
Abstract
Rapid reaction screening and in-depth mechanistic exploration of electroorganic synthesis remain challenging due to low throughput of experimentation and high complexity of electrode and homogenous processes. Here, we report a decoupled electrochemical flow microreactor hyphenated mass spectrometry (namely DEC-FMR-MS) platform for high-throughput reaction screening and intermediate tracking of electrosynthesis. This platform combines in-capillary electrochemical transformation with operando MS interrogation, enabling rapid reactivity survey of a series of electrosynthetic reactions on nanomole scale. Moreover, the spatial decoupling design allows segmented dissection of short-lived intermediates involved pathways in interfacial electrochemical and homogenous chemical events, which are initially interwoven in reaction networks. The utility of this platform is highlighted by 1) discovery and verification of quasi-electrocatalytic pathways in electrooxidative C-H/N-H cross-coupling, 2) kinetic measurements of TEMPOH-mediated dehydrogenation of N-heterocycles, and 3) mapping the landscape of intermediates (alkene radical cation and nitrene) in electrochemical aziridination.
Similar content being viewed by others
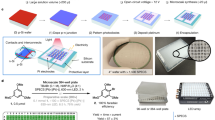


Introduction
Electroorganic synthesis driven by electrons has gained significant attraction in the context of the global development of sustainable and clean energy1,2,3. In parallel with methodological development, electrosynthetic screening and mechanistic investigation also advance electrosynthesis by revealing to what extent the desired products are yielded under different conditions and how the electroactive species are converted at various nodes of reaction network4,5,6,7,8. To ensure efficient screening of reaction conditions, high-throughput experimentation (HTE) was generally designed to synchronously implement multiple electrochemical reactions involving many combinations of variables9,10,11,12,13. However, whether based on batch-type or flow-type electroorganic reactors, most screening strategies rely on follow-up or on-line high performance liquid chromatography (HPLC) to determine the yields, with the analytical rates still far from the requirement of rapid reactivity surveys (Fig. 1a)11. On the other hand, beyond the product analysis at final state, mechanistic studies on electrosyntheses necessitate the monitoring of intermediate processes. Nonetheless, insights into the electrosynthetic mechanisms are commonly gained or deduced from the electroanalytical (e.g. cyclic voltammetry) or spectroelectrochemical characterizations14,15, which lack the capability of directly verifying the identity of key intermediates in the reactions.

a Schematic of conventional electroorganic synthesis monitoring and screening methods based on batch-type electrolysis cells and liquid chromatography-mass spectrometry (LC-MS). b The DEC-FMR-MS platform for mechanistic investigation and reaction screening of electroorganic synthesis. The upper left inset shows the decoupling design in the T-junction for independent control of electrode reactions of each substrate and subsequent homogeneous processes, and the details at the reactant inlets of EC-FMR-1 and EC-FMR-2.
With high sensitivity and specificity, mass spectrometry (MS) offers an ideal analytical terminal for both reaction screening and mechanistic exploration16,17,18. As a high-throughput sample processing platform, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) has shown extraordinary adaptability to large-scale synthetic screening, but the off-line MS interrogation hindered the visualization of transient processes in a confined time window, particularly in electrosynthesis16. Over the past decade, advancements of ambient ionization mass spectrometry (AIMS) afford great opportunities on probing short-lived intermediates and final products even in real time, thus expediting our understanding of chemical transformations at electrochemical interfaces19,20,21. Regarding synthetic electrochemistry, a series of electrochemical mass spectrometry (EC-MS) platforms featuring bipolar electrode micropipettes22,23,24, nanoelectrostatic spray25,26, floating electrolysis27,28, and other interfacing modes17,21,29 have demonstrated their ability to capture fleeting intermediates and monitor their fates, providing molecular evidence for unraveling or verifying the mechanism of electrochemical processes7,28. Despite these merits, most of the current EC-MS techniques still utilize high voltage (~kV) in contact or non-contact manner for ionization, thus bringing difficulties in precise and continuous control on the electrode potential22,30. Moreover, manual preloading of reactants into the reactor (typically also the emitter) is necessary for each single run, limiting the possibility of HTE for electrochemical synthesis.
Another critical point is that electroorganic synthesis involves initial generation of reactive intermediates at heterogeneous electrode-electrolyte interface (EEI) and subsequent transformation to secondary intermediates and products in electrolyte31,32. Compared to merely identifying electrogenerated intermediates by conventional MS snapshot, characterization of dynamic fates of reactive species in the complete pathway is more essential for clarifying the interconnected transformation routes in both electrode and homogenous processes28,29. In particular, the direct identification of individual redox event and how it interact with other reactant/intermediates are long-sought for understanding the reaction mechanism, guiding the catalyst/mediator design, and quenching side reactions33. However, the reactive intermediates (e.g. cationic species) rapidly shuttle between electrode interface and bulk solution, undergoing multi-step gain/loss of electrons and addition of reaction partners34,35. How to distinguish the species produced at the heterogeneous EEI from those born in homogeneous reaction and illustrate their respective roles in the reaction cascades remains elusive.
Here, we propose a decoupled electrochemical flow microreactor hyphenated non-high voltage ionization MS (namely DEC-FMR-MS) to isolate the interfacial electrochemistry from follow-up homogenous reaction for real-time MS interrogation, enabling rapid reaction screening and segmented intermediate dissection of electroorganic synthesis at molecular level (Fig. 1). In this setup, adoption of Venturi sonic-spray ion source eliminates the influence of extra ionization voltage on intrinsic electrochemistry, making possible the flexible tuning of variables (e.g. potential, catalyst and substrate) in electrosynthesis screening. The design of decoupled electrochemical flow reactor allows independent control of electrode reactions of each substrate and subsequent homogeneous processes, helping to clarify individual radical-involved pathway from complicated reaction networks (Fig. 1b). By coupling this MS platform with an electrochemical microplate (ECMP), MS yield-based high-throughput screening of electroorganic synthesis (4 s per sample) on nanomole scale is achieved. The capability of the proposed methodology in reaction screening and mechanistic exploration is demonstrated by fully revealing the reactive intermediates and final products of C-H/N-H radical cross-coupling, electrocatalytic dehydrogenation, and electrochemical aziridination. By probing the respective electrode reactions and homogenous transformation of reactants in a decoupled manner, we uncover the hidden quasi-electrocatalytic pathways in electrooxidative C-H/N-H radical cross-coupling, measure the kinetics of TEMPOH-mediated dehydrogenation reaction of N-heterocycles, and revisit the alkene radical cation- and nitrene-initiated homogenous reaction routes in electrochemical aziridination.
Results
Design and configuration of the DEC-FMR-MS platform
The DEC-FMR-MS platform was devised by merging the flow routes of two EC-FMRs assigned for individual substrate activation into a T-junction, with the mixed fluids continuously directed to Venturi-sonic spray ion source for MS detection (Fig. 1b and Supplementary Figs. 1, 2). The EC-FMR-1 with the configuration similar to our previous work36 was used to generate cationic intermediates of one substrate, while the EC-FMR-2 was split into a capillary with Pt wire working electrode (Pt WE) inside as a screening probe, and an EC microwell with Ag wire quasi-reference electrode (QRE) inserted and graphite plate counter electrode (GP CE) as the bottom (Supplementary Fig. 3) In this manner, anodic events of substrates in a one-pot EC batch reactor were divided into two independent circuits for flexibly modulating the electro-activation of each substrate. The reactive intermediates respectively generated in the two EC-FMRs were aspirated by Venturi effect into the mixing capillary, where homogeneous chemical reactions subsequently occurred (Supplementary Fig. 2). The terminal high voltage-free Venturi-sonic spray ion source avoids the potential influence exerted from in-source electrochemistry, and ensures fast delivery (velocity = 0.27 m/s) and MS readout of initially electrogenerated intermediates, secondary intermediates and final electrosynthetic products in their intrinsic states. Particularly, the reaction in EC-FMR-2 was triggered by dipping the screening probe into the substrate solution in microwells to complete the circuit. This “dip-and-run” mode permits rapid sample injection and switching in the ECMP controlled by a motorized XY-stage (Supplementary Fig. 4), thereby affording the feasibility of HTE in electroorganic synthesis.
Capability of the DEC-FMR-MS platform in mechanistic exploration and yield assessment
First, we employed the electrooxidative C-H/N-H cross-coupling reaction of N, N-dimethylaniline (DMA) and phenothiazine (PTA) as a model reaction to investigate the capability of DEC-FMR-MS in electroorganic reaction yield assessment37,38. To guarantee the normalized MS response of product, the electrochemically inactive acetylcholine (Ach) was mixed with the reactants in ECMP as the internal standard (IS) (Supplementary Fig. 5). According to previous reports from Lei group, C-N bond can be formed through radical-radical cross coupling between DMA radical cation (DMA•+, 5) and nitrogen radical (PTA•, 7)39. Recently, the nitrenium ion-involved cross coupling pathway was also revealed (Fig. 2a)22. In this setup, PTA and DMA in acetonitrile (ACN) were respectively assigned to the EC-FMR-1 and EC-FMR-2, with a potential of 1.5 V (vs Ag QRE) applied to Pt WEs (termed dual electrolysis mode). All the species derived from the reactor were monitored by MS while lifting and dropping the ECMP. Compared to no potential applied on Pt WEs (Supplementary Fig. 6), signals of the product radical cation 3aa′ (m/z 318.1194, see Supplementary Fig. 7 for the origin of this species), IS (m/z 146.1176), the intermediates 4, 5, 7 (m/z 121.0886, 198.0378, 199.0455) and by-product N,N,N′,N′-tetramethylbenzidine (TMB, 9) (m/z 241.1740) were all unambiguously detected (Fig. 2b), indicating the capability of DEC-FMR-MS in capturing short-lived intermediates and monitoring the cross-coupling products (See COMSOL simulation and MS/MS spectra in Supplementary Figs. 8, 9 and Figs. 10, 11). Along with the screening probe hopping from one microwell to another, the extracted ion chromatograms (EICs) of IS and 3aa′ demonstrated a string of sampling event-dependent pulsed signals (each sampling event lasting for 4 s) with a stable intensity ratio of 3aa′ to IS (the relative standard deviation was 9.9%) (Fig. 2c, d). Furthermore, compared to the conventional batch reaction for 1–2 h, the online reaction in DEC-FMR can generate more than 10-fold enhanced product signal, mainly due to the high ratio of electrode surface to electrolyte volume (Fig. 2e and Supplementary Fig. 12). The excellent signal reproducibility and reaction efficiency make the proposed DEC-FMR-MS an ideal platform for HTE and analysis of organic electrosynthesis.

a The mechanism for electrooxidative C-H/N-H cross-coupling between 1a and 2a deduced by the identified intermediates. b–d The positive-ion-mode mass spectrum (b), The extracted ion chromatograms (EICs) of internal standard (IS) (m/z 146.1176) and product 3aa′ (m/z 318.1194) (c) and time-dependent relative intensity ratio of 3aa′ to IS (d) obtained by DEC-FMR-MS for the high-throughput testing of the electrooxidative radical-radical cross-coupling between DMA and PTA in dual electrolysis mode. e Comparison of the relative ion signal intensity ratio of 3aa′ to IS obtained by online reaction in the DEC-FMR-MS platform and conventional batch reaction. The error bars in (e) represent the standard deviations of three replicates.
Reactivity survey and mechanistic dissection of electrooxidative C-H/N-H cross-coupling
Taking the electrooxidative C-H/N-H cross-coupling between electron-rich arenes and diarylamine derivatives as an example (Fig. 3a), we investigated the feasibility of DEC-FMR-MS in electrochemical reaction screening by the dual electrolysis mode (Fig. 3b). In this mode, certain arene or diarylamine was introduced into EC-FMR-1, while the coupling partners with diverse substituents were distributed in the microwells of ECMP containing a fixed amount of IS (Fig. 3c). To minimize the cross contamination, the reactor was washed by ACN three times after each sampling event (Supplementary Fig. 13). Similar to the coupling between DMA and PTA, ion signals of the products (both M•+ and [M + H]+) emerged in the spectra (Supplementary Figs. 14–16), and the transformation efficiency was assessed according to the ion signal ratio of product to IS (namely MS yield) (Fig. 3d). Furthermore, how oxidative potential (from 0.5 V to 2.5 V) affects the product yield was also explored. As shown in Fig. 3e, significant differences in MS yields were observed in the DEC-FMR-MS-based reaction screening across a diversity of arenes, diarylamines and electrooxidative potentials. In principle, due to limited reaction time (around 1 s) in the DEC-FMR, the MS yield was primarily dependent on the initial rate of electrochemical reaction, which can truly reflect the reactivity of substrates. The results showed that with 1a as the coupling partner, PTA bearing electron-withdrawing substituents (2b–2d), phenoxazine (2e) and diarylamines (2f, 2g) could afford the desired products in relative high MS yields. On the other hand, DMA and its derivatives with electron-donating groups at the meta-position (1b, 1c) exhibited high reactivity in the amination. By contrast, other electron-rich arenes including 2,5-dimethylpyrrole (1e) and 2-phenylindole (1f) showed decreased MS yields in this transformation. All the above reactivities obtained by this platform coincided well with the yields calculated by Lei group39, underscoring the reliability of the proposed DEC-FMR-MS in rapid survey of organic electrosynthesis with nanomole sample consumption (8 μL).

a, b Schematics of electrooxidative C-H/N-H cross-coupling reaction (a) and the T-junction of DEC-FMR-MS for studying this reaction in dual electrolysis mode (b). c Arenes and diarylamine derivatives as substrates for the electrooxidative C-H/N-H cross-coupling reaction. d, e Positive-ion-mode mass spectra of representative cross-coupling products 3fa′ and 3ag′ (d), and heat map of MS yields across a variety of arenes, diarylamine derivatives and electrooxidative potentials (e) obtained by DEC-FMR-MS. f, g Schematics of the T-junction of DEC-FMR-MS working in single electrolysis mode with 2a (f) or 1a (g) electrooxidized. h–k Representative positive-ion-mode mass spectra (h, i) and potential-dependent ion signals of m/z 241.1704, 240.1628, 199.0455, 198.0378 and 318.1194 (j, k) for the reaction in which 2a (h, j) and 1a (i, k) was electrooxidized along with a linear potential scan in EC-FMR-1. l, m The proposed quasi-electrocatalytic pathways for C-H/N-H cross-coupling between 1a and 2a mediated by 7 (l) and by-product 10 (m), respectively.
The presented DEC-FMR-MS allows spatial separation between interfacial electrooxidation and homogeneous coupling, thus offering the opportunity for mapping the homogeneous process of individual reactive species generated at EEI. To dissect the mechanism of cross-coupling between PTA and DMA, we employed a single electrolysis mode in which only EC-FMR-1 was turned on to figure out the reaction pathway involving electro-activated single substrate (Fig. 3f, g). Firstly, PTA was loaded in EC-FMR-1 for electrolysis, with the intermediates converging with unoxidized DMA in the T-junction (Fig. 3f). In addition to radical cation and nitrenium ion of PTA (5 and 7), the cross-coupling product 3aa′ was surprisingly detected at an onset potential higher than 5 and 7 (Fig. 3h, j, Supplementary Fig. 17a–d). This observation implied that cross-coupling still took place without the electrooxidation of DMA, going against with the classic understanding that electrochemical radical-radical cross-coupling requires concurrent production of radicals from each substrate at EEI (Fig. 3a). By excluding the probable interference between electrochemical reactions in EC-FMR-1 and EC-FMR-2 (Supplementary Figs. 18–21), we speculate that DMA may undergo oxidation to DMA•+ by the electrogenerated oxidative nitrenium ion 7 in homogeneous electrolyte, and further react with another molecule of 6 to finalize the reaction22 (Fig. 3l). Similar homogenous oxidation process for the generation of DMA•+ could also be seen when replacing PTA with 1-hydroxy-2,2,6,6-tetramethylpiperidine (TEMPOH), whose oxidation potential was close to that of PTA•+ (Supplementary Fig. 22). Based on the above evidence, a quasi-electrocatalytic pathway that electrogenerated nitrenium ion oxidizes DMA to DMA•+ in homogenous system was uncovered, contributing to the C-H/N-H cross-coupling reaction.
Then we exchanged PTA with DMA in the DEC-FMR-MS running in the single electrolysis mode. Apart from the expected homocoupling by-product 9 and its radical cation (TMB•+) 10, the ion signals of 5, 7 and 3aa′ were still observed (Fig. 3i, k), featuring a good correlation with the applied potential steps (Supplementary Fig. 17e–h). The EICs showed that 7 and 3aa′ emerged after the dimerization of DMA, represented by the generation of [TMB + H]+ and TMB•+ (Fig. 3k). Thereby, we considered the likelihood that oxidative or homocoupled DMA species may drive the transformation of PTA to 5 and 740,41. Our guess was confirmed by directly loading TMB instead of DMA in the EC-FMR-1, showing that the electrogenerated 10 was able to oxidize PTA in homogenous system (Supplementary Fig. 23, 24). Therefore, another pathway for the C-H/N-H cross-coupling mediated by the homocoupling product 9 as an electrocatalyst was proposed, in which the oxidative 10 converted PTA to 6 for further coupling with DMA•+ to furnish the final product 3aa′ (Fig. 3m).
Reactivity survey and kinetic measurement of electrocatalytic reactions
Molecular electrocatalysis has recently formed an attractive class of chemical transformations, which leverages redox mediators instead of electron transfer at EEI to indirectly drive the conversion of substrates with higher selectivity and less undesired side reactions (Fig. 4a)15,31,42. This reaction is greatly compatible with the proposed DEC-FMR-MS, in which the electro-activation of redox mediators in EC-FMR-1 is spatially isolated from the merging with the substrates infused from EC-FMR-2 with no voltage applied (termed single electrolysis mode in Fig. 4b). Recruiting TEMPOH-mediated dehydrogenation of 1,2,3,4-tetrahydroquinoldine (THQ, 11a) as example (Fig. 4c)43,44, we observed the apparent electro-conversion of TEMPOH to TEMPO+ in EC-FMR-1 and confirmed that the stepwise oxidation of THQ was solely attributed to TEMPO+ in homogeneous system (Figs. 4d–f, Supplementary Fig. 25, 26). Furthermore, the intermediates in this process including the short-lived radical cation 13 (m/z 133.0896), and the two-step dehydrogenation products 14 (m/z 132.0817) and 12a (m/z 130.0661) were all captured (Fig. 4f and Supplementary Fig. 27). We then used this platform to perform reaction screening of electrocatalytic dehydrogenation of N-heterocycles. By choosing THQ as the substrate, we found that only the catalyst TEMPOH with the ability of both hydrogen abstraction and electron transfer can afford the final product 12a, in comparison with ferrocene (M2) and triphenylamine (M3) that barely possess electron transfer capability (Fig. 4g, h, Supplementary Fig. 28 and 29)45,46. The reactivity survey was implemented among a series of saturated six-membered and five-membered N-heterocycles with different substituent groups, demonstrating diversified potential-dependent MS yields (Fig. 4h). This result also gives a hint that the substrates with strong electro-withdrawing group (11e and 11h) display low reactivity, in line with the conventional yield evaluation43.

a, b Schematics of electrocatalytic dehydrogenation of N-heterocycles (a) and the T-junction of DEC-FMR-MS for studying the molecular electrocatalytic reactions in single electrolysis mode (b). c The proposed mechanism for dehydrogenation of 11a mediated by electroactivated M1. d–f Schematics and positive-ion-mode mass spectra of the reactions with different reactants infused into the DEC-FMR-MS platform. g Redox mediators and substrates selected for electrocatalytic dehydrogenation of N-heterocycles. h Heat map of MS yields among various redox mediators, N-heterocycles and electrooxidative potentials obtained by DEC-FMR-MS. i, j Schematics of electrocatalytic dehydrogenation of 11a mediated by M1 (i) and modified DEC-FMR-MS setup for measuring the total kinetics of homogeneous dehydrogenation (j). k Positive-ion-mode mass spectra for the reaction between electrochemically generated M1+ and 11a operated at different Lcap. l The plot of ln (I 156 / (I 156+I 158)) against treaction for TEMPOH-mediated dehydrogenation of 11a, 11b, and 11c. m Total kinetic constants kdehydrogenation for the secondary homogeneous reactions between M1+ and different N-heterocycles. The error bars in (l) represent the standard deviations of three replicates.
In this electrocatalytic process, the homogeneous reaction kinetics between electrogenerated TEMPO+ and substrates determines the efficacy of whole transformation (Fig. 4i), which yet remains difficult to obtain in a one-pot EC reactor. Our design of DEC-FMR-MS offers the opportunity to spatiotemporally isolate the downstream homogenous reaction for independent measurement. By simply adjusting the length of mixing capillary (Lcap), we could modulate the homogeneous reaction time (treaction) between TEMPO+ and THQ derivatives while recording the resulting products spraying out of capillary by MS (Fig. 4j and Supplementary Fig. 30). With the extension of capillary, the electrogenerated TEMPO+ demonstrated a gradually declined ion signal, meaning the reversion to TEMPOH due to the dehydrogenation of THQ (Fig. 4k). Considering this homogeneous reaction as a pseudo first-order reaction, we plot the natural logarithm of relative intensity of TEMPO+ (I156 / (I156 + I158)) against t, which showed a good linear relationship for the substrate THQ (R2 = 0.943). Thus, the rate constant k of the entire dehydrogenation cascade reaction could be obtained from the slope of the fit line, and the measured value was 1.72 s−1 (Fig. 4l, see details in Supplementary Fig. 31). Similarly, the TEMPO+-driven dehydrogenation of other N-heterocycles also exhibited apparent first-order kinetics, according to the time-dependent linear decline of ln (I 156 / (I 156+I 158)) (Fig. 4l and Supplementary Fig. 32). As indicated in the kinetic measurement, the THQ core substituted by electron-donating groups (-CH3 and -O-CH3) in benzene ring showed faster kinetics than those bearing electron-withdrawing moieties (-Cl and -NO2) (Fig. 4m and Supplementary Fig. 33).
Mechanistic insights into electrochemical aziridination of alkenes
Next, we moved on to apply the DEC-FMR-MS platform to explore the electrochemical aziridination of alkenes3,47,48. These reactions commonly experience alkene radical cation-49,50 or nitrene-mediated pathways51,52, depending on the oxidative potentials of alkene and amine (Fig. 5a). In particular, the alkene activation pathway undergoes sequential electrochemical oxidation of alkene (A) and coupling intermediate radical (C•)49,50,53, so we modified the setup by prolonging the WE of EC-FMR-1 into the mixing capillary (termed sequential electrolysis mode) to allow further oxidation of C• to complete the ring closure (Fig. 5b and Supplementary Fig. 34). As a typical reaction, the aziridination starting from trans-anethole (TA, 15a) and N-aminophthimide (PhtNH2, 16a) demonstrated the product (17aa, m/z 309.1232) and the acetonitrile-trapped carbocation (25, m/z 350.1495)50 in the mass spectrum obtained by the modified DEC-FMR-MS (Fig. 5c and Supplementary Figs. 35, 36). By comparing the relative ion signal of the resulting aziridines (normalized by IS at m/z 146.1176), reactivity investigation was performed among a variety of alkenes (15a–15m) and amines (16a–16d) (Fig. 5d, e and Supplementary Fig. 37, 38). The results showed that TA (15a) and triaryl-substituted alkenes (15h, 15i, 15l) can effectively react with the tested amines, probably due to their relative low oxidation potential and stable radical cations53. With regard to amines, PhtNH2 (16a) proved a more favorable coupling partner than other primary alkyl amines to afford the targeted aziridines, because of its ease to generate nitrene.

a, b Schematics of electrochemical aziridination of alkenes involving alkene and amine activation pathways (a) and the T-junction of DEC-FMR-MS for studying this reaction in sequential electrolysis mode (b). c Positive-ion-mode mass spectrum of the reaction between 15a and 16a obtained by DEC-FMR-MS. d Alkenes and amines as substrates for the electrochemical aziridination. e Heat map of MS yields among different alkenes, amines, and electrooxidative potentials obtained by DEC-FMR-MS in sequential electrolysis mode. f Schematics of electrochemical aziridination operated in the DEC-FMR-MS platform in sequential electrolysis mode with (i) and without (ii) potential applied on WE in EC-FMR-2, and dual electrolysis mode (iii). The applied voltage was 6 V. g Contribution of diverse processes concluded from the comparison of the ion signal intensity ratio of 17aa to IS in setup i-iii. h Reaction map for electrochemical aziridination of alkenes including alkene radical cation- and nitrene-mediated pathways with all the intermediates identified and tracked by DEC-FMR-MS. i, j Single electrolysis mode DEC-FMR-MS and segmented MS setups for unraveling 15a (i) or 16a (j) activation pathways. k, l Positive-ion-mode mass spectra (3 V) and the potential-dependent EICs of m/z 147.0805, 148.0883, 309.1232, and 350.1495 for the aziridination initiated by electroactivated 15a acquired by DEC-FMR-MS (k) and segmented MS monitoring (l). m, n Positive-ion-mode mass spectra (3 V) and the potential-dependent EICs of m/z 161.0345, 162.0424, 309.1232, and 350.1495 for the aziridination initiated by electroactivated 16a acquired by DEC-FMR-MS (m) and segmented MS monitoring (n). The error bars in (g) represent the standard deviations of three replicates.
Taking the aziridination of TA with PhtNH2 as an example, we further dig into the reaction mechanism by flexibly configuring the reactor. First, to clarify the proportion of the two pathways mediated by different intermediates in the whole process, the voltage supply of EC-FMR-2 was cut off to deduct the contribution of activated PhtNH2 (PhtNH2*) (Fig. 5f, setup ii). In comparison with the sequential electrolysis mode (Fig. 5f, setup i), the relative intensity of final product 17aa saw a dramatic decline by 67.0% (Fig. 5g, setup ii, Supplementary Fig. 39b), meaning that PhtNH2* generated in setup i accounted for the majority of the transformation as compared to the activated TA (TA*). Secondly, we attempted to clarify the role of extended WE in the oxidative deprotonation of the coupling intermediate C• in the alkene activation pathway. The WE was thus retracted from the mixing capillary back to the EC-FMR-1 (Fig. 5f, g, setup iii), causing a 16.7% decrease of the final product. This difference implied that the extended section of Pt WE indeed facilitated the electron loss of intermediate C• (intermediate 20) to finalize the reaction (P1) (Fig. 5h and Supplementary Fig. 39c), but it remained uncertain whether the electrooxidation is the only route to convert the intermediate C• to aziridine.
Although the alkene or amine activation pathways have been proposed by organic chemists52,53, it remains challenging to track the evolution of these crucial intermediates in the above two pathways, due to the difficulty in individually deconstructing the two routes with similar oxidation potentials from the intercrossed reaction network (Supplementary Fig. 40). To unscramble the fates of electro-activated TA or PhtNH2 in the reaction with the coupling partners, we employed a single electrolysis mode in which only the voltage supply of EC-FMR-1 was on (Figs. 5i and j, left panel). When TA was introduced into EC-FMR-1, the ion signal of product 17aa and by-product 25 were detected (Fig. 5k), indicating that solely anodic oxidation of TA was able to trigger the aziridination, with the by-product 25 demonstrating the carbocation-involved route (Fig. 5h, green pathway). Without anode inserted into the mixing capillary, the emergence of 17aa proved that there must be some other electron acceptors in homogeneous solution responsible for oxidative deprotonation of the coupling intermediate 20 (P2), rather than anode. To further visualize the intermediates initially born at anode, we segmented this reactor by only retaining the EC-FMR-1 to directly capture the electroactivated species (Fig. 5i, right panel). Unlike the carbocation 19 (m/z 147.0805) detected in both cases (Supplementary Fig. 35), the newly emerged signal of TA•+ (18, m/z 148.0883) was not found in the spectrum obtained by the intact setup (Fig. 5l and Supplementary Fig. 41), corroborating that TA•+ was the key species for the coupling reaction with PhtNH2. After exchanging TA with PhtNH2 in the two reactors, we could still observe the signal of product 17aa (Fig. 5m), which means the anodic oxidation of PhtNH2 can independently initiate the aziridination. By the segmented MS interrogation of the EC-FMR-1, the radical cation 22 (m/z 162.0424) and protonated nitrene 23 (m/z 161.0345) were unambiguously captured and identified when oxidation voltage was applied (Fig. 5n and Supplementary Figs. 35, 36, 41), whereas no activated PhtNH2 species was detected in the presence of TA (Fig. 5m). Moreover, the acetonitrile-trapped carbocation 25 was not visualized here, ruling out the carbocation-involved route possibly starting from the radical cation 2252. According to the above evidence, we ascribed the only way of aziridination mediated by amine activation to the direct transfer of electrogenerated nitrene 23 to alkene (P3). Therefore, by virtue of the decoupling design, the intercrossed alkene radical cation- and nitrene-mediated pathways in aziridination were clearly split up, with the identity and interdependence of all intermediates in the reaction map definitely confirmed, including the anode-independent oxidative transformation P2 (Fig. 5h).
Investigation of paired electrosynthesis and exploration of unknown radical coupling reactions
In addition to above electrooxidation reactions, we further inspected the applicability of the proposed MS platform in paired electrolysis that integrates both anodic and cathodic transformations54,55,56. The electrochemical deoxygenative amination reported by Xu et al. was selected as a model reaction, which combines the cathodic reaction of 6-nitroquinoline (26a) and the anodic reaction of 2-phenylethyl hydrazinecarboxylate (27a)57 to deliver the final product 28aa. Upon applying −5 V or 5 V to the Pt WE in EC-FMR-1 and EC-FMR-2, respectively, the key intermediates reactive 6-nitrosoquinoline (29, m/z 159.0553) and 2-phenylethyl N-iminocarbamate (30, m/z 179.0815) resulting from electroreduction of 26a and electrooxidation of 27a were unambiguously detected (Supplementary Fig. 42). Notably, the ion signal of product 28aa (m/z 249.1392) emerged exclusively when simultaneously applying potentials on both electrodes. The ion signal of products derived from the reactions between 26a and other carbazates (27b and 27c) could also be detected by the DEC-FMR-MS platform (Supplementary Fig. 43), further underscoring its applicability in exploring paired electrosynthesis reactions.
Although the DEC-FMR-MS platform has demonstrated its utility for condition optimization and substrate expansion via reactivity survey, another key advantage of HTE lies in its potential for exploring new electroorganic reactions. 4-Alkyl-1,4-dihydropyridines (DHPs) have recently emerged as versatile alkyl radical donors in thermal radical reactions, photoredox catalysis, and electrochemical reactions58,59,60. The DEC-FMR-MS platform may allow systematic screening of reaction partners that can electrochemically generate radicals for coupling with 4-alkyl-1,4-DHP-derived alkyl radicals, thereby suggesting new electroorganic transformation possibilities. We here recruited a series of 4-alkyl-1,4-DHPs to assess their reactivity with a diverse array of coupling partners under electrochemical conditions by infusing them respectively into the DEC-FMRs for MS interrogation (Supplementary Fig. 44). Mass spectra showed that some tested diarylamines (2a, 2e, and 2g) and alkene (15a) underwent electrochemical activation and cross-coupled with the radical species electrochemically generated from 4-(4-methoxyphenyl)methyl-1,4-DHP (34a), giving the desired products (37, 39, 41, and 42) (Supplementary Figs. 45–49). Moreover, 1,4-DHP bearing other alkyl substitutes (34b and 34c) also exhibited considerable reactivity with diarylamines (2a, 2e, and 2g) (Supplementary Figs. 50 and 51). Equipped with established radical-generation strategies, the reaction screening conducted on the DEC-FMR-MS platform enables further exploration of potential coupling between radical donors and acceptors via electrochemical redox processes.
Discussion
In conclusion, we present a versatile operando mass spectrometry platform (DEC-FMR-MS) for high-throughput reactivity assessment and intermediate tracking of electroorganic synthesis by interfacing a decoupled electrochemical flow microreactor with V-EASI-MS. Different from conventional yield calculation, the DEC-FMR-MS platform leveraged MS yield to accomplish high-efficiency (4 s per sample) electrosynthetic screening on nanomole scale among a diversity of substrates, catalysts and voltages applied, including electrooxidative C-H/N-H cross-coupling, molecular electrocatalysis, and electrochemical aziridination. More importantly, the decoupling design allows for segmented mechanistic investigation of electrode and homogenous processes based on the fate of intermediates. Due to this merit, we discovered the quasi-electrocatalytic pathways mediated by nitrenium ion or radical cation for electrooxidative C-H/N-H cross-coupling, other than the classic electrogenerated radical coupling. Furthermore, the kinetics of the secondary homogeneous process in molecular electrocatalysis, typically TEMPOH-mediated dehydrogenation of N-heterocycles, was successfully measured. Lastly, by spatially separated characterization of alkene and amine activation routes, we mapped the intermediate (alkene radical cation and nitrene) landscape of electrochemical aziridination with upstream and downstream molecular evidence.
Although the decoupling design permits tracing of reaction pathways originating from individual electrogenerated intermediates, there are still some limitations in capturing ultrafast electrogenerated species and assessing multi-step electrosynthetic reactions. Firstly, the successful detection of the short-lived intermediate hinges on its residual concentration at the capillary outlet, which is influenced by the extent of decay occurring during transport. In this setup, the mixing capillary required for homogeneous reactions prolongs the decay time, partially limiting the ability to capture reaction intermediates. Nevertheless, it is also inappropriate to simply regard the in-capillary transfer time (~0.37 s) as the intermediate detectable threshold lifetime for this MS platform. Whether the metastable intermediate can be detected depends on three critical parameters: reactant concentration, formation and depletion reaction kinetics of the intermediate, and detection limit of mass spectrometer. Thus, it is still possible to detect those species with short half-lives but fast formation kinetics. Secondly, while the flow microreactor in the DEC-FMR-MS platform facilitates rapid intermediate transfer and high-throughput reaction analysis, it inherently reduces both electrode and homogeneous reaction time, thereby limiting intermediate retention for subsequent electrochemical conversions. Consequently, it may struggle to capture the intermediates or products from multi-step electrochemical processes and thermodynamically favorable but kinetically slow reactions. These limitations could potentially be mitigated by extending the mixing region and configuring electrodes in series to facilitate multi-step redox transformations. Looking forward, we envision the DEC-FMR-MS platform would be also applicable to parameter optimization and mechanism exploration of other electrosynthetic reactions, such as electrochemical transition-metal catalysis61 or photoelectrochemical catalysis62, with more reaction variables to be adjusted and more interconnected interfacial and homogenous processes to be deconstructed.
Methods
Configuration of DEC-FMR-MS
The DEC-FMR-MS platform is comprised of two EC-FMRs, a mixing capillary, V-EASI source, and an LTQ-Orbitrap MS detector (Supplementary Figs. 1 and 2). In the EC-FMR-1, a fused-silica capillary (I.D. 200 μm, O.D. 360 μm, length 7 cm) with Pt WE (Φ 100 μm) inside was inserted into the PDMS reactant reservoir to form a three-electrode system with the Pt CE (Φ 500 μm) and Ag QRE (Φ 500 μm) inserted into the PDMS reservoir. The extended capillary of EC-FMR-1 was then connected vertically with another capillary (I.D. 200 μm, O.D. 360 μm, length 5 cm) with Pt WE (Φ 100 μm) inside by a PEEK T-junction. Serving as a screening probe, the vertical capillary was coupled with the ECMP to form the EC-FMR-2. The fabrication of DEC-FMR was finalized by further connecting a mixing capillary (I.D. 200 μm, O.D. 360 μm, length 10 cm) to the T-junction as the outlet flow path for homogeneous reaction (Supplementary Fig. 2c). In addition, EC-FMR-1 and EC-FMR-2 was controlled independently by two electrochemical workstations (CHI760E, CH Instruments Inc.) to decouple and regulate the individual electrochemical reactions. To minimize in-source EC reaction, V-EASI was utilized for soft ionization without the need for high voltage. In detail, the mixing capillary in DEC-FMR passed through a stainless-steel T-junction (Swagelok, Solon, Ohio) and then an outer capillary (I.D. 530 μm, O.D. 700 μm, length 1 cm) for introducing nebulizing nitrogen gas. Finally, the electrogenerated species from the EC-FMRs and products formed in the mixing capillary were ionized and monitored by LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The specific details for fabrication of EC-FMRs and ECMP can be seen in Supplementary Information.
High-throughput screening of electrosynthetic reactions
The DEC-FMR-MS allowed for high-throughput and rapid screening for electrosynthetic reactions with the aid of a motorized XY-stage (Supplementary Fig. 4). In the high-throughput experiment, the potentiostatic method was consistently employed to initiate the electrochemical reactions in EC-FMR-1 and EC-FMR-2, while the mass spectrometer continuously acquired the ion signal of products spraying out. In detail, 500 μL of the reactant solution containing 1 mM LiOTf was initially added into PDMS reservoir of EC-FMR-1, while a sequence of solutions (25 μL) containing reaction partners and IS was added into each microwell of the ECMP on the XY-stage. By manipulation of the XY-stage, the screening probe in EC-FMR-2 was sequentially inserted into each microwell (positioned 2 mm above the bottom) to initiate the electrochemical reaction, and kept for a given reaction time before being withdrawn for the next sampling action. Based on the self-extraction of Venturi effect induced by a high-velocity N2 stream (58 psi), reactive intermediates generated in EC-FMR-1 and EC-FMR-2 were rapidly transferred to the mixing capillary for subsequent homogeneous chemical reactions, and the resulting products and IS were timely detected by MS. After each reaction survey in EC-FMR-2, the screening probe was rinsed three times in the microwell containing 25 μL of ACN to mitigate cross-contamination. For surveying the influence of the applied potential on electroorganic synthesis, the applied potential in EC-FMR-1 and EC-FMR-2 was synchronously modulated. The Supplementary Information provides additional details on various electrolysis modes for different electrosynthetic reactions.
Tracking of intermediates in electrosynthetic reactions
To achieve the real-time tracking of the dynamic fate of the fleeting intermediates in electrosynthetic reactions, a higher N2 pressure (75 psi) was employed in V-EASI. The reactants were dissolved in ACN containing 1 mM LiOTf and assigned to EC-FMR-1 and EC-FMR-2. For the MS decoupling of electrosynthetic reactions, voltage was typically applied only on the WE in EC-FMR-1. The potential step and linear sweep experiments were done by using chronoamperometry (CA) and cyclic voltammetry (CV), while the intermediates and products were rapidly delivered to MS inlet for detection. The moment of triggering the EC methods was defined as time zero, and the mass spectra were continuously recorded at the meantime. During CV scan, the scan rate was set as 20 mV/s ranging from 0 V to 1.5 V. The potential step was adjusted using CA by alternately applying 0 V and 1.5 V (for aziridination reaction, the potential was switched between 0 V and 3 V) on the Pt WE.
Mass spectrometry settings and data processing
All MS data were obtained using the LTQ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). The MS ion transfer capillary was maintained at 275 °C, and the resolution of 30,000 was employed for all analyses. All data were analyzed utilizing the Qual Browser feature of the XcaliburTM program (Thermo Fisher Scientific, San Jose, CA). The collision-induced dissociation (CID) and high energy collision dissociation (HCD) were performed for the structural analysis of the intermediates and products from electrooxidative C-H/N-H cross-coupling, TEMPOH-mediated dehydrogenation of THQ, and aziridination reaction.
Data availability
The data generated in this study can be found in the Figshare database (https://doi.org/10.6084/m9.figshare.29608940). Source data are provided with this paper.
References
Leech, M. C. & Lam, K. A practical guide to electrosynthesis. Nat. Rev. Chem. 6, 275–286 (2022).
Kingston, C. et al. A survival guide for the “electro-curious”. Acc. Chem. Res. 53, 72–83 (2020).
Holst, D. E., Wang, D. J., Kim, M. J., Guzei, I. A. & Wickens, Z. K. Aziridine synthesis by coupling amines and alkenes via an electrogenerated dication. Nature 596, 74–79 (2021).
Sheng, H. et al. Autonomous closed-loop mechanistic investigation of molecular electrochemistry via automation. Nat. Commun. 15, 2781 (2024).
Liu, Y. et al. Time-resolved EPR revealed the formation, structure, and reactivity of N-centered radicals in an electrochemical C(sp3)-H arylation reaction. J. Am. Chem. Soc. 143, 20863–20872 (2021).
Dinh, L. P. et al. Persistent organonickel complexes as general platforms for Csp2–Csp3 coupling reactions. Nat. Chem. 16, 1515–1522 (2024).
Cheng, H. et al. Picomole-scale transition metal electrocatalysis screening platform for discovery of mild C-C coupling and C-H arylation through in situ anodically generated cationic Pd. J. Am. Chem. Soc. 144, 1306–1312 (2022).
Nie, W. et al. Ultra-high-throughput mapping of the chemical space of asymmetric catalysis enables accelerated reaction discovery. Nat. Commun. 14, 6671 (2023).
Perera, D. et al. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 359, 429–434 (2018).
Buitrago Santanilla, A. et al. Nanomole-scale high-throughput chemistry for the synthesis of complex molecules. Science 347, 49–53 (2015).
Mo, Y., Rughoobur, G., Nambiar, A. M. K., Zhang, K. & Jensen, K. F. A multifunctional microfluidic platform for high-throughput experimentation of electroorganic chemistry. Angew. Chem. Int. Ed. 59, 20890–20894 (2020).
Wu, L. et al. High-throughput nano-electrostatic-spray ionization/photoreaction mass spectrometric platform for the discovery of visible-light-activated photocatalytic reactions in the picomole scale. Anal. Chem. 93, 14560–14567 (2021).
Chen, S., Wan, Q. & Badu-Tawiah, A. K. Picomole-scale real-time photoreaction screening: discovery of the visible-light-promoted dehydrogenation of tetrahydroquinolines under ambient conditions. Angew. Chem. Int. Ed. 55, 9345–9349 (2016).
Ding, W., Li, M., Fan, J. & Cheng, X. Palladium-catalyzed asymmetric allylic 4-pyridinylation via electroreductive substitution reaction. Nat. Commun. 13, 5642 (2022).
Badalyan, A. & Stahl, S. S. Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature 535, 406–410 (2016).
Lin, S. et al. Mapping the dark space of chemical reactions with extended nanomole synthesis and MALDI-TOF MS. Science 361, eaar6236 (2018).
Surendran, A. K., Pereverzev, A. Y. & Roithová, J. Intricacies of mass transport during electrocatalysis: a journey through iron porphyrin-catalyzed oxygen reduction. J. Am. Chem. Soc. 146, 15619–15626 (2024).
Das, A. et al. On-the-fly mass spectrometry in digital microfluidics enabled by a microspray hole: toward multidimensional reaction monitoring in automated synthesis platforms. J. Am. Chem. Soc. 144, 10353–10360 (2022).
Cooks, R. G., Ouyang, Z., Takats, Z. & Wiseman, J. M. Ambient mass spectrometry. Science 311, 1566–1570 (2006).
Brown, T. A., Chen, H. & Zare, R. N. Detection of the short-lived radical cation intermediate in the electrooxidation of N,N-dimethylaniline by mass spectrometry. Angew. Chem. Int. Ed. 54, 11183–11185 (2015).
Brown, T. A., Chen, H. & Zare, R. N. Identification of fleeting electrochemical reaction intermediates using desorption electrospray ionization mass spectrometry. J. Am. Chem. Soc. 137, 7274–7277 (2015).
Hu, J. et al. Dissecting the clash Chemistry of electrogenerated reactive intermediates by microdroplet fusion mass spectrometry. Angew. Chem. Int. Ed. 60, 18494–18496 (2021).
Hu, J. et al. Coupling a wireless bipolar ultramicroelectrode with nano-electrospray ionization mass spectrometry: insights into the ultrafast initial step of electrochemical reactions. Angew. Chem. Int. Ed. 59, 18244–18248 (2020).
Li, Y. et al. Paper-in-tip bipolar electrospray mass spectrometry for real-time chemical reaction monitoring. Angew. Chem. Int. Ed. 63, e202318169 (2024).
Zhang, X. et al. Identifying metal-oxo/peroxo intermediates in catalytic water oxidation by in situ electrochemical mass spectrometry. J. Am. Chem. Soc. 144, 17748–17752 (2022).
Yu, Z. et al. Revealing the sulfur redox paths in a Li-S battery by an in situ hyphenated technique of electrochemistry and mass spectrometry. Adv. Mater. 34, e2106618 (2021).
Chen, J. et al. In situ probing and identification of electrochemical reaction intermediates by floating electrolytic electrospray mass spectrometry. Angew. Chem. Int. Ed. 62, e202219302 (2023).
Liu, X., Chen, J., Wei, Z., Yi, H. & Lei, A. Deciphering reactive intermediates in electrooxidative coupling of indoles through real-time mass spectrometry. Chem 10, 2131–2146 (2024).
Wan, Q. et al. Elucidating the underlying reactivities of alternating current electrosynthesis by time-resolved mapping of short-lived reactive intermediates. Angew. Chem. Int. Ed. 62, e202306460 (2023).
Cheng, H., Tang, S., Yang, T., Xu, S. & Yan, X. Accelerating electrochemical reactions in a voltage-controlled interfacial microreactor. Angew. Chem. Int. Ed. 59, 19862–19867 (2020).
Novaes, L. F. T. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941–8002 (2021).
Elsherbini, M. & Wirth, T. Electroorganic synthesis under flow conditions. Acc. Chem. Res. 52, 3287–3296 (2019).
McKenzie, E. C. R. et al. Versatile tools for understanding electrosynthetic mechanisms. Chem. Rev. 122, 3292–3335 (2022).
Takumi, M., Sakaue, H. & Nagaki, A. Flash electrochemical approach to carbocations. Angew. Chem. Int. Ed. 61, e202116177 (2022).
Yoshida, J. I., Shimizu, A. & Hayashi, R. Electrogenerated cationic reactive intermediates: the pool method and further advances. Chem. Rev. 118, 4702–4730 (2018).
Zhang, X. et al. Insights into electrochemiluminescence dynamics by synchronizing real-time electrical, luminescence, and mass spectrometric measurements. Chem. Sci. 13, 6244–6253 (2022).
Bariwal, J. & Van der Eycken, E. C–N bond forming cross-coupling reactions: an overview. Chem. Soc. Rev. 42, 9283–9303 (2013).
Morofuji, T., Shimizu, A. & Yoshida, J.-I. Direct C–N coupling of imidazoles with aromatic and benzylic compounds via electrooxidative C–H functionalization. J. Am. Chem. Soc. 136, 4496–4499 (2014).
Liu, K. et al. Electrooxidative para-selective C-H/N-H cross-coupling with hydrogen evolution to synthesize triarylamine derivatives. Nat. Commun. 10, 639 (2019).
Hou, Z.-W., Mao, Z.-Y., Melcamu, Y. Y., Lu, X. & Xu, H.-C. Electrochemical synthesis of imidazo-fused N-heteroaromatic compounds through a C−N bond-forming radical cascade. Angew. Chem. Int. Ed. 57, 1636–1639 (2018).
Feng, E. Q., Hou, Z.-W. & Xu, H.-C. Electrochemical synthesis of tetrasubstituted hydrazines by dehydrogenative N-N bond formation. Chin. J. Org. Chem. 39, 1424–1428 (2019).
Hickey, D. P. et al. Predicting electrocatalytic properties: modeling structure–activity relationships of nitroxyl radicals. J. Am. Chem. Soc. 137, 16179–16186 (2015).
Wu, Y., Yi, H. & Lei, A. Electrochemical acceptorless dehydrogenation of N-heterocycles utilizing TEMPO as organo-electrocatalyst. ACS Catal. 8, 1192–1196 (2018).
Wan, Q., Chen, S. & Badu-Tawiah, A. K. An integrated mass spectrometry platform enables picomole-scale real-time electrosynthetic reaction screening and discovery. Chem. Sci. 9, 5724–5729 (2018).
Nutting, J. E., Rafiee, M. & Stahl, S. S. Tetramethylpiperidine N-oxyl (TEMPO), phthalimide N-oxyl (PINO), and related N-oxyl species: electrochemical properties and their use in electrocatalytic reactions. Chem. Rev. 118, 4834–4885 (2018).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Siu, T. & Yudin, A. K. Practical olefin aziridination with a broad substrate scope. J. Am. Chem. Soc. 124, 530–531 (2002).
Liu, M. S., Du, H. W., Cui, J. F. & Shu, W. Intermolecular metal-free cyclopropanation and aziridination of alkenes with XH2 (X=N, C) by thianthrenation. Angew. Chem. Int. Ed. 61, e202209929 (2022).
Yu, J. & Wu, J. Aziridination of internal alkenes using primary alkyl amines in a microflow electrocell. Chem 7, 18–19 (2021).
Li, J. et al. Electrochemical aziridination by alkene activation using a sulfamate as the nitrogen source. Angew. Chem. Int. Ed. 57, 5695–5698 (2018).
Siu, T., Picard, C. J. & Yudin, A. K. Development of electrochemical processes for nitrene generation and transfer. J. Org. Chem. 70, 932–937 (2005).
Wang, S. et al. Electrochemical flow aziridination of unactivated alkenes. Natl. Sci. Rev. 10, nwad187 (2023).
Ošeka, M. et al. Electrochemical aziridination of internal alkenes with primary amines. Chem 7, 255–266 (2021).
Liu, Z.-R. et al. Synergistic use of photocatalysis and convergent paired electrolysis for nickel-catalyzed arylation of cyclic alcohols. Sci. Bull. 69, 1866–1874 (2024).
Hilt, G. Recent advances in paired electrolysis and their application in organic electrosynthesis. Curr. Opin. Electroche. 43, 101425 (2024).
Nie, L. et al. Linear paired electrolysis enables redox-neutral (3 + 2) annulation of benzofuran with vinyldiazo compounds. J. Am. Chem. Soc. 146, 31330–31338 (2024).
Xu, J. et al. Electrochemical deoxygenative amination of stabilized alkyl radicals from activated alcohols. Nat. Commun. 15, 6116 (2024).
Nakajima, K., Nojima, S. & Nishibayashi, Y. Nickel- and photoredox-catalyzed cross-coupling reactions of aryl halides with 4-alkyl-1,4-dihydropyridines as formal nucleophilic alkylation reagents. Angew. Chem. Int. Ed. 55, 14106–14110 (2016).
Chen, W. et al. Building congested ketone: substituted Hantzsch ester and nitrile as alkylation reagents in photoredox catalysis. J. Am. Chem. Soc. 138, 12312–12315 (2016).
Yan, H., Hou, Z.-W. & Xu, H.-C. photoelectrochemical C−H alkylation of heteroarenes with organotrifluoroborates. Angew. Chem. Int. Ed. 58, 4592–4595 (2019).
Cheng, X. et al. Recent applications of homogeneous catalysis in electrochemical organic synthesis. CCS Chem. 4, 1120–1152 (2022).
Sendeku, M. G. et al. Frontiers in photoelectrochemical catalysis: a focus on valuable product synthesis. Adv. Mater. 36, 2308101 (2024).
Acknowledgements
This research is supported by the National Natural Science Foundation of China (No. 22374069, Q. M.), the Fundamental Research Funds for the Central Universities (No. 020514380345, Q. M.), the funding from State Key Laboratory of Analytical Chemistry for Life Science (No. 5431ZZXM2401, Q. M.), the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2023A1515110224, X. Z.), and the Jiangsu Funding Program for Excellent Postdoctoral Talent (No. 2023ZB442, X. Z.). We also thank Prof. Zhangquan Peng (Dalian Institute of Chemical Physics, CAS) for the idea of naming the MS platform.
Author information
Authors and Affiliations
Contributions
Q.M. and X.Z. conceived the project. X.Z. designed and built the DEC-FMR-MS. X.Z., Y.Z., M.L., Q.Y., and W.L. performed the experiment. X.Z. analyzed and interpreted the experimental data. X.Z. and Q.M. co-wrote the paper. Q.M., X.C., and J.-J.Z. corrected the manuscript and guided the revision. All authors contributed to the discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Abraham Badu-Tawiah, Hai-Chao Xu, and the other anonymous reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Zhang, Y., Li, M. et al. Screening and dissecting electroorganic synthesis by mass spectrometry decoupling of electrode and homogeneous processes. Nat Commun 16, 7452 (2025). https://doi.org/10.1038/s41467-025-62677-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62677-y
