
Abstract
Hearing loss is one of the most prevalent sensory disorders, but no commercial biological treatments are currently available. Here, we identify an East Asia-specific founder mutation, the homozygous c.220 C > T mutation in MPZL2, that contributes to a significant proportion of hereditary deafness cases in our cohort study. We find that the disease-causing mutation can be targetable by adenine base editors (ABEs) that enable A·T-to-G·C base corrections without DNA double-strand breaks. To demonstrate this, we develop a humanized mouse model (hMPZL2Q74X/Q74X) that recapitulates human MPZL2 deafness and leads to progressive hearing loss. A PAM-flexible ABE variant with reduced bystander and off-target effects (ABE8eWQ-SpRY:sgRNA3) is packaged in dual adeno-associated viruses (AAVs) and injected into the inner ear of hMPZL2Q74X/Q74X mice and effectively corrects the mutation. This treatment significantly restores hearing function, improves inner ear structural integrity, and reverses altered gene expression. Base editing may hold therapeutic potential for hereditary deafness, including most cases of MPZL2 deafness.
Similar content being viewed by others


Introduction
Hearing loss is the most common sensory disorder in humans and affects about 466 million people worldwide (World Health Organization, https://who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss). Although no current treatments are capable of fully restoring biological hearing function1,2, advancements in our understanding of the genetic architecture and molecular mechanisms of hearing loss have led to significant progress in inner ear gene therapies3,4. Notably, CRISPR genome editing technologies have been harnessed to directly correct disease-causing mutations for the fundamental treatment of genetic disorders5. In particular, base editors, including cytosine base editors (CBEs) and adenine base editors (ABEs), are engineered genome-editing tools that enable the precise repair of targeted genomic base pairs without double-strand breaks6,7. Presently, ABEs and CBEs have been successfully used in treating hearing loss in hereditary deafness mouse models8,9, suggesting that base editors hold potential as one-time treatments for hereditary deafness caused by pathogenic point mutations.
Recessive loss-of-function mutations represent approximately 80% of hereditary deafness cases10. One of these, non-syndromic autosomal recessive deafness-111 (DFNB111), is a leading cause of mild-to-moderate progressive hearing loss11. Here, we identify an East Asia-specific founder mutation of DFNB111, the homozygous c.220 C > T variant in MPZL2, that contributes to a significant proportion of hereditary deafness cases in our cohort study. To model DFNB111, we generate a humanized MPZL2 c.220 C > T knock-in mouse model that replicates the human condition. For in vivo editing, we test various combinations of ABE variants, four types of Cas9 variants with different PAMs, and single-guide RNA (sgRNA)s to correct the c.220 C > T mutation. Ultimately, we determine the optimal PAM-flexible ABE variant (i.e., ABE8eWQ-SpRY:sgRNA3), which achieves a 60% editing efficiency in vitro with no detectable bystander or off-target effects. The administration into the inner ear of ABE8eWQ-SpRY:sgRNA3 packaged in dual AAV-ie vectors successfully restores both auditory function and the structural integrity of the inner ear. The development of humanized mouse models and the successful correction of the MPZL2 founder mutation using a single PAM-flexible ABE together represent a significant advancement in base editor gene therapy, suggesting great promise for treating hereditary deafness, including most DFNB111 cases.
Results
Clinical significance of the East Asia-specific founder mutation in MPZL2-related DFNB111
We reviewed our sensorineural hearing loss (SNHL) cohort, which consisted of 1437 unrelated families attending the hereditary deafness clinic within the Otorhinolaryngology Division at two tertiary centers (Seoul National University Hospital (SNUH) and the Eye & ENT Hospital, Fudan University (EENT)), between March 2021 and February 2024. Within this cohort, a filtering process yielded 234 pediatric probands ( ≤ 18 years old) with symmetric, mild-to-moderate (range 21–55 dB hearing threshold), non-syndromic SNHL (Fig. 1a). Through comprehensive genetic testing using exome/genome sequencing, we identified disease-causing mutations in 155 probands (65.7%). The detailed genotypes and their pathogenicity are described in Supplementary Data 1. Collectively, 17 deafness genes that were seen in 2 or more probands were identified as disease-causing within these 155 genetically diagnosed families. GJB2 was the most frequently affected gene (36.8%, 57/155), followed by STRC (18.1%, 28/155) and MPZL2 (9.0%, 14/155) (Fig. 1b). In our cohort, we identified a total of 24 MPZL2-associated DFNB111 patients from 20 unrelated families, regardless of age at ascertainment. The pedigrees, genotypes, and hearing loss phenotypes are presented in Fig. 1c, d. The significant contribution of the MPZL2 c.220 C > T mutation to DFNB111 was found. This C → T mutation at position c.220 (c.220 C ⋅G to T⋅A) creates a stop codon (TAG) that replaces the Gln codon (CAG) at p.74 (Q74X), likely resulting in nonsense-meditated mRNA decay or a truncated protein. In our DFNB111 patients, 23 (95.8%) harbored at least one c.220 C > T allele. Notably, 19 of these patients (79.2%) were homozygous for the c.220 C > T mutation (Fig. 1e). The mutational landscape of DFNB111, as documented in the literature and including our cohort, is illustrated in Supplementary Fig. 1a, highlighting the high mutational burden of the c.220 C > T mutation. The c.220 C > T allele recurred frequently, especially in East Asian populations, suggesting a founder mutation in East Asia (Fig. 1f). In contrast, the second most frequently recurrent allele, c.72delC, was found across different genetic ancestries. Auditory function-gene profiles exhibited significant progressive hearing loss over the course of decades (Fig. 1g), specifically an annual progression of hearing loss of 0.61 dB at low frequencies, 0.57 dB at middle frequencies, and 0.61 dB at high frequencies (Supplementary Fig. 1b). Collectively, we hypothesized that a single ABE that corrects the c.220 C > T founder mutation could serve as a “one-and-done” therapy, potentially treating or even curing the majority of DFNB111 patients, which constitute a significant proportion of all hereditary deafness cases.

a Schematic flow diagram illustrating the genetic diagnosis study of symmetric, mild-to-moderate, non-syndromic sensorineural hearing loss (ns-SNHL) in children from two tertiary centers. b Gene signatures of symmetric, mild-to-moderate, ns-SNHL in children. The bar plot shows the frequencies of 17 deafness genes that were seen in 2 or more probands from 155 genetically diagnosed families. c Pedigrees and genotypes of 24 affected patients from 20 unrelated DFNB111 families. Arrows, probands; Black filled circles or rectangles, affected patients. d Serial audiograms of 23 affected DFNB111 patients. Red, right ear; Blue, left ear. e MPZL2 mutational landscape on a Lollipop plot (upper), and the prevalence of MPZL2 in trans mutation combinations in our cohort (lower). f The mutational burden of different MPZL2 mutations depending on the genetic ancestry groups in the Chord diagram. g The natural course of hearing loss in DFNB111 patients over decades across different hearing frequencies. Source data for all relevant panels are provided within the Source Data file.
Generation and characterization of the MPZL2 humanized mouse models
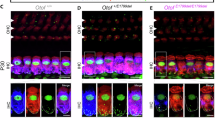
Because the mouse Mpzl2 and human MPZL2 sequences differ near the c.220 C > T mutation, we decided to develop a humanized knock-in mouse model of DFNB111 by inserting 648 bp of human MPZL2 complementary DNA (cDNA) containing the pathogenic c.220 C > T mutation into the mouse Mpzl2 locus (Fig. 2a). The integration of the human cassette and successful on-targeting was verified (Supplementary Fig. 2a). Additionally, the sequence of the hMPZL2Q74X mice was confirmed using Sanger sequencing (Fig. 2b). In the humanized knock-in mouse model of DFNB111 (hMPZL2Q74X/Q74X), the mRNA transcript levels of human MPZL2 and mouse Mpzl2 were undetectable across tissues at P4 and P28, in contrast to the normal expression observed in Mpzl2 wild type (WT) mice (Fig. 2c and Supplementary Fig. 2b). Consistent with previous studies12,13, we found that MPZL2 protein was expressed in the organ of Corti, primarily in outer hair cells (OHCs), inner hair cells (IHCs), Deiters’ cells (DCs), and at the contact between DCs and the basilar membrane, as well as pillar cells, the lateral wall of spiral limbus, basal cells of stria vascularis, fibrocyte type II of spiral ligament, and root cells. These findings were observed during both the neonatal period (P4) and the adult period (P28) in Mpzl2 WT mice (Supplementary Fig. 2c, d). Conversely, the MPZL2 protein expression was absent in hMPZL2 Q74X/Q74X mice at both P4 and P28. At 4 weeks of age, hMPZL2 Q74X/Q74X mice exhibited an elevation of 14.5 dB at 24 kHz (P = 0.02) and 23.5 dB at 32 kHz (P < 0.001) in auditory brainstem response (ABR) thresholds compared to Mpzl2 WT mice (Fig. 2d), indicating the onset of hearing loss beginning at 4 weeks, specifically at higher frequencies. Similar to an Mpzl2 knock-out mouse model13, hMPZL2 Q74X/Q74X mice initially displayed mild-to-moderate hearing loss, which progressively worsened to severe hearing loss. At 8 weeks of age, hMPZL2 Q74X/Q74X mice showed elevated thresholds of 24.5 dB at click (P = 0.001), 16.0 dB at 4 kHz (P = 0.007), 16.0 dB at 8 kHz (P = 0.019), 41.0 dB at 16 and 24 kHz, and 42.0 dB at 32 kHz (16-32 kHz, all P < 0.0001) compared to Mpzl2 WT. At 12 weeks of age, the ABR thresholds in hMPZL2Q74X/Q74X mice exceeded 70 dB across the click and 4–32 kHz frequencies (P < 0.0001), except at 8 kHz. In contrast, hMPZL2Q74X/WT mice retained ABR thresholds comparable to Mpzl2 WT mice (Fig. 2d). We also performed distortion product otoacoustic emission (DPOAE) measurements to assess the function of OHCs. Similarly, DPOAE thresholds in hMPZL2Q74X/Q74X mice were significantly higher than those in hMPZL2Q74X/WT and Mpzl2 WT mice (Fig. 2e), suggesting that the MPZL2 c.220 C > T mutation reduces the functionality of OHCs. In hMPZL2Q74X/Q74X mice, 24 kHz thresholds were elevated by 16.0 dB at 4 weeks (P = 0.01) compared to Mpzl2 WT. At 8 weeks, thresholds increased by 28.0 dB at 16 kHz (P < 0.0001) and 26.0 dB at 24 kHz (P = 0.001). At 12 weeks, elevations were 32.5 dB at 16 kHz (P < 0.0001), 26.5 dB at 24 kHz (P < 0.0001), and 12.5 dB at 32 kHz (P = 0.002) compared to Mpzl2 WT. In hMPZL2Q74X/Q74X mice, a significant loss of OHCs and DCs in the organ of Corti was observed, particularly in the mid- and high-frequency regions of the cochlea, along with cellular disarrangement and a collapsed tunnel of Corti (Fig. 2f and Supplementary Fig. 3a). Quantification of OHCs in the middle and basal turns showed significant differences between Mpzl2 WT and hMPZL2Q74X/Q74X, as well as between hMPZL2Q74X/WT and hMPZL2Q74X/Q74X mice (P < 0.001). DC counts in the basal turn also differed significantly in both comparisons (P < 0.0001). In contrast, no distinct structural abnormalities were observed in the stria vascularis or spiral ligament, and the density of spiral ganglion neurons (SGNs) remained unaffected (Supplementary Fig. 3b–d and Supplementary Fig. 4). To evaluate whether the hearing loss in hMPZL2Q74X/Q74X mice is affected by the insertion of human cDNA into the mouse Mpzl2 locus, we also generated another humanized MPZL2 WT mouse model (hMPZL2WT/WT) using a sequential knock-in strategy (Supplementary Fig. 5a), and we confirmed successful on-target integration and sequence integrity of the human MPZL2 wild-type allele (Supplementary Fig. 5b, c), along with detectable mRNA transcript levels of MPZL2 (Supplementary Fig. 5d). The hMPZL2WT/WT mice exhibited normal thresholds across frequencies at 12 weeks of age, comparable to Mpzl2 WT mice (Supplementary Fig. 6a, b), and displayed normal inner ear structures (Supplementary Fig. 6c, d). These observations indicate that the auditory and inner ear phenotypes in hMPZL2Q74X/Q74X mice are specifically caused by the pathogenic c.220 C > T mutation.

a Targeting strategy for the humanized MPZL2 mouse model (hMPZL2Q74X) using the CRISPR/Cas9 system. b Sequence verification of hMPZL2Q74X mice and Mpzl2 WT mice using Sanger sequencing. c RT-PCR analysis to detect MPZL2 and Mpzl2 expression in P4 hMPZL2Q74X/Q74X and Mpzl2 WT mice. d, e Comparison of ABR and DPOAE thresholds among Mpzl2 WT (red, n = 10), hMPZL2Q74X/WT (green, n = 10), and hMPZL2Q74X/Q74X (blue, n = 10) mice at 4, 8, and 12 weeks of age. Data are presented as the mean ± SEM. Statistical significance was determined using one-way ANOVA with Bonferroni’s multiple comparisons test. Significance levels are indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. f Representative section images (10 μm) from 12-week-old Mpzl2 WT mice (n = 1), hMPZL2Q74X/WT mice (n = 1), and hMPZL2Q74X/Q74X mice (n = 2), immunolabeled with Myosin VIIa (HCs, red) and anti-Sox2 (SCs, green) of the organ of Corti in 12 weeks of age. Arrowheads indicate IHCs and DCs, white arrows point to OHCs, and the white line marks SCs. Asterisks indicate loss of OHCs and DCs. Scale bar, 25 μm. Data are shown as the mean ± SEM. Statistical significance was determined using one-way ANOVA with Bonferroni’s multiple comparisons tests, and significance levels are indicated as ****P < 0.0001. g Schematic diagram of the RNA-sequencing analysis (n = 3). h Heatmap analysis of DEGs (upregulation, red; downregulation, blue). i Volcano plot of DEGs (upregulation, red; downregulation, blue), (n = 3, statistical significance was evaluated using a two-sided test (P < 0.05)). j Gene ontology (GO) enrichment analysis in biological processes (BP). k Top 20 GO BP terms associated with cell adhesion, basement membrane organization, and ECM organization. l Quantitative RT-PCR assay in triplicate to validate the DEGs of interest (n = 3, biological replicates): COL9A1/2/3, EMILIN1, IBSP, POSTN, TNXB, LAMA1/2. Statistical significance was determined using one way ANOVA with Kruskal–Wallis test. Data are presented as the mean ± SEM. Source data for all relevant panels are provided within the Source Data file.
We subsequently conducted RNA-seq analysis to explore the molecular pathways involved in the cochlear histopathology caused by the c.220 C > T mutation. Cochlear membranous labyrinth tissue from hMPZL2Q74X/Q74X and Mpzl2 WT mice were dissected at P28 and used for RNA-seq analysis (Fig. 2g). Principal Component Analysis of the total RNA-seq data revealed a distinct clustered pattern of gene expression, and a heatmap visualized the patterns and clusters of individual variable values between hMPZL2Q74X/Q74X and Mpzl2 WT mice (Fig. 2h). We identified a total of 1254 differentially expressed genes (DEGs) (Fig. 2i), and Gene Ontology (GO) analysis of the DEGs revealed significant enrichment in biological process, cellular component, and molecular function terms (Fig. 2j and Supplementary Fig. 7). REVIGO analysis further highlighted significant enrichment in biological processes related to cell adhesion, extracellular matrix (ECM) organization, and basement membrane integrity (Fig. 2k). We filtered the DEGs using the gEAR (http://umgear.org) and SHIELD databases (http://shield.hms.harvard.edu), and we further refined the list to focus on DEGs associated with cell adhesion, ECM organization, and basement membrane integrity (Supplementary Data 2). The associated transcripts, including COL9A1/2/3, EMILIN1, IBSP, POSTN, TNXB, and LAMA1/2, identified through the RNA-seq analysis were validated by quantitative RT-PCR (Fig. 2l). Collectively, the RNA-seq results suggested that the MPZL2 c.220 C > T mutation disrupts cellular pathways involved in cell adhesion, ECM organization, and basement membrane integrity, thus contributing to the histopathological changes observed in hMPZL2Q74X/Q74X mice.
In vitro selection of the optimal ABE and sgRNA combination to correct the MPZL2 c.220 C > T mutation
To select the best base editing system in vitro, we constructed an endogenous HEK293T cell line that mimics the pathology of human DFNB111 patients by introducing a nucleotide change (c.220 C > T) at the MPZL2 genome locus, resulting in a nonsense mutation (p.Q74X), and used it as a model for base editor screening (Fig. 3a and Supplementary Fig. 8). For successful ABE-based base editing, an appropriate PAM is required to position the target base within the optimal editing window, approximately 3–8 positions from the 5’ end of the protospacer. Thus, we searched for an NGG PAM sequence that the nCas9 of ABE could recognize near the target adenine. However, the target site only had one sgRNA binding site with the only available NGG PAM, which placed the target adenine at spacer position 2 (i.e., A2), which was outside the editing window where ABE works best (Fig. 3b). Given the lack of suitable NGG PAMs, we explored the use of engineered or evolved Cas9 nuclease-based ABE variants targeting non-NGG PAM. Thus, we included four types of Cas9 variants with different PAM requirements: NG-Cas914 (NG), SpRY15 (NNN; NAN, NTN, and NCN), and eNme2-C16(N4NC) in addition to the canonical SpCas917 (NGG). Moreover, we also considered different ABE platforms including ABEmax, ABE8eWQ, and ABE8e18, which exhibited different editing activities and different editing windows. Consequently, we reconstituted a total of 14 ABE:sgRNA combinations using different adenine deaminases and Cas9 variants, and we designed all possible 6 sgRNAs to more optimally target the c.220 C > T mutation in the MPZL2 allele. The target adenine was at position 2, 4, 5, 6 and 7 with the corresponding AGG, AG, TAG, CTA, TCT, and AGGACG PAMs, and these were referred to as sgRNA1–6. All ABE:sgRNA combinations were co-transfected into the HEK293T-MPZL2 mutant cells, and the on-target editing efficiency was analyzed by targeted deep sequencing. The results showed that SpRY-based ABE with sgRNA3 (NAN; TAG PAM) and sgRNA4 (NTN; CTA PAM) induced significantly higher levels of A-to-G editing at the target adenine compared to the other ABE:sgRNA combinations (Fig. 3c). We thoroughly evaluated unintended bystander edit events (e.g., A-to-G or C-to-other (T/G/A) edits). Notably, the combination of ABE8eWQ-SpRY:sgRNA3 showed high precision by inducing less bystander editing and preserving high targeted editing efficiency (Bystander A1: 0.14%, A2: 0.15% and Target A5: 51.8%, Silent A9: 0.84%) (Fig. 3c). While ABE8e-SpRY induced base editing at both the target adenine and bystander adenines (Bystander A1: 0.33%, A2: 1.77% and Target A5: 42.41%, Silent A9: 37.30%). Also, no cytosine bystander event was observed at both AC*N and TC*N motifs present within the targetable window (Fig. 3c, d). This indicates that ABE8e had a wider editing window (3–10 bp) than ABE8eWQ (4–8 bp), and reiterate that ABE-induced cytosine deaminase activity was less significant compared to its adenine deamination activity which is consistent with previous reports19,20. Taken together, our data showed that it would be best to use ABE8eWQ-SpRY and ABEmax-SpRY with sgRNA3 and sgRNA4, respectively, to correct the target c.220 C > T in exon 2 of MPZL2. Then we finalized the combination of ABE8eWQ-SpRY:sgRNA3 for further in vivo study considering that ABE8eWQ is known to have fewer sgRNA-independent RNA off-target effects compared to ABEmax21,22.

a Schematic representation of a correction strategy using PAM-flexible ABEs in HEK293T monoclonal cells harboring the homozygous C > T nonsense mutation (c.220 C > T: p.Q74X) at the MPZL2 locus. b Design of sgRNAs targeting the MPZL2 c.220 C > T mutation. For more effective editing, the target adenine A needs to be within ABE’s preferred targeting window. In the table below, 6 sgRNA candidates featuring different PAMs are listed, each placing the target A (red) at different positions on the protospacer: A2, A4, A5, A6, and A7. c Comparison of the A-to-G editing efficiencies in vitro at the target adenine and bystander adenines for all ABEs used in this study (n = 3 for all conditions, except for ABE8eWQ:sgRNA3, for which n = 4). d C-to-other editing efficiencies at either AC*N and TC*N motifs within the targetable window of the selected SpRY-based ABEs. The data is from c (Mean values, same as in c, n = 3, except for ABE8eWQ:sgRNA3, for which n = 4). e Potential off-target sites identified by Cas-OFFinder and GUIDE-seq. The venn diagram shows the degree of off-target site overlap between the two methods (f) Heatmap showing A-to-G conversion rates at off-target sites predicted using Cas-OFFinder following ABE (sgRNA3 + ) treatment. g Validation of A-to-G conversion rates at GUIDE-seq-detected off-target sites after ABE (sgRNA3 + ) treatment. Sites that showed differences between control (untreated) and ABE (sg3 + ) samples are marked with an asterisk (*) and labeled by genomic locus, and coding region is shown in purple text. A purple asterisk (*) indicates overlapping site detected by both methods. h Targeted RNA off-target effects in HEK293T cells mimicking the MPZL2 c.220 C > T variant with three SpRY ABEs that were expressed using NAN PAM sgRNA. The average frequency of A-to-I transitions in three mRNA transcripts (CCNB1IP1, AARS1, and TOPORS) with each of the SpRY-mediated ABE variants. Source data for all relevant panels are provided within the Source Data file.
Off-target analysis in the human genome
To evaluate potential sgRNA-dependent off-target reactions in HEK293T-MPZL2 mutant cells treated with ABE8eWQ-SpRY:sgRNA3, we screened for genome-wide off-target sites for SpRY using Cas-OFFinder software23, and we performed a PAMless NNN search that allowed for up to 2 mismatches and/or 1 DNA/RNA bulge outside the seed region at position 10–18 from the 5’ end. We identified 18 potential off-target sites (OT1–OT18) in the human genome (Fig. 3e, Supplementary Data 3). In all, base editing rates at the off-target sites were similar to controls, but we only detected a slight increase in the A-to-G conversion rate at position 5 at OT14, which is in the exon regions of the CDC34 gene (Fig. 3f). However, there have been no clinical phenotypes associated with the CDC34 gene (https://www.omim.org/). We were also able to detect new potential off-target sites by GUIDE-seq24, in addition to the previously identified sites. One of these sites overlapped with the off-target sites, OT14; CDC34 gene identified by Cas-OFFinder (Fig. 3e, Supplementary Data 3). To determine whether base-editing events induced by ABE occurred at these sites, we performed targeted deep sequencing on sites that had a GUIDE-seq read count >5. Of these, except for overlapping site, the real off-target activity was observed at 4 sites (OT2, OT3, OT8 and OT11), which are in non-coding regions such as intergenic and intronic regions (Fig. 3g).
We next evaluated sgRNA-independent RNA off-targets of ABE8eWQ-SpRY compared with two other ABE variants, ABEmax-SpRY and ABE8e-SpRY. We co-transfected each ABE variant along with sgRNA3 into HEK293T-MPZL2 mutant cells and analyzed the frequency of A-to-I conversion in three representative RNA transcripts (CCNB1IP1, AARS1, and TOPORS)22,25. Across all three mRNA transcripts, ABE8eWQ-SpRY induced lower RNA off-target effects compared to ABEmax-SpRY and ABE8e-SpRY, and these were similar to controls (Fig. 3h). These data further support that ABE8eWQ-SpRY was the best option in our case.
In vivo correction of the MPZL2 c.220C > T mutation by dual AAV-ie-ABE8eWQ-SpRY:sgRNA3
To efficiently deliver ABE8eWQ-SpRY:sgRNA3 to the cochlea, we used AAV-ie, a variant with superior inner ear tropism and minimal ototoxicity that is known to target cochlear hair cell (HCs) and supporting cell (SCs) more effectively than other AAV serotypes12,26. Given the limitation of AAVs’ small packaging capacity ( ~ 4.7 kb), we used a split intein-mediated AAV system to generate the dual vector of ABE8eWQ-SpRY:sgRNA3. The ABE was split into the amino-terminal (N) and carboxy-terminal (C) halves at amino acid residue 574 of SpRY (Fig. 4a). Dual plasmids encoding C terminal and N terminal of ABE8eWQ-SpRY:sgRNA3 were co-transfected into these two cell lines, and on-target editing efficiency and bystander edits were analyzed before administering the dual AAV system in vivo. The editing efficiency of ABE8eWQ-SpRY:sgRNA3 in endogenous HEK293T-MPZL2 c.220 C > T cell line was: Bystander A1: 0.12%, A2: 0.60% and Target A5: 50.71%, Silent A9: 0.57%, and no cytosine bystander event was observed (Fig. 4b). We also tested editing efficiency and bystander editing events in an exogenous clonal MPZL2 c.220 C > T mutation HEK293T cell line using dual plasmids that encoding the N-terminal and C-terminal of ABE8eWQ-SpRY:sgRNA3, and got the similar results. The editing efficiency of ABE8eWQ-SpRY:sgRNA3 in an exogenous clonal MPZL2 c.220 C > T mutation HEK293T cell line was: Bystander A1: 0.10%, A2: 0.10% and Target A5: 56.1%, Silent A9: 0.67%, C1-to-other: 0%, C2-to-other: 0%, C3-to-other: 0%, and no cytosine bystander event was observed too (Supplementary Fig. 9). Our results indicate that the editing efficiency of ABE8eWQ-SpRY:sgRNA3 in these two cell lines are similar, and the ABE8eWQ-SpRY:sgRNA3 shows high precision without detectable bystander edits and with relevant targeted editing efficiency.

a Schematic representation of the dual AAV constructs utilizing split-intein for ABE delivery in vivo, resulting in intein-mediated assembly of complete ABE:sgRNA complexes. The dual plasmids were packaged into the AAV serotype AAV-ie. b Assessment of the editing efficiencies of the MPZL2 target adenine and other bystander adenines or cytosines using dual AAV vectors that encoded split-intein ABE8eWQ-SpRY and sgRNA3 (n = 2). c Experimental overview of the in vivo base editing. Dual AAV-ie was used to induce split intein assembly, with the N-terminal and C-terminal components each prepared at 5.0 × 1013 vg/mL. These were mixed at a 1:1 ratio, and 2000 nl was injected into the RWM of P2 hMPZL2Q74X/Q74X mice. BioRender was used to make the Figure. “Created in BioRender. 남, 배. (2025) https://BioRender.com/kz4zbsm”. The images of the syringe, cochlea, and postnatal pup used in the figure were created using BioRender. d In vivo efficiency of A ∙ T to G ∙ C editing at on-target sites (P = 0.001) and bystander effects in DNA extracted from the organ of Corti (n = 8, P < 0.0001). Statistical analyses were performed using a two-tailed unpaired Student’s t-test. In the box-and-whisker plot, the whiskers mark the minimum and maximum, the box includes the 25th to 75th percentiles, and the line within the box indicates the median of the data set. Significance levels are indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. e In vivo potential off-target sites relative to the target site. The potential off-target sites were identified using Cas–OFFinder, including those with up to two mismatches and/or up to one DNA/RNA bulge. f RNA off-target test in vivo at 8 weeks after dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 injection. RNA off-target test in the injected mice was performed, and no RNA off-target editing was detected in vivo. n = 2 (Treated) and n = 2 (Control) per group. Source data for all relevant panels are provided within the Source Data file.
After optimizing the injection volume (Supplementary Fig. 10) and injection time point for dual AAV-ie-ABE8eWQ-SpRY:sgRNA3, a total of 2 µL dual AAV (1:1 mixed) at a titer of 5.0 × 1013 vg/mL was injected into the inner ear of neonatal hMPZL2Q74X/Q74X mice at P1-2 via the round window membrane (Fig. 4c) as previously described12. Fourteen days after injection, the organ of Corti was dissected from the cochleae of treated hMPZL2Q74X/Q74X mice. We sequenced the DNA from the cochleae of treated hMPZL2Q74X/Q74X mice and tested the base editing efficiency at the MPZL2 c.220 C > T locus in the organ of Corti of eight treated mice. Our results showed that the on-target editing efficiency of treated hMPZL2Q74X/Q74X mice was 2.04 ± 0.44% (range: 1.28–3.66%, P = 0.001) (Fig. 4d), and the bystander editing efficiencies of A0, A1, A2, and A9 were significantly low (Fig. 4d). Additionally, we performed targeted deep sequencing to measure base editing efficiencies at the RNA level. RNA levels (hereafter referred to as cDNA) were assessed by extracting RNA from the organ of Corti of treated mice and untreated mice at P16, followed by reverse transcription into cDNA. The average cDNA editing efficiency in the organ of Corti of hMPZL2Q74X/Q74X mice injected with dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 was 3.75% (range: 1.87–5.84%) (Supplementary Fig. 11). We further used the Cas-OFFinder software to search for potential off-target sites of SpRY in mice and implemented high-throughput sequencing to analyze six potential off-target sites. In three treated mice aged 16 days, no obvious off-target editing was detected at the six predicted off-target sites compared with untreated mice (Fig. 4e). We also performed RNA off-target test in the injected mice in vivo, and no RNA off-target editing was detected by ABE8eWQ-SpRY:sgRNA3 (Fig. 4f). These results indicated that dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 performed with relatively high precision without detectable bystander edits and with clinically relevant editing efficiency.
In vivo base editing of MPZL2 restored the hearing of hMPZL2 Q74X/Q74X mice
To investigate the in vivo base editing efficacy on auditory function, ABR and DPOAE thresholds were recorded at 4 weeks, 8 weeks, and 12 weeks post-injection. Representative click ABR waveforms at 12 weeks from Mpzl2 WT, untreated hMPZL2Q74X/Q74X, and treated hMPZL2Q74X/Q74X mice are shown in Fig. 5a. At 4 weeks, treated hMPZL2Q74X/Q74X mice exhibited slightly lower ABR thresholds compared to untreated mice, showing reductions of 4.1 dB, 10.4 dB, and 3.2 dB at 16 kHz, 24 kHz, and 32 kHz, respectively, although these reductions were not statistically significant (Fig. 5b). We continued to assess hearing function over time and observed more pronounced improvements at 8 weeks and 12 weeks post-injection. At 8 weeks, the ABR thresholds in the treated ears improved by 2.5 dB to 23.3 dB across all frequencies (Fig. 5c). Specifically, a significant difference in hearing thresholds at 16 kHz was observed between the treated ears of hMPZL2Q74X/Q74X mice and untreated hMPZL2Q74X/Q74X mice (49.2 ± 5.0 dB vs. 72.5 ± 6.1 dB, P = 0.007) (Fig. 5c). By 12 weeks, the treated hMPZL2Q74X/Q74X ears showed greater recovery compared to untreated hMPZL2Q74X/Q74X mice, with threshold improvements from 12.2 dB to 28.6 dB across all frequencies, showing significant differences. Treated hMPZL2Q74X/Q74X mice exhibited significantly reduced thresholds by 20.4 dB at click (P < 0.001), 11.2 dB at 4 kHz (P = 0.012), 26.9 dB at 8 kHz (P < 0.0001), 28.6 dB at 16 kHz (P = 0.001), 14.2 dB at 24 kHz (P = 0.028), and 12.5 dB at 32 kHz (P = 0.022) compared to untreated hMPZL2Q74X/Q74X mice (Fig. 5d). The ABR threshold at 8 kHz in treated hMPZL2Q74X/Q74X mice was comparable to that of Mpzl2 WT mice (Fig. 5d). At 16 weeks, ABR measurements showed that the treated ears exhibited sustained recovery across most frequencies, with improvements ranging from 3.3 dB to 46.48 dB, except at 4 kHz. Specifically, treated hMPZL2Q74X/Q74X mice recovered ABR thresholds by 22.5 dB at click (P < 0.001), 23.7 dB at 8 kHz (P = 0.0001), 35.2 dB at 16 kHz (P < 0.0001), 13.6 dB at 24 kHz (P = 0.04) and 9.1 dB at 32 kHz (P = 0.012) compared to untreated hMPZL2Q74X/Q74X mice (Fig. 5e). At 20 weeks, the treated ears maintained long-term recovery compared to the untreated ears, particularly at click, 8 kHz, 16 kHz, and 32 kHz (P = 0.032) (Fig. 5f). Notably, significant differences between treated and untreated ears ranged from 8.4 dB to 36.6 dB at click (P = 0.001), 9.6 dB to 42.2 dB at 8 kHz (P = 0.001), 17.1 dB to 44.0 dB at 16 kHz (P < 0.0001), and 0.5 dB to 12.9 dB at 32 kHz. These findings confirm the long-term effectiveness and stability of AAV-ie-ABE8eWQ in restoring auditory function in hMPZL2Q74X/Q74X mice. The DPOAE thresholds did not show obvious differences between treated and untreated hMPZL2Q74X/Q74X mice at 4 weeks (Fig. 5g). At 8 weeks, the DPOAE thresholds of the treated ears decreased by 3.3 dB and 7.1 dB at 8 kHz and 16 kHz, respectively (Fig. 5h). At 12 weeks, the decreases were 6.1 dB, 11.64 dB, and 5.0 dB at 8 kHz, 16 kHz, and 24 kHz (Fig. 5i). At 16 weeks, the treated ears exhibited a reduction of 8.8 dB at 8 kHz, 16.2 dB at 16 kHz, and 6.6 dB at 24 kHz compared to the untreated mice, with a persistent significant difference at 8 kHz (P = 0.023) and16 kHz (P = 0.003) (Fig. 5j). At 20 weeks, the treated ears showed a significant reduction of 9.1 dB at 8 kHz compared to the untreated ears (P = 0.048) (Fig. 5k).

a Representative ABR waveforms from three groups of mice (Mpzl2 WT, hMPZL2Q74X/Q74X, and treated hMPZL2Q74X/Q74X) in response to click stimuli recorded at 12 weeks post-injection. The bold line in each group represents the threshold. b–f Comparison of ABR thresholds among Mpzl2 WT (red line), hMPZL2Q74X/Q74X (blue line), and treated hMPZL2Q74X/Q74X (green line) mice at 4 weeks (b), 8 weeks (c),12 weeks (d), 16 weeks (e), and 20 weeks (f). Comparison of DPOAE thresholds among Mpzl2 WT (red line), hMPZL2Q74X/Q74X (blue line), and treated hMPZL2Q74X/Q74X (green line) mice at 4 weeks (g), 8 weeks (h),12 weeks (i), 16 weeks (j), and 20 weeks (k). At 16 weeks, 8 kHz (P = 0.023) and 16 kHz (P = 0.003); at 20 weeks, 8 kHz (P = 0.048) showed significant differences between hMPZL2Q74X/Q74X and treated hMPZL2Q74X/Q74X mice. Statistical significance was calculated using one-way ANOVA with Bonferroni’s correction for multiple comparisons. Significance levels are indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Data are presented as the mean ± SEM. Source Data file. Source data for all relevant panels are provided within the Source Data file.
To confirm the safety of our therapeutic strategy, dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 was injected into the inner ears of Mpzl2 WT mice. The ABR thresholds of injected Mpzl2 WT ears showed no deterioration compared to untreated Mpzl2 WT mice and un-injected (contralateral) Mpzl2 WT ears at 4 weeks, 8 weeks, or 12 weeks (Supplementary Fig. 12a). Additionally, no degeneration of OHCs or DCs was observed in the organ of Corti of treated mice (Supplementary Fig. 12b, c). Thus, in vivo base editing with the dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 system demonstrated a favorable safety profile with no observed ototoxicity. Furthermore, to determine whether editing occurs in non-cochlear tissues, we performed on-target DNA editing analysis in the non-cochlear tissues, including liver, brain, and thigh muscle, collected at P16 following inner ear local injection of the dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 system at P2. The results showed no dissemination in the non-cochlear tissues, with nearly undetectable levels, except for low-level editing (0.4–0.5%) observed in the liver of some treated mice (Supplementary Fig. 13a). However, we observed the absence of potential off-target sites of SpRY in the liver tissues of mice with low-level editing (0.4–0.5%), as predicted by Cas-OFFinder software, consistent with findings in untreated mice (Supplementary Fig. 13b).
In vivo base editing of MPZL2 rescued the MPZL2 expression, inner ear structure, and molecular functions in hMPZL2 Q74X/Q74X mice
The hMPZL2Q74X/Q74X mice treated at P1-2 were euthanized at P28, and RT-PCR and immunofluorescence analyses were performed. Human MPZL2 mRNA expression was significantly restored post-injection compared to untreated mice (Fig. 6a). Consequently, MPZL2 expression in the cochlea, particularly in the organ of Corti of the treated mice, was significantly rescued, reaching levels comparable to those observed in Mpzl2 WT mice (Fig. 6b, Supplementary Fig. 14). These data suggest that normal MPZL2 protein was properly constructed and expressed in the target cells post-injection. Furthermore, at 12 weeks the number of OHCs in middle turn (P = 0.043) and the number of DCs in the middle (P = 0.019) and basal turns (P = 0.016) of treated mice were significantly greater than those in untreated mice (Fig. 6c). Similarly, we observed increased survival of OHCs and DCs, as well as preserved organ of Corti, in treated mice compared with untreated mice (Fig. 6d). Additionally, treated mice exhibited visible hair bundles, particularly in the middle and basal turns (Fig. 6e). We subsequently performed RNA-seq to identify alterations in gene expression profiles in hMPZL2Q74X/Q74X mice treated with dual AAV-ie-ABE8eWQ-SpRY:sgRNA3. Principal Component Analysis of the RNA-seq data revealed a distinct clustering pattern of gene expression between treated and untreated mice (Fig. 6f). A total of 5773 DEGs (3693 upregulated and 2080 downregulated) were identified between treated and untreated mice (Supplementary Fig. 15a). GO analysis of the DEGs revealed significant enrichment in biological processes related to cell adhesion, ECM organization, and immune system process (Fig. 6g). Through a filtering process, we pinpointed the upregulated DEGs in treated hMPZL2Q74X/Q74X mice relative to untreated hMPZL2Q74X/Q74X mice, identifying 80 transcripts that were also significantly downregulated in untreated hMPZL2Q74X/Q74X mice compared with the Mpzl2 WT mice (Fig. 6h). These transcripts were notably enriched in biological processes related to cell adhesion and ECM organization (Supplementary Fig. 15b). Specifically, we identified 14 transcripts associated with cell adhesion and ECM organization, and gene expression profiles in treated hMPZL2Q74X/Q74X mice were restored to the levels observed in Mpzl2 WT mice, in contrast to the downregulated expression in untreated hMPZL2Q74X/Q74X mice (Fig. 6i). Collectively, these findings demonstrate that dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 treatment not only restores MPZL2 mRNA and protein expression levels but also rescues the altered cellular pathways underlying the c.220 C > T mutation, resulting in significant improvements in both hearing function and inner ear structural integrity.

a RT-PCR showed enhanced MPZL2 expression in cochlear membranous tissues at P28 following injection at P2. b Representative section images of MPZL2 expression in organ of Corti at P28 following injection at P2, immunolabeled with an anti-MPZL2 antibody (green). Scale bar: 25 μm. c Representative whole-mount images of the HC and SC layer from Mpzl2 WT, untreated hMPZL2Q74X/Q74X, and treated hMPZL2Q74X/Q74X mice at 12 weeks of age, along with the quantification of surviving OHCs (n = 4) and DCs (n = 4) immunolabeled with anti-Myosin VIIa (HCs, green) and anti-Sox2 (SCs, red). Scale bar: 20 μm. Data are presented as the mean ± SEM. In the bar graphs, Mpzl2 WT is shown in red, untreated hMPZL2Q74X/Q74X in blue, and treated hMPZL2Q74X/Q74X in green. Statistical significance was determined using one-way ANOVA with Bonferroni’s correction for multiple comparisons. Significance levels are indicated as *P < 0.05, **P < 0.01, and ***P < 0.001. d Representative section images of the organ of Corti at 12 weeks of age in Mpzl2 WT (n = 1), untreated hMPZL2Q74X/Q74X (n = 1), treated hMPZL2Q74X/Q74X mice (n = 2) after injection at P2, immunolabeled with anti-Myosin VIIa (HCs, red) and anti-Sox2 (SCs, green) antibodies. Scale bar: 25 μm. e Scanning electron microscope images from Mpzl2 WT, hMPZL2Q74X/Q74X, and treated hMPZL2Q74X/Q74X mice in the middle and basal turns of the cochlea. Scale bar: 10 μm. f Heatmap analysis of DEGs between untreated hMPZL2Q74X/Q74X mice (n = 3) and treated hMPZL2Q74X/Q74X mice (n = 3) (upregulation, red; downregulation, blue). g Biological processes (BP) terms associated with cell adhesion, ECM organization, and immune system processes. h Schematic diagram of RNA-sequencing analysis (n = 3). i Quantitative RT-PCR assay of the 14 genes of interest related to cell adhesion and ECM organization across the three groups (n = 3). Statistical significance was determined using one-way ANOVA with Kruskal–Wallis test for multiple comparisons. Data are presented as the mean ± SEM. Source data for all relevant panels are provided within the Source Data file.
Discussion
Recessive mutations are the most frequent cause of hereditary deafness10. Recessive mutations can be treated with AAV-mediated gene replacement, which delivers wild-type cDNA into inner ear cells to correct the underlying genetic defects27. We recently used the AAV-ie capsid to deliver the mouse Mpzl2 cDNA controlled by a ubiquitous promoter into the inner ear of Mpzl2 knock-out mice and rescued their hearing12. However, because the MPZL2 protein is expressed across multiple cell types within the inner ear, and because there is currently a lack of AAV serotypes and specific promoters that can precisely target these cells, gene replacement therapy may result in both overexpression of MPZL2 in targeted cells and ectopic expression in non-targeted cells, potentially leading to cytotoxic effects28,29. In addition, it is known that AAV-mediated gene therapies often fail to sustain their therapeutic effects over extended periods30,31.
An alternative treatment strategy for hereditary deafness is gene editing, which corrects the mutated DNA bases and addresses the genetic cause of hearing loss at its “root” rather than replacing the mutant gene32. Previously, in vivo CRISPR nuclease-based gene editing was used to disrupt the reading frame of dominant-negative alleles in dominant deafness mouse models33,34,35 and to restore the nonhomologous end joining-mediated frame in the Pcdh15 mouse model of DFNB2336. However, CRISPR-based disruption of the disease locus is ineffective for ameliorating recessive alleles. Moreover, CRISPR-driven double-strand breaks lead to unintended consequences such as large DNA deletions, chromosomal depletion, and p53-driven programmed cell death37,38. To bypass this issue, base editors offer a one-time therapeutic strategy to permanently correct pathogenic mutations without generating double-strand breaks39,40. However, a significant limitation of base editing gene therapy is the need to design distinct sgRNA systems for different mutation sites within the same gene, which demands substantial human and financial resources. In this regard, mutational hotspots or founder mutations present attractive targets for base editors. In DFNB111 patients, the predominant MPZL2 c.220 C > T mutation enables universal application of the dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 system without redesign.
Because inner ear HCs lack regenerative ability in mammals, their degeneration leads to permanent hearing loss. Congenital deafness caused by gene mutations induces disrupted inner ear structural integrity at birth or even during embryonic development, and in such cases gene transfer early in the prenatal period to correct genetic defects and prevent irreversible degeneration is an effective treatment strategy4. However, this strategy is complex and carries significant risks, precluding progress in preclinical research on gene therapies for congenital deafness. Preclinical research and clinical translation have advanced more rapidly for genes like OTOF, where the mutations do not disrupt the structural integrity of the inner ear2,41. Additionally, progressive hereditary deafness is particularly favorable for the development of gene therapy because the target cells in the cochlea remain structurally intact and functionally competent, providing a wider therapeutic window for intervention. Our study, along with previous studies, suggest that Mpzl2 mutants in mice do not interfere with cochlear development or cause the degeneration of normal HCs and SCs at P412,42. In hMPZL2Q74X/Q74X mice, hearing loss and the degeneration of OHCs and DCs both progress from P28 onward, providing a sufficient time window between the onset of hearing loss, the genetic diagnosis, and treatment43. Similarly, children with DFNB111 often experience milder forms of hearing loss during their teenage years, with some even passing newborn hearing screenings13. Notably, the extended therapeutic window for late-onset progressive deafness might reduce the risk of gene editing delivery vectors dispersing into the brain parenchyma, which is mitigated by the progressive occlusion of communication between the cochlear perilymph and cerebrospinal fluid in the human cochlea44.
Key factors—including advanced editing efficacy, safety, and optimized vector tropism—are pivotal in correcting mutant alleles and restoring auditory function. For ABEs, a wild-type tRNA-specific adenosine deaminase (TadA) from Escherichia coli was engineered to operate on DNA instead of RNA, and ABEmax with TadA7.10 and ABE8e with TadA-8e variants were developed to have higher base editing activities. However, both ABEmax and ABE8e also have unwanted C-to-T conversion activities at a preferred motif (TC*N) and have transcriptome-wide sgRNA-independent off-target effects20. To address these issues, we previously developed a novel ABE variant (ABE8eWQ) by introducing two variants (V106W and D108Q) into ABE8e, which resulted in significantly reduced bystander effects and RNA off-target activity20,45. We found that the c.220 C > T mutation could be targeted by ABE8eWQ-SpRY, which demonstrated high editing precision and robust editing efficiency in vitro. SpRY has the potential to broadly target the genome, and this is associated with off-target effects at the DNA level15. However, ABE8eWQ-SpRY exhibited no significant DNA off-targets at predicted sgRNA-dependent sites, along with undetectable RNA off-targets in the human cells. Using the dual AAV-ie-ABE8eWQ-SpRY:sgRNA3 system, we herein achieved high precision without detectable bystander effects and with relevant editing efficiency in vivo. This correlated with the restoration of hearing function, inner ear structures, and the expression of MPZL2 and related genes. Nevertheless, the ABE-sgRNA therapeutic system still requires further optimization to enhance its editing efficiency in vivo, with the goal of restoring higher levels of auditory function and with long term effects.
In summary, we identified an East Asia-specific founder mutation, the homozygous c.220 C > T mutation in MPZL2, as a promising target for ABE-based gene therapy. The development of humanized mouse models and the successful correction of this mutation using a single PAM-flexible ABE may be a step toward clinical translation of base editing gene therapy for treating hereditary deafness, including most cases of MPZL2 deafness.
Methods
Ethics statement
This research complied with all relevant ethical regulations. Animal experiments in this research were approved by the Ethics Committee at Fudan University, China and the Institutional Animal Care and Use Committees (IACUC; MS-2022-01, MS-2023-01) at the GEM Center of Macrogen (Seoul, Republic of Korea) and at Seoul National University Hospital. The human data collection was approved by the Institutional Review Board of Seoul National University Hospital (no. IRB-H-0905-041-281 and IRB-H-2202-045-1298) and by the Eye, Ear, Nose and Throat Hospital affiliated with Fudan University Review Board of the Office of Research Compliance through protocol 2020122-1 and 2024073. We obtained written informed consent from either the parents of the children or from the participants themselves.
Human genetics
In this study, we used a retrospective research design and focused on pediatric participants attending the Hereditary Hearing Loss Clinic within the otorhinolaryngology departments of two institutions. 234 pediatric cases ( ≤ 18 years of age, regardless of sex) with symmetric, mild-to-moderate (hearing threshold range 21–55 dB), non-syndromic SNHL were involved. The demographic data and audiological phenotypes were retrieved from the electronic medical records.
Whole-exome sequencing and Multiplex ligation-dependent probe amplification
Genomic DNA was extracted from peripheral blood samples using a Chemagic 360 instrument (Qiagen, Venlo, Netherlands) or a DNeasy 96 Blood & Tissue Kit (Qiagen, Germany). We first applied whole-exome sequencing to sequence the exonic regions. The target regions were captured using SureSelectXT Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) or CapTruth Human Exome V2.0 (Medical Laboratory of Nantong ZhongKe Co., Ltd., Nantong, China). A library was prepared following the manufacturer’s instructions and was paired-end sequenced using a NovaSeq 6000 sequencing system (Illumina, San Diego, CA, USA) or DNBSEQ-T7 platform (MGI, Shenzhen, China) with an average depth of coverage of 100×. Sequencing reads were aligned to the human reference genome (GRCh38) and processed according to the Genome Analysis Toolkit best-practice pipeline for calling single nucleotide variants and short insertions/deletions (indels)46. The ANNOVAR program was used for variant annotation using the RefSeq gene set and Genome Aggregation Database (gnomAD)47,48. Rare non-silent variants were selected as candidates, including nonsynonymous single nucleotide variants, coding indels, and splicing variants. We also used the Korean Reference Genome Database (KRGDB) and KOVA (Korean Variant Archive; Korean population database) databases for further filtration of ethnic-specific variants49,50. Additionally, the ClinVar and HGMD databases were screened to check whether candidate variants had been previously identified in other patients51,52. For individuals exhibiting non-syndromic, symmetric, mild-to-moderate SNHL, we evaluated copy number variations using the SALSA Multiplex Ligation-dependent Probe Amplification Probemix P461-B1 STRC-CATSPER2-OTOA (MRC-Holland, Amsterdam, the Netherlands)53.
Whole-genome sequencing
Patients who remained undiagnosed after whole-exome sequencing were subjected to whole-genome sequencing. DNA libraries were prepared using TruSeq DNA PCR-Free Library Prep Kits (Illumina) or AngTruth-seq EZ DNA Library Preparation Module (for MGI) v2 (Medical Laboratory of Nantong ZhongKe Co. Ltd., Nantong, China) and sequenced on an Illumina NovaSeq6000 or DNBSEQ-T7 platform with an average depth of coverage of 30×. The obtained genome sequences were aligned to the human reference genome (GRCh38) using the BWA-MEM algorithm. PCR duplicates were removed using SAMBLASTER54. The initial mutation calling for base substitutions and short indels was performed using HaplotypeCaller and Strelka2, respectively55. Structural variations were identified using Delly56. We classified candidate variants according to the American College of Medical Genetics and Genomics and Association for Molecular Pathology guidelines for hearing loss57.
Humanized mice
C57BL/6 N and ICR mice were purchased from Orientbio Inc. (Sungnam, Republic of Korea) and were housed under specific pathogen-free conditions in individually ventilated cages. The temperature and humidity of the breeding environment were maintained at 22 ± 1 °C and 50%, respectively, with a 12-h light:dark cycle. All feed and individually ventilated cages were sterilized, and all air conditioners had filters. The protocols for the generation of hMPZL2Q74X and hMPZL2WT mice were performed according to the Korean Food and Drug Administration (KFDA) guidelines and were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC; MS-2022-01, MS-2023-01) at the GEM Center of Macrogen (Seoul, Republic of Korea) and at Seoul National University Hospital. Female and male mice were randomly chosen for all experiments.
Humanized mouse sgRNA
sgRNA searches and potential off-target analyses were performed using the CRISPR design tools CHOPCHOP58 and CRISPOR59. To generate the hMPZL2Q74X mouse, the sgRNA was searched based on the NCBI database (NC_000075.7, NM_007962.4) within the partial regions of exon 1 and intron 1, including the 5’ UTR-ATG region. For the hMPZL2WT mouse, the search was based on human MPZL2 (hMPZL2) transcript NM_005797.4, within the regions of Exon 2 and 3. The sequences of the sgRNAs used are listed in Supplementary Data 4.
Humanized mouse targeting donor
The hMPZL2Q74X targeting donor was designed based on the target site (GRCm39; chr9: 44,954,049) and incorporated approximately 600-bp homology arms on both sides of the hMPZL2Q74X expression cassette. The 5’ and 3’ homology arms included sequences from the upstream-5’UTR and from intron 1 of the mouse Mpzl2 gene, respectively. The designed donor sequences were synthesized in vector DNA form (Invitrogen, Waltham, MA, USA). For the hMPZL2WT mouse, single-stranded DNA (ssDNA) was used for the donor. The ssDNA was designed with the hMPZL2 mutation site (c.220 T > C:p.X74Q) at the center and 60 bp homology arms on both sides and was synthesized.
Humanized mouse embryo collection
To induce superovulation, 7.5 IU each of pregnant mare serum gonadotropin (PMSG; Dsmbio, Uiwang, Republic of Korea) and human chorionic gonadotropin (hCG; Dsmbio) were intraperitoneally injected for 5–8 weeks in female mice at 48-hour intervals. Subsequently, zygotes were obtained from the oviducts of these female mice after mating with males to produce hMPZL2Q74X mice. To generate hMPZL2WT mice, superovulation was induced in the same manner, and then unfertilized embryos were collected from female mice that had not mated with male mice. The collected embryos were in vitro fertilized using sperm from hMPZL2Q74X homozygous mice to ensure the development of zygotes. Previously reported detailed procedures for in vitro fertilization were referenced60. The zygotes were washed in Quinn’s Advantage Medium with HEPES (ART-1024, Cooper Surgical, Trumbull, CT, USA) and then transferred to droplets of KSOM medium (MR-121-D, Sigma-Aldrich, St. Louis and Burlington, MA, USA) in a CO2 incubator at 37 °C.
Humanized mouse CRISPR mixture and microinjection
sgRNA was synthesized using the GeneArt Precision sgRNA Synthesis Kit (A29377, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Microinjection was performed by injecting a mixture composed of Cas9 protein (Macrogen), purified sgRNAs, and linearized hMPZL2Q74X vector or hMPZL2WT ssDNA into the pronucleus of 1-cell zygotes in Quinn’s Advantage Medium with HEPES at concentrations of 20 ng/µl, 25 ng/µl, and 20 ng/µL, respectively. Microinjected zygotes were incubated at 37 °C for 2 h and were transplanted by surgical methods into the oviducts of pseudopregnant recipient ICR mice at the 1-cell or 2-cell stages.
Humanized mouse genotyping
Genomic DNA for genotyping was extracted from the tails of 2-week-old mice using an Axen Total DNA mini kit (MG-P005-50, Macrogen). The PCR assay was conducted using the EF-Taq DNA Polymerase (SEF16-R250, Solgent, Daejeon, Republic of Korea) and Axen High-Q Taq DNA Polymerase (MG-E-003-250, Macrogen). The PCR was performed in a total volume of 20 µL consisting of 2 µL of 10 × reaction buffer, 4 µL of 5 × GC enhancer, 0.4 µL of 10 mM dNTP mixture, 2 µL each of 10 µM forward and reverse primers, about 200 ng of extracted genomic DNA, and PCR-grade water to make up the final volume. The PCR cycling conditions were an initial denaturation at 95 °C for 5 min followed by 35 cycles at 95 °C for 10 s, 56–60 °C for 20 s, and 72 °C for 30 s/kb. For genotyping hMPZL2WT mice, PCR products using the hMPZL2Q74X allele as a template were added to create heteroduplexes through re-annealing, followed by treatment with T7 endonuclease 1 (M0302S, NEB, Ipswich, MA, USA). For hMPZL2WT mice, PCR cloning (E1203S, NEB) was additionally performed for allele separation, and Sanger sequencing was performed for sequence verification of the two types of humanized mice. The sequences of the primers used for genotyping are listed in Supplementary Data 5.
HEK293T-MPZL2 c.220C > T clonal cells
To generate an endogenous cell line carrying a point mutation at a MPZL2 locus, identical to that found in DFNB11 patients, prime editing was employed. HEK293T (ATCC CRL-3216) cells were seed into a 24-well plate and transfected with PE and the corresponding pegRNA-expressing plasmid. After editing, successfully edited cells were isolated and expanded. Genomic DNAs of single-cell clones were individually purified, and the clone containing intended pathogenic single mutation (MPZL2 c.220 C > T) was validated by Sanger sequencing. The primer pairs used are as follows: Forward primer- ATGGGGCATCTCAGTTTCACT and Reverse primer- CATGGGTTGGAAGGGATCTA.
To generate exogenous clonal MPZL2 c.220 C > T mutation HEK293T cell line, the HEK293T-MPZL2 c.220 C > T stable cell line was established by cloning a 201-bp disease-associated gene fragment containing the C·G-to-T·A mutation from the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) by assembling the fragments into a modified lentivector from lentiCRISPR v2 (#52961), thus yielding the lentivector plasmid Lenti MPZL2-P2A-puro. HEK293T (ATCC CRL-3216) cells were seeded into 6-well plates (Corning) at approximately 85% confluency per well and were co-transfected with 2.25 μg of the lentivector plasmid, 2.25 μg psPAX2 (#12260, encoding the viral packaging proteins), and 1.5 μg pMD2.G (#12259, encoding the VSV-G envelope protein) using Lipofectamine 3000 (L3000015, Thermo Fisher Scientific) following the manufacturer’s instructions. Virus-containing supernatant was collected after 48 h of transfection and centrifuged at 2000 × g for 10 minutes to remove cell debris and filtered through a 0.45-μm polyvinylidene difluoride (PVDF) filter (IPVH00010, Millipore). A total of 150 μL filtered virus-containing supernatant was added to HEK293T cells at approximately 40–50% confluency cultured in 6-well plates. After 24 h of transduction with lentivirus, the cells were split into the wells of a new plate supplemented with puromycin (2.5 μg/ml). After 72 h of puromycin selection, a stable cell line with the hMPZL2 c.220 C > T mutation was successfully established. To ensure single-copy integration, cells with the fewest surviving colonies were collected and subsequently expanded for further transfection procedures.
Cell culture
Cultured cells used in this study, including HEK293T-MPZL2 c.220 C > T and patient-derived cell lines, were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. All culture media consisted of DMEM supplemented with heat-inactivated FBS (10% for HEK293T cells and 20% for patient-derived fibroblasts), 100 units/mL penicillin/streptomycin, and 2 mM L-glutamine. Cells were routinely tested for mycoplasma contamination.
ABE expression plasmid construction
The ABE8e (no.138489, Addgene, Watertown, MA) and NG-ABE8e (no.138491, Addgene) used in this study were separately obtained from previously reported plasmids available from Addgene. Other ABE variant expression plasmids were constructed by replacing the deaminase and Cas-nuclease domains using a Gibson assembly of linearized destination vector and PCR amplicons with Gibson-compatible ends for each assembly junction. PCR was performed using KOD-Multi & Epi (KME-101, TOYOBO).
sgRNA expression plasmid construction
The sgRNA expression plasmids for the various Cas effectors were generated using custom oligonucleotides and restriction enzyme-based classical cloning methods based on pRG2 (no. 104174, Addgene). Briefly, for each guide RNA sequence, a pair of complementary oligos with 4-bp overhangs were annealed and inserted via a cut-ligation reaction with BsaI and T4 DNA ligase in the pRG2-hU6 plasmid. All sgRNA sequences were designed with a G preceding 19-bp or 23-bp spacer targets for transcription initiation from the Polymerase III promoter. To produce a vector expressing sgRNA for the other SpCas9 ortholog, eNm2-C, an sgRNA scaffold was synthesized from Macrogen and integrated with the pRG2-hU6 plasmid. The gRNA was inserted again into the plasmid, where the original sgRNA scaffold was swapped with the scaffold tailored for eNme2-C. The sgRNAs were inserted into the PSK-mU6-sgRNA plasmid between two Bbs1 restriction sites. All plasmids used in this study were purified using Nucleobond Xtra Midi EF (MN, 740420.5) for Midi-prep or Exfection plasmid LE (111-102, GeneAll) for Mini-prep. The sequences of the sgRNAs are listed in Supplementary Data 4.
Targeted deep sequencing
To evaluate the on/off-target editing efficiency, genomic DNA was amplified by a three-step nested PCR using KOD-Multi & Epi (KME-101, TOYOBO). PCR 1–2 steps were performed with primers specifically targeting the genomic region of interest. The total PCR cycles were kept to a minimum to avoid PCR bias. The primers used for PCR step 2 included the minimal adapters for adding barcodes in the following PCR step 3. Finally, the barcoded PCR products were pooled, purified using the Expin PCR SV mini kit (103-102, GeneAll), and sequenced using an Illumina Miniseq according to the manufacturer’s instructions. Sequencing results were analyzed using a BE-Analyzer (http://www.rgenome.net/be-analyzer/)61.
RNA off-target analysis
HEK293T cells were seeded in a 24-well plate, and the next day they were transfected with 390 ng ABE-encoding plasmid and 125 ng sgRNA using JetOPTIMUS® (Polyplus) according to the manufacturer’s protocol. Briefly, the plasmids were mixed with JetOptimus reagent in JetOptimus buffer, then the mixture was incubated for 10 min and added into each well. For RNA extraction, cells were harvested by treating with TRIzol reagent 24 h after transfection. To synthesize cDNA, reverse transcription was performed using ReverTra Ace-α- (FSK-101, TOYOBO) according to the manufacturer’s instructions. The target region was amplified using KOD-Multi & Epi (KME-101, TOYOBO), and the PCR products were analyzed using an Illumina MiniSeq instrument. To obtain the percentage of adenosines edited to inosines, the number of adenosines converted to guanosines was divided by the total number of adenosines in the products. The sequences of the primers are listed in Supplementary Data 5.
AAV plasmid construction
For AAV production, we used the split intein-mediated ABE constructs designed in a previous study62, with some modifications as follows. Dual ABE constructs were amended sequentially in two steps each. The N-terminal vector was first modified by installing an additional A61R mutation (GCG to AGG) in the 5′ half of the Cas9 coding sequence (2–1368 aa) for SpRY using the QuickChange mutagenesis method63. The TadA8eWQ domain and linkers were then subcloned into the N-terminal vector between the NotI and BgIII sites. The C-terminal vector was modified to encode the final selected sgRNA3 (NAN PAM) from the in vitro screening and to contain the remainder of the SpRY mutations. A pair of recombinant vectors contained either inverted terminal repeat (ITR)-CMV promoter-nucleic localization signal (NLS)-ecTadA8eWQ-SpRY(N)-Npu(N)-bGHpA-ITR or ITR-CMV promoter-Npu(C)-SpRY(C)-NLS-1xHA-bGHpA;U6-sgRNA-ITR. The dual viral plasmids for AAV packaging were purified using Nucleobond Xtra Midi EF (MN, 740420.5) for Midi-prep, and the dual viral plasmids were separately packaged into two AAV-ie viruses (PackGene Biotech).
Round window injection into the inner ear of neonatal mice
Neonatal mice of either sex were used for injections, and the mice were randomly assigned to the different experimental groups. All surgical procedures were performed in a clean, dedicated space, and all instruments were thoroughly cleaned with 70% ethanol and autoclaved prior to surgery. P1-2 hMPZL2Q74X/Q74X mice were used for AAV-ie-ABE8eWQ injection. Mice were anesthetized by hypothermia on crushed ice, and a skin incision was made behind the ear of the mouse to expose the tympanic ring and the stapedial artery under an operating microscope (OPMI pico, ZEISS). Glass micropipettes (WPI) held by a Nanoliter 2020 Microinjection System (WPI) were inserted through the round window membrane, which allows access to inner-ear cells. The total injection volume was 2 μl per cochlea for round window membrane injection, and the release rate was 7 nL/s under the control of a MICRO2T SMARTouch microinjection controller (WPI). The skin was closed with a 6-0 nylon suture (NB617P, Ailee).
Tissue homogenization
Isolated tissues, including the Corti, liver, brain and thigh-muscle, were transferred into microtubes and finely minced and weighed. The following samples were then homogenized using a mini homogenizer and incubated in lysis buffer containing Proteinase K until complete lysis is obtained. For DNA extraction, silica column-based kit (NucleoSpin® Tissue, Macherey-Nagel, Germany) was used according to the manufacturer’s protocol.
In vivo gene editing efficiency test
Genomic DNA was purified from the organ of Corti of injected hMPZL2 c.220 C > T mice and untreated hMPZL2 c.220 C > T mice (negative control). Purified DNA was amplified by PCR for each condition using primers for the genomic DNA. HTS amplicon libraries containing an adapter sequence (forward, 5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT-3′; reverse 5′- ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC T -3′) at the 5′ end was prepared by PCR using KOD-Plus-Neo DNA Polymerase (KOD-201, Toyobo). The aforementioned products were processed through another round of PCR amplification with distinct barcode sequences added to the primers. To test cDNA editing efficiency, RNA was extracted from a same tissue using AllspinTM (GeneAll, Korea) after homogenization of cochlear tissues. Reverse transcription to cDNA was performed by using ReverTra Ace qPCR RT master Mix (TOYOBO, FSK-101). Then, 1 μL of cDNA was used for RT-PCR. PCR amplification was conducted using the same conditions as for genomic DNA. The sequences of the primers are listed in Supplementary Data 5. The resulting libraries were pooled and subjected to 150 bp paired-end sequencing on an Illumina HiSeq platform. The A-to-G conversions in the HTS data were analyzed with a BE-Analyzer61.
Audiometric tests
Mice were anesthetized via intraperitoneal injection with a mixture of dexmedetomidine hydrochloride (200 mL/kg) and Zoletil (35 mg/kg). ABR and DPOAE thresholds were measured using a BioSigRZ system (Tucker-Davis Technologies, Alachua, FL, USA) in a soundproof chamber. Anesthetized animals were first recorded to collect ABR, and then moved to a second setup to record DPOAE.
ABR recordings and analysis
ABR signals were collected via subdermal needle electrodes placed at the mastoid portion of the skull, the vertex of the skull, and the dorsal rump as the recording electrode, reference electrode, and ground electrode, respectively. The ABR signals were evoked and filtered through a 300 Hz to 3 kHz passband and averaged at 512 repeats of each sound pressure level (SPL). Click stimuli (90 to 10 dB, 2.5 ms, 21 pps rate) and tone-burst stimuli (4, 8, 16, 24, and 32 kHz, 2.5 ms, 21 pps rate) were applied from 90 to 10 dB SPL in 10 dB steps. The ABR threshold was defined as the lowest sound level at which any waveform peak could be observed.
DPOAE recordings and analysis
The f1 and f2 primary tones were generated at a frequency ratio of 1.2 with f2 levels at 4, 8, 16, 24, and 32 kHz and L1 – L2 = 10 dB SPL. The f2 levels were swept in 5 dB steps from 80 dB to 20 dB SPL. The DPOAE threshold was defined from the average spectra at the f2 levels that produced a DPOAE with a magnitude 5 dB SPL above the noise floor.
Histology
Histological and immunofluorescence analyses were conducted on cochleae from 12–15-week-old mice. After anesthetization via intraperitoneal injection, the mice were perfused with 4% paraformaldehyde (BPP-9004-001L, T&I) and the cochleae were dissected from the temporal bone. For histology, the cochleae were decalcified and embedded in paraffin blocks that were then sectioned at a thickness of 4 μm using a rotary microtome. The tissue sections were mounted on glass slides, deparaffinized in Histo-clear II (HS-202, National Diagnostics) for 10 min, washed in 100%, 95%, 90%, and 70% ethanol, and finally washed for 10 min in tap water. Hematoxylin and eosin staining was performed, with the slides being incubated in hematoxylin for 2 min and then stained with eosin for 20 s. The slides were subsequently mounted using a mounting medium and analyzed for cytoarchitecture evaluation at 4 × 20 × and 100 × magnification using an ECLIPSE Ci microscope (Nikon).
Immunofluorescence
For immunofluorescence assays of 12–15-week-old cochlea, tissue samples were post-fixed with 4% paraformaldehyde overnight at 4 °C. The cochleae were rinsed with PBS and decalcified using 10% ethylene diamine tetraacetic acid (EDTA) overnight at room temperature (RT). The decalcified cochleae were cryoprotected with a sucrose gradient in PBS (10%, 20%, and 30% sucrose) for 1 h at RT, then soaked in 30% sucrose overnight at 4 °C. The specimens were embedded in Tissue-Tek OCT, and mid-modiolar cryosections were made at 10 µm thickness and mounted on glass slides. The tissue sections were permeabilized with 1% Triton X-100 and blocked with 4% BSA in PBS for 20 min at RT and then incubated overnight at 4 °C with the following primary antibodies: rabbit anti-MYO7A (1:400 dilution, PA1-936, Invitrogen), mouse anti-SOX2 (1:400 dilution, sc-365823, Santa Cruz), rabbit anti-MPZL2 (1:300 dilution, 11787-1-AP, Proteintech), and mouse anti-TUBB3 (1:300 dilution, 801202, Biolegend). After washing with PBS, the sections were incubated with the corresponding Alexa-conjugated secondary antibodies for 1 h at RT. After rinsing with PBS, specimens were mounted on microscope glass slides using mounting medium with DAPI (AB104139, Abcam). Images were captured using a Zeiss Axioskop light microscope and a Leica TCS SP5 confocal microscope (Leica, Wetzlar, Germany). For whole-mount immunofluorescence staining, the decalcified cochleae were dissected into three pieces in ice-cold PBS. Samples were permeabilized, blocked, and incubated overnight with rabbit anti-MYO7A and mouse anti-SOX2 or rabbit anti-MPZL2 primary antibodies at 4 °C. Appropriate Alexa-conjugated secondary antibodies were applied for 1 h at RT, and mounting medium with DAPI was used to mount the specimens. Images were obtained using a Leica TCS SP8 confocal microscope (Leica, Wetzlar, Germany). We adjusted the confocal z-stack range (1 to maximum 46) to ensure that staining patterns aligns consistently between whole mounts and cryosection samples. The numbers of MYO7A+ inner hair cells and OHCs and SOX2+ DCs were counted in 100 μm cochlear sections from 10 cryosection images (two mid-modiolus sections per cochlea) obtained from five cochleas across the apical, middle, and basal turns, and the counted cells were analyzed as percentages. The number of SGNs in the apical, middle, and basal turns was counted using ImageJ software. For SGN quantification, TUBB3+ SGNs were counted per 72,900 μm². SGN density in H&E-stained paraffin sections was determined by counting the number of SGNs per 8000 μm².
Scanning electron microscopy
Cochleae were dissected and gently perfused with 2.5% glutaraldehyde (G7651-10ML, Sigma-Aldrich) in PBS through the round and oval windows and into the apex. Samples were fixed in 2.5% glutaraldehyde in PBS overnight at 4 °C. After washing with PBS, the cochleae were decalcified and dissected into three turns. The specimens were post-fixed in 1% osmium tetroxide (19172, EMS) in PBS for 2 hours and dehydrated in an ethanol gradient (50%, 60%, 70%, 80%, 90%, and 100% ethanol) at 10-minute intervals. The samples were then rinsed with a mixture of 100% ethanol and hexamethyldisilazane (HMDS, Sigma-Aldrich) in a 1:1 ratio for 30 min, followed by drying with HMDS only for 30 min. The dried samples were mounted on metal disks and gold sputter-coated (Cressington sputter coater 208HR), and images were obtained at 5.0 kV magnification using an electron microscope (Hitachi S-4700 FESEM).
Reverse transcription (RT)-PCR and quantitative RT-PCR
Cochlea, kidney, and lung tissues were freshly dissected from P4 or P28 wild-type and mutant mice. These tissues were immediately frozen in liquid nitrogen and stored at –80 °C until processing. Total RNA was isolated using TRIzol Reagent (Invitrogen) as per the manufacturer’s instructions. cDNA synthesis was performed from 2 μg of total RNA using Accupower RT-pre-mix (Bioneer, Daejeon, Republic of Korea). PCR was then performed using diluted cDNA and 10 pmol of specific primers to assess the mRNA levels. The thermal cycling conditions consisted of an initial denaturation at 95 °C for 5 min followed by 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min. The amplified DNA was visualized using agarose gel electrophoresis with Loading STAR (DYNE Bio, Seongnam, Republic of Korea). For quantitative RT-PCR reactions, 1/20 diluted cDNA was combined with SYBR qPCR master mix as the reporter dye and 10 pmol of primers to detect mRNA expression of specific genes. The thermal cycling conditions were as follows: 95 °C for 3 minutes for enzyme activation followed by 40 cycles of 95 °C for 10 seconds, 53 °C for 15 seconds, and 72 °C for 30 s. The primer sequences are listed in Supplementary Data 5.
RNA sequencing
RNA sequencing analyses were conducted as previously reported64. Briefly, total RNA was extracted from P28 mouse cochleae (three pairs of cochleae from wild-type and mutant mice) utilizing TRIzol (Invitrogen, USA) following the manufacturer’s protocol. RNA sequencing libraries were prepared using a SMARTER stranded total RNA-seq in accordance with the manufacturer’s protocols (Takara, Japan). After confirming the library size (approximately 300–500 bp) and fluorophore-based quantification of each library concentration (Quantus; Promega, USA), the samples were sequenced on a Nextseq 2000 under paired-end conditions (Illumina). After filtering, fastq files were mapped against the reference genome GRCm39 (ensemble) and pseudo-aligned by Kallisto (version 0.51.0)65. Transcript counts were imported at the gene level using txiimport66, and DEGs were identified using DEseq267. Individual samples were further analyzed to list genes with a fold change greater than 1.5 and a p < 0.05. DAVID GO analysis was performed to examine GO terms for biological processes, cellular components, and molecular functions68. Representative GO terms were summarized and visualized using REVIGO69. DEGs were further compared with the mouse cochlear single-cell RNA-seq database (http://umgear.org) and the SHIELD database (http://shield.hms.harvard.edu) in order to refine the list of genes implicated in specific cellular pathways. The RNA-seq findings were validated by quantitative RT-PCR analysis using specific primers for selected DEGs. The sequences of the primers are listed in Supplementary Data 5.
Statistics and reproducibility
Statistical analyses and visualizations were conducted using R Version 4.2.2. and GraphPad Prism (Version 9.0.2). The code for human genetics plots (Fig. 1 and Supplementary Fig. 1) is available at https://github.com/SNUH-hEARgeneLab/MPZL2. Differences between two groups were analyzed using an unpaired Student’s t-test, while one-way ANOVA with Bonferroni correction and the Kruskal–Wallis test were conducted for comparisons among more than two groups, as appropriate. All data are presented as the mean ± SEM. Statistical significance is indicated in the figures as follows: ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DNA sequencing data reported in this paper cannot be deposited in a public repository because of the patient’s genetic information, the possibility of privacy invasion, psychosocial risks (such as social stigma and discrimination), and the IRB’s restrictions on public data release. To access the data, please submit a request to the lead contact (yilai_shu@fudan.edu.cn, maru4843@hanmail.net). The request will be reviewed and, if approved, the lead contact will work with the requestor on sharing the data, and by adhering to the consent agreements established with the study participants. The RNA-seq data generated in this study have been deposited in the GEO database under accession code GSE299064.
Statistical analyses and visualizations were conducted using R Version 4.2.2 and GraphPad Prism Version 9.0.2, and the corresponding code can be accessed at https://github.com/SNUH-hEARgeneLab/MPZL2. Any additional information required to reanalyze the data reported in this paper is available from the lead contact (maru4843@hanmail.net) upon request.
All other data supporting this work are included in this article, the Supplementary Information, or source data files. Source data are provided with this paper.
References
Wang, H. et al. Bilateral gene therapy in children with autosomal recessive deafness 9: single-arm trial results. Nat. Med. 30, 1898–1904 (2024).
Lv, J. et al. AAV1-hOTOF gene therapy for autosomal recessive deafness 9: a single-arm trial. Lancet 403, 2317–2325 (2024).
Jean, P. et al. Single-cell transcriptomic profiling of the mouse cochlea: An atlas for targeted therapies. Proc. Natl. Acad. Sci. USA 120, e2221744120 (2023).
Petit, C., Bonnet, C. & Safieddine, S. Deafness: from genetic architecture to gene therapy. Nat. Rev. Genet 24, 665–686 (2023).
Sharma, G., Sharma, A. R., Bhattacharya, M., Lee, S. S. & Chakraborty, C. CRISPR-Cas9: a preclinical and clinical perspective for the treatment of human diseases. Mol. Ther. 29, 571–586 (2021).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Gaudelli, N. M. et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Yeh, W. H. et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 12. https://doi.org/10.1126/scitranslmed.aay9101 (2020).
Peters, C. W. et al. Rescue of hearing by adenine base editing in a humanized mouse model of Usher syndrome type 1F. Mol. Ther. 31, 2439–2453 (2023).
Jiang, L., Wang, D., He, Y. & Shu, Y. Advances in gene therapy hold promise for treating hereditary hearing loss. Mol. Ther. 31, 934–950 (2023).
Kim, B. J. et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet Med 22, 1119–1128 (2020).
Jiang, L. et al. Hearing restoration by gene replacement therapy for a multisite-expressed gene in a mouse model of human DFNB111 deafness. Am. J. Hum. Genet https://doi.org/10.1016/j.ajhg.2024.08.008 (2024).
Wesdorp, M. et al. MPZL2, encoding the epithelial junctional protein myelin protein zero-like 2, is essential for hearing in man and mouse. Am. J. Hum. Genet 103, 74–88 (2018).
Nishimasu, H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018).
Walton, R. T., Christie, K. A., Whittaker, M. N. & Kleinstiver, B. P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science 368, 290–296 (2020).
Huang, T. P. et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat. Biotechnol. 41, 96–107 (2023).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Jeong, Y. K., Song, B. & Bae, S. Current status and challenges of DNA base editing tools. Mol. Ther. 28, 1938–1952 (2020).
Jeong, Y. K. et al. Adenine base editor engineering reduces editing of bystander cytosines. Nat. Biotechnol. 39, 1426–1433 (2021).
Kim, H. S., Jeong, Y. K., Hur, J. K., Kim, J. S. & Bae, S. Adenine base editors catalyze cytosine conversions in human cells. Nat. Biotechnol. 37, 1145–1148 (2019).
Grunewald, J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019).
Zhou, C. et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 (2019).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Grunewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019).
Tan, F. et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 10, 3733 (2019).
Hu, S. W. et al. Engineering of the AAV-compatible hair cell-specific small-size Myo15 promoter for gene therapy in the inner ear. Research (Wash. D. C.) 7, 0341 (2024).
Zhao, X. et al. Characterization of promoters for adeno-associated virus mediated efficient Cas9 activation in adult Cas9 knock-in murine cochleae. Hear Res. 394, 107999 (2020).
Aaron, K. A. et al. Selection of viral capsids and promoters affects the efficacy of rescue of Tmprss3-deficient cochlea. Mol. Ther. Methods Clin. Dev. 30, 413–428 (2023).
Minoda, R., Miwa, T., Ise, M. & Takeda, H. Potential treatments for genetic hearing loss in humans: current conundrums. Gene Ther. 22, 603–609 (2015).
Xue, Y. et al. RNA base editing therapy cures hearing loss induced by OTOF gene mutation. Mol. Ther. 31, 3520–3530 (2023).
Kong, C., Yin, G., Wang, X. & Sun, Y. In utero gene therapy and its application in genetic hearing loss. Adv. Biol. (Weinh) e2400193 https://doi.org/10.1002/adbi.202400193 (2024).
Zhu, W. et al. Targeted genome editing restores auditory function in adult mice with progressive hearing loss caused by a human microRNA mutation. bioRxiv https://doi.org/10.1101/2023.10.26.564008 (2023).
Tao, Y. et al. Treatment of monogenic and digenic dominant genetic hearing loss by CRISPR-Cas9 ribonucleoprotein delivery in vivo. Nat. Commun. 14, 4928 (2023).
Cui, C. et al. Precise detection of CRISPR-Cas9 editing in hair cells in the treatment of autosomal dominant hearing loss. Mol. Ther. Nucleic Acids 29, 400–412 (2022).
Liu, L. et al. Template-independent genome editing in the Pcdh15(av-3j) mouse, a model of human DFNB23 nonsyndromic deafness. Cell Rep. 40, 111061 (2022).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Shalem, O., Sanjana, N. E. & Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet 16, 299–311 (2015).
Musunuru, K. et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 593, 429–434 (2021).
Arbab, M. et al. Base editing rescue of spinal muscular atrophy in cells and in mice. Science 380, eadg6518 (2023).
Tang, H. et al. Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum. Genet 142, 289–304 (2023).
Bademci, G. et al. MPZL2 is a novel gene associated with autosomal recessive nonsyndromic moderate hearing loss. Hum. Genet 137, 479–486 (2018).
Nourbakhsh, A. et al. Stem cells and gene therapy in progressive hearing loss: the state-of-the-art. J. Assoc. Res. Otolaryngol. 22, 95–105 (2021).
Li, L., Shen, T., Liu, S., Qi, J. & Zhao, Y. Advancements and future prospects of adeno-associated virus-mediated gene therapy for sensorineural hearing loss. Front Neurosci. 18, 1272786 (2024).
Cha, J. H. et al. Discovery of novel disease-causing mutation in SSBP1 and its correction using adenine base editor to improve mitochondrial function. Mol. Ther. Nucleic Acids 35, 102257 (2024).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 111011–111033 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Jung, K. S. et al. KRGDB: the large-scale variant database of 1722 Koreans based on whole genome sequencing. Database (Oxford) 2020 https://doi.org/10.1093/database/baaa030 (2020).
Lee, J. et al. A database of 5305 healthy Korean individuals reveals genetic and clinical implications for an East Asian population. Exp. Mol. Med. 54, 1862–1871 (2022).
Stenson, P. D. et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet 136, 665–677 (2017).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Kim, B. J. et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet. Med. 22, 1119–1128 (2020).
Faust, G. G. & Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 (2012).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613 (2018).
Labun, K. et al. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018).
Chaigne, A. In vitro fertilization of mouse oocyte. Methods Mol. Biol. 1818, 37–42 (2018).
Hwang, G. H. et al. Web-based design and analysis tools for CRISPR base editing. BMC Bioinforma. 19, 542 (2018).
Jo, D. H. et al. Visual function restoration in a mouse model of Leber congenital amaurosis via therapeutic base editing. Mol. Ther. Nucleic Acids 31, 16–27 (2023).
Liu, H. & Naismith, J. H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91 (2008).
Lee, S. Y. et al. Ramifications of POU4F3 variants associated with autosomal dominant hearing loss in various molecular aspects. Sci. Rep. 13, 12584 (2023).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Supek, F., Bosnjak, M., Skunca, N. & Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6, e21800 (2011).
Acknowledgements
Most analysis of the sequencing data was carried out using the computing server at the Genomic Medicine Institute Research Service Center. This research was supported and funded by Phase III (Postdoctoral fellowship) grant of the SPST (SNU-SNUH Physician Scientist Training) Program (S.-Y.L.), SNUH Kun-hee Lee Child Cancer & Rare Disease Project, Republic of Korea (No.25C-059-0100 to S.-Y.L.), National Research Foundation of Korea (NRF) and funded by the Ministry of Education (No.2022R1C1C1003147 to S.-Y.L.), SNUH Research Fund (No.37-2023-0120 & 04-2021-0670 to S.-Y.L.), Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (No.RS-2025-02213987 to S.-Y.L.), and Seoul R&BD Program (No.BT240056 to S.-Y.L.). This research was supported by NRF (No.2021M3A9H3015389, SRC-NRF2022R1A5A102641311 to S.B.) and by the Korean Fund for Regenerative Medicine (KFRM) grant (No. RS-2024-00332601 to S.B.). The study was also supported by the National Natural Science Foundation of China (grants 82225014, 82171148 to Y.S., grant 82301318 to B.Z., and grant 82301332 to H.T.), the National Key R&D Program of China (grant 2020YFA0908201 to Y.S., and grant 2023YFA0915004 to H.T. and B.Z.), the Science and Technology Commission of Shanghai Municipality (grants 23J31900100 to Y.S.), the Shanghai Municipal Education Commission (grant 2023ZKZD12 to Y.S.), National Key Research and Development Program of China (grant 2020YFA0908201 to Y.S.), Xuhui District Hospital and Site Cooperation Project (Life and Health field, 23XHYD-05, to Y.S.), Shanghai Municipal Health Commission (grant 2024CXJQ02 to Y.S.) and the Chenguang Program of the Shanghai Education Development Foundation and the Shanghai Municipal Education Commission (grant 23CGA08 to B.Z.). We wish to thank Aavatar Therapeutics (Seunghee Cho, Sangho Park, Dongwoo Song, and Beomseok Choi) for their invaluable technical support during this research.
Author information
Authors and Affiliations
Contributions
Y.S., S.-Y.L., and S.B. jointly conceived and supervised the project. S.-Y.L., and S.H.J. performed the sequencing and bioinformatics analysis. S.W.H., H.K., W.H.C., H.S., B.Z., D.M., H.G., Y.Z., H.T., L.C., and X.C. performed the in vitro experiments. S.-Y.L., S.J., L.J., M.G.K., H.B.C., S-Y.L., Z.W., Y.Z., Z.Z., Y.X., and L.G. performed the in vivo experiments and analyzed the data. S.-Y.L., Y.S., S.B., S.W.H., S.J., L.J., and H.S. wrote the manuscript. Y.S., S.-Y.L., S.B., S.W.H., L.J., Z.W., A.C., M.-W.S., M.K.P., J.-J.S., J.H.L., and B.Z. reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, S.W., Jeong, S., Jiang, L. et al. PAM-flexible adenine base editing rescues hearing loss in a humanized MPZL2 mouse model harboring an East Asian founder mutation. Nat Commun 16, 7186 (2025). https://doi.org/10.1038/s41467-025-62562-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62562-8
