
Abstract
The perturbation of protein translocation into the secretory pathway using Sec61 translocon inhibitors is a novel and promising strategy for tackling many pathological situations, including cancer and viral infections. However, a highly sensitive and direct screening platform for selecting Sec61 inhibitors is unavailable. Here, we develop a “resuming luminescence upon translocation interference” (RELITE) assay capable of selecting Sec61 inhibitors in a single round of screening. This assay exploits the inactivation of firefly luciferase, once translocated into the endoplasmic reticulum (ER), and the possibility of diverting and “re-lighting” luciferase into the cytosol by a Sec61 inhibitor. Using this method, we select small molecules capable of hampering the protein expression of the PD-L1 immune checkpoint by interfering with its ER translocation and delivering it for degradation. In conclusion, our screening method will greatly facilitate the selection of Sec61 inhibitors for down-modulating the expression of many disease-relevant proteins.
Similar content being viewed by others

Introduction
About one-third of the human proteome, including secreted signaling molecules, adhesion factors, receptors, ion channels and transporters, are handled by the secretory pathway to fulfill their physiological actions1. Before reaching their functional destination, these proteins are translocated into the endoplasmic reticulum (ER) by the function of specific translocation signal sequences, including N-terminal signal peptides (SPs). Signal sequences target nascent proteins to the Sec61 channel and open this “translocon” for polypeptide transport across the ER membrane2. Perturbation of this pathway can be a powerful approach to interfere with the expression and function of many disease-relevant proteins, thereby being an attractive therapeutic strategy3,4,5. Many natural products have been identified to target the central core of the Sec61 channel3,6 including decatransin7, mycolactone8, apratoxins9, coibamide A10 and ipomoeassin F11. Their non-selective activity, which prevents the ER translocation of most of the Sec61 clients, exerts severe cytotoxic effects, limiting their use in therapies. Cyclotriazadisulfonamide (CADA) compounds are a unique class of small molecule Sec61 inhibitors that have been described to down-modulate only a subset of translocating proteins based on the structures of their unique SPs4,6,12, including the type I integral membrane protein human CD4 (huCD4), reducing its expression on the plasma membrane of T-cells, thereby acting as an HIV entry inhibitor13. According to recently reported Cryo-EM structures, two of these selective ER translocation inhibitors (which we term SERTIs) bind to roughly the same location of the central core of Sec61 as the non-selective inhibitors mentioned above14,15. Therefore, the discovery of such molecules that can decrease the expression of specific proteins in an SP-dependent manner has opened the possibility that SPs may become a valid target for drug design4.
Since Sec61 lacks enzymatic activity, identifying Sec61 inhibitors with low toxicity remains a big challenge. So far, screening strategies are mainly based on reduced expression or secretion of reporter proteins such as Gaussia luciferase (GLuc) or green fluorescent protein (GFP) alone or fused to target proteins16,17,18. Although sensitive, like in the case of GLuc, these approaches are not selective for identifying direct Sec61 inhibitors. Indeed, reduced expression or secretion of the reporter proteins could have several primary causes. Compounds could interfere with any crucial steps driving the proper trafficking of proteins over the secretory pathway, possibly causing many false-positive candidates to be selected.
The programmed death ligand 1 (PD-L1) belongs to the immune checkpoint family and is particularly important in the immune evasion of cancer cells19. Indeed, the interaction with its receptor (PD-1), expressed by several cells of the immune system, causes the activation of an apoptotic suppressive pathway, which contributes to tumor survival and expansion20. In recent decades, the immunotherapy approach has demonstrated that using monoclonal antibodies against either the ligand or the receptor can impede the activation of this suppressive pathway, rendering the cancer cells sensitive to the immune system21. However, this approach has proven its efficacy only on a limited number of tumors and patients, leading many laboratories and companies to invest considerable resources in developing alternative approaches to inhibit this suppressive pathway21. SP-dependent protein down-modulation could represent an alternative valid strategy to hamper PD-L1/PD-1 axis activation, since most tumors are found to highly express PD-L1 or to improve its expression once contacted by the immune system22.
In this work, we develop the luciferase-based assay RELITE (pronounced re-light— REsuming Luminescence upon translocatIon inTErference) to screen Sec61 inhibitors. On this platform, we screen small molecules consisting of CADA and its analogs, and we identify compounds capable of inhibiting the translocation of a protein precursor bearing the PD-L1 SP. These CADA analogs can efficiently reduce PD-L1 expression independently of its expression level under constitutive, induced, or transient over-expression conditions. These findings will open the way for the generation of a new class of drugs for down-modulating PD-L1 expression.
Results
Developing a Luciferase-based assay for selecting Sec61 inhibitors
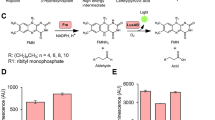
We exploited two distinctive properties of the Firefly luciferase reporter protein to develop a sensitive yet specific assay for selecting Sec61 inhibitors. The first is represented by its sensitivity to post-translational modifications, such as N-glycosylation15. Indeed, the active Firefly luciferase has a spurious N-glycosylation site at the level of asparagine N197 (NxS/T consensus, Supplementary Fig. S1a), which remains not glycosylated when the luciferase is expressed in the cytosol. On the contrary, when equipped with an ER translocation signal, Firefly luciferase accesses the ER lumen, becoming N-glycosylated and inactivated. Accordingly, treatment with N-glycosyltransferase inhibitor Tunicamycin (Tu) has been reported to rescue Firefly luciferase activity15. The second property relates to the folding kinetics of Firefly luciferase that begins with the co-translational formation of an N-terminal 22 kDa sub-domain that drives the subsequent rapid folding of the entire enzymatically active luciferase protein23. Although the pharmacological inhibition of Sec61 translocon has been shown to provoke the proteasomal degradation of the non-translocated protein precursors10,17,24, we assumed that under the inhibited translocation conditions, the rapid folding kinetics of the newly synthesized luciferase could anticipate its subsequent proteasomal degradation, thereby transiently rescuing its enzymatic activity in the cytosol (Fig. 1a). For these reasons, we call this assay REsuming Luminescence upon translocatIon inTErference or RELITE assay, since the possibility of “re-lighting” the luciferase once its translocation is interfered with by a Sec61 inhibitor (Fig. 1a). To test this hypothesis, we generated a bi-cistronic IRES vector whose cassette contains a firefly luciferase (FLuc), equipped with an SP to drive ER translocation, followed by the cytosolic Renilla luciferase (RLuc) used as an internal expression control. An internal ribosomal entry site (IRES) allows the co-expression of the two luciferase enzymes (Fig. 1b). To test and validate the RELITE assay, we chose the SP of CD4 since the Sec61 inhibitors, CADA and its more potent analog CK147, have already been shown to inhibit the ER translocation of CD4 in a SP-dependent manner13,25. A human cell line (HEK293) was transiently transfected with the abovementioned vector and, after 24 h, was treated with increasing concentrations of CADA or CK147. Up to 6 hours of incubation, equal fractions of total lysates were analyzed by the Dual-Glo Luciferase system, which allows consecutive measurement of the FLuc and RLuc enzymatic activity in the same well26. Strikingly, CADA treatment caused an increased FLuc activity (normalized on the RLuc activity), reaching a half-maximal inhibitory concentration (IC50) of about 648 nM compared to the 96 nM reached by its more potent analog CK147 (Fig. 1c). In addition, the treatment with Tunicamycin (Tu)27 caused a significant elevation of the FLuc activity, thereby confirming its sensitivity to modifications by N-glycans addition (Supplementary Fig. S1b). This effect was independent of the ER stress induced by Tu since another well-known ER stress inducer, such as Thapsigargin (Tg) (Supplementary Fig. S1c)28, failed to increase activity (Supplementary Fig. S1b). Notably, co-treatment with the protein synthesis inhibitor cycloheximide (CHX) abrogated the CADA effect (Supplementary Fig. S1b), confirming that CADA exerts its function during the co-translational translocation process of FLuc reporter protein13.

a Schematic view of the RELITE assay. b Schematic view of Bi-cistronic IRES vector. c HEK293 cells were seeded on a 60 mm plate and transiently transfected with IRES-vector bearing the SP of CD4 or a CTRL SP. After 24 h, cells were split in a 96-well plate and treated with increasing concentrations of CADA or CK147 compounds. After 6 h, FLuc and RLuc signals were measured using the Dual-Glo Luciferase system. Data were represented as FLuc/RLuc ratios, and the IC50 values of CADA and CK147 were determined, respectively. d HEK293 cells were grown in a 60 mm plate, transiently transfected, and treated with CADA as in (c). After 6 hours, P100 and S100 fractions were obtained after mechanical lysis and ultracentrifugation, and FLuc signals were measured on each fraction. e HEK293 cells were grown in a 60 mm plate, transiently transfected with the IRES-vector represented in (b) for 8 h and, after that, cells were split in (a) 96-well plate. After 16 h, cells were treated with compounds indicated in the graph, and the FLuc activity was measured over time up to 6 h. f HEK293 cells were handled as in (e) and, after 6 h of treatment with indicated compounds, cells were incubated for an additional 2 h with 100 µg/ml cycloheximide (CHX) before lysis, and FLuc activity was measured. g HEK293 cells were handled as in (e). After treatment, cells were harvested and mechanically lysed by syringe. Cytosolic and membrane fractions were obtained by differential centrifugation and analyzed by western blotting using a specific antibody against the HA-tag. Antibodies against GAPDH (cytosolic marker) and Calnexin (membrane marker) were used as fractionation controls. h HEK293 cells were handled as in (e), and after 6 h of treatment with the indicated molecules, FLuc/RLuc ratios were analyzed from total cell extracts by the Dual-Glo Luciferase system. i HEK293 cells were handled as in (g) after treatments with Eeyarestatin 1 (ES1), MG132, or a combination of them for 6 h. Data are presented as mean values +/− SD. N = 3 independent experiments.
Cell fractionation showed that CADA increased FLuc activity only in the cytosolic S100 fraction (Fig. 1d), suggesting that Sec61 inhibitors prevented the translocation process and re-localized the enzymatically active FLuc into the cytosol. The cytosolic FLuc half-life was estimated at around 30 minutes by extending CADA treatment with CHX (Supplementary Fig. S2). This observation suggested that upon translocation inhibition by CADA treatment, FLuc could be degraded by the proteasome. Accordingly, the treatment with the proteasome inhibitor MG132 caused a time-dependent increment of the FLuc enzymatic activity (Fig. 1e). Interestingly, a combination of MG132 and CADA treatment produced an additive effect (Fig. 1e), indicating that the recovered FLuc activity was the result of a balanced situation between the efficiency of the translocation inhibition necessary to accumulate the active FLuc into the cytosol and the efficiency of the proteasomal degradative pathway involved in its removal. To test this hypothesis, we treated cells with CADA, MG132 or a combination of them for 6 hours, and we measured the activity of FLuc after an additional 2 hours of treatment with CHX (Fig. 1f). Strikingly, CHX treatment caused a significant reduction of the FLuc activity pre-treated with CADA, whereas no reduction was observed in cells pre-treated with MG132 alone or in combination with CADA (Fig. 1f). Finally, to confirm the cytosolic localization of the FLuc, we analyzed, by western blotting and cytosol/membrane fractionation, the distribution of the N-terminally HA-tagged FLuc reporter protein before and after treatment with CADA, MG132, or a combination of them. Western blotting analysis performed on total cell extract showed a significant reduction of the total HA-FLuc protein levels upon CADA treatment for 6 h, confirming that the interfered translocation causes the degradation of the Sec61 client (Fig. 1g, left histogram). Moreover, as shown in Fig. 1g (right histogram), HA-FLuc was mainly detected in the membrane fraction (P100), whereas treatments with CADA alone or in combination with MG132 determined the almost total localization of HA-FLuc into the cytosolic fraction (S100). Notably, all the cytosolic cyto-HA-FLuc proteins migrated differently from and membrane-enclosed counterparts (ER-HA-FLuc), most likely by the lack of N-glycosylation modifications. Accordingly, CADA treatment generated an HA-FLuc form which co-migrated with the one obtained after digestion with Endoglycosidases H (EndoH) (Supplementary Fig. S1d), which selectively removes high mannose oligosaccharides typically added to the protein translocating into the ER29. Interestingly, no additive effect was measured upon the combination of CADA and MG132 treatments by analyzing protein levels (Fig. 1g, right histogram) in comparison to the rescued enzymatic activity (Fig. 1e). However, this apparent discrepancy was resolved analyzing the FLuc enzymatic activity by immunoprecipitating the polyubiquitinated version of FLuc co-transfected with Flag-Ubiquitin construct (Flag-Ub). As shown in Supplementary Fig. S3, upon co-treatment with CADA and MG132, a significant FLuc activity was measured in the pull-down with Flag antibody, thereby indicating that a significant portion of the polyubiquitinated FLuc enzyme was still functional.
Finally, exploiting the shorter incubation time for testing selective Sec61 inhibitors, we challenged the RELITE assay for measuring the activity of known non-selective Sec61 inhibitors. Strikingly, 6 hours of treatment with known non-selective inhibitors, such as Mycolactone (MYCO) and Apratoxin A30, caused a significant elevation of the FLuc/RLuc ratio and the additive effect when co-incubated with the proteasome inhibitor MG132 (Fig. 1h). On the contrary, the treatment with another non-selective Sec61 inhibitor Eeyarestatin 1 (ES1) determined a significant reduction of the FLuc/RLuc ratio. Surprisingly, cytosol/membrane fractionation and western blotting analysis revealed a significant reduction of FLuc protein levels and the appearance of a cytosolic fraction of the protein upon co-incubation of ES1 with the proteasomal inhibitor MG132 (Fig. 1i). These results suggest that the gating into the Sec61 channel, which has been reported to be prevented by ES1 compound31, is required for FLuc refolding before proteasomal degradation.
RELITE assay drives the selection of CADA analogs capable of down-modulating PD-L1 expression in an SP-dependent manner
Next, we challenged the RELITE assay for the selection of Sec61 inhibitors capable of interfering with the expression of a specific disease-relevant protein based on its unique SP. We chose the programmed death ligand-1 (PD-L1) as the main target of our investigation due to its enormous implication in the survival and aggressiveness of many different types of cancer32. Therefore, to identify PD-L1 expression inhibitors, we placed the PD-L1 SP upstream of the FLuc enzyme (Fig. 2a), and we screened a small-molecule library consisting of about 100 CADA analogs in a 96-well plate format. Among them, we selected ten molecules that significantly improved the FLuc/RLuc ratio (Fig. 2b), and the quality of the RELITE assay was determined by calculating the Z-factor, whose value of 0.84 (1 > Z > 0.5) let us categorize it as an excellent assay33.

a Schematic view of the IRES-vector bearing the FLuc equipped with the SP of PD-L1 protein, followed by the cytosolic Renilla luciferase (RLuc). b GB138 cells were grown in 60 mm plates and transiently transfected with the IRES-vector shown in (a). After 24 h, cells were split in 96-well plates and treated with a library of CADA analogs at 20 μM for 6 h. FLuc/RLuc ratio values are shown in the aerogram by using a color scale from white up to magenta (high). c Native (SPn) and mutated (SPm) amino acidic sequences of the PD-L1 SP. d GB138 cells stably expressing C-terminally HA-tagged PD-L1 protein equipped with SPn or SPm were treated with selected CADA analogs (BL458, BL445 or BL390) for 24 h at 20 μM. Total cell extracts were separated by SDS-PAGE, and PD-L1-HA was revealed using an anti-HA antibody. Calnexin (ER marker) protein level was used as a loading control. e, f GB138 cells stably expressing SPnPD-L1-HA were treated with selected CADA analogs (BL458, BL445 and BL390) at scaling concentrations from 0 to 20 μM for 24 h. PD-L1-HA protein levels are shown in the graph, and the IC50 value is indicated with a dashed line. g GB138 cells were treated with 20 μM CADA analogs (BL458 and BL466) for 24 h, and the endogenous PD-L1 protein expression level was analyzed by SDS-PAGE. h Effect of BL458 titration on the endogenous PD-L1 protein level. The IC50 value is shown with a dashed line. i GB138 cells were transiently transfected with SPn- or SPm-PD-L1-HA vectors and treated with BL458 at 20 μM for 24 hours. PD-L1-HA protein levels were analyzed by SDS-PAGE. Calnexin (ER marker) protein level was used as a loading control. Data are presented as mean values +/− SD. N = 3 independent experiments. j Vulcano plots showing significantly down-(blue dots) and up-(red dots) regulated proteins in GB138 cells after treatment with BL458 molecule at 0.2, 2, and 20 µM for 24 h.
As previously shown, a polar residue within CD4 SP played a crucial role in the CADA-dependent CD4 down-modulation13. Notably, PD-L1 contains three polar residues within its SP (T11, Y12, and H14), and to test their involvement in the PD-L1 down-modulation induced by the selected CADA analogs, we replaced them with leucine (L), a hydrophobic and more frequent amino acid found in the model SPs12 (Fig. 2c). To test the efficacy of the selected molecules to down-modulate PD-L1 expression and its dependence on the three polar residues, the glioblastoma cell line (GB138), stably expressing the two versions of PD-L1 equipped with its own (SPn) or mutated (SPm) variant of the SP, were treated with the ten selected CADA analogs. As shown in Fig. 2d, three of them (BL458, BL445 and BL390) were found to strongly reduce the PD-L1 expression when the SPn drove its translocation (SPnPD-L1-HA). On the contrary, the SPm-equipped version of PD-L1 (SPmPD-L1-HA) was more resistant to the treatments. Moreover, dose-response treatments showed that BL458 was more potent than the others, reaching an IC50 of about 263 nM (Fig. 2e, f). Notably, this value was much lower than that calculated by CADA treatment, which reached an IC50 value of about 20 μM, thereby highlighting the potency of chemical modifications in improving the effectiveness of CADA molecules against a selected SP (Supplementary Fig. S4). Moreover, the cell viability assay showed that even at concentrations higher than 20 μM, BL458 was not cytotoxic (Supplementary Fig. S5). To note, the ER marker Calnexin, a bona fide substrate of the Sec61 channel, was not affected by the treatments with selected molecules, thereby highlighting the specificity of such molecules to target the PD-L1 SP-dependent translocation process. Next, we tested the ability of BL458 to down-modulate PD-L1 protein levels under different expression conditions. As shown in Fig. 2g, BL458 significantly reduced the endogenously expressed PD-L1 protein in the GB138, and the dose-response treatment (Fig. 2h) revealed an IC50 (200 nM) comparable to the one obtained under stably expressing conditions. A strong down-modulation effect was also found when SPnPD-L1 was transiently over-expressed in GB138 cells, whereas, under the same experimental conditions, the SPmPD-L1 was almost resistant to the treatment with BL458 (Fig. 2i).
Finally, to analyze the impact of BL458 on the cell proteome, we performed a comparative analysis of the proteome profile of GB138 cells treated with scaling concentrations of BL458 at 0.2, 2, and 20 µM (Supplementary Table 1). Vulcano plot analysis (Fig. 2j) showed a minimal impact of BL458 on the cell proteome, ranging from 0.38% (at 0.2 µM) up to 2.7% (20 µM) of significant expression variation within 6055 analyzed proteins. In addition, we registered only 0.2% of down-modulated proteins at 0.2 µM, 1.25% at 2 µM, up to 1.9% at 20 µM treatment with the BL458 molecule. Furthermore, gene ontology analysis (Supplementary Fig. S6a) revealed that more than 90% of the down-modulated proteins belonged to the secretory pathway protein class, including secretory, plasma membrane, endoplasmic reticulum proteins, thereby confirming the Sec61 channel as the primary target of the BL458 molecule.
CADA analog induces proteasomal degradation of the PD-L1 precursor and exerts its prolonged effect over time
As previously shown13, CADA reduced CD4 expression by generating a stalled translocation situation of the CD4 precursor, which is resolved by the cell extracting and delivering the CD4 polypeptide chain for proteasomal degradation. Therefore, we wondered whether the reduction of PD-L1 protein expression caused by BL458 depended on a similar mechanism. To this end, GB138 cells stably expressing SPnPD-L1-HA were treated with BL458 in the presence or absence of MG132 (Fig. 3a). Treatment with the proteasome inhibitor increased PD-L1 protein levels, while BL458, as expected, reduced its expression, albeit to a lesser degree given the shorter incubation time the cells were subjected to (10 instead of 24 h). We chose this shorter time because of the greater cellular toxicity to the prolonged exposure to MG132. Interestingly, treatment with both compounds caused the appearance of a lower molecular weight form of PD-L1 compatible with the non-glycosylated (NG) version of the protein that did not appear following MG132 treatment alone, confirming the model that translocation inhibition leads to proteasomal degradation of the non-translocated polypeptide chain. This model was further confirmed by cell fractionation, showing that the non-glycosylated form of PD-L1 was found in the cytosolic fraction (S100), whereas all glycosylated forms of the protein, as expected, were found in the membrane fraction (P100) (Fig. 3b). Notably, the non-glycosylated form of PD-L1, rescued by the combined treatment with BL458 and MG132, showed a slightly higher molecular weight than the one generated by endoglycosidase H (EndoH) digestion, which is known to remove the high-mannose sugars of the N-glycosylation modification occurring in the ER lumen29(Fig. 3c). Similar result was obtained treating cells with Tunicamycin, a known N-glycosylation inhibitors, which caused the appearance of the non-glycosylated form of PD-L1 (NG) which migrated slightly higher than the one after co-treatment with BL458 and MG132. Together, these results suggest that PD-L1 forms at a lower molecular weight, represents the untranslocated pre-protein, non-glycosylated but still equipped with SPn (SPn-PD-L1-HA).

a GB138 cells stably expressing SPnPD-L1-HA were treated with 20 μM BL458 or with 5 μM proteasome inhibitor (MG132) or a combination of them for 10 h, and PD-L1-HA protein levels were analyzed by SDS-PAGE. Calnexin and GAPDH were used as fractionation controls (membrane and cytosolic marker, respectively). Fully- and non-glycosylated forms of PD-L1-HA (FG and NG, respectively) are indicated. b GB138 cells stably expressing SPnPD-L1-HA were grown in a 60 mm plate and treated with 20 μM BL458. After 24 h, proteins of the P100 and S100 fractions were separated and analyzed by SDS-PAGE. Glycosylated and non-glycosylated forms of PD-L1-HA (G and NG, respectively) are indicated. c Schematic view of expected PD-L1 forms before and after EndoH digestion, Tunicamycin treatment (10 μg/ml for 6 h), or after translocation inhibition (upper scheme). GB138 cells were transiently transfected with SPn-PD-L1-HA for 24 h and treated with the combination of BL458 (20μM) and MG132 (5 μM) for 8 h. After this incubation, cells were lysed, and an aliquot of mock cells was digested with EndoH or treated with tunicamycin before being analyzed by western blotting, together with non-digested mock cells and BL458 + MG132 treated ones (d) Cells as in (a) were treated with BL458 (20 μM), and after 16 h, cells were washed 3 times before being incubated with fresh media for an additional 10, 24, or 48 h (washout or WO). PD-L1-HA protein levels were analyzed by SDS-PAGE. e Cells as in (a) were treated with 20 μM of BL458 for 1 or 10 h of pulse (P) followed by 9 or 0 h of chase (C), and PD-L1-HA levels were analyzed by SDS-PAGE. f Cells, as in (a), were subjected to a time-course treatment with 20 μM BL458, and PD-L1-HA protein levels were analyzed by SDS-PAGE. g Electron microscopy picture of the Sec61 protein-conducting channel in the “non-inserting” state (5A6U.pdb). The plug domain with the relative R66I mutation is highlighted in the dashed zoomed square. h Naïve HEK293 cells or reconstituted with Sec61α-WT or its mutant Sec61α-R66I were transiently transfected with SPn-PD-L1-HA and treated with increasing concentration of BL458 compound for 16 hours. PD-L1-HA protein level was analyzed by SDS-PAGE. i The histogram reports the quantification of PD-L1-HA protein level upon BL458 titration. IC50 threshold is shown as a dashed line. Data are presented as mean values +/− SD. N = 3 independent experiments.
Next, we asked how long-lasting the effect of BL458 was before PD-L1 expression was fully recovered. Therefore, stably SPnPD-L1-HA-expressing GB138 cells were treated with BL458 for 16 h and, after repeated washes, were incubated for an additional time of up to 48 h (wash-out or WO) in the absence of BL458. As shown in Fig. 3d, treatment with BL458 reduced PD-L1 levels by > 90% while, after 10 h of WO, we recorded only about a 10% recovery of PD-L1 protein expression, and we registered an almost total rescue of PD-L1 expression level only after 48 hours. This result could suggest that BL458 may persist inside the Sec61 channel, thereby exerting its inhibitory effect in a prolonged manner. Therefore, we conducted a pulse and chase experiment to test this hypothesis. Specifically, SPnPD-L1-HA cells were treated for 1 h (pulse) with BL458, subjected to 3 washes with fresh medium, and incubated for a further 9 h (chase) in the absence of BL458. By western blotting, PD-L1 protein levels were compared with those of parallel cells treated with BL458 for the entire 10 h. As shown in Fig. 3e, 1 out of 10 h with BL458 reduced PD-L1 protein expression similar to that recorded after continuous 10 h treatment. However, when analyzed after only 1 h of treatment, as well as up to 4 h, PD-L1 protein levels remained unchanged and started to decrease significantly only after 6 h of treatment (Fig. 3f). These results confirmed that the inhibition of translocation leads to the degradation of the newly synthesized protein precursor and the reduction of the total expression level of a specific investigated protein remains linked to the half-life of the already synthesized mature protein. Moreover, these data suggest that BL458 could reside in the Sec61 channel, where it repetitively inhibits the translocation of multiple PD-L1 precursors, thereby exerting a prolonged inhibitory effect. Finally, to verify that the selected BL458 molecule interfered with PD-L1 translocation by targeting the Sec61 channel, we tested BL458 on cells stably expressing exogenous wild-type (WT) Sec61α or a plug domain mutant (R66I) (Fig. 3g) that was previously shown to confer resistance to CT8 and a cotransin natural product7,34. As shown in Fig. 3h, BL458 potently reduced PD-L1-HA expression in HEK293 cells or reconstituted with Sec61α-WT (IC50 ≈ 250 nM), whereas this effect was reduced in cells expressing Sec61α-R66I mutant (IC50 ≈ 2 μM) (Fig. 3i), thereby suggesting that BL458 could act on Sec61α to interfere with PD-L1 translocation and to cause its degradation.
CADA analog hampers PD-L1 constitutive and inducible levels
Tumor-infiltrating immune cells foster an immunosuppressive tumor microenvironment (TME), including upregulating PD-L1 expression35. Among them, the heterogeneous population of myeloid cells in TME, such as tumor-associated macrophages (TAMs), plays a crucial role in promoting critical processes associated with immunosuppression, among which PD-L1 expression induction22,36. Therefore, we asked whether BL458 could hamper the PD-L1 endogenous up-regulation in the GB138 glioblastoma cell line. To this end, we co-cultivated PBMCs-derived macrophages with GB138 cells in the presence or absence of BL458 for 24 h. GB138 were identified as CD45-/PDL1high while macrophages as CD45+/PDL1low as shown in Fig. 4a. BL458 treatment significantly reduced the percentage of cells (15.13 ± 1.29% to 3.2 ± 0.1%) and the relative expression levels of PD-L1 (1 to 0,57 ± 0.02) on the cell surface in not co-cultured cells (Fig. 4b, c). Strikingly, upon co-culture conditions6, the percentage of PD-L1+ cells increased from 15.13 ± 1.29% to 95.23 ± 0.38%, and the PD-L1 expression levels detected on the cell surface increased up to 5 folds (1 to 4.84 ± 0.16). The treatment with BL458 hampered PD-L1 up-regulation by significantly reducing either the percentage of PD-L1+-cells (95.23 ± 0.38% to 45.63 ± 0.12%) and its endogenous expression level on the cell surface (4.84 ± 0.16 to 0.85 ± 0.05) (Fig. 4b, c). Figure 4c also shows the impact of BL458 in cell lines expressing low/absent levels of the glycoprotein (SF767 glioblastoma cell line and HeLa cervical carcinoma cell line)37. Indeed, the three cancer cell lines exploit various levels of PD-L1 expression, from GB138 with the highest percentage of expression (15.13 ± 1.29%), SF767 with levels more than halved compared to GB318 (2.87 ± 0.61%) and HeLa, which shows deficient PD-L1 levels (0.47 ± 0.21%). PD-L1 mean fluorescence intensity (MFI) was in line with the % of expression (Fig. 4c, left). Treatment with BL458 did not show reduction of the percentage of PD-L1+ cells and MFI on the cell surface in cells with low/absent levels of the protein in not co-cultured cells (Fig. 4c). Upon co-culture, PD-L1 was significantly induced in SF767 (2.87 ± 0.61% to 21.2 ± 1.51%), but not in HeLa cells where the PD-L1 detected on the cell surface did not reach a consistent increase, remaining at very low percentages (3.83 ± 1.17%). In SF767 cells, BL458 counteracted the percentage of PD-L1+-cells (21.2 ± 1.51% to 14.47 ± 1.16%) but not its endogenous expression level on the cell surface (Fig. 4c). Due to the insufficient induction of PD-L1 levels in HeLa cells, the use of the molecule after co-culture does not demonstrate any effect on reducing PD-L1 (Fig. 4c). Representative dot plots of SF767 and HeLa cells are shown in Supplementary Fig. S7a. To further assess the efficacy of the BL458 molecule on PD-L1, we used PBMCs isolated from healthy donors. As shown in Fig. 4d, the BL458 effectively counteracted the percentage of PD-L1+ cells (29.13 ± 1.95% to 14.55 ± 3.99%) or its endogenous expression level on the cell surface of monocytes, an immune population where the protein is highly expressed, while in lymphocytes the levels remained low (Fig. 4f). Upon TCR triggering (CD3/CD28), PD-L1 levels increased in both mono and lymphocytes and were effectively counteracted by BL458, thus strengthening the effectiveness of the molecule even in a more physiological system (Fig. 4d–g). Interestingly, the immunosuppressive population of T regulatory lymphocytes (Tregs: CD4+CD25highCD127dim), whose expansion and suppressive ability is dependent on the PD-L138 was significantly reduced following BL458 stimulation (Supplementary Fig. S7b). Finally, to test the selectivity of the BL458 molecule on PD-L1, we conducted an immunophenotype analysis of PBMCs by flow cytometry. Within the lymphocyte population, other cell surface antigens such as CD3, CD4, CD19, and CD16 did not show any change in their expression after stimulation with the molecule (Supplementary Fig. S7c). However, although not as critical as the reduction of PD-L1 levels, BL458 appeared to influence the expression of CD8; this finding is in line with previous findings demonstrating that PD-L1 is expressed on activated CD8+ T cells to promote survival and their effector function39. Together, these results confirm that BL458 interferes with PD-L1 biogenesis under either constitutive or induced conditions.

a GB138 cells were co-cultured with PBMCs-derived macrophages in DMEM medium for 24 h, treated with either BL458 (20 µM) or DMSO as a control (Mock). Flow cytometry was employed for GB138 and macrophage discrimination based on CD45 and PD-L1 expression. GB138 are identified as CD45-/PDL1high, while macrophages as CD45+/PDL1low. b Flow cytometric histograms of PD-L1 expression in GB138 gated cells not co-cultured (NoCC, left) or co-cultured with macrophages (CC, right) and treated with BL458 (20 µM) or DMSO (Mock). Histograms, shown in overlay, indicate a significant reduction of PD-L1 expression upon BL458 (20 µM) treatment.c Graphical representation of mean values (columns) and standard deviations (bars) obtained from three independent experiments for PD-L1 expression (% of positive cells, left) and PD-L1 mean fluorescence intensities (MFI, represented as fold change, right) assayed by flow cytometry in two glioblastoma cell lines GB138 and SF-767, and cervical carcinoma HeLa cell line. N = 3 independent experiments. Graphical representation of mean values (columns) and standard deviations (bars) obtained from four independent experiments for PD-L1 expression (left) and PD-L1 mean fluorescence intensities (MFI, right) in monocytes (d) and lymphocytes (f) upon activation of PBMCs with Dynabeads Human T-Activator CD3/CD28 for 16 h or no activation and followed by treatment with BL458 (20 µM) or DMSO (Mock control) for 24 h. Representative flow cytometric dot plots of PD-L1 expression in monocytes (e) and lymphocytes (g). Data are presented as mean values +/− SD. N = 4 independent experiments.
Discussion
Assays based on the luciferase reporter gene have long been used for drug discovery due to their high sensitivity and robust signal40,41,42,43,44. Here, we report the development of a highly sensitive cell-based luciferase method suitable for identifying Sec61 inhibitors in a single round of screening in a 96-well plate format. Inhibiting protein translocation into the ER interferes with the biogenesis of newly synthesized transmembrane or secreted proteins, thereby causing the reduction of protein expression and secretion, respectively24. Therefore, currently developed methods for identifying Sec61 inhibitors are usually based on reporter proteins whose folding and activity are compatible with access and trafficking over the secretory pathway, such as green fluorescent protein (GFP and its variants) and Gaussia Luciferase (GLuc)13,17. As such, screening small molecule libraries potentially containing Sec61 inhibitors usually reduces reporter protein signals. However, the secretory pathway is highly druggable, and many libraries contain small molecules capable of interfering with such a pathway at different levels3,45,46,47, thereby generating similar readouts and the selection of false-positive candidates. Therefore, selected molecules need to be subjected to multiple rounds of screening, which renders the entire screening procedure inevitably more expensive and time-consuming. Our method is sensitive, fast (just 6 h of treatment), and more selective since not responding to molecules known to interfere with the secretory pathway (Brefeldin A, BFA), ER homeostasis (Thapsigargin, TG or Hydroxychloroquine, HCQ) and protein synthesis (Cycloheximide, CHX or Puromycin, PURO) (Supplementary Fig. S8b). All these molecules could be selected as false-positive in pharmacological screening using secreted reporter protein such as Gaussia Luciferase (GLuc) (Supplementary Fig. S8a). Moreover, we provide evidence that inhibition of steps upstream the Sec61 inhibition, like molecules capable of interfering with SRP pathway, are not selected as false-positive candidates. Indeed, treatment with a known SRP inhibitor TAS-10348 caused the reduction of both FLuc and RLuc signals (Supplementary Fig. S1e–h) as expected in the case of Regulation of Aberrant Protein Production (RAPP) pathway activation, which senses the defects of SPs recognition by SRP, and specifically targets secretory and membrane protein mRNAs for degradation49. On the other hands, small molecules able to interfere with N-glycosylation, such as tunicamycin, can be selected as false-positive by our RELITE assay (Supplementary Figs. S1b and S7b). However, cell viability assay (Supplementary Fig. S5c) showed that tunicamycin, as well as molecules able to interfere with general cellular processes, including the CADA analog BL466, would be soon excluded by further investigations because of highly toxic for the cells. Moreover, current assays are usually characterized by longer incubation time for treatment, such as 24 or 48 h13,17, which could cause the activation of compensatory cellular mechanisms that can potentially give rise to resistance phenomena or even potentiate them, provoking unexpected effects capable of generating similar readouts confused as primary effects. Our RELITE assay is a method based on the luminescence recovery upon ER translocation interference measurable in a very short time of treatment (within only 6 hours). The success of the RELITE assay is tightly dependent on the fast-folding kinetics of the firefly luciferase coupled with the high protein synthesis rate reached under transient over-expression conditions, which generates a cytosolic accumulation of the reporter protein for a sufficient time to be measured before its removal by the proteasome degradation pathway. Moreover, the RELITE assay is sensitive enough not to be limited to the selection of a Sec61 inhibitor but also to discriminate between more or less potent inhibitors based on the amino acidic composition featuring a specific SP driving the translocation process. This property has let us identify, among a library of 100 CADA analogs, the ones capable of interfering with translocation driven by the PD-L1 SP as well as the key amino acidic residues relevant for such an effect. This result further underlines the importance of the chemical modifications for the generation of new libraries starting from already known Sec61 inhibitors and the possibility of quickly screening them for selecting specific protein expression inhibitors. Indeed, inhibiting the Sec61 channel has broad therapeutic potential, including viral infections, autoimmune disorders, and cancer4,6,44. Although most of the identified Sec61 inhibitors exert a non-selective inhibitory effect that is potentially toxic for the cells, identifying selective Sec61 inhibitors has raised the intriguing possibility of targeting specific disease-relevant proteins with minimal toxic cellular effects. How do these molecules selectively exert their inhibitory effect? This is still an open question, and our work does not clarify it at the molecular level. Our data confirm that the translocation inhibitory activity of the selected molecules is still dependent on the presence of polar residues within the SP sequence, suggesting a possible contribution of these amino acids for direct binding between the molecules and the SP inside the Sec61 channel13. However, a proteomic study has shown that CADA is capable of inhibiting, although to a lesser extent, the translocation of other Sec61 clients bearing SPs containing different amino acidic sequences without any conservation of the abovementioned polar residues50. Therefore, such specificity of action could also depend on the role of SP in the Sec61 channel opening process. As previously shown51, SPs supplant helix 2 of Sec61α and replace its interactions with helices 7 and 8 to stabilize the open channel conformation, thereby causing the opening of the central pore and the lateral gate of the channel. Therefore, the conserved hydrophobicity and length of helix 2 suggest that the biophysical properties of helix 2 may dictate what constitutes a functional SP, a property that is similar but not identical across species because of the uniqueness of the amino acidic composition of the SPs. Interestingly, CryoEM technology showed that several Sec61 inhibitors, including the CADA compound, interfere with the protein translocation process commonly by keeping both lateral and luminal gates of Sec61 in translocation-incompetent conformations14,17. Thus, the selectivity of some inhibitors could depend on the ability of only some SPs to displace a particular inhibitory molecule from the channel, thereby opening it and driving a proper polypeptide translocation14,17. However, our pulse-chase experiment (Fig. 3e) suggests that such molecules could exert a prolonged inhibitory activity. As such, not-targeted SPs could only partially displace the inhibitory molecule, which could remain in the channel or, alternatively, thanks to their hydrophobic nature, immersed into the lipid bilayer and recycled for the same or another Sec61 channel. The displacement property could rely on both the hydrophobicity and secondary conformation of the SP once inserted into the channel. Accordingly, the amino acidic substitution of the three polar residues of the PD-L1 SP with hydrophobic ones significantly reduced the inhibitory effect of our selected compounds BL458, BL445 and BL390. Among them, our data suggest that the BL458 molecule is most effective to down-modulate PD-L1 expression under endogenous, constitutive, induced, and overexpression conditions. Moreover, proteome profile data showed that the BL458 molecule has a very low impact on the cell proteome with a < 3% variation under higher concentration (20 µM). In addition, crossing the results got with the three different concentrations used (0.2, 2, and 20 µM) only six putative proteins (PD-L1 included) seemed to be targeted by BL458 molecule (Supplementary Fig. S6b). However, further investigation and validation tests will be necessary to access the selectivity of the BL458 molecule and which further chemical modifications will be necessary to introduce to further improve it.
In conclusion, our work has provided a fast and reliable tool for screening small-molecule Sec61 inhibitors, highlighting the potency of chemical modification for generating new libraries to be tested against other SPs of medicinal interest.
Methods
Ethics
PBMCs derive from buffy coats from healthy donors who donate to the transfusion center of our University Federico II. All methods were performed in accordance with the relevant guidelines and regulations. The Federico II University Ethics Committee approved research experiments on human blood (protocol n. 12/11/ES19) that were conducted in accordance with the ethical principles of the Declaration of Helsinki. Informed written consent was obtained from all blood donor providers of PBMCs.
Reagents
The solid chemical and liquid reagents were obtained from E. Merck (Darmstadt, Germany), Farmitalia Carlo Erba (Milan, Italy), Serva Feinbiochemica (Heidelberg, Germany), Delchimica (Naples, Italy) and BDH (Poole, United Kingdom). The enhanced chemiluminescence (ECL) (#170-5061; BIO-RAD). Dual-Glo Luciferase system (E1910; Promega). MG132 5 µM (474790; Sigma-Aldrich), Tunicamycin 10 µg/ml (T7765, Sigma-Aldrich), Thapsigargin 3 µM (586005, CALBIOCHEM), Cycloheximide 100 μg/ml (C7698, Sigma-Aldrich), Puromycin 10 µg/ml (P4512, Sigma-Aldrich), Brefeldin A 5 µg/ml (B7651, Merck), Hydroxychloroquine 100 µg/ml (H0915, Sigma-Aldrich).
Mycolactone was kindly provided by Carolina Demangel Lab52, dissolved in DMSO at 200 µM and stored at − 20 °C (working concentration 200 nM). Apratoxin A was kindly provided by Ville Paavilainen Lab9 dissolved in DMSO at 10 mM and stored at − 20 °C (working concentration 10 µM). Eeyarestatin 1 (MedChem, HY-110078) was dissolved in DMSO at 10 mM and stored at − 20 °C (working concentration 10 µM). TAS-103 (MedChem, HY-13758A) was dissolved in DMSO at 10 mM and stored at − 20 °C (working concentration 10 µM).
Cell culture and transfection
GB138 cells, established in 2011 from a resected adult GBM sample, were kindly provided by Bernard Rogister30. Human glioma cell lines SF76737 and the cervical carcinoma HeLa were provided by CEINGE cell bank (Cellular Technology Platform, https://www.ceinge.unina.it/en/cell-cultures) at the Advanced Biotechnology Institute (Naples, Italy). They were routinely grown at 37 °C in Dulbecco’s modified essential medium (DMEM; Sigma-Aldrich), containing 10% fetal bovine serum (FBS; Sigma-Aldrich S), 100 µ/ml Penicillin/Streptomycin, 2 mM L-Glutamine (L-Gln; Sigma-Aldrich).
GB138 cells stably expressing the SPn- and SPm-PD-L1-HA protein were grown in the same medium, supplemented with 400 μg/ml G418 (Gibco). HEK293 cells were pursued from ATCC biobank (ATCC-CRL-1573). HEK293 cells stably expressing Sec61a-WT and Sec61a-R66I were kindly provided by Ville Paavilainen Lab17. They were routinely grown at 37 °C in Dulbecco’s modified essential medium (DMEM; Sigma-Aldrich), containing 10% fetal bovine serum (FBS; Sigma-Aldrich S), 100 µ/ml Penicillin/Streptomycin, 2 mM L-Glutamine (L-Gln; Sigma-Aldrich) and Doxycycline (Sigma-Aldrich, D9891) at a concentration of 1–5 μg ml−1 for at least 48 hours to induce the expression of Sec61a proteins. Cells were transfected by using an X-tremeGENE HP DNA transfection reagent (REF 06366236001, Roche) according to the manufacturer’s instructions.
Antibodies
All the antibody information, including supplier, catalog and lot number, and working dilution, is shown in Supplementary Table 1.
Constructs, cDNA cloning and plasmid construction
PD-L1-turboGFP (Origene) was used as a template for generating PD-L1-HA constructs. SPn-PD-L1-HA was generated by PCR using the following couple of oligos: Fw (EcoRI): 5’-CGGAATTCCCACCATGAGGATATTTGCTGTCTTTATATTC-3’; Rev (XbaI): 5’-GCTCTAGATTAAGCGTAATCTGGAACATCGTATGGGTATCCTCCTCCCCGTCTCCTCCAAATGTGTATCACTTTGC-3’. SPm-PD-L1-HA was generated by PCR using the following couple of oligos: Fw (EcoRI): 5’-CGGAATTCATGAGGATATTTGCTGTCTTTATATTCATGACCCTGTGGCTGTTGCTGAACGCATTTACTGTCACGGTTCCCAAGG-3’; Rev (XbaI): 5’-GCTCTAGATTAAGCGTAATCTGGAACATCGTATGGGTATCCTCCTCCCCGTCTCCTCCAAATGTGTATCACTTTGC-3’. SP and HA tags were included into the oligos sequences.
The plasmid FP5258 encoding for SPn-Luciferase-IRES-Renilla was generated as following: the sequence corresponding to IRES-Renilla was extracted from a synthetic gene (IDT Integrated DNA Technologies) and cloned into a pIRES-puro3 plasmid (Clonetech) using the PmlI and SbfI restriction sites. Once the intermediate plasmid was obtained, the sequence corresponding to SPn-Luciferase was extracted from a synthetic gene (IDT Integrated DNA Technologies) using the AgeI and NotI restriction sites. The plasmid FP5259 encoding for SP-hCD4-Luciferase-IRES-Renilla was generated as following: the sequence corresponding to SP-hCD4 was extracted from a synthetic gene (IDT Integrated DNA Technologies) and cloned into FP5258 using the NheI and SbfI restriction sites. The plasmid FP5585 encoding for SPn-PDL1(aa19-38)-HA-Luciferase-IRES-Renilla was generated as following: the sequence corresponding to SPn-PDL1(aa19-38)-HA was extracted from a synthetic gene (IDT Integrated DNA Technologies) and cloned into FP5258 using the AgeI and SbfI restriction sites. The plasmid FP5586 encoding for SP-hCD4-CD4(aa26-32)-HA-Luciferase-IRES-Renilla was generated as following: the sequence corresponding to SP-hCD4-CD4(aa26-32)-HA was extracted from a synthetic gene (IDT Integrated DNA Technologies) and cloned into FP5258 using the AgeI and SbfI restriction sites. The plasmid Ubiquitin-FLAG (Ub-FLAG) belongs to the lab stock plasmid collection.
RELITE assay for screening CADA analogs library
5 × 105 of GB138 cells were seeded in a 60 mm dish. The day after, cells were transfected with 5 μg of bi-cistronic IRES-vectors coding for the SP-equipped Firefly and the cytosolic Renilla Luciferase by using X-TremeGENE HP DNA Transfection Reagent (REF 06366236001, Roche) and following manufacturer instructions. After 4 h, cells were trypsinized and seeded in 96-well plates (Merck) at 104 cells/well. Cells were incubated overnight before being treated with small molecules at 20 μM final concentration. The small molecules library of CADA analogs has been generated in Thomas Bell’s lab, it meets the community requirements, and contains macrocyclic triamines consisting of 12-13 C and N atoms, bearing arenesulfonamide sidearms and hydrophobic tail groups with structural variations53,54,55,56,57,58,59. All analogs are dissolved in DMSO buffer and stored at room temperature for a short time (months) or freeze at − 20 °C for a longer time (years). After 6 h of incubation with molecules, cells were rinsed 3 times with ice-cold PBS and lysed by 30 min incubation with ice-cold Passive Lysis Buffer (REF E1910, Promega), 50 μL/well. 10 μL of lysate were mixed with 45 μL of Dual-Glo LAR-II Reagent (REF E1910, Promega) and 45 μL of Dual-Glo Stop & Glo Reagent (REF E1910, Promega), according to manufacturer instructions. Luminescence was measured by Biotek Synergy H1 microplate reader by using a white opaque 96-well plate (ThermoFisher).
The quality of this high-throughput screening assay was assessed by means of a Z-factor, calculated with the Eq. (1) below:
Where σs and σc are the standard deviation of sample and control, respectively, and μs and μc are the mean of sample and control, respectively.
Preparation of cell extracts, immunoprecipitation SDS-PAGE and western blotting
Preparation of cell extracts, SDS-PAGE and Western blot analysis were performed as previously detailed60,61. Briefly, total cell extracts were performed using a B-Buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol and 1% Triton-X-100) supplemented with protease inhibitors. The Bradford assay estimated protein concentration. Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were then treated with a blocking buffer (25 mM Tris, pH 7.4, 200 mM NaCl, 0.5%) containing 5% non-fat powdered milk and incubated overnight with primary antibodies. Membranes were finally incubated with an HRP-conjugated secondary antibody, and chemiluminescence was determined using the ECL detection system. Densitometric analysis was performed using the Fiji Image software (Bethesda, MD, USA).
Endoglycosidases digestion
For the deglycosylation digestion, 10 µg of protein extracts were treated according to the manufacturer’s instructions and incubated for 1 h at 37 °C with 500 U of Endo H (New England BioLabs).
LC-MS/MS and data Analysis
Instruments for LC-MS/MS analysis consisted of a NanoLC 1200 coupled via a nano-electrospray ionization source to the quadrupole-based Q Exactive HF benchtop mass spectrometer62. For the chromatographic separation, a binary buffer system consisting of solution A: 0.1% formic acid and B: 80% acetonitrile, 0.1% formic acid was used. The peptides were separated according to their hydrophobicity on an analytical column (75 μm) in-house packed with C18-AQ 1.9 μm C18 resin with a gradient of 7–32% solvent B in 45 min, 32–45% B in 5 min, 45–95% B in 3 min, 95–5% B in 5 min at a flow rate of 300 nl/min. MS data acquisition was performed in DIA (Data Independent Acquisition) mode using 32 variable windows covering a mass range of 300–1650 m/z. The resolution was set to 60’000 for MS1 and 30’000 for MS2. The AGC was 3e6 in both MS1 and MS2, with a maximum injection time of 60 ms in MS1 and 54 ms in MS2. NCE were set to 25%, 27.5%, 30%.
All acquired raw files were processed using Spectronaut software (v. 19.5). For protein assignment, spectra were correlated with Uniprot Human database (UP000005640). Searches were performed with tryptic specifications and default settings for mass tolerances for both MS and MS/MS spectra. The other parameters were set as follows:
The Perseus software (1.6.2.3) was used to logarithmize, group and filter [after filter] the protein abundance. ANOVA and two-sample t-test analysis was performed using FDR = 0.05. Proteins with a difference Log2 Difference > 1 and -log10 p-value > 1.3 were considered significantly enriched (see “Significantly deregulated” in the Excel file).
Cell fractionation
Cells were treated for 10 h with BL458 and the proteasome inhibitor MG132 (5 μM). The cell homogenate was obtained using the homogenate solution (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol) by mechanical lysis with a syringe (ten passages). Homogenates were centrifuged at 2000 r.p.m. for 5 minutes to remove the nuclear fraction. Post-nuclear fractions were ultracentrifuged at 45,000 rpm (100000 G) for 2 h. The soluble fraction (S100) represented by the supernatant was withdrawn, whereas the membrane fraction (P100) was solubilised in B-Buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol and 1% Triton-X-100) in ice for 30 min, followed by centrifugation to remove the insoluble Triton-X-100 fraction. Equal amounts of both S100 and P100 fractions were analyzed by SDS-PAGE.
Cell viability assay
Cell viability was determined by using the CellTiter-Glo® Luminescent Cell Viability Assay. Briefly, GB138 cells were grown in a 96-well plate and incubated for 24 or 48 h with increasing concentrations of BL458, BL445 or BL390. Then, the CellTiter-Glo reagent was added to the culture medium and incubated at room temperature for 2 min with shaking to induce cell lysis. After that, luminescence signals were recorded with a Luminometer Microplate Reader.
PBMC isolation, Co-cultures, and FACS analysis
Peripheral blood mononuclear cells (PBMCs) were separated by differential centrifugation using a Ficoll-Hypaque density gradient (Histopaque-1077; Sigma-Aldrich, St. Louis, MO, USA), washed, and resuspended in 5% fetal bovine serum (FBS)/Roswell Park Memorial Institute (RPMI) 1640 medium (Corning, Glendale, Arizona, USA). PBMCs (5 × 105cells/mL) were then seeded into a six-well plate and differentiated into macrophages for 6 days with M-CSF (Immunotools, Friesoythe, Germany) used at a final concentration of 50 ng/mL63. Next, cancer cells (2.5 × 105 cells/mL) were seeded with PBMCs-derived macrophages and co-cultured in DMEM medium for 24 h with BL458 (20 µM) or DMSO as a control (Mock). PBMCs-derived macrophages and cancer cells were maintained in monocultures as controls in each experiment. For PBMC activation, cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (4.5 μm, ThermoFisher Scientific, Massachussetts, USA) at 37 °C in 5% CO2 at a cell concentration of 1.0 × 106 cells/mL at a bead-to-cell ratio = 1:10. Following 16 h of activation, both non-activated PBMCs and activated PBMCs were treated with BL458 (20 µM) or DMSO (Mock control) for a further 24 h. Co-cultured cancer cells and PBMCs-derived macrophages were harvested by trypsinisation and distinguished by CD45 antigen expression by using anti-human CD45-APC (2D1 clone, Invitrogen, Waltham, MA, USA) or anti-human CD45-BV786 (HI30 clone, BD Biosciences, New Jersey, USA) by flow cytometry. Activated PBMCs were identified among lymphocytes and monocytes using a forward versus side scatter (FSC vs. SSC) gating strategy combined with CD45 antigen expression (anti-human CD45-BV786, HI30 clone, BD Biosciences). PD-L1 expression was assessed using anti-human CD274-PE (MIH1 clone; eBioscience, Thermo Fisher Scientific, Massachussetts, USA) or anti-human CD274-APC-R700 (MIH1 clone, BD Biosciences). All antibodies were used at a concentration of 0.05 μg/ml and incubated for 15 min in the dark at room temperature (20–25 °C). Isotype-conjugated control antibodies were used to assess nonspecific binding. Samples were acquired using a BD AccuriTM C6 Cytometer (Becton, Dickinson and Company BD; Bergen County, NJ, USA) or a three lasers BD FACSCelestaTM Cell Analyzer (BD Biosciences). Data were analyzed using the C6 Accuri software or the BD FACSDiva software (BD Biosciences).
Quantification and statistical analysis
All the results are given as mean ± s.d. obtained from independent experiments. Statistical analysis was performed by Student’s t test and one-way ANOVA. P values are shown as asterisks: **** for P-value < 0.0001, *** for P-value < 0.001, ** for P-value < 0.01, * for P-value < 0.05 and ns for data not statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data is provided with this paper as a Source Data file. Materials, including CADA analogs are available upon request. Full scan blots are provided in the Source Data file. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD063288 and are available as Supplementary Data 1. Source data are provided in this paper.
References
Barlowe, C. K. & Miller, E. A. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193, 383–410 (2013).
Yim, C. et al. Profiling of signal sequence characteristics and requirement of different translocation components. Biochim. Biophys. Acta Mol. Cell Res. 1865, 1640–1648 (2018).
Luesch, H. & Paavilainen, V. O. Natural products as modulators of eukaryotic protein secretion. Nat. Prod. Rep. 37, 717–736 (2020).
Lumangtad, L. A. & Bell, T. W. The signal peptide as a new target for drug design. Bioorg. Med. Chem. Lett. 30, 127115 (2020).
Harant, H. et al. Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol. Pharm. 71, 1657–1665 (2007).
Van Puyenbroeck, V. & Vermeire, K. Inhibitors of protein translocation across membranes of the secretory pathway: novel antimicrobial and anticancer agents. Cell Mol. Life Sci. 75, 1541–1558 (2018).
Junne, T. et al. Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J. Cell Sci. 128, 1217–1229 (2015).
McKenna, M., Simmonds, R. E. & High, S. Mechanistic insights into the inhibition of Sec61-dependent co- and post-translational translocation by mycolactone. J. Cell Sci. 129, 1404–1415 (2016).
Paatero, A. O. et al. Apratoxin kills cells by direct blockade of the sec61 protein translocation channel. Cell Chem. Biol. 23, 561–566 (2016).
Tranter, D. et al. Coibamide A targets sec61 to prevent biogenesis of secretory and membrane proteins. ACS Chem. Biol. 15, 2125–2136 (2020).
Zong, G. et al. Ipomoeassin F binds sec61alpha to inhibit protein translocation. J. Am. Chem. Soc. 141, 8450–8461 (2019).
Gutierrez Guarnizo, S. A. et al. Pathogenic signal peptide variants in the human genome. NAR Genom. Bioinform. 5, lqad093 (2023).
Vermeire, K. et al. Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol. 12, e1002011 (2014).
Itskanov, S. et al. A common mechanism of Sec61 translocon inhibition by small molecules. Nat. Chem. Biol. 19, 1063–1071 (2023).
Bennett, D. C. et al. High-throughput screening identifies aclacinomycin as a radiosensitizer of EGFR-mutant non-small cell lung cancer. Transl. Oncol. 6, 382–391 (2013).
Klein, W. et al. Use of a sequential high throughput screening assay to identify novel inhibitors of the eukaryotic SRP-Sec61 targeting/translocation pathway. PLoS ONE 13, e0208641 (2018).
Rehan, S. et al. Signal peptide mimicry primes Sec61 for client-selective inhibition. Nat. Chem. Biol. 19, 1054–1062 (2023).
Wenzell, N. A. et al. Global signal peptide profiling reveals principles of selective Sec61 inhibition. Nat. Chem. Biol. 20, 1154–1163 (2024).
Kok, V. C. Current understanding of the mechanisms underlying immune evasion from PD-1/PD-L1 immune checkpoint blockade in head and neck cancer. Front. Oncol. 10, 268 (2020).
Ghosh, C., Luong, G. & Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 12, 2735–2746 (2021).
Upadhaya, S., Neftelinov, S. T., Hodge, J. & Campbell, J. Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat. Rev. Drug Discov. 21, 482–483 (2022).
Pu, Y. & Ji, Q. Tumor-associated macrophages regulate PD-1/PD-L1 immunosuppression. Front. Immunol. 13, 874589 (2022).
Frydman, J., Erdjument-Bromage, H., Tempst, P. & Hartl, F. U. Co-translational domain folding as the structural basis for the rapid de novo folding of firefly luciferase. Nat. Struct. Biol. 6, 697–705 (1999).
Pauwels, E., Schulein, R. & Vermeire, K. Inhibitors of the sec61 complex and novel high throughput screening strategies to target the protein translocation pathway. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms222112007 (2021).
Pauwels, E. et al. Structural insights into TRAP association with ribosome-Sec61 complex and translocon inhibition by a CADA derivative. Sci. Adv. 9, eadf0797 (2023).
Mishra, P., Rai, S. & Manjithaya, R. A novel dual luciferase based high throughput assay to monitor autophagy in real time in yeast S. cerevisiae. Biochem. Biophys. Rep. 11, 138–146 (2017).
Yoon, D. et al. Structure-based insight on the mechanism of N-glycosylation inhibition by tunicamycin. Mol. Cells 46, 337–344 (2023).
Dibdiakova, K. et al. Both thapsigargin- and tunicamycin-induced endoplasmic reticulum stress increases expression of Hrd1 in IRE1-dependent fashion. Neurol. Res. 41, 177–188 (2019).
Freeze, H. H. & Kranz, C. Endoglycosidase and glycoamidase release of N-linked glycans. Curr. Protoc. Mol. Biol. https://doi.org/10.1002/0471142727.mb1713as89 (2010).
Kroonen, J. et al. Human glioblastoma-initiating cells invade specifically the subventricular zones and olfactory bulbs of mice after striatal injection. Int. J. Cancer 129, 574–585 (2011).
Cross, B. C. et al. Eeyarestatin I inhibits Sec61-mediated protein translocation at the endoplasmic reticulum. J. Cell Sci. 122, 4393–4400 (2009).
Han, Y., Liu, D. & Li, L. PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res. 10, 727–742 (2020).
Zhang, J. H., Chung, T. D. & Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73 (1999).
Mackinnon, A. L., Paavilainen, V. O., Sharma, A., Hegde, R. S. & Taunton, J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife 3, e01483 (2014).
Jiang, X. et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 18, 10 (2019).
Xu, C. et al. Macrophages facilitate tumor cell PD-L1 expression via an IL-1beta-centered loop to attenuate immune checkpoint blockade. MedComm 4, e242 (2023).
D’Arrigo, P. et al. The splicing FK506-binding protein-51 isoform plays a role in glioblastoma resistance through programmed cell death ligand-1 expression regulation. Cell Death Discov. 5, 137 (2019).
Lin, X. et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer 23, 108 (2024).
Pulko, V. et al. B7-h1 expressed by activated CD8 T cells is essential for their survival. J. Immunol. 187, 5606–5614 (2011).
Tang, N. et al. A living cell-based fluorescent reporter for high-throughput screening of anti-tumor drugs. J. Pharm. Anal. 11, 808–814 (2021).
Xie, W. et al. A luciferase reporter gene system for high-throughput screening of gamma-globin gene Activators. Methods Mol. Biol. 1439, 207–226 (2016).
Xie, X. et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 11, 5214 (2020).
Che, P., Cui, L., Kutsch, O., Cui, L. & Li, Q. Validating a firefly luciferase-based high-throughput screening assay for antimalarial drug discovery. Assay. Drug Dev. Technol. 10, 61–68 (2012).
Chen, K. Y. et al. A highly sensitive cell-based luciferase assay for high-throughput automated screening of SARS-CoV-2 nsp5/3CLpro inhibitors. Antiviral Res. 201, 105272 (2022).
Coulet, M. et al. Identification of small molecules affecting the secretion of therapeutic antibodies with the retention using selective hook (RUSH) system. Cells 12, https://doi.org/10.3390/cells12121642 (2023).
Gomez-Navarro, N. et al. Selective inhibition of protein secretion by abrogating receptor-coat interactions during ER export. Proc. Natl. Acad. Sci. USA 119, e2202080119 (2022).
Yamamoto-Hijikata, S., Suga, K., Homareda, H. & Ushimaru, M. Inhibition of the human secretory pathway Ca(2+), Mn(2+)-ATPase1a by 1,3-thiazole derivatives. Biochem. Biophys. Res. Commun. 614, 56–62 (2022).
Yoshida, M., Kabe, Y., Wada, T., Asai, A. & Handa, H. A new mechanism of 6-((2-(dimethylamino)ethyl)amino)−3-hydroxy-7H-indeno(2,1-c)quinolin-7-one dihydrochloride (TAS-103) action discovered by target screening with drug-immobilized affinity beads. Mol. Pharm. 73, 987–994 (2008).
Karamyshev, A. L. et al. Inefficient SRP interaction with a nascent chain triggers a mRNA quality control pathway. Cell 156, 146–157 (2014).
Pauwels, E. et al. A proteomic study on the membrane protein fraction of T cells confirms high substrate selectivity for the ER translocation inhibitor cyclotriazadisulfonamide. Mol. Cell Proteom. 20, 100144 (2021).
Voorhees, R. M., Fernandez, I. S., Scheres, S. H. & Hegde, R. S. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell 157, 1632–1643 (2014).
Hong, H., Demangel, C., Pidot, S. J., Leadlay, P. F. & Stinear, T. Mycolactones: immunosuppressive and cytotoxic polyketides produced by aquatic mycobacteria. Nat. Prod. Rep. 25, 447–454 (2008).
Bell, T. W. et al. Synthesis and structure-activity relationship studies of CD4 down-modulating cyclotriazadisulfonamide (CADA) analogues. J. Med. Chem. 49, 1291–1312 (2006).
Demillo, V. G. et al. Unsymmetrical cyclotriazadisulfonamide (CADA) compounds as human CD4 receptor down-modulating agents. J. Med. Chem. 54, 5712–5721 (2011).
Chawla, R. et al. Tuning side arm electronics in unsymmetrical cyclotriazadisulfonamide (CADA) endoplasmic reticulum (ER) translocation inhibitors to improve their human cluster of differentiation 4 (CD4) receptor down-modulating potencies. J. Med. Chem. 59, 2633–2647 (2016).
Ali, R. et al. Tsuji-trost cyclization of disulfonamides: Synthesis of 12-membered, 11-membered, and pyridine-fused macrocyclic triamines. ACS Omega 4, 1254–1264 (2019).
Lumangtad, L. A. et al. Syntheses and anti-HIV and human cluster of differentiation 4 (CD4) down-modulating potencies of pyridine-fused cyclotriazadisulfonamide (CADA) compounds. Bioorg. Med. Chem. 28, 115816 (2020).
Berger, K. et al. Reduction of progranulin-induced breast cancer stem cell propagation by sortilin-targeting cyclotriazadisulfonamide (CADA) compounds. J. Med. Chem. 64, 12865–12876 (2021).
Xu, X. et al. Role of Sec61alpha2 translocon in insulin biosynthesis. Diabetes 73, 2034–2044 (2024).
D’Agostino, M. et al. ER reorganization is remarkably induced in COS-7 cells accumulating transmembrane protein receptors not competent for export from the endoplasmic reticulum. J. Membr. Biol. 247, 1149–1159 (2014).
Scerra, G. et al. Early onset effects of single substrate accumulation recapitulate major features of LSD in patient-derived lysosomes. iScience 24, 102707 (2021).
Michalski, A. et al. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell Proteom. 10, M111 011015 (2011).
Yu, T. et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat. Commun. 10, 4353 (2019).
Acknowledgements
We thank Professor Maria Antonietta De Matteis (Tigem, Pozzuoli, IT), Stefano Bonatti, and Tommaso Russo (DMMBM, UNINA, IT) for sharing reagents, providing scientific support, and helping with discussions. We thank Dr. Marialuisa Alessandra Vecchione and the DMMBM flow cytometry facility (facsility.dmmbm@gmail.com). We also thank the TIGEM institute for the Proteomics Facility. This work in the M.D.A. lab has been funded by the Italian Minister for Research and University (PRIN2022, B53D2300249006), the Italian Minister for Research and University (PRIN 20177XJCHX) to M.R., the STAR-Junior Principal Investigator Grants 2018 and the Individual Grant - SIS 2024 (PROJECT NUMBER: 30223) to S.R. The work in F.P. lab has been supported by the Fondation pour la Recherche Médicale (EQU201903007925), by the Agence Nationale de la Recherche (ANR-19-CE13- 0006-03; ANR-20-CE14-0017-02; ANR-19-CE13-0002-03; ANR-11-LABX-0038), and has also received support under the program « Investissements d’Avenir » launched by the French Government and implemented by ANR with the references CelTisPhyBio (11-LBX-0038), ANR-10-IDEX-0001-02 8 PSL. The work of M.C.S. has been supported by grants ANR-11-LABX-0038 and ANR-10-IDEX- 0001-02.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.D.A. Experiment design: G.S., F.V., S.R., M.R., F.P., T.B., and M.D.A. CADA analogs library preparation and Z-factor calculation: A.I. and T.B. Investigation: G.S., F.V., L.M., M.C.S., A.L., R.B., A.D.M., V.C., G.A., P.R., and M.G.C. Funding acquisition: M.D.A. and M.R. Writing original draft: M.D.A. Writing, review, and editing: All the authors read, revised, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
Massimo D’Agostino has an Italian (102024000006772) and European (EP25166272.2) patent pending applications on the luciferase reporter and the RELITE assay for screening Sec61 inhibitors. T.W. Bell and T. Kapri have a U.S. Provisional Application (63/790,874) for PD-L1 down-modulators. The authors declare no additional competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Vitale, F., Scerra, G., Marrone, L. et al. A light-resuming strategy as a screening method for selecting Sec61 inhibitors down-modulating PD-L1 expression. Nat Commun 16, 7243 (2025). https://doi.org/10.1038/s41467-025-62439-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62439-w
