
Abstract
Electron-deficient nitrogen-containing aromatic heterocycles, particularly pyridine derivatives, can be converted into all-carbon aromatic frameworks via the ANRORC (Addition of Nucleophile, Ring-Opening, and Ring-Closing) skeletal editing process, providing a convenient tool for molecular diversification. However, reported methods are mainly limited to nucleophilic modification of ring-opening species. Herein, we report a distinct electrophilic modification method, smoothly converting pyridines into arene-1,3-dialdehydes or naphthalene-1,3-dialdehydes.
Similar content being viewed by others
Introduction
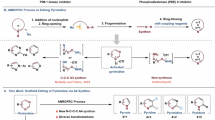
The pyridine ring is a prevalent structural motif in natural products, pharmaceuticals, dyes, pesticides, and functional materials1,2. Its functional modification and derivatization are critical to drug discovery and material science. Numerous well-established methodologies have been developed for pyridine C–H activation3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20, while the skeletal editing of pyridine has recently garnered attention and is recognized as a transformative strategy towards achieving distinctive structural modifications from conventional functional modifications. Pyridine skeletal editing can generally be categorized into two main types (Fig. 1A). One type focuses on the skeletal editing of pyridine into nitrogen-containing heterocycles via ring contraction/expansion or degenerate ring transformation (DRT)21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36. These types of transformations have been extensively studied, and numerous reactions have been reported. On the other hand, while significant advances have been made in nitrogen-atom insertion into heteroarenes37,38 or pyridine construction from cyclic molecules through nitrene-mediated processes39,40, the reverse transformation that converts pyridines into arene architectures via denitrogenation remains relatively underdeveloped41,42,43,44,45,46. One elegant method is a cascade [4+2]/retro-[4+2] pathway (Fig. 1B, top), in which the dearomatization of pyridine via cycloaddition or 1,2-addition reactions generates a cyclic dieneamine intermediate that undergoes a [4+2] cycloaddition with benzyne or substituted alkynes, followed by a retro-[4+2] reaction to reconstruct aromatic ring. Representative examples were reported by Studer47 and Boswell48, respectively. Such a method allows for the introduction of diverse substituents at adjacent positions on the constructed phenyl ring. Another important method is the addition of nucleophile, ring-opening, and ring-closing reaction of pyridinium salt (ANRORC, Fig. 1B, bottom)49,50,51,52,53,54,55,56,57. During this process, a key ring-opening intermediate is formed, including Streptocyanines, Zincke imines, and Zincke aldehydes. These intermediates can be modified by an extra nucleophile at an electrophilic position (Fig. 1C, path a), providing a broad array of substituted arenes. For instance, Kano et al.52,53,54 reported the conversion of pyridines into benzophenones, anilines, or benzaldehydes through the utilization of Streptocyanine intermediates with different functionalized carbon nucleophiles, while Greaney55 used malonates as soft nucleophiles to convert pyridines into benzoates via Zincke imine intermediates. In addition, Glorious56 utilized a Zincke aldehyde in combination with a Wittig reagent to transform pyridine to a benzene ring bearing a variety of versatile functional groups at the ipso position that was originally occupied by the pyridine nitrogen atom.

A Pyridine skeletal editing. B Two general strategies for skeletal editing of pyridine to arene. C Two types of modifications of ring-opening species in ANRORC. D This work: electrophilic modification of ring-opening species.
Despite great progress achieved for the modification of ring-opening intermediates with nucleophilic reagents, electrophilic modification of these intermediates with versatile electrophiles remains an elusive challenge (Fig. 1C, path b). In consideration of the fact that electrophilic modifications will occur at different positions from nucleophilic modifications of ring-opening intermediates, leading to totally different substituted aromatic rings, the development of an electrophilic modification method would be in high demand.
Herein, we report an electrophilic modification of ring-opening intermediates of pyridinium salts for a distinct type of pyridine skeletal editing reaction, converting pyridines into arenes bearing two aldehyde functional groups at 1,3-positions of the formed aromatic ring (Fig. 1D). In the reaction, the use of dual-electrophilic Vilsmeier reagent efficiently modifies nucleophilic sites of ring-opening intermediates derived from 3-aryl or 3-alkenylpyridinium salts.
Results
Optimization studies
We commenced our study, employing 3-phenylpyridinium salt as the model substrate and pyrrolidine as a nucleophilic ring-opening reagent in CHCl3. After the ring-opening step, the solution was added to the prepared Vilsmeier reagent (10.0 equiv.), and the mixture was heated at 60 °C for 10 h. Considering that the electron-deficient pyridine was beneficial to ring-opening, we initially conducted a systematic examination of a series of electron-withdrawing activating groups, including triflyl (Tf), nitroso (NO), 3-nitrophenyl, 2,4-dinitrophenyl (DNP), and 2,6-dinitrophenyl (2,6-DNP) groups. Among them, the 2,4-DNP activator gave the highest yield (80%) of naphthalene-1, 3-dialdehyde 1, while the 3-nitrophenyl group afforded a moderate yield (59%). The 2,6-DNP group showed lower reactivity (18% yield), likely due to steric hindrance. The Tf group gave 13% yield, and no product was observed with the NO group (Fig. 2A). Other secondary amines, such as dipropylamine, piperidine, 4-methylpiperidine, and 1-methylpiperazine, did not give better results than pyrrolidine (Fig. 2B). By increasing the amount of pyrrolidine to 2.2 equiv. (Supplementary Table S3) and reducing the amount of Vilsmeier reagent to 7.0 equiv., the yield can be slightly increased to 83% (Fig. 2C, entry 2). Further optimization found that the preparation time t1 of Vilsmeier reagent was reduced to 0.5 h, and the yield was further increased to 89% (entry 3). Reducing or increasing the preparation time t1 resulted in a decreased yield (Supplementary Tables S4 and S5). Additionally, the change of either the reaction time t2 or the reaction temperature decreased the yield to some extent (entries 4–7). In contrast to the superior performance of CHCl3, THF resulted in a negligible yield (10%), whereas 1,4-dioxane achieved a viable but suboptimal yield of 75% (entries 8–9).

A N-Activating group screening; B Secondary amine evaluation; C Reaction condition optimization; D Investigation into one-pot reactions. aYield was determined by 1H NMR using 1,2-dichloroethane as the internal standard; breaction conditions: 3-phenylpyridinium salt (0.1 mmol), pyrrolidine (0.2 mmol), (COCl)2 (10.0 equiv.), DMF (1.0 mL), CHCl3 (1.0 mL), 60 °C, 10 h; cN-DNP 3-phenylpyridinium salt (0.1 mmol), HNR2 (0.2 mmol) were used. dpyrrolidine (0.02 mmol, 0.2 equiv.) was used; CHCl3 = chloroform, DMF = N, N-Dimethylformamide.
Efforts to establish catalytic conditions failed due to pyrrolidine consumption by excess Vilsmeier reagent. Indeed, nearly quantitative conversion of pyrrolidine to pyrrolidine-1-carbaldehyde (>95%) was observed by 1H NMR analysis upon reaction completion (Supplementary Table S7). Consequently, the optimal reaction condition was as following: The N-DNP 3-phenylpyridinium salt 1a (0.10 mmol) and pyrrolidine (0.22 mmol, 2.2 equiv.) reacted at room temperature in CHCl3 (0.1 M) for 1 h, and the resulting solution was then added into the preformed Vilsmeier reagent (DMF 1.0 mL and 7.0 equiv. of oxalyl chloride reacted at 0 °C for 0.5 h), and then reacted together at 60 °C for 10 h (Fig. 2C, entry 3). Based on the optimal conditions, a one-pot procedure was investigated. Following the ring-opening of 1a with pyrrolidine, oxalyl chloride and DMF were added (instead of preformed Vilsmeier reagent), affording 1 in 60% yield—significantly lower than under standard conditions (Fig. 2D, eq. 1). Notably, when oxalyl chloride and DMF were added prior to 1a and pyrrolidine, the yield dropped significantly to 2% (Fig. 2D, eq. 2). This dramatic decrease is ascribed to the consumption of pyrrolidine (the ring-opening reagent) by the Vilsmeier reagent, thereby suppressing the formation of the ring-opened intermediate—the key species in this transformation (for details, see Supplementary Table S8).
With the optimal conditions in hand, we investigated the substrate scope of the reaction (Fig. 3). A variety of 3-(hetero) aryl pyridinium salts were well compatible with this skeletal editing reaction, providing the corresponding naphthalene-1,3- dialdehydes (Fig. 3A). Pyridinium salts with substituents at the 4-position of the 3-aryl group, including electron-donating group (e.g., alkyl, MeO, PhO, Ph) offered moderate to good yield of desired product (2–7), while those bearing electron-withdrawing groups (e.g., -F, -Cl, -CF3, -CO2Me and -SO2Me) gave lower reactivity (8–12), in general requiring higher temperature and prolonged reaction time, providing 6-substituted naphthalene-1,3-dialdehydes in moderate yields (8–10). When a pyridinium salt with a meta-substituent at the C3-aryl group was used, which has two possible attacking positions available for the Vilsmeier reagent, the reaction occurred preferentially at the less sterically hindered site to deliver the 7-substituted naphthalene-1,3-dialdehyde (13–18) in moderate yield. However, ortho-substituents on the 3-aryl group of pyridine (e.g., Me, F, vinyl) completely suppressed reactivity, likely due to steric hindrance and/or electronic destabilization of intermediates. 3,4-Dimethylphenyl pyridinium salt displayed high efficiency, delivering the corresponding product 19 in 90% yield. 3,4-Disubstituted pyridinium salt also underwent this transformation smoothly, giving a 2-substituted naphthalene-1,3-dicarbaldehyde, which is difficult to access via other synthetic methods (20, 23% yield). Furthermore, functional groups imposing strong electronic effects—including -NO₂, -Ac, -CN, -CHO, and -OH—proved incompatible. These substituents may deactivate the aromatic ring toward electrophilic attack or participate in undesired side reactions (for details, see Supplementary Fig. S7). 3-(Naphthalen-2-yl)pyridinium salt reacted with Vilsmeier reagent exclusively at the α-position, affording phenanthrene-1,3-dicarbaldehyde 21 in 73% yield. However, the pyridinium salts bearing anthracenyl, phenanthracenyl, and pyrenyl groups at the 3-position exhibit nearly no reactivity (for details, see Supplementary Fig. S8). Pyridinium salts with 3-heteroaryls (furan-2-yl, thiophen-2-yl, and benzofuran-2-yl) also reacted well, giving the heteroarenyl fused benzene-1,3-dialdehydes (22–24) in 45-68% yields. Additionally, 3-alkenylpyridinium salts also underwent this skeletal editing reaction to give arene-1,3-aldehyde successfully in moderate to good yields (Fig. 3B, 25–31). Notably, the Z-isomer demonstrates enhanced reactivity relative to the E-isomer of the alkenylpyridinium salts. For example, the Z-28a, Z-29a, and Z-31a consistently afford significantly higher yields of products 28, 29, and 31, respectively, when compared to their E-isomer counterparts.

aStandard conditions: substrate (0.1 mmol, 1.0 equiv.), pyrrolidine (2.2 equiv.), CHCl3 (1.0 mL), DMF (1.0 mL), (COCl)2 (7.0 equiv.), 60 °C, 10 h; bisolated yield; creaction was conducted at 80 °C for 20 h; dreaction was conducted at 60 °C for 20 h; esubstrate was Z configuration; fsubstrate was E configuration.
Substrates scope
The developed method can also be successfully extended to complex bioactive molecules (Fig. 3C). For example, the reaction of bacillamide analogs afforded the corresponding product in 60% yield (32), while Canagliflozin, an SGLT2 inhibitor used in diabetes management, gave a 75% yield (33). Similarly, the method was effective in modifying abiraterone acetate, an androgen biosynthesis inhibitor used in prostate cancer treatment, delivering 50% yield (34). Additionally, the reaction of pirarifenamide analogs, JTK-853 and Estrone, produced the desired products in 58% (35), 37% (36), and 40% (37) yield, respectively. Despite the observed yield variations (ranging from moderate to good, 36%–75%), this methodology introduces a distinct reactivity pattern that could effectively complement current strategies for late-stage modification of pyridine-containing bioactive compounds, thereby expanding options for structure-activity relationship studies and pharmacological optimization.
To demonstrate the scalability of this reaction, a gram-scale experiment of 1a was conducted, providing the desired product 1 without loss of reactivity (Fig. 4, 87% yield). The resultant product, containing two formyl groups, proved to be a versatile synthetic precursor. For example, it can be transformed into a diverse range of functional products, such as naphthalene-1,3-dimethanol 38 through reduction in 90% yield, dinitrile 39 through oxidation amination in 38% yield, and naphthalene-1,3-dicarboxylic acid 40 through oxidation in 48% yield. Furthermore, the product underwent Wittig olefination to form olefins 41 in 55% yield or reacted with dimethyl (1-diazo-2-oxopropyl)phosphonate to offer dialkynes 42 in 80% yield. It was transformed into dioxazole 43 in 68% yield through reaction with TosMIC reagent in the presence of K2CO3. Furthermore, by controlling the reaction conditions, we can also obtain different functionalized monoaldehyde compounds, such as 38a, 38b, 41a, 41b, 42a, 42b, in synthetically useful yields. These compounds would otherwise be difficult to synthesize. The aforementioned results clearly demonstrated the good practicality of our newly developed method.

aIsolated yields. bScale-up reaction. cProduct elaboration and synthetic applications.
Mechanistic studies
A series of experiments were conducted to elucidate the reaction pathway. Streptocyanine 1af as the starting material was dissolved it in CHCl3 with 7.0 equiv. of the Vilsmeier reagent and heated at 60 °C, affording the desired product in 92% yield (Fig. 5A, eq. 1). However, when N-DNP-Zincke imine 1ag was employed as the starting material, the reaction proceeding via a similar procedure gave a significantly decreased yield (18%, Fig. 5A, eq. 2). Kinetic isotope effect (KIE) was measured via parallel experiments of 1a and 1a-D under the optimized reaction conditions, revealing a small KIE (0.66, Fig. 5B, eq. 3). Similarly, a KIE value of 0.67 was observed in a competition reaction between 3-C6D5-pyridine and 3-phenylpyridine (Fig. 5B, eq. 4). These results indicate that C–H bond cleavage might not be the rate-determining step. In addition, a typical inverse secondary kinetic isotope effect (KIE, 0.80) was obtained in a parallel experiments using Vilsmeier reagents that were prepared from DMF-d7 and DMF, respectively (Fig. 5B, eq. 5). This result indicates that the formyl carbon of DMF may undergo a rehybridization process from sp² to sp³ first and then back to sp² during the reaction.

A Identification of key intermediate. B KIE experiments. C The proposed reaction mechanism.
Based on these experiments, a plausible reaction mechanism58,59 was proposed in Fig. 5C. The reaction commences with the nucleophilic attack of a pyrrolidine on the N-DNP 3-phenylpyridinium chloride salt, leading to the ring-opening of the pyridine ring and the formation of a Zincke imine IM-I. This intermediate then further reacts with the second molecular pyrrolidine, generating a streptocyanine IM-II, which then undergoes electrophilic addition with the Vilsmeier reagent. The resulting IM-III eliminates one molecule of HCl to produce IM-IV. Subsequently, a 6π-electrocyclization or intramolecular SEAr pathway occurs, yielding IM-V. The elimination of HNMe2 results in IM-VI, a new aromatic ring bearing two iminium moieties. Finally, hydrolysis of IM-VI produces aromatic or naphthalene dialdehyde compounds.
In summary, a unique electrophilic modification of the ring-opening intermediates of Zincke quaternary ammonium salts by Vilsmeier formylation reagent has been developed, achieving an efficient skeletal reconstruction of 3-aryl/alkenylpyridines into naphthalene dialdehydes or benzenedialdehydes in up to 90% yield. This method represents a significant advance in the field of molecular editing, providing a distinct strategy for the modification of pyridine scaffolds, a structure commonly found in numerous biologically active molecules and pharmaceuticals. The direct transformation of pyridine derivatives into more complex aromatic frameworks demonstrates that the current approach can offer a convenient and rapid tool for the modification of pyridine-containing bioactive molecules, which is crucial for drug discovery and development. Further development of other types of skeletal editing reactions of pyridines is underway in our lab.
Methods
General procedure for the synthesis of Zincke salts
General procedure A
To an oven-dried 100 mL flask, equipped with a magnetic stirrer, was charged with 3-bromopyridine (0.79 mL, 8.0 mmol), substituted phenylboronic acid (10.4 mmol, 1.3 equiv.), Pd(PPh3)4 (0.28 mg, 0.24 mmol, 3%), and Na2CO3 (4.4 g, 32 mmol, 4.0 equiv.). The system was then purged with nitrogen through three cycles of evacuation and backfilling using a balloon. Subsequently, toluene (16.0 mL), water (16.0 mL), and ethanol (5.0 mL) were added to the flask. The reaction mixture was heated under reflux conditions at 110 °C for 24 h. Upon completion of the reaction, the mixture was allowed to cool, followed by filtration and extraction with ethyl acetate (3 × 15 mL). The combined organic extracts were then dried over anhydrous magnesium sulfate (MgSO4), filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 10:1 to 1:1), to yield the desired 3-arylpyridine product.
General procedure B
Under a nitrogen atmosphere, a solution (c = 1.0 M) of potassium tert butoxide (1.1 g, 10.4 mmol, 1.3 equiv.) in tetrahydrofuran was dropped into a solution (c = 1.0 M) of phosphorus ylides (9.6 mmol, 1.2 equiv.) in tetrahydrofuran at 0 °C. The mixture was stirred at 0 °C for 30 min, and then a solution (c = 1.0 M) of pyridine-3-carbaldehyde (0.78 mL, 8.0 mmol, 1.0 equiv.) in tetrahydrofuran was dropped at 0 °C in 15 min. After 16 h of reaction at room temperature, saturated NH4Cl aqueous solution (10.0 mL) was added and stirred for 15 min. The organic phase was separated, and the aqueous phase was extracted three times with ethyl acetate. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Using a mixture of petroleum ether/ethyl acetate (20:1) as the eluent, the crude product was purified by silica gel column chromatography to obtain the desired 3-vinylpyridine.
General procedure C
To the reaction vessel was added the appropriate pyridine (5.0 mmol), 1-halo-2,4-dinitrobenzene (1.6 g, 8.0 mmol,1.6 equiv.), and acetone (15.0 mL) or ethanol (15.0 mL), and the resulting mixture was heated at 60–80 °C for 24 h. The mixture was allowed to cool to room temperature, and concentrated in vacuo to yield the crude product, then washed with ethyl acetate to yield the Zincke salt. Alternatively, EA (ethyl acetate) and DCM/EtOH (1:1) could be used as the eluent; the crude product was purified by silica gel column chromatography to obtain Zincke salt.
General procedure for the synthesis of aryldialdehydes
General procedure D
To a tube 1 equipped with a stir bar was added DMF (1.0 mL). Oxalyl chloride (59.2 μL, 0.7 mmol, 7.0 equiv.) was added dropwise, and the resulting solution was reacted at 0 °C (ice water bath) for 0.5 h. To a tube 2 equipped with a stir bar was added the appropriate Zincke Salt and CHCl3 (0.1 M). Pyrrolidine (18.0 μL, 0.22 mmol, 2.2 equiv.) was added dropwise, and the resulting solution was reacted at 25 °C for 1 h. Then the streptocyanine from tube 2 was added to tube 1 and reacted at 60 °C for 10 h. After cooling to room temperature, the reaction solution was concentrated and purified by thin layer chromatography (silica gel, petroleum ether/ethyl acetate = 5:1) to obtain pure products.
Further experimental details are provided in the Supplementary Information.
Data availability
The details of experimental procedures and the data about the findings of this study are available within the article and its supplementary information. All data are available from the corresponding authors upon request.
References
Sahu, D. et al. Advances in synthesis, medicinal properties and biomedical applications of pyridine derivatives: a comprehensive review. Eur. J. Med. Chem. 12, 100210 (2024).
Mohammad Abu-Taweel, G. et al. Medicinal importance and chemosensing applications of pyridine derivatives: a review. Crit. Rev. Anal. Chem. 54, 599–616 (2022).
Ye, M., Gao, G. & Yu, J. Ligand-promoted C-3 selective C-H olefination of pyridines with Pd catalysts. J. Am. Chem. Soc. 133, 6964–6967 (2011).
Zeng, Y. et al. Direct C-H functionalization of pyridine via a transient activator strategy: synthesis of 2,6-diarylpyridines. Org. Lett. 19, 1970–1973 (2017).
Yin, C. et al. C6-selective direct arylation of 2-phenylpyridine via an activated N-methylpyridinium salt: a combined experimental and theoretical study. Adv. Synth. Catal. 360, 3990–3998 (2018).
Proctor, R. S. J. & Phipps, R. J. Recent advances in minisci-type reactions. Angew. Chem. Int. Ed. 58, 13666–13699 (2019).
Li, W. et al. Stereodivergent synthesis of alkenylpyridines via Pd/Cu catalyzed C-H alkenylation of pyridinium salts with alkynes. Org. Lett. 22, 7814–7819 (2020).
Yang, X. et al. Regioselective direct C-H trifluoromethylation of pyridine. Org. Lett. 22, 7108–7112 (2020).
Li, S. et al. Selective direct C-H polyfluoroarylation of electron-deficient N-heterocyclic compounds. Org. Chem. Front. 7, 3887–3895 (2020).
Zhang, T. et al. A directive Ni catalyst overrides conventional site selectivity in pyridine C-H alkenylation. Nat. Chem. 13, 1207–1213 (2021).
Zhou, X., Zhang, M., Liu, Z., He, J. & Wang, X. C3-selective trifluoromethylthiolation and difluoromethylthiolation of pyridines and pyridine drugs via dihydropyridine intermediates. J. Am. Chem. Soc. 144, 14463–14470 (2022).
Boyle, B. T., Levy, J. N., Lescure, L. D., Paton, R. S. & McNally, A. Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science 378, 773–779 (2022).
Cao, H., Cheng, Q. & Studer, A. Radical and ionic meta-C-H functionalization of pyridines, quinolines, and isoquinolines. Science 378, 779–785 (2022).
Yang, X. et al. Chichibabin-type phosphonylation of 2-(hetero)aryl pyridines: selective synthesis of 4-phosphinoyl pyridines via an activated N-benzylpyridinium salt. Adv. Synth. Catal. 364, 2387–2394 (2022).
Liu, Z. et al. Borane-catalyzed C3-alkylation of pyridines with imines, aldehydes, or ketones as electrophiles. J. Am. Chem. Soc. 144, 4810–4818 (2022).
Sun, G. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67–72 (2023).
Tang, J. et al. Copper-mediated and palladium-catalyzed cross-coupling of indoles and N-methylpyridinium salts: a practical way to prepare 3-(pyridin-2-yl)indoles. Org. Lett. 25, 5203–5208 (2023).
Wang, H. & Greaney, M. F. Regiodivergent arylation of pyridines via Zincke intermediates. Angew. Chem. Int. Ed. 63, e202315418 (2024).
Tang, J. et al. Radical meta-C-H halogenation of azines via N-benzyl activation strategy. Org. Lett. 26, 5899–5904 (2024).
Li, C. et al. Regioselective synthesis of 4-functionalized pyridines. Chem. 10, 628–643 (2024).
Kost, A. N., Gromov, S. P. & Sagitullin, R. S. Pyridine ring nucleophilic recyclizations. Tetrahedron 37, 3423–3454 (1981).
Alekseeva, ΝV. & Yakhontov, L. N. Reactions of pyridines, pyrimidines and 1,3,5-triazines with nucleophilic reagents. Russ. Chem. Rev. 59, 514–530 (1990).
Duan, X. et al. Facile synthesis of 2-methylnicotinonitrile through degenerate ring transformation of pyridinium salts. J. Org. Chem. 87, 7975–7988 (2022).
Zhang, J. et al. Superoxide radical anion triggered dual functioanlization of pyridine through degenerate ring transformation. CCS Chem. 1–10 https://doi.org/10.31635/ccschem.025.202405265 (2025).
Bartholomew, G. L. et al. 14N to 15N isotopic exchange of nitrogen heteroaromatics through skeletal editing. J. Am. Chem. Soc. 146, 2950–2958 (2024).
Feng, M. et al. Pyridine-based strategies towards nitrogen isotope exchange and multiple isotope incorporation. Nat. Commun. 15, 6063 (2024).
Tolchin, Z. A. & Smith, J. M. 15NRORC: an azine labeling protocol. J. Am. Chem. Soc. 146, 2939–2943 (2024).
Weber, H. & Rohn, T. 2H-Azirine und 2-(ω-Cyanalkyl)furane als neuartige photoprodukte aus [n](2,6)pyridinophan-N-oxiden und ihre bedeutung für den reaktionsverlauf. Chem. Ber. 122, 945–950 (1989).
Feng, Z., Allred, T. K., Hurlow, E. E. & Harran, P. G. Anomalous chromophore disruption enables an eight-step synthesis and stereochemical reassignment of (+)-marineosin A. J. Am. Chem. Soc. 141, 2274–2278 (2019).
Hurlow, E. E. et al. J. Am. Chem. Soc. 142, 20717–20724 (2020) .
Xu, K. et al. Synthesis of 2-formylpyrroles from pyridinium iodide salts. Org. Lett. 22, 6107–6111 (2020).
Tang, J. et al. Tandem ring-contraction/regioselective C-H iodination reaction of pyridinium salts. J. Org. Chem. 88, 2809–2821 (2023).
Inui, H., Sawada, K., Oishi, S., Ushida, K. & McMahon, R. J. Aryl nitrene rearrangements: spectroscopic observation of a benzazirine and its ring expansion to a ketenimine by heavy-atom tunneling. J. Am. Chem. Soc. 135, 10246–10249 (2013).
Babra, J. S., Russell, A. T., Smith, C. D. & Zhang, Y. Combining C-H functionalisation and flow photochemical heterocyclic metamorphosis (FP-HM) for the synthesis of benzo[1,3]oxazepines. Tetrahedron 74, 5351–5357 (2018).
Mailloux, M. J., Fleming, G. S., Kumta, S. S. & Beeler, A. B. Unified synthesis of azepines by visible-light-mediated dearomative ring expansion of aromatic N-Ylides. Org. Lett. 23, 525–529 (2021).
Boudry, E., Bourdreux, F., Marrot, J., Moreau, X. & Ghiazza, C. Dearomatization of pyridines: photochemical skeletal enlargement for the synthesis of 1,2-diazepines. J. Am. Chem. Soc. 146, 2845–2854 (2024).
Zhang, X. et al. Asymmetric dearomative single-atom skeletal editing of indoles and pyrroles. Nat. Chem. 17, 215–225 (2025).
Zhang, X. et al. Copper-catalyzed enantioselective skeletal editing through a formal nitrogen insertion into indoles to synthesize atropisomeric aminoaryl quinoxalines. Angew. Chem. Int. Ed. 64, e202420390 (2025).
Xu, Y., Xiang, S., Che, J., Wang, Y. & Tan, B. Skeletal editing of cyclic molecules using nitrenes. Chin. J. Chem. 42, 2656–2667 (2024).
Reisenbauer, J. C., Ori, G., Franchino, A., Finkelstein, P. & Morandi, B. Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104–1109 (2022).
Schmerling, L. & Toekelt, W. G. Direct conversion of pyridine to benzoic acid. J. Am. Chem. Soc. 86, 1259–1259 (1964).
Sagitullin, R. S., Gromov, S. P. & Kost, A. N. Alkylamino group exchange upon recyclization of pyridinium salts into anilines. Tetrahedron 34, 2213–2216 (1978).
Hamada, Y., Morishita, K. I., Ozawa, I., Takeuchi, I. & Hirota, M. Reaction of diazaphenanthrenes and their N-oxides with methylsulfinyl carbanion. Chem. Pharm. Bull. 27, 1535–1543 (1979).
Fout, A. R., Bailey, B. C., Tomaszewski, J. & Mindiola, D. J. Cyclic denitrogenation of N-heterocycles applying a homogeneous titanium reagent. J. Am. Chem. Soc. 129, 12640–12641 (2007).
Cabrera-Pardo, J. R., Chai, D. I. & Kozmin, S. A. Silver-promoted benzannulations of siloxyalkynes with pyridinium and isoquinolinium salts. Adv. Synth. Catal. 355, 2495–2498 (2013).
Zhu, C., Kuniyil, R. & Ackermann, L. Manganese(I)-catalyzed C-H activation/Diels-Alder/retro-Diels-Alder domino alkyne annulation featuring transformable pyridines. Angew. Chem. Int. Ed. 58, 5338–5342 (2019).
Cheng, Q. et al. Skeletal editing of pyridines through atom-pair swap from CN to CC. Nat. Chem. 16, 741–748 (2024).
Boswell, B. R., Zhao, Z., Gonciarz, R. L. & Pandya, K. M. Regioselective pyridine to benzene edit inspired by water displacement. J. Am. Chem. Soc. 146, 19660–19666 (2024).
Hamada, Y. & TakeUchi, I. Reaction of benzo[f or h]quinolines and their N-oxides with methylsulfinyl carbanion. J. Org. Chem. 42, 4209–4213 (1977).
Falcone, N. A., He, S., Hoskin, J. F., Mangat, S. & Sorensen, E. J. N-oxide-to-carbon transmutations of azaarene N-oxides. Org. Lett. 26, 4280–4285 (2024).
Wang, T. et al. Skeletal editing of pyridine and quinoline N-oxides through nitrogen to carbon single atom swap. CCS Chem. 7, 392–402 (2025).
Morofuji, T., Kinoshita, H. & Kano, N. Connecting a carbonyl and a π-conjugated group through a p-phenylene linker by (5+1) benzene ring formation. Chem. Commun. 55, 8575–8578 (2019).
Morofuji, T., Inagawa, K. & Kano, N. Sequential ring-opening and ring-closing reactions for converting para-substituted pyridines into meta-substituted anilines. Org. Lett. 23, 6126–6130 (2021).
Morofuji, T., Nagai, S., Watanabe, A., Inagawa, K. & Kano, N. Streptocyanine as an activation mode of amine catalysis for the conversion of pyridine rings to benzene rings. Chem. Sci. 14, 485–490 (2023).
Conboy, A. & Greaney, M. F. Synthesis of benzenes from pyridines via N to C switch. Chem. 10, 1940–1949 (2024).
Wu, F. et al. Nitrogen-to-functionalized carbon atom transmutation of pyridine. Chem. Sci. 15, 15205–15211 (2024).
Li, S. et al. C3 Selective chalcogenation and fluorination of pyridine using classic Zincke imine Intermediates. Nat. Commun. 15, 7420 (2024).
Chen, X., Zhao, H., Chen, C., Jiang, H. & Zhang, M. Hydrogen-transfer-mediated α-functionalization of 1,8-naphthyridines by a strategy overcoming the over-hydrogenation barrier. Angew. Chem. Int. Ed. 56, 14232–14236 (2017).
Zhao, H. et al. Intermolecular diastereoselective annulation of azaarenes into fused N-heterocycles by Ru(II) reductive catalysis. Nat. Commun. 13, 2393 (2022).
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos. 22072099 and 21871187 to H.F.) and the Sichuan Science and Technology Program (No. 2024YFFK0016 to H.F.; No. 2024ZYD0099 to X.Z.). We also thank Chunchun Zhang from the Centre of Analysis & Testing, Dongyan Deng, and Jing Li from the College of Chemistry, Sichuan University, for NMR, HRMS measurements.
Author information
Authors and Affiliations
Contributions
M.Y. and Y.S. initiated and conducted the primary experiments and analyzed data, with M.Y. taking primary responsibility for figure preparation and manuscript formatting; C.L., S.H., S.L., and J.Z. contributed to experiments and characterization of products; D.Y. elucidated product structures. Z.S., W.J., W.X., J.X., X.Z., and R.L. contributed to the discussion on the study; H.F. drafted the initial version of the manuscript and, together with H.C., supervised the research and provided critical revisions. All authors contributed to this research project through in-depth discussions, data analysis, and result interpretation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dong-Liang Mo, Lvyin Zheng, and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yan, M., Shi, Y., Lv, C. et al. Skeletal editing of pyridines to aryldialdehydes. Nat Commun 16, 7133 (2025). https://doi.org/10.1038/s41467-025-62627-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62627-8
