
Abstract
Netrin-4 is a tumor suppressor that interferes with formation of the laminin lattice. We employed cryo-electron microscopy to determine a structure of the protein complex consisting of the N-terminal fragments from netrin-4 and laminin γ1. The structure reveals that netrin-4 binds laminin γ1 at the molecular interface where laminin β1 would have bound, thus inhibiting the assembly of the heterotrimeric laminin polymer nodes consisting of α1, β1, and γ1 subunits, and their polymerization into the extracellular lattice. The four orders of magnitude higher affinity of the netrin-4-laminin γ1 interaction results from the larger buried surface area than the one formed by β1 and γ1 laminins and greater electrostatic surface complementarity. Our findings, supported by site-directed mutagenesis, solid-phase binding analysis, laminin polymerization, and Schwann cell assays, collectively demonstrate that, in addition to inhibiting laminin polymerization, netrin-4 disassembles the pre-existing laminin lattice. The structure has the potential to facilitate the development of novel therapies for cancer treatment.
Similar content being viewed by others


Introduction
Basement membranes (BM) are extracellular biopolymers essential for cell differentiation and polarization, which function not only as structural scaffolds supporting cell anchorage and mechano-signaling platforms orchestrating cell behavior, but also as physical barriers preventing cancer cells traversing tissue boundaries1,2,3. During BM assembly, laminins (Lm) polymerize to form a planar lattice on cell surfaces4,5. Failures in Lm polymerization manifest in a diverse group of human disorders that we collectively defined as Lm N-terminal domain lamininopathies (LN-lamininopathies)6,7.
The Lm polymer node, which constitutes the repeating unit of the Lm lattice, is a flexible N-glycosylated heterotrimer consisting of one of each thirteen8 identified to date α, β, and γ subunits that assemble together in at least seventeen different combinations9. Crystal structures of monomeric α510, β111, and γ111 fragments, along with sequence analysis, revealed that Lm isoforms are elongated multidomain proteins comprised of the N-terminal (LN) domains with the seven- or eight-stranded antiparallel β-sheet jelly-roll motifs, followed by tandem laminin epidermal growth factor-like (LE) domains, and an extended α-helical region12. Individual Lm subunits heterotrimerize through their so-called, N-terminal short arms13,14,15 consisting of the LN and several LE domains, while the C-terminal long arm is a coiled-coil formed by three α-helices each contributed by α, β, and γ subunits. From the coiled-coil extends a cluster of five laminin-type globular (LG) domains, present only at the C-termini of α subunits, anchored to the cytoskeleton through cell surface receptors, such as integrins and α-dystroglycan3,16,17,18.
Netrins (Net) are a family of extracellular matrix proteins structurally homologs to Lm, which function as guiding cues during nervous system development and angiogenesis19,20,21,22. Netrin-4 (Net4) is a tumor suppressor23,24,25,26 that regulates the BM stiffness19,27,28. A key pathophysiological role of Net4 is its inhibitory effect on cancer growth and metastases reported for several tumor types, including breast, colorectal, and other cancers23,24,25,26. Net4 expression is significantly reduced in patients with invasive breast cancer29. In contrast, cancer cell lines with overexpressed Net4 display a decreased vascularization in vivo23,24, the effect inversely associated with cancer progression23,24. Similarly to Lm, Net4 consists of the N-terminal LN domain, four centrally-located LE domains (LEa1-4), and the unique C-terminal netrin-like domain (NTR)30 connected to LEa4 via an extended linker. The X-ray structure of the N-terminal fragment from Net4 lacking the NTR (Net4ΔC) revealed an elongated molecule with the LN domain containing a seven-stranded antiparallel β-sheet, followed by four LE domains, each containing the EGF-like fold19.
We have recently employed cryo-EM to determine a 3.7 Å structure of the trimeric complex consisting of Lm 56 kDa α1, 64 kDa β1, and 52 kDa γ1 (Lm111)6,7. The asymmetric Lm polymer node resembles a triskelion with centrally located LN domains and three rod-like structures projecting outwards, each containing one or two LE domains. The inter-subunit interfaces are formed by two sets of interacting regions contributed by each of the neighboring subunits6,7. The structure is stabilized by a network of multiple intra-subunit disulfide bonds, along with inter-subunit electrostatic and hydrophobic interactions. Interestingly, the cryo-EM map revealed the presence of eight extended densities representing N-glycans attached to protein surfaces, as determined by LC-MS6.
The anti-angiogenic and anti-metastatic properties of Net4 appear to be directly related to its impact on BM assembly, and particularly to its ability to inhibit formation of the Lm lattice27,31,32. It has been proposed that Net4 disrupts Lm polymer nodes by binding to γ1 and displacing β119,31,32. However, due to the intrinsic dynamics of Net4-γ1 and the consequent lack of structural information, the detailed molecular basis underlying this proposed interaction is not known. Here, we present a cryo-EM structure of the dimeric complex consisting of Net4ΔC and the N-terminal fragment from Lm γ1 containing the LN and LEa1 domains (γ1ΔC) and investigate the Net4-dependent mechanisms for the disassembly of the Lm lattice in vitro and in cell culture.
Results and discussion
Strategies employed for cryo-EM structure determination of the Net4ΔC-γ1ΔC complex
Recent advances in cryo-EM provide an unprecedented insight into structures of macromolecular complexes6,7,33,34,35,36,37,38,39,40, however, structure determination of intrinsically dynamic sub-100 kDa biomolecules continues presenting considerable difficulties. Due to its relatively small size (97 kDa complex consisting of 45 kDa Net4ΔC and 52 kDa Lm γ1ΔC), asymmetry, structural features mainly comprising loops (>80%) (Fig. 1b, Supplementary Fig. 8, 9), the presence of preferred orientations (Supplementary Fig. 3b) and continuous structural heterogeneity (Supplementary Figs. 6, 7), Net4ΔC-γ1ΔC presents a challenging target for cryo-EM structure determination. We have successfully mitigated the abovementioned issues by employing several data processing and acquisition strategies, along with the AI algorithms outlined below and detailed in the Methods section.

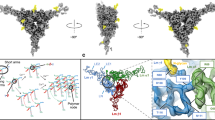
a Different views of a color-coded cryo-EM map. b Analogous views of the molecular model. Net4ΔC, γ1ΔC, N-glycans, and disulfide bonds are colored in orange, blue, pink, and yellow, respectively. Different structural elements are labeled in the figure. c Four specifically N-glycosylated asparagine residues6,19 colocalize with four unique Coulomb densities representing N-glycans in the map confirming the correctness of the model. Seven N-Acetylglucosamine (NAG 1-7) moieties were included in the model.
Firstly, the preferred orientation issue was addressed by systematically acquiring a large data set of 45,515 movies at six microscope stage tilts41 (i.e. no tilt, and 100, 200, 250, 300 and 400 tilts, Supplementary Fig. 3b), careful adjustment of the protein concentration4, and by performing multiple rounds of iterative 2D rebalancing steps and non-uniform refinements in cryoSPARC 4 (Supplementary Figs. 1, 2). We monitored convergence of the protocol by assessing the conical FSC area ratio (cFAR)41,42, the sampling compensation factor (SCF)42,43 and the high average relative signal amount within azimuth-elevation viewing regions (relative signal)43, as all three parameters are sensitive to the extent of sampling in Fourier space. During the procedure, cFAR values increased from 0.52 to 0.73 while the quality of calculated Coulomb maps improved, and then the cFAR stopped changing, indicative of convergence (Supplementary Fig. 4b). In addition, the respective SCF (Supplementary Fig. 5b) and relative signal (Supplementary Fig. 5c, d) values of 0.947 and of 0.8–0.82 indicate sufficient Fourier sampling, with only residual remaining directional anisotropy (Supplementary Fig. 4b).
Secondly, to minimize effects of continuous structural heterogeneity38 evidenced by 3D variability analysis (Supplementary Fig. 6) and 3D flexible refinement (3D Flex)44 presented in Supplementary Fig. 7 and Supplementary Movie 1, we combined several software packages for data processing including cryoSPARC 445 and Relion 446. An extensive 3D classification without alignment in Relion 4 significantly improved the map quality. We tested a wide range of temperature (T) parameters for scaling the Bayesian likelihood function, along with a varying number of calculated 3D classes. The Relion step was preceded and succeeded by two rounds of 3D Flex refinement in cryoSPARC 4 (Supplementary Fig. 2), the AI algorithm that employs graph neural networks for modeling flexible motions in biomolecules. This strategy considerably decreased continuous heterogeneity in the data and improved map quality, as quantitatively demonstrated by changes in the 3D variability analysis. Specifically, the procedure reduced variations in calculated eigenvectors (Supplementary Fig. 6a, c) and resulted in more compact distributions of reaction coordinates (Supplementary Fig. 6b, d).
Thirdly, the molecular model was built and validated by employing three independent approaches, all of which yielded nearly identical solutions (Supplementary Tables 3–5 and Supplementary Figs. 8, 9). In the first approach, we first docked the previously determined cryo-EM structure of γ1ΔC (PDB ID: 8DMK)6 and the X-ray structure of Net4ΔC (PDB ID: 4WNX)19 into the Coulomb density map using UCSF ChimeraX47, and then iteratively refined the model using a combination of Phenix 1.2148 and COOT 1.149. To quantitatively assess the map vs. model agreement at each step of the refinement, we calculated mean Q-scores in ChimeraX47 (Supplementary Fig. 10d and Supplementary Table 2) and monitored the validation statistics in Phenix 1.2148 (Supplementary Table 1) including FSC model vs. map (Supplementary Fig. 10a) and the model vs. map cross-correlation coefficients calculated for each individual residue (Supplementary Fig. 10b, c). The refinement was carried out until the abovementioned parameters stopped improving. In the second method, we employed DeepMainmast50, an integrated deep learning pipeline with the U-shaped network architecture (UNet3 + ) for unsupervised de novo model building from Coulomb maps. Finally, in the third approach, we utilized AlphaFold2 (AF2)51 for structure prediction of the Net4ΔC-Lm γ1ΔC complex. The RMSD values calculated between the three models range from 1 Å to 1.2 Å for pruned atoms (132-217 out of 260 residues) and from 1.6 Å to 3.9 Å for all atoms (Supplementary Table 3 and Supplementary Figs. 8, 9). The first group of atoms contains the interacting regions from the LN domains (Fig. 2). The latter group includes 257-260 out of 260 residues, and it consists of more heterogenous LEa1 domains and the tip of the LN domain in Net4ΔC (Fig. 1a, b). The presence of continuous structural heterogeneity38 in these regions is reflected by the results of the 3D Flex analysis (Supplementary Movie 1), and by the gradient of local resolutions across the map (Supplementary Fig. 4c).

a Net4ΔC and γ1ΔC are displayed in orange and blue, respectively. The intermolecular interface mapped onto the surfaces of Net4ΔC and γ1ΔC are shown in blue and orange, respectively. The protein-protein interface is formed by both the LN (regions N1, N2, and N3) and LEa1 (N4) domains in Net4ΔC, and by the tip of the LN domain in γ1ΔC (regions G1, G2, and G3) interacting with one another, as indicated by the arrows. Some of the structural details from each of the interacting regions (Supplementary Table 4) are presented in the following figures: b the N4-G3 interaction, c N3-G2, d, e N2-G2, and f, g the N1-G1 interaction.
In addition, to further improve map quality and prevent map oversharpening, we systematically tested several different sharpening methods, including tools relying on global Guinier plot-based sharpening, either automatic (Relion PostProcessing, PhenixAutoSharpen)52 or manual (CryoSPARC sharpening tool)53, local automatic Wiener filter-based methods (LocalDeblur and CryoSPARC Local Resolution Estimation/Filtering)54, and the convolutional neural network-based algorithm (DeepEMhancer)55. We also performed map phasing with Phenix Density Modification48. The model was then refined against each of the sharpened maps in Phenix/Coot, and the map vs. model agreement was quantitatively assessed by calculating mean Q-scores in ChimeraX47 (Supplementary Table 2). Best results were obtained by employing the global Guinier plot-based manual sharpening with the B-factor of −100 Å2 and by phasing map density in Phenix. Both procedures increased the mean Q-scores from 0.4 to 0.42. These values, calculated at the relevant sigma 0.6 (legend to Supplementary Table 2), reflect the Q-score values expected for a Coulomb map solved at the nominal resolution of ~3.9 Å56, and they are consistent with the reported local resolutions in Net4ΔC-γ1ΔC ranging from 3.4 Å to 5.6 Å (Supplementary Fig. 4c). We also calculated the nominal resolution of 3.64 Å in cryoSPARC 445 (Supplementary Fig. 4a). However, we think that this value might have been overestimated by the program, and the best estimate is provided by the range of local resolutions. The nominal resolution of the map is approximately 3.9 Å56, the fact reflected by mean Q-scores calculated in ChimeraX47 (Supplementary Table 2 and Supplementary Fig. 10c), and consistent with the map vs. model FSC obtained in Phenix 1.2148 with 0.143 of 3.9 Å (Supplementary Fig. 10a).
Taken together, by developing and employing the protocols described above, we have successfully determined the structure of Net4ΔC-γ1ΔC. Details of the single particle cryo-EM workflows and validation are presented in the Supplementary Figs. 1, 2 and Supplementary Table 1, respectively.
Cryo-EM structure of the Net4ΔC-γ1ΔC complex
The asymmetric structure of the Net4ΔC-γ1ΔC complex resembles the letter “Y” (Fig. 1a, b). Both, Net4ΔC and γ1ΔC, are foot-like shaped molecules with regions resembling the heel and the toe (Fig. 1a) that are stabilized by the structurally conserved seven-stranded jelly-roll motifs located in their respective N-terminal LN domains, followed by the C-terminal LEa1 domains (Fig. 1b). Strands β7, β2 and β5 form the front inner faces, while strands β1, β6, β3 and β4 form back outer faces of the jelly-roll motifs (Fig. 1b). The 3D Flex analysis of the cryo-EM data revealed the planar swinging motion of the LEa1 rod in γ1ΔC at the LN-LEa1 junction (Supplementary Movie 1 and Supplementary Fig. 7a). We applied a similar analysis to the cryo-EM data previously collected during structure determination of the related Lm polymer node6,7. Likewise, the analysis revealed the planar swinging at the LN-LE1 junction not only in γ1ΔC but also in α1ΔC and β1ΔC (Supplementary Movie 2). Hydrophobic cores of Net4ΔC and γ1ΔC are sandwiched between front and back faces of jelly-roll motifs. Both proteins also contain several short α helices inserted between individual β-strands6,7,19 (Fig. 1b). Additionally, Net4ΔC and γ1ΔC are stabilized by a network of intramolecular disulfide bonds. There are eight and seven disulfide bridges in Net4ΔC and γ1ΔC, respectively (Supplementary Figs. 1b and 12a). A superposition of the cryo-EM structure of Net4ΔC-γ1ΔC with the structure of the Lm polymer node6 consisting of γ1ΔC, and structures of monomeric γ1ΔC11 and Net4ΔC19 highlight apparent structural homologies with minor structural differences related to the presence or absence of a specific binding partner (Supplementary Fig. 13). The cryo-EM structure of Net4ΔC-γ1ΔC significantly differs from the previously proposed SAXS model of Net4ΔC-γ1ΔC19. In the model, the proteins are aligned in an antiparallel fashion along their long axes, and they appear to be interacting through the tip of the LN domain in Net4ΔC and the tip of the loop spanning residues Y109-T116 in γ1ΔC. We do not observe such an interaction in the cryo-EM structure.
The Net4ΔC-γ1ΔC complex is N-glycosylated
The cryo-EM map reveals the presence of four extended densities attached to its surface (Fig. 1c). These densities colocalize with four unique N-glycosylated asparagine residues in the model, namely N56 and N163 in Net4ΔC, along with N60 and N134 in γ1ΔC. This fact confirms the correctness of the model, as such a scenario would not be possible in any other arrangement of protein subunits in the Coulomb map. We have previously employed the LC-MS glycopeptide analysis to confirm the identity and investigate the composition of two out of four N-glycans, namely attached to N60 and N134, in the γ1ΔC sample used for cryo-EM structure determination of Net4ΔC-γ1ΔC6. In addition, the crystal structure of the Net4ΔC monomer19 revealed the presence of the remaining two unique N-glycans attached to N56 and N163 on its surface, the pattern confirmed by the cryo-EM structure. Consequently, we added seven N-acetylglucosamine (NAG) moieties to the model of Net4ΔC-γ1ΔC (Fig. 1c). The added NAG moieties show a good agreement with the map density, the fact reflected by the validation statistics (Supplementary Table 1).
Net4ΔC and γ1ΔC coordinate calcium ions in the complex
The cryo-EM structure of the Lm polymer node6,7 containing γ1ΔC, and X-ray structures of monomeric Net4ΔC19 and γ1ΔC11 revealed that both Net4ΔC and γ1ΔC coordinate one calcium ion in their LN domains (Supplementary Fig. 13a–c). Residues D108 and T116 are involved in calcium coordination in γ1ΔC6,7,11, whereas A93, D96, T104, W105, and Y254 chelate one calcium ion in Net4ΔC19. Although excess calcium was added to the protein samples used for cryo-EM structure determination, careful inspection of the Coulomb map did not allow unambiguous assignment of densities corresponding to the two calcium ions. This fact is consistent with the reported resolution (Supplementary Fig. 4). Hence, to directly measure calcium content in the protein samples used for cryo-EM, we employed Inductively Coupled Plasma-Mass Spectrometry (ICP-MS). Prior to the ICP-MS analysis, both proteins were extensively dialyzed against the metal free buffer. The ICP-MS revealed a nearly stoichiometric content of calcium in Net4ΔC and γ1ΔC, as indicated by the amounts of detected 43Ca and 44Ca (Supplementary Table 7). In contrast, the control sample of α1ΔC that is not expected to coordinate calcium ions revealed the presence of only trace amount of calcium.
In addition, we compared conformations of the two loops from Net4ΔC-γ1ΔC expected to bind calcium with analogous regions showed to chelate calcium in the previously determined structures. We superposed seventeen amino-acids (P90-W106) representing the calcium-binding loop in the previously solved X-ray structure of the Net4ΔC monomer (PDB ID: 4WNC)19 with a correspondent region from Net4ΔC-γ1ΔC. The backbone RMSDs are 1.03 Å/1.66 Å (pruned 13/all 17 pairs). We also superposed thirty-three amino-acid loop (H90-T122) consisting of the calcium-chelating residues in the previously determined crystal structure of γ1ΔC monomer (PDB ID: 4AQT)11 with an analogous region from Net4ΔC-γ1ΔC. The resultant backbone RMSDs are 0.95 Å calculated for both pruned and all 33 pairs of residues. A similar superposition of thirty-three residues from the γ1ΔC subunit engaged in formation of the Lm polymer node (PDB ID: 8DMK) 6 and Net4ΔC-γ1ΔC yielded backbone RMSDs of 1.13 Å/1.19 Å (pruned 32/all 33 pairs, respectively). Taken together, results of the ICP-MS analysis, along with the arrangement of protein loops that is expected for calcium-binding, confirm the presence of calcium in Net4ΔC-γ1ΔC.
The Net4ΔC-γ1ΔC binding interface
The dimeric Net4ΔC-γ1ΔC complex is stabilized by a network of electrostatic and hydrophobic interactions (Supplementary Fig. 12b, c). The protein-protein interface is formed by the tip of the LN domain in γ1ΔC, and by both the LN and LEa1 domains in Net4ΔC (Fig. 1a, b). Specifically, the interface involves the following four regions in Net4ΔC (N1-4) that interact with three regions in γ1ΔC (G1-3), (Fig. 2a). (i) The N1 region that contains the loop connecting the helix 1 (α1) and β-strand 1 (β1), and consists of S98, F99, R100 and F101, interacts with the G1 region in γ1ΔC involving the loop linking β-strands βa and βb (Fig. 2f, g). The G1 contains, for instance, T80, G81, and S88. (ii) The N2 and N3 are engaged in another intermolecular interaction with the G2 from γ1ΔC (Fig. 2c–e). The N2 involves the inner face of the jelly-roll motif (β2, β5, β7), along with the loop linking α2 and β5, whereas the N3 consists of the loop connecting β5 and α3 in Net4ΔC. These two regions consist of, for example, T128, I131, M132, N184, Y185, F198, D206, and other residues listed in the Supplementary Table 4. The matching G2 interface in γ1ΔC involves loops connecting β3 with β4, and β5 with β6, the latter containing α2 (residues E158, I183, N184, R191, D211, S213, and others). (iii) Lastly, the N4 region from Net4ΔC consisting of LEa1 loops a and b interacts with the G3 from γ1ΔC spanning the loop linking strands β7 with β8 and containing α4 (Fig. 2b, e). These regions involve, for instance: F263, D269, Q70 in N4, and F259, D261, V263, F264, Y273 in G3 (Supplementary Table 4).
All three models of Net4ΔC-γ1ΔC that we independently derived using different methods, namely Phenix 1.2148/COOT 1.149, DeepMainmast50 and AF251 revealed identical binding interfaces. These models have been described in the first section of the Results and Discussion, and they are displayed in Supplementary Figs. 8, 9. Furthermore, up to ~57.8 % interface residues located within a 5 Å distance of each other are identical in all three models, and up to ~86.7 % residues are identical in two out of three models (Supplementary Table 5). The interface residues identified in each model are listed in the Supplementary Table 4.
The binding of α1 with β1-γ1 dimers to form the Lm polymer node requires the presence of calcium57. The cryo-EM structure of the Lm polymer node revealed that the calcium-binding loop from γ1 forms a part of the interface with α16,7 (see below and Supplementary Fig. 17b). Consequently, we previously proposed that in the absence of a calcium ion the loop in γ1 is not structured, hence the α1-γ1 interface cannot be formed, explaining the calcium dependence for the assembly of a trimeric Lm polymer nodes6,7. In contrast, the cryo-EM structure of Net4ΔC-γ1ΔC unveiled that the analogous γ1 loop is not engaged in formation of the protein-protein interface, while explaining the calcium-independent association of Net4ΔC with γ1ΔC31. The crystal structure of Net4ΔC19, along with our structural and ICP-MS analyses, confirmed that Net4ΔC coordinates a calcium ion. Interestingly, the recent study, in which calcium binding to the LN domains from γ1 and netrin-1 was measured using isothermal titration calorimetry and thermal shift assay, revealed a nearly 20-fold difference in metal binding affinities displayed by the two proteins (i.e. 1.36 μM for netrin-1-Ca2+ at 42 °C, and 22.4 μM for γ1-Ca2+ at 47 °C). The study concluded that, unlike a reversible binding of the metal ion in γ1 that facilitates the formation of the Lm polymer nodes, the binding of calcium to the LN domains in netrins is irreversible under physiological conditions, and it plays a structural role by stabilizing the LN domain.
In summary, the cryo-EM structure of Net4ΔC-γ1ΔC revealed regions engaged in formation of the intermolecular interface and provided an insight into the calcium-independent formation of Net4ΔC-γ1ΔC.
Mutational analysis of the Net4ΔC-γ1ΔC interface
We employed a site-directed mutagenesis and a solid-phase binding assay to investigate the interaction of Net4ΔC with γ1ΔC and its several altered variants, namely: Y147R, R149E, S213R, D261R, D266R, D285R (Supplementary Fig. 14). First, we measured the dissociation constant (Kd) for the Net4ΔC-γ1ΔC binding (Kd=2.93 ± 0.51 nM); (Supplementary Table 6). We then sampled the N2-G2 interface (Fig. 2). The cryo-EM structure revealed that S213 from γ1ΔC is located at the protein-protein interface (Fig. 2d). The S213R substitution abolished the binding (Supplementary Table 6). Next, we sampled the N4-G3 interface by changing D261 into arginine. Likewise, the D261R mutation abolished the complex formation (Supplementary Table 6). We also investigated the D266R substitution. The mutation of D266 that is located at the tip of the LN domain, however, away from the protein-protein interface, resulted in the Kd of 4 ±0.4 nM, the value comparable to the Kd measured for the truncated wild-type proteins. In addition, we engineered two recombinant γ1ΔC variants harboring amino acid substitutions at the protein region that mediates the γ1-α1 interaction, as revealed by the cryo-EM structure of the Lm polymer node6. The Y147R and R149E mutations had no significant effect on the Net4ΔC-γ1ΔC interaction with the Kd of 2.28 ± 0.43 nM and 1.89 ± 0.2 nM, respectively. Likewise, a substitution of D285R located at the LEa1 domain of γ1ΔC had only a minimal effect on the protein-protein binding with the Kd of 1.57 ± 0.21 nM (Supplementary Fig. 14 and Supplementary Table 6).
Furthermore, the cryo-EM structure explains results of the previously conducted site-directed mutagenesis of the Net4ΔC surface19. The microscale thermophoresis assay demonstrated that a double mutant protein consisting of E195A and R199A substitutions reduces the Net4ΔC-γ1ΔC binding affinity from 2 nM to 66 nM19. The cryo-EM structure revealed that both E195 and R199 are located in the β5 of the Net4ΔC’s jelly-roll motif that faces γ1ΔC. While R199 directly contributes to the N2-G2 interface (Supplementary Table 4), the G193-E195 segment lacks clear Coulomb density in the cryo-EM map; hence, we removed it from the model. Interestingly, AF2 predicted that E195 is in a hydrogen bonding distance from γ1ΔC’s S213, which substitution to arginine abolished the protein-protein interaction (Supplementary Fig. 14 and Supplementary Table 6). Another previously reported alternation of Net4ΔC’s sequence, namely a deletion of a so-called “KAPGA” motif consisting of four residues from the LE1’s loop b (K281-A285), reduces the Net4ΔC-γ1ΔC binding affinity from 2 nM to 50 nM19. While, as this is also the case in the X-ray structure of Net4ΔC19, we cannot unambiguously assign the density representing KAPGA, the motif is directly preceded by F286 (Supplementary Fig. 14b). The F286, in turn, forms a part of the N4-G3 interface (Supplementary Table 4). Hence, we hypothesize that shortening of the loop b in Net4ΔC likely affects the N4-G3 interaction.
In summary, the mutagenesis studies confirmed the protein-protein interface visualized by the cryo-EM structure (Fig. 2), indicated that Net4ΔC-γ1ΔC adopts “the-heel-to-toe” organization previously observed in the cryo-EM structure of the Lm polymer node (Fig. 1)6,7,58, and supported the mechanistic insight into the Net4-dependent inhibition of Lm lattice formation described in the following sections.
Molecular basis underlying the high-affinity interaction between Net4ΔC and γ1ΔC
The affinity of the Net4ΔC-γ1ΔC interaction measured in the microscale thermophoresis assay is 2 nM19. In contrast, a size-exclusion chromatography (SEC) analysis of Lm complexes revealed that the β1-γ1 dimerization occurs with more than four orders of magnitude weaker binding affinity (Kd = 22 μM)59. Consequently, it was first suggested31 and later confirmed by SEC19 that Net4ΔC can displace β1ΔC from Lm polymer nodes assembled in vitro with the N-terminal fragments of α1, β1, and γ1. What is the reason for such a significant discrepancy in binding affinities measured for the two complexes?
First, we superposed Net4ΔC from Net4ΔC-γ1ΔC with β1ΔC from the structure of the Lm polymer node6, (Supplementary Fig. 13d). Net4ΔC and β1ΔC share significant structural homology, the fact reflected by the RMSD of 1.32 Å calculated for 93 pruned residues consisting of the LN domain, and 5.3 Å calculated for all 256 residues containing both the LN and LEa1 domains. Likewise, we superposed the structure of Net4ΔC from Net4ΔC-γ1ΔC with the X-ray structure of a monomeric β1ΔC11 (Supplementary Fig. 13e). The RMSDs are 1.34 Å/6.72 Å (pruned 96/all 252 residues).
In the next step, we overlapped cryo-EM structures of Net4ΔC-γ1ΔC and the Lm polymer node6 such that γ1ΔC subunits were superposed (backbone RMSDs of 1.24 Å/2.34 Å calculated for 168 pruned/all 257 residues; Supplementary Fig. 13b), and then carefully compared the Net4ΔC-γ1ΔC and β1ΔC-γ1ΔC binding interfaces. The detailed comparison of these two interfaces revealed the molecular basis underlying the high-affinity Net4ΔC-γ1ΔC interaction. The Fig. 3a shows protein regions engaged in formation of the β1-γ1 and the α1-γ1 interfaces in the cryo-EM structure of the Lm polymer node6,7, mapped onto the surface of γ1ΔC from Net4ΔC-γ1ΔC. The Fig. 3b also displays an additional Net4ΔC-γ1ΔC interface, mapped onto the same structure of γ1ΔC. The Net4ΔC-γ1ΔC interaction involves most residues from the β1ΔC-γ1ΔC interface, for example: T80, T83-K87, D211-S213, F259-D261, F264 and others. These residues form parts of the N1-G1, N2/N3-G2, and N4-G3 interacting regions (Fig. 2 and Supplementary Table 4). However, the Net4ΔC-γ1ΔC interface is larger, as it contains several other residues that are not engaged in β1ΔC-γ1ΔC binding, for instance: G180, E183, N184, R191, and D297 from N2/N3-G2. In addition, some of the amino acids from the α1ΔC-γ1ΔC interface visualize by the cryo-EM structure of the Lm polymer node6,7, are also involved in Net4ΔC binding to γ1ΔC, for example E208. Consequently, the buried surface area of Net4ΔC-γ1ΔC is 23% larger than the one of β1-γ1 (i.e. 1397 Å2 vs. 1138 Å2, respectively), as it encompasses additional protein regions.

a The molecular surface of γ1ΔC shown in blue with color-coded residues involved in binding to β1ΔC and α1ΔC in the Lm polymer node6. These residues are displayed in red and green, respectively. b Identical as above except for γ1ΔC residues involved in binding to Net4ΔC shown in orange. The γ1ΔC forms 23% larger intermolecular interface with Net4ΔC, than it does with β1ΔC (1397 Å2 vs. 1138 Å2, respectively)6. The following figures highlight the electrostatic surface potential of protein regions involved in formation of intermolecular interfaces in c γ1ΔC, d Net4ΔC, and e β1ΔC. The four orders of magnitude higher binding affinity of Net4ΔC to γ1ΔC vs. β1ΔC is due to the larger buried surface area and greater electrostatic complementarity of the Net4ΔC and γ1ΔC binding surfaces. The surface of Net4ΔC displays a strong positive electrostatic potential, while the surfaces of β1ΔC and γ1ΔC have net negative electrostatic potentials.
We also analyzed the electrostatic properties of the Net4ΔC-γ1ΔC and β1ΔC-γ1ΔC interfaces (Fig. 3c–e). The Fig. 3c shows that the binding surface from γ1ΔC has a strong net negative electrostatic potential. Similarly, the matching molecular surface in β1ΔC is negatively charged (Fig. 3e), explaining the necessity of α1 binding to the β1-γ1 dimer with the Kd of 2 μM (i.e. an order of magnitude higher than the Kd measured for the β1-γ1 dimerization) to stabilize the Lm polymer node structure58. In contrast, the complementary surface from Net4ΔC has a strong matching net positive electrostatic potential (Fig. 3d).
Taken together, the electrostatic complementarity of the interacting surfaces (Fig. 3c, d), in combination with the larger buried surface area (Fig. 3a, b), collectively result in the high affinity of binding between Net4ΔC and γ1ΔC, while providing the rationale for efficient inhibition of Lm polymerization by Net4ΔC described in the following sections.
The mechanism for the Net4-dependent inhibition of Lm polymerization
The Lm lattice is a dynamic biopolymer, which structure and composition undergo constant remodeling1. At equilibrium, Lm polymer nodes continuously disassemble and reassemble1. The Surface Plasmon Resonance (SPR)31,59 revealed that the low-affinity β1ΔC-γ1ΔC dimerization (Kd of 22 μM) and the α1ΔC binding to the β1ΔC-γ1ΔC dimer (Kd of 2 μM) are both associated with fast dissociation rates. Consequently, we propose that Net4ΔC inhibits Lm polymerization by replacing β1ΔC through its high-affinity interaction with γ1ΔC (Kd of 2 nM)19, while the β1ΔC-γ1ΔC dimers and Lm polymer nodes undergo rapid off-rate transitions.
In support of this notion, previous biochemical experiments, including SPR, SEC, and solid-phase binding studies, explicitly demonstrated that Net4ΔC binds to γ1ΔC; however, it does not bind to α1ΔC and β1ΔC19,31,32,59. We confirmed these observations in context of the preassembled Lm matrix by employing the solid phase binding assay to investigate the interaction of Net4ΔC with Lm111 and its three truncated variants, in which the LN and LEa1-4 domains were removed from α1 (Lm111Δα1), β1 (Lm111Δβ1) and γ1 (Lm111Δγ1); (Supplementary Fig. 15). Net4ΔC binds to Lm111, Lm111Δα1 and Lm111Δβ1 with the Kd of 0.39 ± 0.07 nM, 0.56 ± 0.06 nM and 0.48 ± 0.06, respectively. In contrast, Net4ΔC does not specifically bind to Lm111Δγ1, directly confirming the Net4ΔC-γ1ΔC interaction.
The SEC analysis59 with Net4ΔC, α1ΔC and γ1ΔC indicated that the Net4ΔC-γ1ΔC interaction excludes the possibility of α1ΔC-γ1ΔC binding, as α1ΔC has to be stabilized by both the α1ΔC-γ1ΔC and α1ΔC-β1ΔC interfaces to form the Lm polymer node6. To reveal the molecular basis underlying these mutually exclusive interactions, we superposed Net4ΔC from Net4ΔC-γ1ΔC and β1ΔC from the Lm polymer node and inspected β1ΔC residues interacting with α1ΔC (Supplementary Fig. 16a)6. Likewise, we investigated the analogous surface on Net4ΔC (Supplementary Fig. 16b). Twenty-six residues contribute to the α1ΔC-β1ΔC interface in the Lm polymer node (Supplementary Fig. 18)6. However, only four of these residues are conserved in the Net4ΔC structure visualized by cryo-EM (Supplementary Fig. 16b, 18), indicating that the toe of Net4ΔC cannot support α1ΔC binding.
In addition, Net4ΔC and β1ΔC molecular surfaces display little structural homology in the toe regions, and their electrostatic potentials differ significantly (Supplementary Fig. 16c–e), further emphasizing the incompatibility of Net4ΔC binding to α1ΔC. Overlaying structures of Net4ΔC-γ1ΔC and the Lm polymer node6,7 such that γ1ΔC subunits are superposed unveils that the protein region located at the tip of the LN domain in Net4ΔC sterically clashes with α1ΔC in the structure of the Lm polymer node (Supplementary Fig. 17). This occluding region consists of four loops stapled together by five disulfide bonds and it contains two N-glycosylated asparagine residues, namely N56 and N163 (Fig. 1c). The four loops are: (i) the N-terminal loop spanning residues C62-C77, (ii) the loop connecting strands β2 and β3 (residues K135-A140), (iii) a part of the loop connecting strands β4 and β5 (residues T162-N-162 and S186-G193), and (iv) the loop connecting strands β6 and β7 (amino acids R231-Y251).
In summary, we propose that Net4ΔC inhibits Lm polymerization through its high-affinity interaction with γ1ΔC by replacing β1ΔC in Lm polymer nodes that undergo rapid off-rate transitions.
Net4 disrupts the preassembled Lm lattice in vitro
Our structure-derived mechanism underlying the high-affinity interaction between Net4ΔC and γ1ΔC, and the resultant inhibition of Lm polymerization57, reconciles previously reported results, while providing the mechanistic explanation of their molecular basis.
Firstly, the Lm polymerization assay showed that Net4ΔC can inhibit the process of Lm polymerization19. Secondly, the Atomic Force Microscopy (AFM) analysis of a polymerized Matrigel matrix revealed that stiffness of the Lm lattice is weakened after co-incubating Lm with Net4ΔC (i.e. the stiffness decreased from 645.51 ± 2.97 Pa to 191.41 ± 1.36 Pa19), again implying that Net4ΔC interferes with Lm polymerization. Interestingly, the addition of the Net4ΔC variant harboring E195A and R199A mutations to the Lm lattice did not alter its stiffness19. This observation is consistent with the fact that the two residues are located at the intermolecular interface (Supplementary Table 4), hence their substitutions render the altered Net4ΔC inactive in the AFM experiment.
To further support the above observations, we performed the Lm polymerization assay (Fig. 4a). In addition to confirming that Net4ΔC inhibits Lm polymerization (quantification of lanes 3 and 5 on the gel indicates 37% reduction in the amount of polymerized Lm), our results for the first time directly demonstrate the ability of Net4ΔC to dissolve the pre-existing Lm111 lattice (lanes 7 vs. 9). The 1-hour incubation of Lm111 with the 1.3-fold molar excess of Net4ΔC, leads to a significant reduction in the amount of the observed Lm lattice (i.e. by 30%), the result comparable to the one from lane 5 on the gel.

a The Lm polymerization assay demonstrates that Net4ΔC blocks Lm polymerization (lane 5) and disrupts the pre-existing Lm111 (lane 9) in vitro. Lm111 was incubated in the absence (-) or presence (+) of Net4ΔC. Net4ΔC was either co-incubated with Lm111 from the beginning of the polymerization reaction (T0) or added to the reaction following 1 h Lm111 assembly (T1). The “S” and “P” denote the supernatant and the pelleted fractions from the reaction. The latter fraction reflects the portion of Lm that polymerized into the Lm111 lattice. Individual protein bands are labeled in the figure. b–f Results of the Schwann cells (SC) assay revealed the inhibitory effect of Net4 on Lm polymerization and Lm lattice formation in cell culture. Formation of the Lm EHS63,64 lattice was visualized by fluorescent staining with polyclonal antibodies directed against α1 coupled with Alexa 647. The scale bar is 50 μm. b Control SC cultured for 1 h. c, e SC cultured with Net4ΔC and Net4 from the beginning of the incubation (T0), respectively. d, f SC cultured for 1 h prior to addition of Net4ΔC and Net4, respectively. Before netrin-4 variants were added (+), Lm EHS not associated with the cell surface was either washed away (w) or remained in the cell culture dishes (nw). SC were then incubated with Net4ΔC or Net4 for additional 1 or 2 h. g Quantified results of the SC assay that included the 1 h incubation (T1) with netrin-4 variants. h Quantified results of the SC assay that included the 2 hincubation (T2) with netrin-4 variants. Net4 is more efficient than Net4ΔC in dissolving the preexisting Lm EHS lattice. Fluorescence intensities were normalized to cell counts based on DAPI staining i.e. antibody fluorescence summed intensities were divided by the number of DAPI-stained nuclei (mean ± S.D., n ≥ 7 per condition). Black dots indicate individual measurements, whereas scale bars indicate standard deviations from the mean.
To recapitulate, the cryo-EM structure of Net4ΔC-γ1ΔC provides the detailed mechanistic insight into the molecular basis underlying the disruption of the preassembled Lm lattice by Net4ΔC through its high-affinity interaction with γ1ΔC, the fact supported by multiple lines of evidence gathered using the in vitro experiments. Next, we investigated the ability of Net4 and Net4ΔC to dissolve the Lm matrix in cell culture.
Net4 interferes with Lm lattice formation on cell surface
We have monitored both the inhibitory effect of Net4 on Lm polymerization and the Net4-dependent disassembly of the Lm lattice on the surface of Schwann cells (SC). SC provides a convenient model to study the BM14. The Lm lattice derived from the mouse Engelbreth-Holm-Swarm sarcoma60 (Lm EHS) was assembled on SC, as evidenced by fluorescent staining with polyclonal antibodies directed against the C-terminal globular domains of α1 (Fig. 4b and bars 1 in Fig. 4g, h). To test the Net4-dependent inhibition of Lm polymerization, the 5-fold molar excess of Net4ΔC (Fig. 4c) or a full-length Net4 (Net4) containing the C-terminal NTR domain (Fig. 4e), was added at the beginning of SC incubation with Lm EHS. In both cases, netrin-4 variants prevented Lm polymerization, as evidenced by the lack of fluorescent staining of SC (bars 2 and 5 in Fig. 4g, h). In contrast, to investigate the role of netrin-4 in dissolving the pre-existing Lm lattice, Net4ΔC (Fig. 4d) or Net4 (Fig. 4f) were added to the preassembled Lm111 on the SC surface. The SC were then further cultured for additional 1 or 2 h in the presence of either one of netrin-4 variants (Fig. 4g and h, respectively). Lm EHS not associated with the cell surface was washed away prior to addition of netrin-4 variants (bars 3 and 6 in Fig. 4g, h) or Lm EHS remained in cell culture dishes, such that it could potentially continue polymerizing on SC surface during incubation with either one of netrin-4 variants (bars 4 and 7 in Fig. 4g, h).
Our results clearly demonstrate that both Net4ΔC and Net4 disassemble the pre-existing Lm111 in cell culture. After 1-hour incubation, Lm111 content on SC was reduced to ~75–80%, regardless of whether Lm EHS was removed or remained in the mixture (bars 3, 6 or 4, 7 in Fig. 4g, respectively). Interestingly, Net4 is more efficient than Net4ΔC in dissolving the pre-existing Lm lattice, as evidenced by approximately 90% vs. 60% reduction of Lm111 content on SC following an extended 2 h incubation (bars 6, 7 vs. bars 3, 4 in Fig. 4h, respectively). Although the binding affinities of γ1 to Net4 and γ1 to Net4ΔC are nearly identical18, this observation raises the possibility that the C-terminal NTR domain of Net4 can participate in the disassembly of the Lm lattice either directly (for instance, by transiently interacting with Lm polymer nodes) or indirectly (for example, through contacts with cell surface receptors).
Taken together, in support of our structure-derived mechanistic models, we demonstrated that Net4 and Net4ΔC block Lm polymerization and disassemble the pre-existing Lm lattice in cell culture.
Molecular basis of the Net4-dependent disassembly of the Lm lattice
In conclusion, we have determined a cryo-EM structure of Net4ΔC-γ1ΔC. The structure reveals fundamental molecular mechanisms governing the Net4-dependent inhibition of Lm polymerization and the resultant disassembly of the Lm lattice (Fig. 5). Net4 can interfere with the process of β-γ dimerization (Fig. 5a), prior to a calcium-dependent joining of the β-γ dimer and α into a functional trimer (Fig. 5b). The high affinity Net4-γ binding blocks this process, effectively inhibiting Lm polymerization (Fig. 5c). Alternatively, Net4 can interfere with the pre-existing Lm lattice by capturing β subunits that transiently dissociated from Lm polymer nodes (Fig. 5d), subsequently leading to the disassembly of the lattice (Fig. 5e). One of the intriguing open questions is the possible involvement in this process of the NTR from Net4 that was not visualized in the cryo-EM and X-ray19 structures (Fig. 5e).

The Lm lattice is a dynamic biopolymer, which undergoes constant remodeling through Lm polymerization and dissociation of pre-existing Lm polymer nodes in the presence of Net4. Lm polymerization is a two-step process involving (a) a calcium-independent dimerization of β and γ through their N-terminal short arms, followed by (b) a calcium-dependent trimer formation, in which α joins the β-γ dimer, and subsequent extension of the Lm lattice. c Net4 can interfere in this process by binding to γ, thus inhibiting the β-γ dimer formation. Alternatively, d Net4 can interfere with the pre-existing Lm lattice by capturing γ, which transiently dissociates from the Lm polymer node, subsequently leading to (e) the disassembly of the Lm matrix. f The C-terminal domain of Net4 may transiently participate in this process, although its exact role is not known. Calcium ions are displayed as white circles. A trimeric Lm polymer node consisting of Lm short arms from α, β, and γ, along with the β-γ and Net4ΔC-γ dimers, is highlighted in the figure by black ovals.
The calcium-dependent formation of the Lm polymer node and its calcium-independent disassembly by Net4 are an example of how flexible intermolecular interfaces can be differentially controlled by metal ions, while providing the additional level of tunability of the protein-protein interactions. The rapidly emerging model for the protein-protein binding within the BM highlights the importance of divalent cations, which, in addition to balancing the Net4-γ1 and Lm polymer node interactions6, also govern the binding of the C-terminal domain of α2 with matriglycan61, and interactions involving the C-terminal fragment of Lm511 with the α6β1 integrin headpiece62. Because Net4 is an important tumor suppressor, which acts by interfering with formation of the Lm matrix, the structural insight has the potential to facilitate the development of novel therapies for the treatment of cancer.
Methods
Protein expression and purification
The recombinant proteins were purified from human embryonic kidney cells (HEK293, ATCC crl-1573 tm) stably expressing mouse Net4ΔC and human γ1ΔC58. Mouse and human Net4ΔC share 91.5% sequence identity (Supplementary Fig. 19). The HEK293 cells were cultured in DMEM (Invitrogen, 11995-081) supplemented with 10% Fetal Bovine Serum (Atlanta Biological, S11150), 200mM L-Glutamine and Penicillin-Streptomycin (1000 μg/ml Penicillin and 1000 μg/ml Streptomycin, Invitrogen, 15140122) as well as 1 μg/ml Puromycin (Invitrogen, J67236XF), 100 ug/ml Zeocin (Invitrogen, R25001) or 500 μg/ml G418 (Invitrogen, 11811023), respectively. Both proteins were purified from media using anti-FLAG M2-agarose (Sigma, A2220), concentrated in an Amicon Ultra-15 filter (30 K MWCO), (Millipore UFC903024), and dialyzed in TBS50 (90 mM NaCl, 50 mM Tris pH 7.4, 0.125 mM EDTA). The non-tagged Net4ΔC used in ELISA was a generous gift from the Koch lab19.
Site-directed mutagenesis of the inter-subunit interface in Net4ΔC-γ1ΔC
The following γ1ΔC variants: Y147/R, R149/E, S213/R, D261/R, D266/R, D285/R were cloned, expressed, and purified as described above58. Two-step overlapping PCR was used to generate a C-terminally flag-tagged mouse Net4ΔC pcDNA3.1 G418 plasmid. In brief, 5’ (960 bp) and 3’ (330 bp) initial product was produced from Net4 cflag xba-1F 5’-GTTCAAGTCTCCCAGGCCTGC-3’ and NetDC flag-2R 5’-TCGTCGTCCTTGTAGTCTCTGCAGCCGTAGTCT-3’ and NetDC flag-2F 5’-AGACTACGGCTGCAGAGACTACAAGGACGACGA-3’ and γ1 LNLEa Apa-1R 5- GACACCTACTCAGACAATGCGATGC-3 using the pcR4 TOPO NTN4 (Genomics ABIN4006770) and gLNNd pcDNA3.1 Zeocin58, respectively. The DNA fragments were joined together with the Net4 c flag xba-1F and g1 LNLEa Apa-1R primers to generate a 1.26 kb product. Following XbaI and KpnI digestion, an 835 bp insert was ligated into the similarly digested pcR Topo mouse netrin-4 G418 vector (5088 bp) for final product of 5923 bp. Net4ΔC open reading frame (ORF) was additionally moved to a pcDNA3.1(-) zeocin vector for ease of cell cloning and protein production. The ORF was excised with NotI and KpnI, blunted with Klenow (Thermo Fisher EP0051), and inserted into a MssI digested pcDNA3.1 (-) zeocin vector.
Solid-phase binding assay
Ninety-six-well high protein–binding Costar plates were coated with non-tagged Net4ΔC (2.5 μg/ml). The wells were washed and blocked with the buffer (PBS, 1% BSA, 0.06% Triton X-100). Two-fold serial dilutions of C-terminally flag-tagged gLNLEa proteins58 were prepared in PBS buffer containing 1% BSA and 1% NP40, and applied (0.1 ml) to the Net4 coated wells. After 2-hour incubation at room temperature, unbound protein was removed, the wells washed and incubated with α1-flag antibody coupled to HRP (1:2000, Sigma A8592), washed again, and then incubated with TMB (3,3′,5,5′-tetramethylbenzidine, Thermo Fisher 34028) for 2 min. Absorbance was measured at A450 following the addition of sulfuric acid to halt further color development. In an analogous experiment, the Net4ΔC-coated Costar plates were incubated with recombinant mouse laminin 111 (Lm111) and its three truncation variants, in which LN-LEa1-4 domains in α1 (Lm111Δα1), β1 (Lm111Δβ1), and γ1 (Lm111Δγ1) were removed.
Lm polymerization assay
The stock of Lm111 Englebreth-Holm-Swarm63,64 (Lm EHS) was diluted to the concentration of 0.34 mg/ml in a buffer containing 50 mM Tris pH 7.4, 90 mM NaCl, and 2 mM CaCl2). Reactions were incubated for 2 h at 37 °C with 0.1% Triton X-100 and Bovine Serum Albumin (BSA; 0.6 μg/ml) and then centrifuged at 11,000 × g, followed by dissolution of supernatant and pelleted fractions in SDS14. Net4ΔC was added (0.032 mg/ml) either at the start of the incubation or 1 h post-incubation. Supernatant and pelleted fractions were analyzed by 8% SDS-PAGE and stained with Coomassie blue. The density of gel bands was measured with the Image Lab software (BioRad).
Schwann cells assay
Schwann cells isolated from neonatal Sprague-Dawley rats (courtesy of Dr. James Salzer, New York University). High passage rat Schwann cells (SC), seeded at 65,000 cells per well, were expanded in culture for 10 passages and maintained in Dulbecco’s modified Eagle’s medium, 10% fetal calf serum (Gemini Bioproducts), neuroregulin (0.5 μg/ml, Sigma), forskolin (0.2 μg/ml, Sigma), 1% glutamine, and penicillin-streptomycin14. Lm EHS derived from the mouse sarcoma extracts was added at the concentration of 30 μg/ml and incubated with SC for 45 min in the absence or presence of the 5-fold excess of Net4 or Net4ΔC. Duplicate wells also contained the 5-fold excess of Net4 or Net4ΔC, however netrin-4 variants were added after 1-hour incubation of Lm EHS. Excess of Lm EHS was either removed by replacing the cell medium or left in the cell culture dishes prior to addition of netrin-4 variants. SC were then washed with phosphate buffered saline (PBS), fixed with 3% paraformaldehyde, and blocked with PBS consisting of 5% goat serum. Fluorescent labeling was performed with the primary anti-rabbit α1-E3 antibody14 added at the concentration of 1 μg/ml, and the secondary anti-α1-rabbit antibody coupled with Alexa 647 (Invitrogen A-21245). SC were also labeled with DAPI for normalization. Fluorescent readings were quantitated from digital images (n ≥ 7 per condition, each 1300 × 1030 pixels, 437 × 346 μm) recorded using a ×20 microscope objective with IPLab 3.7 software (Scanalytics). A segmentation range was chosen to subtract background and acellular immunofluorescence. The sum of pixels and their intensities in highlighted cellular areas of fluorescence were measured and normalized by dividing by the number of cells determined by a count of DAPI-stained nuclei for each image. Data were expressed as the mean ± S.D. of normalized summed intensities with the data analyzed by one-way analysis of variance with Holm-Sidak comparisons in SigmaPlot 9.01 and SigmaStat 3.1 (Systat) 14.
Negative-stain EM
Samples of Net4ΔC-γ1ΔC were screened by negative-stain EM at the wide range of concentrations obtained by diluting the protein stock (6 mg/ml) in 90 mM NaCl, 50 mM Tris pH 7.4, and 125 μM EDTA. The 3 µl aliquots were applied to carbon-coated 300-mesh copper grids, which were previously rendered hydrophilic with PELCO easiGlow glow discharger. Grids were stained with 1.5% (w/v) uranyl formate containing 0.125% trehalose for 30 s at room temperature. After staining, grids were air-dried and inserted into the microscope. Images were recorded using Philips CM-12 electron microscope operating at 80 kV acceleration voltage at a magnification of 60,000 or 75,000 times corresponding to the pixel size of 1.95 Å/px or 2.44 Å/px, respectively57. Recorded images were analyzed in the program Eman 2.265.
Inductively coupled plasma-mass spectrometry
ICP-MS was used to measure calcium content in protein samples. Prior to the analysis, aliquots of purified α1ΔC (1 mg/ml), γ1ΔC (1 mg/ml), and Net4ΔC (0.35 mg/ml) were dialyzed against the metal-free phosphate buffered saline (PBS), and then additionally washed three times in the same buffer using Amicon Ultra columns (Millipore). Approximately 200 ng of each protein was dried down in a Teflon vial, incubated with 0.5 mL of concentrated HNO3 for 24 h, dried again, resuspended in 2 ml of 2% HNO3 solution, and analyzed using an iCAP-Q (Thermo Fisher Scientific) running the software Qtegra 2.11 (Thermo Fisher Scientific). The following acquisition parameters were used: channels 3 per peak; dwell time 0.01 s; 10 sweeps; RF forward power 1550 W; sample gas 1.14 L/min; cool gas 14 L/min; aux gas 0.8 L/min; measured in pulse counting. To reduce effects of the isobaric interference from the argon plasma, we measured 43Ca and 44Ca. Metal-free PBS and 2% HNO3 were used as controls. The data were analyzed with the Qtegra 2.11 (Thermo Fisher Scientific). Counts per second (cps) were converted to parts per billion (ppb) using a 4-point single element synthetic standard diluted from a High Purity Standards 1000 ppm Ca solution (ZeptoMetrix). The limit of detection (LOD) was 0.726 ppb (43Ca) and 1.075 ppb of 44Ca. Calibrations were forced through the blank, which was the acid matrix the samples and standards were diluted with (43Ca: f(x) = 168.1 + /- 2.7 + 149, r2 = 0.999; 44Ca: 2701.4 + /-30 + 23134, r2 = 0.999). Relative Standard Deviations (RSD) were calculated for each sample measurement. The ICP-MS revealed a nearly stoichiometric content of calcium in Net4ΔC and γ1ΔC, as indicated by the amounts of detected 43Ca/44Ca (163.1 ± 3.2 ng/170.2 ± 2.4 ng and 132.48 ± 2.33 ng/134.31 ± 1.35 ng, respectively). In contrast, α1ΔC revealed only a trace amount of both isotopes (19.54 ± 5.7 ng 43Ca; 25.55 ± 1.99 ng 44Ca).
Cryo-EM sample preparation, screening and data collection
Samples of Net4ΔC-γ1ΔC were prepared by mixing both proteins at the equimolar ratio in a buffer consisting of 90 mM NaCl, 50 mM Tris pH 7.4, and 125 μM EDTA at concentrations ranging from 60 μg/ml to 540 μg/ml. Excess calcium (1.5 mM CaCl2) was then added, and the protein mix was incubated for 10 min at room temperature prior to grid preparation. Samples were adsorbed onto freshly glow-discharged 300 mesh UltrAuFoil R1.2/1.3 (Millipore Sigma, TEM-Q350AR13A) or gold Quantifoil R2/1 (SPI Supplies, 4330G-CF) grids with PELCO easiGlow glow discharger and flash frozen in liquid ethane using either Vitrobot Mark IV dual-blotting plunger or Leica GP Climate controlled sample plunger with a controlled temperature and humidity. The screening and the data acquisition were performed at the Pacific Northwest Center for Cryo-EM (PNCC). The grids were extensively screened with a 200 kV Thermo Fisher Scientific Talos Arctica electron microscope equipped with a Gatan BioQuatum energy filter and Gatan K2 Summit direct electron detector using the software SerialEM 4.066 for automated data collection. Typical acquisition parameters were as follows: dose rate of 4.88 e-/px/s in a counting mode, magnification of 130,000 times corresponding to the pixel size of 1.037 Å/px with a defocus range −0.5 to −2.5 μm. We collected 32 frames with 250 ms per frame exposure, a total exposure of 8 s, and an accumulated dose of 39.04e−. The screening procedure also involved pre-processing, 2D classification, and class-averaging of the acquired data sets in cryoSPARC 4. Cryo-EM grids, in which most particles yielded 2D classes displaying different views of the complex (concentrations 120 μg/ml–180 μg/ml), were used for acquisition of six data sets at different stage tilts using a 300 kV Thermo Fisher Scientific Titan Krios equipped with an energy filter and Gatan K3 direct electron detector. We collected a total of 45,515 movies including the following data sets: no stage tilt (8594 movies), 100 stage tilt (5799), 200 stage tilt (7839), 250 stage tilt (6720), 300 stage tilt (8903), 400 stage tilt (7660). The data were acquired using software SerialEM 4.066 and the following acquisition parameters: dose rate of 18 e-/px/s in a super-resolution mode, magnification of 130,000 times corresponding to the pixel size of 0.324 Å/px with a defocus range -0.8 μm to -3 μm. Defoci varied depending on the stage tilt i.e. −0.8 to −2.2 μm for 100–300 tilts and no stage tilt data sets, and -1.5 μm to -3 μm for 400 stage tilts. We collected 30 frames with 0.6 – 0.8 electrons per pixel per frame, a total exposure of 1.2 s–1.4 s, and an accumulated dose of 50–52 e-/A2.
Structure determination
The structure of Net4ΔC-γ1ΔC was calculated using a combination of cryoSPARC 445, Relion 446, and Scipion 367. We employed two structure calculation strategies (Supplementary Fig. 1, 2) to systematically improve the map quality and resolution by iteratively minimizing effects of continuous structural heterogeneity and overcoming issues related to the presence of preferred orientations. The following description outlines details of the protocol that yielded final three maps deposited to the Electron Microscopy Data Bank (Supplementary Table 2). Data collected at different stage tilts (45,515 movies) were combined for structure calculations. Movies were patch motion- and patch CTF-corrected at the pixel size of 0.648 Å/px. Following inspection of pre-processed averaged micrographs, 4028 out of 44,518 micrographs were discarded from further processing. We manually picked 2000 particles from the selected micrographs to create 2D templates for automated particle picking. Particles were then automatically picked from a set of 40,490 curated and denoised micrographs using the template picker with templates representing a combination of not only predominant front and back views, but also less prevalent side and tilted views of the complex. The template picker automatically selected 2,388,081 particle images. Selected particles were inspected and extracted from averaged micrographs with a box size of 384 pixels and then subjected to Fourier cropping with the resultant pixel size of 1.296 Å/px. Particle stacks were subsequently subjected to five rounds of 2D classification and averaging. During the 2D analysis, artifacts and particles not converging to stable classes were removed from the data set. The resultant set of 1,263,630 particle images was then used for ab-initio modeling, followed by one round of homogenous refinement. To address the preferred orientation issue, we subsequently performed four rounds of iterative 2D rebalancing steps and non-uniform refinements during which the convergence of the protocol was monitored by assessing the conical FSC area ratio (cFAR)41,42, the sampling compensation factor (SCF)42,43 and the high average relative signal amount within azimuth-elevation viewing regions (relative signal)43. All three parameters are sensitive to the extent of sampling in Fourier space. During the protocol, cFAR values increased from 0.52 to 0.73, and then they stopped changing, indicative of the convergence, while the size of the stack of particle images was reduced to 254,953. The respective SCF and relative signal values of 0.947 and of 0.8-0.82 indicated sufficient Fourier sampling. Next, we minimized effects of continuous structural heterogeneity38 evidenced by the results of the 3D variability analysis. The set of 254,953 particles was used as an input to 3D flexible refinement44 (3D Flex) i.e. the deep-learning algorithm that employs graph neural networks for modeling flexible motions in biomolecules. In the following step, the resultant 3D map and the set of 254,000 particles were transferred to Relion 446 using the PyEM script (csparc2star.py) and then used as inputs for extensive 3D classification without alignment. A wide range of temperature (T) parameters for scaling the Bayesian likelihood function was tested, along with a variable number of calculated 3D classes. The best results were obtained with 5 classes. The 3D map and the particle stack representing the most populated class (71,890 particle images corresponding to 28.1 % of all particles) were then imported from Relion 446 to cryoSPARC 445, and subjected to an additional round of 3D Flex that resulted with the final 3D map (71,000 particle images). All 3D maps were calculated with the C1 symmetry. To further improve map quality, we systematically tested several different sharpening methods, including tools relying on global Guinier plot-based sharpening, either automatic (Relion PostProcessing, PhenixAutoSharpen)52 or manual (CryoSPARC sharpening tool)53, local automatic Wiener filter-based methods (LocalDeblur and CryoSPARC Local Resolution Estimation/Filtering)54, and the convolutional neural network-based algorithm (DeepEMhancer)55. We also performed map phasing with Phenix Density Modification48. The model was then refined against each of the sharpened maps using a combination of Phenix 1.2148 and COOT 1.149 (please see the following Methods section), and the map vs. model agreement was quantitatively assessed by calculating mean Q-scores in ChimeraX47. Best results were obtained by employing the global Guinier plot-based manual sharpening with the B-factor of -100 Å2 and by phasing map density in Phenix. Both procedures increased the mean Q-scores from 0.4 to 0.42. These values, calculated at the relevant sigma (σ = 0.6; for details, please see legend to Supplementary Table 2), reflect the Q-score values expected for a Coulomb map solved at the nominal resolution of ~3.9 Å56. The Q-score-based estimate of resolution is consistent with the calculated local resolutions ranging from 3.4 Å to 5.6 Å. We also calculated the nominal resolution of 3.64 Å in cryoSPARC 442. However, we think that this value might have been overestimated by the program, and the best estimate is provided by the range of local resolutions. The nominal resolution of the map is ~3.9 Å56, the fact reflected by mean Q-scores calculated in ChimeraX47, and consistent with the map vs. model FSC obtained in Phenix 1.2148 with 0.143 of 3.9 Å.
Model building
The molecular model was built and validated by employing three independent approaches described below, all of which yielded nearly identical solutions (Supplementary Table 3), namely: (i) a combination of COOT 1.149 and Phenix 1.2148 (ii) DeepMainmast50 (Supplementary Fig. 8), and (iii) AF251 (Supplementary Fig. 9). (i) The initial model of Net4ΔC-γ1ΔC was obtained by docking previously determined cryo-EM structure of γ1ΔC (PDB ID: 8DMK [https://www.wwpdb.org/pdb?id=pdb_ 00008dmk] 6 and the X-ray structure of Net4ΔC (PDB ID: 4WNX [https://www.wwpdb.org/pdb?id=pdb_ 00004wnx] 19 into the Coulomb density map using UCSF ChimeraX47. The model was inspected in COOT 1.149. The initial model of Net4ΔC-γ1ΔC was then iteratively refined using a combination of COOT 1.147 and Phenix 1.2148 with imposed NCS, secondary structure, and Ramachandran restrains. We systematically improved the quality of the model by addressing specific issues, such as for instance: interatomic clashes, violations of Ramachandran and other geometry restrains, including errors in sidechain rotamers. Each specific violation was addressed in COOT 1.1 between subsequent Phenix 1.21 refinement runs. To quantitatively assess the map vs. model agreement at each step of the refinement, we calculated mean Q-scores in ChimeraX47 and monitored the validation statistics using Phenix 1.21 (FSC model vs. map and the model vs. map cross-correlation coefficients calculated for each individual residue). Residues characterized by low Q-score values were first visually inspected in ChimeraX47 and COOT 1.149. Residues, which we were not able unambiguously assign to the map were then removed from the model. In this process, we removed from the model parts of the four surface loops (three in Net4ΔC and one in γ1ΔC), namely: Net4ΔC (G169-E175, G193-E195, P273-T285), and in Lm γ1ΔC (R195-E200, S313-P620), as well as parts of the N- and C-termini in both proteins. Although CaCl2 was added to the protein samples, we were not able to unambiguously assign Coulomb densities representing two calcium ions, hence they were omitted from the model. However, the ICP-MS revealed a nearly stoichiometric content of calcium in both Net4ΔC and γ1ΔC used to prepare cryo-EM samples. The refinement strategy outlined in this paragraph was carried out for forty iterations involving COOT 1.149 and Phenix 1.2148 until the Q-score values and Phenix validation statistics stopped improving. The final model displays many structural details, some of which are displayed in Fig. 2 including four N-glycosylated6,19 asparagine residues (N56 and N163 in Net4ΔC, along with N60 and N134 in γ1ΔC). The four specific asparagine residues colocalize with four unique Coulomb map densities representing N-glycans. This fact confirms the correctness of the model, as such a scenario would not be possible in any other arrangement of protein subunits within the cryo-EM map. Consequently, seven N-Acetylglucosamine (NAG) moieties were added to the model (Fig. 1c). The final model was validated using MolProbity 4.02, and it displays good validation statistics presented in the Supplementary Table 1. (ii) In the second method, we employed DeepMainmast50, an integrated deep learning pipeline with the U-shaped network architecture (UNet3 + ) for unsupervised de novo model building from the Coulomb map.
The following parameters were used for model building: path training time (t = 6000), fragment assembly time (T = 6000), number of CPUs (C = 20), number of threads (M = 20). In addition, the full-atom building and refinement were carried out using PULCHRA68 and Rosetta-CM69. The model obtained in DeepMainmast50, and the one built using a combination of COOT 1.149 and Phenix 1.2146 are nearly identical (Supplementary Fig. 8). The RMSD values (Supplementary Table 3) calculated between the two models are: γ1ΔC (1 Å for 217 out of 257 pruned residues involving the interacting regions from the LN domain, and 1.6 Å for all atoms), Net4ΔC (1.2 Å for 133 out of 260 pruned residues that also involve the interacting regions from the LN domain, and 3.9 Å for all atoms consisting of the LEa1 domain and the tip of the LN domain that are more heterogenous). (iii) We also employed AlphaFold251 for structure prediction of the Net4ΔC-γ1ΔC complex. We used ColabFold: AlphaFold2 equipped with the sequence search module MMseq2. Structural modeling was run using “Mmseqs2_unirev_env” and “unpaired_paired” pairing strategy with a combination of pair sequences from the same species and unpaired MSA. The paired MSA was executed by searching the UniRef100 database and by pairing the best hits sharing the same NCBI taxonomic identifiers. The unpaired MSA was executed by searching the UniRef100 database and the environmental sequences. Five models were generated. The final structural models were relaxed using Amber2470 and ranked by pLDDT. The models obtained using AlphaFold251, and a combination of COOT 1.147 and Phenix 1.2146 are nearly identical (Supplementary Fig. 9). The RMSD values (Supplementary Table 3) calculated between the two models are: γ1ΔC (1.2 Å for 162 out of 257 pruned residues involving the interacting regions from the LN domain, and 2.4 Å for all atoms from 257 residues), Net4ΔC (1.2 Å for 132 out of 260 pruned residues that also involve the interacting regions from the LN domain, and 2.8 Å for all atoms consisting of the LEa1 domain and the tip of the LN domain that are more heterogenous, Supplementary Movie 1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under the accession code EMD-44008. The unsharpened map of Net4ΔC-γ1ΔC was deposited as the main entry, along with two accessory maps, namely the sharpened map and the density-modified map. The atomic coordinates of Net4ΔC-γ1ΔC have been deposited in the Protein Data Bank (PDB) under the accession code 9AZ3. Atomic coordinates of previously determined cryo-EM and X-ray structures used in this study are available in the PDB under the following accession codes: 8DMK (Lm polymer node), [https://www.wwpdb.org/pdb?id=pdb_00008dmk]6, and 4WNX (Net4ΔC), [https://www.wwpdb.org/pdb?id=pdb_00004wnx]19. The source data underlying Fig. 4a, g, h, and Supplementary Fig. 4a, 10a–c, 14, 15a, b are provided as a Source Data file. Source data are provided with this paper.
References
Yurchenco, P. D. & Kulczyk, A. W. Polymerizing laminins in development, health and disease. J. Biol. Chem. 300, 107429–107449 (2024).
Yurchenco, P. D. & Patton, B. L. Developmental and pathogenic mechanisms of basement membrane assembly. Curr. Pharm. Des. 15, 1277–1294 (2009).
Yurchenco, P. D. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 3, a004911 (2011).
Yurchenco, P. D., Tsilibary, E. C., Charonis, A. S. & Furthmayr, H. Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity. J. Biol. Chem. 260, 7636–7644 (1985).
Yurchenco, P. D., Cheng, Y. S. & Colognato, H. Laminin forms an independent network in basement membranes. J. Cell. Biol. 117, 1119–33 (1992).
Kulczyk, A. W. et al. Cryo-EM reveals the molecular basis of laminin polymerization and LN-lamininopathies. Nat. Commun. 14, 317–324 (2023).
Kulczyk, A. W. Artificial intelligence and the analysis of cryo-EM data provide structural insight into the molecular mechanisms underlying LN-lamininopathies. Sci. Rep. 13, 17825 (2023).
Yurchenco, P. D. Integrating activities of laminins that drive basement membrane assembly and function. Curr. Top. Membr. 76, 1–30 (2015).
Macdonald, P. R., Lustig, A., Steinmetz, M. O. & Kammerer, R. A. Laminin chain assembly is regulated by specific coiled-coil interactions. J. Struct. Biol. 170, 398–405 (2010).
Hussain, S. A., Carafoli, F. & Hohenester, E. Determinants of laminin polymerization revealed by the structure of the alpha5 chain amino-terminal region. EMBO Rep. 12, 276–82 (2011).
Carafoli, F., Hussain, S. A. & Hohenester, E. Crystal structures of the network-forming short-arm tips of the laminin beta1 and gamma1 chains. PLoS One 7, e42473 (2012).
Burgeson, R. E. et al. A new nomenclature for the laminins. Matrix Biol. 14, 209–211 (1994).
Yurchenco, P. D. & Cheng, Y. S. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J. Biol. Chem. 268, 17286–17299 (1993).
McKee, K. K., Harrison, D., Capizzi, S. & Yurchenco, P. D. Role of laminin terminal globular domains in basement membrane assembly. J. Biol. Chem. 282, 21437–47 (2007).
Hohenester, E. & Yurchenco, P. D. Laminins in basement membrane assembly. Cell Adh. Migr. 7, 56–63 (2013).
Sun, Z., Guo, S. S. & Fassler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 215, 445–456 (2016).
Jayadev, R. & Sherwood, D. R. Basement membranes. Curr. Biol. 27, R207–R211 (2017).
Leclech, C., Natale, C.F. & Barakat, A.I. The basement membrane as a structured surface - role in vascular health and disease. J. Cell. Sci. 133, jcs239889(2020).
Reuten, R. et al. Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes. Nat. Commun. 7, 13515 (2016).
Finci, L. I. et al. The crystal structure of netrin-1 in complex with DCC reveals the bifunctionality of netrin-1 as a guidance cue. Neuron 83, 839–849 (2014).
Xu, K. et al. Neural migration. Structures of netrin-1 bound to two receptors provide insight into its axon guidance mechanism. Science 344, 1275–9 (2014).
Grandin, M. et al. Structural decoding of the netrin-1/UNC5 interaction and its therapeutical implications in cancers. Cancer Cell 29, 173–85 (2016).
Eveno, C. et al. Netrin-4 delays colorectal cancer carcinomatosis by inhibiting tumor angiogenesis. Am. J. Pathol. 178, 1861–9 (2011).
Lejmi, E. et al. Netrin-4 inhibits angiogenesis via binding to neogenin and recruitment of Unc5B. Proc. Natl. Acad. Sci. USA 105, 12491–6 (2008).
Nacht, M. et al. Netrin-4 regulates angiogenic responses and tumor cell growth. Exp. Cell Res. 315, 784–94 (2009).
Larrieu-Lahargue, F., Welm, A. L., Thomas, K. R. & Li, D. Y. Netrin-4 induces lymphangiogenesis in vivo. Blood 115, 5418–26 (2010).
Reuten, R. et al. Basement membrane stiffness determines metastases formation. Nat. Mater. 20, 892–903 (2021).
Chaudhuri, O. et al. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 13, 970–8 (2014).
Esseghir, S. et al. Identification of NTN4, TRA1, and STC2 as prognostic markers in breast cancer in a screen for signal sequence encoding proteins. Clin. Cancer Res. 13, 3164–73 (2007).
Banyai, L. & Patthy, L. The NTR module: domains of netrins, secreted frizzled related proteins, and type I procollagen C-proteinase enhancer protein are homologous with tissue inhibitors of metalloproteases. Protein Sci. 8, 1636–42 (1999).
Schneiders, F. I. et al. Binding of netrin-4 to laminin short arms regulates basement membrane assembly. J. Biol. Chem. 282, 23750–8 (2007).
Staquicini, F. I. et al. Discovery of a functional protein complex of netrin-4, laminin gamma1 chain, and integrin alpha6beta1 in mouse neural stem cells. Proc. Natl. Acad. Sci. USA 106, 2903–8 (2009).
Kulczyk, A. W. Cryo-electron microscopy studies of biomolecular structure and dynamics. Micromachines 15, 1092–1095 (2024).
Yang, C. Y. et al. Deciphering DED assembly mechanisms in FADD-procaspase-8-cFLIP complexes regulating apoptosis. Nat. Commun. 15, 3791 (2024).
Kulczyk, A. W. et al. Cryo-EM structure of Shiga toxin 2 in complex with the native ribosomal P-stalk reveals residues involved in the binding interaction. J. Biol. Chem. 299, 102795 (2022).
Kulczyk, A. W., Moeller, A., Meyer, P., Sliz, P. & Richardson, C. C. Cryo-EM structure of the replisome reveals multiple interactions coordinating DNA synthesis. Proc. Natl. Acad. Sci. USA 114, E1848–E1856 (2017).
DiIorio, C. & Kulczyk, A. W. Novel artificial intelligence-based approaches for ab initio structure determination and atomic model building for cryo-electron microscopy. Micromachines 14, 1674–1700 (2023).
DiIorio, C. & Kulczyk, A. W. Exploring the structural variability of dynamic biological complexes by single-particle cryo-electron microscopy. Micromachines 14, 118–142 (2023).
DiIorio, C. & Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-EM) processing workflow with cryoSPARC, RELION and Scipion. J. Vis. Exp. 179, e63387 (2022).
Burley, S.K. et al. Electron microscopy holdings of the Protein Data Bank: the impact of the resolution revolution, new validation tools, and implications for the future. Biophys. Rev. 1-21 (2022).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Baldwin, P. R. & Lyumkis, D. Non-uniformity of projection distributions attenuates resolution in Cryo-EM. Prog. Biophys. Mol. Biol. 150, 160–183 (2020).
Baldwin, P. R. & Lyumkis, D. Tools for visualizing and analyzing Fourier space sampling in Cryo-EM. Prog. Biophys. Mol. Biol. 160, 53–65 (2021).
Punjani, A. & Fleet, D. J. 3DFlex: determining structure and motion of flexible proteins from cryo-EM. Nat. Methods 20, 860–870 (2023).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 11, e83724 (2022).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sc. i 30, 70–82 (2021).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–21 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Terashi, G., Wang, X., Prasad, D., Nakamura, T. & Kihara, D. DeepMainmast: integrated protocol of protein structure modeling for cryo-EM with deep learning and structure prediction. Nat. Methods 21, 122–131 (2024).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Terwilliger, T. C., Sobolev, O. V., Afonine, P. V. & Adams, P. D. Automated map sharpening by maximization of detail and connectivity. Acta Crystallogr. D. Struct. Biol. 74, 545–559 (2018).
Rosenthal, P. B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–45 (2003).
Ramirez-Aportela, E. et al. Automatic local resolution-based sharpening of cryo-EM maps. Bioinformatics 36, 765–772 (2020).
Sanchez-Garcia, R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021).
Pintilie, G. et al. Measurement of atom resolvability in cryo-EM maps with Q-scores. Nat. Methods 17, 328–334 (2020).
Yurchenco, P. D., Tsilibary, E. C., Charonis, A. S. & Furthmayr, H. Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity. J. Biol. Chem. 260, 7636–44 (1985).
McKee, K. K., Hohenester, E., Aleksandrova, M. & Yurchenco, P. D. Organization of the laminin polymer node. Matrix Biol. 98, 49–63 (2021).
Purvis, A. & Hohenester, E. Laminin network formation studied by reconstitution of ternary nodes in solution. J. Biol. Chem. 287, 44270–7 (2012).
Paulsson, M. et al. Laminin-nidogen complex. Extraction with chelating agents and structural characterization. Eur. J. Biochem. 166, 11–19 (1987).
Briggs, D. C. et al. Structural basis of laminin binding to the LARGE glycans on dystroglycan. Nat. Chem. Biol. 12, 810–4 (2016).
Arimori, T. et al. Structural mechanism of laminin recognition by integrin. Nat. Commun. 12, 4012 (2021).
Engel, J. et al. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J. Mol. Biol. 150, 97–120 (1981).
Timpl, R. et al. Laminin-a glycoprotein from basement membranes. J. Biol. Chem. 254, 9933–7 (1979).
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Sorzano, C. O. S. et al. A new algorithm for high-resolution reconstruction of single particles by electron microscopy. J. Struct. Biol. 204, 329–337 (2018).
Rotkiewicz, P. & Skolnick, J. Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 29, 1460–5 (2008).
Song, Y. et al. High-resolution comparative modeling with RosettaCM. Structure 21, 1735–42 (2013).
Case, D. A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183–6191 (2023).
Acknowledgements
We thank Linda Godfrey (Department of Earth and Planetary Sciences MS facility at Rutgers) for acquiring the ICP-MS data. We thank Maria Voigt (RCSB PDB) for help in preparing materials for Fig. 5. We thank Iwona Bizukojc (cryo-EMcorp and Waters Corporation) for reading of the manuscript. We are grateful to Marcelo de Farias, Craig Yoshioka, and Claudia Lopez for their assistance in data collection at the PNCC. A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. This study was supported by a PNCC-51312 grant to A.W.K., a NIH grant R01-DK36425 to P.D.Y., and a start-up grant from Rutgers University to A.W.K.
Author information
Authors and Affiliations
Contributions
A.W.K. conceptualized the Net4ΔC-γ1ΔC project along with P.D.Y. and K.K.M. A.W.K. conceptualized the cryo-EM project, collected and analyzed cryo-EM data, calculated the structure, built and refined the structural models, analyzed data, wrote the original manuscript, and revised the manuscript with input from K.K.M and P.D.Y. K.K.M. prepared the Net4ΔC construct, expressed and purified the protein, performed γ1ΔC mutagenesis, Lm polymerization and Schwann cells assays, and conducted the solid-phase binding analysis. P.D.Y. performed negative-stain EM analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kevin Campbell who co-reviewed with Soumya Joseph, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kulczyk, A.W., McKee, K.K. & Yurchenco, P.D. Cryo-EM reveals molecular mechanisms underlying the inhibitory effect of netrin-4 on laminin matrix formation. Nat Commun 16, 7256 (2025). https://doi.org/10.1038/s41467-025-62814-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-62814-7
