
Abstract
The demand for mining metals via electrodeposition drives the need for high-performance electrodes. Traditional adsorbents are electrically insulating, limiting efficiency due to poor electrical contact. We overcome this by infiltrating conductive polymers into adsorbent pores, enhancing electrical connectivity and aligning chelators. This improves electrical pathways, enabling rapid nucleation and high space-time efficiency. Electrochemical uranium uptake from spiked seawater reaches 26.5 g uranium per gram of adsorbents, four times higher than electrodes mixing adsorbents with carbon black, and two orders of magnitude higher than physicochemical methods. The system also achieves uranium mining from natural seawater at 17.4 mg g−1 with an enrichment index of 1.1 × 10⁷. This strategy offers a blueprint for designing electrodes with better electronic access to active sites, boosting performance in electrically driven processes.
Similar content being viewed by others
Introduction
Resource mining from seawater is known for its high complexity1,2,3. Electrodeposition has attracted immense attention over the past century as it can mitigate the inherent limitations associated with physicochemical adsorption methods, such as slow diffusion and limited uptake capacity due to the inadequate driving force and finite active sites4,5. In principle, the imposed electric field not only facilitates ion migration but also entails the electric current to reduce the deposition of dissolved metal cations on the electrode without saturation. Traditionally, bare metal foils have been used as working electrodes6,7. However, due to the lack of nucleation sites and low surface area, these systems suffer from sluggish initial kinetics, thereby leading to energy waste. To combat this issue, the use of a high-surface-area electrode system containing specific binding sites is desired. The physical mixture of an adsorbent material and electrically conductive blend materials is an emerging strategy for the construction of electrode materials4,5,8. However, phase separation is inevitable if two bulky materials are directly mixed, resulting in a part of the functionalities being electrochemically inaccessible and thus unable to participate in the electrochemical process. Therefore, there is a need to develop new methods for the construction of electrode materials to realize molecule-level junctions between two electrode elements for full use of the chelator under applied potentials.
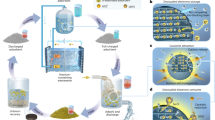
Known for their molecule-level tunability in terms of structure and function, covalent organic frameworks (COFs)9,10,11,12,13,14,15,16,17,18 have found wide-ranging applications in fields such as catalysis and environmental remediation19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34. The ordered pore channels of COFs make them attractive as host materials for guest encapsulation to integrate multiple components. The association of the functional space of COFs with organic polymers has proven to be a suitable strategy to prepare composite materials with synergistic combination properties of each component35,36,37,38,39. Indeed, accommodating a conducting polymer into one-dimensional channels of redox-active COF films has been proven to promote electron transport between electrodes and redox-active sites on the COF (Fig. 1)40,41.

a The physical mixture of the COF and carbon black, whereby the phase separation occurs, resulting in a part of the functionalities being electrochemical inaccessible and low electrodeposition efficiency. b The confinement of conductive polymers within the pore channels of COFs via a host-guest assembly strategy, thus the convenient electron transport is constructed, promising rapid nucleation and high electrodeposition efficiency.
Results
Synthesis and characterization
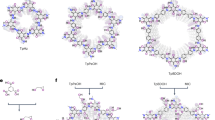
As an electrodeposition application, we focus on seawater uranium mining because nuclear energy is considered sustainable, producing virtually no air pollution. With the nuclear energy further development, a stable supply of fission fuel must be secured. Estimates suggest that at the current rate of consumption, nonrenewable uranium remaining in terrestrial sources can barely last for 100 years. Oceans around the world contain around three parts per billion of uranium, with a total amount of 4.5 billion tons, enough to power nuclear plants for generations42,43,44. While uranium extraction from seawater using physicochemical sorbents has advanced in affinity and reusability, practical application remains limited by performance loss after multiple cycles and poor operability in real marine environments. Traditional sorbents, which rely on passive adsorption of UO22+ onto functional groups in porous polymers or supramolecular networks, often suffer from pore blockage and inefficient desorption, leading to reduced extraction capacity and material degradation over time. In contrast, electrodeposition strategies that integrate porous materials functionalized with uranyl-specific binding groups offer an active and tunable approach driven by an applied potential. This method provides several advantages: it enhances selectivity by combining chemical recognition with electrochemical control, enabling targeted reduction of uranium species over competing ions such as Ca2+ or Mg2+ in seawater; accelerates extraction kinetics through enhanced ion transport and interfacial charge transfer; and prevents pore clogging, as uranium is deposited on the surface rather than trapped inside the pore network. Additionally, the electrode can be readily regenerated through simple soaking and rinsing, thereby circumventing the labor-intensive cleaning and filtration steps typically associated with conventional adsorbents. These features make the approach well-suited for continuous operation and scalable deployment in real seawater, overcoming key limitations of conventional sorption-based methods. Therefore, technology development for mining uranium from seawater is critical. To fulfill this task and to implement the aforementioned host-guest assembly strategy, a host COF material was designed with two goals: first, high density of binding sites aligned on the pore channel, and second, fully π-conjugated structure to ensure a high degree of electronic communication throughout the framework. To this end, a C = C-linked COF (sp2c-COF) synthesized via the Knoevenagel condensation of 1,4-phenylenediacetonitrile and tetrakis(4-formylphenyl)pyrene was chosen45,46,47,48, wherein the nitrile groups can be readily converted into amidoxime, the state-of-the-art uranyl chelator for uranium extraction (Fig. 2). Its formation was confirmed by powder X-ray diffraction (PXRD) and N2 sorption analyses. The intense reflections in the PXRD pattern indicate the high crystallinity of the resulting material. To elucidate the structure, models using Materials Studio were constructed, which revealed that the experimental PXRD pattern matched well with the proposed model with symmetry of C2/m in an eclipsed orientation (Supplementary Fig. 1). sp2c-COF exhibited reversible type I isotherms, indicative of its uniform microporous structure. Fitting the adsorption isotherm gave a Brunner-Emmet-Teller (BET) surface area of 806 m2 g−1 and pore size distribution of 1.8 nm (Supplementary Fig. 2). The successful conversion of cyano groups into amidoxime after being treated with NH2OH was validated by Fourier transform infrared spectroscopy (FT-IR), which revealed the appearance of C = N (1659 cm−1), C-N (1384 cm−1), and N-O (902 cm−1) peaks, characteristic of the amidoxime group, coupled with a low-intensity peak related to nitrile stretching (2216 cm−1, Supplementary Fig. 3), and by solid-state 13C nuclear magnetic resonance (NMR) analysis, which did not show the peak related to cyano group at 116.4 ppm, but showed a peak attributed to the open-chain amidoxime group at 174.2 ppm (Supplementary Fig. 4), indicative of the high conversion degree44. PXRD and N2 sorption measurements (Supplementary Fig. 1) confirmed the retention of crystallinity and porosity after the amidoximation process, which implies the accessibility of the amidoxime, albeit decreased surface area in comparison with that observed for the pristine COF (806 m2 g−1 versus 297 m2 g−1).

The concept of enhancing the electronic accessibility of the amidoxime groups on the pore channel of the COF by inserting conductive polymers and schematic illustration of the synthesis of COF-based sorbent and the corresponding chemical structures.
With an amidoxime-functionalized COF material available, we proceeded to incorporate conductive polymers within the pores of the COF. To demonstrate the host-guest assembly principle, poly(3,4-ethylenedioxythiophene) was the conductive polymer of our choice as it has been proven to be easily available and highly conductive49. To facilitate this process, sp2c-COF-AO was soaked in a 3,4-ethylenedioxythiophene (EDOT)-CH3CN solution, with a mass ratio of sp2c-COF-AO to EDOT of 1, overnight, and then CH3CN was slowly evaporated under vacuum to push the monomer ingress into the pores through capillarity. Following this, an ammonium persulfate aqueous solution was introduced into the system to induce polymerization (see details in the Supplementary Method), resulting in the formation of PEDOT@sp2c-COF-AO, during which the color of the COF turned from light orange to dark green.
The confinement of PEDOT within the sp2c-COF-AO framework was confirmed by thermogravimetric analysis (TGA), X-ray photoelectron spectroscopy (XPS), solid-state 13C NMR, N2 sorption analysis, FT-IR spectroscopy, wide-angle X-ray scattering (WAXS), scanning electron microscopy (SEM), transmission electron microscopy (TEM), and elemental mapping. TGA profiles revealed that a greater weight loss for PEDOT@sp2c-COF-AO below 400 °C compared to sp2c-COF and sp2c-COF-AO (Supplementary Fig. 5), suggesting the incorporation of PEDOT. XPS analysis of PEDOT@sp2c-COF-AO displayed distinct S 2p peaks at 162.0 and 163.1 eV (Supplementary Fig. 6), absent in the pristine sp2c-COF-AO, directly confirming the presence of PEDOT. Solid-state 13C NMR further validated successful PEDOT incorporation, with a prominent peak at 64.8 ppm assigned to ether oxygen in PEDOT moieties (Supplementary Fig. 4).
A marked reduction in the BET surface area of PEDOT@sp2c-COF-AO (88 m2 g−1) relative to sp2c-COF-AO (297 m2 g−1) indicated pore occupation by PEDOT chains (Supplementary Fig. 7). FT-IR analysis showed attenuated characteristic PEDOT peaks in PEDOT@sp2c-COF-AO compared to its physical mixture (Supplementary Fig. 8). Notably, shifts in absorption bands suggested electronic interactions (e.g., π–π stacking or charge transfer) between PEDOT and the COF framework, consistent with molecular-level confinement rather than surface deposition. These interactions likely enhance interfacial compatibility and electronic coupling within the porous structure. WAXS measurements confirmed the retention of the crystalline integrity of sp2c-COF-AO after PEDOT incorporation, with minimal disruption to its long-range order (Supplementary Fig. 9). SEM and TEM imaging further corroborated preserved framework morphology post-PEDOT loading (Supplementary Figs. 10–12), while elemental mapping verified homogeneous sulfur distribution (Supplementary Fig. 13). Collectively, these results confirm that PEDOT resides predominantly within the COF pores. Despite reduced porosity (PEDOT@sp2c-COF-AO: 88 m2 g−1), sufficient pore accessibility for chelators was retained. Elemental analysis quantified the PEDOT loading at 36.2 wt% (8.16 wt% sulfur). Conductivity measurements via a four-probe method revealed a remarkable conductivity of 135 S m−1 for PEDOT@sp2c-COF-AO, comparable to carbon-based materials, whereas pristine sp2c-COF-AO and sp2c-COF exhibited negligible conductivity. This underscores the critical role of encapsulated PEDOT in enabling charge transport within the hybrid framework.
Uranium sorption studies
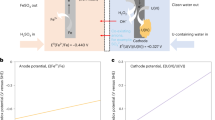
Having addressed the question related to the conductivity of the COF, we then asked if the current responses of the COF could be enhanced. To answer this question, cyclic voltammetry measurements were conducted to compare the differences between PEDOT@sp2c-COF-AO and sp2c-COF-AO in responses to UO2(NO3)2. Figure 3a shows the cyclic voltammograms of seawater spiked with 400 ppm uranium at a scan rate of 50 mV s−1 at various potential ranges. The association of PEDOT with sp2c-COF-AO led to a significant improvement in current responses, with the generated current intensity being one order of magnitude higher than that of the parent COF. The voltammogram exhibited two distinctive Faradaic peaks over the capacitive background. The peaks at around −0.62 V and 0.20 V versus SHE (Standard Hydrogen Electrode) represent the reduction of U(VI) to U(V) and the oxidation of U(V) to U(VI), respectively. The greater magnitude of the reduction peak than that of the reverse oxidation peak can be attributed to the disproportionation of U(V) to U(IV), implying the possibility of electrochemical extraction of uranium from seawater50,51,52,53,54.

a Cyclic voltammograms of simulated seawater using PEDOT@sp2c-COF-AO coated glassy carbon electrode as a working electrode (black) and uranium spiked simulated seawater (400 ppm) using sp2c-COF-AO (olive) and PEDOT@sp2c-COF-AO (orange) coated glassy carbon as working electrodes, respectively. b Schematic illustration of uranium extraction processes in this work. In step I, under the current condition, UO22+ or H+ gets reduced into UO2 or H2 on the cathode; meanwhile, negative Cl− or OH− is oxidized to Cl2 or O2 on the anode side. In step II, under the electroless condition, spontaneous redox reactions occurred, whereby the generated Cl2 or O2 was reduced at the graphite rod to compensate for the oxidation of the UO2 into UO3, forming a galvanic cell (yellow, U4+; organ, U6+; red, O; olive, Cl; white, H).
Next, we demonstrate the superior performance of infiltrated conductive polymer over carbon black in enhancing the current response of insulating materials for uranium electrodeposition. To systematically evaluate this, PEDOT@sp2c-COF-AO was benchmarked against other electrode systems. Uranium uptake capacities were normalized to the sp2c-COF-AO content in each material for direct comparison (Table 1). A two-electrode configuration was employed (Fig. 3b and Supplementary Fig. 14a), featuring a coated platinum foil as the working electrode and a graphite rod as the counter electrode. To mitigate gas entrapment from water electrolysis and extend nucleation time, an alternating pulse current (400 Hz, duration ratio 1:4) was applied, cycling between −5 V and 0 V (Fig. 3b). PEDOT@sp2c-COF-AO achieved a uranium extraction capacity of 720 mg g−1 in 12 h using deionized water containing 20 ppm uranium, outperforming physical mixtures of sp2c-COF-AO with PEDOT (560 mg g−1) or carbon black (480 mg g−1) under identical conditions (Table 1). Visible uranium electrodeposition on the adsorbent-coated electrode confirmed successful uranyl ion capture (Supplementary Fig. 14b and c). Control experiments with a bare Pt foil revealed dominant water splitting and gas evolution, with no detectable uranium deposition even at reduced potentials (−2.5 to 0 V). Given identical amidoxime group content and the low intrinsic electrodeposition efficiency of sp2c-COF, sp2c-COF-AO, and PEDOT alone, these results suggest a synergistic mechanism. The pore-confined PEDOT chains act as molecular wires, electrically connecting the chelating sites on COF to the electrode, thereby amplifying uranium extraction under an applied field. This contrasts with physical mixtures, where coiled PEDOT chains fail to fully access chelating sites, limiting electrical connectivity. The critical role of electrochemical accessibility was further evidenced by the sensitivity of performance to the PEDOT-to-COF ratio. Excessively low PEDOT content (PEDOT@sp2c-COF-AO (1:2) left channels partially unwired, reducing chelator utilization (432 mg g−1), while excessive PEDOT (PEDOT@sp2c-COF-AO (2:1)) caused pore blockage (480 mg g−1).
To highlight the advantage of electrodeposition over physicochemical adsorption, we compared the uptake capacities obtained using these two methods under various initial uranium concentrations of 20–400 ppm. Physicochemical adsorption resulted in saturated capacities of 240–309 mg g−1. In contrast, electrochemical extraction showed no saturation, and uptake capacities greatly increased with an increase in the initial uranium concentration (Supplementary Fig. 15). The discrepancy in the uptake capacity became more significant as the initial uranium concentration increased. An extraction capacity of 14.2 g g−1 was achieved by electrodeposition after 24 h using a 400 ppm UO2(NO3)2 aqueous solution as the electrolyte, which is 46 times higher than that obtained by physicochemical adsorption. To validate the binding mechanism, density functional theory (DFT) calculations on a sp2c-COF-AO fragment revealed a strong adsorption energy (Eads = –16.45 eV) for uranyl ions (UO22+), confirming robust interactions with the amidoxime-functionalized framework (Supplementary Fig. 16).
To evaluate the chemical stability of the COF-coated electrode, PXRD analysis was first conducted after immersion in strongly acidic and basic solutions, which confirmed the structural integrity of sp2c-COF, sp2c-COF-AO, and PEDOT@ sp2c-COF-AO under these harsh conditions (Supplementary Figs. 1b, 9, and 17a–d). In addition, a series of uranium solutions at a concentration of 400 ppm with pH values ranging from 2 to 9 were prepared for further testing. Subsequent electrochemical extraction revealed a maximum capacity at pH 6, while consistently high capacities were maintained across a wide pH range. These results confirm the robust stability and effectiveness of PEDOT@sp2c-COF-AO for uranium extraction under diverse pH conditions (Supplementary Fig. 17e).
Electrodeposition of uranium from simulated seawater
Encouraged by these results, we turned our attention to extracting uranium from simulated seawater, and a solution containing 25.6 g L−1 of NaCl and 0.198 g L−1 of NaHCO3 was used as a background solution. The PEDOT@sp2c-COF-AO-coated Pt foil electrode afforded an exceptional uptake capacity of 26.5 g g−1 from the simulated seawater spiked with 400 ppm uranium. This is one of the highest mass-normalized adsorbent uptake capacities reported to date for uranium extraction (Fig. 4 and Supplementary Table 1)55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89 Surprisingly, it was found that the uranium uptake capacity from the simulated seawater was approximately two times higher than that obtained using deionized water as a background solution, which is contradicted to that of traditional physical adsorption. As it is well known that three carbonates are bound to uranyl ions in the presence of NaHCO3, the functional groups on the adsorbent material must overcome the competitive binding of uranyl with carbonates, thus decreasing the extraction efficiency (Supplementary Fig. 18).

Equilibrium uptake capacities of PEDOT@sp2c-COF-AO and representative benchmark sorbents reported under comparable conditions, illustrating the significantly enhanced adsorption performance achieved through the integration of conductive polymers.
Investigation of uranium electrodeposition process
To gain insight into the mechanism involved in seawater uranium electrodeposition, we characterized the extracted uranium species in order to map the electrochemical reaction cycles. SEM analysis revealed that the formed precipitate exhibited a uniform plate structure (Supplementary Fig. 19). The composition of the deposits was determined by XPS. As shown in Supplementary Fig. 20, only uranium and oxygen were detected. Peaks at 392.5 eV and 381.5 eV were assigned to the U 4f5/2 and U 4f7/2 core levels of hexavalent uranium ions, respectively. Through XRD characterization, the deposited particles were identified as uranium trioxide UO3·xH2O (JCPDS 43-0364, Supplementary Figs. 21–22). The appearance of four Raman peaks at 323, 434, 545, and 726 cm−1, which are characteristic of UO3·xH2O, coincided with the XRD result (Supplementary Fig. 23)90,91. These results agree with the principle of electrodeposition that metal ions precipitate out in the form of charge-neutral oxide species. However, according to a previous study, uranyl ions were proposed to be electrodeposited in the form of (UO2)O2·xH2O60. This inconsistency motivated further exploration of the involved deposition mechanism. To simplify the discussion, the cathode and anode were separated and connected by a salt bridge. At the cathode compartment, in addition to the visualized UO3·xH2O deposition on the electrode, the pH value of the solution decreased from 4.9 to 4.4, whereas no detachable changes were observed at the anode compartment. To unravel a more detailed reaction mechanism, a set of control experiments was performed. To identify the role of NaCl, Na2SO4 was used to sustain the electrical conductance in the reaction medium as it did not participate in the electrolysis. A decreased extraction efficiency was observed, suggesting that Cl− ions were involved in the entire electrodeposition process. To determine the role of dissolved O2, the system was bubbled with N2, and no obvious change in extraction efficiency was detected, indicating that the oxygen from the air was not involved in the electrodeposition process (Supplementary Fig. 24). Furthermore, XPS and PXRD results revealed that the chemical valences of the extracted uranium species harvested under various conditions were identical (Supplementary Figs. 25 and 21). Considering these experimental phenomena and the basic principles associated with brine electrolysis, the electrochemical reactions that occur on an electrode can be described as follows (Fig. 5).

a Schematic illustration of possible transformation of uranium species on the electrode. b Possible reactions occurred on the cathode and anode during uranium electrodeposition.
Under the applied conditions, the positively charged UO22+ and H+ migrate to the cathode, where they are reduced to UO2 or H2, respectively, while Cl⁻ or OH⁻ is oxidized to Cl2 or O2 at the anode. However, the deposited uranium species were characterized as having a + 6 valence state (UO3), conflicting with the expected +4 oxidation state (UO2) based on these reactions. Notably, the absence of gas evolution at the anode led us to propose spontaneous redox reactions forming a galvanic cell: the generated Cl2 or O2 is reduced at the graphite rod, compensating for the oxidation of UO2 to UO3 (Fig. 5). To test this hypothesis, in-situ current detection was performed using the bias waveform shown in Supplementary Fig. 26. A measurable current persisted during the bias-off period, supporting the galvanic cell mechanism. We attribute the enhanced extraction efficiency in NaCl-containing systems to the higher oxidation potential of Cl2 compared to O2, both of which drive UO2 oxidation to UO3.
Our analysis further suggests that the reduction of UO22+ to UO2 plays a pivotal role: this step promotes uranium solidification, improving extraction efficiency and enabling subsequent oxidation to UO3 even in Cl2-free systems. The solidified UO2 acts as a trapped intermediate, facilitating its eventual conversion to UO3. However, direct detection of UO2 proved challenging due to its rapid oxidation during electrodeposition. In-situ Raman spectroscopy of the PEDOT@sp2c-COF-AO-coated electrode confirmed UO22+ adsorption and final UO3 formation but showed no discernible UO2 signals (Supplementary Fig. 27). Crucially, HAADF-STEM imaging revealed UO2 nanoparticles within the adsorbent, with lattice spacings (0.32, 0.28, 0.19, and 0.17 nm) matching the (111), (200), (220), and (311) planes of crystalline UO2 (Supplementary Fig. 28). This nanoscale evidence reconciles the proposed mechanism: UO2 forms transiently before full oxidation to UO3. The overall process involves a galvanic cell that repurposes side products (O2 or Cl2) to oxidize UO2—a previously unreported pathway (Supplementary Figs. 29–30). Further supporting the role of conductive PEDOT, energy-dispersive X-ray spectroscopy (EDX) coupled with HRTEM revealed uniformly dispersed uranium species in PEDOT@sp2c-COF-AO, contrasting with aggregated particles in sp2c-COF-AO after electroextraction (Supplementary Figs. 31–32). This highlights how PEDOT infiltration enhances electronic accessibility to amidoxime groups, optimizing charge transfer and uranium immobilization.
Electrodeposition of uranium from natural seawater
To explore the utility of this system for seawater uranium mining, we compared the uranium extraction efficiencies of electrodeposition and physicochemical adsorption by using uranium-spiked seawater (400 ppm). The uptake capacity for PEDOT@sp2c-COF-AO was 260 mg g−1, representing a removal efficiency of ∼3.5%. Under otherwise identical conditions, >99.0% of uranium species can be extracted under a bias voltage (Fig. 6). Notably, PEDOT@sp2c-COF-AO exhibits a significantly higher efficiency than those of other leading systems used in this field of study. For example, the uptake capacity obtained using PEDOT@sp2c-COF-AO for uranium extraction from spiked uranium seawater is four times that obtained using an electrochemical method with an alternating current (7774 mg g−1 versus 1932 mg g−1), even at a lower initial uranium concentration (400 ppm versus 1000 ppm)60. PEDOT@sp2c-COF-AO achieves a uranium extraction capacity of 17.4 mg g−1 from natural seawater, demonstrating record electrocatalytic performance after 56 days of continuous exposure to 10 liters of seawater55,56,57,58,59,60,61,62,92,93, revealing that more than 50 % of uranium species dissolved in natural seawater can be enriched by PEDOT@sp2c-COF-AO under an alternating current. Accordingly, the enrichment index was calculated to be 1.1 × 107, which is almost one order of magnitude higher than the state-of-the art extraction system (Supplementary Table 2). In addition to a considerably higher capacity, electrochemical extraction gave faster kinetics, overcoming the conventional drawbacks of physicochemical adsorption. Moreover, given the strong binding affinity of amidoxime with most useful metal ions, PEDOT@sp2c-COF-AO exhibits comparable extraction efficiency for UO22+ with Cu2+, and Fe3+ ions (Supplementary Table 3). Importantly, the electrode made from PEDOT@sp2c-COF-AO can be readily regenerated by the treatment in HNO3 and NaHCO3 aqueous solution in sequence, with fully maintained electrochemical extraction efficiency for at least eight consecutive cycles (Supplementary Table 4).

Uranium extraction performance from spiked seawater by PEDOT@sp2c-COF-AO coated Pt foil electrode using the electrodeposition method with an initial uranium concentration of 400 ppm (20 mL).
Discussion
In summary, we illustrate an alternative strategy to prepare high-performance electrode materials for electrodeposition by confining conductive polymers within the pore channels of an amidoxime-functionalized COF material. The molecule-level junction between these two electrode elements significantly improved the electronic accessibility of the chelator. As a result, the composite exhibited significantly enhanced electrochemical extraction efficiency in comparison with that obtained using a physical mixture of carbon black and adsorbent materials, which also overcomes long-standing challenges associated with the physicochemical adsorption in terms of limited uptake capacity and slow kinetics, achieving record high uranium uptake capacity and enrichment index. As this strategy is applicable to various conductive polymers and porous materials, the presented host-guest assembly strategy offers a general approach for preparing broad classes of composites with high electron accessibility of functionalities for various applications involving electricity. For example, a similar synergistic effect was observed when polyaniline (PANI) was introduced in a COF-based adsorbent for gold extraction (Supplementary Fig. 33). Considering the range of processes involved with electricity, where the electronic accessibility of functionality plays a critical role, we anticipate that the conjugation of a porous functional material and a conductive polymer will inspire the design and synthesis of next-generation electrode materials.
Methods
Materials and measurements
Solvents and other commercially available reagents were purchased from Sigma-Aldrich and used without further purification. UO2(NO3)2·6H2O was purchased from Electron Microscopy Sciences. The uranium stock solution was prepared by dissolving the appropriate amount of UO2(NO3)2·6H2O in deionized water. The Powder X-ray diffraction (PXRD) data were collected on a Bruker AXS D8 Advance A25 Powder X-ray diffractometer (40 kV, 40 mA) using Cu Kα (λ = 1.5406 Å) radiation. Nitrogen sorption isotherms at 77 K were measured using Micromeritics ASAP 2020 M and Tristar system. The samples were outgassed for 12 h at 373 K before the measurements. Scanning electron microscopy (SEM) images were collected using a Hitachi SU 1510 and SU 4800. Cs corrected High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were collected on a FEI Spectra 300 double Cs-corrected electron microscope operated at 300 kV, equipped with a convergence semi-angle of 24.1 mrad. Transmission electron microscopy (TEM) images and Energy-dispersive X-ray spectroscopy (EDS) studies were performed at a Talos 200S TEM equipped with dual-detector super EDS system. EDS mapping images were collected at 200 kV with a convergence semi-angle of 22.5 mrad. X-ray photoelectron spectroscopy (XPS) spectra were performed on a Thermo ESCALAB 250 with Al Kα irradiation at θ = 90° for X-ray sources, and the binding energies were calibrated using the C1s peak at 284.9 eV. IR spectra were recorded on a Nicolet Impact 410 FTIR spectrometer. Inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was measured with a Perkin-Elmer plasma 40 emission spectrometer. 13C (125 MHz) cross-polarization magic-angle spinning (CP-MAS) was recorded on a Bruker Avance 500 spectrometer equipped with a magic-angle spin probe in a 4-mm (13C) ZrO2 rotor. 1H NMR (400 MHz) and 13C NMR spectra were recorded on a Bruker Avance-400 spectrometer. Chemical shifts are expressed in ppm downfield from TMS at δ = 0 ppm, and J values are given in Hz. Wide-angle X-ray scattering (WAXS) experiments were carried out using Xenocs Xeuss 3.0 equipped with a Cu anode X-ray generator and a Dectris Pilatus 2D detector. The 2D data was then radially averaged to 1D. Careful background correction was performed before data analysis. Conductivity properties were tested on an SZT-2A Four Point Probe Tester at ambient temperature. In situ Raman spectra were collected from the PEDOT@sp2c-COF-AO-coated electrode using a Renishaw inVia confocal Raman microscope equipped with a 532 nm excitation laser. Cyclic voltammograms (CVs) and square-wave voltammograms (SWVs) were collected on a Pine WaveDrive 10 potentiostat.
Synthesis of PEDOT@sp2c-COF-AO
The parent sp2c-COF material was synthesized via the Knoevenagel condensation of 1,4-phenylenediacetonitrile and tetrakis(4-formylphenyl)pyrene, and then treated with trimethylamine-methanol solution of NH2OH·HCl at 70 °C for 6 h to afford sp2c-COF-AO. To infiltrate conductive polymers in the COF channels, sp2c-COF-AO was dispersed in acetonitrile followed by the addition of 3,4-ethylenedioxythiophene. After slowly evaporating acetonitrile, during which the 3,4-ethylenedioxythiophene molecules were driven into the COF channels, an ammonium persulfate aqueous solution was introduced into the mixture to induce the polymerization of 3,4-ethylenedioxythiophene inside the channel of sp2c-COF-AO. After being further stirred overnight at room temperature, the resulting dark green powder was isolated by filtration and dried at 50 °C under vacuum to yield PEDOT@sp2c-COF-AO (Supplementary Method).
Electrode preparation
As a typical procedure, 5 mg of PEDOT@sp2c-COF-AO (1:1) and 30 µL Nafion solution were suspended in 0.5 mL of EtOH. After being sonicated for 30 min, 1.0 mL of the resulting solution was taken out and coated on one side of the platinum foil (1 × 1 cm2). After drying under the air, the platinum foil was connected to the Pt electrode holder. The uncoated surface of the platinum foil was wrapped by EVA (ethylene-vinyl acetate copolymer), which was a type of dielectric polymer to suppress the electrolysis of water by preventing the bare platinum foil from contacting with the aqueous solution.
Uranium sorption experiments
The aqueous solutions with different uranium concentrations were obtained by diluting the stock UO2(NO3)2 ∙ 6H2O solution with the proper amount of distilled water unless otherwise indicated. The concentrations of uranium during all the experiments were detected by inductively coupled plasma-optical emission spectroscopy (ICP-OES). All the adsorption experiments were performed at ambient conditions. The concentration of uranium without being treated by sorbent material was analyzed for each sorption experiment as a negative control.
Uranium sorption performance tests from distilled water
Uranium aqueous solutions (80 mL, 9.5 ~ 398 ppm) were introduced into an electrolytic cell equipped with coated Pt foil and graphite rod, which were used as the working electrode and counter electrode, respectively. An alternating pulse current with a frequency of 400 Hz with an optimized duration ratio of 1 to 4 was applied, during which the voltage alternated between a negative value and zero (−5 − 0 V). At appropriate time intervals, aliquots (0.5 mL) were taken from the solution. The uranium concentrations in the resulting solutions were analyzed by ICP-OES. The adsorption capacity at different intervals was calculated as follows:
where V is the volume of the treated solution (mL) and m is the amount of used adsorbent (mg), and Ci and Ct are the initial concentration and the concentration of uranium at t (min), respectively.
Uranium sorption performance tests from simulated seawater, seawater, or Na2SO4 solution
The electrodeposit procedures are similar to that from distilled water, expect that simulated seawater (25.6 g L−1 NaCl and 0.198 g L−1 NaHCO3), seawater (Tampa Bay area), or Na2SO4 aqueous solution (41.5 g L−1 of Na2SO4 and 0.198 g L−1 of NaHCO3) was used as a background solution.
Selectivity tests
The electrodeposit procedures are similar to that from distilled water, except that an 80 mL aqueous solution spiked with U, Cu, and Fe at a concentration of ca. 10 ppm, respectively, was used as a background solution. The remained concentrations of U, Cu, and Fe after 24 h were analyzed by ICP-OES.
Uranium sorption kinetics by physicochemical method
160 mL of uranium aqueous solution (20 ppm) and sp2c-COF-AO (1 mg) were introduced into an Erlenmeyer flask with a magnetic stir bar. The resulting mixture was stirred at room temperature for 12 h. At appropriate time intervals, aliquots (5 mL) were taken from the mixture, and the adsorbents were separated by syringe filter (0.45 μm membrane filter). The uranium concentrations in the resulting solutions were analyzed by ICP-OES. The adsorption capacity at different intervals was calculated as follows:
where V is the volume of the treated solution (mL) and m is the amount of used adsorbent (mg), and Ci and Ct are the initial concentration and the concentration of uranium at t (min), respectively.
Electrochemistry tests
Cyclic voltammetry measurements were performed on a WaveDrive 10 potentiostat with a standard three-electrode configuration using glassy carbon (1 mm), platinum wire, and saturated silver chloride electrode as the working electrode, counter electrode, and reference electrode, respectively. Inks were prepared by combining 5 mg of COFs, 0.5 mL of ethanol solvent, and 30 µL of Nafion solution. The resulting mixture was ultrasonicated for 30 min. 20 µL ink was taken out and deposited on the glassy carbon working electrode. The experiments were performed using simulated seawater as a background solution, with initial uranium concentrations of 0 ppm and 400 ppm at room temperature, with a scan rate of 50 mV s−1.
Square-wave voltammetry measurements were performed on a WaveDrive 10 potentiostat with a standard three-electrode configuration using glassy carbon (1 mm), platinum wire, and saturated silver chloride electrode as the working electrode, counter electrode, and reference electrode, respectively. Inks and working electrodes coating preparation have followed the procedure described above. The experiments were performed in simulated seawater and sodium sulfate solution with initial uranium concentrations of 0 ppm and 400 ppm, respectively. The initial square-wave voltage was set at 0 V with an amplitude of −5 V and a frequency of 25 Hz.
DFT calculation
To investigate the interaction forces between COFs and UO22+, we employed DFT software to calculate the adsorbing energy. When using ORCA 5.0.4 software94 for structure optimization, we employ the PBE0 functional95. For light atoms (C, H, O, N), the def2-SVP basis set is used, while for the heavy atom U, the SARC-ZORA-TZVP basis set96,97,98 suitable for lanthanide and actinide metals is applied. After optimization, the single-point energy of the resulting geometry is calculated under the same functional, but with the def2-TZVP basis set99 used for the light atoms. Combining the calculated energy, we calculated the adsorption energy between COF and UO22+ using the following equation:
Here, ECOF-ion complex denotes the total energy of the COF-ion complex, ECOF and Eion are the energy of the isolated COF, or isolated ion, respectively.
The differential charge density map was calculated from molden file using ORCA5.0.4, recalculated by Multiwfn software100, and plotted using VMD 1.9.3 software101.
Data availability
The authors declare that all the data supporting the findings of this study are available within the article (and Supplementary Information files). Additional data are available from the corresponding author upon request.
References
Diallo, M. S., Kotte, M. R. & Cho, M. Mining critical metals and elements from seawater: opportunities and challenges. Environ. Sci. Technol. 49, 9390–9399 (2015).
Yang, S., Zhang, F., Ding, H., He, P. & Zhou, H. Lithium metal extraction from seawater. Joule. 2, 1648–1651 (2018).
Liu, C. et al. Lithium extraction from seawater through pulsed electrochemical intercalation. Joule. 4, 1459–1469 (2020).
Xu, J. et al. Remediation of heavy metal contaminated soil by asymmetrical alternating current electrochemistry. Nat. Commun. 10, 2440 (2019).
Wu, T. et al. Amidoxime-functionalized macroporous carbon self-refreshed electrode materials for rapid and high-capacity removal of heavy metal from water. ACS Cent. Sci. 5, 719–726 (2019).
Chou, W.-L. & Huang, Y.-H. Electrochemical removal of indium ions from aqueous solution using iron electrodes. J. Hazard. Mater. 172, 46–53 (2009).
Miller, A. T., Hassler, B. L. & Botte, G. G. Rhodium electrodeposition on nickel electrodes used for urea electrolysis. J. Appl. Electrochem. 42, 925–934 (2012).
Su, X. et al. Electrochemically-mediated selective capture of heavy metal chromium and arsenic oxyanions from water. Nat. Commun. 9, 4701 (2018).
Côté, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Ma, T. et al. Single-crystal x-ray diffraction structures of covalent organic frameworks. Science 361, 48–52 (2018).
Geng, K. et al. Covalent organic frameworks: design, synthesis, and functions. Chem. Rev. 120, 8814–8933 (2020).
Lohse, M. S. & Bein, T. Covalent organic frameworks: structures, synthesis, and applications. Adv. Funct. Mater. 28, 1705553 (2018).
Bisbey, R. P. & Dichtel, W. R. Covalent organic frameworks as a platform for multidimensional polymerization. ACS Cent. Sci. 3, 533–543 (2017).
Kandambeth, S., Dey, K. & Banerjee, R. Covalent organic frameworks: chemistry beyond the structure. J. Am. Chem. Soc. 141, 1807–1822 (2019).
Jin, Y., Hu, Y. & Zhang, W. Tessellated multiporous two-dimensional covalent organic frameworks. Nat. Rev. Chem. 1, 0056 (2017).
Rodríguez-San-Miguel, D. & Zamora, F. Processing of covalent organic frameworks: an ingredient for a material to succeed. Chem. Soc. Rev. 48, 4375–4386 (2019).
Qian, C. et al. Toward covalent organic frameworks bearing three different kinds of pores: the strategy for construction and COF-to-COF transformation via heterogeneous linker exchange. J. Am. Chem. Soc. 139, 6736–6743 (2017).
Song, Y., Sun, Q., Aguila, B. & Ma, S. Opportunities of covalent organic frameworks for advanced applications. Adv. Sci. 6, 1801410 (2019).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213 (2015).
Ding, S.-Y. et al. Construction of covalent organic framework for catalysis: Pd/COF-LZU1 in Suzuki-Miyaura coupling reaction. J. Am. Chem. Soc. 133, 19816–19822 (2011).
Chen, R. et al. Designed synthesis of a 2D porphyrin-based sp2 carbon-conjugated covalent organic framework for heterogeneous photocatalysis. Angew. Chem. Int. Ed. 58, 6430–6434 (2019).
Zhang, J., Han, X., Wu, X., Liu, Y. & Cui, Y. Multivariate chiral covalent organic frameworks with controlled crystallinity and stability for asymmetric catalysis. J. Am. Chem. Soc. 139, 8277–8285 (2017).
Pachfule, P. et al. Diacetylene functionalized covalent organic framework (COF) for photocatalytic hydrogen generation. J. Am. Chem. Soc. 140, 1423–1427 (2018).
Sun, Q. et al. Pore environment control and enhanced performance of enzymes infiltrated in covalent organic frameworks. J. Am. Chem. Soc. 140, 984–992 (2018).
Yang, Y. et al. Surface pore engineering of covalent organic frameworks for ammonia capture through synergistic multivariate and open metal site approaches. ACS Cent. Sci. 4, 748–754 (2018).
Ding, S.-Y. et al. Thioether-based fluorescent covalent organic framework for selective detection and facile removal of mercury (II). J. Am. Chem. Soc. 138, 3031–3037 (2016).
Huang, N., Zhai, L., Xu, H. & Jiang, D. Stable covalent organic frameworks for exceptional mercury removal from aqueous solutions. J. Am. Chem. Soc. 139, 2428–2434 (2017).
Kandambeth, S. et al. R. Selective molecular sieving in self-standing porous covalent-organic-framework membranes. Adv. Mater. 29, 1603945 (2017).
Lu, Q. et al. Postsynthetic functionalization of three-dimensional covalent organic frameworks for selective extraction of lanthanide ions. Angew. Chem. Int. Ed. 57, 6042–6048 (2018).
Sun, Q. et al. Covalent organic frameworks as a decorating platform for utilization and affinity enhancement of chelating sites for radionuclide sequestration. Adv. Mater. 30, 1705479 (2018).
Furukawa, H. & Yaghi, O. M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications. J. Am. Chem. Soc. 131, 8875–8883 (2009).
Yuan, C. et al. Nanochannels of covalent organic frameworks for chiral selective transmembrane transport of amino acids. J. Am. Chem. Soc. 141, 20187–20197 (2019).
Jhulki, S. et al. Humidity sensing through reversible isomerization of a covalent organic framework. J. Am. Chem. Soc. 142, 783–791 (2020).
Ascherl, L. et al. Solvatochromic covalent organic frameworks. Nat. Commun. 9, 3802 (2018).
Guo, Z. et al. Fast ion transport pathway provided by polyethylene glycol confined in covalent organic frameworks. J. Am. Chem. Soc. 141, 1923–1927 (2019).
Xie, Z. et al. Stable 2D heteroporous covalent organic frameworks for efficient ionic conduction. Angew. Chem. Int. Ed. 58, 15742–15746 (2019).
Karmakar, A. et al. Thermo-responsive MOF/Polymer composites for temperature-mediated water capture and release. Angew. Chem. Int. Ed. 59, 2–9 (2020).
Sun, Q. et al. Reaction environment modification in covalent organic frameworks for catalytic performance enhancement. Angew. Chem. Int. Ed. 58, 8670–8675 (2019).
Sun, Q., Aguila, B., Perman, J., Nguyen, N. & Ma, S. Flexibility matters: cooperative active sites in covalent organic framework and threaded ionic polymer. J. Am. Chem. Soc. 138, 15790–15796 (2016).
Vitaku, E. et al. Phenazine-based covalent organic framework cathode materials with high energy and power densities. J. Am. Chem. Soc. 142, 16–20 (2020).
Mulzer, C. R. et al. Superior charge storage and power density of a conducting polymer-modified covalent organic framework. ACS Cent. Sci. 2, 667–673 (2016).
Abney, C. W., Mayes, R. T., Saito, T. & Dai, S. Materials for the recovery of uranium from seawater. Chem. Rev. 117, 13935–14013 (2017).
Lu, Y. Uranium extraction: coordination chemistry in the ocean. Nat. Chem. 6, 175–177 (2014).
Xie, Y. et al. Uranium extraction from seawater: material design, emerging technologies and marine engineering. Chem. Soc. Rev. 52, 97–162 (2023).
Jin, E. et al. Two-dimensional sp2 carbon-conjugated covalent organic frameworks. Science 357, 673–676 (2017).
Bai, S. et al. Two-dimensional semiconducting covalent organic frameworks via condensation at arylmethyl carbon atoms. Nat. Commun. 10, 2467 (2019).
Zhao, Y. et al. Fully conjugated two-dimensional sp2-carbon covalent organic frameworks as artificial photosystem I with high efficiency. Angew. Chem. Int. Ed. 58, 5376–5381 (2019).
Cui, W.-R. et al. Regenerable and stable sp2 carbon-conjugated covalent organic frameworks for selective detection and extraction of uranium. Nat. Commun. 11, 436 (2020).
Kayser, L. V. & Lipomi, D. J. Stretchable conductive polymers and composites based on PEDOT and PEDOT:PSS. Adv. Mater. 31, 1806133 (2019).
Hennig, C., keda-Ohno, A. I., Emmerling, F., Kraus, W. & Bernhard, G. Comparative investigation of the solution species [U(CO3)5]6− and the crystal structure of Na6[U(CO3)5]·12H2. O. Dalton Trans. 39, 3744–3750 (2010).
Ikeda, A. et al. Comparative study of uranyl (VI) and -(V) carbonato complexes in an aqueous solution. Inorg. Chem. 46, 4212–4219 (2007).
Guin, S. K., Ambolikar, A. S. & Kamat, J. V. Electrochemistry of actinides on reduced graphene oxide: craving for the simultaneous voltammetric determination of uranium and plutonium in nuclear fuel. RSC Adv. 5, 59437–59446 (2015).
Kim, Y. K., Lee, S., Ryu, J. & Park, H. Solar conversion of seawater uranium (VI) using TiO2 electrodes. Appl. Catal. B Environ. 163, 584–590 (2015).
Gupta, R., Jayachandran, K. & Aggarwal, S. K. Single-walled carbon nanotube (SWCNT) modified gold (Au) electrode for simultaneous determination of plutonium and uranium. RSC Adv. 3, 13491–13496 (2013).
Chen, D. et al. Self-standing porous aromatic framework electrodes for efficient electrochemical uranium extraction. ACS Cent. Sci. 9, 2326–2332 (2023).
Yan, H. K., Kou, Z. H., Li, S. X. & Zhang, T. Synthesis of sp2 carbon-conjugated covalent organic framework thin-films via copper-surface-mediated Knoevenagel polycondensation. Small 19, 2207972 (2023).
Ye, H. et al. Amyloid-like coatings decorated electrodes boost the uranium electro-adsorption from seawater. Chem. Eng. J. 451, 138615 (2022).
Wang, C. et al. Uranium in situ electrolytic deposition with a reusable functional graphene-foam electrode. Adv. Mater. 33, 2102633 (2021).
Zhang, C.-R. et al. A novel 3D sp2 carbon-linked covalent organic framework as a platform for efficient electro-extraction of uranium. Sci. China Chem. 66, 562–569 (2023).
Liu, C. et al. A half-wave rectified alternating current electrochemical method for uranium extraction from seawater. Nat. Energy 2, 17007 (2017).
Wang, Z. et al. Constructing an ion pathway for uranium extraction from seawater. Chem 6, 1683–1691 (2020).
Wang, Z. et al. Constructing uranyl-specific nanofluidic channels for unipolar ionic transport to realize ultrafast uranium extraction. J. Am. Chem. Soc. 143, 14523–14529 (2021).
Vivero-Escoto, J. L., Carboni, M., Abney, C. W., deKrafft, K. E. & Lin, W. Organo-functionalized mesoporous silicas for efficient uranium extraction. Micropor. Mesopor. Mater. 180, 22–31 (2013).
Xie, S. et al. Electrospun nanofibrous adsorbents for uranium extraction from seawater. J. Mater. Chem. A 3, 2552–2558 (2015).
Das, S. et al. Extracting uranium from seawater: promising AF series adsorbents. Int. Eng. Chem. Res. 55, 4110–4117 (2016).
Gunathilake, C., Gόrka, J., Dai, S. & Jaroniec, M. Amidoxime-modified mesoporous silica for uranium adsorption under seawater conditions. J. Mater. Chem. A. 3, 11650–11659 (2015).
Wang, L. et al. Loading actinides in multilayered structures for nuclear waste treatment: the first case study of uranium capture with vanadium carbide MXene. ACS Appl. Mater. Interfaces 8, 16396–16403 (2016).
Li, Y. et al. Pore-free matrix with cooperative chelating of hyperbranched ligands for high-performance separation of uranium. ACS Appl. Mater. Interfaces 8, 28853–28861 (2016).
Chatterjee, S. et al. Synthesis of naphthalimidedioxime ligand-containing fibers for uranium adsorption from seawater. Int. Eng. Chem. Res. 55, 4161–4169 (2016).
Sun, Q. et al. Spatial engineering direct cooperativity between binding sites for uranium sequestration. Adv. Sci. 8, 2001573 (2021).
Ling, L. & Zhang, W. -x Enrichment and encapsulation of uranium with iron nanoparticle. J. Am. Chem. Soc. 137, 2788–2791 (2015).
Song, Y. et al. Nanospace decoration with uranyl-specific “hooks” for selective uranium extraction from seawater with ultrahigh enrichment index. ACS Cent. Sci. 7, 1650–1656 (2021).
Sun, Q. et al. Bio-inspired nano-traps for uranium extraction from seawater and recovery from nuclear waste. Nat. Commun. 9, 1644 (2018).
Yuan, Y. et al. Ultrafast and highly selective uranium extraction from seawater by hydrogel-like spidroin-based protein fiber. Angew. Chem. Int. Ed. 58, 11785–11790 (2019).
Zhao, S. et al. A dual-surface amidoximated halloysite nanotube for high-efficiency economical uranium extraction from seawater. Angew. Chem. Int. Ed. 58, 14979–14985 (2019).
Yuan, Y. et al. A bio-inspired nano-pocket spatial structure for targeting uranyl capture. Angew. Chem. Int. Ed. 59, 4262–4268 (2020).
Yan, B., Ma, C., Gao, J., Yuan, Y. & Wang, N. An ion-crosslinked supramolecular hydrogel for ultrahigh and fast uranium recovery from seawater. Adv. Mater. 32, 1906615 (2020).
Yuan, Y. et al. DNA nano-pocket for ultra-selective uranyl extraction from seawater. Nat. Commun. 11, 5708 (2020).
Yu, B. et al. De novo synthesis of bifunctional conjugated microporous polymers for synergistic coordination mediated uranium entrapment. Nano Res. 14, 788–796 (2021).
Zhang, L. et al. Skeleton engineering of homocoupled conjugated microporous polymers for highly efficient uranium capture via synergistic coordination. ACS Appl. Mater. Interfaces 12, 3688–3696 (2020).
Aguila, B. et al. Design strategies to enhance amidoxime chelators for uranium recovery. ACS Appl. Mater. Interfaces 11, 30919–30926 (2019).
Li, B. et al. Functionalized porous aromatic framework for efficient uranium adsorption from aqueous solutions. ACS Appl. Mater. Interfaces 9, 12511–12517 (2017).
Hao, M. et al. Modulating uranium extraction performance of multivariate covalent oganic frameworks through donor-acceptor linkers and amidoxime nanotraps. JACS Au. 3, 239–251 (2023).
Chen, Z. et al. Tuning excited state electronic structure and charge transport in covalent organic frameworks for enhanced photocatalytic performance. Nat. Commun. 14, 1106 (2023).
Yang, H. et al. Tuning local charge distribution in multicomponent covalent organic frameworks for dramatically enhanced photocatalytic uranium extraction. Angew. Chem. Int. Ed. 62, e202303129 (2023).
Hao, M. et al. Converging cooperative functions into the nanospace of covalent organic frameworks for efficient uranium extraction from seawater. CCS Chem. 4, 2294–2307 (2022).
Colmenero, F., Bonales, L. J., Cobos, J. & Timón, V. Density functional theory study of the thermodynamic and raman vibrational properties of γ-UO3 polymorph. J. Phys. Chem. C. 121, 14507–14516 (2017).
Liu, X. et al. Secondary metal ion-induced electrochemical reduction of U(VI) to U(IV) solids. Nat. Commun. 15, 7736 (2024).
Mollick, S. et al. Benchmark uranium extraction from seawater using an ionic macroporous metal-organic framework. Energy Environ. Sci. 15, 3462–3469 (2022).
More, Y. D. et al. Nanotrap grafted anionic MOF for superior uranium extraction from seawater. Small 20, 2302014 (2024).
Sweet, L. E. et al. Investigation of the polymorphs and hydrolysis of uranium trioxide. J. Radioanal. Nucl. Chem. 296, 105–110 (2013).
Yang, H. et al. Functionalized iron-nitrogen-carbon electrocatalyst provides a reversible electron transfer platform for efficient uranium extraction from seawater. Adv. Mater. 33, 2106621 (2021).
Liu, X. et al. Highly efficient electrocatalytic uranium extraction from seawater over an amidoxime-functionalized In-N-C catalyst. Adv. Sci. 9, 2201735 (2022).
Neese, F. et al. WIREs Computational Molecular Science, 12, e1606 (2022).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Rolfes, J. D., Neese, F. & Pantazis, D. A. All-electron scalar relativistic basis sets for the elements Rb–Xe. J. Comput. Chem. 41, 1842–1849 (2020).
van Lenthe, E., Baerends, E. J. & Snijders, J. G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 99, 4597–4610 (1993).
van Lenthe, E., Baerends, E. J. & Snijders, J. G. Relativistic total energy using regular approximations. J. Chem. Phys. 101, 9783–9792 (1994).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
This work was supported by the DOE Office of Nuclear Energy’s Nuclear Energy University Program (Grant No. DE-NE0008281 to S.M.); partial financial support from the US National Science Foundation (CBET-1706025 to S.M.) and the Robert A. Welch Foundation (B-0027 to S.M.) is also acknowledged. The authors also extend their appreciation to the Deanship of Scientific Research at Princess Nourah Bint Abdulrahman University for partially funding this work through the Visiting Researcher Program.
Author information
Authors and Affiliations
Contributions
Q.S. and S.M. conceived and designed the research. Y.S., L.H., and P.C.L. performed the synthesis and carried out the adsorption tests. X.Z. Conducted density functional theory (DFT) calculations. J.L., J.L., and D.Z. performed and analyzed the HAADF-STEM images. Z.D. and T.A. participated in discussing the test results. All authors participated in drafting the paper and gave approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The Authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pradip Pachfule and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Song, Y., Hou, L., Lan, P.C. et al. Creating electrochemical accessibility in covalent organic frameworks for uranium extraction via electrodeposition. Nat Commun 16, 7093 (2025). https://doi.org/10.1038/s41467-025-62501-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62501-7
