
Abstract
Glucose is a vital energy source essential for life and human health. Sodium-glucose cotransporter 2 (SGLT2) is a sodium-glucose symporter that utilizes the electrochemical gradient of sodium to reabsorb glucose from kidney filtrate back into circulation. SGLT2 plays a crucial role in maintaining blood glucose homeostasis and is an important drug target for type 2 diabetes. Despite its significance, the mechanisms by which SGLT2 recognizes and releases substrates during its transport cycle remain largely unknown. Here, we present structures of human SGLT2 in complex with a glucose analogue in the occluded conformation at 2.6 Å resolution, revealing a detailed hydrogen bonding network at the substrate binding site that governs substrate recognition. Additionally, structures of SGLT2 in both the substrate-bound inward-facing conformation and the substrate-free inward-facing conformations illustrate the structural changes that occur during substrate release into cytosol. Our structural analysis, combined with mutagenesis results, identifies specific polar interactions that are essential for maintaining the outer and inner gates in their closed conformations.
Similar content being viewed by others


Introduction
Sodium-glucose cotransporter 2 (SGLT2) is a secondary glucose transporter predominantly found in the brush-border membranes of renal proximal tubules1,2. It plays a critical role in reabsorbing glucose from kidney filtrate back into the blood circulation1,2. Pharmacological inhibition of SGLT2 promotes glucose excretion and is widely utilized to reduce blood glucose levels in the treatment of type 2 diabetes3,4.
SGLT2 transports glucose against its concentration gradient by utilizing the electrochemical gradient of sodium ions2,5. In each transport cycle, SGLT2 moves one glucose molecule and one sodium ion from the extracellular space into the cytosol2,5. This process involves a concerted conformational change in the protein5. In the outward-open conformation, the substrate binding site is accessible to the extracellular environment. Upon binding of sodium and glucose, SGLT2 transitions to an occluded conformation. Subsequently, it isomerizes to the inward-open conformation, facilitating the release of glucose and sodium into the cytosol5,6.
MAP17 is a crucial auxiliary protein that enhances the function of human SGLT2 (hSGLT2) by promoting its surface expression7,8. Structural studies of the hSGLT2-MAP17 complex in the presence of the anti-diabetic drug empagliflozin reveal that hSGLT2 consists of 14 transmembrane helices (TM0-TM13) with a LeuT fold9. The last helix, TM13, interacts with the single transmembrane helix of MAP179. Empagliflozin occupies both the substrate binding site and extracellular vestibule, locking hSGLT2 in an outward-open conformation9. Subsequent structural studies on several other hSGLT2 inhibitors, including TA1887, sotagliflozin, dapagliflozin, and canagliflozin, have confirmed these findings and elucidated the geometry of sodium coordination in the outward-facing conformation10,11. Further investigations into hSGLT2 in the occluded and inward-facing conformations have highlighted global “rocking-bundle-like” structural changes during glucose transport, identifying “moving” and “less-mobile” regions within the transporter8.
Despite these advancements, due to the limited resolution of previous study on structures with substrates8, the precise mechanism of substrate recognition, how the global conformational changes facilitate substrate release, and residues involved in the conformational transition of SGLT2 remain not completely understood. Here, we present a series of high-resolution structures of hSGLT2, including the substrate-bound occluded conformation (hSGLTSO), the substrate-bound inward-facing conformation (hSGLTSI), and the substrate-free inward-facing conformations in the presence of either sodium (hSGLTINa) or potassium (hSGLTIK) to elucidate these mechanisms.
Results
Structure determination of hSGLT2 in the presence of substrate
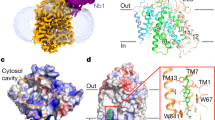
In previous studies, we identified a mouse monoclonal antibody, mAb90, that specifically binds to an extracellular epitope of hSGLT28. Here, we demonstrate that mAb90 binding does not affect the transport activity of hSGLT2 (Fig. 1a). This suggests that mAb90 does not lock hSGLT2 in a specific conformation, making it a potentially valuable fiducial marker for studying the cryo-EM structures of hSGLT2 in various functional states. 18F-Me-4-deoxy-4-fluoro-D-glucopyranoside is a promising radioactive PET tracer that specifically targets SGLT, enhancing our understanding of glucose metabolism in diabetes and oncology12. We found its analog 4-deoxy-4-fluoro-D-glucopyranoside (4FDG) (Supplementary Fig. 1a)13 inhibits the uptake activity of hSGLT2 in a dose-dependent manner (Fig. 1b), indicating that 4FDG is also a hSGLT2 substrate competing with the fluorescent substrate 1-NBD-glucose for transport into the cell. For cryo-EM sample preparation, we expressed and purified the hSGLT2-Fab90 complex in the presence of 4FDG (Supplementary Fig. 1). Subsequent image analysis revealed two distinct 3D classes within the sample (Supplementary Figs. 2,3), both of which were substrate-bound (Supplementary Fig. 4). One 3D class, with a map resolution of 2.6 Å, exhibited a closed outer gate and a closed inner gate (Figs. 1c, d, 2a and Supplementary Table 1), which we named the substrate-bound occluded conformation (hSGLTSO). The other population, with a resolution of 2.47 Å, displayed a closed outer gate and a partially open inner gate (Fig. 3a and Supplementary Figs. 2-4), which we named the substrate-bound inward-facing conformation (hSGLTSI). We did not observe the 4FDG-bound outward-open states, probably due to the absence of negative membrane potential in the cryo-EM conditions14.

a, The 1-NBD-Glucose uptake assay for cells expressing empty vector and hSGLT2cryo-EM constructs (data are shown as the means ± standard deviations; n = 3 biologically independent samples). b, The inhibition curve of the hSGLT2-MAP17 complex by 4FDG in the 1-NBD-glucose uptake assay (data are shown as means ± standard deviations; n = 3 biologically independent experiments). The 1-NBD-glucose uptake is normalized to the uptake of hSGLT2-MAP17 without 4FDG inhibition. Source data are provided as a Source Data file. c, Cryo-EM density map of the hSGLT2-MAP17 complex. hSGLT2 is colored in blue. MAP17 is colored in yellow. The light chain and heavy chain of Fab90 variable regions are colored in dark pink and light pink. The detergent micelle is colored in light gray. d, The cut-open view of the hSGLT2-MAP17 complex shows the binding site of 4FDG substrate inside hSGLT2. 4FDG is colored in orange. The hSGLT2 is colored the same as in c.

a The estimated surface of the hSGLT2-MAP17 complex. 4FDG and Na are shown as balls and sticks. b The electron density of the Na2 site and nearby residues, the distance between Na ion and residues are shown as yellow dashed lines, and the distances are labeled in black. c The electron density of 4FDG and nearby residues, the 4FDG is colored in orange, and the water molecules are colored in dark gray. d Interactions between 4FDG and hSGLT2. 4FDG and its interacting residues are shown as balls and sticks. Putative hydrogen bonds are depicted as black dashed lines. e A 90° rotated view of d.

a Superposition of the hSGLT2OO and hSGLT2SO. The mobile helices are shown as pink (hSGLT2OO) and blue(hSGLT2SO) cylinders respectively, and the less-mobile helices are shown as gray cylinders. The obvious movements of helices are shown as black arrows. b The rotation of a. The obvious movements of helices are shown as black arrows. c–h, Changes of polar interactions between the hSGLT2OO and hSGLT2SO, the color is the same as a. Helices are shown as cylinders. c, e, g, Extracellular polar interactions that maintain the occluded conformation of SGLT2. Dashed lines indicate hydrogen bonds and their distances. d, f, h The residues in the outward-facing conformation correspond to those in c, e, g. Dashed lines indicate the distances between residues that form hydrogen bonds in c, e, g, i, 1-NBD-glucose uptake (n ≥ 3 biologically independent experiments) and surface expression of hSGLT2 mutants in the presence of MAP17 (n = 2 biologically independent experiments). Data are normalized to WT hSGLT2-MAP17 (data are shown as means ± standard deviations). j 1-NBD-glucose uptake of hSGLT2-MAP17 mutants normalized by surface expression. The data are calculated using the mean of 1-NBD-glucose uptake divided by the mean of surface expression and normalized to WT hSGLT2-MAP17. Source data are provided as a Source Data file.
Sodium and glucose binding sites of hSGLT2 in the occluded conformation
The structure of hSGLTSO is overall similar to the previously determined structure of AMG-bound occluded hSGLT2-MAP17 at 3.3 Å8 (PDB ID: 7YNJ, Supplementary Fig. 5a), but with much-enhanced features. In the structure of hSGLTSO, we observed strong density at the putative sodium binding site (Na2 site) (Fig. 2a, b). The sodium ion is coordinated by the main chain carbonyl groups of I76, A73, and A389, as well as the side chain hydroxyl groups of S392 and S393 (Fig. 2b). The distances between the sodium ion and the coordinating oxygen atoms range from 2.2 Å to 2.4 Å, indicating tight binding of the sodium ion (Fig. 2b). This binding mode of sodium is reminiscent of that observed in LeuT (PDB ID: 3TT1) and hSGLT2 in the presence of inhibitors9,10 (PDB ID: 7VSI, Supplementary Fig. 5b, c), contrasting with the looser binding mode seen in vSGLT at the inward-facing conformation15 (PDB ID: 3DH4, Supplementary Fig. 5d).
At the substrate binding site, we detected clear density for 4FDG, along with several putative water molecules (Fig. 2c). 4FDG predominantly adopts the α-D-glucopyranose configuration and forms extensive polar interactions with amino acids in the substrate binding site (Fig. 2d, e). Specifically, the C1-OH group forms a hydrogen bond with H80, while the C2-OH group establishes hydrogen bonds with the side chains of H80, N75, E99, and K321 (Fig. 2d, e). The C3-OH forms hydrogen bonds with W291 and K321 (Fig. 2d, e), and the C4-F group forms hydrogen bonds with the side chain of W291 (Fig. 2d, e). Additionally, the C6-OH group engages in hydrogen bond interactions with Q457, S460, and a water molecule (Fig. 2d, e). Furthermore, 4FDG is sandwiched between the aromatic side chains of W291 and F98 (Fig. 2d, e). We observed three ordered water molecules participating in the hydrogen bonding network at the substrate binding site, potentially stabilizing the local structures (Fig. 2d, e).
Although both SGLT1 and SGLT2 mediate glucose transport, SGLT1 uniquely exhibits the capacity to transport galactose5. To elucidate the molecular determinants underlying this functional divergence, we analyzed the sequence variation around the substrate-binding site between SGLT1 and SGLT2 (Supplementary Fig. 5e). The residues of interest include S287(T) and L283(M) (with SGLT1 residues indicated in brackets), as well as V105(L), which is close to S287 and also differs between the two proteins. We generated four mutants: S287T, S287T + V105L, S287T + L283M, and S287T + L283M + V105L. However, none of these variants exhibited measurable 1-NBD glucose uptake activity for reasons that remain unclear (Supplementary Fig. 5f), which precluded further functional characterization.
Structural transition of SGLT2 from the outward-open to the occluded conformation
SGLT2 inhibitors stabilize SGLT2 in the outward-open conformation11. To understand how SGLT2 isomerizes from the outward-open conformation to the occluded conformation, we compared the empagliflozin-bound outward-open conformation of hSGLT2 (hSGLT2OO, PDB: 7VSI)9 with the hSGLT2SO structure (Fig. 3). This comparison revealed that the extracellular portion of the transmembrane helices TM0, TM3, TM4, TM5, TM8, TM9, and TM10 move toward the center of hSGLT2 during transition (Fig. 3a, b, and Supplementary Video 1). Specifically, TM0, TM4, TM9, and TM10 exhibited angular rotations of 8.5°, 13.7°, 12.2°, and 10.5°, respectively (Fig. 3a, b). These conformational changes effectively close the outer gate of the transporter, consistent with our previous observations8. Moreover, we observed a more compact substrate binding site in the occlude conformation (Supplementary Fig. 5g, h), suggesting that occluded conformation might have higher affinity for substrate, compared to the inward-facing conformation.
The high-resolution structure of hSGLT2SO enabled us to analyze the changes in intramolecular interactions during this structural transition. We identified several putative polar interactions that contribute to the stability of the outer gate. The side chains of S362 and N363 on EL4Hb form hydrogen bonds with the carbonyl oxygens of G450 and L452 in the loop between TM9 and TM10, respectively (Fig. 3c). Similarly, Q451 and D454 on TM10 form hydrogen bonds with T87 on TM1 and Y526 on TM12 respectively (Fig. 3e). D25 on TM0 makes electrostatic interactions with R379 on TM8 (Fig. g). However, in hSGLT2OO, the outward movement of TM0, TM9, and TM10 disrupts these polar interactions (Fig. 3d, f, h). To assess the contribution of these interactions to the transport activity of SGLT2, we mutated the relevant residues to alanine and measured both the uptake activity and surface expression of the mutants (Fig. 3i). Our results showed that when the uptake activities were normalized to surface expression levels, the mutants Q451A, S362A, N363A, D454A, and R379A exhibited significantly reduced transport activity, with N363A nearly losing function entirely (Fig. 3j). These findings suggest that the interactions between S362-G450, T87-Q451, D454-Y526, N363-G450, and D25-R379 are crucial for the stability of the outer gate, particularly highlighting the dominant role of N363-G450 pair.
Reduced substrate and sodium binding of SGLT2 in the inward-facing conformation
In the structure of hSGLT2SI (Fig. 4a), we observed a density around the putative sodium binding site (Fig. 4b). However, only three oxygen atoms—the main chain carbonyl groups of A73 and I76, and the hydroxyl group on the side chain of S393—are in proximity to this density. The distances between this density and the surrounding oxygen atoms range from 3.3 to 3.9 Å (Fig. 4b), which are too large for stable sodium ion coordination. Therefore, we conclude that the sodium binding site in hSGLT2SI is disrupted, and the identity of this density remains uncertain. In comparison to hSGLT2SO, we found that TM1 and TM8 have moved apart, causing the sodium-binding residues at the Na2 site to lose their optimal positioning for sodium coordination (Fig. 4c and Supplementary Video 2).

a The cut-open view of the hSGLT2-MAP17 complex in the surface representation. 4FDG is shown as sticks. b The electron density of the Na2 site and nearby residues, the distances between the density center and related residues are shown as yellow dashed lines, and the distances are labeled in black. c Structural changes of Na2 site from the occluded conformation (blue) to the inward-facing conformation (green). d The electron density of 4FDG and nearby residues, the 4FDG is colored in orange, and the water molecules are colored in dark gray. e Interactions between 4FDG and hSGLT2. 4FDG and its interacting residues are shown as balls and sticks. f A 90° rotated view of e.
At the substrate-binding site, we observed strong density for 4FDG (Fig. 4d), which interacts with hSGLT2 in a manner similar to that seen in hSGLT2SO (Fig. 4e, f). However, detailed analysis revealed small but noticeable outward movements of several residues, including F98, Y290, and N75 (Fig. 4e, f, and Supplementary Video 3). These shifts lead to an enlarged substrate-binding site and a weakened interaction with 4FDG (Fig. 4e, f). These observations suggest that hSGLT2 in the inward-facing conformation exhibits lower affinities for sodium and substrate compared to the occluded conformation.
Structure transition from the occluded conformation to the inward-facing conformation
A comparison of hSGLTSO and hSGLTSI revealed outward movements in TM0, TM3, TM4, TM5, TM8, TM9, and TM10 near the intracellular side, resulting in the opening of the inner gate (Fig. 5a, b, and Supplementary Video 1), consistent with our previous observations8. Notably, TM5, TM8, and TM9 exhibited significant changes, with TM8 moving outward by 6.1 Å and TM5 and TM9 rotating by 7.1° and 15.4°, respectively (Fig. 5a, b).

a Superposition of the hSGLT2SO and hSGLT2SI. The mobile helices are shown as blue (hSGLT2SO) and green (hSGLT2SI) cylinders respectively, and the less-mobile helices are shown as gray cylinders. The obvious movements of helices are indicated with black arrows. b The 120° rotation of a. c–f, Changes of polar interactions between hSGLT2SO and hSGLT2SI, the color is the same as a. Helices are shown as cylinders. c, e Intracellular polar interactions that maintain the occluded conformation of SGLT2. Dashed lines indicate hydrogen bonds and their distances. d, f The residues in the substrate-bound inward-facing conformation correspond to those in c, e. Dashed lines indicate the distances between residues that form hydrogen bonds in c, e. g 1-NBD-glucose uptake (n ≥ 3 biologically independent experiments) and surface expression of hSGLT2 mutants in the presence of MAP17 (n = 2 biologically independent experiments). Data are normalized to WT hSGLT2-MAP17 (data are shown as means ± standard deviations. h 1-NBD-glucose uptake of hSGLT2-MAP17 mutants normalized by surface expression. The data are calculated using the mean of 1-NBD-glucose uptake divided by the mean of surface expression and normalized to WT hSGLT2-MAP17. Source data are provided as a Source Data file.
We compared high-resolution structures to analyze these changes in interactions at the amino acid level. In hSGLTSO, the hydroxyl group of T205 on TM5 forms a hydrogen bond with the carbonyl oxygen of A69 on TM1 (Fig. 5c). Similarly, S396, T403, and D408 on TM8 form hydrogen bonds with R300, Q299 on TM6, and T124 in the loop between TM2 and IL1H, respectively (Fig. 5c, e). These polar interactions are absent in the inward-facing conformation due to the outward movement of TM5 and TM8 (Fig. 5d, f). To further investigate these interactions, we generated alanine mutations to disrupt them. We found that the T124A, T403A, and S396A mutations greatly reduced 1-NBDG uptake activity (Fig. 5g, h). These findings suggest that the interactions between Q299-T403, R300-S396, and T124-D408 contribute to the stability of the inner gate in the occluded conformation.
Widening of the intracellular vestibule in the presence of potassium
To visualize the structural changes associated with substrate release, we resolved the cryo-EM structure of hSGLT2 in the absence of substrate but in the presence of sodium (hSGLT2INa). The structure, with the resolution of 2.92 Å (Supplementary Figs. 1, 6, 8 and Supplementary Table 1), is overall similar to the one determined previously8 (PDB ID: 7YNK, Supplementary Fig. 9a) but with many enhanced side chain features. Comparing the structure of hSGLT2INa with that of hSGLT2SI, we observed conformational changes in TM1a, TM6, and the gating helix TM5. Specifically, TM1a and TM5 rotated by 11.0° and 5.7°, respectively (Fig. 6a–b). Notably, at the substrate binding site, the side chain of N75 shifted by 3.4 Å, disrupting the optimal substrate-binding geometry (Fig. 6c). At the Na2 site, the Cα of I76 moved upward by 2.6 Å, indicating a significant positional shift (Fig. 6d). Collectively, these observations suggest that substrate release is associated with further disruption of both the substrate and sodium-binding sites.

a Superposition of hSGLT2SI and hSGLTINa. The mobile helices are shown as green (hSGLT2SI) and purple (hSGLTINa) cylinders, respectively, and the less-mobile helices are shown as gray cylinders. The obvious movements of helices are shown as black arrows. b The bottom view of (a). c Changes of residues at the substrate binding site, the color is the same as (a). The substrate is colored in orange. Helices are shown as cylinders. The obvious movements of residues are shown as black arrows. d Changes of residues at the Na2 site, the color is the same as a. The obvious movement of residue is indicated by black arrows. e Superposition of the hSGLTINa and hSGLTIK. The mobile helices are shown as purple (hSGLTINa) and orange (hSGLTIK) cylinders, respectively, and the less-mobile helices are shown as gray cylinders. f The bottom view of (e). The obvious movements of helices are shown as black arrows.
SGLT2 releases glucose and sodium into the cytosol, which has low sodium but high potassium concentrations. To mimic the intracellular environment, we determined the cryo-EM structure of SGLT2 in the presence of potassium (hSGLT2IK), achieving 3.16 Å (Supplementary Figs. 1, 7, 8 and Supplementary Table 1). In the presence of potassium, the densities of TM1a, TM6b, and the intracellular loops spanning residues 551–572 appeared either blurry or completely absent in the sharpened map, indicating that they are highly mobile. Moreover, when comparing the structures of hSGLT2INa and hSGLT2IK (Fig. 6e, f), we noted further movement of the intracellular region of hSGLT2, with TM2, TM6a, and TM7 expanding outward by 11.6°, 8.7°, and 6.8°, respectively (Fig. 6e, f). These conformational changes and increased structural mobility in the presence of potassium might further facilitate substrate release into the cytosol.
Discussion
The structures of hSGLT2 in complex with 4FDG provide a high-resolution view of how hSGLT2 recognizes substrates, which also sheds light on its closely related counterpart hSGLT1. Particularly, hydrophilic hydrogen bonding plays a dominant role in this substrate recognition. The 4C-F group of 4FDG forms a hydrogen bond with S287 of hSGLT2, and the equivalent residue in hSGLT1, T287, likely forms a similar hydrogen bond with 4FDG. In contrast, the corresponding residue in hSGLT3 is A287, which cannot form a hydrogen bond with the 4C–F group of 4FDG, correlating with the fact that 4FDG is an effective substrate for hSGLT1 and hSGLT2, but its affinity for hSGLT3 is much lower16. Moreover, we also identified an additional cavity connected to the substrate binding site, which is present in all observed conformations (Supplementary Fig. 9b–d). This cavity is located near the C1-OH group of 4FDG, indicating that SGLT2 may transport substrates with modifications at the C1-OH position of glucose. This finding aligns with the established ability of SGLT2 to transport 1-NBD-glucose9,17(Supplementary Fig. 1a).
Alongside the previously determined inhibitor-bound outward-open structure of hSGLT29,10, the high-resolution structures presented here provide a series of near-atomic snapshots along the transport reaction pathway of hSGLT2. Notably, we observed both global and local structural remodeling of hSGLT2 upon the dissociation of sodium and substrate (Supplementary Videos 1–3). The release of sodium results in an enlargement of the substrate-binding pocket, likely decreasing substrate affinity. Further dissociation of the substrate is associated with additional disruption of this pocket. Among these structures, we identified the substrate-bound and sodium-released inward-facing structure (hSGLT2IS) and the sodium-released inward-facing structure (hSGLT2INa), but no sodium-bound inward-facing structure, suggesting that sodium dissociation might occur prior to the release of 4FDG substrate in hSGLT2. Additionally, when sodium is replaced by potassium, hSGLT2 exhibits increased structural mobility on the intracellular side, facilitating the release of substrate.
It has been reported that intracellular potassium ions enhance the transport activity of neurotransmitter sodium symporter DAT18, SERT19, and LeuT20 through direct binding, and potassium binding has been observed at the Na1 site of NET21 (PDB ID: 8WGR). However, the Na1 site is not conserved in hSGLT2, and we did not observe the potassium ion binding site, probably due to the limited resolution of hSGLT2IK. Therefore, we speculate that potassium binding occurs at an unidentified site, enhancing the structural mobility of hSGLT2 on the intracellular side, and thereby promoting substrate release.
In conclusion, this study provides insights into the structural mechanisms of SGLT2. By elucidating the detailed structures of SGLT2 in various conformations, including its complex with a glucose analog, the research highlights a detailed hydrogen bonding network that facilitates substrate recognition. The findings also underscore the critical polar interactions that stabilize the gate of the transporter, enhancing our understanding of the structural plasticity of SGLT2 during glucose transport.
Methods
Cell culture
HEK293F suspension cells (Thermo Fisher Scientific) were cultured in FreeStyle 293 medium (Thermo Fisher Scientific) or SMM 293-TI medium (Sino Biological) supplemented with 1% fetal bovine serum (FBS) at 37 °C with 5% CO2 and 70% humidity. AD293 adherent cells (Agilent Technologies) were cultured in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% FBS at 37 °C with 5% CO2 and 70% humidity. The cell lines were routinely checked to be negative for mycoplasma contamination, but have not been authenticated.
Constructs
hSGLT2GFP-MAP17GST was generated by fusing loop-tagged GFP between 586 and 587 amino acids of hSGLT2 and fusing the GST tag to the C-terminus of MAP17. The ORF of hSGLT2 and MAP17 were cloned into the modified BacMam expression vector22.
hSGLT2GFP–MAP17nb complex was generated as described previously9, by fusing GFP between 586 and 587 amino acids of hSGLT2 and fusing anti-GFP nanobody to the C-terminus of MAP17. The ORF of hSGLT2 and MAP17 were cloned into the modified BacMam expression vector22.
Neither fusing GFP to SGLT2 nor fusing GST/nanobody to MAP17 affects its normal function, compared to the WT hSGLT2–MAP17 complex (Supplementary Fig. 1b). Point Mutations on hSGLT2GFP were introduced using Quikchange PCR.
Uptake assay
1-NBD-glucose uptake assay was used to measure the glucose-transporting function of SGLT9. AD293 cells cultured in 12-well plates were transfected with wild-type hSGLT2GFP or its mutants with wild-type MAP17. One day post-transfection, cells were seeded into 96-well plates coated with poly-D-lysine. After attachment, cells were washed with 200 μl per well PBS (10 mM Na2HPO4, 2 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl) twice, followed by incubation at 37 °C with 5% CO2 for 1 h in uptake buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 1 mM CaCl2, and 1 mM MgCl2) supplemented with 600 μM 1-NBD-glucose and 0.3% bovine serum albumin (BSA). Subsequently, cells were washed three times with 200 μl per well PBS to stop the uptake. Then the cells were lysed with 150 μl per well TBS buffer containing 1% SDS for 30 min at 25 °C. The lysates were transferred to a clear-bottom black 96-well plate for 1-NBG-glucose detection with excitation at 445 nm, and emission at 525 nm on an Infinite 200Pro imager (Tecan Life Sciences). Protein concentration was determined in a 96-well plate using a BCA protein assay kit (CWBIO). The 1-NBD-glucose fluorescence signals in each well were normalized to the protein concentration. The specific 1-NBD-glucose uptake of each well was calculated by subtracting 1-NBD-glucose fluorescence signals in wells transfected with empty vectors.
Surface labeling using antibodies
The monoclonal antibody was generated by injecting purified hSGLT2-MAP17 protein into mice, followed by hybridoma screening8. mAb90 was generated by Proteintech Group, IL, USA. The surfacing labeling was carried out as described previously23. AD293 cells were plated onto a poly-D-lysine-treated 24-well plate and transfected with wild-type hSGLT2GFP or its mutants with MAP17 and incubated for 40–48 h. The cells were washed with 500 μl PBS per well twice, fixed using 300 μl 4% formaldehyde in PBS for 30 min, and washed with 500 μl PBS per well twice again. For surface expression, the cells were blocked with 300 μl 3% goat serum in PBS per well for 30 min and labeled with 150 μl primary antibody mAb90 (diluted 3350 times in blocking buffer) per well for 1 h. After washing with PBS three times, cells were incubated with horseradish-peroxidase (HRP) labeled goat anti-mouse IgG secondary antibody (#31444; Invitrogen, the antibody was diluted 2500 times in blocking buffer) for 30 min. After extensive washing, the cells were incubated with High-Sig ECL Western Blotting Substrate (Tanon) for 2 min, and chemiluminescence signals were measured with an Infinite M Plex plate reader (Tecan).
Protein expression and purification
The mAb90 was digested to generate Fab fragments using papain, and the reaction was terminated with iodoacetamide. The antibody was tested for complete cleavage by SDS-PAGE. After digestion, the mixture was mixed with loop-tagged GFP fused TP1170 and further purified Fab90-TP1170 complex with Streptactin Beads 4FF (Smart Lifesciences) column. The eluate was loaded onto HiTrap Q HP (GE Healthcare), and the Fab90-TP1170 complex was separated from aggregates with a linear gradient from 0 mM NaCl to 1000 mM NaCl in buffer containing 20 mM Tris, pH 8.0, at 4 °C. The fractions containing the Fab90-TP1170 complex were collected.
The expression and purification of the hSGLT2-MAP17 complex was carried out as described previously with minor modification9,24. For protein expression, HEK293F cells cultured in SMM 293-TI or FreeStyle 293 medium at a density of 2.5\(\times {10}^{6}\) cells per ml were infected with 10% volume of P2 virus. Sodium butyrate (10 mM) was added to the culture 12 h after transfection to promote protein expression, and the cells were transferred to a 30 °C incubator for another 36 h before harvesting. Cells were collected by centrifugation at 3999g (JLA-8.1000, Beckman Coulter) for 10 min at 4 °C, and washed once with TBS buffer (20 mM Tris, pH 8.0 at 4 °C, 150 mM NaCl) containing 2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin. The cells were then flash-frozen and stored at −80 °C.
To prepare the sample for hSGLTSO and hSGLTSI, the membrane pellets of hSGLT2GFP-MAP17GST corresponding to 1 liter culture were resuspended and homogenized in 15 ml TBS buffer containing protease inhibitor (2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 1 mM phenylmethanesulfonyl fluoride (PMSF)), 10 mM MgCl2, 0.7 μg/ml benzonase. 5 mM 4FDG (Toronto Research Chemicals) was added to the crude membrane solution. The mixture was incubated at 37 °C for 1 h for 4FDG binding. Digitonin (BioSynth) was added to a final concentration of 1% (w/v) after cooling the membrane to 4 °C, and the mixture was stirred for 1 h to solubilize membrane proteins. The insoluble debris was removed by centrifugation at 193,400g (Ti50.2, Beckman Coulter) for 30 min. Subsequently, the supernatant was loaded onto a 5 ml Streptactin Beads 4FF (Smart Lifesciences) column and washed with 50 ml wash buffer 1 (TBS buffer plus 40 μM glyco-diosgenin (GDN), 5 mM 4FDG, 10 mM MgCl2 and 1 mM adenosine triphosphate (ATP)) to remove contamination of heat shock proteins. Then, the column was extensively washed with 50 ml wash buffer 2 (TBS buffer supplemented with 40 μM GDN and 5 mM 4FDG). The target protein was eluted with elution buffer containing 50 mM Tris, pH 8.0 at 25 °C, 150 mM NaCl, 40 μM GDN, 5 mM 4FDG, and 10 mM D-desthiobiotin (IBA). The eluate was loaded onto HiTrap Q HP (GE Healthcare), and the hSGLT2-MAP17 complex was separated from aggregates with a linear gradient from 0 mM NaCl to 1000 mM NaCl in buffer containing 20 mM Tris, pH 8.0, at 4 °C, 40 μM GDN, and 5 mM 4FDG. The fractions containing the hSGLT2–MAP17 complex were collected, concentrated, and subjected to a Superose 6 increase 10/300 GL column (GE Healthcare) in buffer that contained 20 mM HEPES pH 7.5, 150 mM NaCl, 40 μM GDN, and 5 mM 4FDG. The peak fractions corresponding to the hSGLT2–MAP17 complex were collected and incubated with Fab90–TP1170 at a molar ratio of 1:1.2. The mixture was concentrated and subjected to a Superose 200 increase 10/300 GL column (GE Healthcare) in buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 40 μM GDN, and 5 mM 4FDG. The peak fractions corresponding to the hSGLT2–MAP17–Fab90 complex were collected for cryo-EM sample preparation.
To prepare the sample for hSGLTINa and hSGLTIK, the membrane pellets of hSGLT2GFP–MAP17nb corresponding to 750 ml culture were resuspended and homogenized in 15 ml TBS buffer containing protease inhibitor (2 μg/ml aprotinin, 2 μg/ml pepstatin, 2 μg/ml leupeptin, and 1 mM phenylmethanesulfonyl fluoride (PMSF)), 10 mM MgCl2, 0.7 μg/ml benzonase. GDN was added to a final concentration of 1% (w/v) to the mixture, and the solution was stirred at 4 °C for 1 h to solubilize membrane proteins. The insoluble debris was removed by centrifugation at 193,400g (Ti50.2, Beckman Coulter) for 30 min. Subsequently, the supernatant was loaded onto a 5 ml Streptactin Beads 4FF (Smart Lifesciences) column and washed with 50 ml wash buffer 1 (TBS buffer plus 40 μM GDN, 10 mM MgCl2 and 1 mM adenosine triphosphate (ATP)) followed by 50 ml wash buffer 2 (TBS buffer supplemented with 40 μM GDN). The target protein was eluted with elution buffer containing 50 mM Tris, pH 8.0 at 25 °C, 150 mM NaCl, 40 μM GDN and 10 mM D-desthiobiotin (IBA). The eluate was loaded onto HiTrap Q HP (GE Healthcare), and the hSGLT2-MAP17 complex was separated from aggregates with a linear gradient from 0 mM NaCl to 1000 mM NaCl in buffer containing 20 mM Tris, pH 8.0, at 4 °C, 40 μM GDN. The fractions containing the hSGLT2–MAP17 complex were mixed with Fab90 at a molar ratio of 1:1.2. The mixture was concentrated and subjected to a Superose 6 increase 10/300 GL column (GE Healthcare) in buffer that contained 20 mM HEPES, pH 7.5, 150 mM NaCl, and 40 μM GDN for hSGLTINa. As for hSGLTIK, the mixture buffer was changed to 20 mM HEPES pH 7.5, 100 mM KCl, and 40 μM GDN during size-exclusion chromatography. The peak fractions corresponding to the hSGLT2–MAP17–Fab90 complex were collected for cryo-EM sample preparation.
Cryo-EM sample preparation and data collection
The surface of Quantifoil Au 300 mesh R 0.6/1.0 grids or Quantifoil Cu 300 mesh R 1.0/1.0 grids was coated with graphene oxide25. Aliquots of 2.5 µl of the hSGLT2–MAP17–Fab90 complex in the presence of 0.5 mM fluorinated octyl-maltoside (FOM, Anatrace) were applied to the grids. After incubation at 4 °C under 100% humidity for 60 s, the grids were blotted for 4 s using a blot force of 4, and then plunge-frozen into liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific). The samples without graphene oxide film were prepared by applying 2.5 µl of the hSGLT2–MAP17–Fab90 to the grids. After incubation at 4 °C under 100% humidity for 2 s, the grids were blotted for 2 s using a blot force of 5, and then plunge-frozen into liquid ethane using a Vitrobot Mark IV. The grids were transferred to a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV. EPU was used for automated data collection. Movies from the dataset of the hSGLT2–MAP17 were recorded on a K3 Summit direct detector (Gatan) mounted post a quantum energy filter (slit width 20 eV) in super-resolution mode with a defocus range of −1.5 to −1.8 μm and a magnification of 105,000× or 81,000×, corresponding to a calibrated pixel size of 0.417 Å or 0.5335 Å, respectively. Each stack of 32 frames was exposed for 8 s or 3.73 s, with an exposure time of 0.25 s or 0.117 s per frame at a dose rate of 4.7 or 21.46 electrons per Å−2 per second, respectively.
Cryo-EM image processing
For hSGLTSO and hSGLTSI, a total of 4549 micrographs were collected. Beam-induced drift was corrected using MotionCor226 and 2× binned to a pixel size of 0.834 Å. The contrast transfer function (CTF) parameters of dose-weighted micrographs were estimated by Gctf27. Micrographs were manually screened, and a total of 3967 micrographs were used for the following process. A total of 1,473,971 particles were auto-picked using Gautomatch-0.56 (developed by K. Zhang). All particles were extracted with a box size of 120 and 2× binned (pixel size 1.668 Å) in Relion-3.128. These particles were subjected to 2D classification with cryoSPARC-3.1.029. After two rounds of 2D classification, 479,998 particles from classes exhibiting recognizable hSGLT2 features were selected for further processing. A batch size of 50,000 particles was used to generate an initial 3D reference model. The initial model was low-pass filtered to resolutions of 8 Å, 15 Å, 25 Å and 35 Å. The low-pass filtered maps were used as references for a heterogeneous refinement without symmetry imposed. Subsequently, 403,040 particles from the best classes were re-extracted using a box size of 240 at 0.834 Å per pixel and re-centered in Relion-3.1 and then refined with NU-refinement and local refinement subsequently in cryoSPARC-3.1.030, resulting in a reconstruction with an overall resolution of 3.44 Å. Further, seed-facilitated 3D classification31 was performed in cryoSPARC-3.1.0, and a set of 962,982 particles was enriched. NU-refinement and local refinement of these particles yielded a reconstruction at 2.60 Å. This model was low-pass filtered to resolutions of 4 Å, 8 Å, 12 Å, 16 Å and 20 Å. The maps were used as references for a multi-reference 3D classification with an adapted mask. The classes of the last 12 iterations with the best features of hSGLT2 and Fab90-scFv in 3D classification were combined, and the duplicated particles were removed. The final 493,652 particles were re-extracted using a box size of 240 and a pixel size of 0.834 Å in Relion-3.1. These particles were subjected to NU-refinement and local refinement in cryoSPARC-3.1.0, yielding a reconstruction at 2.45 Å. The Clustered 3D Variability was performed, and the particles were divided into two classes, and two maps were obtained with resolutions of 2.60 Å and 2.45 Å.
For the hSGLTINa, 966 super-resolution movie stacks were collected in data set 1. Beam-induced drift was corrected using MotionCor226 and 2× binned to a pixel size of 1.067 Å. The contrast transfer function (CTF) parameters of dose-weighted micrographs were estimated by Gctf27. Micrographs were manually screened, and a total of 874 micrographs were used for the following process. A total of 1,015,597 particles were auto-picked using Gautomatch-0.56 (developed by K. Zhang). All particles were extracted with a box size of 120 and 2x binned (pixel size 2.134 Å) in Relion-3.128. These particles were subjected to 2D classification with cryoSPARC-3.1.029. After several rounds of 2D classification, 150,747 particles from classes exhibiting recognizable hSGLT2 features were selected to generate initial 3D reference models. These models were used as references for a heterogeneous refinement with no symmetry imposed. Subsequently, 246,748 particles from the best classes were further subjected to NU-refinement, resulting in a reconstruction with an overall resolution of 4.44 Å. To further improve the resolution, another 2117 super-resolution movie stacks were collected in data set 2 with the same parameters. A total of 4,574,560 particles were auto-picked using Gautomatch-0.56 and Topaz32. Seed-facilitated 3D classification31 and resolution gradient 3D classification were performed in cryoSPARC-3.1.0 with a final set of 207,961 particles. NU-refinement and local refinement of these particles yielded a reconstruction at 2.92 Å.
For the hSGLTIK, 768 super-resolution movie stacks were collected in data set 1. Beam-induced drift was corrected using MotionCor226 and 2× binned to a pixel size of 1.067 Å. The contrast transfer function (CTF) parameters of dose-weighted micrographs were estimated by Gctf27. Micrographs were manually screened, and a total of 756 micrographs were used for the following process. A total of 738,726 particles were auto-picked using Gautomatch-0.56 (developed by K. Zhang). All particles were extracted with a box size of 120 and 2× binned (pixel size 2.134 Å) in Relion-3.128. These particles were subjected to 2D classification with cryoSPARC-3.1.029. After several rounds of 2D classification, 101,053 particles from classes exhibiting recognizable hSGLT2 features were selected to generate initial 3D reference models. These models were used as references for a heterogeneous refinement with no symmetry imposed. Subsequently, 65,880 particles from the best classes were further subjected to NU-refinement, resulting in a reconstruction with an overall resolution of 6.04 Å. To further improve the resolution, another 4171 super-resolution movie stacks were collected in data set 2 with the same parameters. A total of 9,429,036 particles were auto-picked using Gautomatch-0.56 and Topaz32. Seed-facilitated 3D classification31 and resolution gradient 3D classification were performed in cryoSPARC-3.1.0 with a final set of 486,883 particles. NU-refinement and local refinement of these particles yielded a reconstruction at 3.16 Å.
The resolution was estimated using the Fourier shell correlation (FSC) = 0.143 criterion. Local resolution was calculated using cryoSPARC-3.1.0. ThreeDFSC curves were calculated using the 3DFSC server33(https://3dfsc.salk.edu/).
Model building
The homology models of class 1 and class 2 of the hSGLT2–MAP17–Fab90 complex with 4FDG bound were from the hSGLT1–MAP17 complex with LX2761 (PDB ID: 7WMV). The models of the hSGLT2–MAP17–Fab90 complex in 100 mM KCl and in 150 mM NaCl were from the model of the hSGLT2-MAP17 complex in the apo state (PDB ID: 7YNK). The models were fitted into the cryo-EM maps using UCSF Chimera34 and manually adjusted using Coot-0.8.9.335. The models were further refined against the maps using Phenix-1.1936. The RMSDs between different states were calculated with Pymol-1.7.0.5.
Quantification and statistical analysis
Global resolution estimations of cryo-EM density maps are based on the 0.143 Fourier Shell Correlation criterion37. The local resolution was estimated using Relion-3.128. The number of independent reactions (N) and the relevant statistical parameters for each experiment (such as mean or standard deviation) are described in the figure legends. No statistical methods were used to pre-determine sample sizes.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon request. Cryo-EM maps and atomic coordinates of the hSGLT2–MAP17–Fab90 complex in the 4FDG-bound occluded state (hSGLTSO) have been deposited in the EMDB and PDB database under the ID codes EMDB: EMD-62395 and PDB: 9KKQ, respectively. Cryo-EM maps and atomic coordinates of the hSGLT2–MAP17–Fab90 complex in the 4FDG-bound inward-facing state (hSGLTSI) have been deposited in the EMDB and PDB database under the ID codes EMDB: EMD-62439 and PDB: 9KMN, respectively. Cryo-EM maps and atomic coordinates of the hSGLT2-MAP17–Fab90 complex in the substrate-free inward-facing state in the presence of sodium (hSGLTINa) have been deposited in the EMDB and PDB database under the ID codes EMDB: EMD-62396 and PDB: 9KKW, respectively. Cryo-EM maps and atomic coordinates of the hSGLT2–MAP17–Fab90 complex in the substrate-free inward-facing state in the presence of potassium (hSGLTIK) have been deposited in the EMDB and PDB database under the ID codes EMDB: EMD-62398 and PDB: 9KL0, respectively. PDB entries (2XQ2, 3TT1, 7VSI, 7WMV, 7YNJ, 7YNK, 8WGR) used in this study were downloaded from the Protein Data Bank. The source data underlying Fig. 1a, b, Fig. 3i, j, Fig. 5g, h, Supplementary Fig. 1b, and Supplementary Fig. 5f are provided as a Source Data file. Source data are provided with this paper.
References
Wells, R. G. et al. Cloning of a human kidney cDNA with similarity to the sodium-glucose cotransporter. Am. J. Physiol. 263, F459–F465 (1992).
Kanai, Y., Lee, W. S., You, G., Brown, D. & Hediger, M. A. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Invest. 93, 397–404 (1994).
Vallon, V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med. 66, 255–270 (2015).
Dharia, A., Khan, A., Sridhar, V. S. & Cherney, D. Z. I. SGLT2 Inhibitors: The Sweet Success for Kidneys. Annu. Rev. Med. 74, 369–384 (2023).
Wright, E. M., Loo, D. D. & Hirayama, B. A. Biology of human sodium glucose transporters. Physiol. Rev. 91, 733–794 (2011).
Drew, D. & Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 85, 543–572 (2016).
Coady, M. J. et al. MAP17 Is a Necessary activator of renal Na+/glucose cotransporter SGLT2. J. Am. Soc. Nephrol. 28, 85–93 (2017).
Cui, W., Niu, Y., Sun, Z., Liu, R. & Chen, L. Structures of human SGLT in the occluded state reveal conformational changes during sugar transport. Nat. Commun. 14, 2920 (2023).
Niu, Y. et al. Structural basis of inhibition of the human SGLT2-MAP17 glucose transporter. Nature 601, 280–284 (2021).
Hiraizumi, M. et al. Transport and inhibition mechanism of the human SGLT2-MAP17 glucose transporter. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-023-01134-0 (2023).
Sun, Z., Cui, W. & Chen, L. Structures reveal how SGLT inhibitors work. Trends Pharmacol. Sci. 45, 760–763 (2024).
Klimek, K. et al. PET imaging of sodium-glucose cotransporters (SGLTs): unveiling metabolic dynamics in diabetes and oncology. Mol. Metab. 90, 102055 (2024).
Diez-Sampedro, A., Wright, E. M. & Hirayama, B. A. Residue 457 controls sugar binding and transport in the Na(+)/glucose cotransporter. J. Biol. Chem. 276, 49188–49194 (2001).
Paz, A. et al. Conformational transitions of the sodium-dependent sugar transporter, vSGLT. Proc. Natl Acad. Sci. USA 115, E2742–E2751 (2018).
Faham, S. et al. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 (2008).
Voss, A. A., Diez-Sampedro, A., Hirayama, B. A., Loo, D. D. & Wright, E. M. Imino sugars are potent agonists of the human glucose sensor SGLT3. Mol. Pharmacol. 71, 628–634 (2007).
Chang, H. C. et al. Development of a novel non-radioactive cell-based method for the screening of SGLT1 and SGLT2 inhibitors using 1-NBDG. Mol. Biosyst. 9, 2010–2020 (2013).
Schmidt, S. G. et al. The dopamine transporter antiports potassium to increase the uptake of dopamine. Nat. Commun. 13, 2446 (2022).
Hellsberg, E. et al. Identification of the potassium-binding site in serotonin transporter. Proc. Natl Acad. Sci. USA 121, e2319384121 (2024).
Schmidt, S. G., Nygaard, A., Mindell, J. A. & Loland, C. J. Exploring the K(+) binding site and its coupling to transport in the neurotransmitter:sodium symporter LeuT. Elife. https://doi.org/10.7554/eLife.87985 (2024).
Tan, J. et al. Molecular basis of human noradrenaline transporter reuptake and inhibition. Nature 632, 921–929 (2024).
Li, N. et al. Structure of a pancreatic ATP-sensitive potassium channel. Cell 168, 101–110.e110 (2017).
Kang, Y. & Chen, L. Structure and mechanism of NALCN-FAM155A-UNC79-UNC80 channel complex. Nat. Commun. 13, 2639 (2022).
Niu, Y. et al. Structural mechanism of SGLT1 inhibitors. Nat. Commun. 13, 6440 (2022).
Patel, A., Toso, D., Litvak, A. & Nogales, E. Efficient graphene oxide coating improves cryo-EM sample preparation and data collection from tilted grids. bioRxiv (2021).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife https://doi.org/10.7554/eLife.42166 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Wang, N. et al. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 184, 370–383.e313 (2021).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013).
Acknowledgements
We thank Chen Lab members for their kind help. Cryo-EM data collection was supported by the Electron Microscopy Laboratory and the Cryo-EM Platform of Peking University, with the assistance of Xuemei Li, Zhenxi Guo, Changdong Qin, Xia Pei, and Guopeng Wang. Part of the structural computation was also performed on the Computing Platform of the Center for Life Science and the High-performance Computing Platform of Peking University. We thank the National Center for Protein Sciences at Peking University in Beijing, China, for assistance with negative stain EM. The work is supported by grants from the National Natural Science Foundation of China (32430050 and 32225027 to L.C.) and the Center for Life Sciences (CLS to L.C.).
Author information
Authors and Affiliations
Contributions
L.C. initiated the project. L.C., W.C., and Z.S. wrote the paper draft. W.C. and Z.S. purified the protein and prepared the cryo-EM sample. W.C., Z.S., J.X., X.L., and Y.K. collected the cryo-EM data. W.C., Z.S., and L.C. processed the data and built the model. Z.S. made mutations and carried out functional experiments. All authors contributed to the paper preparation. The order of co-first authors is determined by a coin toss.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cui, W., Sun, Z., Xu, J. et al. Mechanism of substrate recognition and release of human SGLT2. Nat Commun 16, 7140 (2025). https://doi.org/10.1038/s41467-025-62421-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62421-6
