo efficacy of paenimicin (Fig. 4e). Given paenimicin’s distinct antimicrobial mechanism and the increasing prevalence of colistin-resistant MDR pathogens, we further investigated its in vivo efficacy using mouse models infected with carbapenem and/or colistin-resistant XDR pathogens (Fig. 4e). At a dose of 20 mg/kg once daily, paenimicin reduced the bacterial burden by 3.42 log10 in colistin resistant E. coli and 3.12 log10 in colistin- and carbapenem- resistant XDR K. pneumoniae, while colistin showed no activity to the strains tested (Fig. 4e). To evaluate the efficacy of paenimicin against intrinsic colistin-resistant pathogens, a vaginal infection model infected with colistin intrinsic-resistant N. gonorrhoeae was established (Fig. 4f). Paenimicin exhibited a dose-dependent reduction, achieving a reduction of 2.83 log10 at a dose of 20 mg/kg per day via s.c. administration. Furthermore, in the treatment of MDR Gram-positive bacteria, paenimicin showed dose-dependent activity against MRSA in both the neutropenic thigh infection model and the wound infection model (Fig. 4e and g). At 20 mg/kg once daily via s.c. administration, paenimicin reduced the bacterial burden by 1.79 log10 and 5.90 log10, respectively, comparable to the reductions achieved with vancomycin. The low nephrotoxicity, favorable pharmacokinetic profile, and potent in vivo efficacy collectively suggest that paenimicin is a promising drug candidate for treating difficult-to-manage infections encountered in clinical settings, particularly those caused by colistin-resistant MDR pathogens.
Discussion
Infections caused by MDR bacterial pathogens represent a critical threat to global public health. The growing prevalence of MDR pathogens, coupled with the declining rate of novel antibiotic discovery, underscores the urgent need for innovative therapeutic strategies. Paenibacillaceae are a rich source of antimicrobial natural products and leveraging these natural resources is expected to offer solutions for combating resistant infections. Applying the culture-independent synBNP approach combined with activity-guided screening, we synthesized and screened 74 peptides whose structures were predicted based on the adenylation domain specificity of Paenibacillaceae NRP BGCs. This effort led to the discovery of a unique class of BNP37 peptides exhibiting potent, broad-spectrum inhibitory activity against MDR pathogens. Subsequent structural optimization of the BNP37 peptides led to the identification of paenimicin, an 11-mer depsi-lipopeptide featuring five positively charged Dab moieties and an N-acylated myristic acid chain, and with enhanced broad-spectrum antibacterial activity and reduced serum-binding affinity. Mechanistic studies revealed that paenimicin interacts not only with the phosphate groups of lipid A but also uniquely targets the hydroxyl group of glucosamine in lipid A, a key interaction enabling its potency against both intrinsic and acquired colistin-resistant pathogens in both in vitro and in vivo settings. Furthermore, paenimicin’s ability to bind the phosphate groups of teichoic acids in Gram-positive bacteria contributes to its broad-spectrum efficacy. Its unique mechanism of action, broad-spectrum activity, absence of detectable resistance, favorable pharmacokinetic profile, low nephrotoxicity, and excellent in vivo efficacy collectively position paenimicin as a promising candidate for treating infections caused by MDR pathogens, particularly those resistant to carbapenems, methicillin, and colistin. The unique macrocyclic structure of paenimicin complicates its synthetic accessibility, particularly during the macrocyclization step, where the synthetic yield rate was remarkably decreased. In the current study, this resulted in a low synthetic yield of ~3% after purification, which may affect the scalability of paenimicin synthesis for future clinical applications. Further optimization of the synthetic route is necessary to improve the overall reaction yield and reduce the percentage of side products. This includes screening for peptide coupling catalysts and modifying the synthetic pathway. Due to the limited genomic data available for Paenibacillaceae bacteria, our study does not encompass all the types of potential NRPs encoded by this family. Furthermore, the structures of peptides derived from some NRP BGCs cannot be accurately predicted due to the presence of one or more unusual amino acids that current algorithms are unable to reliably forecast. Future work will focus on expanding the genomic data for Paenibacillaceae bacteria and advancing computational methods to more accurately predict unusual amino acids in NRPs, both of which are expected to enhance efforts in discovering new antibiotics to combat MDR infections.
Methods
Ethics statement
All animal studies were conducted strictly followed the Regulations for the Care and Use of Laboratory Animals issued by the Ministry of Science and Technology of China. The experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at China Pharmaceutical University (Approved number: 2024-08-078/2024-10-112) and Pharmaron (Approved number: PK-R-06012025).
Identification and bioinformatic analysis of the paenimicin (pae) BGC
To comprehensively explored the non-ribosomal peptides encoded in the family of Paenibacillaceae bacteria, all assembled sequences from publicly available genomic database which encompassed 1245 genomes, as well as 11 sequencing data derived from the sequencing of collected strains were retrieved. The Raw sequenced reads were assembled into contigs using SPAdes (version 3.13.0), with default parameters. Contigs were refined using Ragtag (version 2.1. 0) to obtain scaffold-level genomic data. A total of 17037 BGCs were retrieved using antiSMSH (version 7.0. 0) software analysis for all genomic data. An in-house Python script using a json module from Python was written to filter BGCs. In this analysis, the Python script used to read each JSON file produced by antiSMASH one at a time, gathering information on the kind of BGC, the quantity of A domains, and the presence or absence of a TE domain. We focused exclusively on NRPS BGCs containing a TE domain and 5 to 15 A domains for our study. The resulting 879 BGCs were further analyzed by clustering their GenBank files using Big-Scape (version 1.1.5), and the Clustering results were visualized using Cytoscape (version 3.10.0).
Peptide synthesis
All candidate peptides were synthesized using standard solid-phase peptide synthesis (SPPS) methods on 2-chlorotrityl chloride resin. 2-chlorotrityl chloride resin (0.3 g, 0.1 mmol) was swollen in dichloromethane (DCM) for 30 min at room temperature. Each of the first Fmoc-protected amino acid (0.3 mmol) was loaded using 0.3 mL 2,4,6-collidine in 8 mL DCM for 10 h, and then capping the unreacted carbocations on the resin with 10 mL mixed reagents (Methanol: N,N-Diisopropylethylamine (DIPEA): DCM = 1: 0.5: 9) for 45 min at room temperature. Fmoc deprotection was carried out using 20% piperidine in N,N-Dimethylformamide (DMF) (3 mL) for 7.5 min (2X) and then the resin was washed thoroughly with DMF (3 mL, 5X). Coupling reactions were carried out using Fmoc-protected amino acids (0.3 mmol) pre-mixed with O-(7-Azabenzotriazollyl)-N,N,N’,N’-tetramethyluronium hexafluophosphate (HBTU) (0.3 mmol) and DIPEA (0.3 mmol) in DMF. Each coupling reaction was carried out for at least 1 h and then the resin was washed with DMF (3 mL, 5X). The coupling steps were repeated as described above until the last amino acid (or a lipid acid) in the sequence was loaded.
-
1)
Amide bond formation: Amide bonds were formed between the α-amino group of the N-terminal amino acid or amino acids with an animo group on the side chain and α-carboxyl group of C-terminal amino acid. Alloc-protected animo acid was used to selectively form the amide bond at the desired position. The resin-bound linear peptide was washed with DCM (3 mL, 5X) and treated with phenylsilane (1.5 mmol) and tetrakis (triphenylphosphine) palladium (0) (0.05 mmol.) in DCM and shake 2 h at room temperature, followed by washing with 10% sodium diethyldithiocarbamate trihydare in DMF (50 mL) and DCM (6 mL, 5X).
-
2)
Peptide cyclization: Linear peptides were cleaved from resin by treating with 1% Trifluoroacetic acid (TFA) in DCM for 5 min and collected in a falcon tube. Then the cleaved peptides were mixed with benzotriazol-1-yl-oxytripyrrolidino-phosphonium hexafluorophosphate (PyBOP) (0.8 mmol) and DIPEA (3 mmol.) in 45 mL DCM and shaken overnight.
-
3)
Ester bond formation: Ester bonds were formed between the unprotected hydroxyl group of serine or threonine and the α-carboxyl group of the C-terminal animo acid. The resin-bound peptides were mixed with amino acid (2 mmol), DIPEA (4 mmol.), benzoyl chloride (2 mmol) and 4-(dimethylamino)-pyridine (0.08 mmol) in 15 mL DCM and rock for 48 h.
-
4)
Final cleavage: 5 mL of cleavage cocktail (95% TFA, 2.5% triisopropylsilane and 2.5% water) was added to the air-dried peptide mixture and shaken for 2 h. Then the solution was added to a cold mixture of diethyl ether:hexane (1:1) and kept at -20 °C for 1 h to precipitate the crude peptides.
Minimum inhibitory concentration (MIC) assay
MIC assays were conducted following the protocol recommended by the Clinical and Laboratory Standards Institute (CLSI). Culture conditions are detailed in supplementary table S4. All synthetic compounds were dissolved in sterile dimethyl sulfoxide (DMSO) (SCR, CN) to a stock concentration of 12.8 mg/ml. Colistin, ceftizoxime and ciprofloxacin were used as positive controls for Gram-negative bacteria, while vancomycin and methicillin were used as positive controls for Gram-positive bacteria. Compound stock solutions were serially diluted across 96-well plates using a 2-fold dilution to give a concentration up to 64 μg/ml in a volume of 50 μL per well. Overnight cultures of each assayed strains were diluted 5000-fold in fresh medium and 50 μL of those dilutions were added into each well. The lowest concentration with no visible growth of bacteria after 16 h was recorded as the MIC value. All assays were performed in duplicate and repeated three independent times.
Cell lysis assay
A single colony of S. aureus BNCC 186335 was inoculated overnight at 37 °C with shaking at 220 rpm. Cells were collected, resuspended in sterile PBS (pH 7.4) and then diluted to an OD600nm of 0.35. 900 μL of this bacteria suspension was mixed with 100 μL of 17 μM SYTOX green nucleic acid stain (Thermo Fisher, USA). The mixture was incubated in the dark at 37 °C for 5 min, and then transferred to a 384-well flat bottom black microtiter plate (30 μL per well). The initial fluorescence intensity of each well was measured using a microplate reader (Infinite 200 Pro, Tecan, USA) at 9 s time intervals for 5 min (Excitation/Emission = 488/523 nm). The same volume of paenimicin solutions at concentration of 1, 2, 4, 8, 10×MIC were added into each well, and melittin and DMSO were used as positive and negative controls, respectively. The fluorescence intensity was then continually monitored for 25 min and plotted by Prism 9.0. Experiments were performed three independent times (n = 3).
Membrane depolarization assay
A single colony of S. aureus BNCC 186335 was cultured overnight at 37 °C with shaking at 220 rpm. Cells were harvested, washed in 5 mM HEPES buffer, and resuspended in 5 mM HEPES buffer with 20 mM glucose (OD600nm = 0.1). 2 μL of 500 μM 3,3’-Dipropylthiadicarbocyanine Iodide [DiSC3(5)] dye (Macklin, CN) was added to 1 mL of the cell suspension and incubated in the dark at 37 °C for 30 min. 100 μL of this mixture was then added to a 384-well flat bottom black microtiter plate. The initial fluorescence intensity of each well was recorded using an Spectra Max ID5 (Molecular devices, USA) (Excitation/Emission = 620/670 nm) at 15 s time intervals for 5 min. 1 μL of paenimicin solutions at concentration of 1, 2, 4, 8, 10× MIC were added into each well, and Triton X-100 and DMSO were used as positive and negative controls, respectively. The fluorescence intensity was continually monitored for 20 min and plotted by Prism 9.0. Experiments were performed three independent times (n = 3).
Potassium ion release assay
The release of potassium ion was monitored using an Orion Dual Star pH/ISE Benchtop Meter (Thermo Fisher, USA). A single colony of S. aureus BNCC 186335 was inoculated and incubated overnight in 50 mL LB medium at 37 °C with shaking at 220 rpm. Cells were harvested, washed twice with buffer (10 mM Tris-acetate, 100 mM NaCl, pH 7.4). and then resuspended in the same buffer and adjusted to an OD600nm of 1.0. The concentration of extracellular potassium ions was monitored at 5 s intervals. Paenimicin at concentrations of 1, 2, 4, 8×MIC and gramicidin at 8 × MIC were added into the suspension respectively after 60 s and the measurement was continued until 8 min. Data were plotted by Prism 9.0. The experiments were performed in triplicate (n = 3).
Time-dependent killing assay
An overnight culture of S. aureus BNCC 186335 and E. coli ATCC 25922 was prepared by inoculating and incubating a single colony at 37 °C with shaking at 220 rpm, respectively. The cultures were diluted to a final concentration of 1 × 106 CFU/mL. 4 mL bacteria dilutions were then challenged with 1, 4 and 8 × MIC of paenimicin, 8× MIC of vancomycin or 8× MIC of colistin. After 1, 2, 4, 8 and 16 h, 100 μL aliquots of each culture were collected, serially 10-fold diluted and plated on LB agar plates. As for E. coli, additional time intervals at 0.25, 0.5, 0.75 were added. Colonies were counted after overnight growth at 37 °C. The experiments were performed in three biological replicates (n = 3).
Scanning electron microscopy
Overnight cultures of S. aureus BNCC 186335 and E. coli ATCC 25922 were diluted 1:200 in fresh medium and grown to OD600nm of 0.8. Bacteria were treated with 8×MIC of paenimicin for 0, 1 and 4 h, respectively. Cells were harvested and washed twice with PBS buffer (pH 7.4), and then fixed with 2.5% glutaric dialdehyde solution at 4 °C for 12 h and rinsed three times with PBS buffer. The samples were postfixed with 1% osmium tetroxide solution for 2 h at room temperature. After removing the solution, the samples were rinsed with PBS buffer three times. The samples were dehydrated in a graded series of ethanol concentrations (30%, 50%, 70%, 80%, 90%, 95%) for 15 min each and two times in 100% ethanol for 20 min each and put into a critical point dryer (Quorom k850, UK) to dry out. Subsequently, the samples were fixed on the sample stage using conductive carbon glue and sprayed Pt with the ion type sputtering instrument (Hitachi MC1000, JP) for 120 s. Imaging was carried out in a Hitachi Regulus8100 at 3.0 kv.
Serial passaging assay
A single colony of S. aureus BNCC 186335 and E. coli ATCC 25922 was inoculated and incubated at 37 °C with shaking at 220 rpm overnight, respectively. The overnight cultures were diluted 1:5,000 into fresh medium and the MIC values of paenimicin, ciprofloxacin and bacitracin for S. aureus and paenimicin, ciprofloxacin and tetracycline for E. coli were tested using the MIC method and the MIC values were recorded after 24 h. For bacitracin, the LB medium was supplemented with 50 μg/mL ZnCl2. On the next day, the bacteria cultures of the sub-MIC well of each antibiotic were diluted into fresh medium in a 1:500 ratio and mixed with serial diluted antibiotics. Then the MICs were tested following the method described above. This serial passage process lasted for 28 days. The experiments were conducted in triplicates.
Resistance frequency assay
A single colony of S. aureus BNCC 186335 and E. coli ATCC 25922 was inoculated and incubated at 37 °C with shaking at 220 rpm overnight,respectively. Then the bacterial suspensions were diluted and adjusted to 109 CFU/mL. Following the addition of paenimicin at concentrations of 2, 4, 16 × MIC, 100 μL aliqoutes of suspensions were dispensed into each well of 96-well microplates. The plates were incubated at 37 °C for 24 h.
Feeding assay
The effect of bacteria cell components on paenimicin’s antibacterial activity was evaluated using S. aureus BNCC 186335 and E. coli ATCC 25922. Cell components of Gram-positive and Gram-negative bacteria including peptidoglycan, total protein, genomic DNA, LPS and LTA were dissolved in water to a stock concentration of 5 mg/mL. Lipid A was dissolved in chloroform and premixed with paenimicin for 15 min and then the premixed solutions were evaporated by air. Each solution was added to individual wells of 96-well plates, and serially diluted to final concentrations of 0.0078 to 0.5 mg/mL. The MIC values were evaluated using the same method described for the MIC assay. Fold changes in MIC were calculated based on the formula: Fold change in MIC = Final MIC/Original MIC. The experiments were conducted in duplicate and repeated three independent times.
Isothermal calorimetry (ITC) assay
The binding of paenimicin and LPS and LTA were measured using ITC. The ITC experiments were performed on PEAQ-ITC (Malvern, UK) instrument at 25 °C using a solution of 5 mM of paenimicin or 1 mM of colistin along with 100 µM LPS or LTA in 5 mM HEPES buffer (pH 7.4). The titration process involved an initial injection of 0.23 μL, followed by 18 injections of 2 μL at 80-s intervals, with continuous stirring at 500 rpm. Data were analyzed using PEAQ-ITC software, and the thermodynamic parameters [enthalpy (ΔH), entropy (ΔS), and the equilibrium binding constant (Kd)] were calculated using a one-binding site model.
Knockout of WaaC and WaaG in Escherichia coli MG1655
Genome editing was conducted as described previously40. The N20 sequence was inserted into ptargetF by Gibson assembly yielding the plasmids pTargetT-ΔWaaC and pTargetT-ΔWaaG. two fragments, one upstream and one downstream of the targeted gene, were PCR-amplified from the genomic DNA of Escherichia coli MG1655 using primer pairs WaaC_KOUF/ WaaC_KOUR and WaaC _KODF/ WaaC _KODR for WaaC and WaaG_KOUF/ WaaG _KOUR and WaaG _KODF/ WaaG _KODR for WaaG, respectively. Each pair of PCR products was constructed into a PCR fragment by overlapping PCR, yielding the donor DNA. E. coli MG1655 competent cells were prepared for harboring pCas. Arabinose (10 mM final concentration) was added to the culture for λ-Red induction. For electroporation, 50 μL of cells was mixed with 100 ng of pTargetT series DNA and 400 ng of donor DNA. Cells were recovered at 30 °C for 1 h before being spread onto LB agar containing kanamycin (50 mg/L) and spectinomycin (50 mg/L) and incubated overnight at 30 °C. Transformants were identified by colony PCR and DNA sequencing. For the curing of pTarget series DNA, the edited colony harboring both pCas and pTarget series was inoculated into 2 ml of LB medium containing kanamycin (50 mg/L) and IPTG (isopropyl-β-D-thiogalactopyranoside; 0.5 mM). The culture was incubated for 8−16 h, diluted, and spread onto LB plates containing kanamycin (50 mg/L). The colonies were confirmed as cured by determining their sensitivity to spectinomycin (50 mg/L). The colonies cured of pTarget series DNA were used in a second round of genome editing. pCas was cured by growing the colonies overnight at 37 °C non selectively. The primer and gRNA sequences were provided in the Supplementary Data 3.
BC displacement assay
The affinity of paenimicin to lipid A was assessed by evaluating its ability to displace BODIPY TR cadaverine (BC) from lipid A-BC mixture. 20 μg/mL LPS and 20 μM BC were premixed in 5 mM HEPES (pH7.4) and incubated at 37 °C for 30 min. Subsequently, the mixture was transferred into a 96-well flat bottom black microtiter plate (50 μL per well). 50 μL of paenimicin in 5 mM HEPES were added at final concentrations ranging from 0.0 μg/mL to 1.0 μg/mL. Colistin and kanamycin were used as positive and negative controls, respectively. After incubating in the dark for 5 min, the fluorescence intensity was measured using a microplate reader (Infinite 200 Pro,Tecan) (Excitation/Emission = 580/620 nm) and plotted by Prism 9.0.
Lipid A extraction and feeding assay
Lipid A and pEtN modified lipid A were extracted from E. coli MG1655 and E. coli MG1655-mcr-1 following the previously described method41,42. Briefly, chemical lysis of bacterial cells was performed using a mixture of chloroform, methanol and water (Bligh-Dyer) solvent, and the LPS was pelleted by centrifugation. Then a combination of mild-acid hydrolysis and solvent extractions were used to extract lipid A from the pellet mixture. Lipid A can be defined as the chloroform soluble portion of LPS after mild-acid hydrolysis. The lyophilized crude lipid A was dissolved in chloroform and added into 96-well plates containing 4×MIC of paenimicin or colistin and diluted E. coli MG1655 solution following the feeding assay method. Then the OD600nm was measured using Multiscan SkyHigh at 37 °C for 16 h and plotted using Prism 9.0. The experiments were performed in triplicates (n = 3).
Molecular Dynamic (MD) simulations
Lipid A and colistin structures were extracted from PDB 1QFF and 8DEV, respectively43,44. Lipoteichoic acid (LTA) and paenimicin 3D model was optimized from 2D structure using MOE. Initial complex model was obtained by placing ligand close to receptor and fulfills the ionic charge interactions between Receptor (Lipid A or LTA) and Ligand (Colistin or paenimicin). The complex model was then sent for MD simulation with Desmond (Desmond/Maestro noncommercial version 2022.1). Specifically, 1,2-dipalmitoyl-phosphatidylcholine (DPPC) membrane model was first added automatically, then the DPPC orientation was modified to accommodate hydrophobic tail of the ligand to the center of membrane plane. Complexes were neutralized with Na+ or Cl- ions, and solvation was conducted with a 10 Å orthorhombic TIP3P water buffer box containing 0.15 M NaCl using default parameters in Desmond. MD simulation includes a 5-step minimization with restraints released gradually, and a 200 ns production run without restraints in NPgT ensemble. The first step of the minimization is to restrain solute heavy atoms at 10 K in NVT ensemble for 100 ps. The second is to restrain membrane in z-axis and protein atoms at 100 K in NPT ensemble for 20 ps. The third is to restrain membrane in z-axis and protein atoms at 100 K in NPgT ensemble for 100 ps. The fourth is to heat up from 100−300 K in NPgT ensemble for 150 ps. The fifth is removing all restraints in NVT ensemble for 100 ps. The 200 ns production simulation was conducted at 300 K under 1 bar in NPgT ensemble, and repeated 3 times, trajectory coordinates were saved every 100 ps. The initial and final configuration files of the simulations are provided in Supplementary Data 1 and Supplementary Data 2.
Wall teichoic acid extraction and binding assay
Wall teichoic acid was extracted from S. aureus BNCC 186335 as previously described45. Briefly, wall teichoic acid was isolated from the crude peptidoglycan sacculus after completely washing with 4% SDS to remove lipoteichoic acid. Then hydrolysis with trichloroacetic acid was used to liberate water soluble WTA from peptidoglycan. The lyophilized crude WTA was dissolved in water and diluted to 1 mg/mL, 2 mg/mL, 3 mg/mL, 4 mg/mL and 5 mg/mL and then mixed with 1 mg/mL paenimicin for 0:1, 1:1, 2:1, 3:1, 4:1 and 5:1 w/w ratios, respectively. Mixtures were incubated for 1 h at room temperature and centrifuged for 5 min at 15,000 g to pellet insoluble material. The content of paenimicin in the supernatant was detected by UPLC (Waters, US). Peak aera change rate was calculated and plotted by Prim 9.0.
Cytotoxicity assay
The cytotoxicity of paenimicin and its analogs was tested using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. HepG2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum. Cells were seeded into a 96-well plate with a density of 5,000 cells per well and cultured at 37 °C with 5% CO2 for 24 h. Then the medium was aspirated and replaced with 100 μL fresh medium containing compounds at a final concentration ranging from 64 μg/mL to 0.25 μg/mL, and amphotericin B was used as control. After 48 h of incubation, the medium was removed and 110 μL of freshly prepared MTT solution (0.5 mg/mL) was added to each well. The plate was incubated for another 3 h at 37 °C to allow crystal formation. Subsequently, precipitated formazan crystals were dissolved by addition of 100 μL of solubilization solution (40% DMF, 16% SDS and 2% acetic acid in H2O). The absorbance of each well was measured at OD570nm using a microplate reader (Multiscan SkyHigh, Thermo Fisher). IC50 values were calculated using Prism 9.0 as the concentration of each compound required to inhibit 50% of cell growth. All the experiments were performed in triplicates (n = 3) and repeated two independent times.
Hemolytic assay
Fresh sterile defibrated sheep blood was centrifuged and resuspended in PBS (pH 7.4) and diluted to a concentration of 1 × 109 cells/mL. Serial dilutions of Paenimicin were prepared at concentrations ranging from 100 μM to 0.38 μM and mixed with the blood cell suspensions to yield a final volume of 500 μL. 1% Triton X-100 and 1% DMSO were used as positive and negative controls respectively. After a 3 h incubation time at 37 °C, the mixture was centrifuged at 900 g for 20 min and the supernatant was collected and transferred into a 96-well plate. The hemolysis efficacy was determined by measuring the absorbance at OD540nm using Multiscan SkyHigh and calculated using Prism 9.0. All the experiments were performed in biological triplicates (n = 3).
Pharmacokinetic study
Six to eight-week-old SD male rats (200-300 g) were used in pharmacokinetic studies. Paenimicin was administered via intravenous (IV, 5 mg/kg) and subcutaneous (SC, 10 mg/kg) injection. Blood timepoints were taken after dosing at pre, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h and 48 h for IV dosing, and pre, 1 h, 3 h, 8 h, 24 h, 32 h, 48 h, 56 h, 72 h and 96 h for SC (10 mg/kg) dosing. Aliquots of 0.1 mL of blood were taken by puncture of the jugular vein from each rat (n = 3 per route and dose) at each timepoint. Samples were transferred into plastic micro centrifuge tubes with EDTA-K2 and stored on ice. The blood samples were centrifuged at 4000 g for 5 min at 4 °C to obtain plasma within 30 min. The plasma samples were stored at -80 °C for further analysis. All studies were approved by Pharmaron’s IACUC. (Approved number: PK-R-06012025).
HPLC-MS/MS pharmacokinetic analysis: paenimicin (l mg/mL, in DMSO) was serially diluted in ACN: water (50:50) to prepare the standard curves and quality control (QC) spiking solutions. Standards and QCs were created by adding 5 μL of spiking solutions to 50 μL of drug free plasma (SD K2EDTA Rat, Vital River). 55 μL of standards, 55 μL of QC samples and 55 μL of unknown samples (50 μL of rat plasma with 5 μL of blank solution) were added to 200 μL of acetonitrile containing 10 ng/mL of the internal standard (IS) Dexamethasone (Shanghai Aladdin Biochemical Technology Co.,Ltd) for precipitating protein respectively.
Extracts were vortexed for 1 min and centrifuged at 1,500 g for 15 min. The supernatant was diluted 3 times with water. 20 µL of diluted supernatant was injected into the LC/MS/MS system for quantitative analysis. HPLC-MS/MS analysis was performed on a Sciex Applied Biosystems Triple Quad 5500+ or 6500+ mass spectrometer coupled to a Shimadzu Nexera Series System Controller CBM-40 UPLC (Ultra-High- Performance Liquid Chromatography) system to quantify each drug in the plasma. Chromatography was performed on a Raptor Biphenyl column (3 × 30 mm; particle size, 2.7 μm) using a reversed phase gradient. 5% acetonitrile in ultrapure water with 0.1% formic acid was used for the aqueous mobile phase and 95% acetonitrile in ultrapure water with 0.1% formic acid was used for the organic mobile phase. Multiple-reaction monitoring (MRM) of parent/daughter transitions in electrospray positive-ionization mode was used to quantify the analytes. The following MRM transitions were used for paenimicin (467.00/461.50) and dexamethasone (393.06/373.00). Sample analysis was accepted if the concentrations of the quality control samples were within 20% of the nominal concentration. Data processing was performed using Analyst software (v1.7.3; Applied Biosystems Sciex).
Nephrotoxicity assay
Pathogen-free female ICR mice (Hangzhou Medical College, China), aged 6 weeks and weighing between 23−27 g, were used in the study. The mice were randomly grouped into six individual cages with three mice per group. Mice were acclimatized for 3 days prior to the experiments. Paenimicin was formulated in a solution containing 10% DMSO and 0.5% Tween 80 in 0.9% saline. Colistin or a solvent without any antibiotics was used as the positive control and placebo, respectively. The compounds were administered by subcutaneous injection at a dosage of 40 mg/kg once daily for 1 day or 7 consecutive days. Serum was collected from blood samples 24 h after the last dose. The concentration of nephrotoxicity-related biomarkers including kidney injury molecule-1 (KIM-1), tissue inhibitor of metalloproteinase-1 (TIMP-1), lipocalin-2 (LCN-2), and secreted phosphoprotein 1 (SPP-1) were measured using the commercial kits (Cloud-Clone Corp, China) and the ELISA assays were conducted following the manufacturer’s instructions. All animals were euthanized, and the kidney tissues were collected, fixed, dissected, and subjected to H&E staining (Approved number: 2024-08-078).
Neutropenic thigh infection model
Pathogen-free female ICR mice (Hangzhou Medical College, China), aged 6 weeks and weighing between 23−27 g, were used in this study. The mice were housed in individually ventilated cages and acclimatized for 3 days before the experiments. Then the mice were rendered neutropenic via intraperitoneal injections with 150 mg/kg and 100 mg/kg of cyclophosphamide on day -4 and day -1, respectively. On day 0, mice were infected by intramuscular administration of 50 μL of S. aureus ATCC BAA-44, E. Coli MG1655-mcr-1, A. baumannii ATCC BAA-1605 and K. pneumoniae BNCC 353393 bacteria suspensions in 0.9% saline. This provides a challenge inoculum of ~1×106 cells per thigh. At 2 h post infection, mice were given 100 μL of each antibiotic at different concentrations via SC injection. At 24 h post infection, the mice were euthanized and their thigh muscles were aseptically removed, weighed, homogenized, and enumerated for bacterial burden by colony forming unit (CFU) counts. All graphical data were presented as data points (n = 3 mice/n = 6 thighs) by group and were statistically analyzed using Prism 9.0. All animal study procedures were approved by the Animal Ethics Committee of China Pharmaceutical University (Approved number: 2024-08-078).
Neutropenic skin infection model
BALB/c mice weighing 20−22 g were used in this study, half male and half female (Hangzhou Medical College, China). The mice were randomly grouped in individual cages and acclimatized for 3 days before the experiments. To induce neutropenia, the mice were injected with 50 mg/kg of cyclophosphamide via intraperitoneal injection on the third and first days before infection46,47. Subsequently, mice were anesthetized with 4% chloral hydrate (80 μL/10 g body weight) via intraperitoneal injection. Then the hair on the mice’s backs were removed using a razor, and full-thickness skin perforations were made on the dorsal skin using a biopsy puncher with a diameter of 1.0 cm. A suspension of S. aureus ATCC BAA-44 at a concentration of 2.4 × 106 CFU/mL was evenly dripped on the wound (20 μL/ mice). The mounds were topically treated with vancomycin (20 mg/kg) and paenimicin (1 mg/kg, 10 mg/kg, 20 mg/kg and 40 mg/kg), respectively. All compounds were administered via subcutaneous injection once daily for five consecutive days. Photographic records were taken on days 1, 3, 5, 8, 10, 12 and 14 post infection. On day 15, the wound tissues were collected and the bacterial burden was recorded using the plate counting method. All graphical data were presented as data points (n = 5 mice) by group and were statistically analyzed using Prism 9.0. All animal study procedures were approved by the Animal Ethics Committee of China Pharmaceutical University (Approved number. 2024-10-112).
Neutropenic vaginal infection model
Female BALB/c mice weighing 22−25 g were used in this study (Hangzhou Medical College, China). The mice were randomly grouped (n = 5, each group) and acclimatized for 3 days before the experiment. The mice were subcutaneously injected with 17-β-estradiol (25 mg/kg) for 3 days before Neisseria gonorrhoeae bacteria challenge48,49,50. On the first day of 17-β-estradiol administration, mice were injected with cyclophosphamide (50 mg/kg, i.p.). For the subsequent 2 days, antibiotics (Vancomycin + Streptomycin) were administered intraperitoneally once daily to suppress the overgrowth of commensal bacteria in the murine reproductive tract. On the day of infection, physical damage to the vaginal mucosa was induced using a cotton swab. Then, 50 μL of Neisseria gonorrhoeae ATCC 31426 suspension (5 × 108 CFU/mL) was inoculated into the vaginal cavity using a pipette and subjected tail suspension for 5 min. This infection procedure was repeated for three consecutive days.
On the second day after the last infection, mice were subcutaneously injected with paenimicin at varying doses (1 mg/kg, 5 mg/kg, 10 mg/kg and 20 mg/kg) once daily for three consecutive days. On the second day after the last administration, mouse vaginal secretions were smeared and examined under a microscope. Additionally, vaginal lavage fluid were collected (about 200 μL), and the bacterial burden was recorded. All graphical data were presented as data points by group and were statistically analyzed using Prism 9.0. All animal study procedures were approved by the Animal Ethics Committee of China Pharmaceutical University (Approved number. 2024-10-112).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genomic sequence of Paenibacillus caseinilyticus GW78 is available in GenBank under accession number NZ_JAQAGY010000015.1 [https://www.ncbi.nlm.nih.gov/nuccore/NZ_JAQAGY010000015]. HPLC, HRMS and NMR spectra for paenimicin are presented as Supplementary Information. All data are available in the main text or the supplementary materials. The raw sequencing data of the Paenibacillaceae genome sequences collected in the lab in this study have been deposited in the China National Center for Bioinformation Genome Sequence Archive (GSA) with project number of CRA022965. The Source Data is provided with this paper as a Source Data file.
References
Crofts, T. S., Gasparrini, A. J. & Dantas, G. Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol. 15, 422–434 (2017).
Okeke, I. N. et al. The scope of the antimicrobial resistance challenge. Lancet (Lond., Engl.) 403, 2426–2438 (2024).
Global burden of bacterial antimicrobial resistance 1990-2021: a systematic analysis with forecasts to 2050. Lancet (London, England) 404, 1199−1226 (2024).
Wang, Z., Koirala, B., Hernandez, Y. & Brady, S. F. Discovery of paenibacillaceae family gram-negative-active cationic lipopeptide antibiotics using evolution-guided chemical synthesis. Org. Lett. 24, 4943–4948 (2022).
Scherlach, K. & Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 12, 3864 (2021).
Bauman, K. D., Butler, K. S., Moore, B. S. & Chekan, J. R. Genome mining methods to discover bioactive natural products. Nat. Prod. Rep. 38, 2100–2129 (2021).
Shen, Y., Liu, N. & Wang, Z. Recent advances in the culture-independent discovery of natural products using metagenomic approaches. Chin. J. Nat. Med. 22, 100–111 (2024).
Hemmerling, F. & Piel, J. Strategies to access biosynthetic novelty in bacterial genomes for drug discovery. Nat. Rev. Drug Discov. 21, 359–378 (2022).
Liu, W.-Q. et al. Cell-free biosynthesis and engineering of ribosomally synthesized lanthipeptides. Nat. Commun. 15, 4336 (2024).
Chu, J. et al. Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 12, 1004–1006 (2016).
Chu, J. et al. Synthetic-bioinformatic natural product antibiotics with diverse modes of action. J. Am. Chem. Soc. 142, 14158–14168 (2020).
Chu, J., Vila-Farres, X. & Brady, S. F. Bioactive synthetic-bioinformatic natural product cyclic peptides inspired by nonribosomal peptide synthetase gene clusters from the human microbiome. J. Am. Chem. Soc. 141, 15737–15741 (2019).
Wang, Z., Koirala, B., Hernandez, Y., Zimmerman, M. & Brady, S. F. Bioinformatic prospecting and synthesis of a bifunctional lipopeptide antibiotic that evades resistance. Science 376, 991–996 (2022).
Wang, Z. et al. A naturally inspired antibiotic to target multidrug-resistant pathogens. Nature 601, 606–611 (2022).
Sabnis, A. et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife 10, e65836 (2021).
Poirel, L., Jayol, A. & Nordmann, P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 30, 557–596 (2017).
Miano, T. A. et al. Attributable risk and time course of colistin-associated acute kidney injury. Clin. J. Am. Soc. Nephrolo. 13, 542−550 (2018).
Gai, Z., Samodelov, S. L., Kullak-Ublick, G. A., Visentin, M. Molecular mechanisms of colistin-induced nephrotoxicity. Molecules (Basel, Switzerland) 24, 653 (2019).
Stachelhaus, T., Mootz, H. D. & Marahiel, M. A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505 (1999).
Throckmorton, K. et al. Directed evolution reveals the functional sequence space of an adenylation domain specificity code. ACS Chem. Biol. 14, 2044–2054 (2019).
Linne, U., Doekel, S. & Marahiel, M. A. Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry 40, 15824–15834 (2001).
Alanjary, M., Cano-Prieto, C., Gross, H. & Medema, M. H. Computer-aided re-engineering of nonribosomal peptide and polyketide biosynthetic assembly lines. Nat. Prod. Rep. 36, 1249–1261 (2019).
Strieker, M. & Marahiel, M. A. The structural diversity of acidic lipopeptide antibiotics. Chembiochem. A. Eur. J. Chem. Biol. 10, 607–616 (2009).
Liu, Y. Y. et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168 (2016).
Gogry, F. A., Siddiqui, M. T., Sultan, I. & Haq, Q. M. R. Current update on intrinsic and acquired colistin resistance mechanisms in bacteria. Front. Med. 8, 677720 (2021).
Wang, R. et al. The global distribution and spread of the mobilized colistin resistance gene mcr-1. Nat. Commun. 9, 1179 (2018).
Bertani B., Ruiz N. Function and biogenesis of lipopolysaccharides. EcoSal Plus 8, 10.1128/ecosalplus.ESP-0001-2018 (2018).
Raetz, C. R., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 (2007).
Manioglu, S. et al. Antibiotic polymyxin arranges lipopolysaccharide into crystalline structures to solidify the bacterial membrane. Nat. Commun. 13, 6195 (2022).
Zheng, E. J. et al. Discovery of antibiotics that selectively kill metabolically dormant bacteria. Cell Chem. Biol. 31, 712–728.e719 (2024).
McPhee, J. B. et al. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J. Bacteriol. 188, 3995–4006 (2006).
González-Fernández, C., Bringas, E., Oostenbrink, C. & Ortiz, I. In silico investigation and surmounting of Lipopolysaccharide barrier in gram-negative bacteria: how far has molecular dynamics come?. Comput. Struct. Biotechnol. J. 20, 5886–5901 (2022).
Kesireddy, A. et al. Modeling of specific lipopolysaccharide binding sites on a gram-negative porin. J. Phys. Chem. B 123, 5700–5708 (2019).
Percy, M. G. & Gründling, A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu. Rev. Microbiol. 68, 81–100 (2014).
Brown, S., Santa Maria, J. P. Jr. & Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 67, 313–336 (2013).
Sabra, R. & Branch, R. A. Amphotericin B nephrotoxicity. Drug Saf. 5, 94–108 (1990).
Brown, P. et al. Design of Next Generation Polymyxins with Lower Toxicity: The Discovery of SPR206. ACS Infect. Dis. 5, 1645–1656 (2019).
Roberts, K. D. et al. A synthetic lipopeptide targeting top-priority multidrug-resistant gram-negative pathogens. Nat. Commun. 13, 1625 (2022).
Lepak, A. J., Wang, W. & Andes, D. R. Pharmacodynamic evaluation of MRX-8, a novel polymyxin, in the neutropenic mouse thigh and lung infection models against gram-negative pathogens. Antimicrob. Agents Chemother. 64, e01517-20 (2020).
Jiang, Y. et al. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 81, 2506–2514 (2015).
Henderson, J. C., O’Brien, J. P., Brodbelt, J. S., Trent, M. S. Isolation and chemical characterization of lipid A from gram-negative bacteria. J. Vis. Exp. JoVE 16, e50623 (2013).
Luther, A. et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 576, 452–458 (2019).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129–1143 (2018).
Pristovsek, P. & Kidric, J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: an NMR and molecular modeling study. J. Med. Chem. 42, 4604–4613 (1999).
Kho, K. & Meredith, T. C. Extraction and analysis of bacterial teichoic acids. Bio. Protoc. 8, e3078 (2018).
Malachowa, N., Kobayashi, S. D., Lovaglio, J. & DeLeo, F. R. Mouse model of staphylococcus aureus skin infection. Methods Mol. Biol. (Clifton, NJ) 1960, 139–147 (2019).
Huang, H. N. et al. Use of the antimicrobial peptide epinecidin-1 to protect against MRSA infection in mice with skin injuries. Biomaterials 34, 10319–10327 (2013).
Raterman, E. L. & Jerse, A. E. Female mouse model of neisseria gonorrhoeae infection. Methods Mol. Biol. (Clifton, NJ) 1997, 413–429 (2019).
Abutaleb, N. S., Elhassanny, A. E. M. & Seleem, M. N. In vivo efficacy of acetazolamide in a mouse model of Neisseria gonorrhoeae infection. Microb. Pathog. 164, 105454 (2022).
Shaughnessy, J. et al. A novel factor H-Fc chimeric immunotherapeutic molecule against Neisseria gonorrhoeae. J. Immunol. (Baltim., Md: 1950) 196, 1732–1740 (2016).
Acknowledgements
We are grateful to Professor Wenjun Li from Sun Yat-sen University for providing 11 isolated Paenibacillaceae strains. This study was supported in part by: National Natural Science Foundation of China (22307145, 82473821), Scientific Research Innovation Capability Support Project for Young Faculty (ZYGXQNJSKYCXNLZCXM-H21), National key Research and Development Program of China (2023YFC3402302), Jiangsu Province Major Science and Technology Projects (BG2024046), Jiangsu Province Distinguished Professor Program.
Author information
Authors and Affiliations
Contributions
Z.Q.W. conceived the project; Z.Q.W., W.Y.H., X.T.H. contributed to manuscript writing, review, and editing; Z.Q.W., W.Y.H. contributed to data analysis. W.Y.H. performed the biochemical experiments. X.T.H. performed the peptide synthesis. Q.S.D. contributed to the ITC experiments and nephrotoxicity tests. Y.C.L. contributed to the genetic knockout. T.C. performed the data collection and bioinformatic analysis. W.X. contributed to the MD simulation. W.Y.H., X.T.H. and L.M.M. contributed to the animal study. Y.J.C., Z.S., and N.L. contributed to manuscript review. All authors contributed to data analysis and interpretation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. A patent covering the structure and activity of all synthetic BNP37 derivates and paenimicin has been filed by China Pharmaceutical University (202510010246.5), Z.Q.W, W.Y.H, and X.T.H are listed in the patent.
Peer review
Peer review information
Nature Communications thanks Octavio Franco and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, W., Huan, X., Li, Y. et al. A broad-spectrum antibiotic targets multiple-drug-resistant bacteria with dual binding targets and no detectable resistance. Nat Commun 16, 7048 (2025). https://doi.org/10.1038/s41467-025-62407-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62407-4
