
Abstract
N-Cyano amides are pivotal in agrochemicals, biologically active compounds, and nitrogen-containing heterocycles synthesis. Here, we present an umpolung cyanation strategy for the synthesis of N-cyano amides. By applying the readily accessible O-tosyl hydroxamates as nitrogen electrophiles, direct nucleophilic cyanation can be achieved with trimethylsilyl cyanide (TMSCN) under mild and transition metal-free conditions. This protocol features excellent functional group tolerance, easy scalability, and broad substrate scope (over 70 examples). Moreover, preliminary experimental studies and density functional theory (DFT) calculations support an SN2-type mechanism of this reaction. Our work not only provides efficient, robust, and practical approaches to a variety of useful and complex N-cyano amide molecules but also expands the concept and scope of SN2-type reaction at the non-anomeric amide electrophilic nitrogen center.
Similar content being viewed by others

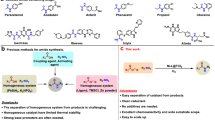

Introduction
N-Cyanoamides are important structural motifs commonly found in a wide array of agrochemicals1,2,3 and bioactive molecules4,5,6 (Fig. 1A). Moreover, they are versatile intermediates in synthetic organic chemistry for accessing biologically active nitrogen-containing heterocycles, such as luotonin A, guanidines, and vasicinone and its derivatives7,8,9,10,11,12,13. As such, novel synthetic methods to produce N-cyanoamides from robust and commercially available starting materials would be of general interest and important synthetic value. Historically, the classical methods for accessing N-cyanoamides, pioneered by Harrison, Courillon, Malacria, and others heavily relied on using cyanogen halides (e.g., BrCN) to furnish electrophilic cyanation reactions with amines or amides as nucleophiles (Fig. 1B and Eq. (1))7,14,15,16. However, these protocols generally suffer from significant drawbacks and limitations, such as limited substrate scope, harsh reaction conditions, and the use of highly volatile and toxic cyanogen halides17,18,19. An umpolung strategy that uses electrophilic amide precursors presents an attractive alternative as it allows for using more easily handled and less toxic nucleophilic trimethylsilyl cyanide (TMSCN) as a reaction partner under mild conditions. Very recently, an elegant study by Norton and co-workers has demonstrated the umpolung synthesis of N-cyanoamides by the reaction of in situ generated copper nitrenoid with TMSCN followed by copper-catalyzed alkylation with alkyl halides (Fig. 1C and Eq. (2))20. Despite this important advance, this protocol still required the use of a highly reactive nitrene intermediate, a transition metal catalyst, and a strong base. In this context, a general, efficient, and operationally straightforward method to prepare N-cyanoamides continues to be of considerable synthetic interest and remains a significant challenge. Our previous investigation on nucleophilic substitution reactions at the non-anomeric amide nitrogen center21 led us to hypothesize that O-tosyl hydroxamates bearing electrophilic amide nitrogen could provide unexplored opportunities for direct N-centered umpolung synthesis of N-cyanoamides (Fig. 1D and Eq. (3)).

A Selected examples of N-cyanoamide-containing bioactive molecules. B Classic routes to N-cyanoamides. C Previous Cu-catalyzed nitrene transfer & N-alkylation to form N-cyanoamides. D Our design and this work: N-centered umpolung enabled N-cyanoamides synthesis. Boc tert-butyloxycarbonyl, Me methyl, TMS trimethylsilyl, t-Bu tert-butyl, Ts tosyl.
Herein, we report an operationally simple protocol for the umpolung preparation of N-cyanoamides using readily accessible O-tosyl hydroxamates and TMSCN under mild and transition metal-free conditions (Fig. 1D and Eq. (3)). This reaction features good functional-group compatibility (over 70 examples), scalability, and late-stage approved drug and complex molecule modifications, thus enabling efficient and robust synthesis of structurally diverse N-cyanoamides. Moreover, this method expands the concept and scope of SN2-type reaction at the non-anomeric amide electrophilic nitrogen center22,23,24.
Results
Optimization of the reaction conditions
We commenced our study using N-methyl-N-(tosyloxy)benzamide 1 and TMSCN as the model system to identify suitable reaction parameters (Table 1, for more details see Supplementary Table 1). We were pleased to find that employing CsF as a base in MeCN at 0 °C afforded product 2 in 94% GC yield (entry 1, 90% isolated yield). A control experiment without the use of CsF did not produce any product, with 95% of the starting material recovered (entry 2), indicating that alkali-metal fluoride as the base played a vital role in promoting the reaction. Indeed, replacing CsF with a less soluble KF as the base, which may result in a low concentration of cyanide anion, only gave the product 2 in 48% yield (entry 3). Moreover, the reaction did not proceed when poorly soluble NaF was applied instead of CsF, with 90% recovery of the starting material (entry 4)25. Notably, using RbF as the base could give the desired product 2 in 86% yield, likely due to its similar solubility to CsF in MeCN (entry 5). Nevertheless, its higher cost led us to prefer the conditions outlined in entry 1. Other solvents, such as DMSO, DMF, and THF, were also tested but resulted in lower yields (entries 6–8). Increasing the reaction temperature to 25 °C and 60 °C, the yield of 2 dropped to 73% and 33%, respectively (entries 9 and 10). The mass balance in entry 10 was primarily a byproduct 2’ (13% isolated yield) derived from 1. To highlight the practical utility of current reaction conditions, the reaction was conducted at a 10 mmol scale, delivering 2 in 83% isolated yield (entry 11). Finally, control experiments confirmed tosylate (–OTs) is the best leaving group; specifically, the use of mesylate (–OMs, entry 12) and nosylate (–ONos, entry 13) generated 2 in 71% and 33% yields, respectively. In contrast, acetoxy (–OAc, entry 14), triflate (–OTf, entry 15), and methoxy (–OMe, Weinreb amide, entry 16) resulted in no formation of 2 under the optimized conditions.
Substrate scope
With the optimal conditions established, we first examined the scope of the umpolung N-cyanation reaction with regard to the substituents on the carbonyl group in O-tosyl hydroxamates (Fig. 2A). We were pleased to observe that aromatic rings with electro-donating and electro-withdrawing groups smoothly underwent N-cyanation reactions (3–15). Various functional groups such as nitrile (6), trifluoromethyl (7), halogens (Br, Cl, F; 8–10), ethynyl (11), and ester (12) were well tolerated in the reaction, resulting in N-cyanoamide products in moderate to good yields. Moreover, heteroaromatic rings, including furan (16), thiophene (17), and benzo[b]thiophene (18) were also compatible with this reaction. The structure of 18 (CCDC 2380286) was confirmed by X-ray crystallography. With respect to aliphatic substituents on the carbonyl group in O-tosyl hydroxamates, simple alkyl chains and rings (19–24) as well as functionalized alkyl chains, such as olefin (25), and pyridine (26), were tolerated. In addition, the styryl group (27) was also compatible well with the reaction.

A Variation of substituents on the carbonyl group in O-tosyl hydroxamates. B Variation of N-substitution in O-tosyl hydroxamates nitrogen. C Chemoselective N-cyanation. D Unsuccessful substrates. Reaction conditions: O-tosyl hydroxamate (0.1 mmol, 1.0 equiv.), TMSCN (1.5 equiv.) and CsF (2.0 equiv.) in MeCN (0.05 M), air, 0 °C, 8–20 h. aTMSCN (2.5 equiv.) and CsF (3.0 equiv.) were used in MeCN (0.05 M), air, 50 °C, 2–7 h. b0.3 mmol scale. c2.0 equiv. of 18-crown-6 was used. brsm based on recovered starting material.
We then focused on the variation of N-substitution in O-tosyl hydroxamates nitrogen, which could be easily achieved through Mitsunobu reaction or reductive amination (Fig. 2B)26,27,28,29,30,31. Simple primary alkyl chain substituents (28–33) were amenable to the reaction. Notably, a variety of synthetically useful unsaturated carbon–carbon bonds, including internal and terminal alkynes (34 and 35), olefins (36 and 37), and dienes (38), which provide further opportunities for structural diversification, all delivered the desired products in good yields.
Moreover, this reaction exhibited excellent functional group compatibility in its tolerance of fluoro (39), trifluoromethyl (40), esters (43 and 44), sulfonamides (46–48, 50), carbamate (57), and phthalimide (51). Of note, medicinally relevant heterocyclic scaffolds32,33,34, such as dioxolane (41 and 42), quinoline (43), tetrahydrofuran (44), thiophene (45), piperidine (46 and 48), tetrahydroisoquinoline (47), tetrahydropyran (49), and indole (50), were also successfully engaged under these reaction conditions. Importantly, the methyl-d3 group35,36, readily obtained from deuterated methanol, was also compatible well, providing the isotopically N-cyanoamide 52 in 89% yield. Secondary alkyl group substituents were represented by isopropyl (53), cyclohexyl (54), tetrahydropyranyl (55), cyclobutyl (56), azetidine (57), and, albeit with moderate yields of N-cyanoamides. In the cases of compounds 21, 23, 27, 41, 42, and 53–55, we found that full conversion of the corresponding O-tosyl hydroxamates was achieved using modified conditions (2.5 equiv. TMSCN, 3.0 equiv. CsF, and 50 °C) to ensure good reactivity.
We then evaluated the compatibility of functional groups during cyanation using O-tosyl hydroxamates possessing sterically unhindered alkyl C–X bonds (X = Cl, OTs, OMs, Br, I) (Fig. 2C). The cyanation occurred selectively at the amide N–OTs group, resulting in excellent yields of N-cyanoamides 58 and 59, while preserving the C–Cl bonds. Importantly, the more reactive alkyl tosylates, mesylates, bromides, and even iodides did not change the inherent chemoselectivity of this reaction. We successfully obtained N-cyanoamides 60–63 from the corresponding amide N–OTs in 38–50% yield, and no cyanation products derived from the reaction of the alkyl C–X bonds were detected. This chemoselectivity indicates that amide N–OTs exhibit higher nucleophilic substitution activity compared to the primary sp³ C–X bonds, which is presumably due to the lower bond dissociation energies (BDEs) of amide N–O bond (∼40 kcal mol−1) with respect to that of primary alkyl C–X bonds (C–Cl: ∼84 kcal mol−1, C–Br: ∼71 kcal mol−1, and C–I: ∼56 kcal mol−1)37,38.
Finally, the reaction demonstrated sensitivity to the steric effects associated with the N-substituents of O-tosyl hydroxamates. For instance, when the nitrogen atom of O-tosyl hydroxamates carried a tert-butyl or phenyl substituent, no desired N-cyanation product was produced (Fig. 2D). This is likely due to the significant steric hindrance presented by these groups. Overall, broad substituent tolerance to an extensive range of functionalities in O-tosyl hydroxamates was realized.
Synthetic utilities
To explore the utility of this new approach with substrates to drug discovery, we next examined the compatibility of pharmaceutical- or biologically active molecule-derived O-tosyl hydroxamates in this reaction (Fig. 3A). We were pleased to find that substrates derived from approved drugs and complex molecules, such as febuxostat (64, X-ray CCDC 2380287), gemfibrozil (65), ibuprofen (66), ketoprofen (67), naproxen (68), probenecid (69), adapalene (70), rimonabant acid (71), isoxepac (72), pregabalin (73), and estrone (74) successfully yielded the corresponding N-cyanoamide in 51–90% yield. Moreover, tepoxalin 75, a nonsteroidal anti-inflammatory drug that has recently been repurposed for anti-tumor studies39,40,41,42, can be effectively converted to its cyanide analog 76 via a two-step modification by applying (1) O-tosylation chemistry (4.6 mmol scale, 73% yield), and (2) the present umpolung N-cyanation (1.0 mmol scale, 93% yield) methodology (Fig. 3B). The structure of compound 76 was confirmed by X-ray crystallography (CCDC 2401552). Again, the robustness of the umpolung N-cyanation reaction was demonstrated by scale-up experiments (Fig. 3C). For instance, several randomly selected reactions were tested for their effectiveness, resulting in the formation of N-cyanoamide products: 4 (1.77 grams, 82%), 6 (1.66 grams, 60%), 28 (2.27 grams, 70%), and 45 (1.35 grams, 75%). To further illustrate the scalability of this reaction, a 100-gram scale synthesis of compound 8 was conducted (Fig. 3C). Encouragingly, an eight-fold increase in reaction concentration allowed a 10,000-fold (1.0 mol) scale-up reaction, and the desired product 8 was obtained in 66% isolated yield (157.7 grams) by recrystallization (for more details see Supplementary Fig. 2).

A Derivatization of approved drugs and complex molecules. B Post-modification of tepoxalin. C Scale-up reactions.
Mechanistic studies
To gain insight into the mechanism, we conducted a series of experiments and performed density functional theory (DFT) calculations. First, to understand the kinetic profile of the umpolung N-cyanation reaction, in situ three-dimensional Fourier transform infrared (FTIR) spectroscopy was employed to monitor the conversion of O-tosyl hydroxamate (1) to N-cyanoamide (2) under the optimized conditions (Fig. 4A). It was found that the C = O stretching vibration peak of compound 1 at 1704 cm−1 rapidly consumed upon the addition of TMSCN, while a new C = O stretching vibration peak for compound 2 appeared at 1710 cm−1. Moreover, a strong linear correlation between the log(kR/kH) values and Hammett σR constants was observed for the reactions of O-tosyl hydroxamates (R = p-OMe, p-Me, H, p-CO2Me, and p-CN) with TMSCN (Fig. 4B)43. The small positive ρ value (+0.91) indicates that a partial negative charge forms in the transition state, suggesting that the tosylate (TsO−) behaves as an anionic leaving group during the turnover-limiting step21,44,45. Furthermore, we determined the reaction order with respect to the reaction components, O-tosyl hydroxamate (1) and TMSCN, via the initial rates method (Fig. 4C). The kinetic data indicated that the reaction was first order in both 1 and TMSCN, suggesting that these two components participate in the turnover-limiting step. These results are consistent with an SN2 process. Additionally, a secondary β-deuterium KIE (0.93) was observed by independently measured rate constants in the parallel experiments (Fig. 4D). The Cβ–D bonds are shorter than the Cβ–H bonds, which allows the CN nucleophile to approach the non-anomeric amide electrophilic nitrogen center with less hindrance. This small and inverse secondary β-deuterium kinetic isotope effect further supports an SN2 mechanism46,47. Finally, in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 0.1–1.0 equiv.) as a radical scavenger in the model reaction, no influence of the outcome of product 2 was detected (Fig. 4E). Moreover, if the reaction involved an amide radical, it could potentially lead to the formation of a cascade radical cyclization product. However, no radical cyclization product 78 was observed; instead, the reaction produced the N-cyanoamide product 77 in 67% yield (Fig. 4E). These radical trapping experiments suggest that a radical mechanism is not involved in this reaction.

A In situ FT-IR profile. B Hammett plot. C Reaction orders. D KIE studies. E Radical trapping experiments.
Next, we performed DFT calculations to investigate the SN2 pathway on the amide nitrogen, as illustrated in Fig. 5. In pathway I, an isomerization of 1 via TS1 (14.2 kcal mol−1) yields the thermodynamically disfavored 1-I (0.9 kcal mol−1). Subsequently, the nucleophilic attack by the cyano ion occurs through TS2 (20.7 kcal mol−1) as the rate-determining step, ultimately forming the N-cyanoamide 2 (−60.2 kcal mol−1). Notably, the C(cyano)–N(amide) distance in TS2 is 2.33 Å, which is 0.15 Å shorter than the C(cyano)–C(amide) distance, indicating that the carbonyl group might direct nucleophilic attack toward the nitrogen. The H(methyl)-O(carbonyl) distance is 2.44 Å, with a C–H···O angle of 117°. These values fall slightly outside the ideal range for a strong hydrogen bond but are consistent with a weak, non-classical C–H···O interaction for transition state stabilization (Supplementary Fig. 10). In contrast, pathway II features a direct nucleophilic attack on the amide nitrogen of 1. However, the steric effect between the methyl group and the benzene ring increases the energy barrier of TS3 to 25.5 kcal mol−1. The resulting product 2-I ( − 58.4 kcal mol−1) can then isomerize to amide 2 via TS4 ( − 49.6 kcal mol−1) with an 8.8 kcal mol−1 energy barrier. A similar direction effect by the carbonyl group is observed in TS3, where the C(cyano)–C(amide) and C(cyano)–N(amide) distances are 2.44 Å and 2.21 Å, respectively. Overall, the isomerization of amide 1 followed by the SN2 pathway on amide nitrogen showed a lower reaction energy barrier and the finalized mechanism for the formation of N-cyanoamide 2 from 1 is summarized at the bottom of Fig. 5.

Free energy profiles (kcal mol−1) of the formation of 2 at the TPSS-D3(BJ)/def2-TZVP (SMD, acetonitrile) //TPSS-D3(BJ)/def2-SVP (SMD, acetonitrile) level.
Discussion
In conclusion, we have developed an efficient and straightforward method for preparing N-cyanoamides. This method utilizes umpolung N-cyanation reactions to convert readily accessible O-tosyl hydroxamates into N-cyanoamides with TMSCN under mild and transition metal-free conditions. Our approach demonstrates remarkable generality, robustness, and scalability (up to 100 gram-scale). These features allow for compatibility with a wide range of substrates (over 70 examples), including pharmaceuticals and biologically active molecules, thereby enabling the synthesis of structurally diverse N-cyanoamides. A combination of kinetic, radical trapping, and spectroscopic mechanistic investigations supports an SN2 mechanism while ruling out the radical pathway, which is further supported by DFT calculations. Studies to investigate the biological activity of the N-cyanoamides prepared by this method, as well as to develop new types of SN2 reactions at the non-anomeric amide nitrogen electrophiles, are ongoing in our laboratories.
Methods
General. For the 1H NMR, 13C NMR and 19F NMR spectra of compounds in this Article, details of the synthetic procedures and details of the computational study, see the Supplementary Information.
General procedure A for the umpolung N-cyanation reaction
To a 15 mL oven-dried vial equipped with a magnetic stir bar was added O-tosyl hydroxamate (0.1 mmol, 1.0 equiv.), TMSCN (0.15 mmol, 1.5 equiv.), CsF (0.2 mmol, 2.0 equiv.), and MeCN (2.0 mL) under an air atmosphere. The vial was then tightly capped and placed in an ice-water bath (0 °C) with vigorous stirring for 8–20 h (TLC monitored the conversion of O-tosyl hydroxamates). After the time indicated for each reaction, the solvent was removed in vacuo, and the crude material was purified by flash column chromatography.
General procedure B for the umpolung N-cyanation reaction
To a 15 mL oven-dried vial equipped with a magnetic stir bar was added O-tosyl hydroxamate (0.1 mmol, 1.0 equiv.), TMSCN (0.25 mmol, 2.5 equiv.), CsF (0.3 mmol, 3.0 equiv.), and MeCN (2.0 mL) under an air atmosphere. The vial was then tightly capped and placed in a hotplate pre-heated to 50 °C with vigorous stirring for 2–7 h (TLC monitored the conversion of O-tosyl hydroxamates). After the time indicated for each reaction, the reaction mixture was cooled to room temperature, the solvent was removed in vacuo, and the crude material was purified by flash column chromatography.
Data availability
The characterization of products, experimental protocols and the computational studies data generated in this study are provided in the Supplementary Information. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition numbers CCDC 2380286 (18), CCDC 2380287 (64), CCDC 2401552 (76) and CCDC 2401553 (S50). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Brough, D. G. Preparation of N-acyl cyanamides as agrochemical fungicides. GB 2226815, A (1990).
Liu, W. et al. Preparation of cyanamide amino acid derivatives useful as reversible inhibitors of cysteine proteases. WO 2003086325, A2 (2003).
Yang, W. C., Li, J., Li, J., Chen, Q. & Yang, G. F. Novel synthetic methods for N-cyano-1H-imidazole-4-carboxamides and their fungicidal activity. Bioorg. Med. Chem. Lett. 22, 1455–1458 (2012).
Shirota, F. N., Nagasawa, H. T., Kwon, C. H. & DeMaster, E. G. N-acetylcyanamide, the major urinary metabolite of cyanamide in rat, rabbit, dog, and man. Drug Metab. Dispos. 12, 337–344 (1984).
Rönn, R. et al. Evaluation of a diverse set of potential P1 carboxylic acid bioisosteres in hepatitis C virus NS3 protease inhibitors. Bioorg. Med. Chem. 15, 4057–4068 (2007).
Larraufie, M. H. et al. The cyanamide moiety, synthesis and reactivity. Synthesis 44, 1279–1292 (2012).
Servais, A., Azzouz, M., Lopes, D., Courillon, C. & Malacria, M. Radical cyclization of N-acylcyanamides: total synthesis of luotonin A. Angew. Chem. Int. Ed. 46, 576–579 (2007).
Larraufie, M. H., Ollivier, C., Fensterbank, L., Malacria, M. & Lacôte, E. Radical synthesis of guanidines from N-acyl cyanamides. Angew. Chem. Int. Ed. 49, 2178–2181 (2010).
Ziedan, N. I., Stefanelli, F., Fogli, S. & Westwell, A. D. Design, synthesis and pro-apoptotic antitumour properties of indole-based 3,5-disubstituted oxadiazoles. Eur. J. Med. Chem. 45, 4523–4530 (2010).
Larraufie, M.-H. et al. Radical migration of substituents of aryl groups on quinazolinones derived from N-acyl cyanamides. J. Am. Chem. Soc. 132, 4381–4387 (2010).
Maestri, G. et al. Rearrangements of N-acyl isothioureas. Alternate access to acylguanidines from cyanamides. Org. Lett. 14, 5538–5541 (2012).
Baguia, H., Deldaele, C., Romero, E., Michelet, B. & Evano, G. Copper-catalyzed photoinduced radical domino cyclization of ynamides and cyanamides: a unified entry to rosettacin, luotonin A, and deoxyvasicinone. Synthesis 50, 3022–3030 (2018).
Jin, J.-K., Zhang, F.-L., Zhao, Q., Lu, J.-A. & Wang, Y.-F. Synthesis of diverse boron-handled N-heterocycles via radical borylative cyclization of N-allylcyanamides. Org. Lett. 20, 7558–7562 (2018).
Cockerill, A. F. et al. An improved synthesis of 2-amino-1,3-oxazoles under basic catalysis. Synthesis 1976, 591–593 (1976).
Kumar, V., Kaushik, M. P. & Mazumdar, A. An efficient approach for the synthesis of N-1 substituted hydantoins. Eur. J. Org. Chem. 2008, 1910–1916 (2008).
Yamasaki, R. et al. Structure and additive-free transamidation of planar N-cyanoamides. J. Org. Chem. 88, 5704–5712 (2023).
Luttrell, W. E. Cyanogen bromide. J. Chem. Health Saf. 16, 29–30 (2009).
Morris, J., Kovács, L. & Ohe, K. Cyanogen bromide. Encyclopedia of Reagents for Organic Synthesis. (2015).
Glotz, G., Lebl, R., Dallinger, D. & Kappe, C. O. Integration of bromine and cyanogen bromide generators for the continuous-flow synthesis of cyclic guanidines. Angew. Chem. Int. Ed. 56, 13786–13789 (2017).
Wan, Y. et al. Copper-catalyzed C–N cross-coupling for construction of alkylated N-cyanamide derivatives via nitrogen umpolung. Chem 10, 2538–2549 (2024).
Fang, W. et al. SN2 reaction at the amide nitrogen center enables hydrazide synthesis. Angew. Chem. Int. Ed. 63, e202317570 (2024).
Glover, S. A. Anomeric amides—structure, properties and reactivity. Tetrahedron 54, 7229–7271 (1998).
Glover, S. A. SN2 reactions at amide nitrogen—theoretical models for reactions of mutagenic N-acyloxy-N-alkoxyamides with bionucleophiles. Arkivoc 2001, 143–160 (2001).
Glover, S. A. & Rosser, A. A. Heteroatom substitution at amide nitrogen—resonance reduction and HERON reactions of anomeric amides. Molecules 23, 2834 (2018).
Wynn, D. A., Roth, M. M. & Pollard, B. D. The solubility of alkali-metal fluorides in non-aqueous solvents with and without crown ethers, as determined by flame emission spectrometry. Talanta 31, 1036–1040 (1984).
Paudyal, M. P. et al. Dirhodium-catalyzed C-H arene amination using hydroxylamines. Science 353, 1144–1147 (2016).
Munnuri, S. et al. Catalyst-controlled diastereoselective synthesis of cyclic amines via C–H functionalization. J. Am. Chem. Soc. 139, 18288–18294 (2017).
Farndon, J. J., Ma, X. & Bower, J. F. Transition metal free C–N bond forming dearomatizations and aryl C–H aminations by in situ release of a hydroxylamine-based aminating agent. J. Am. Chem. Soc. 139, 14005–14008 (2017).
Farndon, J. J., Young, T. A. & Bower, J. F. Stereospecific alkene aziridination using a bifunctional amino-reagent: an aza-Prilezhaev reaction. J. Am. Chem. Soc. 140, 17846–17850 (2018).
Falk, E., Gasser, V. C. M. & Morandi, B. Synthesis of N-alkyl anilines from arenes via iron-promoted aromatic C–H amination. Org. Lett. 23, 1422–1426 (2021).
Zhu, Y., Smith, M. J. S., Tu, W. & Bower, J. F. A Stereospecific alkene 1,2-aminofunctionalization platform for the assembly of complex nitrogen-containing ring systems. Angew. Chem. Int. Ed. 62, e202301262 (2023).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA-approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Delost, M. D., Smith, D. T., Anderson, B. J. & Njardarson, J. T. From oxiranes to oligomers: architectures of U.S. FDA-approved pharmaceuticals containing oxygen heterocycles. J. Med. Chem. 61, 10996–11020 (2018).
Marshall, C. M., Federice, J. G., Bell, C. N., Cox, P. B. & Njardarson, J. T. An update on the nitrogen heterocycle compositions and properties of U.S. FDA-approved pharmaceuticals (2013–2023). J. Med. Chem. 67, 11622–11655 (2024).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Di Martino, R. M. C., Maxwell, B. D. & Pirali, T. Deuterium in drug discovery: progress, opportunities and challenges. Nat. Rev. Drug Discov. 22, 562–584 (2023).
Davies, J., Svejstrup, T. D., Fernandez Reina, D., Sheikh, N. S. & Leonori, D. Visible-light-mediated synthesis of amidyl radicals: transition-metal-free hydroamination and N-arylation reactions. J. Am. Chem. Soc. 138, 8092–8095 (2016).
Luo, Y. R. Comprehensive handbook of chemical bond energies. 225–249, (CRC Press, Chapter 5 BDEs of C–halogen bonds, 2007).
Willburger, R. E., Wittenberg, R. H., Schmidt, K., Kleemeyer, K. S. & Peskar, B. A. Antiinflammatory effect of tepoxalin Blood and synovial tissue studied in patients with knee arthrosis. Acta Orthop. Scand. 69, 295–300 (1998).
Pereira-Leite, C., Nunes, C., Jamal, S. K., Cuccovia, I. M. & Reis, S. Nonsteroidal anti-inflammatory therapy: a journey toward safety. Med. Res. Rev. 37, 802–859 (2017).
McQuerry, J. A., Chen, J., Chang, J. T. & Bild, A. H. Tepoxalin increases chemotherapy efficacy in drug-resistant breast cancer cells overexpressing the multidrug transporter gene ABCB1. Transl. Oncol. 14, 101181 (2021).
Sousa, S. M., Xavier, C. P., Vasconcelos, M. H. & Palmeira, A. Repurposing some of the well-known non-steroid anti-inflammatory drugs (NSAIDs) for cancer treatment. Curr. Top. Med. Chem. 23, 1171–1195 (2023).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991).
Brown, H. C. & Okamoto, Y. Substituent constants for aromatic substitution1-3. J. Am. Chem. Soc. 79, 1913–1917 (1957).
Mita, T., Sugawara, M., Hasegawa, H. & Sato, Y. Synthesis of arylglycine and mandelic acid derivatives through carboxylations of α-amido and α-acetoxy stannanes with carbon dioxide. J. Org. Chem. 77, 2159–2168 (2012).
Westaway, K. C. Using kinetic isotope effects to determine the structure of the transition states of SN2 reactions. Adv. Phys. Org. Chem. 41, 217–273 (2006).
Fang, Y. R. et al. Experimental and theoretical multiple kinetic isotope effects for an SN2 reaction. An attempt to determine transition-state structure and the ability of theoretical methods to predict experimental kinetic isotope effects. Chem. Eur. J. 9, 2696–2709 (2003).
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21871074, J.J.D.), the Anhui Provincial Natural Science Foundation (Grant No. 2308085MB39, J.J.D.), and the Fundamental Research Funds for the Central Universities (Grant No. JZ2025HGTG0260, J.J.D.). We thank Min Zhuge from Shanghai Jiao Tong University for assistance with in situ FTIR experiments. We thank Wenmin Pang from the NMR Laboratory, Instruments Center for Physical Science, University of Science and Technology of China for assistance with NMR experiments and helpful discussions. We thank the Rosen Center for Advanced Computing (RCAC) at Purdue University for providing High-Performance Computing resource. We also thank Dr. Sicong Chen for additional computational guidance. We further thank the Hefei University of Technology for its continued support of our research.
Author information
Authors and Affiliations
Contributions
J.J.D. conceived the project and designed the experiments. H.Y., F.Y.C., Y.H.Z., and Y.P. performed the experiments and analyzed the data. X.Z. and Y.C.W. performed the density functional theory calculations. M.D. and J.J.D. directed the project and wrote the manuscript. All of the authors participated in the discussion and preparation of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
A patent application (No. CN202510219922.X) has been submitted by Hefei University of Technology, with J.J.D., H.Y., Y.H.Z. and F.Y.C. as inventors based on the umpolung N-cyanation method and products. X.Z., Y.P., Y.C.W. and M.D. declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Minyan Wang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H., Zhang, X., Cao, FY. et al. Reverse polarity of amide nitrogen enables expedient access to N-cyano amides. Nat Commun 16, 7706 (2025). https://doi.org/10.1038/s41467-025-63052-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63052-7
