
Abstract
Reconstruction of catalysts by reaction environments represents a viable approach to create highly performed active sites. Herein, we develop a reaction-induced regioselective reconstruction of Ni-doped Ce(OH)3/CeO2 nanorods to form dual-active sites composed of carburized Ni clusters and frustrated Lewis pairs (FLPs), delivering exceptional activity, selectivity and stability for reverse water-gas shift reaction. Ni aggregation in the Ce(OH)3 region, coupled with in-situ carbonization by catalytically generated CO during reaction, induces the formation of the carburized Ni clusters, which effectively promoted H2 dissociation. Additionally, Ni doping in the CeO2 region and Ce(OH)3-to-CeO2 phase transition introduce more oxygen vacancies and thereby generated FLPs in CeO2, which facilitate CO2 adsorption and subsequent hydrogenation by spilled *H species from the carburized Ni clusters. Weak CO adsorption on both the carburized Ni clusters and FLPs significantly suppresses the methanation side-reaction. This reaction-induced regioselective reconstruction strategy provides a new avenue for designing highly performed catalysts.
Similar content being viewed by others

Introduction
Development of advanced catalysts to achieve exceptional activity and selectivity is an ambitious but challenging topic for addressing the global challenges of climate, energy security, and environmental sustainability1,2,3. Currently, the cutting-edge efforts have been focused on acquiring precisely defined active sites with the meticulously pre-designed geometric and/or electronic structures4,5,6,7,8. However, their structures (e.g., sizes, phases, local coordination environments) can be significantly affected by the reaction conditions, generally leading to decayed catalytic performance9,10. Meanwhile, recent advances have revealed that the reconstruction of catalysts induced by adsorbed species and/or catalytic environments can progressively produce new active sites with dramatically improved catalytic performance11,12,13,14,15,16,17. Practically, those reconstructions are challenging to attain through conventional preparation methods. Unfortunately, one irresistibly drawback of the reaction-induced reconstruction strategy is the lack of precise controllability on the pre-designed active sites of catalysts into the anticipated new ones with high performance.
Since a reaction generally involves multiple reactants, another fundamental paradox arises from the entire surface region of heterogeneous catalysts under the reconstruction in response to the reaction environments, which may be unpropitious to the simultaneous co-adsorption and activation of multiple reactants18,19,20,21,22. Motivated by the existential concerns of the reaction-induced reconstruction and encouraged by prior cognitions of the contributions of multiple active sites for co-activation of several molecules on the improved catalytic performance, a novel methodology is anticipated to achieve the precisely controllable and/or regioselective reconstruction of a pre-catalyst under a specific reaction environment. Specifically, this approach can enable in-situ creation of multiple active sites with distinct local environments, facilitating the effective co-activation of all reactants.
Herein, we demonstrate the reaction-induced regioselective reconstruction of the Ni-doped Ce(OH)3/CeO2 nanorods (Ni-Ce(OH)3/CeO2) to construct the dual-active sites of the creatively carburized Ni clusters and precisely designed frustrated Lewis pairs (FLPs), enabling exceptionally active, selective, and robust catalytic performance for the reverse water-gas shift (RWGS) reaction. During RWGS, the thermal decomposition of Ce(OH)3 into CeO2 triggered the selective aggregation of the Ni dopants into clusters in the region of Ce(OH)3, which simultaneously underwent carbonization by the catalytically generated CO to create new carburized Ni clusters. Additionally, the Ce(OH)3-to-CeO2 phase transition generated more structural defects of oxygen vacancy in the reconstructed Ce(OH)3 region. Combining the precisely designed defective Ni-doped CeO2, these high levels of oxygen vacancies in the regioselective reconstructed catalysts resulted in the formation of abundant FLP sites. The synergistic effect of the carburized Ni clusters and FLP sites delivered high capability for the adsorption and activation of H2 and CO2, respectively. Simultaneously, the weak CO adsorption on the FLPs and carburized Ni clusters with the downward shift of the d-band effectively suppressed the methanation side reaction. Hydrogen spillover from the carburized Ni clusters to the FLPs sites enabled a CO generation rate of 27.3 mol gNi−1 h−1 with a selectivity of >99.9% for the reconstructed Ni-Ce(OH)3/CeO2 catalysts at 550 °C. Moreover, the Ni-Ce(OH)3/CeO2 catalysts operated stably and continuously for at least 1000 h.
Results
Theoretical analysis
Recently, we have demonstrated both theoretically and experimentally the effective CO2 activation even at temperatures below 100 °C by the constructed FLPs sites of (Ce3+/Ce3+…O2−) on the defective CeO2(110) surface with two adjacent oxygen vacancies (CeO2(110)−2OV), consisting of the lattice O2− as Lewis base and two neighboring Ce3+ as Lewis acid (Fig. 1a)23,24. Density functional theory (DFT) calculations reveal the much stronger CO2 adsorption on FLPs through a bridged configuration compared to that on the ideal CeO2(110) surface (Fig. 1b and Supplementary Figs. 1 and 2). Conversely, the relatively weak H2 adsorption is theoretically profiled on both ideal CeO2(110) and FLPs sites in comparison to the strong CO2 adsorption on the same sites (Fig. 1b and Supplementary Figs. 1 and 2). Therefore, the competitive adsorption of H2 and CO2 on the defective CeO2(110) indicates the failure of their simultaneous activation, leading to the poor RWGS activity of CeO2 with FLPs alone in our previous report25.

a Scheme of the FLPs site on the defective CeO2(110) surface. When two adjacent oxygen vacancies are present on CeO2(110), the FLPs sites are formed, wherein lattice O2− acts as a Lewis base and two neighboring Ce3+ ions as Lewis acids. b The adsorption behaviors of CO2 and H2 on the CeO2(110) surface with various active sites. c PDOS analysis of the Ni4/CeO2(110) and carburized Ni4/CeO2(110). d Scheme of RWGS on the dual-active sites of the carburized Ni clusters and FLPs.
Recently, the Ni-doped or Ni-anchored CeO2 catalysts have been extensively investigated for H2 dissociation and subsequent CO2 hydrogenation26,27,28,29. To address the impact of Ni, we theoretically investigated the chemical-doping of Ni in CeO2 by replacing one of the Ce3+ of FLPs (CeO2(110)−2OV–Nisurf) and one lattice Ce in the subsurface (CeO2(110)−2OV–Nisub). As revealed from the DFT calculations (Fig. 1b and Supplementary Figs. 3 and 4), the adsorption energy of CO2 is still much higher than that of H2 on the FLPs sites of both CeO2(110)−2OV–Nisurf and CeO2(110)−2OV–Nisub.
Subsequently, a Ni4 cluster supported on the surface of CeO2(110)−2OV (Ni4/CeO2(110)−2OV) is constructed. It is noteworthy that the Ni4 cluster exhibits a directly dissociative adsorption for H2 (−1.26 eV) in comparison to CO2 adsorption (−1.03 eV, Fig. 1a and Supplementary Fig. 5). Theoretically, the dual-active sites of Ni clusters and FLPs sites hold promise to enable the highly performed RWGS reaction. However, experimental results demonstrated the enhanced activity of the Ni clusters deposited on PN-CeO2 (Nicluster/PN-CeO2) with FLPs at the expense of CO selectivity (Supplementary Fig. 6). DFT calculations reveal a strong adsorption energy of *CO on Ni4 cluster of Ni4/CeO2(111)−2OV (−2.16 eV, Supplementary Fig. 7), thereby leading to subsequent over-hydrogenation of CO into CH430,31,32,33,34,35,36,37.
According to the d-band theory, the binding strength between metal and guest molecules is determined by the filling degree of the antibonding state38,39. Introducing carbon atoms into metal clusters can effectively tailor the d-band center of metals15,40,41. Carbon has been demonstrated to be highly miscible in the Ni lattice, followed by the surface diffusion and formation of carbide-like phases42. Motivated by this recognition, the density of states (DOS) of Ni 3d orbital in both Ni4/CeO2(110)−2OV and carburized Ni4C1/CeO2(110)−2OV (Note: A carbon atom was introduced into the surface of the Ni4 cluster) were investigated. It was evident that the presence of a carbon atom on the Ni4 cluster modulates both spin-up and spin-down states, causing a downward shift of the d-band center of Ni from −1.07 eV to −1.86 eV (Fig. 1c). Therefore, the incorporation of C atoms into Ni clusters results in a decrease in the CO adsorption strength. Specifically, the adsorption energies of CO on Ni4C1/CeO2(110)−2OV, Ni4C2/CeO2(110)−2OV and Ni4C3/CeO2(110)−2OV, which contain one, two and three C atoms, respectively, are −1.92 eV, −1.62 eV and −0.89 eV (Supplementary Fig. 7). Comparatively, the carburized Ni cluster in Ni4C1/CeO2(110)−2OV exhibits a spontaneous dissociation of H2 into two *H species, with an adsorption energy of −1.59 eV (Fig. 1b and Supplementary Fig. 8). Meanwhile, CO2 adsorption on the same carburized Ni cluster is relatively weak with an adsorption energy of −1.03 eV (Fig. 1b and Supplementary Fig. 8). Therefore, the carburized Ni clusters exhibit stronger dissociative adsorption ability for H2 compared to the CO2 adsorption, and also benefit the desorption of generated CO.
Based on the theoretical analysis, the highly performed RWGS reaction can be realized by the rationally designed dual-active sites of the carburized Ni clusters and FLPs on CeO2 surface, which involves (Fig. 1d): (I) the spontaneous dissociation of H2 on the carburized Ni clusters to generate *H species; (II) the efficient activation of CO2 on FLPs and subsequent hydrogenation into CO by the spilled *H. Analogous to the carburized Ni clusters, the FLP sites also exhibit the significantly weaker adsorption strength of *CO compared to CO2 (Fig. 1b and Supplementary Fig. 9), thereby effectively suppressing the methanation side reaction. However, the carburized Ni clusters anchored on the CeO2 surface with FLPs can hardly be synthesized through various conventional methods. Recently, the in-situ reactant/product-induced reconstruction has emerged as a promising avenue to generate new highly efficient carbide-like active sites15,40. Inspired by these findings, it stimulates our efforts to develop a new methodology to synthesize a pre-catalyst, which is capable of facilitating the reaction-induced reconstruction to give the carburized Ni clusters while simultaneously ensuring the precisely designed FLP sites under the catalytic environment of RWGS.
Synthesis and characterization of Ni-doped Ce(OH)3/CeO2 nanorods
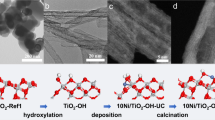
Considering the facile synthesis of Ni-doped CeO2, the reaction-driven Ni aggregation and subsequently reconstituted the carburized Ni clusters might provide an approach to construct the dual-active sites during RWGS owing to the high temperature and abundant carbon resources (CO, CO2, etc.). However, the migration of Ni in Ni-doped CeO2 is highly energy-consuming due to the strong confinement of the Ni dopant within the CeO2 lattice, leading to the difficulty in driving the Ni aggregation. To overcome this challenge and achieve the theoretically proposed dual-active sites, herein, the Ni-doped Ce(OH)3/CeO2 (Ni-Ce(OH)3/CeO2) pre-catalysts were synthesized through a low-pressure hydrothermal method at 100 °C, incorporating varying levels of Ni loading. Different from the stable Ni-doped CeO2, the unstable Ce(OH)3 component undergoes a regioselective reconstruction into CeO2 through the thermal decomposition and oxidation during RWGS, accompanied by the formation of Ni clusters, which could in-situ form the carburized Ni clusters by the catalytically generated CO (Fig. 2a). Importantly, the FLPs sites within the stable CeO2 component could remain unaffected. On this occasion, the dual-active sites of the creatively carburized Ni clusters and precisely designed FLPs would come into being through the in-situ regioselective reconstruction of the Ni-Ce(OH)3/CeO2 pre-catalysts.

a Scheme of in-situ regioselective reconstruction of Ni-Ce(OH)3/CeO2 to form the dual-active sites of the creatively carburized Ni clusters and precisely designed FLPs. b XRD patterns of the Ni-Ce(OH)3/CeO2 pre-catalysts and spent Ni-Ce(OH)3/CeO2 catalysts. c High-resolution TEM images of the Ni-Ce(OH)3/CeO2 pre-catalysts. d CO yields of the RWGS reaction. e Comparison of CO generation rates of Ni-Ce(OH)3/CeO2 (this work) and other state-of-the-art Ni catalysts through RWGS reaction under similar reaction conditions (see Supplementary Table 1 for more details). f Catalytic stability of Ni-Ce(OH)3/CeO2. Reaction conditions: Ni-Ce(OH)3/CeO2 (50 mg), WHSV (72,000 mL gcat−1 h−1), H2:CO2:N2 of 3:1:8 and 550 °C. All catalysts were subjected to in-situ treatment at 600 °C for 2 h under reaction conditions prior to performance evaluation. The spent Ni-Ce(OH)3/CeO2 catalysts referred to the Ni-Ce(OH)3/CeO2 pre-catalysts that had undergone the 1000 h stability test.
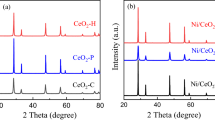
Powder X-ray diffraction (XRD) analysis of the Ni-Ce(OH)3/CeO2 pre-catalysts revealed the formation of a mixed phase of Ce(OH)3 and CeO2 with a molar ratio of 0.51:0.49 (Fig. 2b). The replacement of relatively larger Ce ions by smaller Ni ions induced a slight shift of the (111) plane of Ce(OH)3 in comparison with that of Ce(OH)3/CeO2 (synthesized by the same process without Ni precursor, Supplementary Figs. 10 and 11). Furthermore, transmission electron microscopy (TEM) images of the Ni-Ce(OH)3/CeO2 pre-catalysts did not detect any nickel oxide or metallic nickel species (Fig. 2c and Supplementary Fig. 12). Notably, the high-resolution TEM image of Ni-Ce(OH)3/CeO2 revealed the lattice fringes of 0.30 nm and 0.19 nm (Fig. 2c), which corresponded to the (101) crystal face of Ce(OH)3 and (220) crystal face of CeO2, respectively.
Catalytic performance of Ni-Ce(OH)3/CeO2 for RWGS
Subsequently, the catalytic performances of RWGS were evaluated in a fixed-bed reactor using a feed gas mixture of H2:CO2 (3:1) with a weighted hourly space velocity (WHSV) of 72,000 mL gcat−1 h−1. The Ni-Ce(OH)3/CeO2 pre-catalysts underwent in-situ treatment in the reaction atmosphere at temperatures of 400 °C, 500 °C, and 600 °C for 2 h before their catalytic performance was assessed. The optimal in-situ treatment temperature was determined to be 600 °C (Supplementary Fig. 13). Furthermore, the catalytic performances of Ni-Ce(OH)3/CeO2 with varying Ni contents indicated that the optimal Ni content was 0.5 wt.% (Supplementary Fig. 14). Unless otherwise specified, Ni-Ce(OH)3/CeO2 mentioned later in this study was referred to as the catalyst with a Ni doping of 0.5 wt.%. To highlight the catalytic performance of the Ni-Ce(OH)3/CeO2 pre-catalysts, the Ce(OH)3/CeO2 (Supplementary Fig. 10) and Ni-doped CeO2 catalysts with Ni loading of 0.5 wt.% (Ni-CeO2, Supplementary Fig. 15) were also prepared.
Specifically, the Ce(OH)3/CeO2 catalysts with only FLP sites exhibited the lowest CO2 conversion, which could be attributed to its poor capability for the co-activation of H2 and CO2 (Supplementary Fig. 16). Introduction of Ni dramatically boosted the catalytic activity of Ni-Ce(OH)3/CeO2 and Ni-CeO2. Notably, the Ni-Ce(OH)3/CeO2 catalysts exhibited a significantly higher CO2 conversion than that of Ni-CeO2. Similar to Ce(OH)3/CeO2, high selectivity towards CO (>99.9%) was impressively observed for both Ni-Ce(OH)3/CeO2 and Ni-CeO2, even at very high conversions of CO2 (Supplementary Fig. 16). Consequently, with the highest CO2 conversion and near 100% CO selectivity, the Ni-Ce(OH)3/CeO2 catalysts undoubtedly achieved the highest CO yield (Fig. 2d).
More importantly, the CO yield of Ni-Ce(OH)3/CeO2 reached 55.0% at 550 °C, which closely approached the equilibrium CO yield of 55.1% for RWGS under the operation conditions (Fig. 2d). Consequently, the Ni-Ce(OH)3/CeO2 catalysts exhibited an exceptionally high CO generation rate of 27.3 mol gNi−1 h−1, surpassing the previously reported Ni-based catalysts by at least one order of magnitude with a similar CO selectivity (Fig. 2e and Supplementary Table 1). Although a few catalysts in the literature exhibited a comparable activity in the CO generation rate, their CO selectivity was significantly lower than that of Ni-Ce(OH)3/CeO2 (Fig. 2e and Supplementary Table 1). Additionally, the Ni-Ce(OH)3/CeO2 catalysts delivered remarkable stability for RWGS even at a high temperature of 550 °C. The CO yields remained nearly constant and approached the thermodynamic equilibrium yield for at least 1000 h of reaction (Fig. 2f and Supplementary Fig. 17). Furthermore, each Ni stie achieved an impressively high turnover frequency of >4,500,000. In addition to evaluating the catalysts at high temperatures, we further assessed the stability of Ni-Ce(OH)3/CeO2 at 400 °C. This temperature ensured that the CO2 conversions remained within the kinetic interval of less than 20%. After the reconstruction stage, the Ni-Ce(OH)3/CeO2 catalysts demonstrated the well-maintained CO2 conversion and CO selectivity (Fig. S18), thereby further substantiating their remarkable stability.
Identification of the reaction-driven reconstruction of Ni-Ce(OH)3/CeO2
Considering the ease of the Ce(OH)3 decomposition, the phase of the spent Ni-Ce(OH)3/CeO2 catalysts after 1000 h stability tests was examined through XRD analysis. The observed phase transformation from the mixed Ce(OH)3/CeO2 phase into pure CeO2 strongly suggested the in-situ regioselective reconstruction of the Ni-Ce(OH)3 component (Fig. 2b and Supplementary Fig. 11). Consequently, it became imperative to conduct a comprehensive analysis of the evolution of the surface properties of Ni-Ce(OH)3/CeO2 under the operation, understanding the reconstruction during RWGS and identifying the key factor for their exceptional catalytic performance in comparison with the Ni-CeO2 catalysts and other state-of-the-art catalysts (Fig. 2e).
To illustrate the occurrence of the reaction-driven reconstruction process, the RWGS reaction was conducted by transferring the Ni-Ce(OH)3/CeO2 pre-catalysts at room temperature into the pre-heated reactor with the desired temperatures (400, 500, and 600 °C). An obvious activation period was experimentally observed, as evidenced by their gradually improved CO2 conversions during the initial 8.5 h at 400 °C (Fig. 3a). Notably, the activation periods were significantly shortened from 8.5 h to 1.5 h with the increased reaction temperatures from 400 °C to 600 °C. This observed activation period unequivocally demonstrated the formation of new active sites through a reaction-driven reconstruction of Ni-Ce(OH)3/CeO2 pre-catalysts to form the reconstructed Ni-Ce(OH)3/CeO2 catalysts.

a The CO2 conversions vs. reaction time for RWGS catalyzed by the Ni-Ce(OH)3/CeO2 pre-catalysts. b HAADF-STEM and c partially enlarged HAADF-STEM images of the reconstructed Ni-Ce(OH)3/CeO2 catalysts. d XANES spectra of the Ni foil, Ni-Ce(OH)3/CeO2, reconstructed Ni-Ce(OH)3/CeO2 and NiO. e The k3 weighted Fourier-transformed spectra derived from the EXAFS spectra. f Wavelet-transform plots by using k3 space. Time-dependent SRPES measurements of g Ni L-edge and h C K-edge for the reconstructed Ni-Ce(OH)3/CeO2 catalysts under Ar treatment. i In-situ DRIFTS analysis of the Ni-Ce(OH)3/CeO2 pre-catalysts treated by various gases. The reconstructed Ni-Ce(OH)3/CeO2 catalysts were obtained by in-situ treatment of the Ni-Ce(OH)3/CeO2 pre-catalysts under a H2:CO2 ratio of 3:1 at 600 °C for 2 h.
Then, the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images illustrated the well-preserved nanorod structure with the evolution of mesoporous morphology of the reconstructed Ni-Ce(OH)3/CeO2 catalysts, which were pre-treated under a H2:CO2 ratio of 3:1 at 600 °C for 2 h (Fig. 3b). The measured lattice spacing of 0.190 nm also indicated the preservation of (220) crystal face of CeO2 (Fig. 3c). Importantly, small clusters were observed on the surface of the reconstructed catalysts (Fig. 3c). Moreover, the X-ray photoelectron spectroscopy (XPS) analysis on Ni 2p orbital electrons revealed the fraction of Ni0 species from virtually zero in the pre-catalysts to 44.3% in the reconstructed ones, strongly suggesting the occurrence of the reaction-driven reconstruction of the Ni dopant in the Ni-Ce(OH)3/CeO2 pre-catalysts into Ni clusters (Supplementary Fig. 19). In contrast, no observed Ni0 species in the treated Ni-CeO2 catalysts (pre-treated under H2:CO2 of 3:1 at 600 °C for 2 h) indicated the aggregation of Ni dopants was effectively prevented in CeO2 region, confirming the regioselective reconstruction as proposed in Fig. 2a.
Next, X-ray absorption fine structure (XAFS) spectroscopy was employed to examine the reaction-driven reconstruction of Ni-Ce(OH)3/CeO2. The adsorption edge of the Ni-Ce(OH)3/CeO2 pre-catalysts exhibited a negative displacement compared to the NiO foil (Fig. 3d), suggesting the electron transfer from Ce to Ni sites. Comparatively, the electronic structures of the reconstructed Ni-Ce(OH)3/CeO2 catalysts closely resembled those of Ni foil, indicating the formation of metallic Ni during RWGS. The EXAFS spectra and wavelet-transform (WT) analysis revealed the presence of Ni–Ni bonds at a distance of 2.18 Å in the reconstructed Ni-Ce(OH)3/CeO2 catalysts, consistent with the peak position of Ni−Ni observed in the Ni foil (Fig. 3e, f). Notably, the presence of Ni−O bonds is still evident on the reconstructed Ni-Ce(OH)3/CeO2 catalysts (Fig. 3e, f and Table 1), which can be attributed to the doped Ni atoms within the stable CeO2 region retaining its original structure during the treatments. This observation further suggested that the metastable Ce(OH)3 selectively reconstructs to give Ni clusters and undergoes the subsequent carburization without altering the initial structure of the stable CeO2 region. Therefore, combining TEM, XRD, and XPS results, the XAFS spectra provided crucial evidence to illustrate the reaction-driven reconstruction of the Ni dopants in the specific region of Ce(OH)3 of the pre-catalysts into Ni clusters.
Characterizations of the carburized Ni clusters
Based on the above comprehensive analysis, the reaction-driven reconstruction of Ni-Ce(OH)3/CeO2 created the Ni clusters on the CeO2 surface. Impressively, the reconstructed catalysts exhibited a dramatically high selectivity towards CO, suggesting that the local environments of these reaction-driven reconstructed Ni clusters in Ni-Ce(OH)3/CeO2 were significantly distinct from those of commonly deposited Ni clusters and/or nanoparticles on various supports in previous studies43,44. As evidenced by the XPS profile of the reconstructed Ni-Ce(OH)3/CeO2 catalysts, in addition to the presence of Ni0 species, a small fraction of nickel carbide was detected at 850.3 eV (Supplementary Fig. 19a). Furthermore, the presence of Ni−C species was also revealed from C 1s spectrum with a peak at 283.8 eV (Supplementary Fig. 19b). Considering the carbon-rich environment conducive to a partial carbonization of metal clusters during RWGS15,41,45, the XPS profiles strongly supported the formation of the carburized Ni clusters during the reconstruction process.
The time-dependent synchrotron radiation photoelectron spectroscopy (SRPES) was conducted on the reconstructed Ni-Ce(OH)3/CeO2 catalysts to examine the chemical states of the Ni clusters by Ar plasma etching. The Ni L-edge of the reconstructed Ni-Ce(OH)3/CeO2 catalysts indicated the co-existence of Ni0 and Ni2+ species (Fig. 3g), again confirming the formation of the Ni clusters. The C species as revealed from C K-edge gradually diminished with the continuously prolonged Ar plasma treatment, indicating the etching of carbonaceous species (Fig. 3h). Furthermore, the significant shift towards the lower binding energy of Ni0 species was observed as the decrease of C species, indicating a strong interaction between C species and Ni clusters through the electron transfer from Ni to C. Additionally, the higher binding energy of Ni2+ species suggested the change in coordination from C to O on the CeO2 surface, accompanied by the etching of C species. Therefore, the SRPES spectra analysis unambiguously demonstrated the environmental-induced reconstruction and carbonization to give the carburized Ni clusters.
Investigation of the mechanism of reaction-induced regioselective reconstruction
Typically, the transformation of Ce(OH)3 to CeO2 requires the oxidation of Ce3+ into Ce4+, a process that could potentially be enhanced by the oxidizing capability of CO2. We utilized in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to investigate the reconstruction of the Ni-Ce(OH)3/CeO2 pre-catalysts under a CO2 atmosphere at 100 °C. The experiments were performed at a low temperature to slow down the transformation process for better observation. Under N2 atmosphere, the slow oxidation of Ce3+ to Ce4+ was evidenced by the increased intensity of the characteristic peaks corresponding to Ce4+-OH (Type I at 3730 cm−1 and Type II at 3680 cm−1) and the simultaneous decrease in Ce3+-OH peaks (Supplementary Fig. 20). This oxidation process could be attributed to the high oxygen storage capacity of CeO2. Upon introducing CO2, a prompt intensification in the Ce4+-OH peaks and a significant reduction in the Ce3+-OH peaks were observed. Additionally, the characteristic peaks of CO species emerged (Supplementary Fig. 20). Notably, the peaks of CO species remained largely unchanged upon further introduction of CO2. These findings conclusively demonstrate the oxidization of Ce3+ to Ce4+ and the concurrent reduction of CO2 to CO during the reconstruction of the Ni-Ce(OH)3/CeO2 pre-catalysts.
Afterwards, in-situ DRIFTS was utilized under various gaseous environments to investigate how the reconstruction occurred to form the carburized Ni clusters. The DRIFTS profile exhibited a twin adsorption of Ni-(CO)2/3 on the surface of Ni-Ce(OH)3/CeO2 pre-catalysts (Fig. 3i), indicating the atomically dispersed Ni. Following the in-situ treatment of the Ni-Ce(OH)3/CeO2 pre-catalysts with H2/CO2 at 350 °C, a noticeable reduction in the peak corresponding to Ni-(CO)2/3 was observed (Fig. 3i). Simultaneously, the emerging peaks at 1930 cm−1 and 1840 cm−1 were attributed to the bridged adsorption of CO with three Ni atoms (Ni3−CO) and the carbonization Ni species (NiC−CO), respectively46,47. Although the characteristic peak of Ni3−CO remained under the H2 treatment, the absence of NiC−CO peaks suggested that carburized Ni clusters did not form. In contrast, no Ni clusters were observed when the Ni-Ce(OH)3/CeO2 pre-catalysts were treated under a CO2 atmosphere (Fig. 3i). Given the presence of CO in RWGS, the Ni-Ce(OH)3/CeO2 pre-catalysts were treated under a CO atmosphere at 600 °C for 2 h. As anticipated, both Ni3−CO and NiC−CO peaks were evident in the in-situ DRIFTS spectra. However, due to the lack of CO2 to facilitate the oxidation of Ce(OH)3, the reconfiguration process was less complete in the CO or H2 + CO atmospheres compared to the H2/CO2 atmosphere. Consequently, the existence of CO is necessary for the formation of carburized Ni clusters.
Subsequently, DFT calculations were conducted to elucidate the formation mechanism of carburized Ni clusters. On the Ni4/CeO2(110)-2OV surface, H2 dissociation occurs readily, enabling a *H-assisted pathway for CO dissociation: *CO + *H → *COH → *C + *OH, rather than direct CO dissociation (Supplementary Fig. 21a). This process exhibits an energy barrier of 1.55 eV, substantially lower than the *CO desorption energy of 2.16 eV (Supplementary Fig. 7), indicating that the H-assisted CO dissociation is energetically more favorable than the CO desorption on the Ni clusters. Importantly, CO decomposition on the N4C1 cluster of Ni4C1/CeO2(110)-2OV requires a significantly higher energy barrier of 2.04 eV, exceeding the *CO desorption energy of 1.92 eV (Supplementary Fig. 21b). The high energy barrier for the *COH formation on the carburized Ni clusters kinetically favors CO desorption, thereby inhibiting their further carbonization.
Based on the aforementioned analysis, the Ni-doped Ce(OH)3 component underwent the decomposition and oxidation by CO2 to give CeO2 as well as the aggregated Ni clusters. These Ni clusters were subsequently transformed into the carburized Ni clusters through a *H-assisted CO dissociation. Consequently, achieving appropriate RWGS reaction conditions was essential for the in-situ regioselective reconstruction of the Ni-Ce(OH)3/CeO2 pre-catalysts. In addition to the structural differences, the Ni-Ce(OH)3/CeO2 pre-catalysts treated under reaction conditions demonstrated superior CO2 conversion and CO selectivity, compared to those treated solely with H2, CO2, or CO (Supplementary Fig. 22). Furthermore, XPS data revealed a slight enhancement in oxygen vacancy concentration of reconstructed Ni-Ce(OH)3/CeO2 compared to the pre-catalysts (Supplementary Fig. 23), indicating the preservation of FLPs sites23,24. Additionally, both CO2 conversion and CO selectivity of Ni-Ce(OH)3/CeO2 remained nearly unchanged at 550 °C for at least 1000 h (Supplementary Fig. 17), indicating the stability of the reaction-driven formation of the carburized Ni clusters and FLPs during RWGS.
Carburized Ni clusters for the elimination of the competitive adsorption
As proposed from theoretical analysis, the reaction-driven reconstruction of Ni-Ce(OH)3/CeO2 during RWGS created the carburized Ni clusters and FLPs sites, which selectively activated H2 and CO2, respectively and thereby significantly mitigated their pronounced co-adsorption and activation (Fig. 1b). The experiments for determining the reaction orders of H2 and CO2 at 300 °C and 500 °C were performed within the kinetic range to confirm the alleviated competitive adsorption on the constructed dual-active sites (Supplementary Fig. 24). Theoretically, as the reaction order of a reactant increases, its coverage on the catalyst surface is expected to decrease48,49. The presence of carburized Ni clusters and FLP sites in reconstructed Ni-Ce(OH)3/CeO2 significantly enhanced the adsorption of CO2 and H2 molecules (Supplementary Fig. 25), in comparison to the Ni-CeO2 and reconstructed Ce(OH)3/CeO2 catalysts (Fig. 4a, b). Thus, the increase in the adsorption capacity of H2 and CO2 further enhanced the activity of the reconstructed Ni-Ce(OH)3/CeO2 catalysts.

Reaction orders of a H2 and b CO2 for the RWGS reaction catalyzed by the reconstructed Ni-Ce(OH)3/CeO2, Ni-CeO2, and reconstructed Ce(OH)3/CeO2 catalysts. c Isotope effects of the reconstructed Ni-Ce(OH)3/CeO2 and Ni-CeO2 for the RWGS reaction. d Ln k derived from CO generation rates as a function of 1/T and the derived Ea values. e d-band centers of the reconstructed Ni-Ce(OH)3/CeO2 and H2-treated Ni-Ce(OH)3/CeO2 catalysts. f CO-TPD for the reconstructed Ni-Ce(OH)3/CeO2 and H2-treated Ni-Ce(OH)3/CeO2 catalysts. The reconstructed Ni-Ce(OH)3/CeO2 and Ce(OH)3/CeO2 catalysts were obtained by in-situ treatment of the Ni-Ce(OH)3/CeO2 and Ce(OH)3/CeO2 pre-catalysts under reaction conditions at 600 °C for 2 h, respectively.
On the reconstructed Ni-Ce(OH)3/CeO2 catalysts, the carburized Ni clusters served as active sites to generate the active H* species, which then spilled to the FLPs sites on CeO2 supports (Fig. 1d). H2-temperature programmed reduction (H2-TPR) measurements of the reconstructed Ni-Ce(OH)3/CeO2 catalysts revealed the enhanced reduction of both Ni−O and Ce−O compared to the reconstructed Ce(OH)3/CeO2 and Ni-CeO2 catalysts (Supplementary Fig. 26), indicating their high capability for H2 activation and the occurrence of hydrogen spillover from the carburized Ni clusters. In addition, the higher HD yield of the reconstructed Ni-Ce(OH)3/CeO2 catalysts in the H2−D2 exchange experiments also proved its higher capacity for the H2 activation (Supplementary Fig. 27).
Then, the H2/D2 kinetic isotope effect (KIE) was further examined to explore the involvement of H2 activation and/or spillover for RWGS (Supplementary Fig. 28). The kH/kD value of Ni-CeO2 was 1.5 as a result of the zero-point energy difference between isotopic isomers (Fig. 4c). Comparatively, a significantly higher kH/kD values of 3.8 was observed for reconstructed Ni-Ce(OH)3/CeO2 (Fig. 4c), indicating that this process should be accompanied by the formation and dissociation of O−Hδ+ bonds on the surface of CeO250,51. The comparative values of kH/kD directly confirmed the occurrence of hydrogen spillover on the surface of the reconstructed Ni-Ce(OH)3/CeO2 catalysts.
The elimination of the competitive adsorption between CO2 and H2 through the carburized Ni clusters would significantly decrease the activation energy of RWGS. Derived from the kinetic experiments (400~600 °C, note: CO2 conversions <20%, Supplementary Fig. 29), these catalysts followed a good linearity between Ln k and 1/T, and the corresponding slope of the plot yielded the activity energy (Ea, Fig. 4d). Due to the strong competitive adsorption of CO2 to H2 on FLPs, the reconstructed Ce(OH)3/CeO2 catalysts exhibited the highest value of Ea (53.7 kJ mol−1), thereby leading to the lowest catalytic activity for RWGS. Although the competitive adsorption was not eliminated, the doped Ni in Ni-CeO2 still delivered a lower Ea (48.6 kJ mol−1), owing to the enhanced CO2 adsorption in comparison to reconstructed Ce(OH)3/CeO2 with similar oxygen vacancies (Supplementary Fig. 23), as revealed from their CO2-temperature programmed desorption (CO2-TPD) patterns (Supplementary Fig. 25). For the reconstructed Ni-Ce(OH)3/CeO2 catalysts, the competitive adsorption was eliminated by introducing the carburized clusters, thereby yielding the lowest Ea (41.4 kJ mol−1) for RWGS and achieving exceptional catalytic activity.
Carburized Ni clusters for the suppressed methanation
Previous studies have experimentally demonstrated that poor CO adsorption on FLPs prevents the over-hydrogenation of CO into CH425,52. Complementary DFT calculations confirm that *CO adsorption strength is significantly reduced on carburized Ni clusters under vacuum conditions. To assess practical relevance, we examined the Gibbs free energy of CO desorption on both pristine and carburized Ni clusters under representative operational conditions. Increasing carbonization levels in Ni clusters correlate with gradually reduced Gibbs free energies for CO desorption (Supplementary Fig. 30), indicating that CO release becomes increasingly favorable from carburized clusters under operational conditions.
DFT calculations demonstrate that carburization of Ni clusters induces a downward shift in the d-band center (Fig. 1c), significantly reducing CO adsorption strength. This theoretical insight aligns with experimental observations from high-resolution valence band (VB) spectroscopy. Compared to H2-treated Ni-Ce(OH)3/CeO2, a noticeable displacement of d-band center for reconstructed Ni-Ce(OH)3/CeO2 away from VBM revealed a downward shift of the antibonding orbitals and a higher orbital occupancy rate for the carburized Ni clusters (Fig. 4e). CO-temperature programmed desorption (CO-TPD) directly demonstrated the weaker adsorption of CO on the reconstructed Ni-Ce(OH)3/CeO2 catalysts compared to that on the H2-treated Ni-Ce(OH)3/CeO2 catalysts (Fig. 4f). Therefore, both the carburized Ni clusters via the reaction-driven reconstruction of Ni-Ce(OH)3/CeO2 and the FLPs sites with the unique spatial configuration contribute to the weakened CO adsorption on catalyst surface, enabling the high CO selectivity for RWGS.
Catalytic pathway of dual-active sites
Finally, in-situ DRIFTS experiments were conducted to monitor the intermediates and explore the catalytic pathway. A flow of H2 was introduced to generate *H species on the catalyst surface at 450 °C. After 30 min, the gas feed was switched to a CO2 flow at the same temperature. All catalysts followed the formate pathway for RWGS, as evidenced by the sequential transformation of bicarbonate intermediates (1610 cm−1 and 1310 cm−1) into formate intermediates (2845 cm−1 and 2950 cm−1), and ultimately into *CO (Fig. 5)40,53. DFT calculations also reveal that the *H-assisted CO2 hydrogenation via *HCOO as intermediate on FLPs exhibits significantly reduced reaction energy and energy barrier compared to the direct dissociation route (Supplementary Fig. 31). However, the structural optimizations reveal the intrinsic instability of *HCO, spontaneously recombining with surface *O species to regenerate *HCOO intermediates (Supplementary Fig. 31). This thermodynamic instability conclusively demonstrates that the *HCOO dissociation proceeds through a direct cleavage to *CO rather than via the *HCO formation, consistent with the limited *HCO detection (~1760 cm−1) during in-situ DRIFTS monitoring.

In-situ DRIFTS spectra of a reconstructed Ni-Ce(OH)3/CeO2, b Ni-CeO2, and c H2-treated Ni-Ce(OH)3/CeO2 under the flowing of CO2. The catalysts underwent pre-treatment by a flow of 50% H2/Ar for 1 h. After removing excess H2 by an Ar flow, a flow of 50% CO2/Ar was introduced. The DRIFTS signals were collected at 450 °C every 20 s during a period of 15 min. The reconstructed Ni-Ce(OH)3/CeO2 catalysts were obtained by in-situ treatment of the Ni-Ce(OH)3/CeO2 pre-catalysts under reaction conditions at 600 °C for 2 h. The H2-treated Ni-Ce(OH)3/CeO2 catalysts were obtained by in-situ treatment of the Ni-Ce(OH)3/CeO2 pre-catalysts under H2/Ar (5%) atmosphere at 600 °C for 2 h.
Notably, due to the weak adsorption of CO on the FLPs sites and carburized Ni clusters, no methane (~3016 cm−1) was detected on either the reconstructed Ni-Ce(OH)3/CeO2 or Ni-CeO2 catalysts. However, the reconstructed Ni-Ce(OH)3/CeO2 catalysts exhibited significantly lower signals of *HCO3 species (~1600 cm−1) and the stronger signals of *HCOO (~2845 cm−1) and *CO (~2175 cm−1) species, in comparison with those of Ni-CeO2 (Fig. 5a, b). These comparative results suggested the much weaker ability of Ni-CeO2 with only FLP sites for H2 dissociation. Subsequently, the CO2 flow was cut off, and the catalysts were treated in a flow of H2/N2 at 450 °C to regenerate the *H species. The disappearance rates of *HCO3 and *HCOO peaks on the reconstructed Ni-Ce(OH)3/CeO2 catalysts were significantly faster than those on Ni-CeO2 (Supplementary Fig. 32), further revealing the pivotal roles of the carburized Ni clusters in facilitating H2 dissociation.
The direct cleavage *HCOO to *CO leaves the residual *O species in FLPs. In-situ DRIFTS experiments demonstrated that the characteristic peak of Ce4+-OH at ~3735 cm−1 (Type I, terminal OH) and ~3690 cm−1 (Type II, bridging OH) significantly enhanced along with the reduction of Ce3+-OH at ~3646 cm−1 under CO2 flow (Supplementary Fig. 33). Upon introducing H2, the Ce4+-OH peak gradually diminished, while the Ce3+-OH peak re-emerged, indicating that the residual *O species was removed by the spilled H* species for the recovery of the FLPs sites. Therefore, the surface-adsorbed *H species serve dual functions: not only facilitating CO₂ dissociation through this H-assisted mechanism, but also recovering FLPs sites through *O removal.
The roles of the carburized Ni clusters can be examined by comparing the reconstructed Ni-Ce(OH)3/CeO2 and H2-treated Ni-Ce(OH)3/CeO2 catalysts. Due to the presence of Ni clusters to provide sufficient *H species, these catalysts exhibited obvious characteristic peaks of *CO under CO2 flow, indicating the successful CO2 hydrogenation. It was noteworthy that the weak interaction between *CO and FLPs resulted in the absence of *CO peaks on CeO2 at ~2150 cm−1. Subsequently, the desorbed CO molecules from FLP sites could be captured by Ni clusters according to the presence of peaks at 2054 cm−1 and 2120 cm−1 (Fig. 5a, c). However, due to the weak interaction between CO and carburized Ni clusters, the gas phase CO (~2175 cm−1) was also observed on reconstructed Ni-Ce(OH)3/CeO2. Consequently, there was no transformation of the captured *CO into CH4, as evidenced by the absence of CH4 characteristic peaks (~3016 cm−1, Fig. 5a). In contrast, due to the strong adsorption of *CO on Ni clusters, the H2-treated Ni-Ce(OH)3/CeO2 catalysts exhibited a rapid conversion of *CO intermediates into CH4, as evidenced by the clear characteristic CH4 peaks at 3016 cm−1.
Discussion
In summary, the dual-active sites of the carburized Ni clusters and FLPs sites have been created through the reaction-driven regioselective reconstruction of the Ni-Ce(OH)3/CeO2 catalysts with the mixed phases of CeO2 and Ce(OH)3. During RWGS, the Ni dopants in the Ce(OH)3 phase of Ni-Ce(OH)3/CeO2 undergo the reconstruction to create the carburized Ni clusters, while preserving the integrity of FLP sites in the CeO2 phase. The reconstructed Ni-Ce(OH)3/CeO2 catalysts with the dual-active sites eliminated the competitive adsorption between H2 and CO2, thereby achieving exceptional performance for the RWGS reaction. The carburized Ni clusters with the downshift of the d-band center and FLPs exhibited a weak interaction with CO and, thereafter, dramatically suppressed the methanation side reaction, realizing satisfactory CO selectivity. This finding provides a successful case of overcoming the incompatibility between the precisely designed active sites and the generation of multiple active sites through a regioselective reconstruction. This specific process opens new avenues for the design of highly performing heterogeneous catalysts.
Methods
Synthesis of the Ni-Ce(OH)3/CeO2 pre-catalysts
Initially, a mixture containing Ce(NO3)3 (0.8 mmol mL−1) and Ni(NO3)2 (0.016 mmol mL−1), with a total volume of 5 mL, was introduced into 75 mL of NaOH solution (6.4 mmol mL−1) under continuous stirring at room temperature. After 30 min, this mixture was transferred into a Pyrex bottle (100 mL) for a subsequent hydrothermal process at 100 °C for 24 h. Finally, the Ni-Ce(OH)3/CeO2 catalysts were obtained by alternately washing with H2O and ethanol three times and drying overnight at 60 °C.
Catalytic tests of catalysts
The RWGS reactions were carried out in a fixed-bed reactor operating at atmospheric pressure. The experimental procedure involved the loading of 50 mg of catalysts into a straight quartz tube, with temperature sensors placed both inside and outside the quartz tube. A H2/CO2/N2 mixture gas (8.3 vol.% CO2, 25 vol.% H2, and 66.6 vol.% N2) was introduced into the reactor with a total flow of 60 mL min−1. The gas products were analyzed online using a gas chromatograph equipped with both TCD and FID detectors.
The conversion of CO2 is calculated as the following:
The selectivity of CO and CH4 is calculated as the following:
The yield of CO is calculated as follows:
Where CO2in, CO2out, COout and CH4out represent the moles of CO2, CO and CH4 in the effluent, respectively.
The CO generation rates are calculated as the following:
Where YieldCO is the yield of CO and mcat is catalyst mass.
Data availability
The source data generated in this study are provided in the Source Data file. The authors declare that the main data supporting the findings are available within the article and supplementary information from the corresponding author upon request. Source data are provided with this paper.
References
Li, Z. et al. Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites. Chem. Rev. 120, 623–682 (2019).
Schlogl, R. Heterogeneous catalysis. Angew. Chem. Int. Ed. 54, 3465–3520 (2015).
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Zhang, L., Ren, Y., Liu, W., Wang, A. & Zhang, T. Single-atom catalyst: a rising star for green synthesis of fine chemicals. Natl. Sci. Rev. 5, 653–672 (2018).
Crawley, J. W. M. et al. Heterogeneous trimetallic nanoparticles as catalysts. Chem. Rev. 122, 6795–6849 (2022).
Ji, S. et al. Chemical synthesis of single atomic site catalysts. Chem. Rev. 120, 11900–11955 (2020).
Jing, W. et al. Surface and interface coordination chemistry learned from model heterogeneous metal nanocatalysts: from atomically dispersed catalysts to atomically precise clusters. Chem. Rev. 123, 5948–6002 (2023).
Li, H. et al. Synergetic interaction between neighbouring platinum monomers in CO2 hydrogenation. Nat. Nanotechnol. 13, 411–417 (2018).
Hu, S. & Li, W.-X. Sabatier principle of metal-support interaction for design of ultrastable metal nanocatalysts. Science 374, 1360–1365 (2021).
Martín, A. J., Mitchell, S., Mondelli, C., Jaydev, S. & Pérez-Ramírez, J. Unifying views on catalyst deactivation. Nat. Catal. 5, 854–866 (2022).
Nie, L. et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 358, 1419–1423 (2017).
Daelman, N., Capdevila-Cortada, M. & Lopez, N. Dynamic charge and oxidation state of Pt/CeO2 single-atom catalysts. Nat. Mater. 18, 1215–1221 (2019).
Zhang, S. et al. In-situ reconstruction of single-atom Pt on Co3O4 for hydrogenation. Nano Res. 16, 6507–6511 (2023).
Su, C. et al. Engineering single-atom dynamics with electron irradiation. Sci. Adv. 5, eaav2252 (2019).
Xin, H. et al. Reverse water gas-shift reaction product driven dynamic activation of molybdenum nitride catalyst surface. Nat. Commun. 15, 3100 (2024).
Zeng, L. et al. Stable anchoring of single rhodium atoms by indium in zeolite alkane dehydrogenation catalysts. Science 383, 998–1004 (2024).
Liu, L. et al. Dealuminated Beta zeolite reverses Ostwald ripening for durable copper nanoparticle catalysts. Science 383, 94–101 (2024).
Lin, L. et al. Low-temperature hydrogen production from water and methanol using Pt/alpha-MoC catalysts. Nature 544, 80–83 (2017).
Wang, P. et al. Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation. Nat. Chem. 9, 64–70 (2017).
Zhang, S., Tian, Z., Ma, Y. & Qu, Y. Adsorption of molecules on defective CeO2 for advanced catalysis. ACS Catal. 13, 4629–4645 (2023).
Santen, R. A. V., Neurock, M. & Shetty, S. G. Reactivity theory of transition-metal surfaces: a Brønsted−Evans−Polanyi linear activation energy−free-energy analysis. Chem. Rev. 110, 2005–2048 (2010).
Jiang, L. et al. Facet engineering accelerates spillover hydrogenation on highly diluted metal nanocatalysts. Nat. Nanotechnol. 15, 848–853 (2020).
Zhang, S. et al. Solid frustrated-Lewis-pair catalysts constructed by regulations on surface defects of porous nanorods of CeO2. Nat. Commun. 8, 15266 (2017).
Zhang, S. et al. Interfacial frustrated Lewis pairs of CeO2 activate CO2 for selective tandem transformation of olefins and CO2 into cyclic carbonates. J. Am. Chem. Soc. 141, 11353–11357 (2019).
Li, W. et al. Platinum and frustrated Lewis pairs on ceria as dual-active sites for efficient reverse water-gas shift reaction at low temperatures. Angew. Chem. Int. Ed. 62, e202305661 (2023).
Riley, C. et al. Design of effective catalysts for selective alkyne hydrogenation by doping of ceria with a single-atom promotor. J. Am. Chem. Soc. 140, 12964–12973 (2018).
Barreau, M. et al. Ionic nickel embedded in ceria with high specific CO2 methanation activity. Angew. Chem. Int. Ed. 62, e202302087 (2023).
Winter, L. R. et al. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B Environ. 245, 360–366 (2019).
Zhou, S., Wan, Q., Lin, S. & Guo, H. Acetylene hydrogenation catalyzed by bare and Ni doped CeO2(110): the role of frustrated Lewis pairs. Phys. Chem. Chem. Phys. 24, 11295–11304 (2022).
Xie, Y. et al. Frustrated Lewis pairs boosting low-temperature CO2 methanation performance over Ni/CeO2 nanocatalysts. ACS Catal. 12, 10587–10602 (2022).
Lozano-Reis, P., Gamallo, P., Sayos, R. & Illas, F. Comprehensive density functional and kinetic Monte Carlo study of CO2 hydrogenation on a well-defined Ni/CeO2 model catalyst: Role of Eley-Rideal reactions. ACS Catal. 14, 2284–2299 (2024).
Adhikari, D., Whitcomb, C. A., Zhang, W., Zhang, S. & Davis, R. J. Revisiting the influence of Ni particle size on the hydrogenation of CO2 to CH4 over Ni/CeO2. J. Catal. 438, 115708 (2024).
Pu, T. et al. Dependency of CO2 methanation on the strong metal-support interaction for supported Ni/CeO2 catalysts. J. Catal. 413, 821–828 (2022).
Wang, D. D. et al. Ni single atoms confined in nitrogen-doped carbon nanotubes for active and selective hydrogenation of CO2 to CO. ACS Catal. 13, 7132–7138 (2023).
Vogt, C. et al. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 1, 127–134 (2018).
Zhang, X. et al. Reversible loss of core–shell structure for Ni–Au bimetallic nanoparticles during CO2 hydrogenation. Nat. Catal. 3, 411–417 (2020).
Kattel, S., Liu, P. & Chen, J. G. Tuning selectivity of CO2 hydrogenation reactions at the metal/oxide interface. J. Am. Chem. Soc. 139, 9739–9754 (2017).
Bai, L. et al. Explaining the size dependence in platinum-nanoparticle-catalyzed hydrogenation reactions. Angew. Chem. Int. Ed. 55, 15656–15661 (2016).
Zhang, S. et al. Sustainable production of hydrogen with high purity from methanol and water at low temperatures. Nat. Commun. 13, 5527 (2022).
Khoshooei, M. A. et al. An active, stable cubic molybdenum carbide catalyst for the high-temperature reverse water-gas shift reaction. Science 384, 540–546 (2024).
Chen, H. et al. Dynamic phase transition of iron oxycarbide facilitated by Pt nanoparticles for promoting the reverse water gas shift reaction. ACS Catal. 11, 14586–14595 (2021).
Galhardo, T. S. et al. Optimizing active sites for high CO selectivity during CO2 hydrogenation over supported nickel catalysts. J. Am. Chem. Soc. 143, 4268–4280 (2021).
Song, C. et al. Engineering MOx/Ni inverse catalysts for low-temperature CO2 activation with high methane yields. Nat. Chem. Eng. 1, 638–649 (2024).
Pham, C. Q. et al. Carbon dioxide methanation on heterogeneous catalysts: a review. Environ. Chem. Lett. 20, 3613–3630 (2022).
Chen, J. et al. Benchmarking promoters of Fe/activated carbon catalyst for stable hydrogenation of CO2 to liquid hydrocarbons. Appl. Catal. B Environ. 325, 122370 (2023).
Millet, M.-M. et al. Ni single atom catalysts for CO2 activation. J. Am. Chem. Soc. 141, 2451–2461 (2019).
Bertolini, J. C. & Tardy, B. Vibrational EELS studies of CO chemisorption on clean and carbided (111), (100) and (110) nickel surfaces. Surf. Sci. 102, 131–150 (1981).
Rioux, R. M., Song, H., Hoefelmeyer, J. D., Yang, P. & Somorjai, G. A. High-surface-area catalyst design: synthesis, characterization, and reaction studies of platinum nanoparticles in mesoporous SBA-15 silica. J. Phys. Chem. B 109, 2192–2202 (2005).
Motagamwala, A. H. & Dumesic, J. A. Microkinetic modeling: a tool for rational catalyst design. Chem. Rev. 121, 1049–1076 (2021).
Liu, P. et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 352, 797–801 (2016).
Zhang, S. et al. Boosting selective hydrogenation through hydrogen spillover on supported-metal catalysts at room temperature. Appl. Catal. B Environ. 297, 120418 (2021).
Li, W., Gan, J., Guo, Q., Zhang, S. & Qu, Y. Describing H* adsorption on dual-active sites of single-atom-metals and frustrated-Lewis-pairs for reverse water-gas shift reaction. Sci. China Chem. 68, 1159–1168 (2025).
Graciani, J. et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 345, 546–550 (2014).
Acknowledgements
This work was supported by the Guangdong Basic and Applied Basic Research Foundation (2022B1515020092) and the Shenzhen Science and Technology Program (JCYJ20220530161600002). The research is also supported by the Key Projects of Natural Science Basic Research Program of Shaanxi Province (2024JC-TBZC-16).
Author information
Authors and Affiliations
Contributions
Y.Q. and S.Z. designed the studies and proposed the research direction. W.L. prepared the catalysts, conducted catalytic measurements, characterizations, and in-situ experiments. B.L. carried out the DFT calculations. Q.G. and W.G. performed characterizations and data analysis. All authors contributed to the data interpretation and manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Pablo Lozano-Reis and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, W., Liu, B., Guo, Q. et al. Reaction-induced regioselective reconstruction of Ni-doped Ce(OH)3/CeO2 enables exceptional activity and selectivity for reverse water-shift reaction. Nat Commun 16, 7335 (2025). https://doi.org/10.1038/s41467-025-62771-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62771-1
