
Abstract
Broadly neutralizing antibodies (bNAbs) offer a promising strategy for HIV prevention. Subcutaneous (SC) administration is more feasible than intravenous delivery but may be limited by prolonged administration times and multiple injections. Here we report a pharmacokinetic (PK) modelling study, an unspecified exploratory analysis that involved 57 HIV-negative African women (median age 25 years; BMI range 18.1–39.3 kg/m²) enrolled in the CAPRISA 012B trial (PACTR202003767867253, total participants n = 76). A predefined sub-analysis directly comparing the 20 mg/kg dose level of ENHANZE™ drug product (EDP) versus no-EDP was conducted in a subset of participants (n = 5 with EDP, n = 5 without). CAP256V2LS and VRC07-523LS—potent HIV-1 bNAbs targeting conserved envelope epitopes—were administered SC with and without EDP. The primary outcome of this sub-analysis was duration of administration. Secondary outcomes included PK and safety. Among the subset of participants (n = 10), EDP significantly reduced median administration time from 49.5 to 10.0 minutes and reduced injections per dose from 3 to 1. CAP256V2LS and VRC07-523LS concentrations at 24 weeks post-dose, were 4.8- and 3.0-fold higher, respectively, with EDP. CAP256V2LS exposure (AUC) increased by 40%, despite a 30% decrease in Cmax. EDP was well tolerated with no safety concerns. These findings support EDP-enhanced SC delivery as a scalable and simplified strategy for long-acting antibody-based HIV prevention.
Similar content being viewed by others

Introduction
The global HIV epidemic remains a major challenge, with 1.3 million new infections in 2023, about half of which occurred in sub-Saharan Africa (https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf). Long-acting pre-exposure prophylaxis like 6-monthly lenacapavir1 is needed, particularly in high-burden settings like South Africa. Broadly neutralizing antibodies (bNAbs) have emerged as a promising approach for HIV prevention, following the Antibody Mediated Prevention trial, which showed that while VRC01 did not provide overall efficacy, it was 75% effective against susceptible viral strains2. Combinations of bNAbs targeting multiple epitopes need to be investigated to address this shortcoming of a single antibody and may improve efficacy.
The efficacy of bNAbs depends on maintaining serum trough concentrations above their therapeutic thresholds. Intravenous administration provides higher serum concentrations and better bioavailability3 compared to subcutaneous administration. The World Health Organization (WHO) has outlined a set of Preferred Product Characteristics for bNAbs aimed at HIV prophylaxis (https://www.who.int/publications/i/item/9789240045729). Among these guidelines, WHO prioritizes subcutaneous and intramuscular injections as the preferred routes of administration. Subcutaneous administration of antibodies, which is easier to implement than intravenous administration, takes longer and can require multiple injections when substantial volumes are required. The subcutaneous extracellular matrix, made up of hyaluronan and collagen, limits injectable volumes to 1–2 mL, with larger volumes causing tissue distortion and pain4. This poses a challenge for bnAb administration, as weight-based dosing often exceeds this limit, requiring multiple injections to reach therapeutic levels, which can be time-consuming and uncomfortable for patients.
EDP uses a proprietary recombinant human hyaluronidase referred to as rHuPH20 (Halozyme Inc., San Diego, CA) to facilitate the subcutaneous delivery of co-administered therapies. EDP works by temporarily breaking down hyaluronan in the interstitial space, allowing for larger volumes of product to be administered in the subcutaneous space5.
Duration of administration, number of injections, and pharmacokinetic profiles were assessed when both CAP256V2LS and VRC07-523LS were administered subcutaneously with and without EDP in the CAPRISA 012B phase 1 trial6. Systemic antibody concentrations were assessed using the Meso Scale Discovery (MSD) platform. A population pharmacokinetic model that stratified individual exposure parameters within a virtual population of 1000, grouped by EDP use, was developed. These simulations included both intravenous and subcutaneous routes to ensure a comprehensive comparison of pharmacokinetic model outputs for the same population under both administration routes.
In this work, we showed that hyaluronidase-enhanced subcutaneous delivery using EDP offered a practical and scalable solution for administering bNAbs for HIV prevention.
Results
The study was approved by the University of KwaZulu-Natal Biomedical Research Ethics Committee (BREC) and the South African Health Products Regulatory Authority (SAHPRA) and was conducted at the CAPRISA eThekwini Clinical Research Site in Durban. The cohort comprised a subgroup of HIV-uninfected African women (n = 57), who were enrolled in the CAPRISA 012 trial (total number of participants, n = 76)6. The median age in the subset of participants (n = 10) was 27.5 years (IQR: 24.0–30.0), with a BMI range of 26.4–29.2 kg/m2 (Table 1).
Safety
Safety analysis of EDP was conducted in the subgroup of HIV-uninfected participants (n = 10). EDP use was safe. There were no serious adverse events or dose-limiting toxicities reported. Solicited reactogenicity events ranged from mild to moderate and resolved within the 72-h reactogenicity assessment period. No anti-drug antibodies (ADAs) were detected for CAP256V2LS, VRC07-523LS, or EDP across multiple sampling points.
Pharmacokinetics
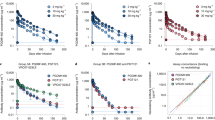
Subcutaneous administration of CAP256V2LS with EDP resulted in consistently higher median concentrations compared to administration without EDP. At months 1, 3, 4, and 6, concentrations were 0.86, 3.29, 2.99, and 4.75 times higher when EDP was used. Similarly, VRC07-523LS concentrations with EDP were also elevated compared to administration without EDP, with median concentrations 1.41, 2.78, 2.62, and 3.04 times higher at months 1, 3, 4, and 6 (Fig. 1). For the dose-normalized PK comparison, a subset of 42 participants was included—comprising individuals who received EDP (n = 20) or non-EDP regimens (n = 22) (Supplementary Table 1). EDP increased CAP256V2LS overall bioavailability as exposure displayed a 40% increase in area under the curve (AUC; p < 0.035). However, Cmax showed a 30% decrease (p = 0.046).

Error bars represent the 95% confidence intervals (CI). An inset for the first ~12 days is shown above.
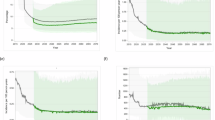
A pharmacokinetic model, based on data from study CAPRISA 012B6 (subgroup of 57 HIV-uninfected participants), was used to further explore the impact of EDP co-administration through simulations. The AUC over a 24-week period was simulated using the model for three weight-based dosing scenarios (5, 10, and 20 mg/kg). Initially, the route of administration—intravenous versus subcutaneous—significantly impacts exposure levels, particularly following a single dose (Supplementary Fig. 1). This difference, however, becomes negligible at later time points once steady state is achieved, where intravenous administration provides greater exposure than subcutaneous, regardless of whether EDP is co-administered (Fig. 2). Notably, co-administration of EDP consistently enhances PK exposure across all simulated dosing scenarios (Fig. 3).

In each case, groups with EDP have higher concentrations for equivalent doses, although it occurs in later weeks for the single dose profile. This results in more consistent differences following multiple doses. Solid lines represent median values of 1000 replicates from the model, while the ribbon represents the 95% confidence interval of these replicates. The pharmacokinetic model was based on the results from the complete CAPRISA 012B trial6.

Median values (and 95% CI) of 1000 simulated replicates from the pharmacokinetic model are presented. The model was based on the results from the complete CAPRISA 012B trial6. In each case, groups with EDP have higher exposure. The model is based on simulated data derived from HIV-uninfected participants who received CAP256V2LS alone or in combination and have completed follow-up to date.
The median antibody infusion duration was 10.0 (IQR: 6.0–15.0) min with EDP compared to 49.5 (IQR: 26.0–52.5) min without EDP (p = 0.003). A total of three manual syringe needles were used for the administration of the study product without EDP, compared to one injection with EDP.
Discussion
Subcutaneous antibody co-administration with EDP was safe, had a shorter duration, fewer injections, and an enhanced pharmacokinetic profile, which had a slightly lower peak concentration but higher concentrations and bioavailability up to 24 weeks post-administration.
The pharmacokinetic enhancement observed with EDP is driven by the enzymatic degradation of hyaluronan in the extracellular matrix, thereby enhancing tissue permeability and promoting faster and more efficient absorption of bNAbs from the subcutaneous injection site into systemic circulation. This mechanism ensures rapid diffusion and increased bioavailability from subcutaneous administration to levels comparable to intravenous delivery7,8,9. The effects on the extracellular matrix are localized, temporary, and reversible8. Hyaluronidase-mediated degradation of hyaluronan reduces depot retention time and accelerates mAb entry into circulation. In an in vitro SC injection chamber model, hyaluronidase addition significantly increased mAb diffusion from the injection site10,11. These in vitro ADME models, including diffusion chambers and 3D tissue constructs, are widely used to predict SC bioavailability and absorption profiles of biologics. Preclinical data support this mechanism, showing that co-formulated hyaluronidase increases absolute mAb bioavailability. For instance, in rats, hyaluronidase increased SC cetuximab bioavailability from ~67% to ~80%, improving systemic uptake12. While its impact varies depending on the mAb (e.g., minimal effect on trastuzumab, which already has high subcutaneous absorption), in vitro and in vivo studies consistently confirm that hyaluronidase enhances subcutaneous mAb pharmacokinetics9,13.
Previous studies demonstrated the suboptimal bioavailability and lower serum concentrations of the bNAbs VRC01LS, VRC07-523LS, PGT121, and PGDM1400 when administered subcutaneously compared to intravenous dosing14,15,16,17. In contrast, in this study, the co-administration of EDP significantly improves the pharmacokinetic profile of subcutaneous CAP256V2LS and VRC07-523LS.
SC administration did not appear to change the observed t1/2, with or without EDP. As demonstrated with trastuzumab and rituximab, EDP does not alter antibody pharmacokinetics, metabolism, or structural integrity, and is unlikely to directly impact elimination kinetics5,13,18. Therefore, the mechanism for the significant impact of EDP on systemic clearance, in addition to absolute bioavailability, is not clear, but may be the result of confounding factors on covariate assessment due to the lack of crossover in the study design. Immunogenicity testing in this study confirmed the absence of ADAs against EDP, CAP256V2LS, or VRC07-523LS, further supporting the pharmacokinetic stability of these antibodies.
Of note, CAP256V2LS showed a greater relative increase in AUC and a 37.8% reduction in clearance compared to VRC07-523LS, likely due to differences in molecular characteristics, Fc receptor interactions, and glycan composition19. Higher and more sustained plasma concentrations of bNAbs could improve protection against HIV acquisition, while the prolonged presence of therapeutic antibody levels might allow for less frequent dosing. Importantly, the capacity to administer larger volumes subcutaneously opens up opportunities for combining 2, 3, or 4 bnAbs, addressing concerns related to viral escape and incomplete neutralization. The use of EDP also reduced infusion administration time, which could result in shorter clinic visits and an improved patient experience.
Administration of bNAbs with EDP has important advantages in real-world settings, where administration time and number of injections can markedly influence whether an intervention is practically feasible for scale-up. In our controlled study, bNAbs were co-mixed with EDP in the research pharmacy and administered using a subcutaneous pump. To enhance feasibility and accessibility in diverse healthcare environments, alternative delivery strategies are available. One promising approach is the sequential administration of rHuPH20 followed by bNAbs. This method simplifies the process by eliminating the need for co-mixing or specialized formulation, significantly reducing the amount of rHuPH20 required—by more than tenfold. Additionally, sequential administration allows for manual syringe-based delivery, which can be performed by healthcare workers or trained caregivers, making it particularly suitable for decentralized or resource-limited settings where flexibility is critical.
Several monoclonal antibodies used in cancer treatment have now been licensed as co-formulated products utilizing EDP technology20. HyQvia is an immune globulin infusion (10%) with rHuPH20 that is approved for primary immunodeficiency and allows for single-site injection21, designed to be self-administered22,23. The CAPRISA 012B trial is the first to evaluate EDP with anti-HIV bNAbs6. The use of EDP is currently being assessed with N6LS in a phase 1 trial (NCT03538626), and there are ongoing explorations for its application with long-acting injectable cabotegravir.
While our study provides valuable insights, the relatively small sample size and focus on young South African women may limit the generalizability of our findings to other populations. Prior bNAb trials have demonstrated variability in PK outcomes across diverse demographics, including differences in BMI, immune activation, and comorbidities24. Future studies should aim to validate these findings in larger and more diverse populations to ensure the broader applicability of EDP-facilitated bNAb administration. Our study needed to categorize the impact of EDP on Cmax and the post-administration peak better, but this was not possible due to the limited number of early time points. Although the mechanisms governing the subcutaneous absorption of large molecules like bNAbs remain unclear, external factors such as needle insertion depth and infusion rate likely influence bioavailability and may contribute to the observed variability in absorption.
In confronting the challenges of the HIV epidemic, particularly in high-burden settings, EDP-facilitated bNAb administration offers a promising enhancement to a potential long-acting HIV prevention strategy. This approach could make the implementation of bNAb-based HIV prevention easier and practically viable, especially in resource-limited settings. Further research, including mechanistic studies, is needed to refine subcutaneous administration protocols for practical implementation in diverse real-world settings.
Methods
Study design and participants
This phase 1 trial was conducted at the CAPRISA eThekwini Clinical Research Site in Durban, South Africa. The protocol was reviewed and approved by the University of KwaZulu-Natal BREC and the SAHPRA and is available at https://www.caprisa.org/Pages/EDCTP-funded%20studies. The trial was registered on the Pan African Clinical Trial Registry, PACTR202003767867253. Volunteers were recruited from Durban and surrounding areas within KwaZulu-Natal using BREC-approved study materials. Written informed consent was obtained from all participants prior to their enrollment in the study, in accordance with ethical guidelines and regulatory requirements. Following successful eligibility assessments, 57 participants (median age 25 years; BMI range 18.1–39.3 kg/m2) were enrolled into one of four groups (Supplementary Table 2)6.
The data included in this study are derived exclusively from HIV-uninfected participants (n = 57). At the time of this analysis, the HIV-positive groups remain in follow-up, and no complete datasets from these participants were available for inclusion. This analysis was exploratory in nature and focused specifically on evaluating the pharmacokinetic impact of co-administering recombinant human hyaluronidase (EDP) among HIV-negative individuals. The objective was to better understand how EDP influences the pharmacokinetic profile of the administered bNAbs.
Fifty-seven HIV-negative women aged 18–40 years were included in this study and received bNAbs formulated with ENHANZE™ drug product (EDP) and without EDP (Supplementary Table 2). Because abdomen-site limitations required equivalent injection volumes, only the 20 mg kg cohorts were dosed with identical antibody combinations ± EDP, permitting a like-for-like comparison. Five participants in each 20 mg/kg cohort (total n = 10), therefore, comprised the pre-specified sub-analysis used for direct EDP versus no-EDP evaluation. All other HIV-uninfected groups (n = 57) contributed to population PK modeling (Fig. 4).

The dosing groups evaluating different administration strategies for bNAbs. CAP256V2LS and VRC07-523LS were delivered subcutaneously, both with and without recombinant human hyaluronidase (ENHANZE™ Drug Product, or EDP).
Pharmacokinetic analysis cohort: PK model and dose-normalized comparison
A total of 57 participants were enrolled across all study arms, and plasma concentration-time data from all 57 participants were included in the development of the population PK model. For the dose-normalized PK comparison, a subset of 42 participants was included—comprising individuals who received EDP (n = 20) or non-EDP regimens (n = 22) (Supplementary Table 1). Participants in Groups 1A and 1B were originally assigned to the non-EDP arm at 5 mg/kg and 10 mg/kg IV dosing, respectively. However, only three participants from these groups were included in the dose-normalized PK analysis. Two participants—one from Group 1A and one from Group 1B—were excluded due to end-of-infusion sampling times occurring later than 1 h post-dose. Repeat dosing groups from Group 2 were also excluded. These exclusions were made a priori to maintain consistency in PK parameter estimation.
Study product administration
CAP256V2LS and VRC07-523LS were individually mixed with EDP in the clinic pharmacy, and each antibody was provided as a single individual dose. Each antibody was administered sequentially at different sites of the abdomen to allow distinction of local reactogenicity. For the subcutaneous groups that utilized EDP, the product was administered into the abdomen at a single site via an infusion pump at a rate of 1 ml per minute. For the subcutaneous group without EDP, the product was given as a manual syringe injection technique with a maximum of 2.5mls per syringe, or the product was administered into the abdomen at a single site via an infusion pump at a rate of 15 ml per hour.
Determination of bNAb serum levels
A quantitative electrochemiluminescence sandwich immunoassay technique was performed on the MSD platform to individually determine CAP256V2LS and VRC07-523LS concentrations in plasma samples. The amount of CAP256V2LS and VRC07-523LS sandwiched by the anti-ID and anti-human IgG antibodies was directly proportional to the concentration of reactive CAP256V2LS and VRC07-523LS in each sample. Measurements were taken from distinct samples. Sample concentrations were interpolated from standard curves using Excel and GraphPad Prism Software 9.2.0 (GraphPad Software, La Jolla, CA, USA). A non-compartmental analysis on the PK was performed using WinNonLin (Certara, Princeton, NJ).
Immunogenicity testing for the detection of anti-rHuPH20 antibodies
This study employed a validated electrochemiluminescence (ECL) immunoassay to detect and quantify anti-rHuPH20 antibodies in plasma samples, conducted at Labcorp Bioanalytical Services. A total of 173 primary samples and 289 backup samples were received, stored at −60 to −80 °C, and handled according to the study protocol. Controls, including positive, negative, and immunodepletion controls, were used across all runs. All controls were validated within known stability parameters and prepared under controlled conditions. The assay was conducted on a 96-well streptavidin-coated plate platform, using biotin-conjugated rHuPH20 as the primary detection reagent and SULFO TAG-conjugated rHuPH20 for secondary detection. The ECL assay was conducted on the MSD platform with Sulfo-tagged reagents for precise antibody detection through relative light unit (RLU) readings. In the screening phase, samples with RLU values above the set threshold were flagged as potential positives. Confirmatory testing involved reassaying positive samples with additional controls to validate the results. Samples that confirmed positive were subsequently titered to quantify the concentration of anti-rHuPH20 antibodies. Eleven analytical runs were accepted, while three runs were rejected due to control criteria not being met. Data from rejected runs were not included in the final analysis. Nineteen samples were reassayed due to normal tier escalation. Fourteen of the nineteen initially screened positive samples confirmed positive. The analysis confirmed a low incidence of ADA development.
Pharmacokinetics analysis
A two-compartment model was developed using NONMEM version 7.5 (ICON Clinical Research LLC, Blue Bell, PA, USA) for population pharmacokinetic analysis. The non-linear mixed effect model (NLME) approach was utilized to account for inter-individual variability. R® V4.4 (or higher), along with a comprehensive R archive network and Certara packages, was used for dataset preparation, exploratory data analysis, visualization, and calculation of individual Bayesian posterior exposure levels.
Population pharmacokinetic model
The pharmacokinetic model development for CAP256V2LS involved analyzing data from participants who had received at least one dose of CAP256V2LS and had one measurable concentration sample (n = 57, Fig. 4). Concentrations below the lower limit of quantitation (BLQ) were excluded. Initially, data exploration guided the structural model development, using standard statistical criteria such as the −2 log-likelihood and graphical diagnostics, including plots of observed vs. predicted values. A stepwise covariate analysis was performed to identify factors influencing CAP256V2LS PK. Continuous covariates were scaled relative to the median value, while categorical covariates were referenced against the most common group. The stepwise forward addition and backward elimination procedure with predefined significance levels (0.05 for inclusion and 0.01 for removal) helped refine the model, ensuring physiologically and pharmacologically relevant covariates were included. The final model was assessed using standard diagnostic plots and visual predictive checks, simulating concentration-time profiles based on 1000 replicates for comparison with observed data to validate the model’s predictive accuracy.
Anti-drug antibody assay
A three-tiered approach was used to evaluate ADAs using an MSD ECL bridging assay (tiers 1 and 2) and a neutralization assay (tier 3). Briefly, for the tier 1 assay, we incubated serially diluted sera with a fixed amount of SULFO TAG CAP256V2LS or VRC07-523LS and corresponding biotinylated CAP256V2LS or VRC07-523LS. The mixture was added to streptavidin-coated MSD plates and analyzed with an MSD Sector instrument. ECL intensity higher than the pre-determined positivity cut-off point was considered positive. Positive sera were then evaluated in the tier 2 assay, where samples were pre-incubated with and without unlabeled CAP256V2LS or VRC07-523LS and evaluated for a reduction of ECL intensity. We used a tier 3 confirmatory HIV-1 neutralization assay to functionally characterize samples that were tier 2 positive. For this assay, we added unlabeled CAP256V2LS or VRC07-523LS to the serum sample at the antibody IC80 concentration of the DU156·12 pseudovirus. Any neutralization readout lower than 50% neutralization by the spiked sample was reported to have functional ADA.
Statistics and reproducibility
The CAPRISA 012B study was divided into four distinct groups evaluating different administration strategies for bNAbs (Fig. 4). CAP256V2LS and VRC07-523LS were delivered subcutaneously, both with and without recombinant human hyaluronidase (ENHANZE™ Drug Product, or EDP).
Group 1 involved a dose-escalation of CAP256V2LS administered intravenously. Group 2 assessed subcutaneous dose-escalation of CAP256V2LS. Group 3 evaluated the subcutaneous co-administration of CAP256V2LS and VRC07-523LS in a dose-escalation format. Detailed findings for these groups have been reported by Mahomed et al.6
In Group 4, participants received fixed doses of antibodies with or without EDP, divided into three subgroups: 1200 mg CAP256V2LS or placebo (4a), 1200 mg VRC07-523LS or placebo (4b), and 1200 mg CAP256V2LS followed by 1200 mg VRC07-523LS or placebo (4c). Each subgroup included five participants in a 4:1 ratio of active treatment to placebo.
Participants were excluded based on factors relating to retention, clinical decision, and declined enrollment (Fig. 4).
Summary statistics were presented as medians and interquartile ranges for continuous variables, while categorical variables were summarized using frequencies and percentages.
Data availability
The data generated in this study are provided in the Supplementary Information/Source data files Source data are provided with this paper.
References
Bekker, L. G. et al. Twice-yearly lenacapavir or daily F/TAF for HIV prevention in cisgender women. N. Engl. J. Med. 391, 1179–1192 (2024).
Corey, L. et al. Two randomized trials of neutralizing antibodies to prevent HIV-1 acquisition. N. Engl. J. Med. 384, 1003–1014 (2021).
Bittner, B., Richter, W. & Schmidt, J. Subcutaneous administration of biotherapeutics: an overview of current challenges and opportunities. BioDrugs 32, 425–440 (2018).
Sequeira, J. A. D. et al. Subcutaneous delivery of biotherapeutics: challenges at the injection site. Expert Opin. Drug Deliv. 16, 143–151 (2019).
Locke, K. W., Maneval, D. C. & LaBarre, M. J. ENHANZE®drug delivery technology: a novel approach to subcutaneous administration using recombinant human hyaluronidase PH20. Drug Deliv. 26, 98–106 (2019).
Mahomed, S. et al. Safety and pharmacokinetics of escalating doses of neutralising monoclonal antibody CAP256V2LS administered with and without VRC07-523LS in HIV-negative women in South Africa (CAPRISA 012B): a phase 1, dose-escalation, randomised controlled trial. Lancet HIV 10, e230–e243 (2023).
Jackisch, C., Müller, V., Maintz, C., Hell, S. & Ataseven, B. Subcutaneous administration of monoclonal antibodies in oncology. Geburtshilfe Frauenheilkd. 74, 343–349 (2014).
Kang, D. W. & Tannenbaum, R. ENHANZE® drug delivery technology: advancing subcutaneous drug delivery using recombinant human hyaluronidase PH20 (Karger Medical and Scientific Publishers, 2022).
Nolan, R. P. & Printz, M. A. Modeling the subcutaneous pharmacokinetics of antibodies co-administered with rHuPH20. Clin. Transl. Sci. 17, 13788–e13788 (2024).
Kinnunen, H. M. et al. A novel in vitro method to model the fate of subcutaneously administered biopharmaceuticals and associated formulation components. J. Control. Release 214, 94–102 (2015).
Bown, H. K. et al. In vitro model for predicting bioavailability of subcutaneously injected monoclonal antibodies. J. Control. Release 273, 13–20 (2018).
Kagan, L., Turner, M. R., Balu-Iyer, S. V. & Mager, D. E. Subcutaneous absorption of monoclonal antibodies: role of dose, site of injection, and injection volume on rituximab pharmacokinetics in rats. Pharm. Res 29, 490–499 (2012).
Frost, G. I. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 4, 427–440 (2007).
Gaudinski, M. R. et al. Safety and pharmacokinetics of the Fc-modified HIV-1 human monoclonal antibody VRC01LS: A Phase 1 open-label clinical trial in healthy adults. PLoS Med. 15, e1002493 (2018).
Gaudinski, M. R. et al. Safety and pharmacokinetics of broadly neutralising human monoclonal antibody VRC07-523LS in healthy adults: a phase 1 dose-escalation clinical trial. Lancet HIV 6, e667–e679 (2019).
Stephenson, K. E. et al. Safety, pharmacokinetics and antiviral activity of PGT121, a broadly neutralizing monoclonal antibody against HIV-1: a randomized, placebo-controlled, phase 1 clinical trial. Nat. Med. 27, 1718–1724 (2021).
Sobieszczyk, M. E. et al. Safety, tolerability, pharmacokinetics, and immunological activity of dual-combinations and triple-combinations of anti-HIV monoclonal antibodies PGT121, PGDM1400, 10-1074, and VRC07-523LS administered intravenously to HIV-uninfected adults: a phase 1 randomised trial. Lancet HIV 10, e653–e662 (2023).
Rosengren, S. et al. Clinical immunogenicity of rHuPH20, a hyaluronidase enabling subcutaneous drug administration. AAPS J. 17, 1144–1156 (2015).
Zhang, B. et al. Engineering of HIV-1 neutralizing antibody CAP256V2LS for manufacturability and improved half life. Sci. Rep. 12, 17876 (2022).
Bittner, B. et al. ENHANZE® Drug Delivery Technology. (2021).
Wasserman, R. L. Clinical practice experience with HyQvia in adults using alternative dosing regimens and pediatric patients: a retrospective study. Adv. Ther. 37, 1536–1549 (2020).
Angelotti, F. et al. Long-term efficacy, safety, and tolerability of recombinant human hyaluronidase-facilitated subcutaneous infusion of immunoglobulin (Ig) (fSCIG; HyQvia(®)) in immunodeficiency diseases: real-life data from a monocentric experience. Clin. Exp. Med. 20, 387–392 (2020).
Ravasio, R. & Ripoli, S. Cost-minimization analysis of HYQVIA® in the treatment of primary immunodeficiency disease (PID) and secondary immunodeficiency disease (SID) in Italy. AboutOpen 10, 69–77 (2023).
Bensalem, A. & Ternant, D. Pharmacokinetic variability of therapeutic antibodies in humans: a comprehensive review of population pharmacokinetic modeling publications. Clin. Pharmacokinet. 59, 857–874 (2020).
Acknowledgements
We thank the study team and the study participants for their contribution to the CAPRISA 012B trial and for furthering HIV prevention research. We thank Halozyme Therapeutics, Incorporated, for the provision of EDP, and we thank Mayank Patel and Kenny Israni for their collaboration. We thank the NIAID VRC Vaccine Production Program, Office of Regulatory Science, Clinical Trials Program, and Vaccine Immunology Program, and the VRC Pilot Plant, operated by the Vaccine Clinical Materials Program (VCMP), Leidos Biomedical Research, Inc., for study product provision. We thank Labcorp for their collaboration on the immunogenicity testing. We thank the CAPRISA 012B protocol and study team, the CAPRISA 012B Protocol Safety Review Team, and the CAPRISA 012B Data and Safety Monitoring Board members for providing trial oversight. This study was supported principally by the European and Developing Countries Clinical Trials Partnership (EDCTP Grant number: RIA2017S (PI: S.A.K.)). Funding was also provided by the South African Medical Research Council with funds from the South African Department of Science and Innovation and the Department of National Health through its Special Initiative on HIV Prevention Technology (PI: S.A.K.). Funding to the VRC for this study was provided by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. VRC07-523LS and CAP256V2LS were manufactured and supplied by the Vaccine Research Center of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. The study investigators had complete control over the design of the study; collection, analysis, and interpretation of data; and writing the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication. The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Author information
Authors and Affiliations
Contributions
S.A.K. is the Principal Investigator of CAPRISA 012B. Q.A.K. and S.M. are Co-Principal investigators. F.O., M.B., and R.H.O. conducted the statistical analysis, P.K. analysis, and modeling. S.M. contributed to clinical investigations and sample collection. S.S.A.K., Q.A.K., and S.M. contributed to the planning and conduct of the trial. K.C. and R.K. were part of the antibody development. J.W., M.M., K.L., S.N., B.L., M.C., and L.S. conducted the laboratory assays. S.M. wrote the first draft of the manuscript. All authors reviewed the final draft of the manuscript and approved the final version. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
S.S.A.K. is listed on patent applications involving CAP256V2LS. The mentioned patent, WO 2015/128846 A1, pertains to a family of broadly neutralizing monoclonal antibodies (bNAbs) derived from donor CAP256, specifically those targeting the V1V2 region of the HIV-1 envelope glycoprotein. The patent covers: the antibodies themselves, including specific members of the CAP256-VRC26 lineage (e.g., CAP256-VRC26.01 through CAP256-VRC26.33); nucleic acid sequences encoding these antibodies; pharmaceutical compositions comprising the antibodies or their encoding sequences; vectors and host cells used for expression of these antibodies; methods of treatment and prophylaxis for HIV-1 infection using the antibodies or encoded sequences, and diagnostic and kit-based applications using these antibodies for detecting HIV infection or immune responses. The invention is co-owned by several institutions, including the U.S. Department of Health and Human Services (NIH), Columbia University, the University of Witwatersrand, the National Health Laboratory Service, and CAPRISA, with inventors including SSAK. There are no restrictions on the publication of scientific data derived from research involving these antibodies, provided such data are generated under appropriate ethical and legal frameworks. Researchers are generally required to acknowledge the source and intellectual property rights of the originating institutions when publishing. The patent does not impede the publication of neutralization data, pharmacokinetic or immunogenicity studies, or findings related to public health relevance. However, commercial exploitation of the antibody (e.g., manufacturing or selling derived products) would require a licensing agreement. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mahomed, S., Osman, F., Beliveau, M. et al. Hyaluronidase-enhanced subcutaneous delivery of bNAbs: a phase 1 randomized controlled clinical trial in HIV-uninfected women. Nat Commun 16, 8177 (2025). https://doi.org/10.1038/s41467-025-63051-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63051-8
