
Abstract
Helicobacter pylori, a globally significant pathogen, plays a central etiological role in diverse gastric pathologies ranging from chronic gastritis and peptic ulcers to gastric adenocarcinoma. Although conventional antibiotics effectively inhibit or kill growing helical H. pylori, metabolically dormant coccoid forms of H. pylori exhibit considerable tolerance, posing a persistent and clinically significant challenge. Here, we report napabucasin (2-acetylfuro-1,4-naphthoquinone) as a redox-cycling antibiotic with potent bactericidal activity against both drug-resistant helical and coccoid forms of H. pylori. Notably, napabucasin does not induce acquired resistance in vitro and demonstrates superior efficacy compared to standard triple therapy in murine infection models. Mechanistic studies reveal that napabucasin acts through 2-oxoglutarate:acceptor oxidoreductase (OOR)-catalyzed futile redox cycling, generating bactericidal levels of reactive oxygen species (ROS). Compared to menaquinone 6, a proposed physiological electron acceptor, napabucasin exhibits enhanced oxidative capacity. Structural, biochemical, and microbiological analyses identify Leu44 and Lys46 within the OorD subunit as critical residues for napabucasin recognition and catalysis. These findings establish OOR-mediated redox cycling as a robust antimicrobial strategy that sustains endogenous ROS production to combat refractory H. pylori infections.
Similar content being viewed by others

Introduction
Helicobacter pylori, a Gram-negative and spiral-shaped bacterium with microaerophilic characteristics, colonizes the human gastric epithelium1,2. With an estimated global infection rate affecting nearly half of the world’s population, H. pylori is a leading etiological agent of chronic gastritis and peptic ulcer disease3. Furthermore, persistent infection is implicated in the progression to severe complications, including gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma, which develop in ~1% of infected individuals4. Consequently, major gastroenterological societies advocate for the eradication of H. pylori in all infected individuals. The standard triple therapy (STT), which combines two antibiotics (typically clarithromycin, amoxicillin, metronidazole, and levofloxacin) with a proton pump inhibitor (PPI), is the most commonly recommended treatment regimen5. However, refractory H. pylori infections, characterized by persistently positive non-serologic test results after one or more treatment attempts6, present a significant clinical challenge. This resistance is primarily driven by increasing antibiotic resistance rates, suboptimal patient adherence to treatment, and the bacterium’s unique adaptive mechanisms. Notably, its ability to morphologically transition to metabolically dormant coccoid forms and to form biofilms on gastric mucosa surfaces is a major contributor to refractory infection7,8,9. Moreover, these forms have been shown to avoid immune responses and to promote carcinogenesis10,11. These factors underscore the urgent need to develop novel anti-H. pylori agents with distinct mechanisms of action, capable of circumventing cross-resistance to existing therapies and effectively combating refractory H. pylori infections.
The generation of endogenous reactive oxygen species (ROS), in particular hydroxyl radicals (OH•), has been suggested as a common killing mechanism of bactericidal antibiotics12,13. This proposal suggests that these antibiotics act through ROS-mediated cell death rather than drug-target interaction. However, the model has been challenged14,15, as certain antibiotics have been shown to exert their killing effect even in the absence of ROS production. Despite this, ROS-generating redox-based agents, clofazimine16 and the quinolinequinone molecule QQ8c17, have emerged as promising candidates for anti-tuberculosis treatment. Given that H. pylori is capable of producing ROS18,19, enhancing the production of endogenous ROS to a bactericidal level may offer a novel and effective antimicrobial strategy for treating H. pylori infections.
Natural products have historically formed the cornerstone of antibiotic discovery20,21. Revisiting natural sources through mining and repositioning represents a viable short-term strategy to replenish the antibiotic pipeline20. Notably, natural products containing 1,4-naphthoquinone scaffolds, which possess redox cycling properties, have been recognized for their diverse biological activities, including antimicrobial, anti-inflammatory, anticancer, and anti-diabetic effects22,23,24,25. Several 1,4-naphthoquinone derivatives, such as menadione26, α-Lapachone27, 2-(hydroxymethyl)anthraquinone28, 2-methoxy-1,4-naphthoquinone29 and synthetic 2-methylnaphtho(2,3-b)furan-4,9-dione30, have been reported to exhibit potent in vitro anti-H. pylori activity. Furthermore, FDA-approved or clinically advanced 1,4-naphthoquinones, including napabucasin (2-acetylfuro-1,4-naphathoquinone)31, atovaquone32, and anthracyclines33, exhibit favorable safety and pharmacokinetic profiles. Thus, 1,4- naphthoquinone scaffolds are well-positioned as promising candidates for the accelerated development of anti-H. pylori drugs.
In this study, we repurpose napabucasin, an anticancer agent currently in phase III clinical trials, as the most potent drug candidate through the exploration of 1,4-naphthoquinone derivatives for antibacterial activity against H. pylori. This work establishes significant anti-H. pylori activity for napabucasin, extending beyond its previously reported anti-biofilm activity against oral Streptococcal species34 and anti-tubercular activity against Mycobacterium tuberculosis35. Our findings reveal that napabucasin effectively kills both drug-resistant helical and coccoid forms of H. pylori with an undetectably low frequency of resistance development, and demonstrates excellent in vivo efficacy in three mouse models of H. pylori infections. Moreover, napabucasin is bioactivated directly by the tricarboxylic acid (TCA) cycle enzyme, 2-oxoglutarate (α-ketoglutarate):acceptor oxidoreductase (OOR), leading to futile redox cycling and sustained generation of bactericidal levels of endogenous ROS.
Results
In vitro antimicrobial activity of napabucasin
The anti-H. pylori potential of 24 1,4-naphthoquinone derivatives was initially evaluated using a broth microdilution assay against four standard and clinical drug-resistant strains. Napabucasin (Fig. 1a) exhibited the most potent activity, with minimal inhibitory concentration (MIC) values ranging from 0.016 to 0.031 μg/mL (Supplementary Table 1). Consequently, we prioritized napabucasin following the screening. In vitro susceptibility testing against H. pylori strain G27 revealed that napabucasin (10 ng/disk) produced an inhibition zone with a diameter of 4.87 ± 0.57 cm, comparable to clarithromycin (4.57 ± 0.49 cm). In contrast, levofloxacin, metronidazole, and amoxicillin did not produce detectable inhibition zones under identical conditions (Fig. 1b). These results indicate superior antibacterial activity of napabucasin against H. pylori G27 compared to these established antibiotics. Further evaluation against a panel of 34 H. pylori strains (including 30 single antibiotic-resistant and multidrug-resistant clinical isolates) confirmed potent activity, with MIC50/90 values of 0.016/0.031 μg/mL (range: 0.008 to 0.063 μg/mL) (Fig. 1c and Supplementary Table 2), indicating a lack of cross-resistance. The antimicrobial spectrum of napabucasin was subsequently determined against a panel of Gram-positive and Gram-negative bacteria (Supplementary Table 3). It showed moderate antibacterial activity against specific bacterial species, including Moraxella catarrhalis, Neisseria gonorrhoeae, Campylobacter jejuni, Streptococcus pneumoniae, and several Staphylococcus and Bacillus species.

a Chemical structure of napabucasin (NPB). b Disc diffusion assay against H. pylori G27. Results from a disk diffusion assay using 10 ng of NPB or clinical antibiotics per disk are shown to evaluate the inhibitory effect on H. pylori G27. LVX, levofloxacin; MTZ, metronidazole; CLR, clarithromycin; AMX, amoxicillin. c Antibacterial activities of NPB and clinical antibiotics against H. pylori standard strains and drug-resistant clinical isolates. d Kill kinetics of H. pylori G27 by NPB. Data are represented as mean ± SEM (n = 3 biological replicates). e Development of resistance to NPB and MTZ in H. pylori G27. f CFU counts in the stomach of H. pylori NSH57-infected mice, euthanatized 48 h after the last treatment with vehicle (0.5% CMC-Na plus 0.2% Tween-80), triple therapy (omeprazole [OPZ] + AMX + CLR [AC]), omeprazole plus 14 mg/kg napabucasin [OPZ + NPB]). Data are represented as mean ± SEM. n = 8 mice/group. If no colonies were detected, the limit of detection (102 CFU/g) was used for calculations. g Alpha-diversity estimate calculated from the sequenced data using Shannon indices. Stool samples were collected two days after the last treatment and sequenced for the 16S rRNA gene. Each point represents the Shannon Index for an individual mouse. The five lines, from bottom to top, are the minimum value, the first quartile, median, third quartile, and maximum values. n = 8 mice/group. h Principal Component Analysis (PCA) of the three groups based on OTU abundance. The number in brackets represents the contributions of principal components to differences among samples. Each dot represents an individual sample. n = 8 mice/group. i CFU counts in the stomach of H. pylori BHKS159-infected mice, euthanatized 48 h after the last treatment with vehicle, triple therapy (OPZ + AC), omeprazole plus 28 mg/kg napabucasin, omeprazole plus 7 mg/kg napabucasin, 28 mg/kg napabucasin alone, and 7 mg/kg napabucasin alone. Data are represented as mean ± SEM. n = 10 mice/group. Statistical significance was calculated using a one-way ANOVA with Tukey’s post-hoc test. Source data are provided as a Source Data file.
Napabucasin exhibited significantly greater and more rapid bactericidal activity than metronidazole, with a reduction of more than three log10 in colony-forming units (CFUs) observed within 8 h at concentrations of 1–4× MIC (Fig. 1d). To assess the potential for resistance development to napabucasin, 1010 CFUs of H. pylori G27 were plated on agar containing 2×, 4×, or 8× MIC of napabucasin. In two independent experiments, no napabucasin-resistant mutants were obtained. Similarly, serial passage in liquid medium containing napabucasin for 15 transfers yielded no putative mutants, whereas metronidazole exhibited a 128-fold increase in MIC (Fig. 1e). These results suggest that napabucasin is likely to minimize resistance development.
In vivo efficacy
Napabucasin was then tested in a mouse model of gastric infection with a mouse-adapted H. pylori strain NSH5736 to assess its therapeutic efficacy in vivo. The combination therapy of napabucasin (14 mg/kg) plus omeprazole significantly reduced the bacterial burden (P < 0.0001) 48 h after the final treatment, and demonstrated superior efficacy in eradicating H. pylori compared to STT (omeprazole plus 14 mg/kg amoxicillin and 7 mg/kg clarithromycin [OPZ + AC]) (Fig. 1f). Given the narrow-spectrum antibacterial activity of napabucasin, we characterized its impact on the gut microbiome. Fecal samples from 8 mice per group were collected 2 days post-treatment and analyzed via 16S rRNA gene sequencing. Operational taxonomic unit (OTU) analysis revealed no significant differences in either diversity metrics or community composition between the vehicle control and napabucasin dual-therapy groups, based on the calculated alpha diversity (Shannon index) and beta diversity (principal component analysis, PCA) indices (Fig. 1g, h). In contrast, the STT group exhibited substantial alterations in microbial structure, evidenced by significant PCA ordination shifts and reduced Shannon diversity (P < 0.0001).
Additionally, we assessed the in vivo efficacy against a mouse-adapted multidrug-resistant BHKS15937, which mimics clinical infections by drug-resistant H. pylori in humans. We found that mice treated with napabucasin both as a monotherapy and dual therapy showed a >1-log reduction in bacterial burden compared with the vehicle group (Fig. 1i). The combination of napabucasin (28 mg/kg) and omeprazole caused a statistically significant (P < 0.0001) reduction in bacterial burden compared to STT. No significant differences were observed between the dual therapy of a lower dose (7 mg/kg) of napabucasin and STT. Importantly, monotherapy with napabucasin (28 mg/kg) demonstrated more potent in vivo bacterial clearance efficacy than STT (Fig. 1i).
Killing by napabucasin is linked to generation of bactericidal levels of ROS
Napabucasin has been shown to induce high levels of ROS for cancer cell death as a substrate of NAD(P)H dehydrogenase 1 (NQO1)38,39. We then determined whether napabucasin boosts ROS production in H. pylori cells. Treatment with napabucasin upregulated intracellular ROS levels in a concentration-dependent manner, whereas treatment with any of the four conventional antibiotics did not significantly affect ROS accumulation (Fig. 2a, b). ROS, including the superoxide anion (O2• −), hydrogen peroxide (H2O2), and the hydroxyl radical (OH•) (Fig. 2c), are formed as natural byproducts of normal oxygen metabolism. We then measured oxygen consumption rate (OCR) of H. pylori cells to assess the respiratory outcome of napabucasin treatment. Treatment of H. pylori with napabucasin demonstrated significant elevation in OCR compared to that with DMSO (Fig. 2d), indicating that napabucasin-induced cellular death is linked to accelerated respiration. In addition, we determined whether napabucasin treatment elevated intracellular levels of OH• and free ferrous iron, which acts as a catalyst in the Fenton reaction to generate OH•. Both of increase was indeed observed (Supplementary Figs. 1a and 2a). As a control, free Fe2+ levels were increased significantly in the fur mutants that induce iron acquisition systems (Supplementary Fig. 2b).

a, b ROS production in H. pylori G27 treated with indicated concentrations of napabucasin (NPB) or antibiotics for 4 h. ROS levels were quantified using a Cell ROS Assay kit (a) and monitored by fluorescent microscopy (b). LVX, levofloxacin; MTZ, metronidazole; CLR, clarithromycin; AMX, amoxicillin. c Schematic representation of reactive oxygen species production and antioxidant defense enzymes in H. pylori. e−, electron, O2• −, superoxide anion; OH•, hydroxyl radical; NO−, nitric oxide; ONOO−, peroxynitrite anion. d Measurement of oxygen consumption rates (OCR) in H. pylori G27 treated with 1× MIC of NPB (0.031 μg/ ml), MTZ (2 μg/ml), AMX (0.063 μg/ml), carbonyl cyanide 3-chlorophenylhydrazone (CCCP; 0.5 μg/ml) or 1% DMSO, using the MitoXpress® Xtra-oxygen probe. Relative OCR was calculated using the slope of the linear portion of fluorescence profiles for each treatment. e Killing efficacy of NPB, MTZ, and AMX under microaerobic and anaerobic conditions. H. pylori G27 cultures were split, diluted, and grown in BHI broth containing 10% FCS treated with 1× MIC of napabucasin, MTZ, and AMX, and incubated either microaerobically in a tri-gas incubator or anaerobically in an anaerobic chamber. Samples were removed, washed, diluted, and plated for colony counts at the indicated time points. f Susceptibility of H. pylori G27 and four mutant strains to NPB and antibiotics. The MICs were determined by broth microdilution assays, as described in Methods. g Viability of H. pylori G27 and four mutant strains treated with the indicated concentrations of NPB. If no colonies were present, calculations were made using the limit of detection (1 × 101 CFU/ml). h ROS were quantified in cells of H. pylori G27 and three mutant strains treated, for 4 h, with 1× MIC of NPB or MTZ. Statistical significance was calculated using a one-way ANOVA with Tukey’s post-hoc test. All data are represented as mean ± SEM (n = 3 biological replicates) except for b, c. Source data are provided as a Source Data file.
The role of ROS in bactericidal antibiotic-mediated killing remains actively debated. While some evidences support their involvement, other studies challenge their necessity or universality14,15,40. We then reasoned whether H. pylori killing by napabucasin depends on ROS. To examine this relationship directly, we assessed the efficacy of napabucasin under anaerobic conditions, where the absence of O₂ prevents ROS formation. As shown in Fig. 2e, killing of H. pylori cells was not observed when an anaerobic growth condition was applied during napabucasin treatment, indicating an O2/ROS-dependent killing. In contrast, metronidazole exhibited enhanced killing under anaerobic conditions compared to microaerobic conditions (Fig. 2e). This is consistent with the requirement of anaerobic conditions for the metronidazole-reductase activity of RdxA, which is essential for metronidazole activation in H. pylori41,42. To further validate the detrimental role of ROS in the action of napabucasin, we examined the impact of ROS scavenging on the bactericidal effect of napabucasin. As expected, the addition of ROS quenchers, thiourea and N-acetyl-L-cysteine (4 mM and 10 mM), to growing culture of H. pylori together with napabucasin significantly increased the survival rate of H. pylori (Supplementary Fig. 3). This protective effect further confirms the contribution of ROS to napabucasin-induced bacterial killing.
The observation that killing by napabucasin correlates with generation of bactericidal levels of ROS led us to hypothesize that disabling the major antioxidant defense systems would potentiate the potency of napabucasin. Upon napabucasin treatment, we would expect to see stimulation of oxidative stress defense systems. Indeed, transcriptome analysis revealed that napabucasin broadly activated genes encoding the antioxidant defense enzymes, including superoxide dismutase (SodB), catalase (CatA), alkyl hydroperoxide reductase (AhpC), and the neutrophil-activating protein (NapA) (Supplementary Fig. 4). We then sought to determine whether disrupting the antioxidant defense enzymes potentiates the efficacy of napabucasin by elevating ROS levels. Viability assays showed that four mutant strains (∆napA, ∆sodB, ∆catA, and ∆ahpC; Supplementary Fig. 5) exhibited much increased susceptibility to napabucasin when compared with the wild type strain, with the ∆ahpC strain showing the highest susceptibility (Fig. 2f, g). Consistent with this, napabucasin-treated cells of the mutant strains accumulated higher levels of ROS, OH•, and free Fe2+ than the wild type strain (Fig. 2h and Supplementary Figs. 1b, c and 2c, d). Note that no significant differences in susceptibility to four common antibiotics were observed between these mutants and the wild-type strains (Fig. 2f), indicating that ROS may not play a role in killing of H. pylori by these antibiotics.
In bacteria, sustained production of ROS is proposed to induce apoptosis-like death, characterized by DNA fragmentation, chromosome condensation, phosphatidylserine exposure, and membrane depolarization43,44, which are hallmarks of eukaryotic mitochondrial apoptosis. As expected, all these characteristic events were observed in H. pylori cells treated with napabucasin. In contrast, metronidazole elicited only minimal changes. The signal intensities detected in the metronidazole-treated group were comparable to those of the DMSO control and markedly weaker than those induced by napabucasin (Supplementary Fig. 6).
Napabucasin targets 2-oxoglutarate:acceptor oxidoreductase (OOR)
To investigate the mode of action of napabucasin, we performed a drug affinity responsive target stability (DARTS) assay45,46, a label-free small-molecule target identification strategy, to identify potential targets. Using a DARTS-based quantitative proteomics approach via mass spectrometry, we analyzed the proteomes following treatment with three increasing concentrations of napabucasin compared with a DMSO control and clustered trends in protein abundance using fuzzy c-means clustering. The abundance patterns across 20 protein clusters are illustrated using colored trend lines (Supplementary Fig. 7), with Cluster 18 (Supplementary Table 4) showing a trend of progressively increasing protein abundance in response to escalating doses of napabucasin. Given that napabucasin functions as a redox cycling agent, the oxidoreductases identified in Cluster 18, including 2-oxoglutarate:acceptor oxidoreductase (OOR) subunit OorD, were selected for target validation. Based on DARTS results, napabucasin was shown to stabilize OorD against pronase digestion in a dose-dependent manner (Fig. 3a). As a control, the band intensity of bovine serum albumin (BSA) treated with napabucasin was comparable to that with DMSO, indicating that napabucasin specifically binds to OorD. Moreover, microscale thermophoresis (MST) confirmed the strong binding of napabucasin to OorD, with an equilibrium dissociation constant (Kd) of 23.57 ± 5.54 nM (Fig. 3b).

a Representative image of DATRS assay to test the binding between napabucasin (NPB) and OorD. The OorD protein together with BSA was treated with NPB (10, 100, and 1000 μg/ml) and DMSO, proteolyzed by pronase, and then detected by silver staining. The gel is representative of three independent experiments. b MST measurements of the OOR/OorD proteins binding with NPB at several concentrations (0.488 nM–8 μM). Normalized fluorescence (ΔFnorm) from three independent experiments, with SEM shown, was plotted against the concentration of napabucasin or metronidazole. Data analysis was performed with the MO Affinity Analysis software (NanoTemper Technologies) using the Kd model. c NPB inhibits menaquinone 6 (MK-6) reduction by OOR. The IC50 value for inhibition of MK-6 reduction activity was determined using OOR assays in vitro. d High performance liquid chromatography (HPLC) chromatograms show production of succinyl-CoA (S-CoA) by the OOR reactions with NPB and MK-6 as the electron acceptors, as seen by the peak at 22.4 min in the samples. e Kinetic characterization of OOR catalysis with NPB (red) and MK-6 (blue). The curves were fitted to the Michaelis–Menten equation. f Cell viability of H. pylori strain G27 transformed with pTM117 (Vec), pBHKP500 [p(oorDABC)] or pBHKP501 [p(oorD)], performed in Columbia blood agar plates supplemented with or without NPB. Growth with/without metronidazole was used as a control experiment. The figure is representative of 3 independent experiments. g Detection of ROS levels in H. pylori cells treated with NPB after 4 h of incubation. Statistical significance was calculated using a one-way ANOVA with Tukey’s post-hoc test. h Proposed MoA of NPB against H. pylori. i The anti-H. pylori activities of 1,4-naphthoquinone derivatives correlate with their efficiency in OOR-catalyzed reduction. MK-6 was used as a control and showed no antibacterial activity against H. pylori without inducing ROS generation. N.D., not detected. Data are represented as mean ± SEM (n = 3 biological replicates) in (b, c, e, g, i). Source data are provided as a Source Data file.
H. pylori OOR is a four-subunit enzyme complex (OorDABC) that catalyzes the conversion of α-ketoglutarate and coenzyme to succinyl-CoA, a key intermediate in the tricarboxylic acid (TCA) cycle. We successfully purified this multienzyme complex to homogeneity using E. coli expression system (Supplementary Fig. 8a) and confirmed a strong binding affinity between napabucasin and OOR, with a Kd of 21.3 ± 8.41 nM (Fig. 3b). Of the four subunits of OOR, OorD binds to napabucasin (Supplementary Fig. 8b). We then assessed the effect of napabucasin on enzymatic activity of OOR. As the physiological electron acceptor for OOR reaction has not been identified yet, we tested eight potential acceptors for their reductase activities. Aside from an artificial chromogenic electron acceptor methylviologen, only menaquinone 6 (MK-6), the primary respiratory quinone in H. pylori47, was found to support the OOR in vitro reaction (Supplementary Fig. 9). Along with the high binding affinity of OOR for menaquinone 6 (Kd = 116.34 ± 6.68 nM; Supplementary Fig. 10), we presume that menaquinone 6 may act as a physiological electron acceptor in OOR reaction. Furthermore, we found that napabucasin exhibited potent inhibitory activity against menaquinone 6 reduction by OOR, with an IC50 of 14.55 ± 2.76 nM (Fig. 3c). The production of succinyl-CoA greatly increased when napabucasin was used as the electron acceptor, compared to menaquinone 6 (Fig. 3d). The kinetic efficiency of OOR in catalyzing napabucasin reduction (kcat/Km of 1.486 μM−1 s−1) is approximately two-fold greater than that with menaquinone 6 (kcat/Km of 0.773 μM−1 s−1) (Fig. 3e), indicating that OOR prefers napabucasin rather than menaquinone 6 as a substrate. Therefore, napabucasin functions as a more efficient and oxidizing electron acceptor than menaquinone 6, the potential physiological acceptor of the OOR reaction. This trend was also observed when comparing NPB to methyl viologen in terms of OOR reductase activity (Supplementary Fig. 11).
To further explore the direct relationship between napabucasin and OOR in the context of H. pylori cell death, we measured changes in napabucasin sensitivity in H. pylori cells overexpressing this protein complex. As expected, overexpression of OOR rendered H. pylori cells more susceptible to napabucasin (Fig. 3f). The increased killing efficacy of napabucasin towards the overexpression strains was positively correlated with elevated levels of ROS, OH•, and free Fe2+ induced by napabucasin treatment (Fig. 3g and Supplementary Fig. 1d and 2e). Note that overexpression of OOR did not affect metronidazole sensitivity (Fig. 3f), consistent with the lack of metronidazole binding to OOR (Fig. 3b). Linking the ROS-dependent killing, we proposed that OOR catalyzes the enzymatic reduction of the para-quinone moiety in napabucasin, resulting in the production of an unstable hydroquinone intermediate, which subsequently reacts with oxygen to generate O2•− in return for the regeneration of the quinone (Fig. 3h).
To evaluate whether the anti-H. pylori activities of 1,4-naphthoquinone derivatives correlate with their efficiency in OOR-catalyzed reduction, we compared four compounds: napabucasin, 2-methoxy-1,4-naphthoquinone (MeONQ), 2-(hydroxymethyl)anthraquinone, and 1,3-dihydroxy-2-methylanthraquinone (rubiadin) listed in descending order of anti-H. pylori activities. We observed a consistent pattern in the oxidative properties of bioactivation by OOR and in the levels of induced endogenous ROS: napabucasin > MeONQ > 2-(hydroxymethyl)anthraquinone > rubiadin (Fig. 3i). Additionally, the redox potentials of these compounds were indicative of their activity, with a strong correlation between lower redox potentials and higher anti-H. pylori activity. These findings suggest the redox-cycling capability of napabucasin, mediated by OOR reduction, contributes to its potent anti-H. pylori effect.
OorD Leu44 and Lys46 mediate interaction of OOR with napabucasin
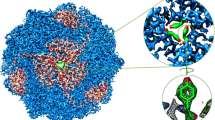
To elucidate the molecular mechanism of napabucasin bioactivation by OOR at a molecular level, we sought to determine the structures of apo and napabucasin-bound OorDABC complexes. Size exclusion chromatography revealed that the apo-form OOR may exist as a dimer (Supplementary Fig. 8c), which was confirmed by subsequent cryo-EM imaging (Supplementary Fig. 12). The structure of complete OOR was resolved by cryo-EM method at 2.95 Å, employing C1 symmetry to avoid the overfitting (Fig. 4a and Supplementary Fig. 12). Each well-resolved protomer comprises four subunits: OorD, OorA, OorB, and OorC (Supplementary Fig. 13a). The dimerization interface is predominantly formed by the OorA and OorB subunits, while OorC and OorD from opposing protomers remain separated. These structural and functional module arrangements are similar with those of the thiamine pyrophosphate (TPP)-dependent oxalate oxidoreductase from Moorella thermoacetica48. In this structure, TPP is situated at the interface between OorA and OorB, contributing to complex stability. OorB contains a proximal [4Fe-4S] cluster (SF4), forming four coordination bonds with the nearby OorBCys19, OorBCys22, OorBCys53, and OorBCys202 (Supplementary Fig. 13b). Another two iron-sulfur clusters, the medial SF4 and the distal SF4, bind with OorD, which could also be identified as the ferredoxin domains. These iron-sulfur clusters, facilitating the transfer of low-potential electrons generated by TPP to downstream electron carriers, are unambiguously localized through well-defined electron densities (Supplementary Fig. 13b). The distances between adjacent clusters are 10.0 Å, 13.1 Å, and 9.7 Å, respectively (Fig. 4b), enabling effective electron transfer to subsequent biochemical pathways.

a Cryo-EM structure of the apo-form OOR. The images present both the front and top views of the cryo-EM map, as well as the cartoon representation of OOR in its apo state. Each protomer consists of four subunits: OorA (dark blue), OorB (dark green), OorC (purple), and OorD (chocolate). Protomer B is illustrated in lighter colors to facilitate differentiation from protomer A. b Electron transfer pathway within protomer A. TPP is shown in sticks while proximal SF4, medial SF4, and distal SF4 are shown as spheres. The inter-ligand distances are indicated with dotted lines, and the respective measurements are annotated. c Cryo-EM structure of OOR in complex with NPB. d Interactions of NPB with the nearby residues. The threshold of the local mesh map of NPB is set to 0.25. Distances between NPB and proximal SF4, medial SF4 are marked. e MST measurements of the OOR mutant proteins binding to NPB at various concentrations (0.488 nM-8 μM). Normalized fluorescence (ΔFnorm) was plotted against the concentration of NPB or metronidazole (MTZ). The Kd values are shown in each plot. f Relative NPB or menaquinone 6 (MK-6) reductase activity of the OOR mutant proteins. Data are represented as mean ± SEM. g Resistant spot dilution assays for the parent strain BHKHpS42 (OOR), BHKHpS43 (L44A), BHKHpS44 (K46A), and BHKHpS45 (L44A K46A) on Columbia blood agar plates containing indicated concentrations of NPB. MTZ addition was used as a control. Data are represented as mean ± SEM (n = 3 biological replicates) in (e, f, h). h Detection of ROS levels in the parent strain BHKHpS42 (OOR), BHKHpPS43 (L44A), BHKHpS44 (K46A), and BHKHpS45 (L44A K46A) after treatment with NPB for 4 h. Statistical significance was calculated using a one-way ANOVA with Tukey’s post-hoc test. Source data are provided as a Source Data file.
To further characterize napabucasin binding to OOR at the molecular level, we determined the cryo-EM structure of the napabucasin-bound OOR complex at a resolution of 3.03 Å with the C1 symmetry (Supplementary Fig. 12). Although napabucasin decreases the stability of the protein complex, the overall assembly remains consistent with the apo-form structure (Fig. 4c). Consequently, in the final density map, protomer A exhibits superior density compared to the other protomer, prompting its use for further analysis. The local density of napabucasin, while moderate due to partial occupancy, was sufficiently resolved to localize its binding site at the interface of OorD, OorB, and OorC subunits, where it engages in van der Waals contacts with adjacent residues (Fig. 4d and Supplementary Fig. 13c). Compared with the apo structure, no obvious conformational changes are observed at the napabucasin binding site, with the exception of OorBGlu214, whose side chain undergoes a twist to avoid the spatial clashes (Supplementary Fig. 13c). Notably, the side chain amino group of OorDLys46 could form a hydrogen bond with the acetyl group of napabucasin, while OorDLeu44 could stabilize napabucasin binding through hydrophobic interactions (Fig. 4d). The distances from the naphthoquinone group to the proximal and medial SF4 clusters are 11.8 Å and 13.0 Å, respectively, distances conducive to electronic transitions (Fig. 4d). The positions of key prosthetic groups, including TPP, proximal SF4, medial SF4, and distal SF4, remain largely unchanged from the apo structure. This suggests that napabucasin can efficiently accept electrons from the proximal or medial SF4 clusters, more effectively than the physiological electron acceptor, menaquinone 6, in agreement with the previous in vitro enzymatic experiments.
To ascertain the importance of OorDLeu44 and OorDLys46 in the napabucasin-OOR interaction observed via cryo-EM, we constructed three OOR mutants: OorD single mutations (L44A and K46A) and the double mutation (L44A/K46A) (Supplementary Fig. 8d). MST assays showed that these mutations reduced napabucasin binding affinity by 6.2-fold, 3.9-fold, and 9.3-fold respectively (Fig. 4e), leading to 4.9-fold, 4.1-fold, and 7.7-fold decreases in napabucasin reductase activity (Fig. 4f). Notably, the mutations in these residues had minimal impact on the binding ability of OOR to the substrate menaquinone 6 (Supplementary Fig. 10) and on the reductase activity towards this substrate (Fig. 4f). These findings indicate that OorDLeu44 and OorDLys46 are crucial for mediating the interaction of OOR with napabucasin, but not with menaquinone 6. Furthermore, we measured changes in napabucasin sensitivity in H. pylori cells carrying these mutations. Spot dilution assays showed that these amino acid substitutions significantly rendered H. pylori cells more resistant to napabucasin in vivo (Fig. 4g). In agreement, H. pylori transformants with L44A, K46A, or L44A/K46A mutations exhibited decreased ROS levels when treated with napabucasin compared to the parental strain (Fig. 4h). These observations suggest that the interaction mediated by OorDLeu44 and OorDLys46 plays a pivotal role in the recognition and catalysis of napabucasin by OOR.
Napabucasin kills coccoid forms of H. pylori
The coccoid form of H. pylori is a morphological manifestation of cell adaptive response to extreme environmental conditions, which survive antibiotic treatment7,49,50. We then evaluated the bactericidal activity of napabucasin against coccoid forms of H. pylori strain G27 using the LIVE/DEAD Bacterial Viability kit to quantitatively assess viability. Napabucasin at 4×MIC (0.125 μg/ml) completely eliminated the coccoid forms from 4-day aging cultures of H. pylori G27 after 12 h of incubation (Fig. 5a). Given the centrality of amoxicillin in H. pylori eradication regimens and its ability to induce a morphological transition to coccoid forms51,52, we further examined the bactericidal activity of napabucasin against amoxicillin-generated coccoid H. pylori cells. Treatment with napabucasin at 4×MIC for 12 h resulted into complete killing of these coccoid forms (Fig. 5b). In contrast, the four common antibiotics, even at 64×MIC, exhibited no bactericidal activities. Scanning electron microscopy (SEM) analysis of the coccoid forms treated with napabucasin revealed severe membrane damage and fragmentation, resembling the morphology of isopropanol-treated dead bacteria, where the cells formed dense clusters (Fig. 5c). In contrast, the spherical morphology of coccoid forms was preserved when exposed to the conventional antibiotics.

Cell viability of coccoid forms from aging H. pylori G27 (a) and amoxicillin-generated H. pylori G27 (b), following treatment with the indicated concentrations of napabucasin (NPB) or antibiotics for 1 h or 12 h. Live cell percentages after treatment were quantitatively assessed using the LIVE/DEAD BacLight viability stain. #, not detected. c SEM images of coccoid forms from amoxicillin-generated H. pylori G27, after treatment with the indicated concentrations of NPB or antibiotics for 6 h (Scale bar, 1 μm). “Coccoid” represents morphology of coccoid forms before treatments. Isopropanol-treated cells serve as a positive control for cell death. Antibiofilm activity of napabucasin determined by crystal violet staining (d), viable colony count method (e), and LIVE/DEAD BacLight viability stain (f). DMSO and MTZ (metronidazole) served as negative and positive controls, respectively. All images in c, f are representative of two independent experiments. Data are represented as mean ± SEM (n = 3 biological replicates) in (a, b, d, e). g Schematic diagram for assessing the efficacy of napabucasin against H. pylori coccoid forms in a mouse model infected with the mouse-adapted strain, MSD132. h CFU counts in the stomach of H. pylori MSD132-infected mice, euthanatized 48 h after the last treatment with vehicle, triple therapy (OPZ + AC), omeprazole plus 10 mg/kg napabucasin (OPZ + NPB), omeprazole plus 25 mg/kg napabucasin, respectively. Data are represented as mean ± SEM. n = 8 mice/group. Statistical significance was calculated using a one-way ANOVA with Tukey’s post-hoc test. Source data are provided as a Source Data file.
Since the coccoid forms are also present within H. pylori biofilms, we determined whether napabucasin has antibiofilm activity against H. pylori. Crystal violet assays showed that napabucasin at 1× MIC eradicated 54% of mature biofilms of H. pylori G27 (Fig. 5d). The killing of biofilm-encased H. pylori was confirmed by viable cell counts and LIVE/DEAD staining. A significant ~1.1 to 4.3-log10 decrease in CFU/ml was observed in H. pylori biofilms treated with 0.016 to 0.063 μg/ml napabucasin compared to vehicle-treated controls (Fig. 5e), in accordance with the results of the LIVE/DEAD staining assays (Fig. 5f). As a control, metronidazole (MTZ) exhibited moderate antibiofilm activity but was less effective than napabucasin.
To assess the in vivo activity of napabucasin against coccoid forms of H. pylori, we established a third mouse model mimicking the failure of clinical treatments in humans. The model utilized H. pylori strain MSD132, which can colonize the mouse stomach for at least 28 weeks53. Two weeks after inoculation, infected mice were randomly divided into four groups and treated with omeprazole plus amoxicillin at three different dosages or vehicle (control group) (Supplementary Fig. 14a). One or three weeks after the last treatment, mice receiving the two low-dosage therapies showed high levels of bacterial colonization (~106 CFU units/g tissue), as determined by both viable colony count method and quantitative real-time PCR (Supplementary Fig. 14b–f). Moreover, SEM images revealed that the morphology of surviving H. pylori cells had transitioned into coccoid forms (Supplementary Fig. 14g), indicating the successful establishment of the animal model for amoxicillin-induced coccoid H. pylori infection. We then used this mouse model, induced by amoxicillin plus omeprazole, to confirm the in vivo efficacy of napabucasin (Fig. 5g). As shown in Fig. 5h, treatments with omeprazole in combination with 10 mg/kg and 25 mg/kg napabucasin resulted in 4.1-fold and 2083-fold decreases in bacterial burden, respectively. Notably, mice treated with the higher dose of napabucasin exhibited 224-fold (P < 0.0001) lower bacterial load compared to those receiving STT treatment.
Discussion
In recent years, the efficacy of current therapies for H. pylori infections has been significantly compromised by the rapid emergence and spread of drug resistance in H. pylori and inadequate patient compliance54,55. Adding to these issues, the morphological transition from spiral to coccoid forms represents an independent mechanism of therapeutic failure7,50. This dormant state induces profound metabolic downregulation and structural alterations, including cell wall thickening and biofilm formation, that confer tolerance to antibiotics targeting active cellular processes. Furthermore, coccoid forms evade immune detection via surface antigen modulation, facilitating persistent infection and relapse56. These insights underscore the necessity of developing novel antimicrobial approaches capable of eliminating both morphological forms to achieve successful eradication. In this study, we present napabucasin, a compound that has shown superior efficacy against drug-resistant growing helical and metabolically dormant coccoid forms of H. pylori in both in vitro and in vivo settings, outperforming the four conventional antibiotics currently utilized in H. pylori treatment protocols. Mechanistic studies reveal that napabucasin exerts its antibacterial effects through a distinctive pathway involving reduction of its para-quinone moiety, leading to generation of bactericidal levels of ROS. Notably, we have identified a determining role for the TCA enzyme, 2-oxoglutarate:acceptor oxidoreductase OOR, in the bioactivation and redox cycling of napabucasin. Thus, our findings suggest an effective antimicrobial strategy for the eradication of coccoid H. pylori cells—OOR-catalyzed redox cycling of 1,4-naphthoquinone derivatives, rather than conventional inhibition.
This study provides substantial evidence supporting the bioactivation of napabucasin by the TCA cycle enzyme OOR, which leads to the generation of bactericidal levels of ROS. OOR catalyzes the oxidative decarboxylation of 2-oxoglutarate in the presence of CoA and our identified potential electron acceptor, menaquinone 6, to produce succinyl-CoA and CO2. Here, we show that napabucasin binds with high affinity to OorD and acts as a more efficient electron acceptor than menaquinone 6. The catalytic reduction of napabucasin by OOR yields an unstable hydroquinone intermediate, which undergoes nonenzymatic oxidation by O2, thereby generating ROS. Thus, we describe a distinct pathway for endogenous ROS generation in H. pylori, coupled to the redox cycling of napabucasin, which is fueled by OOR and O2. Importantly, we have identified OorDLeu44 and OorDLys46 as two key residues for the recognition and catalysis of napabucasin by OOR, bringing us one step closer to fully elucidating the molecular mechanism of napabucasin bioactivation. Note that napabucasin is also effective against several specific pathogens, including M. catarrhalis, N. gonorrhoeae, S. pneumoniae, and various Staphylococcus and Bacillus species. In these organisms, the conventional 2-oxoglutarate dehydrogenase multienzyme complex effectively substitutes for OOR to produce succinyl-CoA. Therefore, napabucasin can target other bacterial oxidoreductases, broadening its antimicrobial spectrum.
Napabucasin, initially characterized as an inhibitor of cancer stemness57, demonstrates significant anticancer efficacy across diverse tumor models based on extensive in vitro and in vivo evidence. Here we have repurposed this compound as a potent anti-H. pylori agent. Although both its anticancer and antibacterial mechanisms involve sustained ROS generation, napabucasin seemed to exhibit selectivity for H. pylori cells over host cancer cells. Reported half-maximal inhibitory concentration (IC50) values for napabucasin ranged from 0.291 to 1.249 μM across 17 stemness-high human cancer cell lines57 and 0.3 to 11 μM across 51 cancer cell lines39. In this study, we also evaluated napabucasin cytotoxicity in nine cancer cell lines and observed consistent IC50 values of 0.17–1.123 μM (Supplementary Table 5). Critically, calculated selectivity indices (SI = IC50/MIC50) ranged from 10.88 to 71.87, indicating favorable selectivity of napabucasin for H. pylori over human cancer cells. This selectivity likely arises from napabucasin’s higher binding affinity for H. pylori OOR relative to its human target NQO1, combined with the more robust antioxidant defense systems (antioxidant enzymes and non-enzymatic antioxidants) in human cells that tolerate higher compound concentrations.
Napabucasin holds promising therapeutic potential due to its low tendency to acquire resistance. This is likely attributed to its specific mode of action-the generation of bactericidal levels of ROS. Overall, excess ROS production can induce irreversible damage to cellular components (DNA, proteins, and lipids), overwhelm the cellular antioxidant defense system, and ultimately lead to cell death. As such, the ability of H. pylori to acquire resistance to napabucasin is highly limited. This differs from previously reported ROS-induced antibiotic resistance, where sublethal levels of the antibiotics, rifampicin58 and ciprofloxacin59, facilitate the emergence of genetic resisters to antibiotics. This may result from the moderate level of ROS, which activates drug efflux pumps and/or sigma-S (σS) general stress response59, and promotes mutagenesis by induction of the SOS system60.
H. pylori with a coccoid lifestyle can establish chronic infections, which are refractory to antibacterial therapies and promotes the recurrence of H. pylori within one year after treatment7,61. Therefore, the development of novel antibacterial agents that target and kill coccoid forms of H. pylori is highly required. Here we demonstrate that napabucasin effectively clears this troublesome pathogen in a newly established mouse model of coccoid H. pylori infections. Notably, oral doses of 480 mg/day (half the dose used in Phase III clinical trials) of napabucasin produced plasma Cmax values of 194 to 652 ng/ml and AUC0-24 values of 2018 to 6184 ng h/m62, which are more than 12 and 126 times the MIC50, respectively. In addition to its favorable pharmacokinetics, napabucasin demonstrates an excellent safety profile63,64. This opens the attractive possibility of napabucasin as a safe and effective candidate for the treatment of refractory H. pylori infections. The high-resolution structures of apo-OOR and napabucasin-bound OOR also provide a valuable starting point for the further development of anti-H. pylori agents.
Methods
Ethics statement
All animal experiments in this study were performed in accordance with the guidelines for the Care and Use of Laboratory Animals issued by the Ministry of Science and Technology of the People’s Republic of China. The experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing Medical University (Approval No: IACUC-2005036).
Bacterial strains and culture conditions
The bacterial strains used in this study are summarized in Supplementary Table 6. All H. pylori strains, including 4 standard and 30 clinical strains, were cultured in Columbia agar base (OXOID, UK) or Brain Heart Infusion (BHI) broth (OXOID) containing 10% fetal calf serum (FCS). All plates or media were incubated at 37 °C for 48 ~ 72 h under microaerophilic conditions (85% N2, 10% CO2, 5% O2, and 90% relative humidity) using a tri-gas CO2 incubator (Binder, model CB160; Germany). H. pylori clinical strains were identified based on colonial morphology, Gram staining, and 16S rDNA sequencing. Gram-positive bacteria, including Staphylococcus aureus ATCC25923 and USA300, Staphylococcus haemolyticus ATCC 29970, Staphylococcus epidermidis ATCC 12228, Bacillus subtilis 168, Bacillus cereus ATCC 14579, Listeria monocytogenes EGD-e, Enterococcus faecium ATCC 19434, Enterococcus faecalis ATCC 29212, and Gram-negative bacteria including Pseudomonas aeruginosa PAO1, Salmonella enterica subsp. enterica serovar Typhimurium ATCC 14028, Acinetobacter baumannii ATCC 19606, Proteus mirabilis ATCC 29906, Enterobacter cloacae ATCC 13047, Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 35657, Morganella morganii ATCC 25830, Stenotrophomonas maltophilia ATCC 51331, and Shigella dysenteriae Sd197 were cultured aerobically in BHI broth at 37 °C. Moraxella catarrhalis ATCC 25238 and Streptococcus pneumoniae ATCC 49619 were cultured in Mueller-Hinton (MH) broth at 37 °C with 5% CO2. Haemophilus influenzae ATCC 49766 was grown in BHI broth supplemented with 10 μg/ml hemin and 10 μg/ml NAD at 37 °C with 5% CO2. Neisseria gonorrhoeae ATCC 19424 was grown in BHI broth containing 5% FCS at 37 °C with 5% CO2. Campylobacter jejuni NCTC 11168 was cultured as H. pylori. Propionibacterium acnes ATCC 11827, Bifidobacterium longum ATCC 15697, Prevotella intermedia ATCC 25611, Fusobacterium nucleatum ATCC 25586, Bacteroides Fragilis ATCC 25285, and Akkermansia muciniphila ATCC-BAA-835 were anaerobically cultured in BHI medium containing 10% FCS in an anaerobic chamber (AW 200SG; Electrotek, UK) at 37 °C.
MIC assays
The MICs against H. pylori were determined using the broth microdilution assay in 96-well microtiter plates as described previously37. H. pylori strains were grown overnight at 37 °C with shaking under microaerophilic conditions. Each culture was diluted to an approximate cell density of 5 × 105 CFU/ml in BHI broth supplemented with 10% FCS. Drug plates were prepared by twofold serial dilutions of the test compounds into clear-bottom 96-well plates. Bacterial cultures were added to each 96-well drug plate with a final volume of 200 μL. After 3-day incubation in a microaerophilic atmosphere at 37 °C, the plates were examined visually, and the MIC was determined to be the lowest concentration that resulted in no turbidity.
The antimicrobial spectrum of napabucasin against a panel of reference strains was determined as described previously in ref. 65. Briefly, aerobic and anaerobic strains from liquid cultures were diluted into the assay plate to achieve an approximate cell density of 1 × 105 CFU/ml. Assay plates were prepared by 2-fold dilution of compound across the plate and included a positive growth control. After incubating the aerobes at 37 °C for 24 h and the anaerobes for 24–48 h, the MIC was determined as the lowest concentration of compound that inhibits bacterial growth, as detected by the unaided eye. All MIC assays were repeated with at least three independent experiments.
Time-dependent killing
The exponential cultures of H. pylori G27 in BHI broth containing 10% FCS were prepared from an overnight culture to yield an initial cell density of ~106 CFU/ml. Cultures were challenged with napabucasin at concentrations of 1×, 2×, 4×, or 8× the MIC, or an equivalent amount of DMSO (as a vehicle control). Cultures were grown at 37 °C with shaking under microaerophilic conditions. At intervals, 100-μl aliquots were removed from each culture, serially diluted, and plated onto Columbia blood agar plates. After 4 days of incubation, the viable cells were counted, and CFU/ml was calculated.
The effect of ROS scavengers on the bactericidal efficiency of napabucasin
This study employed the CFU counting method to investigate the impact of ROS scavengers, N-acetyl-L-cysteine and thiourea, on the bactericidal effect of NPB. H. pylori strain G27 was cultured in BHI broth supplemented with 10% FCS until it reached the mid-exponential phase. The culture was then adjusted to an initial cell density of ~1 × 106 CFU/ml and exposed to 1× MIC of napabucasin (0.031 μg/ml) in a microaerophilic environment for 8 h. This treatment was carried out either in the presence or absence of NAC and thiourea. Following the treatment, each sample was thoroughly washed, serially diluted, spread onto solid media, and CFUs were counted. The survival rate (%) was calculated using the formula: [CFU(NPB + scavenger)/CFU(scavenger)] × 100. The assays were performed with three independent experiments.
Resistance studies
For single-step resistance, 1010 CFU of H. pylori G27 were plated onto Columbia blood agar base containing 2×, 4×, or 8× MIC of napabucasin. After 5 d of incubation at 37 °C, no resistant colonies were detected. The development of drug resistance by sequential passaging was investigated using a previously reported method37. Briefly, H. pylori G27 cells at exponential phase were diluted to a final concentration of ~2 × 107 CFU/ml in BHI supplemented with 10% FCS and containing 0.25×, 0.5×, 1×, 2×, or 4× the MIC of napabucasin. After a 3-day microaerobic incubation at 37 °C under continuous shaking, the cultures were checked for growth, and cells from the second highest concentration showing visible growth were used to inoculate the subsequent culture. This procedure was repeated for up to 15 cycles (45 days), and alterations in MICs after every 2 cycles during the course of continued exposure were determined by the broth microdilution assay. For comparative analyses, the antibiotic metronidazole was also tested with each batch of napabucasin.
DNA manipulation and construction of H. pylori mutant strains and H. pylori strain overexpressing OOR
Plasmid and chromosomal DNAs were extracted with the QIAprep spin miniprep kit (Qiagen). Oligonucleotide primers (listed in Supplementary Table 6) were synthesized, and the cloned genes were verified by sequencing performed by GeneScript Co. (China). H. pylori knockout mutants were constructed by natural transformation via allelic exchange as described previously in ref. 66. The ahpC knockout in the chromosome of H. pylori G27 was achieved via allelic exchange with the introduction of the DNA fragment, a sandwich fusion in which the aphA3 cassette was flanked by the upstream and downstream regions of the ahpC gene, amplified by fusion PCR. The resulting strain, BHKHpS32 (G27 ∆ahpC), was isolated on Columbia blood agar plates supplemented with kanamycin (25 μg/ml). Deletion of ahpC in the genome was confirmed by PCR using appropriate primers, followed by sequencing of the PCR products. Strains BHKHpS34 (G27 ΔcatA), BHKHpS35 (G27 ΔsodB), BHKHpS31 (G27 ΔnapA), and BHKHpS33 (G27 ∆fur) were constructed using the same method.
Strains BHKHpS42 (G27 IR203::oorDABC ∆oorDABC), BHKHpS43 (G27 IR203::oorDL44AABC ∆oorDABC), BHKHpS44 (G27 IR203::oorDK46AABC ∆oorDABC), and BHKHpS45 (G27 IR203::oorDL44AK46AABC ∆oorDABC) were constructed first by inserting the wild-type oorDABC and its mutants oorDL44A, oorDK46A, or oorDL44A K46A into an improved pIR203C04 complementation system67, respectively. The four regions were synthesized and ligated to pBHKP252 carrying a sandwich fusion in which the catGC cassette was flanked by the upstream and downstream regions of the hp0203–204 intergenic region, to generate pBHKP537, pBHKP538, pBHKP539, and pBHKP545, respectively. The four plasmids were then introduced into strain G27 by natural transformation via allelic exchange, and strains BHKHpS17, BHKHpS18, BHKHpS19, and BHKHpS20 were isolated on Columbia blood agar plates supplemented with chloramphenicol (10 μg/ml). The oorDABC knockout in the chromosome of the four strains was achieved via another allelic exchange with the introduction of a sandwich PCR fusion in which the aphA3 cassette was flanked by the upstream and downstream regions of the oorDABC gene. Finally, strains BHKHpS42, BHKHpS43, BHKHpS44, and BHKHpS45 were isolated on Columbia blood agar plates supplemented with kanamycin (25 μg/ml) and chloramphenicol (10 μg/ml). Insertion and deletion of oorDABC or each mutant in the genome were confirmed by PCR using appropriate primers, followed by sequencing of the PCR products.
The OOR-overexpressing strain BHKHpS37 (G27/oorDABC) was constructed by introducing the expression plasmid pBHK500, an E. coli-H. pylori shuttle plasmid pTM11768 expressing OOR, into the wild-type strain G27 using natural transformation. For the construction of the expression plasmid, genomic DNA from strain G27 was used as a template to amplify the target gene oorDABC via PCR. The recombinant plasmid pBHK500 was then constructed by ligating the digested DNA fragment with the digested pTM117 vector using restriction endonucleases KpnI and XbaI and T4 DNA ligase. The construct was verified by colony PCR and DNA sequencing. The OorD-overexpressing strain BHKHpS38 (G27/oorD) and the control strain BHKHpS36 (G27/vector) were constructed by introducing the recombinant plasmid pBHK501 and the empty vector pTM117 into strain G27, respectively.
Measurement of ROS and hydroxyl radical production
ROS levels were quantitatively measured using a Cell (GMS10016.13 v. A) ROS Assay kit (Genmed Scientifics, Inc.; Wilmington, DE, USA), which employs 5- (and-6)-chloromethyl-2′,7′-dichlorofluorescin diacetate (CM-H2DCFDA) as a fluorescent probe. ROS detected by this probe include all primary species (O2• −, H2O2, OH•, and nitrogen dioxide). CM-H2DCFDA freely permeates the plasma membrane and is hydrolyzed by intracellular esterases in the cytosol to form nonfluorescent 2′,7′-dichlorodihydrofluorescein (DCFH), which is subsequently oxidized to highly fluorescent DCF in the presence of ROS. Briefly, ~5 × 107 H. pylori cells were incubated with napabucasin or antibiotics at 0.25×, 1×, and 4× the MIC for 4 h at 37 °C. After treatment, aliquots of the GENMED Reagent B were added to the test cultures to achieve a final concentration of 10 μM. Cultures were then incubated at 37 °C for another 30 min in the dark. The fluorescence intensity was then measured on a Cell Imaging Multimode Microplate Reader (Cytation 5; Biotek, USA) with excitation and emission wavelengths of 495 and 520 nm, respectively. ROS production was also measured by fluorescence microscopy. Glass slides were viewed in FITC using a fluorescence microscope (LSM710; Carl Zeiss, Oberkochen, Germany).
To assess intracellular OH• production in response to napabucasin or metronidazole treatment, we employed the fluorescent reporter dye 3′-(p-hydroxyphenyl) fluorescein (HPF; Sigma-Aldrich, St. Louis, USA). After treatment, HPF was added to each culture to achieve a final concentration of 1 μM. Cultures were then incubated at 37 °C for another 30 min in the dark. The fluorescence intensity was then measured on the Microplate Reader with excitation and emission wavelengths of 490 and 515 nm, respectively. Images were captured from three randomly selected fields under a fluorescence microscope (Axio Imager A2; Zeiss, Germany). These experiments were performed in duplicate from three independent biological experiments.
Measurement of intracellular free Fe2+ production
Intracellular Fe2+ levels were quantitatively measured using the fluorescent probe FerroOrange (Dojindo Laboratories; Kumamoto, Japan). Briefly, ~5 × 107 H. pylori cells were incubated with napabucasin or metronidazole for 4 h at 37 °C. After treatment, FerroOrange was added to each culture to achieve a final concentration of 1 μM. Cultures were then incubated at 37 °C for another 30 min in the dark. The fluorescence intensity was then measured on the Microplate Reader with excitation and emission wavelengths of 532 and 572 nm, respectively. Images were captured from three randomly selected fields under a fluorescence microscope (Axio Imager A2; Zeiss, Germany). These experiments were performed in duplicate from three independent biological experiments.
Oxygen consumption assays
The MitoXpress Xtra oxygen consumption assay was used to measure oxygen consumption in H. pylori. H. pylori G27 was grown to an OD600 of 0.3 in BHI broth containing 10% FCS. Cultures were preincubated with 1×MIC of napabucasin or metronidazole or 1% DMSO (solvent control) for 2 h. One hundred and fifty microliters of each culture were then transferred to black, clear-bottom 96-well plates containing 10 μL of the MitoXpress probe (Luxcel Biosciences). Each well was overlaid with high-sensitivity mineral oil to prevent oxygen diffusion. Time-resolved fluorescence reading (Ex380 nm/Em650 nm) was recorded on a Cell Imaging Multimode Microplate Reader (Cytation 5; Biotek) for 90 min. Lifetime calculation of each condition was computed, and the OCR was determined from the slope of the lifetime graphs. These experiments were performed in duplicate from three independent biological experiments.
Transcriptome analysis
For the challenge experiments, 3 ml of H. pylori at an OD600 of 0.4 was exposed to 0.125 μg/ml napabucasin for 1 h and 6 h in biological triplicate, respectively. Treated and untreated cells were pelleted at 4 °C by centrifugation in 1-ml aliquots. The supernatants were removed, and samples were immediately frozen in liquid nitrogen at −80 °C until processed for total RNA isolation. RNA was extracted using the TRIzol method (Invitrogen, CA, USA) and treated with RNase-free DNase I (Takara, Kusatsu, Japan). RNA degradation and contamination were monitored on 1% agarose gels. RNA concentration and purity were measured using a NanoDrop spectrophotometer (Thermo Scientific, DE, USA). RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). rRNAs were removed to yield only mRNA using Ribo-off rRNA Depletion Kit (Bacteria) (Vazyme, Nanjing, China) for library construction. rRNA-depleted samples were analyzed on an Agilent Bioanalyzer to ensure that more than 99.5% of rRNAs were depleted in each sample. Then, 2–5 ng of the rRNA-depleted samples was used as the input material to construct each cDNA library for RNA sequencing using the NEBNext UltraTM RNA library Prep Kit (NEB, USA). Library quality was checked using the Agilent high-sensitivity DNA assay on the Agilent Bioanalyzer 2100 system. The library preparations were sequenced on an Illumina Novaseq 6000 platform by Beijing Allwegene Technology Company Limited (Beijing, China), and paired-end (PE) 150 bp reads were generated. PE reads assembly, and quality control were performed based on the methods described previously69. These reads were mapped to H. pylori G27 by Bowtie2 v2.2.6. HTSeq v 0.5.4 p3 was used to count the reads numbers mapped to each gene. Gene expression levels were estimated by fragments per kilobase of transcript per million fragments mapped (FPKM). The differentially expressed genes (DEGs) were analyzed with edgeR (v3.16.5) with default screening conditions of FDR ≤ 0.05 and |log2(fold change)| ≥ 1. Volcano plots were created using plot_volcano from soothsayer (https://github.com/jolespin/soothsayer) in Python v.3.6.6. Heat maps were generated using seaborn, and operon plots were created with Matplotlib. Cluster Profiler (v3.4.4) was used to analyze the functions of the DEGs to determine the roles of these genes in H. pylori cells and the metabolic pathways involved. The annotation databases used were Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG).
Protein subcloning, expression, and purification
The oorDABC gene amplified from H. pylori strain G27 genomic DNA was inserted into vector pET-28b to give the plasmid pBHK518 with a N-terminal hexahistidine (His) tag. OorDABC (OOR) was expressed in Rosetta (DE3) pLysS grown at 37 °C in LB medium under microaerobic conditions. At an OD600 of 0.8, the cultures were induced with 0.15 mM isopropyl-β -D-thio-D-galactoside (IPTG) and grown at 30 °C for an additional 5 h prior to harvest. The cells were collected, resuspended in lysis buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, 1 mM dithiothreitol, pH 8.0), lysed by French pressure cell treatment, and centrifuged. The clarified bacterial supernatant was loaded onto a nickel-ion affinity column (Qiagen). The column was washed with a 50 mM NaH2PO4 (pH 8.0) buffer containing 300 mM NaCl, 40 mM imidazole, and 1 mM dithiothreitol, to remove contaminant proteins. The His-tagged OOR protein was eluted in the same buffer containing 200 mM imidazole. The protein was concentrated by ultrafiltration (30 kDa cutoff) and exchanged into a sodium phosphate buffer (50 mM NaH2PO4, 150 mM NaCl, 1 mM dithiothreitol, pH 8.0). The protein purity was visualized by gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The solution structure of OOR was analyzed by size exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare) using an AKTA Purifier 10 at 0.4 ml/ min in phosphate running buffer (135 mM NaCl, 2.7 mM KCl, 1.5 mM Na2HPO4, and 8 mM K2HPO4, 10% glycerol, pH 7.6). The oorD gene amplified from H. pylori strain G27 genomic DNA was inserted into vector pQE-2 to give plasmid pBHK519 with a N-terminal hexahistidine (His) tag. The expression and purification of OorD were constructed by the same method described above. The final concentrations of the proteins were quantified by the Bradford Reagent (Bio-Rad).
Microscale thermophoresis (MST)
MST was conducted using a Monolith NT.115 (NanoTemper Technologies). The MST power and LED excitation power were both set at 20%. OOR or its variant was labeled using a His-Tag Labeling Kit RED-tris-NTA (NanoTemper Technologies), followed by the manufacturer’s protocol. The concentration of RED-tris-NTA labeled OOR was maintained at a constant 25 nM, and the test compounds were serially diluted twofold (15–16 points per curve). Measurements were carried out in PBST buffer (14 mM NaCl, 8 mM Na2HPO4, 2 mM KH2PO4, 1 mM KCl, 0.05% Tween 20, pH 7.4) and standard capillaries. Binding curves were obtained by plotting the normalized fluorescence [Fnorm (%) = Fhot/Fcold; interval time = 20 s] versus the logarithm of compound concentrations. Experiments were performed in triplicate, and data analysis was performed with the MO Affinity Analysis software (Nanotemper) using the Kd model, a standard fitting model derived from law of mass action.
DARTS assay
For unbiased target identification, H. pylori G27 cells were lysed using Bacterial Protein Extraction Reagent (B-PER, Thermo Scientific, 78243), supplemented with Halt Cocktail protease/phosphatase inhibitor (Thermo Scientific, 78440). After centrifugation, the supernatant was mixed with DARTS assay buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 10 mM CaCl2), and protein concentration was then determined using the Bradford Assay. The lysates were equally divided into four tubes for incubation with either vehicle (2% DMSO) or varying concentrations of napabucasin (10, 100, and 1000 μg/ml) for 1 h at room temperature. The four incubated samples, designated as I (DMSO), II (10 μg/ml napabucasin), III (100 μg/ml napabucasin), and IV (1000 μg/ml napabucasin), were then digested with 25 ng/μL Pronase (Roche, 10165921001) at room temperature for 30 min. Proteolysis by pronase was stopped using protease inhibitors (Roche, 11697498001), and samples were stored at −80 °C for SDS–PAGE and proteomics analysis.
Tandem mass tag (TMT) proteomics
Proteins from the aforementioned samples were extracted, and the protein concentrations were determined by BCA Protein Assay Kit (Bio-Rad, USA). An equal amount of proteins (100 µg) for each sample was reduced with DTT (Sigma-Aldrich), alkylated with iodoacetamide (Sigma-Aldrich), and digested with sequencing grade modified trypsin (Promega) using the Filter-Aided Sample Preparation (FASP) method as described previously in ref. 70. After digestion, the peptides of each sample were desalted on Empore™ SPE C18 Cartridges (Sigma-Aldrich) according to manufacturer’s instructions, concentrated by vacuum centrifugation, and reconstituted in 40 µl of 0.1% (v/v) formic acid. The peptide mixture of each sample (100 μg) was then labeled using TMT reagent (Thermo Scientific) according to the manufacturer’s instructions. The labeled peptides were fractionated by SCX chromatography using the AKTA Purifier system (GE Healthcare). The dried peptide mixture was reconstituted and acidified with buffer A (10 mM KH2PO4 in 25% of ACN, pH 3.0) and loaded onto a PolySULFOETHYL column (5 µm, 200 Å, PolyLC Inc; Maryland, USA). The peptides were eluted at a flow rate of 1 ml/min using a gradient: 0% buffer B (500 mM KCl, 10 mM KH2PO4 in 25% of ACN, pH 3.0) for 25 min, 0–10% buffer B during 25–32 min, 10%–20% buffer B during 32–42 min, 20–45% buffer B during 42–47 min, 45%–100% buffer B during 47–52 min, 100% buffer B during 52–60 min, and buffer B was reset to 0% after 60 min. The elution was monitored by absorbance at 214 nm, and fractions were collected every minute. The collected fractions were desalted on Empore™ SPE C18 Cartridges (Sigma-Aldrich) and concentrated by vacuum centrifugation. All samples were stored at −80 °C until required for LC-MS/MS analysis.
LC-MS/MS analysis was performed on a Q Exactive mass spectrometer (Thermo Fisher Scientific, USA) interfaced with the Proxeon Biosystems Easy-nLCTM system (Thermo Fisher Scientific) for 60/90 min. The peptides were loaded onto a reverse phase trap column (Acclaim PepMap100, nanoViper C18; Thermo Scientific) connected to the C18-reversed phase analytical column (Thermo Fisher Scientific Easy Column) in buffer A (0.1% Formic acid). The peptide mixtures were separated with a linear gradient of buffer B (84% acetonitrile and 0.1% Formic acid) at a constant flow rate of 300 nL/min. The mass spectrometer was operated in positive ion mode with the following parameters: electrospray voltage, 2.0 kV; dynamic exclusion duration, 40.0 s; automatic gain control (AGC), 3e6; survey scans were acquired at a resolution of 70,000; resolution for HCD spectra, 17,500 at m/z 200; isolation width, 2 m/z. The MS raw data for each sample were searched using the MASCOT engine (Matrix Science, London, UK; version 2.2) embedded into Proteome Discoverer 1.4 software (Thermo Fisher Scientific) for identification and quantitation analysis. Differentially expressed proteins (DEPs) with fold change >1.2 and FDR-adjusted p < 0.05 were subjected to bioinformation analysis. Soft clustering analysis was conducted using the Fuzzy C-Means algorithm71 in the Mfuzz package72 to identify the trend of gradually increasing protein abundance with increasing napabucasin dose.
OOR assays and kinetics characterization
The enzymatic activity of OOR for methyl viologen (MV) reduction was determined with the artificial electron acceptor MV, as described previously in ref. 73 with modifications. The reaction mixtures contain 50 mM Tris-HCl (pH 8.0), 6 μg OOR, 0.1 mM CoA, 0.5 mM MV, and 0.5 mM 2-oxoglutarate in a total volume of 100 μL. Reactions were carried out with 96-well microtiter plates under anaerobic conditions and were initiated by the addition of OOR. The plates were sealed by Microplate Sealing Films, and the reduction of MV was monitored spectrophotometrically at an optical density of 600 nm (OD600) using a Synergy HTX multimode microplate reader (BioTek Instruments, Winooski, USA) at 30 °C. For inhibition studies, varying concentrations of napabucasin (1 nM–250 μM) or DMSO (negative control) were added to the reaction mixtures. After a 2-min incubation, the OD600 was measured using the microplate reader. The enzymatic activities were expressed in units per mg of protein, where one unit of activity equals the reduction of 1 μmol of MV/min. Nonlinear regression curve fits were used to calculate the IC50 of napabucasin for inhibition of MV reduction activity.
When menaquinone 6 (MK-6) replaced MV as an electron acceptor, varying concentrations of napabucasin (1 nM–250 μM) or DMSO (negative control) were added to the above reaction mixtures (with MK-6 replacing MV) for inhibition studies. High-performance liquid chromatography (HPLC) was used to detect the consumption of MK-6 and to calculate the IC50 of napabucasin for inhibition of MK-6 reduction activity. Chromatographic separation was performed using a C18 HPLC column (KromasiL 100-5-C18 column, 250 × 4.6 mm, Sweden) at room temperature, and the volume of each injection was 60 μL. The mobile phase comprising A (acetonitrile) and B (isopropanol) was used under gradient conditions: 0–5 min, A 100%, B 0%; 5–40 min, A 30%, B 70%; 40–45 min, A 0%, B 100%; 45–50 min, A 0%, B 100%; 60–70 min, A 100%, B 0%; and the flow rate was 1.0 ml/min. The consumption of MK-6 was determined by the standard curve, which was plotted with the concentration of menaquinone 6 as the abscissa and the peak area as the ordinate. Nonlinear regression curve fits were used to calculate the IC50 of napabucasin for inhibition of MK-6 reduction activity.
The enzymatic kinetics were determined using varying concentrations of napabucasin, MK-6, or MV (0.244–62.5 μM) as the electron acceptor. To ensure the consistency of the experimental procedures involving them, OOR activity here was assayed using Ellman’s reagent [5,5′-dithiobis(2-nitrobenzoic acid)74, to detect the decrease in the concentration of CoA thiol groups resulting from succinyl-CoA synthesis. Reactions (volume of 100 μL as described above) were quenched with 50 μL of 1.5 mM Ellman’s reagent (Sigma) in 0.1 M sodium phosphate (pH 7.2) at time zero point or 5-min intervals. Loss of reduced CoA, determined spectrophotometrically (decrease in absorbance at λ = 412 nm; ε = 14,150 M−1 cm−1), over time was used to calculate the initial velocity (V0) for each electron acceptor concentration. All enzyme kinetics experiments were conducted in triplicate, and Michaelis-Menten kinetic parameters were calculated by GraphPad Prism software 9.5.
Anti-H. pylori efficacy in vivo
Six-week-old specific-pathogen-free female C57BL/6 mice were obtained from the Animal Core Facility of Nanjing Medical University, and housed at the same place in a 12-h light–dark cycle with ambient temperature and humidity maintained at 68–74 °F and 30–70%, respectively. All experimental procedures were performed in a biosafety level two laminar flow hood. All animals were healthy upon receipt and were weighed and monitored during a 7-day acclimation period before the start of the study. Mice were randomly assigned to experimental groups of 8–10 animals.
Each mice received 0.3 ml of 1 × 109 CFU/ml H. pylori NSH57 in BHI broth through oral gavage every 2 days, repeated five times (on days 6, 8, 10, 12, and 14, respectively), and the infection was allowed to develop for 2 weeks. The mice were randomly assigned to three groups (n = 8) to receive vehicle (0.5% CMC-Na plus 0.2% Tween-80), napabucasin with omeprazole (a proton pump inhibitor), or triple therapy. Mice were first administered omeprazole through oral gavage at a dose of 400 μmol/kg, followed by a lag time of 30 min before administration of the assigned treatments. Napabucasin (14 mg/kg) and triple-therapy formulation (amoxicillin 14 mg/kg and clarithromycin 7 mg/kg) were administered once daily for a consecutive 3 days by oral gavage. Forty-eight hours after the last treatment, mice were killed and the stomachs were removed from the abdominal cavity, cut along the greater curvature, and rinsed in saline to remove the stomach contents. At the same time, feces were also collected from each mouse for partial 16S rDNA gene sequencing of the extracted community. For H. pylori colonization, gastric tissue was suspended in 1 mL BHI broth and homogenized for bacterial recovery. The homogenate was serially diluted and spotted onto Columbia blood agar plate containing vancomycin (10 μg/ml), amphotericin B (2 μg/ml), and bacitracin (20 μg/ml). The plates were then incubated at 37 °C under microaerobic conditions for 5 d, and bacterial colonies were enumerated and expressed as CFU per gram of tissue.
A second mouse model was also constructed using the mouse-adapted multi-drug resistant BHKS159 strain37 to further assess anti-H. pylori efficacy in vivo. The mice were randomly assigned to six treatment groups (n = 10) to receive with vehicle, triple therapy (28 mg/kg amoxicillin plus 14 mg/kg clarithromycin), omeprazole plus 28 mg/kg napabucasin (OPZ + NPB), omeprazole plus 7 mg/kg napabucasin, 28 mg/kg napabucasin alone and 7 mg/kg napabucasin alone, respectively. Five-day drug treatment (orally administration once daily for 5 consecutive days) was used in this model to determine if napabucasin has superior in vivo killing efficacy to the triple therapy approach, in the case of increasing times of administration. Forty-eight hours after the last treatment, mice were killed, and the stomachs were removed from the abdominal cavity. The stomachs were cut into two longitudinal sections, and each section was weighed. The sections were used for assessment of bacterial colonization.
Bacterial community composition and diversity
The microbial community DNA was extracted from mouse stool samples using MagPure Stool DNA KF kit B (Magen; China) following the manufacturer’s instructions. The composition of the fecal bacterial community was determined by partial 16S rDNA gene sequencing of the extracted community DNA. The V4 hypervariable regions of the 16S rDNA gene were amplified with the degenerate primers 515-F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806-R (5′-GGACTACHVGGGTWTCTAAT-3′). These primers were tagged with Illumina adapters, pads, and linker sequences. PCR reactions were performed in triplicate for each sample in a total reaction volume of 50 μL containing 30 ng of template. The PCR cycling conditions were as follows: 95 °C for 3 min, 30 cycles of 95 °C for 45 s, 56 °C for 45 s, 72 °C for 45 s and final extension for 10 min at 72 °C for 10 min. The PCR products were purified using Agencourt AMPure XP beads and eluted in Elution buffer. Libraries were qualified by the Agilent Technologies 2100 bioanalyzer. The validated libraries were sequenced on Illumina HiSeq 2500 platform (BGI, Shenzhen, China) following the standard pipelines of Illumina, generating 2 × 250 bp paired end reads. Raw reads were filtered to remove adaptors, low-quality, and ambiguous bases. Paired-end reads were then merged using the Fast Length Adjustment of Short reads program (FLASH, v1.2.11)75 to obtain the tags. The tags were clustered into operational taxonomic units (OTUs) with a cutoff value of 97% using UPARSE software (v7.0.1090)76. Chimera sequences were detected by comparing them to the Gold database using UCHIME software (v4.2.40)77. Then, OTU representative sequences were taxonomically classified using Ribosomal Database Project (RDP) Classifier v.2.2 with a minimum confidence threshold of 0.6, and the classification was trained on the Greengenes database (v201305) using QIIME v1.8.078. The USEARCH_global79 tool was used to compare all Tags back to the OTUs to generate the OTU abundance statistics table of each sample. Alpha and beta diversity were estimated by MOTHUR (v1.31.2)80 and QIIME (v1.8.0)78 at the OTU level, respectively. Sample cluster was conducted by QIIME (v1.8.0)78 based on UPGMA. Heatmaps were generated with the package “gplots” implemented in the software “R” package v3.4.1.
Cryo-EM data collection and processing
For the apo OorDABC, 3 μL of protein solution (1.5 mg/ml) was applied to 300-mesh Quantifoil R0.6/1 grid after a 20-s H2/O2 glow discharge. The grids were blotted with 0 blot force for 3 s at 4 °C and 100% humidity, and plunge-frozen in liquid ethane using Vitrobot IV. The cryo-EM data were collected using a 300 keV Titan Krios electron microscope, equipped with a K3 direct electron detector. The movie stacks were recorded at 29,000× with a calibrated pixel size of 0.82 Å/pixel. The exposure time was set to 2.4 s, and the total cumulative dose was 60 e−/ Å2. SerialEM was used to automatically record all images, and the defocus range was set from −1.8 μm to −1.2 μm. In total, 6132 movies were captured for this dataset. For the OorDABC complex with napabucasin, the sample preparation and the data collection parameters are similar to the apo OorDABC. In total, 3552 movies were captured for this dataset.
For the apo OorDABC, all movie stacks were motion-corrected using Relion 3.03 with a 5 × 4 patch and a twofold binning81. The contrast transfer functions (CTF) were estimated by cryoSPARC Patch CTF Estimation82. All particles were blob-picked with a range from 100 Å to 200 Å and extracted with a box size of 320 pixels. After 2D classification, clear classes were selected as references for template picking. A total of 2,498,096 particles were extracted to conduct for 2D classification. Classes with obvious characteristics and clear morphology were selected, resulting in 521,453 particles for initial 3D reconstruction into four classes using Ab initio methods. The four classes of volumes were used as templates for the heterogeneous refinement. The class with distinct shape and dimer characteristics contained 186,894 particles. All 3D classifications and refinements were accomplished in cryoSPARC. After non-uniform Refinement83, the final resolution reached 2.95 Å. According to the golden-standard Fourier shell correlation (FSC) with threshold of 0.143, the local resolution ranges were analyzed by cryoSPARC. For OorDABC with napabucasin, the image processing was performed with a similar pipeline with a box size of 320 pixels. The final resolution was 3.03 Å. 3DFSC calculation was performed to assess the potential preferred orientation84. The detailed information was shown in Supplementary Fig. 12 and Supplementary Table 7.
Model building and validation
The two atomic models were manually built in Coot 0.8.985 using the templates of predicted structures generated by CryoNet (https://cryonet.ai). The SMILES string of napabucasin was generated and copied from ChemDraw (PerkinElmer). Napabucasin was manually built into the densities. Real-space refinements and validation were performed using Phenix 1.1686. The figures related with structural information were created using UCSF ChimeraX-0.987 or PyMOL 2.0.1 (Schrödinger, LLC).
Analysis of DNA fragmentation, DNA condensation, and phosphatidylserine exposure
These analyses were performed as described previously43 with minor modifications. Briefly, ~5 × 107 H. pylori G27 cells were incubated with 1×MIC of napabucasin or metronidazole for 4 h at 37 °C. Following treatment, DNA fragmentation was performed using terminal deoxynucleotide transferase dUTP nick end labeling (TUNEL). One Step TUNEL Apoptosis Assay Kit (Beyotime, Shanghai, China) was used for DNA fragmentation experiments. Fluorescent staining of DNA was performed with the DNA-specific stain, Hoechst 33342 (Invitrogen), according to kit instructions. For phosphatidylserine (PS) exposure experiments, cells were labeled with fluorescein isothiocyanate (FITC)-conjugated Annexin V using the Vybrant Apoptosis Assay Kit (Invitrogen). After staining, the cells were analyzed with a fluorescence microscope (Axio Imager A2; Zeiss, Germany). Images were captured from three randomly selected fields under the microscope.
Membrane depolarization assay
Cell membrane potential was monitored by the membrane potential sensitive dye 3,3′-dipropylthiadicarbocyanine iodide (DiSC3(5)) (Invitrogen, USA). Approximately 5 × 107 H. pylori G27 cells in BHI broth containing 10% FCS were incubated with 1 µM DiSC3(5) in black 96-well microtiter plates for 1 h to ensure maximal uptake of the dye. The cells were then treated with varying concentrations of napabucasin or metronidazole. After treatment for 2 h or 8 h, fluorescence intensity was then measured on a Cell Imaging Multimode Microplate Reader (Cytation 5; Biotek, USA) with excitation and emission wavelengths of 622 and 670 nm, respectively. These experiments were performed in duplicate from three independent biological experiments.
Coccoid form of H. pylori killing assay
As has been previously demonstrated, H. pylori coccoid forms can be induced by aging cultures88 or exposure to sub-MIC of amoxicillin51. In this study, 4-day aging and amoxicillin-induced H. pylori G27cells were used to model the coccoid forms. Briefly, ~5 × 107 H. pylori G27 cells in Brucella broth containing 10% FCS were cultivated for 4 days at 37 °C. Additionally, H. pylori G27 cells were also incubated with amoxicillin (0.063 µg/ml) and cultivated for an additional 3 days at 37 °C. Both cultures were then centrifuged at 600 × g for 5 min to pellet any remaining spiral form of the bacteria. The supernatants were then centrifuged at 11,000 × g for 5 min, and the resulting pellets, primarily containing the induced coccoid forms, were confirmed by light microscopy. To test the bactericidal activity of napabucasin against the coccoid forms of H. pylori, 100 μL of coccoid G27 cells at an OD600 of 0.3 was exposed to 4×MIC of napabucasin, 64×MIC of each antibiotic, or DMSO (as a vehicle control) in BHI broth containing 10% FCS. The samples were incubated in a 96-well plate at 37 °C with shaking for 1 and 12 h. After incubation, bacterial viability in the suspension was quantitatively measured by LIVE/DEAD BacLight Bacterial Viability Kit (ThermoFisher Scientific, L7012), which consists of two fluorescent probes, propidium iodide (PI) and SYTO 9. In brief, the bacteria were centrifuged, resuspended with 1 μM SYTO-9 and 2.5 μM PI diluted with PBS, and incubated at 37 °C for 30 min. The collected bacteria were then washed and resuspended in PBS. The fluorescence intensity of SYTO-9 (green) was then measured on a Cell Imaging Multimode Microplate Reader (Cytation 5; Biotek, USA) using excitation and emission wavelengths of 485 and 530 nm, respectively. The fluorescence intensity of PI (red) was measured using excitation and emission wavelengths of 485 and 630 nm, respectively. The green/red fluorescence ratios (RatioG/R) were then calculated, and the percentage of live coccoid forms for each sample was acquired based on a standard curve. Each of the samples and the standard curve was tested in triplicate.
Scanning Electron Microscopy (SEM) analysis
The morphology of coccoid forms of H. pylori G27 treated with napabucasin was examined with SEM. After centrifugation, the slides dipped in bacterial solution were fixed in 2.5 % fresh glutaraldehyde at 4 °C overnight. The slides were then soaked three times with PBS for 10 min each. Dehydration was performed by sequential treatments with increasing concentrations of ethanol: 50% for 20 min, 75% for 20 min, 85% for 20 min, 95% for 20 min, and two treatments of 100% ethanol for 20 min each. The dehydrated samples were dried under vacuum overnight, mounted on carbon tape, and coated with gold-platinum for imaging using a Scanning Electron Microscope (JSM-7900F; JEOL, Japan).
Antibiofilm assay
The antibiofilm activity of napabucasin against H. pylori was assessed as described previously in ref. 65. Briefly, an overnight culture of H. pylori G27 was diluted with Brucella broth supplemented with 10% FCS to OD600 of 0.15. The diluted culture was then incubated in a 96-well polystyrene plate at 37 °C for 3 days to allow biofilm formation. After biofilm formation, the medium was then aspirated, and the plates were washed twice with PBS. Fresh brucella broth supplemented with 7% FCS and various concentrations of napabucasin, metronidazole, or DMSO (as a vehicle control) was added to the wells for another 24 h. The dispersal of mature biofilms was measured using the crystal violet staining method89.
Bacterial viability within the biofilm was measured using a Live/Dead BacLight Bacterial Viability Kit (ThermoFisher Scientific, L7012). Following incubation for 24 h under microaerobic conditions as described above, nonadherent cells were washed three times with PBS and then stained with the two fluorescent dyes at 37 °C in the dark. After rinsing, the images were observed using a confocal laser scanning microscope (LSM710; Carl Zeiss, Germany), and three randomly selected fields were captured for analysis. The effect of napabucasin on the cell viability of preformed biofilms was also investigated using colony count methods as described previously in ref. 65. Viable bacterial colonies on plates were enumerated and expressed as CFU/ml. All data points are expressed as means ± standard deviations (SD) from three independent experiments performed in triplicate.
Establishment of a mouse model of coccoid H. pylori infection
A third mouse model of H. pylori infection was established to assess the in vivo activity of napabucasin against coccoid forms of H. pylori. This model used strain MSD132, which is capable of colonizing the mouse stomach for at least 28 weeks53. Using appropriate doses of amoxicillin treatment, we expected this model to achieve successful colonization of amoxicillin-induced coccoid forms of strain MSD132, which mimics failure of a clinical treatment for H. pylori infection in humans. Six-week-old specific-pathogen-free female C57BL/6 mice were used for this study. Each mice received 0.3 mL of 1 × 109 CFU/ml H. pylori MSD132 in BHI broth through oral gavage every 2 days, repeated five times (on days 6, 8, 10, 12, and 14, respectively), allowing the infection to develop for 2 weeks. The mice were then randomly assigned to four groups (n = 10) to receive either vehicle (0.5% CMC-Na plus 0.2% Tween-80), three doses of napabucasin with omeprazole (24.84 mg/kg OPZ plus 5.04 mg/kg AMX, 41.4 mg/kg OPZ plus 8.4 mg/kg AMX, and 63 mg/kg OPZ plus 14 mg/kg AMX), and were administered once daily for a consecutive 5 days by oral gavage. One week and three weeks after the last treatment, mice were euthanized, and the stomachs were removed from the abdominal cavity. The stomachs were cut into two longitudinal sections, and each section was weighed. The two sections were used for assessment of bacterial colonization and morphology analysis using SEM, as described above. The bacterial load was determined by both viable colony count method and quantitative real-time PCR as described previously in ref. 90. The total DNA from infected tissue was extracted using E.Z.N.A. Universal Pathogen Kit (Omega Bio-tek, D4035-01). Real-time PCR was performed with primers targeting the vacA (qvacA-F/qvacA-F) or cagA gene (qcagA-F/qcagA-F) using SYBR green Supermix (Bio-RAD). H. pylori count number was normalized to standard curve synthesized from a known copy number of MSD132 cells. If the mouse stomach showed high level of coccoid H. pylori colonization (>105 CFU units/g tissue), the model was considered successfully constructed.
In vivo activity against coccoid forms of H. pylori
The in vivo activity of napabucasin against coccoid forms of H. pylori was evaluated using the mouse model described above. The study protocol including H. pylori MSD132 inoculation and infection development in C57BL/6 mice, followed by the induction of amoxicillin (8.4 mg/kg) plus omeprazole (41.4 mg/kg) was presented in Fig. 5g. Two weeks after the last treatment, the mice were randomly assigned to four treatment groups (n = 10) to receive with vehicle, triple therapy (omeprazole plus 25 mg/kg amoxicillin plus 10 mg/kg clarithromycin), omeprazole plus 10 mg/kg napabucasin, and omeprazole plus 25 mg/kg napabucasin, respectively. Mice were first administered omeprazole through oral gavage at a dose of 41.4 mg/kg, followed by a 30-min lag time before administration of the assigned treatments. Each formulation was administered once daily for a consecutive 5 days by oral gavage. Forty-eight hours after the last treatment, mice were euthanized, and the stomachs were excised and rinsed in saline to remove stomach contents. The stomachs were then weighed and used for the assessment of bacterial colonization by viable colony count method.
Quantification and statistical analysis
All microbiological and biochemical assays show biological replicates and were repeated with at least three independent experiments. Data are presented as means ± SEM. To compare means between more than two groups, a oneway ANOVA with Tukey’s post-hoc test was conducted. Statistical analysis, as indicated in each figure legend, was performed using GraphPad Prism software version 9.5. P < 0.05 was considered to be statistically significant. All figures were prepared with Adobe Illustrator v29.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequencing data of the mouse fecal 16 s rRNA and the transcriptomic dataset generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession numbers PRJNA884387 and PRJNA884367, respectively. The proteomic dataset has been deposited in the iProX database (https://www.iprox.cn) and can be accessed via the following projectID: IPX0010331000. The accession numbers for cryo-EM density map of apo OorDABC, and in complex with napabucasin in this study are EMD-39556 and EMD-39557, respectively. The accession numbers for the coordinates for the apo OorDABC and in complex with napabucasin in this study are PDB: 8YS5 and PDB: 8YS6, respectively. Source data are provided with this paper.
References
FitzGerald, R. & Smith, S. M. An overview of Helicobacter pylori infection. Methods Mol. Biol. 2283, 1–14 (2021).
Suerbaum, S. & Michetti, P. Helicobacter pylori infection. N. Engl. J. Med. 347, 1175–1186 (2002).
NIH Consensus Conference. Helicobacter pylori in peptic ulcer disease. NIH consensus development panel on Helicobacter pylori in peptic Ulcer disease. JAMA 272, 65–69 (1994).
Covacci, A., Telford, J. L., Del Giudice, G., Parsonnet, J. & Rappuoli, R. Helicobacter pylori virulence and genetic geography. Science 284, 1328–1333 (1999).
Argueta, E. A. & Moss, S. F. Treatment of Helicobacter pylori. Curr. Opin. Gastroenterol. 35, 544–550 (2019).
Shah, S. C., Iyer, P. G. & Moss, S. F. AGA Clinical Practice Update on the Management of Refractory Helicobacter pylori Infection: Expert Review. Gastroenterology 160, 1831–1841 (2021).
Ierardi, E. et al. The puzzle of coccoid forms of Helicobacter pylori: beyond basic science. Antibiotics 9, 293 (2020).
Cellini, L. Coccoid forms of Helicobacter pylori. J. Infect. Dis. 173, 1288–1289 (1996).
Hathroubi, S., Servetas, S. L., Windham, I., Merrell, D. S. & Ottemann, K. M. Helicobacter pyloribiofilm formation and its potential role in pathogenesis. Microbiol. Mol. Biol. Rev. 82, e00001–e00018 (2018).
Chaput, C. et al. Role of AmiA in the morphological transition of Helicobacter pylori and in immune escape. PLoS Pathog. 2, e97 (2006).
Loke, M. F., Ng, C. G., Vilashni, Y., Lim, J. & Ho, B. Understanding the dimorphic lifestyles of human gastric pathogen Helicobacter pylori using the SWATH-based proteomics approach. Sci. Rep. 6, 26784 (2016).
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A. & Collins, J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007).
Van Acker, H. & Coenye, T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 25, 456–466 (2017).
Keren, I., Wu, Y., Inocencio, J., Mulcahy, L. R. & Lewis, K. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339, 1213–1216 (2013).
Liu, Y. & Imlay, J. A. Cell death from antibiotics without the involvement of reactive oxygen species. Science 339, 1210–1213 (2013).
Yano, T. et al. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 286, 10276–10287 (2011).
Heikal, A. et al. Activation of type II NADH dehydrogenase by quinolinequinones mediates antitubercular cell death. J. Antimicrob. Chemother. 71, 2840–2847 (2016).
Nagata, K. et al. Helicobacter pylori generates superoxide radicals and modulates nitric oxide metabolism. J. Biol. Chem. 273, 14071–14073 (1998).
Nakamura, A. et al. Oxidative cellular damage associated with transformation of Helicobacter pylori from a bacillary to a coccoid form. Free Radic. Biol. Med. 28, 1611–1618 (2000).
Miethke, M. et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 5, 726–749 (2021).
Wencewicz, T. A. New antibiotics from Nature’s chemical inventory. Bioorg. Med. Chem. 24, 6227–6252 (2016).
Kapoor, N., Kandwal, P., Sharma, G. & Gambhir, L. Redox ticklers and beyond: naphthoquinone repository in the spotlight against inflammation and associated maladies. Pharm. Res. 174, 105968 (2021).
Ravichandiran, P., Sheet, S., Premnath, D., Kim, A. R. & Yoo, D. J. 1,4-naphthoquinone analogues: potent antibacterial agents and mode of action evaluation. Molecules 24, 1437 (2019).
Angulo-Elizari, E., Henriquez-Figuereo, A., Moran-Serradilla, C., Plano, D. & Sanmartin, C. Unlocking the potential of 1,4-naphthoquinones: a comprehensive review of their anticancer properties. Eur. J. Med. Chem. 268, 116249 (2024).
Silva, L. R. et al. Computer-aided design of 1,4-naphthoquinone-based inhibitors targeting cruzain and rhodesain cysteine proteases. Bioorg. Med. Chem. 41, 116213 (2021).
Lee, M. H. et al. Inhibitory effects of menadione on Helicobacter pylori growth and Helicobacter pylori-induced inflammation via NF-kappaB inhibition. Int. J. Mol. Sci. 20, 1169 (2019).
Kong, Y. et al. Enzymatic characterization and inhibitor discovery of a new cystathionine {gamma}-synthase from Helicobacter pylori. J. Biochem. 143, 59–68 (2008).
Park, B. S. et al. Antibacterial activity of Tabebuia impetiginosa Martius ex DC (Taheebo) against Helicobacter pylori. J. Ethnopharmacol. 105, 255–262 (2006).
Wang, Y. C. et al. In vitro activity of 2-methoxy-1,4-naphthoquinone and Stigmasta-7,22-diene-3beta-ol from Impatiens balsamina L. against Multiple Antibiotic-Resistant Helicobacter pylori. Evid. Based Complement Altern. Med. 2011, 704721 (2011).
Nagata, K., Hirai, K. I., Koyama, J., Wada, Y. & Tamura, T. Antimicrobial activity of novel furanonaphthoquinone analogs. Antimicrob. Agents Chemother. 42, 700–702 (1998).
Shao, Z., Wang, H., Ren, H., Sun, Y. & Chen, X. The anticancer effect of napabucasin (BBI608), a natural naphthoquinone. Molecules 28, 5678 (2023).
Nixon, G. L. et al. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 68, 977–985 (2013).
Minotti, G., Menna, P., Salvatorelli, E., Cairo, G. & Gianni, L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharm. Rev. 56, 185–229 (2004).
Kuang, X. et al. Repurposing napabucasin as an antimicrobial agent against oral streptococcal biofilms. Biomed. Res. Int. 2020, 8379526 (2020).
Li, C. et al. Discovery of napabucasin derivatives for the treatment of tuberculosis. Medchemcomm 10, 1635–1640 (2019).
Baldwin, D. N. et al. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75, 1005–1016 (2007).
Huang, Y. et al. In vitro and in vivo activities of zinc linolenate, a selective antibacterial agent against Helicobacter pylori. Antimicrob. Agents Chemother. 63, e00004–e00019 (2019).
Froeling, F. E. M. et al. Bioactivation of napabucasin triggers reactive oxygen species-mediated cancer cell death. Clin. Cancer Res. 25, 7162–7174 (2019).
Guo, G. et al. NQO1 is a determinant for cellular sensitivity to anti-tumor agent Napabucasin. Am. J. Cancer Res. 10, 1442–1454 (2020).
Smirnova, G., Muzyka, N., Lepekhina, E. & Oktyabrsky, O. Roles of the glutathione- and thioredoxin-dependent systems in the Escherichia coli responses to ciprofloxacin and ampicillin. Arch. Microbiol. 198, 913–921 (2016).
Olekhnovich, I. N., Goodwin, A. & Hoffman, P. S. Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. FEBS J. 276, 3354–3364 (2009).
Martinez-Julvez, M. et al. Structure of RdxA-an oxygen-insensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. FEBS J. 279, 4306–4317 (2012).
Dwyer, D. J., Camacho, D. M., Kohanski, M. A., Callura, J. M. & Collins, J. J. Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis. Mol. Cell 46, 561–572 (2012).
Wyllie, A. H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555–556 (1980).
Huang, L., Wang, D. & Zhang, C. Drug affinity responsive target stability (DARTS) assay to detect interaction between a purified protein and a small molecule. Methods Mol. Biol. 2213, 175–182 (2021).
Lomenick, B. et al. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. USA 106, 21984–21989 (2009).
Marcelli, S. W. et al. The respiratory chain of Helicobacter pylori: identification of cytochromes and the effects of oxygen on cytochrome and menaquinone levels. FEMS Microbiol. Lett. 138, 59–64 (1996).
Gibson, M. I. et al. The structure of an oxalate oxidoreductase provides insight into microbial 2-oxoacid metabolism. Biochemistry 54, 4112–4120 (2015).
Kadkhodaei, S., Siavoshi, F. & Akbari Noghabi, K. Mucoid and coccoid Helicobacter pylori with fast growth and antibiotic resistance. Helicobacter 25, e12678 (2020).
Cheng, T. & Boneca, I. G. The shapeshifting Helicobacter pylori: from a corkscrew to a ball. Mol. Microbiol. 121, 260–274 (2024).
DeLoney, C. R. & Schiller, N. L. Competition of various beta-lactam antibiotics for the major penicillin-binding proteins of Helicobacter pylori: antibacterial activity and effects on bacterial morphology. Antimicrob. Agents Chemother. 43, 2702–2709 (1999).
Bode, G., Mauch, F. & Malfertheiner, P. The coccoid forms of Helicobacter pylori. Criteria for their viability. Epidemiol. Infect. 111, 483–490 (1993).
Dorer, M. S., Cohen, I. E., Sessler, T. H., Fero, J. & Salama, N. R. Natural competence promotes Helicobacter pylori chronic infection. Infect. Immun. 81, 209–215 (2013).
Zanotti, G. & Cendron, L. Structural aspects of Helicobacter pylori antibiotic resistance. Adv. Exp. Med. Biol. 1149, 227–241 (2019).
Tshibangu-Kabamba, E. & Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance - from biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 18, 613–629 (2021).
Sijmons, D., Guy, A. J., Walduck, A. K. & Ramsland, P. A. Helicobacter pylori and the role of lipopolysaccharide variation in innate immune evasion. Front. Immunol. 13, 868225 (2022).
Li, Y. et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 112, 1839–1844 (2015).
Paul, A., Nair, R. R., Jakkala, K., Pradhan, A. & Ajitkumar, P. Elevated levels of three reactive oxygen species and Fe(II) in the antibiotic-surviving population of mycobacteria facilitate de novo emergence of genetic resisters to antibiotics. Antimicrob. Agents Chemother. 66, e0228521 (2022).
Pribis, J. P. et al. Gamblers: an antibiotic-induced evolvable cell subpopulation differentiated by reactive-oxygen-induced general stress response. Mol. Cell 74, 785–800.e787 (2019).
Kohanski, M. A., DePristo, M. A. & Collins, J. J. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 37, 311–320 (2010).
Sun, Y. & Zhang, J. Helicobacter pylori recrudescence and its influencing factors. J. Cell Mol. Med. 23, 7919–7925 (2019).
Kawazoe, A. et al. Phase 1 study of napabucasin, a cancer stemness inhibitor, in patients with advanced solid tumors. Cancer Chemother. Pharm. 85, 855–862 (2020).
Taniguchi, H. et al. Phase I study of napabucasin in combination with FOLFIRI + bevacizumab in Japanese patients with metastatic colorectal cancer. Int. J. Clin. Oncol. 26, 2017–2024 (2021).
Shah, M. A. et al. Randomized, double-blind, placebo-controlled phase III study of paclitaxel +/- napabucasin in pretreated advanced gastric or gastroesophageal junction adenocarcinoma. Clin. Cancer Res. 28, OF1-OF9 (2022).
Luo, P. et al. Dihydrotanshinone I is effective against drug-resistant Helicobacter pylori in vitro and in vivo. Antimicrob. Agents Chemother. 65, e01921–20 (2021).
Jiang, X. et al. The cyclopropane fatty acid synthase mediates antibiotic resistance and gastric colonization of Helicobacter pylori. J. Bacteriol. 201, e00374–19 (2019).
Langford, M. L., Zabaleta, J., Ochoa, A. C., Testerman, T. L. & McGee, D. J. In vitro and in vivo complementation of the Helicobacter pylori arginase mutant using an intergenic chromosomal site. Helicobacter 11, 477–493 (2006).
Carpenter, B. M. et al. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl. Environ. Microbiol 73, 7506–7514 (2007).
Bokulich, N. A. et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59 (2013).
Wisniewski, J. R. Filter-aided sample preparation: the versatile and efficient method for proteomic analysis. Methods Enzymol. 585, 15–27 (2017).
Schwammle, V. & Jensen, O. N. A simple and fast method to determine the parameters for fuzzy c-means cluster analysis. Bioinformatics 26, 2841–2848 (2010).
Kumar, L. & M, E. F. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 (2007).
Hughes, N. J., Clayton, C. L., Chalk, P. A. & Kelly, D. J. Helicobacter pylori porCDAB and oorDABC genes encode distinct pyruvate:flavodoxin and 2-oxoglutarate:acceptor oxidoreductases which mediate electron transport to NADP. J. Bacteriol. 180, 1119–1128 (1998).
Ellman, G. L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 (1959).
Magoc, T. & Salzberg, S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963 (2011).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. Advances, interactions, and future developments in the CNS, Phenix, and Rosetta structural biology software systems. Annu Rev. Biophys. 42, 265–287 (2013).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Cellini, L., Allocati, N., Di Campli, E. & Dainelli, B. Helicobacter pylori: a fickle germ. Microbiol. Immunol. 38, 25–30 (1994).
Hathroubi, S., Zerebinski, J. & Ottemann, K. M. Helicobacter pylori biofilm involves a multigene stress-biased response, including a structural role for flagella. mBio 9, e01973–18 (2018).
Lee, A. et al. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112, 1386–1397 (1997).
Acknowledgements
We thank Prof. Nina Salama and Prof. Karen Ottemann for providing H. pylori strains. We would like to express our gratitude to the Bio-Electron Microscopy Facility of ShanghaiTech University for the technical support. We would also like to thank for the support of Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine, ShanghaiTech University. This work was supported by grants from the National Natural Science Foundation of China (Nos. 82473974 and 82073899 to H.B.; No. 82302542 to Y.G.), and Shanghai Rising-Star Program (No. 23QA1406400 to Y.G.).
Author information
Authors and Affiliations
Contributions
H.B. and Y.G. supervised the study. H.B., Y.G. and X.H. designed experiments. X.H., W.L., Y.H., H.H., Z.M., L.Z., T.S., Y.B., Z.Y., S.H., J.W., L.D., Q.T. and J.J. performed experiments. X.H., Y.G., W.L., Y.H., H.H., L.Z., T.S., Q.X. and S.B. analyzed data. H.B., Y.G., X.H. and W.L. wrote the manuscript. All authors reviewed/approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
S.B., Y.H. and X.H. are co-inventors on an approved China patent (ZL2021 1 0303156.7) titled “Pharmaceutical application of Napabucasin”. All the other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Nagendran Tharmalingam, Giuseppe Zanotti, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hang, X., Lan, W., Yanqiang, H. et al. 2-oxoglutarate:acceptor oxidoreductase-catalyzed redox cycling effectively targets coccoid forms of Helicobacter pylori. Nat Commun 16, 6965 (2025). https://doi.org/10.1038/s41467-025-62477-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62477-4
