
Abstract
The cerebellum is critical for motor timing control and error-driven motor learning. To reveal how the cerebellum transmits these process-relevant signals to the premotor cortex, we conducted two-photon calcium imaging of cerebellar-thalamocortical axons in the premotor cortex in male mice during a self-timing lever-pull task that required 1–1.7 s of waiting after cue onset. In non-expert sessions with many lever-pulls being made before the 1-s waiting, the axons of thalamic neurons that received cerebellar outputs exhibited larger transient activity immediately after the cue onset in post-error (i.e., post-non-rewarded) trials than in post-success trials, and the waiting time and success rate were greater in post-error trials than in post-success trials. In expert sessions, the post-error-specific activity or behavior was absent. Instead, ramping activity toward lever-pull onset that did not depend on the waiting time shortened in expert sessions in comparison with non-expert sessions. Our results suggest that the cerebellum emits the reward-based post-error signal for waiting time adjustment during learning, and the well-tuned motor timing signal after learning.
Similar content being viewed by others

Introduction
Whatever movement we execute, when to start it is as critical as how to perform it. For motor timing control, passing of time from a sensory cue presentation should be monitored by the brain, and motor preparation should start before the appropriate motor initiation timing. The cerebellum and basal ganglia (BG) are assumed to be crucial for monitoring elapsed time in sub-seconds and supra-seconds, respectively1,2,3,4,5,6,7,8,9. The cerebellum is required for learning of the eyeblink reflex with sub-second interval timing10,11. Temporal discrimination (e.g., perceptual timing) over sub-second intervals also requires the cerebellar cortex12. In a self-timing saccade task in the macaque, the striatum showed ramping activity that occurred immediately after the cue presentation, and its slope became slower as the waiting time increased6. By contrast, the dentate nucleus (DN), which is the lateral nucleus of the deep cerebellar nuclei (DCN), shows sharp ramping activity immediately before the saccade onset and its slope does not depend on the waiting time6,13. Inactivation of DN was shown to increase the latency of the self-timing saccade6, and in accord with this, motor timing is impaired in patients with DN lesions14,15. The activity of the lateral cerebellum becomes synchronized as the motor initiation timing stabilizes16,17. Thus, control of precise motor timing is one of the major functions of the cerebellum.
Cerebellar and BG outputs project to the motor thalamus, and neurons in the motor thalamus project their axons to the motor cortex18,19,20,21. Inactivation of the motor thalamus delays self-timing22,23. The cerebellar-thalamus-cortical pathway synchronizes motor cortical activity, and perturbation of it disrupts motor timing24. For duration estimation, the premotor cortex, supplementary motor area (SMA), and cerebro-cerebellar interaction are also important9,25,26,27. The rodent secondary motor cortex (M2), which is a homolog of the primate premotor cortex and SMA, is a crucial area for preparation and initiation of well-learned goal-directed movements28,29,30,31. However, it remains unclear whether the timing-related activity in the cerebellum is transmitted to M2 through the motor thalamus, and how it emerges during learning.
The cerebellum is also crucial for motor learning based on error signals32,33,34,35. Strong error-related signals are observed during learning, but as proficiency improves, these signals decrease36. Recently, reward-related and reward expectation-related activities were found in the cerebellum37,38, suggesting that not only sensory prediction error signals, but also reward prediction error signals, are involved in motor learning in the cerebellum33,38,39,40. However, it is unclear whether these error-related signals occur in learning of motor timing control and are transmitted to M2 through the motor thalamus, and how signals regarding failure in a trial are utilized in subsequent trials.
Here, we developed a self-timing lever-pull task for head-fixed mice and conducted two-photon calcium imaging of thalamocortical axons in layer 1 (L1) of M2 during performance of the task. We used the anterograde-tracing property of adeno-associated virus (AAV) serotype 1 (AAV1)41,42 to specifically label the axons of thalamic neurons that received outputs from the lateral part of the DCN (LCN), which included DN and a part of the interposed nucleus. We also imaged layer 5 (L5) neurons in M2. Our results suggest that in the early phase of learning, the LCN transmits post-error-specific signals to M2 at the start of the elapsed time monitoring to change the following M2 L5 population activity, thereby increasing the waiting time and success rate.
Results
Both the DCN and M2 are related to self-timing control
We developed a self-timing lever-pull task that required motor timing control based on a sensory input (Fig. 1a). To obtain a water reward, head-fixed mice needed to wait for 1–1.71 s after an auditory cue (10 kHz continuous sound) presentation started, and then pull a lever. Since there was no sign indicating 1 s, the mouse needed to estimate the elapsed time before initiating the lever pull. If the lever was pulled within 1.71 s, the cue sound disappeared at the lever pull; otherwise, the cue sound disappeared at 1.71 s from the cue onset. When the lever was pulled more than 1 s after the cue onset, the cue in the next trial was presented at least 1.5 s after the lever was returned to the natural position. If the lever was pulled less than 1 s after the cue onset (early pull), timing of the next cue presentation was extended as a penalty (see Methods for details). Before the self-timing task started, the mice underwent training to wait before pulling, which was performed in three stages: the first stage was to pull the lever in response to a go cue (300-ms pink noise) sound without waiting (simple go cue task), the second stage was to pull the lever in response to a go-cue sound that was presented 1 s or 1.35 s after the cue sound started, and the third stage was to pull the lever 1–1.71 s after the cue onset with increasing rates of omission of the go-cue sound presentation (Fig. 1b and Supplementary Fig. 1a–e, see Methods for details). In the self-timing task, pull latency (the time from the cue onset to the lever-pull onset) showed a distribution with a large peak around 1 s and a small peak around 0.2 s (Fig. 1b, c). The proportion of trials with a pull latency of less than 500 ms was 6.6%, and these trials probably reflected an immediate lever-pull response to the cue sound without waiting. In fact, this proportion decreased during the second and third stages of training (Supplementary Fig. 1d).

a Schematic of the self-timing lever-pull task, which required mice to wait for 1–1.71 s before the lever pull. b Lever trajectories in the fourth session (simple go-cue task) and twentieth session (self-timing task) in a representative mouse. Black lines show the mean lever trajectories and gray lines show all individual trajectories (the first 50 trials in each session). c Distribution of the pull latencies in the self-timing task in mice that were used for two-photon calcium imaging of thalamocortical axons (5069 trials from 8 mice). The latency was 1115.7 ± 6.2 ms (mean ± SEM). The number of non-pull trials (429, 8.46% of all trials) is shown on the right. d Schematic of inhibition of the LCN (roughly the area indicated by the oval) using muscimol. ACSF on the first day and muscimol on the second day were injected before the task performance. DN dentate nucleus, IN interposed nucleus, FN fastigial nucleus. e, f Pull latency (e) and pull rate (f) in the sessions in which ACSF or muscimol was administered to the LCN (n = 7 mice). Black lines indicate mean ± SEM. Pull latency, *P = 0.0156; pull rate, P = 0.156, Wilcoxon signed-rank test, two-sided. g Schematic of the inhibition of M2 or M1. ACSF on the first day and muscimol on the second day was injected into M2 before the task performance started, and the same procedure was repeated for the M1 injection on the third and fourth days. h Pull rate in the sessions in which ACSF or muscimol was administered to M2 (left) and M1 (right). Black lines indicate mean ± SEM. M2, *P = 0.0117, n = 9 mice; M1, P = 1, n = 7 mice, Wilcoxon signed-rank test, two-sided. Source data are provided as a Source Data file.
To investigate whether the cerebellum contributes to motor timing control, we inhibited the activity of the LCN by injecting 10 ng of muscimol (a low dose for LCN inhibition) 60 minutes before the self-timing task started (Fig. 1d and Supplementary Fig. 2a, b). The pull latency was significantly longer in the muscimol-injected session than in an artificial cerebrospinal fluid (ACSF)-injected session (Fig. 1e). The pull rate (the number of trials with lever-pulls within 3 s after the cue onset over the total number of trials) tended to show a decrease in the muscimol-injected session, but the difference was not significant (Fig. 1f).
Given that motor timing control is necessary for motor initiation that requires M2, we hypothesized that the signals controlling the motor timing would be mainly transmitted to M2, rather than to the primary motor cortex (M1). To test this, we injected 100 ng of muscimol (a low dose for M2 or M1 inhibition28) into either M2 or M1 (Fig. 1g and Supplementary Fig. 2c). In the current study, we refer to the rostral forelimb area as M2 and the caudal forelimb area as M128,43,44. When M2 was weakly inhibited, the pull latency was longer than when ACSF was injected into M2, and the difference in the pull rate was not significant although the pull rate tended to decrease (Fig. 1h and Supplementary Fig. 2d). By contrast, when M1 was weakly inhibited, the pull latency did not change, although the pull rate decreased (Fig. 1h and Supplementary Fig. 2d). These results suggest that LCN and M2 are important for timing control in the self-timing lever-pull task.
It is possible that the delay in the latency observed in the weak LCN inhibition reflected impairment in the lever-pull execution, rather than impairment in the timing control for pull initiation. Therefore, we examined the effect of the LCN inactivation on performance in the simple go-cue task (Supplementary Fig. 3a–d). When 100 ng of muscimol (a high dose for LCN inhibition) was used, the lever pull was largely inhibited (Supplementary Fig. 3e). Thus, the sound-triggered lever-pull movement requires the LCN activity, even without the self-timing control. When 10 ng of muscimol (a low dose for LCN inhibition) was injected as in the self-timing task, neither the pull rate nor the reaction time (from the go-cue onset to the lever-pull onset) differed significantly between the ACSF and muscimol sessions (Supplementary Fig. 3f, g). The reaction time delay induced by the weak inhibition of LCN was much shorter than the delay in the pull latency in the self-timing task (Supplementary Fig. 3h). These results suggest that the weak inhibition of LCN largely affected the timing control of motor initiation, rather than motor execution.
Anatomical pathways from LCN to M2 through the thalamus
Next, we examined anatomical pathways from LCN to M2 through the thalamus. First, we introduced an AAV that expresses green fluorescent protein (GFP) into the LCN. By examining axonal GFP expression in the thalamus, we confirmed that the LCN neurons projected to the ventromedial nucleus (VM) and ventrolateral nucleus (VL), which are included in the motor thalamus (Supplementary Fig. 4a, b). Second, we injected AAV1-human Synapsin1 promotor (syn)-Cre41 into the LCN in Ai162D mice in which GFP expression is Cre-dependent45. Subsets of VM and VL neurons expressed GFP, indicating that these neurons received synaptic inputs from the LCN (Supplementary Fig. 4c). By contrast, when AAV1-syn-Cre was injected into the substantia nigra par reticulata (SNr) in Ai162D mice, GFP was expressed in VM neurons, but not VL neurons (Supplementary Fig. 4d). Thus, in the VM, although the neurons that receive SNr outputs are dominant20,21, there are a subset of neurons that convey LCN signals as previously demonstrated21. Third, to investigate whether the LCN projects to M2 via the VM and VL, we injected AAV1-syn-Cre into the LCN, and AAV-CAG-flex-GFP into the VM or VL (Fig. 2a and Supplementary Fig. 4e). In this case, VM and VL neurons that incorporated both types of AAVs should express GFP via a Cre-dependent FLEx switch41,42. We found that GFP-expressing VM and VL neurons mainly projected to L1 and deep layers in M2, respectively (Fig. 2b and Supplementary Fig. 4e, f). The distinct cortical layers of VM and VL projections are also consistent with other anatomical studies18,20,46.

a Schematic of transsynaptic labeling of mThLCN→M2 axons by injections of AAV1-syn-Cre into LCN (roughly the area indicated by the oval) and AAV-CAG-flex-GFP into VM. IN, interposed nucleus; FN, fastigial nucleus. b Cre-positive DCN, GFP-positive VM, GFP-positive M2, and GFP fluorescence profile along the cortical depth direction. The second-to-right panel corresponds to the yellow boxed area in the middle panel. Scale bars from left to right, 200 μm, 400 μm, 400 μm, and 200 μm. c Schematic of two-photon calcium imaging of mThLCN→M2 axons (top) and a representative image (bottom). We repeated this experiment 42 times from eight mice. d Examples of motion-corrected calcium traces (ΔF/F) in the five regions of interest (ROIs) shown in (c). The lever trajectory is shown at the top (Lever). e Successful trial-averaged activities of mThLCN→M2 axons aligned to the cue onset (left) and lever-pull onset (right) during the self-timing task. Topmost: pseudo-colored normalized trial-averaged activity of each active axonal bouton, ordered by the time of peak activity. Second to top, mean activity of all boutons. Second to bottom, averaged lever trajectory. Bottommost, lick rate. Shading indicates ± SEM. n = 42 sessions from 8 mice. f Histogram of pairwise correlation coefficients in the activity between axonal boutons in mThLCN→M2 axons. The pairs of boutons whose correlation coefficients were above the points indicated by the arrows were assumed to originate from the same axons. n = 128 708 pairs from 42 sessions from 8 mice. g, h Top, calcium traces of two representative axonal boutons. g A cue-preferring axon and its traces aligned to the cue onset. h A pull-preferring axon and its traces aligned to the pull onset. Trials were divided into three groups according to the pull latency (green, 700–1000 ms; blue, 1000–1300 ms; purple, 1300–1600 ms). Bottom, the corresponding lever trajectories. i Proportions of cue-preferring and pull-preferring axons and the axons that were classified into both types (n = 42 sessions). The mean ± SEM are also shown. Source data are provided as a Source Data file.
Cue- and pull-related activities are transmitted from LCN to M2 through the thalamus
To detect the activity of cerebellar-thalamocortical axons, we induced expression of a genetically encoded calcium indicator (GECI), jGCaMP7f 47, in thalamic neurons that received projections from the LCN. This was done by introducing AAV1-syn-Cre into the LCN and AAV1-syn-flex-jGCaMP7f into the motor thalamus (Fig. 2c and Supplementary Fig. 4g). Then, we conducted two-photon calcium imaging of jGCaMP7f-expressing axons in M2 L1 (mThLCN→M2 axons) during the self-timing lever-pull task (Fig. 2c). We conducted off-line motion correction20,42 and removed the imaging sessions that still showed large frame-by-frame displacements even after the correction (Supplementary Fig. 5a, b). Then, we extracted active putative presynaptic boutons, and we refer to their calcium transients as the axonal activity (Fig. 2d, e). Boutons showing high correlation were combined into one, as in our previous study (Fig. 2f)20,42. We analyzed the axonal imaging data from 42 sessions in 8 mice and focused on the axonal activity from the cue onset to the lever-pull onset, which should reflect signals related to the motor timing control.
The axon-averaged activity showed a first transient peak immediately after the cue onset and ramped up toward the lever-pull onset (Fig. 2e). We defined the axons with peak activity around the cue onset in successful trials as cue-preferring axons, and their activity 0.2 s after the cue onset as cue activity (Fig. 2g). In addition, we defined the axons with peak activity around the lever-pull onset in successful trials as pull-preferring axons, and their activity at pull onset as pull activity (Fig. 2h). The pull-preferring axons showed ramping activity toward the lever pull. The proportions of cue-preferring and pull-preferring axons were approximately 10% and 25%, respectively, with only approximately 2% of axons being classified as both (Fig. 2i).
Ramping activity of mThLCN→M2 axons did not depend on the pull latency
Cue-preferring and pull-preferring axons showed peak activity at the beginning and end of the waiting, respectively, and the latter showed ramping activity. Therefore, we hypothesized that these activities were related to the pull latency. First, we examined the relationships between these axonal activities and the pull latency in the same trials. To reveal whether the activity of these axons depended on the pull latency, trials with a pull latency of 700–1600 ms (pull trials) were divided into three groups with different latencies: 700–1000 ms, 1000–1300 ms, and 1300–1600 ms (Fig. 3a). The activity of cue-preferring axons was similar between the three groups (Fig. 3b). The starting points of the activity of pull-preferring axons apparently differed according to the pull latency (Fig. 3c). However, when the activity of pull-preferring axons was sorted according to the lever-pull onset, the activity pattern was similar among the three latency groups (Fig. 3c). The cue activity and the activity of cue-preferring axons 0.5 s after the cue onset (cue+0.5s activity) did not differ between the three groups (Fig. 3d). The pull activity and the activity of pull-preferring axons 0.3 s before the pull (pull–0.3s activity) did not differ largely between the three groups (Fig. 3d). This indicates that the activity of cue-preferring and pull-preferring mThLCN→M2 axons did not represent the pull latency in that trial.

a–c Trial-averaged calcium traces of all axons (a), cue-preferring axons (b), and pull-preferring axons (c) aligned to the cue onset (left) and lever-pull onset (right). The corresponding trial-averaged lever trajectories are also shown. For each session, trials were divided into three groups according to the pull latency (green, 700–1000 ms; blue, 1000–1300 ms; purple, 1300–1600 ms). Shading indicates ± SEM. n = 42 (a), 34 (b), and 41 (c). d Cue, cue+0.5s, pull–0.3s, and pull activities. Trials were divided into three groups according to the pull latency (700–1000 ms, green; 1000–1300 ms, blue; 1300–1600 ms, purple). The mean ± SEM are also shown. n = 34 (left) and 41 (right). For each type of activity, the activity was compared using Wilcoxon signed-rank tests with Bonferroni correction (α = 0.0167), two-sided. **P = 3.40 × 10–4 (n = 41 sessions). e Cue, pull–0.3s, and pull activities in four groups of trials with different pull latencies (cyan, 500–1000 ms; magenta, 1000–1710 ms; dark gray, 2000–3000 ms) and without pulling (light gray). The mean ± SEM are also shown. Sessions with at least one of each of the four types of trials were used (n = 18 for cue-preferring axons and n = 23 for pull-preferring axons). For each type of activity, the activity was compared between the groups of trials using Wilcoxon signed-rank tests with Bonferroni correction (α = 0.00833 for cue-preferring axons and 0.0167 for pull-preferring axons), two-sided. Cue activity, *P = 0.00285 and **P = 0.00159. Pull activity, *P = 0.00350 and **P = 0.00192. f Schematic of the time courses of the combined activity of cue-preferring and pull-preferring axons in five groups of trials with different pull latencies (green, 700–1000 ms; blue, 1000–1300 ms; purple, 1300–1600 ms; dark gray, 2000–3000 ms) and with no pulling (light gray). Source data are provided as a Source Data file.
We also compared the activity of cue-preferring and pull-preferring axons between non-rewarded trials with a pull latency of 0.5–1 s, rewarded trials (pull latency of 1–1.71 s), non-rewarded trials with a pull latency of 2–3 s (late pulls), and non-pull trials (Fig. 3e). The cue activity was substantially smaller in the late-pull trials and non-pull trials than in the rewarded trials. The pull activity in the late-pull trials was smaller than in the trials with a pull latency of 0.5–1 s and the rewarded trials (Fig. 3e). Thus, in the late-pull trials, both cue and pull activities of the mThLCN→M2 axons were weak. These differences between the trials with different lever-pull latencies were not detected in the lick rate (Supplementary Fig. 6a, b). In addition, there was no significant difference in the lick rate between 0.4–0.2 s before the pull onset and 0.1 s before to 0.1 s after the lever-pull onset (Supplementary Fig. 6b). Thus, the activity of cue-preferring and pull-preferring axons did not primarily reflect the lick rate. Substantially lower cue activity in mThLCN→M2 axons in the late-pull trials might be related to a slow down to reach the threshold of M2 activity for pull initiation, and in some trials, the lever might not be pulled (Fig. 3f). This might explain the results of the weak inhibition of LCN and M2 (Fig. 1e, h and Supplementary Fig. 2d).
Ramping activity of mThLCN→M2 axons is shortened from non-expert to expert sessions
Next, we examined whether the cue and pull activities depended on the learning level. In the second and third stages of the training sessions, the mice needed to learn to wait sufficiently long (>1 s) without making an early pull. Therefore, we used the early-pull rate as an appropriate indicator of the learning level in the self-timing task. The imaging sessions for mThLCN→M2 axons were classified into three types according to the early-pull rate: those with a high early-pull rate (more than the 67th percentile; H sessions), those with a middle early-pull rate (33rd–67th percentiles; M sessions), and those with a low early-pull rate (less than the 33rd percentile; L sessions) (Fig. 4a). The H sessions showed shorter pull latencies than the M and L sessions, and showed lower success rates than the L sessions (Fig. 4b). The trial-to-trial correlation of the lever-pull trajectory did not significantly differ between these sessions, although it did tend to increase from H to L sessions (Fig. 4b). We also refer to H and L sessions as non-expert and expert sessions, respectively, in the self-timing task.

a, b Early-pull rate (a), pull latency (left in b), success rate (middle in b), and lever trajectory correlation (right in b) in 14 H, 14 M, and 14 L sessions (from eight mice). The same color indicates sessions from the same mouse. Bars indicate mean ± SEM. ***P = 7.38 × 10–6 between H and M sessions, 7.38 × 10–6 between H and L sessions, and 7.47 × 10–6 between M and L sessions for early-pull rate, ***P = 1.50 × 10–4 between H and M sessions, 7.47 × 10–6 between H and L sessions, and 5.81 × 10–5 between M and L sessions for pull latency, ***P = 9.25 × 10–6 between H and L sessions and 5.80 × 10–5 between M and L sessions for early-pull rate, Wilcoxon rank sum test with Bonferroni correction (α = 0.0167), two-sided. c Proportions of cue-preferring and pull-preferring axons, and axons that were classified into both types. The mean ± SEM are also shown. In two-sided Wilcoxon rank sum tests with Bonferroni correction (α = 0.0167), no significant differences were detected for each type. n = 14 for H, M, and L sessions. d Cue, pull–0.3s, and pull activities. The mean ± SEM are also shown. Cue activity: n = 12, 12, and 10 for H, M, and L sessions, respectively. Pull–0.3s and pull activity: n = 13, 14, and 14 for H, M, and L sessions, respectively. In pull–0.3s activity, ++P = 0.00244 and +++P = 0.000244 in comparison with zero, Wilcoxon signed-rank tests with Bonferroni correction (α = 0.0167), two-sided. e Cue and pull–0.3s activities in trials grouped by pull latency (green, 700–1000 ms; blue, 1000–1300 ms; purple, 1300–1600 ms). The mean ± SEM are also shown. Wilcoxon signed-rank tests with Bonferroni correction (α = 0.0167, two-sided) and Wilcoxon rank sum tests with Bonferroni correction (α = 0.0167, two-sided) were used for comparisons between trials with different pull latencies and between different sessions, respectively. In cue activity, *P = 0.00507. When pull–0.3s activity was compared to zero (Wilcoxon signed-rank test with Bonferroni correction, α = 0.0167, two-sided), ++P = 0.00171 for 700–1000 ms and 0.00244 for 1000–1300 ms in H sessions, and ++P = 6.10 × 10–4 for 700–1000 ms, 3.66 × 10–4 for 1000–1300 ms, and 0.00305 for 1300–1600 ms in M sessions. Cue activity: n = 12, 12, and 10 for H, M, and L sessions, respectively. Pull–0.3s activity: n = 13, 14, and 14 for H, M, and L sessions, respectively. f Schematic of the time courses of combined activity of cue-preferring and pull-preferring axons in trials grouped by pull latency (green, 700–1000 ms; blue, 1000–1300 ms; purple, 1300–1600 ms). Line thickness represents trial frequency. Source data are provided as a Source Data file.
The proportions of cue-preferring and pull-preferring axons did not significantly differ between these sessions (Fig. 4c). The cue activity and pull–0.3s activity tended to decrease from H to L sessions, with the pull–0.3s activity in L sessions being near zero (Fig. 4d and Supplementary Fig. 7a, b). In these activities, there was no significant difference between the three pull-latency groups (700–1000 ms, 1000–1300 ms, and 1300–1600 ms) (Fig. 4e). The pull activity was also not significantly different between H, M, and L sessions (Fig. 4d and Supplementary Fig. 7b). In the averaged activity of those axons that were neither cue-preferring nor pull-preferring, and in the averaged activity of all axons, the ramping activity toward the lever-pull onset was not apparent (Supplementary Fig. 7c–f). These results suggest that in the expert sessions, regardless of the pull latency, the cue activity of cue-preferring axons was smaller than in the non-expert sessions, and the ramping activity of pull-preferring axons occurred closer to the lever-pull onset and rose more rapidly than in the non-expert sessions (Fig. 4f).
Cue activity of mThLCN→M2 axons and pull latency increase in post-error trials in non-expert sessions
Cerebellar Purkinje cells show error signals immediately after failed actions38. Therefore, we suspected that the high cue activity in the non-expert sessions might be related to the failure in the earlier trials. We first examined whether the behavior differed between the trials immediately after successful trials (post-success trials) and trials immediately after failed trials with an early pull (post-error trials). In thalamocortical axonal imaging, both pull latency and success rate were higher in the post-error trials than in post-success trials in H sessions, but not in M and L sessions (Fig. 5a). This indicates that in the non-expert sessions, the error in a trial improved the behavior in the next trial. Similarly, in H sessions, the cue activity was higher in the post-error trials than in the post-success trials (Fig. 5b, d and Supplementary Fig. 8a–d). By contrast, the pull–0.3s activity and pull activity were not significantly different between post-success and post-error trials (Fig. 5c, d and Supplementary Fig. 8e–h). These results suggest that the cue activity in H sessions strongly represented the post-error signal (Fig. 5e).

a Pull latency and success rate of post-success (pS) and post-error (pE) trials in H (n = 14), M (n = 14), and L (n = 14) sessions of mThLCN→M2 axon imaging. Gray lines indicate individual sessions and black lines indicate the mean ± SEM. *P = 0.0295 for pull latency, 0.0494 for success rate, n = 14, Wilcoxon signed-rank test, two-sided. b Activity of cue-preferring axons in post-success (blue) and post-error (orange) trials in H, M, and L sessions (n = 12, 12, and 10, respectively) aligned to the cue onset. The corresponding lever trajectories are also shown. Shading indicates ± SEM. The black lines and dots indicate the period in which the difference in activity between post-success and post-error trials was significant (P < 0.05, Wilcoxon signed-rank test, false discovery rate [FDR]-adjusted). c Activity of pull-preferring axons in post-success (blue) and post-error (orange) trials in H, M, and L sessions (n = 13, 14, and 14, respectively) aligned to the pull onset. The corresponding lever trajectories are also shown. Shading indicates ± SEM. d Cue, pull–0.3s, and pull activities in post-success (pS; blue) and post-error (pE; orange) trials in H, M, and L sessions. The mean ± SEM are also shown. **P = 0.00244, *P = 0.0161 for the comparison between post-success and post-error trials, *P = 0.00920 for the comparison between H, M, and L sessions, Wilcoxon signed-rank tests with Bonferroni correction (α = 0.0167), two-sided. Cue activity, n = 12, 12, and 10 for H, M, and L sessions, respectively. Pull−0.3s and pull activities: n = 13, 14, and 14 for H, M, and L sessions, respectively. e Schematic of the time course of the combined activity of mThLCN→M2 cue-preferring and pull-preferring axons in H sessions. Blue traces indicate post-success trials. Orange traces indicate post-error trials. Source data are provided as a Source Data file.
We further examined the properties of the post-error cue activity in H sessions. When we divided the trials into those with pull latencies of 700–1000 ms and those with latencies of 1000–1300 ms, the post-error cue activity was not significantly different between the two groups (Supplementary Fig. 8i). Thus, the post-error cue activity was not related to how long the pull latency was extended in the corresponding trial. When the early-pull trials were divided into those with pull latencies of 1–500 ms and those with latencies of 500–1000 ms, the cue activity was not significantly different between these two groups of previous early-pull trials (Supplementary Fig. 8j). Thus, the post-error cue activity also did not represent how early the lever was pulled in the one-trial-back trial. Furthermore, the cue activity was not affected by the success or error in the two-trial-back trial (Supplementary Fig. 8k). Considering that the mice would desire the reward more strongly after two consecutive errors than after one error, the post-error-specific cue activity should not reflect the reward craving level. Since the timeout was set after the early-pull trials, the stimulus-onset asynchrony (SOA) before post-error trials (i.e., the time interval between the cue onset in a trial and that in the next trial) was longer than that before post-success trials (Supplementary Fig. 8l). Thus, the long SOA might just increase the cue activity. However, the cue activity in the post-success trials was not significantly different between trials with SOA of <6 s and those with SOA of >9 s (Supplementary Fig. 8m). The cue activity in the post-error trials was also not significantly different between trials with SOA of <10 s and those with SOA of >12 s (Supplementary Fig. 8n). Thus, the large post-error cue activity in H sessions represented the absence of the reward in the one-trial-back trial.
Large post-error cue activity of mThLCN→M2 axons in non-expert sessions did not primarily reflect changes in non-task associated body and orofacial movements
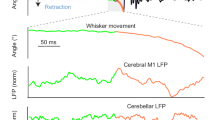
It is possible that the large post-error cue activity in H sessions might also be related to some body and/or orofacial movements that were not directly related to the instructed movement28,48,49,50. Therefore, in addition to the lick rate, we made comparisons between post-success and post-error trials of movement of the right whisker pad that was assumed to correspond to whisking, jaw movement, and movement of the abdomen (Supplementary Fig. 9a–c). In H sessions, the movement of the whisker pad and jaw 0.1–0.3 s after the cue onset was significantly greater in the post-error trials than in the post-success trials (Supplementary Fig. 9d). In addition, the same was true in L sessions (Supplementary Fig. 9d). The abdominal movement 0.1–0.3 s after the cue onset was significantly greater in the post-error trials than in the post-success trials in M sessions, and tended to be greater in H and L sessions as well (Supplementary Fig. 9d). The lick rate 0.1–0.3 s after the cue onset did not differ between post-success and post-error trials in H, M, and L sessions (Supplementary Fig. 9c, d). These results differed substantially from those for the cue activity of mThLCN→M2 axons (Fig. 5d).
Then, we constructed generalized linear models to fit five variables (one-trial-back trial outcome [success or early pull] and the four movements) to the cue activity to determine the unique contribution of each variable28,48,51 (see Methods for details). In H and M sessions, the unique contribution of the one-trial-back trial outcome was larger than that of any of the movement variables in six out of eight comparisons (Supplementary Fig. 9e). In L sessions, the unique contribution of the one-trial-back trial outcome was not larger in any comparison. These results suggest that the outcome of the previous trial had a stronger influence on the cue activity of mThLCN→M2 axons than any of the movements examined in H and M sessions.
For the whisker pad and jaw, the movements 0.4–0.2 s before the pull onset were not apparently smaller than those around the pull onset (0.1 s before to 0.1 s after), as was the case for the lick rate (Supplementary Fig. 9f, g), and the abdominal movement 0.4–0.2 s before the pull onset did not tend to decrease from H and M sessions to L sessions (Supplementery Fig. 9f). These properties differed from those of pull–0.3s and the pull activities of mThLCN→M2 axons (Figs. 4d and 5d). Furthermore, when any of the four types of movements occurred in the inter-trial interval, the activity of the cue-preferring and pull-preferring axons did not change (Supplementary Fig. 9h, i).
All these results suggest that the activity of cue-preferring and pull-preferring mThLCN→M2 axons did not primarily reflect licking or any movement of the whisker, jaw, or abdomen.
Population activity pattern after the cue onset differs between post-success and post-error trials in the non-expert sessions
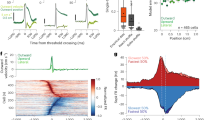
So far, we have mainly focused on the activities of cue-preferring axons and pull-preferring axons. However, the other axons, as well as the total axons, also showed some difference in their average activity between post-success and post-error trials in H sessions (Supplementary Fig. 10a–d). Therefore, we examined how the dynamics of the population activity differed between post-success and post-error trials in H sessions, without using the classification of cue-preferring and pull-preferring axons. First, we conducted principal component analysis (PCA) on the trial-averaged cue-aligned activity and pull-aligned activity of each axon for H sessions. The trajectories of the top three PCs (PC1–PC3) of mThLCN→M2 axons in post-success and post-error trials were apart around the cue onset (Fig. 6a). This difference might mainly reflect the difference in the pull latency. However, when the trials were divided into three groups according to pull latency (700–900 ms, 900–1100 ms, and 1100–1300 ms), the difference within the same group of trials was apparent at the cue onset (Fig. 6b). Assuming that the top five PCs (PC1–PC5) represented the major components (explained variance: 44.6%; Supplementary Fig. 10e, f), we quantified the differences in their trajectories by calculating the Euclidean distance between post-success and post-error trials at four time points (0.4 s before cue onset [cue–0.4s] and 0.2 s after cue onset [cue+0.2s] for the cue-aligned activity, and 0.3 s before pull onset [pull–0.3s] and pull onset for the pull-aligned activity). At the cue+0.2s time point, the difference was large and significant in all groups (Fig. 6c). However, the difference decreased at the pull–0.3s and pull-onset time points (Fig. 6c). Thus, in the non-expert sessions, strong post-error-specific activity of the major components of the population appeared at cue onset, irrespective of the pull latency, and weakened toward the pull onset.

a, b Trajectory of PC1–PC3 of mThLCN→M2 axonal activity in post-success (blue) and post-error (orange) trials in H sessions. Open circles, filled stars, and filled squares show cue onset, cue+0.2s, and average pull-onset time points, respectively. Axons in sessions with at least one of each of the six types of trials were used (316 axons from 14 sessions). b Trials were divided into three groups according to pull latency (left, 700–900 ms; middle, 900–1100 ms; right, 1100–1300 ms). c Euclidean distance in the trajectory of the top five PCs between post-success and post-error trials at cue–0.4s, cue+0.2s, pull–0.3s, and pull-onset time points. Trials were divided into three groups according to pull latency (green, 700–900 ms; blue, 900–1100 ms; purple, 1100–1300 ms). Asterisks indicate values above the 99th percentile of chance levels (dashed lines), which were estimated by shuffling the trial assignments and performing the same calculation. d Prediction accuracy of the population activity for classifying the trials into post-success and post-error trials. n = 14 sessions. Black bars, the mean ± SEM. **P = 0.00122, compared with the value at the cue–0.4s time point, Wilcoxon signed-rank test with Bonferroni correction (α = 0.0167), two-sided. +++P = 1.22 × 10–4 for cue-0.4s, cue+0.2s, pull−0.3s, and pull-onset time points, compared with 0.5, Wilcoxon signed-rank test with Bonferroni correction (α = 0.0125), two-sided. e Matrices of Pearson correlation coefficients for the population vectors (316 axons from 14 H sessions) that represented the one-back trial outcome between each time. The vectors were aligned to the cue onset (left) and pull onset (right). f Proportions of the axons that were modulated by the one-back trial outcome at each time point and at both of the two time points. Asterisks indicate statistically significant proportions (more than the 99th percentile of the shuffled data). g Schematic of transitions of the one-back trial outcome-representing axons (red arrows). Black arrows indicate the axons that did not represent it. Thick gray arrows indicate the axons that represented the one-back trial outcome across different time points. Source data are provided as a Source Data file.
The information on the outcome of the one-trial-back trial might not be necessarily included in only the major components of the population. Therefore, we used a support vector machine to determine the extent to which the population including all axons predicted whether the one-trial-back trial was a successful trial or an early-pull trial in H sessions. The population significantly predicted the outcome of the one-trial-back trial at almost all time points, including the cue–0.4s point (Fig. 6d and Supplementary Fig. 10g). The population vector52 that represented the difference in the activity between post-success and post-error trials showed high correlations across time before the cue onset, but this situation changed immediately after the cue onset (Fig. 6e). The proportion of axons whose activity differed between post-success and post-error trials at both cue–0.4s and cue+0.2s time points was not significantly higher than the proportion that was calculated when the difference at the cue–0.4s time point in each axon was combined with the difference at cue+0.2s time point in a randomly chosen axon (Fig. 6f). Thus, it was unlikely that a significant number of axons retained the information on the outcome of the one-trial-back trial both before and after the cue onset. These results suggest that although the signals regarding the outcome of the trial were transferred to the next trial, the major components of the population activity and the combination of neurons carrying these signals showed change immediately after the cue onset in the next trial in H sessions (Fig. 6g).
Although L sessions were fewer than H sessions and thus suffered from lower statistical power, we conducted population activity analyses and found no apparent differences in the trajectories of the top five PCs between the post-success and post-error trials, the ability of the population activity to predict the one-trial-back trial outcome, or the change in the combination of axons carrying the one-trial-back trial outcome signals immediately after the cue onset (Supplementary Fig. 11a–i). These results suggest that the population activity of mThLCN→M2 axons did not strongly represent the difference in the whisker pad and jaw movements between the post-success and post-error trials in L sessions.
L5 neurons in M2 increased pull activity in the post-error trials in the non-expert sessions
To investigate how the mThLCN→M2 pathway affected downstream activity, we conducted two-photon imaging of layer 5 (L5) pyramidal neurons in M2 (M2L5 neurons) of mice in which another GECI, jRGECO1a53, was expressed in M2 (Fig. 7a–d and Supplementary Fig. 12a–c). Compared with the mThLCN→M2 axons, the proportion of cue-preferring neurons was low (<10%), whereas the proportion of pull-preferring neurons was high (>30%) (Fig. 7e). In contrast to mThLCN→M2 axons, the rise time of the pull-preferring activity that was sorted to the lever-pull onset appeared to be faster in the trials with shorter pull latency (Fig. 7d). Although the amplitude of the pull activity was not significantly different between the three latency groups, the pull–0.3s activity differed (Supplementary Fig. 12c). In this case, the difference in pull–0.3s activity should reflect the difference in the slope of the ramping activity. This is consistent with a previous study that found that M2 shows elapsed time-related activity8. When the sessions were divided into H, M, and L sessions in a manner similar to the case for mThLCN→M2 axons (Supplementary Fig. 12d), the dependency of the pull–0.3s activity on the pull latency was observed in all sessions (Fig. 7f, g).

a Schematic of two-photon calcium imaging of M2L5 neurons expressing jRGECO1a (top) and a representative two-photon image (bottom). b Successful trial-averaged activities of M2L5 neurons aligned to the cue onset (left) and lever-pull onset (right) during the self-timing task. Conventions are the same as in Fig. 2e. Shading indicates ± SEM. n = 37 sessions from five mice. c, d Trial-averaged calcium traces of all neurons aligned to the cue onset (c) and lever-pull onset (d). Conventions are the same as in Fig. 3b, c. Shading indicates ± SEM. n = 37 sessions. e Proportions of M2L5 neurons classified into cue-preferring neurons, pull-preferring neurons, and both neuron types in H (n = 24), M (n = 9), and L (n = 4) sessions. The mean ± SEM are also shown. In Wilcoxon rank sum tests with Bonferroni correction (α = 0.0167, two-sided), no statistical difference was detected for any of the types. f Cue, pull–0.3s, and pull activities. Conventions are the same as in Fig. 4e. The mean ± SEM are also shown. *P = 0.00814 between 700–1000 ms and 1000–1300 ms, 0.0106 between 700–1000 ms and 1300–1600 ms in H sessions, *P = 0.00391 between 700–1000 ms and 1000–1300 ms, 0.00781 between 700–1000 ms and 1300–1600 ms in M sessions, Wilcoxon signed-rank test with Bonferroni correction (α = 0.0167), two-sided. Cue-preferring neurons, n = 18, 5, and 2 for H, M, and L sessions, respectively. Pull-preferring neurons, n = 23, 9, and 4 for H, M, and L sessions, respectively. g Schematic of the time course of the combined activity of cue-preferring and pull-preferring neurons. Conventions are the same as in Fig. 4f. Source data are provided as a Source Data file.
Next, the trials were divided into post-success and post-error trials. In this dataset, the pull latency in H sessions was higher in post-error trials than in post-success trials, but the difference was not statistically significant (Supplementary Fig. 12e), while the success rate was significantly higher in post-error trials than in post-success trials. The cue activity of M2L5 neurons in H sessions was not significantly different between post-success and post-error trials (Fig. 8a and Supplementary Fig. 12f). By contrast, the post-error pull activity was higher than the post-success pull activity in H sessions (Fig. 8a and Supplementary Fig. 12g). Thus, the post-error-specific activity of cue-preferring and pull-preferring neurons in H sessions differed considerably between mThLCN→M2 axons and M2L5 neurons (Figs. 5e and 8b).

a Cue, pull–0.3s, and pull activities of M2L5 neurons in post-success (pS; blue) and post-error (pE; orange) trials. The mean ± SEM are also shown. Wilcoxon signed-rank tests with Bonferroni correction (α = 0.0167, two-sided) were used for comparisons. Cue activity: n = 19, 5, and 2 for H, M, and L sessions, respectively. Pull–0.3s and Pull activity: n = 24, 9, and 4 for H, M, and L sessions, respectively. **P = 3.96 × 10–4. b Schematic of the time course of the combined activity of cue-preferring and pull-preferring M2L5 neurons in post-success (blue) and post-error (orange) trials in H sessions. c Time from the time point at which the lever exceeded 10% of its maximum distance (0.5 mm from the natural position) to the lever-pull onset in post-success (pS; blue) and post-error (pE; orange) trials. The mean ± SEM are also shown. H sessions, **P = 8.29 × 10−4, n = 24 sessions, Wilcoxon signed-rank test, two-sided. Source data are provided as a Source Data file.
To clarify whether the post-error-specific activity of M2L5 neurons at pull onset was reflected in the behavior in H sessions, we took a closer look at the lever trajectory. In H sessions, the rise time at the beginning of the lever pull was slightly but significantly shorter in post-error trials than in post-success trials (Fig. 8c). These results suggest that the post-error-specific increase in the pull activity of M2L5 neurons in H sessions was related to the decrease in the initial rise time of the lever pulling.
We also analyzed the population activity in H sessions (Supplementary Fig. 13a–d). In all groups, the difference in its major components, in particular PC1, was substantial at the pull onset in H sessions (Fig. 9a, b and Supplementary Fig. 13e). The difference in the activity trajectory of the top five PCs (the explained variance was 54.2%) between post-success and post-error trials was small but significant in all three groups at the cue+0.2s time point, and in two groups at the pull–0.3s time point, with the difference then sharply increasing at pull onset (Fig. 9c and Supplementary Fig. 13e, f). Even when the top three PCs (the explained variance was 46.7%, which was similar to that of the top five PCs in mThLCN→M2 axons) were used, the difference was apparent at the pull onset (Supplementary Fig. 13g). The information on the outcome of the one-trial-back trial was maintained in the population activity from before to after the cue onset (Fig. 9d and Supplementary Fig. 13h). In contrast to mThLCN→M2 axons, the post-cue change in the population vector that represented the one-back trial outcome was gradual (Fig. 9e). A subset of neurons that possessed information on the outcome of the one-trial-back trial at the cue–0.4s time point retained this information at the cue+0.2s and pull–0.3s time points (Fig. 9f, g). The change in the population vector that represented the one-back trial outcome clearly occurred approximately 200 ms before the lever-pull onset (Fig. 9e), although a subset of neurons retained the one-trial-back trial information at both pull–0.3s and pull time points (Fig. 9f, g). In L sessions, there was no apparent difference in the population activity between the post-success and post-error trials (Supplementary Fig. 14a–i). These results indicate that the dynamics of the population activity in the non-expert sessions also differed between the mThLCN→M2 axons and M2L5 neurons. The transition in the mThLCN→M2 axon activity preceded that in M2L5 neurons, but the time courses of their transitions partially overlapped.

a, b Trajectory of the top three PCs of M2L5 neuron activity in post-success (blue) and post-error (orange) trials in H sessions. The conventions are the same as in Fig. 6a, b. Neurons in sessions with at least one of each of the six types of trials were used (772 neurons from 23 sessions). c Distance in the trajectory of the top five PCs between post-success and post-error trials. The conventions are the same as in Fig. 6c. d Prediction accuracy of the population activity for classifying the trials into post-success and post-error trials. n = 24 sessions. The mean ± SEM are also shown. **P < 0.00333 compared with the value at the cue–0.4s time point, Wilcoxon signed-rank test with Bonferroni correction (α = 0.0167), two-sided. +++P < 2.5 × 10–4 compared with 0.5, Wilcoxon signed-rank test with Bonferroni correction (α = 0.0125), two-sided. e Matrices of Pearson correlation coefficients for the population vectors (798 neurons from 24 sessions) that represented the one-back trial outcome between each time in H sessions. The conventions are the same as in Fig. 6e. f Proportions of the neurons that were modulated by the one-back trial outcome at each time point and at both of the two time points. Asterisks indicate statistically significant proportions (more than the 99th percentile of the shuffled data). Yellow asterisks indicate pairs of the two time points that showed significant proportions in M2L5 neurons, but not mThLCN→M2 axons. g Schematic of transitions of the one-back trial outcome-representing neurons (red circles). Open circles indicate the neurons that did not represent it. Some of the neurons maintained the one-back trial outcome representation across different time points (gray arrows). Source data are provided as a Source Data file.
Discussion
In this study, we found reward-based post-error-specific activities in mThLCN→M2 axons and M2L5 neurons in non-expert sessions at the single-neuron and population levels, and the relevant behavioral changes (Fig. 10). These results suggest that the temporal evolution of activity changes occurring in cerebellum and M2 was based on the reward-based error monitoring and was associated with behavioral improvement in the early stage of learning. By contrast, in the expert sessions, neither the activity of these two types of neurons nor the pull latency were different between the post-success and post-error trials. Pull latency-independent ramping activity of mThLCN→M2 axons was shorter and sharper in the expert sessions than in the non-expert sessions. These results imply that the dynamics of the cerebellar-thalamus-cortical activity in the self-timing task varied depending on the level of learning.

In the post-error trials in the non-expert sessions, the motor thalamus (mTh) that received the cerebellar output transmitted larger signals to M2 immediately after the cue onset (orange solid line), the M2 neurons showed larger signals at the lever-pull onset (blue triangles), and the initial lever-pull speed was faster than in the post-success trials (black arrow). The dashed lines indicate signal flows that we propose on the basis of the results of this and other studies: the post-error signal from the ACC and other brain regions with the sensory cue may activate the cerebellum (cer), and the cerebellum increases the activity that is sent to the motor thalamus (orange dotted lines). The post-error-specific change in the cerebellar activity may also change M2 and BG activities that are responsible for monitoring the elapsed time to elongate the pull latency (green dotted lines). The interaction between the cerebellum and BG may also be mediated by other pathways (black dotted line). The change in M2 activity may affect activity downstream, including M1 and the spinal cord (blue dotted lines).
Prominent increase in the cue activity of the mThLCN→M2 pathway in post-error trials in the non-expert sessions
Purkinje cells respond to reward-expecting cues54,55, and their activity depends on the size of the reward56. Our results also suggest that the cue activity of mThLCN→M2 axons partly reflected the reward expectancy and/or attention to the following lever-pull movement because it was small in the late lever-pull trials and non-pull trials. In previous reports, a subset of Purkinje cells in the mid-lateral cerebellum changed their activity after the error action (i.e., non-rewarded action) in the early stage of learning, and this error-specific activity decreased as the learning progressed17,38. Although these results are similar to our current results, in that post-error activity changes in the early stages of learning, the increase in the population-averaged activity specific to the cue presentation in the post-error trials has not previously been reported. The whisker-pad and jaw movements immediately after the cue onset were substantially affected by the one-trial back outcome, but did not depend on the learning stage. These movements might simply reflect some internal state related to reward expectation28,50. Our results suggest that the post-error-specific cue activity in the non-expert sessions represented a cognitive signal, rather than a sensory error signal, the degree of reward craving, or body/orofacial movement.
Possible mechanisms that increase the post-error cue activity of mThLCN→M2 axons in the non-expert sessions
The decoding analysis showed that the mThLCN→M2 axons and M2L5 neurons transferred information regarding the outcome of the trial to the next trial. In M2, information regarding the outcome of a trial is reported to be retained into the next trial51,57. In a mouse reaching task, a subset of L5 neurons in M1 changed their activity after the failed movement, and this change in baseline activity was maintained until the cue onset in the next trial, although these neurons did not increase their activity at the start of the post-error trial58. Consistent with this report, M2L5 neurons did not show the post-error-specific cue activity. By contrast, the population of mThLCN→M2 axons that carried information regarding the outcome of the one-back trial showed a large change and increased immediately after the cue onset. Thus, other brain areas might promote this change. The anterior cingulate cortex (ACC) also showed increased activity immediately after the error trial, and this increased activity was maintained during the inter-trial interval59. In the mouse, ACC neurons projecting to the visual cortex modulate visual cortical activity during the inter-trial interval after the post-error trial, which may increase the post-error attention60. In the Stroop test in humans, ACC shows an increased response specifically in post-error trials61. Thus, the post-error cue response in the ACC, or the enhanced cue response in the sensory cortex mediated by ACC, might also be transmitted to the cerebellum, and the post-error-specific cue activity of mThLCN→M2 axons might emerge (Fig. 10). This could be tested by measuring the activity in ACC and auditory cortex in the self-timing task.
Possible mechanisms for post-error-specific increase in the pull latency in the non-expert sessions
The increase in the cue activity of mThLCN→M2 axons in the post-error trials in the non-expert sessions might have affected the change in the population activity of M2L5 neurons around the cue onset, even though the averaged activity did not increase and the change in the population activity was small. The post-error-specific activity of mThLCN→M2 axons might induce feedforward inhibition in the motor cortex through parvalbumin-positive neurons62. Then, the activity state in M2 might become slightly different from that without the post-error-specific input. In elapsed time encoding, the striatum shows activity dynamics as the upstream area of M28. Thus, after the cue onset, the cerebellar cue activity, or another signal, might be transferred to the striatum by other pathways (black dotted line in Fig. 10)63,64,65. The cortico-BG-thalamo-cortical loop might cooperatively maintain the post-error-specific neuronal state during the waiting time, and this cooperative activity might decrease the speed of the M2 ramping activity to increase the pull latency (green dotted lines in Fig. 10). The post-error-specific neuronal state in M2 and/or other input to M2 around the pull onset might increase the pull activity of M2L5 neurons. As a result, pull-related activity of M1 might increase to speed up the initial lever pull. Alternatively, the M2 signal might be directly transmitted to the spinal cord66 to assist in increasing the initial pull speed (blue dotted lines in Fig. 10).
The post-error elongation of the pull latency in the non-expert sessions might be related to the post-error slowing (PES) in a stop signal task, which is well known as a general cognitive response in humans67. However, in PES, as the number of error trials increases, the frequency of PES decreases68. This is in contrast to the current result. In human PES, the task-related activity in the motor cortex decreases69; however, we did not detect a decrease in M2L5 activity in the post-error trials. It is unknown whether human PES depends on the level of learning. Therefore, it is unclear whether the post-error elongation of the pull latency in the non-expert sessions reflects the same phenomenon as PES. In humans, the ventrolateral prefrontal cortex (vlPFC) shows higher activity in post-error trials with increased reaction time than in post-error trials with non-increased reaction time70. Granger causality analysis suggested that the cerebellum, thalamus, and SMA mediate post-error processing for vlPFC activation during PES. In addition, the SMA and cerebellum are activated in duration (particularly around 1 s) recognition in humans4,9,27. Thus, the cerebellum→motor thalamus→M2 pathway that we found in the mouse may be the primitive circuit for both PES and perception timing of the short-time range in the human. It should be clarified whether the post-error-specific cue activity occurs even in the timing cognition task that does not require the motor timing control.
Establishment of stable cue and pull activity in the expert sessions
Post-error-specific cue or pull activity was not detected in mThLCN→M2 axons or M2L5 neurons in the expert sessions. Even at the population level, there was little difference between the post-success and post-error trials. These results suggest that the mice used the same neuronal ensemble for the motor timing after the cue onset, irrespective of whether the previous trial succeeded or failed. In the expert sessions, the mean pull latency was approximately 1.3 s and the mean success rate was approximately 0.7. In these sessions, the ramping activity that originated from LCN was sharp, and the ramping activity in M2L5 neurons appeared to depend on the pull latency. In a macaque self-timing saccade task with a waiting time of >1 s, after sufficient training, the slope of the ramping activity of DN did not depend on the waiting time, whereas that of the caudate putamen did depend on it6. Thus, in both species, motor timing would be similarly regulated by the cerebellum (for the sub-second range) and striatum (for the supra-second range). In the expert sessions, the threshold for the start of the cerebellar ramping activity may be accurately set immediately before the motor initiation, whereas the striatum may increase the frequency with which activity reaches the threshold after the appropriate latency. These steep and incremental activities might be integrated in M2 to prepare the appropriate cortical activity for the pull movement.
Our results also suggest that the steep ramping activity of LCN is formed through learning. Considering that the cerebellum is necessary for timing control of <1 s in eyeblink conditioning10, the LCN ramping activity in the non-expert sessions might be directly related to control of the waiting time of <1 s, and then after the error trials, a different population of the striatum and M2 might be engaged to elongate the waiting time toward 1 s. By contrast, in the expert sessions, the mice might recognize that this task required monitoring of a time duration >1 s, and came to separately use striatum activity for waiting of >1 s and cerebellar activity for fine tuning of motor initiation timing. They no longer adjusted the pull latency or the cerebellar cue activity in the post-error trials, and might have pulled the lever on the basis of internal clock9 and timing models, which fulfilled the requirement for reward acquisition.
Reward-based error-driven learning of motor timing control
Our results suggest that the cerebellum might play a critical role in error-driven learning of motor timing. This is consistent with the cerebellum showing error-related signal during learning, and with this signal decreasing as performance improves17,36,38. The internal model for the self-timing control might be generated through the cerebellum32,33. It remains unexamined whether learning of self-timing control requires error-driven synaptic plasticity in the cerebellum32,34,71. This will be addressed using transgenic mouse lines with impairment of cerebellar plasticity11,72,73. In addition, the current task should require the learning of accurate monitoring of the elapsed time through the BG1,8. The post-error-specific cue activity represented only the reward absence in the previous trial, not the sensory error regarding how long the waiting time was. Reward-based learning can occur in both areas33,38,39,40,74; thus, during learning of the motor timing control, these areas might cooperatively optimize their models of the internal clock and timing through their own reward-based error and dopaminergic signals.
Limitations
The number of muscimol inactivation experiments (7–9 mice in each condition) was insufficient to compare the inactivation effect between H, M, and L sessions. In the imaging of M2L5 neurons, the session number with cue-preferring and pull-preferring neurons in L sessions was too low (four) to permit non-parametric statistical tests. In the current study, the function of the activity at each time point from the cue onset to the pull onset was not directly demonstrated. To address this issue, experiments with transient optogenetic activation and inactivation are necessary.
Methods
Animals
All animal experiments were approved by the Animal Experimental Committee of the University of Tokyo. Wild-type C57BL/6 mice (male, 2–3-months-old; Japan SLC) and Ai162D transgenic mice (B6.Cg-Igs7tm162.1(tetO−GCaMP6s,CAG−tTA2)Hze/J, JAX stock #031562, three males and one female, 12-months-old) were used for the experiments. For behavioral experiments, only male mice were used to prevent potential behavioral variations caused by the estrous cycle. All mice were housed in a 12:12 hour light-dark cycle (light cycle; 8 AM–8 PM) and were provided with adequate water and food, except during behavioral task training. The ambient temperature and humidity were 22 °C–25 °C and 40–80%, respectively.
Surgery
Mice were anesthetized by intraperitoneal administration of ketamine (74 mg/kg) and xylazine (10 mg/kg), fixed in a stereotaxic apparatus (SR-5M-HT, Narishige, Tokyo, Japan), and the skull exposed through a head skin incision. The skull was then drilled under a stereomicroscope (M60, Leica Biosystems, Nussloch, Germany) using a disposable drill tip (SD-101, Narishige). The solution of AAV or drug was filled into a glass pipette filled with mineral oil and locally injected using a Nanoject III microinjector (3-000-207, Drummond Scientific Co., PA). The hole was closed with silicon elastomer (Kwik-Cast, World Precision Instruments, FL), and the exposed skull was covered with dental resin cement (Super Bond, Sun Medical, Shiga, Japan), except when the glass window for two-photon imaging was attached. After the surgery, the anti-inflammatory analgesic carprofen (6 mg/kg), and the antibiotics sulfadiazine (24 mg/kg), and trimethoprim (4.8 mg/kg), were administered intraperitoneally. After the surgery, the mice were returned to their home cages and warmed on a heat pad until the next morning.
For transsynaptic tracing of cerebellar-thalamocortical axons, a mixture of AAV1-syn-Cre41 (1.3 × 1013 genome copies [GC]/mL; Addgene, #105553-AAV1) and AAV1-CAG-tdTomato (9.5 × 1011 GC/mL; Addgene, #59462-AAV1) or AAV5-CAG-tdTomato (5.0 × 1011 GC/mL; Addgenee, #59462-AAV5) was introduced into the LCN (6.1 mm posterior [AP: −6.1 mm] and 2.3–2.4 mm lateral [ML: 2.3–2.4 mm] to the bregma, and 2.1–2.2 mm ventral to the cortical surface [DV: 2.1–2.2 mm]; 30–50 nL at a speed of 10 nL/min). Within one week from the day of these introductions, 30–50 nL of AAV1-CAG-flex-GFP (1.6–3.1 × 1013 GC/mL; Addgene, #51502-AAV1) was similarly introduced into the thalamus VL (AP: −1.0 mm, ML: 1.0 mm, DV: 3.5 mm) or VM (AP: from −1.15 to −1.2 mm, ML: 0.8 mm, DV: 4.0 mm). Although AAV1 that carried the gene encoding the fluorescent protein would also be transynaptically transfected to cells in non-injected regions, we expected the transsynaptic transfection rate to be much lower (1–5%)75 than the rate of directly transfected neurons in the VL or VM. Therefore, even if cells in non-injected regions further took up AAV1-syn-Cre, we considered that the expression of the fluorescent protein would be much lower than that in VM or VL neurons, and could be considered to be negligible. The mice were allowed to recover in their home cages for 4 weeks to allow for virus expression.
For anterograde tracing of cerebellar-thalamic pathways, 100 nL of AAV9-syn-EGFP (2.0 × 1013 GC/mL; #Addgene, 50465-AAV9) was introduced into the LCN, and the brains were fixed 2 weeks later. For the experiments to visualize the projection target in the motor thalamus from the LCN and SNr (Supplementary Fig. 4), 100 nL of AAV1-syn-Cre was introduced into the LCN or SNr (AP: −3.2 mm, ML: 1.5 mm, DV: 4.3 mm) of Ai162D mice, and their brains were fixed 4 weeks later.
To prepare for task training under a head-fixed condition, a metal head plate44 (Tsukasa Giken, Shizuoka, Japan) was attached to the exposed skull and was glued with dental resin cement (Estecem II, Tokuyama Dental, Tokyo, Japan; Fuji lute BC, GC, Tokyo, Japan). The exposed skull was covered with other dental cement (Super Bond, Sun Medical). After the surgery, the mice were allowed to recover in cages for one week before starting water restriction.
For in vivo two-photon imaging of mThLCN→M2 axons, a mixture (140 nL) of AAV1-syn-Cre (1.3 × 1013 GC/mL) and AAV1-CAG-tdTomato (2.1–9.5 × 1011 GC/mL) was injected into the LCN (AP: −6.1 mm, ML: 2.3–2.4 mm, DV: 2.1–2.2 mm) of the mice and the head plate was attached on the same day. From the next day to 3 days later, a mixture of AAV1-syn-flex-jGCaMP7f (1.2 × 1013 GC/mL; Addgene, #104492-AAV1) and AAV1-CAG-tdTomato (2.8–4.8 × 1011 GC/mL) was injected into the VL and VM (100 nL per site). For in vivo two-photon imaging of L5 neurons in M2, AAV1-syn-NES-jRGECO1a (1.7 × 1013 GC/mL; Addgene, #100854-AAV1) was injected into two sites in M2 (AP: 2.5 mm, ML: 0.8 mm, DV: 1.0 mm; and AP: 2.0 mm, ML: 0.8 mm, DV: 1.0 mm; 200 nL per site) the day after the head plate was attached.
Task training (described below) started 1 week after the surgery. After completing the training, a glass window was attached to the skull using the following procedures. First, carprofen (6 mg/kg), sulfadiazine (24 mg/kg), trimethoprim (4.8 mg/kg), and dexamethasone (1.32 mg/kg) were administered intraperitoneally 1 hour before surgery. For M2 imaging, an area of 3.2–0.9 mm AP and 0–2.3 mm ML was craniotomized at least 3 days before the imaging experiments. A 2 × 2-mm cover glass (thickness No. 5, Matsunami Glass, Osaka, Japan) and 3 × 3-mm thin cover glass (0.08–0.12 mm, Matsunami Glass) were laminated and placed over the craniotomized area. The glass window was 0.2 mm thicker than those previously used16, to reduce the vertical motion artifact of the brain during imaging. In addition, the glass window was placed before performing the imaging experiment, rather than immediately after the virus injection, to maintain strong adhesion between the glass window and the skull. Training was suspended for 2 days after the surgery.
Histology
Mice were deeply anesthetized by intraperitoneal administration of ketamine (74 mg/kg) and xylazine (10 mg/kg) and were transcardially injected with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA, 09154-85, Nacalai Tesque, Kyoto, Japan). Whole brains were removed and postfixed overnight in 4% PFA with shaking. The brain was sliced into 100-μm-thick sections using a vibratome (VT1000S, Leica Biosystems). For mice in which sections were prepared from both the cerebellum and motor cortex, the whole brain was embedded in 4% agarose (01162-15, Nacalai Tesque) dissolved in PBS and sliced. For nuclear staining, DAPI (1:1000, D523, Dojindo, Kumamoto, Japan), NeuroTrace Blue (1:200, N21479, Invitrogen, MA), NeuroTrace Green (1:200, N21480, Invitrogen), or propidium iodide (2 μg/mL, 29037, Nacalai Tesque) was diluted in 0.3% Triton X/PBS and reacted with the tissue section for 30 minutes. Then, the tissue sections were washed with 0.3% Triton for 10 minutes and with PBS for 1 hour, before being mounted with Mowiol 4-88 (81381-250 G, Sigma-Aldrich, MO). The sections were observed and imaged using a fluorescence microscope (BX53, Olympus, Tokyo, Japan). We used ImageJ (National Institute of Health, MD) to measure the fluorescence intensity of the cerebral cortex, and standardized the maximum intensity to 1. When Ai162D mice were used (Supplementary Fig. 4c, d), the brain sections were washed in PBS-X (0.5% triton-X in PBS) containing 10% normal goat serum, and then incubated with the primary antibodies (1:500, rabbit anti-GFP; A6455, polyclonal, lot: 2901490; Invitrogen) overnight at 4 °C. Afterwards, sections were washed in 0.3% PBS-X and then incubated for 2 h with Alexa Fluor-488 conjugated secondary antibody (1:500, Goat anti-rabbit IgG; A11034, polyclonal, lot: 1705912; Invitrogen). Then, the sections were washed twice, the cell nuclei were stained with propidium iodide, and the sections were mounted on glass slides.
Training of the behavioral task
The behavioral task used the lever-pull task device previously used in our laboratory20,44,76, with a lever at the right forelimb, an armrest at the left forelimb, a spout and infrared lick sensor near the mouth, a cylindrical retainer around the body trunk, and a sound-generating speaker in the outer box (O’Hara, Tokyo, Japan). We designed the task and controlled the task device using LabVIEW software (National Instruments, TX). The lever pulling was limited to 5.0 mm from the natural position, and the lever-pull threshold was set to one-third of the range (1.67 mm). For every trial, mice were rewarded with water (2–6 µL for the self-timing task and the last stage of the training task, 4 µL for the other tasks) if the performance was successful. During the training, the outer box was closed and the movements of the mouse were observed using an infrared camera. The training took place during the light period.
To habituate the mice to the task apparatus, the day after water restriction started the mice were placed on an experimenter’s hand and manually fed with water through a spout until they stopped licking. Training was performed once a day for up to 60 minutes, with body weight being measured before and after training, with water or agar (Transport Agar, Oriental Yeast, Tokyo, Japan) being given to maintain at least 85% of the original body weight. Head-fixed mice were trained for two days to lick the spout within 1 s after a go-cue onset in which pink noise was presented for 300 ms. The go cue was presented at least 5–6 s after the previous presentation and 1.5–2.5 s after the last lick. The lever-pull training consisted of the following three stages.
Stage 1 (simple go-cue task)
Head-fixed mice were trained to pull the lever with the right forelimb within 1 s after the go-cue onset and hold it for more than 100 ms. For each trial, the SOA (the time interval in the tone onset between the previous trial and the current trial) was determined as follows: a value randomly chosen from 5–6 s was defined as SOA1, and a value randomly chosen from 1.5–2.5 s was added to the time interval between the previous cue onset and the timing of the last lever return (including the return of spontaneous lever-pull movement) and was defined as SOA2. Then, if SOA1 was longer than SOA2, the SOA was SOA1. Otherwise, the SOA was SOA2. Training was continued for more than 2 days until the success rate was more than 80%. Then, the training moved to the next stage.
Stage 2 (1-s waiting task with go cue)
A continuous tone of 10 kHz (tone cue) was presented at the onset of each trial. A water drop was delivered if the mice pulled the lever and held it for at least 100 ms between 1 s and 1.71 s after the cue onset. The cue sound pressure was approximately 66 dB, a level at which the mice did not give an immediate physical response. The go cue was presented 1 s after the onset of the tone cue for less than six sessions, and was then presented 1.35 s after the tone cue onset for four to nine sessions. Even if the mice pulled the lever before the go cue, they obtained the reward as long as more than 1 s had elapsed after the tone cue onset. If the lever was pulled earlier than 1 s after the tone cue onset (early pull), the cue sound stopped and the go cue was not presented. If the mice did not pull the lever within 1.71 s after the tone cue onset, they did not obtain the reward and were moved on to the next trial. For each trial after a trial with a lever-pull latency of >1 s or a non-pull, the SOA was determined in the same way as in stage 1. For each trial after the early-pull trial, a value randomly chosen from 9 to 10 s was added to the pull latency in the early-pull trial and was defined as SOA3. SOA2 was defined in the same way as described above. Then, if SOA3 was longer than SOA2, the SOA was SOA3. Otherwise, the SOA was SOA2. The fact that the SOA was generally longer after early-pull trials than after rewarded trials means that penalty time was given in the early-pull trials.
Stage 3 (1-s waiting go cue-omitted task)
In the following sessions, after 10 trials of presenting the go cue at the beginning of each session, the go cue was omitted if successful trials continued. If more than T minutes had elapsed since the last reward (T was changed in the order of 0.3, 0.5, and 1) and the lever was not pulled in the trial at the end of T minutes, the go cue was presented again. The reward amount was 2 µL when the latency from the tone cue onset to the lever pull (pull latency) was 1–1.28 s, 4 µL when it was 1.28–1.43 s, and 6 µL when it was 1.43–1.71 s. The stage 3 training was ended after mice made more than 80 successful trials without a go cue in 1 hour.
For imaging experiments, the mice were allowed to acclimatize to the task device under two-photon microscopy (FVMPE-RS, Olympus) by performing the 1-s waiting go cue-omitted task with T of 1 under the microscope for 1–3 days. Then, two-photon imaging was conducted while the mice performed the self-timing task, in which the tone cue was presented at the same timing as in the stage 3 training, but the go cue was not presented at all. Some other groups of the mice were used for pharmacological experiments after the stage 3 training. For the self-timing task, we calculated the success rate as the number of rewarded trials divided by the total number of trials in which the lever was pulled, and the early-pull rate as the number of early-pull trials divided by the total number of trials during the session. In the imaging experiments, the number of trials during the imaging period was used. Lever trajectory correlation was calculated as the pairwise correlation of trajectories during the period from 0.3 s before to 0.15 s after the pull onset.
Pharmacological inhibition using muscimol
The head plate was modified to provide space for drug injection into the DCN. After mice had learned the self-timing task or the simple go-cue task, a 0.4-mm diameter hole was created by the craniotomy at each injection site (from the bregma to the LCN, AP: −6.1 mm, ML: 2.4 mm) the day before the experiment, under 1–1.5% isoflurane. The hole was covered with silicon (Kwik-Cast, World Precision Instruments) and dental resin cement (Super Bond). On the days of the experiment, the silicon coating was removed under isoflurane anesthesia, a glass pipette was inserted to a depth of 2.2 mm (3 degrees from the dorsoventral axis to caudal side), and 100 nL of ACSF was administered over 5 minutes using a Nanoject III on the first day, and the same amount of 0.1 mg/mL muscimol hydrobromide (G019, Sigma-Aldrich) was administered on the second day. The pipette was left inserted for 5 minutes after completion of the injection. After removing the pipette, the hole was covered with silicon and cement and the animal was placed on a heat pad for 60 minutes to recover from anesthesia, before being moved to the task device.
For inhibition of the motor cortex, a normal head plate was attached to the mouse skull and training began 1 week later. After the stage 3 training, a craniotomy with a 0.4-mm-diameter hole was performed at the injection sites of both M2 (AP: 2.5 mm, ML: 0.8–1.2 mm) and M1 (AP: 0.5 mm, ML 1.0 mm) the day before the experiment. The holes were covered with the silicon and resin cement. On the day of the experiment, the silicon coating was removed under isoflurane anesthesia, a glass pipette was inserted into the M2 hole to a depth of 0.3 mm, and 200 nL of ACSF was administered on the first day over 10 minutes using a Nanoject III, and the same amount of 0.5 mg/mL muscimol hydrobromide (G019, Sigma-Aldrich) was administered on the second day. On the third and fourth days, the same procedure was applied to M1. The pipette was left inserted for 5 minutes after completion of the injection. After removing the pipette, the hole was covered with Kwik-Cast and Super Bond, and the animal was placed on a heat pad for 60 minutes.
The muscimol dosage was determined on the basis of past reports77,78. Because our purpose was to analyze the timing of the lever pull in the self-timing task, we used lower doses of muscimol (10 ng for the cerebellar nucleus and 100 ng for the motor cortex) than previously reported as being required to reduce the lever-pull rate44,78, in order to prevent the mice from skipping the lever pull because of strong inhibition. The difference in the muscimol amount between M2 (or M1) and the LCN was to account for the volume difference between these areas. However, in the M1 inhibition experiment with the low concentrations of muscimol, two mice showed less than half of their pull rate in the control session, and were therefore excluded from the analysis. In the analysis of the remaining mice, the pull rate also significantly decreased (Supplementary Fig. 2d). These results suggest that M1 activity is more intimately associated with movement execution than is M2 activity28. In the simple go-cue task, we also used a high dose of muscimol (100 ng; concentration of 1 mg/mL) to determine whether the LCN was necessary for lever-pull execution.
We analyzed the data from 30 minutes of trials from the beginning of the session in the self-timing lever-pull task, or 15 minutes of trials in the simple go-cue task. After the experiment, 50 nL of Vybrant DiI Cell-Labeling Solution (V22885, Invitrogen) was locally injected into the same site, the brain was fixed, and tissue sections were prepared to confirm the injection site. Pharmacological inhibition experiments were conducted between 10 AM and 5 PM.
In vivo two-photon imaging
Two-photon images were acquired using an FVMPE-RS system (Olympus) with a broadly-tunable ultrafast laser (InSight DS-OL, Spectra Physics, CA) tuned to 940 nm. Fluorescence emission was collected using a GaAsP photomultiplier tube (Hamamatsu Photonics, Shizuoka, Japan). Mice were head-restrained under an XLPLN25XWMP lens (numerical aperture [NA] 1.05, Olympus) during the self-timing lever-pull task. For axonal imaging, a field of view (FOV) of 127.28 × 127.28 µm (512 × 512 pixels) was imaged at a frame rate of 30 Hz for 10–20 min. The depth below the cortical surface of the imaged fields was 10–20 μm. To separate fluorescence signals, a 570-nm dichroic mirror (Olympus) was used with bandpass filters of 495–540 nm and 575–630 nm for jGCaMP7f and tdTomato, respectively. Imaging timing and behavioral data were synchronized in real time using LabVIEW. Sessions with 30 or more rewarded trials during recording were analyzed. For imaging of L5 neurons in M2, the laser wavelength was tuned to 1100 nm and fluorescence images were acquired in a single channel for jRGECO1a. The FOV was 509.12 × 509.12 µm (512 × 512 pixels) at a depth of 420–650 µm from the cortical surface.
Video monitoring
Two high-speed video cameras (Scout scA640-70gm, Basler, Germany, with an M0814-MP fixed-focus lens of 8-mm focal length, Computer, NC) were used to capture the face and anterior trunk of the mice, including the thoracic and abdominal regions. The cameras were synchronized with a pulse signal generated by the data acquisition board used for the task acquisition, and the images were acquired at 70 Hz. An infrared LED light source (940 nm, 60 LEDs) was positioned at the back of each camera to illuminate the video-monitored areas without affecting the two-photon imaging. The video recording failed in three of the 42 sessions (one M session and two L sessions) for two-photon imaging of mThLCN→M2 axons, and these sessions were excluded from the video analysis.
Data analysis
Identification of axonal boutons
MATLAB (2019a, 2020a, 2021a, 2022a, 2023a, 2024a; MathWorks) and ImageJ (National Institute of Health) were used for analyses. TurboReg79 and NoRMCorre80 were used for motion correction of images. In TurboReg, each pixel of the entire FOV was moved by the same distance, whereas in NoRMCorre, a 512 × 512-pixel image was divided into 64 × 64-pixel images and the pixels were moved at different distances between the divided images. By combining the two motion correction methods, both the displacement of the entire field and local distortions were eliminated from most sessions. Motion correction was performed on the red channel data showing the fluorescence of tdTomato, and the shifts calculated for the correction were applied to the green channel data showing the fluorescence of jGCaMP7f. To estimate the image displacements that remained after the motion correction, we calculated frame-by-frame displacements of the motion-corrected calcium imaging movies using the MATLAB function imregtform. In the distribution of the 99th percentiles of the displacements in individual sessions, six sessions were considered to be outliers. In addition, to exclude the sessions with large displacements even if the displacements were rarer, we also calculated the 99.9th percentile of displacements for each session. In the distribution of the 99.9th percentiles of displacements, seven sessions were considered outliers. Out of the 50 imaging sessions, eight sessions that were outliers in at least one of the distributions for the 99th and 99.9th percentiles were removed from the following analyses.
Using the motion-corrected images, axonal boutons were detected as regions of interest (ROIs) using CaImAn81. Furthermore, using a modified CaImAn code to extract active axonal boutons82, we defined the average luminance in non-ROI areas as the luminance of the neuropil (the value calculated as the “Background” in CaImAn), and calculated the luminance time series for each ROI by subtracting the luminance of the neuropil from the average luminance within each ROI82. Detrended relative fluorescence changes (ΔF/F) were calculated for 15 percentile values over an interval of ±30 s around each sample time point. Spike timing was also inferred from the ΔF/F values by spike estimation using the “constrained nonnegative matrix factorization” algorithm81 implemented in CaImAn. The inferred firings were resampled to 100 Hz using linear interpolation (the MATLAB function interp1) and were synchronized with the behavioral data and frame triggers of the high-speed video and microscopy for later analysis.
The activity correlation between ROIs was calculated, and those ROIs with correlation above 0.40 for mThLCN→M2 axons, which represented the trough of the distribution in the histogram of the correlation of all ROI pairs (Fig. 2f), were taken as originating from the same axon, and the average activity between these ROIs was used for the analysis.
Analysis of neuronal activity
For each axonal bouton (or neuronal soma), the activity for 1 s before to 2 s after the cue onset was used for the analysis of cue-aligned activity, and the activity for 2 s before to 1 s after the lever-pull onset (time point of the lever-pull threshold) was used for the analysis of lever pull-aligned activity. The activity values were trial-averaged. For the classification, we used the activity in successful trials, and defined the cue-preferring axons (neurons) as those whose activity peaked for 0.1–0.3 s after the cue onset, and defined pull-preferring axons (neurons) as those whose activity peaked during the period from 0.2 s before to 0.2 s after the lever-pull onset. Cue activity and cue+0.5s activity were defined as the average activity of cue-preferring axons (neurons) during 0.1 to 0.3 s after the cue onset and during 0.4 to 0.6 s after the cue onset, respectively. Pull–0.3s activity and pull activity were defined as the average activity of pull-preferring axons (neurons) during 0.2 to 0.4 s before the pull onset and from 0.1 s before to 0.1 s after the pull onset, respectively. Except for the analyses in which trials were classified according to the pull latency, cue activity was calculated from all trials and pull–0.3s activity and pull activity were calculated from trials with lever-pulls.
To clarify whether the neural activity was related to the lever-pull latency (Fig. 3), trials were divided into three groups according to lever-pull latency: 700–1000 ms, 1000–1300 ms, and 1300–1600 ms, and the trial-averaged activity in each group was calculated. The lower bound of 700 ms was set not only to exclude the activity associated with premature responses that had very short waiting times, but also to minimize contamination of the pull–0.3s activity of the pull-preferring axons by the decay component of the activity immediately after the cue onset in a small subset of these axons that were also cue-preferring (Figs. 2i and 3c). Since the ratio of trials in which the lever position did not exceed the lever-pull threshold for more than 100 ms to all the pulled trials was small (0.056 ± 0.0082, 42 sessions from 8 mice), all trials with the lever-pull latency of 700–1600 ms were used. To examine the dependency of the neural activity on the level of learning of the task, the thalamocortical axon imaging sessions were divided into three groups according to the early-pull rate: those with an early-pull rate of greater than the 67th percentile, those between the 33rd–67th percentile, and those less than the 33rd percentile of all sessions with two-photon imaging of thalamocortical axons, were classified as high (H), middle (M), and low (L) sessions, respectively. For the session classification in M2L5 neurons, the early-pull rates that separated H and M sessions and M and L sessions of the thalamocortical axon imaging were used. However, in the latter case, the number of L sessions was only two. In the sessions with the third and fourth lowest early-pull rates, the values were closer to the first and second lowest early-pull rate values than to the fifth lowest value (Supplementary Fig. 12d). Therefore, we grouped these four sessions together as L sessions.
For the analysis of the effects of the previous outcome on the target trials, previous early-pull trials and rewarded trials were used as post-error trials and post-success trials, respectively. Target trials with a pull latency of >2000 ms (the ratio of these trials to the total trials was 0.023 ± 0.0039, n = 42 sessions from 8 mice) were removed because the mice might have shown low motivation and/or low attention in them, and the neuronal activity and behavior in them might not be related to the previous outcome.
Analysis of population activity
We used PCA to analyze the differences in the major components of population activity between post-success and post-error trials and between trials with different pull latencies. We used the neuronal activity in trials with a lever-pull latency of 700–1300 ms (these trials were further divided into those with pull latencies of 700–900 ms, 900–1100 ms, and 1100–1300 ms) and whose previous trials were early-pull trials or successful trials. Axons (or neuronal somata) in sessions with at least one of each of the six types of trials were used. For each of the H, M, and L sessions, the average cue-aligned activity and pull-aligned activity of each neuron were concatenated and subjected to PCA processing. We calculated the Euclidean distance of the top five PC trajectories between post-success and post-error trials in each pull-latency group at four time points (0.4 s before the cue onset [cue–0.4s] and 0.2 s after the cue onset [cue+0.2s] from the cue-aligned activity, and 0.3 s before the pull onset [pull–0.3s] and pull onset from the pull-aligned activity) using the MATLAB function pdist. Chance levels were calculated by shuffling the trial assignments and performing the same process 1000 times. The significance of the distance was determined according to the 99th percentile of chance levels (Figs. 6c and 9c).
Classification analysis of the one-back-trial outcomes was performed with a linear support vector machine (SVM) classifier (MATLAB’s fitcsvm function) with 10-fold cross-validation28. The classifier was trained to solve a two-class classification problem to predict one-back trial types (successful or early-pull trials) from neuronal population activity. Sessions with at least 20 successful and 20 early-pull trials were used and 20 trials of each were randomly chosen. The time series of neuronal activity from 0.1 s before to 0.1 s after each of the four time points (cue–0.4s, and cue+0.2s, pull–0.3s, and pull onset) were used. Ten-fold cross-validation was performed by splitting the data into ten segments using stratified partitioning. The classifier was then trained on the combined data of nine segments and tested on the remaining segment, with the process being repeated until all segments were tested. The SVM hyperparameters were set to the MATLAB default value, with a box constraint of 1, a linear kernel function, and a kernel scale of 1. The predicted trial types for all segments were combined. The percentage of agreement between the predicted one-back trial outcomes and the actual one-back trial outcomes was then used as the prediction accuracy. The subsampling of trials was repeated 100 times and the mean of the values was used.
To analyze the dynamics of the population activity representing the one-back trial outcome, we defined the decoding vector at each time point, t, as follows:
where N is the number of neurons and vi is the post-error-trial-averaged activity at time t in the ith neuron, minus the post-success-trial-averaged activity at time t in the ith neuron. To visualize the similarities of the decoding vectors between different time points (from 0.8 s before to 0.7 s after the cue onset, or from 0.8 s before to 0.5 s after the pull onset), we calculated the Pearson’s correlation coefficient of the decoding vectors for all combinations of two time points52.
We also analyzed to what extent the neuronal population representing the outcome of the one-trial back trials was maintained across the four time windows that were used in the PCA (cue–0.4s, and cue+0.2s, pull–0.3s, and pull onset). For each axon (or neuron), the mean activity during the time window was calculated for each trial and the mean activities were compared between the post-success and post-error trials. If they were significantly different (Wilcoxon rank sum test, P < 0.05), the axon (or neuron) was defined as being one-back trial outcome-modulated at that time window. The proportion of the one-back trial outcome-modulated axons (or neurons) was calculated for each time window. In addition, the proportions of the axons (or neurons) that were one-back trial outcome-modulated in both of the two time windows were determined. To calculate the chance level for the proportion of the one-back trial outcome-modulated axons (or neurons), we separately shuffled the order of the axons (or neurons) at the two targeted time windows, and then conducted the calculation in the same way. This procedure was repeated 1000 times and the 99th percentile value was determined as the threshold of significance.
Motion extraction from high-speed videography
Videos obtained from the high-speed camera were processed with FaceMap package49,51 (https://github.com/MouseLand/FaceMap) to quantify the movement of body parts. ROIs were set on the right whisker pad, jaw, and abdomen, as shown in Supplementary Fig. 9a, and the motion energy (absolute frame-to-frame difference) of each ROI was calculated. This motion energy was standardized to a maximum range of 0–1 within each session. We only used the front-view videos because most of the mouse’s face was obscured by the infrared lick sensor in the side-view videos.
Generalized linear model analysis
To determine to what extent each motion variable was related to the cue activity of mThLCN→M2 axons in each trial, we used the MATLAB function fitglm to construct generalized linear models with one-trial-back trial outcome (or previous trial outcome, pO), lick rate, and motion energy of whisker pad, jaw, and abdomen as predictor variables, and the cue activity of each cue-preferring axon as the response variable. The data from all trials were used. In the full model, all predictor variables were used, whereas in the other models one of the predictor variables was excluded in turn. For each axon, we calculated the coefficient of determination (R2) of each model as a measure of the fitting accuracy. The difference in the fitting accuracy between the full model and each of the other models was calculated (ΔR2) to reflect the unique contribution of the removed variable.
Relationship between neuronal activity and body movement during inter-trial intervals
For each ROI, the increase in motion energy from 0.1 s before each time point was calculated throughout the entire session. The lick rate was averaged over 1 s centered at each point, and the increase in the averaged lick rate from 0.1 s before each time point was calculated throughout the entire session. After excluding a period of 3 s from the cue onset of each trial from the entire session, the time points in which the motion increase exceeded the top one percentile were chosen for each session. To examine the relationship between the motion increase and the activity of cue-preferring and pull-preferring mThLCN→M2 axons, the average activity of cue-preferring axons and pull-preferring axons within ±0.1 s of the chosen time point was compared with that from 0.6 to 0.4 s before that point.
Statistical analysis
Statistical analyses were performed using MATLAB statistics and machine learning toolbox. Data are presented as the mean ± standard error of the mean (SEM), and error bars in graphs represent the SEM. The Wilcoxon rank sum test with Bonferroni correction, Wilcoxon signed-rank test with Bonferroni correction, and Wilcoxon signed-rank test followed by false discovery rate (FDR) were used for statistical comparisons. When data shuffling was used for the comparison, the 99th percentile value was set as the significance level. Statistical methods were not employed to predetermine sample sizes; however, sample sizes were estimated according to previous methodically comparable laboratory experiments and are similar to those generally employed in the field. No blinding or randomization was performed in this study. In the pharmacological experiments, control and inhibition experiments were conducted on the same animals. Information on all statistical tests is provided as a Source Data file (https://doi.org/10.6084/m9.figshare.27915756).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper (https://doi.org/10.6084/m9.figshare.27915756). Raw data from immunohistochemistry and imaging experiments are too large and heterogeneous to be deposited in a usable form; however, any additional requests for information can be directed to, and will be fulfilled by, the corresponding authors.
Code availability
There is no code based on an original algorithm.
References
Mello, G. B. M., Soares, S. & Paton, J. J. A scalable population code for time in the striatum. Curr. Biol. 25, 1113–1122 (2015).
Buhusi, C. V. & Meck, W. H. What makes us tick? Functional and neural mechanisms of interval timing. Nat. Rev. Neurosci. 6, 755–765 (2005).
Coull, J. T., Cheng, R. K. & Meck, W. H. Neuroanatomical and neurochemical substrates of timing. Neuropsychopharmacology 36, 3–25 (2011).
Lewis, P. A. & Miall, R. C. Brain activation patterns during measurement of sub- and supra-second intervals. Neuropsychologia 41, 1583–1592 (2003).
Tanaka, M. et al. Roles of the cerebellum in motor preparation and prediction of timing. Neuroscience 462, 220–234 (2021).
Kunimatsu, J., Suzuki, T. W., Ohmae, S. & Tanaka, M. Different contributions of preparatory activity in the basal ganglia and cerebellum for self-timing. Elife 7, e35676 (2018).
Mauk, M. D. & Buonomano, D. V. The neural basis of temporal processing. Annu. Rev. Neurosci. 27, 307–340 (2004).
Zhou, S., Masmanidis, S. C. & Buonomano, D. V. Neural sequences as an optimal dynamical regime for the readout of time. Neuron 108, 651–658 (2020).
Ivry, R. B. & Spencer, R. M. C. The neural representation of time. Curr. Opin. Neurobiol. 14, 225–232 (2004).
Gerwig, M. et al. Trace eyeblink conditioning in patients with cerebellar degeneration: Comparison of short and long trace intervals. Exp. Brain Res. 187, 85–96 (2008).
Koekkoek, S. K. E. et al. Cerebellar LTD and learning-dependent timing of conditioned eyelid responses. Science 301, 1736–1739 (2003).
Grube, M., Cooper, F. E., Chinnery, P. F. & Griffiths, T. D. Dissociation of duration-based and beat-based auditory timing in cerebellar degeneration. Proc. Natl. Acad. Sci. USA 107, 11597–11601 (2010).
Ohmae, S., Kunimatsu, J. & Tanaka, M. Cerebellar roles in self-timing for sub- and supra-second intervals. J. Neurosci. 37, 3511–3522 (2017).
Ivry, R. B., Keele, S. W. & Diener, H. C. Dissociation of the lateral and medial cerebellum in movement timing and movement execution. Exp. Brain Res. 73, 167–180 (1988).
Spencer, R. M. C., Zelaznik, H. N., Diedrichsen, J. & Ivry, R. B. Disrupted timing of discontinuous but not continuous movements by cerebellar lesions. Science 300, 1437–1439 (2003).
Tsutsumi, S. et al. Purkinje cell activity determines the timing of sensory-evoked motor initiation. Cell Rep. 33, 1–15 (2020).
Hoang, H. et al. Dynamic organization of cerebellar climbing fiber response and synchrony in multiple functional components reduces dimensions for reinforcement learning. Elife 12, e86340 (2023).
Kuramoto, E. et al. Two types of thalamocortical projections from the motor thalamic nuclei of the rat: A single neuron-tracing study using viral vectors. Cereb. Cortex 19, 2065–2077 (2009).
Aoki, S., Coulon, P. & Ruigrok, T. J. H. Multizonal cerebellar influence over sensorimotor areas of the rat cerebral cortex. Cereb. Cortex 29, 598–614 (2019).
Tanaka, Y. H. et al. Thalamocortical axonal activity in motor cortex exhibits layer-specific dynamics during motor learning. Neuron 100, 244–258 (2018).
Alonso-Martínez, C., Rubio-Teves, M., Casas-Torremocha, D., Porrero, C. & Clascá, F. Cerebellar and basal ganglia inputs define three main nuclei in the mouse ventral motor thalamus. Front. Neuroanat. 17, 1–18 (2023).
Van Donkelaar, P., Stein, J. F., Passingham, R. E. & Miall, R. C. Temporary inactivation in the primate motor thalamus during visually triggered and internally generated limb movements. J. Neurophysiol. 83, 2780–2790 (2000).
Tanaka, M. Inactivation of the central thalamus delays self-timed saccades. Nat. Neurosci. 9, 20–22 (2006).
Nashef, A., Cohen, O., Harel, R., Israel, Z. & Prut, Y. Reversible block of cerebellar outflow reveals cortical circuitry for motor coordination. Cell Rep. 27, 2608–2619 (2019).
Shih, L. Y. L., Yeh, T. C., Kuo, W. J., Tzeng, O. J. L. & Hsieh, J. C. Effect of temporal difficulty on cerebrocerebellar interaction during visual duration discrimination. Behav. Brain Res. 207, 155–160 (2010).
Aso, K., Hanakawa, T., Aso, T. & Fukuyama, H. Cerebro-cerebellar interactions underlying temporal information processing. J. Cogn. Neurosci. 22, 2913–2925 (2010).
Smith, A., Taylor, E., Lidzba, K. & Rubia, K. A right hemispheric frontocerebellar network for time discrimination of several hundreds of milliseconds. Neuroimage 20, 344–350 (2003).
Terada, S. I., Kobayashi, K. & Matsuzaki, M. Transition of distinct context-dependent ensembles from secondary to primary motor cortex in skilled motor performance. Cell Rep. 41, 111494 (2022).
Murakami, M., Vicente, M. I., Costa, G. M. & Mainen, Z. F. Neural antecedents of self-initiated actions in secondary motor cortex. Nat. Neurosci. 17, 1574–1582 (2014).
Chabrol, F. P., Blot, A. & Mrsic-Flogel, T. D. Cerebellar contribution to preparatory activity in motor neocortex. Neuron 103, 506–519 (2019).
Gao, Z. et al. A cortico-cerebellar loop for motor planning. Nature 563, 113–116 (2018).
Ito, M. Cerebellar circuitry as a neuronal machine. Prog. Neurobiol. 78, 272–303 (2006).
Kawato, M., Ohmae, S., Hoang, H. & Sanger, T. 50 years since the Marr, Ito, and Albus models of the cerebellum. Neuroscience 462, 151–174 (2021).
Herzfeld, D. J., Kojima, Y., Soetedjo, R. & Shadmehr, R. Encoding of error and learning to correct that error by the Purkinje cells of the cerebellum. Nat. Neurosci. 21, 736–743 (2018).
Shadmehr, R., Smith, M. A. & Krakauer, J. W. Error correction, sensory prediction, and adaptation in motor control. Annu. Rev. Neurosci. 33, 89–108 (2010).
Imamizu, H. et al. Human cerebellar activity reflecting an acquired internal model of a new tool. Nature 403, 192–195 (2000).
Wagner, M. J., Kim, T. H., Savall, J., Schnitzer, M. J. & Luo, L. Cerebellar granule cells encode the expectation of reward. Nature 544, 96–100 (2017).
Sendhilnathan, N., Semework, M., Goldberg, M. E. & Ipata, A. E. Neural correlates of reinforcement learning in mid-lateral cerebellum. Neuron 106, 188–198 (2020).
Kostadinov, D. & Häusser, M. Reward signals in the cerebellum: origins, targets, and functional implications. Neuron 110, 1290–1303 (2022).
Hoang, H. et al. Predictive reward-prediction errors of climbing fiber inputs integrate modular reinforcement learning with supervised learning. PLoS Comput. Biol. 21, e1012899 (2025).
Zingg, B. et al. AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47 (2017).
Yoshida, E. et al. Whether or not to act is determined by distinct signals from motor thalamus and orbitofrontal cortex to secondary motor cortex. Nat. Commun. 16, 3106 (2025).
Tennant, K. A. et al. The organization of the forelimb representation of the C57BL/6 mouse motor cortex as defined by intracortical microstimulation and cytoarchitecture. Cereb. Cortex 21, 865–876 (2011).
Hira, R. et al. In vivo optogenetic tracing of functional corticocortical connections between motor forelimb areas. Front. Neural Circuits 7, 1–10 (2013).
Daigle, T. L. et al. A suite of transgenic driver and reporter mouse lines with enhanced brain-cell-type targeting and functionality. Cell 174, 465–480 (2018).
Kuramoto, E. et al. Ventral medial nucleus neurons send thalamocortical afferents more widely and more preferentially to layer 1 than neurons of the ventral anterior-ventral lateral nuclear complex in the rat. Cereb. Cortex 25, 221–235 (2015).
Dana, H. et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 16, 649–657 (2019).
Musall, S., Kaufman, M. T., Juavinett, A. L., Gluf, S. & Churchland, A. K. Single-trial neural dynamics are dominated by richly varied movements. Nat. Neurosci. 22, 1677–1686 (2019).
Stringer, C. et al. Spontaneous behaviors drive multidimensional, brainwide activity. Science 364, 255 (2019).
Li, W. R. et al. Neural mechanisms underlying uninstructed orofacial movements during reward-based learning behaviors. Curr. Biol. 33, 3436–3451 (2023).
Kondo, M. & Matsuzaki, M. Neuronal representations of reward-predicting cues and outcome history with movement in the frontal cortex. Cell Rep. 34, 108704 (2021).
Inagaki, H. K. et al. A midbrain-thalamus-cortex circuit reorganizes cortical dynamics to initiate movement. Cell 185, 1065–1081 (2022).
Dana, H. et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 5, e12727 (2016).
Kostadinov, D., Beau, M., Pozo, M. B. & Häusser, M. Predictive and reactive reward signals conveyed by climbing fiber inputs to cerebellar Purkinje cells. Nat. Neurosci. 22, 950–962 (2019).
Heffley, W. & Hull, C. Classical conditioning drives learned reward prediction signals in climbing fibers across the lateral cerebellum. Elife 8, e46764 (2019).
Larry, N., Yarkoni, M., Lixenberg, A. & Joshua, M. Cerebellar climbing fibers encode expected reward size. Elife 8, e46870 (2019).
Sul, J. H., Jo, S., Lee, D. & Jung, M. W. Role of rodent secondary motor cortex in value-based action selection. Nat. Neurosci. 14, 1202–1208 (2011).
Levy, S. et al. Cell-type-specific outcome representation in the primary motor cortex. Neuron 107, 954–971 (2020).
Danielmeier, C. et al. Acetylcholine mediates behavioral and neural post-error control. Curr. Biol. 25, 1461–1468 (2015).
Norman, K. J. et al. Post-error recruitment of frontal sensory cortical projections promotes attention in mice. Neuron 109, 1202–1213 (2021).
Fu, Z. et al. Single-neuron correlates of error monitoring and post-error adjustments in human medial prontal cortex. Neuron 101, 165–177 (2019).
Nashef, A., Cohen, O., Perlmutter, S. I. & Prut, Y. A cerebellar origin of feedforward inhibition to the motor cortex in non-human primates. Cell Rep. 39, 110803 (2022).
Washburn, S. et al. The cerebellum directly modulates the substantia nigra dopaminergic activity. Nat. Neurosci. 27, 497–513 (2024).
Ichinohe, N., Mori, F. & Shoumura, K. A di-synaptic projection from the lateral cerebellar nucleus to the laterodorsal part of the striatum via the central lateral nucleus of the thalamus in the rat. Brain Res. 880, 191–197 (2000).
Hoshi, E., Tremblay, L., Féger, J., Carras, P. L. & Strick, P. L. The cerebellum communicates with the basal ganglia. Nat. Neurosci. 8, 1491–1493 (2005).
Wang, X. et al. Deconstruction of corticospinal circuits for goal-directed motor skills. Cell 171, 440–455 (2017).
Ullsperger, M., Danielmeier, C. & Jocham, G. Neurophysiology of performance monitoring and adaptive behavior. Physiol. Rev. 94, 35–79 (2014).
Notebaert, W. et al. Post-error slowing: an orienting account. Cognition 111, 275–279 (2009).
Danielmeier, C., Eichele, T., Forstmann, B. U., Tittgemeyer, M. & Ullsperger, M. Posterior medial frontal cortex activity predicts post-error adaptations in task-related visual and motor areas. J. Neurosci. 31, 1780–1789 (2011).
Ide, J. S., Li, C. & shan, R. A cerebellar thalamic cortical circuit for error-related cognitive control. Neuroimage 54, 455–464 (2011).
Halverson, H. E., Khilkevich, A. & Mauk, M. D. Relating cerebellar Purkinje cell activity to the timing and amplitude of conditioned eyelid responses. J. Neurosci. 35, 7813–7832 (2015).
Kakegawa, W. et al. Optogenetic control of synaptic AMPA receptor endocytosis reveals roles of LTD in motor learning. Neuron 99, 985–998 (2018).
Ichise, T. et al. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science 288, 1832–1835 (2000).
Watabe-Uchida, M., Zhu, L., Ogawa, S. K., Vamanrao, A. & Uchida, N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873 (2012).
Hutson, T. H., Kathe, C. & Moon, L. D. F. Trans-neuronal transduction of spinal neurons following cortical injection and anterograde axonal transport of a bicistronic AAV1 vector. Gene Ther. 23, 231–236 (2016).
Terada, S. I., Kobayashi, K., Ohkura, M., Nakai, J. & Matsuzaki, M. Super-wide-field two-photon imaging with a micro-optical device moving in post-objective space. Nat. Commun. 9, 3550 (2018).
Giovannucci, A. et al. Cerebellar granule cells acquire a widespread predictive feedback signal during motor learning. Nat. Neurosci. 20, 727–734 (2017).
Dacre, J. et al. A cerebellar-thalamocortical pathway drives behavioral context-dependent movement initiation. Neuron 109, 2326–2338 (2021).
Thevenaz, P., Ruttimann, U. E. & Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41 (1998).
Pnevmatikakis, E. A. & Giovannucci, A. NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94 (2017).
Pnevmatikakis, E. A. et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron 89, 285–299 (2016).
Yoshida, E. et al. In vivo wide-field calcium imaging of mouse thalamocortical synapses with an 8 K ultra-high-definition camera. Sci. Rep. 8, 1–15 (2018).
Acknowledgements
We thank M. Nishiyama for animal care, Y.H. Tanaka and Y.R. Tanaka for useful advice on anatomical experiments, and S. Ishii for helpful discussion. This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas (17H06309 to M.M.), for Transformative Research Areas (A) (22H05160 to M.M.), and for Scientific Research (A) (19H01037 and 23H00388 to M.M.), and a JSPS Fellowship (20J12547 to R.A.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; AMED (JP22dm0207001 and JP22dm0107150 to M.M.).
Author information
Authors and Affiliations
Contributions
R.A. and M.M. designed the experiments. R.A. conducted all experiments and data analyses. S.-I.T. conducted preliminary data analyses. R.A. and M.M. wrote the paper, with comments from S.-I.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Andrei Khilkevich and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ako, R., Terada, SI. & Matsuzaki, M. A cerebello-thalamo-cortical pathway transmits reward-based post-error signals for motor timing correction during learning in male mice. Nat Commun 16, 7663 (2025). https://doi.org/10.1038/s41467-025-62831-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-62831-6
