
Abstract
CO2-assisted oxidative dehydrogenation of light alkane is a promising and innovative technology for light olefin production; however, the interference of side reactions and sluggish reactivity of CO2 limit olefin yields. This paper describes an economically viable tandem catalytic system by coupling alkane dehydrogenation and the reverse water gas shift (RWGS) reaction, employing PtSn/SiO2 as ethane dehydrogenation (EDH) sites and nano-CaCO3 as the hydrogen acceptor for sequent RWGS. This tandem catalytic system significantly surpasses commercial CrOx- and Pt-based catalytic systems, and breaks the EDH thermodynamic equilibrium limitation, reaching 142% of the nominal equilibrium ethylene yield of non-oxidative EDH process with 96.7% selectivity under industrially relevant conditions. Experimental characterization and theoretical analysis confirm that CaCO3 mediates the pathway of hydrogen spillover that originates from adjacent PtSn/SiO2, which effectively facilitates the RWGS reaction and thus shifts the EDH toward ethylene. This tandem catalytic strategy assisted by carbonates potentially expands the palette of catalytic systems pertaining to hydrogen transfer mechanisms in CO2-involved hydrogenation or dehydrogenation reactions.
Similar content being viewed by others

Introduction
Ethylene is a crucial foundational feedstock in the chemical industry1,2. Traditional ethylene production processes, including naphtha steam cracking, Fischer-Tropsch synthesis of olefins and methanol to olefins, are afflicted by high energy consumption, numerous operating units, and low atom utilization3,4,5,6. Ethane dehydrogenation (EDH) presents significant cost advantages in the direct conversion of ethane from shale gas into ethylene6,7. However, the single-pass yield of ethylene in the EDH process is limited by thermodynamic equilibrium (Supplementary Fig. 1)8,9,10,11. Taking into consideration of Le Chatelier’s principle, prompt removal of H2 from the product mixture can shift the reaction equilibrium to enhance ethylene yield12,13,14. Addition of the oxidant, such as O2 is a universal method for removing H215,16,17. Nevertheless, O2 co-feeding inevitably leads to excessive oxidation of products and the risk of explosion5.
CO2, which is considered a mild oxidant, is capable of avoiding the over-oxidation of products and it also holds great significance in consuming H2 through the reverse water gas shift (RWGS) reaction18,19,20,21,22,23. CO2-assisted oxidative dehydrogenation of ethane (CO2-ODHE) provides a higher theoretical yield of ethylene than EDH (Fig. 1a and Supplementary Figs. 2–4)14,24. Nevertheless, the dry reforming of ethane (DRE) is thermodynamically more favorable than CO2-ODHE (Supplementary Fig. 2: ΔGDRE < ΔGCO2-ODHE), which results in low ethylene selectivity and yield from ODHE due to the thermodynamic competition from DRE (Supplementary Fig. 3)25,26,27,28,29. Chen et al. recently demonstrated that a PtSn/SiO2 catalyst exhibits excellent propylene selectivity in CO2-assisted oxidative dehydrogenation of propane (CO2-ODHP)30. However, the rate-determining RWGS reaction still limited the single-pass yield of propylene30, in which the inert CO2 molecules displayed finite removal of the H2 generated from alkane dehydrogenation. Consequently, effective activation of CO2 molecules is crucial for tuning hydrogen consumption and mediating the tandem process of EDH and RWGS reactions.

a Thermodynamic equilibrium compositions of CO2-ODHE (DRE not allowed) at different temperature. The gray dashed line represented the thermodynamic equilibrium composition of ethylene in EDH. b, c Catalytic performance of PtSn/SiO2 + CaCO3 and commercialized catalytic systems. Conversion and selectivity (b), yield and carbon balance (c). Reaction conditions: 600 °C, a weight hourly space velocity (WHSV) of C2H6 equal to 8.14 h−1. EDH: C2H6: Ar = 1:1, CO2-ODHE: C2H6: CO2 = 1:1. d Ratio of ethylene yield to traditional EDH process thermodynamic equilibrium ethylene yield for different catalytic systems. e Comparison of tandem catalyst with previously reported EDH and CO2-ODHE catalysts, shown as pentagrams, hollow triangles, and circles, respectively. Numbers correspond to the entries in Supplementary Table 2. f Ethylene yield at different temperatures. g Long-term cyclic stability evaluation of PtSn/SiO2 + CaCO3, PtSn/SiO2, and PtSn/γ-Al2O3 systems. Reaction conditions: T = 600 °C, catalyst: 200 mg, total flow rate of 20 mL/min, Ar balanced (50% C2H6, 50% CO2; 50% C2H6, 10% CO2; 50% C2H6, 25% CO2; 75% C2H6, 25% CO2).
CaCO3 is a low-cost and common material in integrated CO2 capture and utilization, which can act as a reversible carrier for gas-solid carbonate hydrogenation in the calcium looping process31,32,33,34,35. Thermodynamically, the decomposition of CaCO3 is apparently expedited in the presence of H2 (Supplementary Fig. 5), inferring that CaCO3 might be a potential hydrogen acceptor. Moreover, solid carbonates can heuristically serve as intermediate regulators that are favorable to CO2-involved reactions. For instance, Xia et al. observed that the chemical activation of CO2 molecules in the form of carbonates on PbO facilitated the CO2 reduction reaction toward HCOOH36. Here, we describe a tandem process of alkane dehydrogenation and the RWGS reaction over CaCO3 coupled with PtSn/SiO2 catalysts to enhance ethylene yield (Supplementary Fig. 6). In the tandem catalytic system, PtSn/SiO2 converts ethane into ethylene and hydrogen species (H*), while the adjacent CaCO3 induces H* to migrate from the dehydrogenation sites to react with solid carbonates, generating H2O and CO via the RWGS reaction. Simultaneously, the CO2 molecules are trapped by vacancies in CaCO3 to complete the redox cycle. Under near-realistic and challenging conditions of high space velocity and high ethane concentration, the tandem catalyst achieves 31.9% ethylene single-pass yield with 96.7% ethylene selectivity, exceeding the equilibrium yield of the non-oxidative EDH process and closely approaching the equilibrium of CO2-ODHE. The ethylene space-time yield (STY) is respectively 112- and 1.6-fold higher than that of commercial CrOx- and Pt-based catalysts under equivalent conditions. In addition, energy consumption and net CO2 emissions of the tandem catalytic system were also significantly lower than those of commercial PtSn/γ-Al2O3 catalytic EDH system.
Results
Catalytic performance in CO2-ODHE
The catalytic performances of different catalytic systems were tested on stream of 50 vol.% C2H6 balanced with either Ar or CO2 at 600 °C. In the absence of CO2, PtSn/SiO2 attained high ethylene selectivity (98.5%) at 22.4% ethane conversion that approached thermodynamic equilibrium of EDH (22.5%), comparable to commercial PtSn/γ-Al2O3 (Fig. 1b and Supplementary Fig. 7). After co-feeding CO2 with ethane, PtSn/SiO2 presented 26.2% ethylene yield with 96.0% ethylene selectivity, breaking the thermodynamic equilibrium limit of EDH (Fig. 1c). When coupling PtSn/SiO2 with CaCO3, we noted that the H2 yield decreased at the increased conversions of ethane and CO2, and the highest ethylene yield (selectivity) of 31.9% (96.7%) was obtained (Fig. 1b, c). This reached 142% of the nominal equilibrium ethylene yield of EDH (Fig. 1d), outperforming the commercial CrOx- and Pt-based catalytic systems, as well as other previous reported catalysts (Fig. 1e and Supplementary Table 2). Moreover, the ethylene yields of PtSn/SiO2 + CaCO3 tandem catalyst were near the thermodynamic equilibrium values of CO2-ODHE across different temperatures (450–650 °C) and ethane volume fractions (10–50%) (Fig. 1f and Supplementary Fig. 8). The carbon balance in CxHy and total carbon balance of PtSn/SiO2 + CaCO3 tandem catalyst reached 99.1% and 99.0%, respectively (Supplementary Fig. 9), demonstrating the negligible influences of DRE and deep dehydrogenation side reactions during CO2-ODHE. We found an induction period of C2H6 and CO2 conversions over PtSn/SiO2 + CaCO3 tandem catalysts in the initial 30 min of reaction, and in situ X-ray diffraction (in situ XRD) confirmed that this stage corresponds to the transformation of CO2 into carbonate (Supplementary Fig. 10). CaCO3 alone produced 0.9% ethylene yield at 600 °C (Fig. 1f), implying CaCO3 is inclined to function as a hydrogen acceptor rather than an EDH site.
Concerning the evaluation of ethylene productivity, ethylene STY was calculated to reach 7.81 mol C2H4 gmetal−1 h−1 (78.1 mmol C2H4·gcat−1·h−1) upon tandem catalyst, which was 112 and 1.6 times higher than those of the commercial CrOx- and Pt-based catalytic systems, respectively (Fig. 1d). The STY was significantly higher than those reported catalysts for CO2-ODHE, EDH, and O2-ODH (Supplementary Table 2). In addition, for CO2-ODHP, the tandem catalyst exhibited exceptional catalytic activity with 94.5% propylene selectivity and 55.0% single-pass propylene yield (Supplementary Fig. 11), which moved beyond the thermodynamic equilibrium of direct propane dehydrogenation (PDH, 48.0%) at 550 °C (Supplementary Fig. 4), surpassing the values reported in the previous literature (Supplementary Fig. 12 and Supplementary Table 3).
For the long-term cyclic stability at 600 °C, the tandem catalyst showed a slightly declined ethane conversion that remained higher than the equilibrium of non-oxidative EDH process (22.5%), whereas the commercial PtSn/γ-Al2O3 and K-CrOx/γ-Al2O3 showed rapid deactivation during 10 h at high ethane concentration and weight hourly space velocity (50 vol.% C2H6, WHSV = 8.14 h−1) (Fig. 1g and Supplementary Fig. 13). Notably, the deactivation constant (kd) of PtSn/SiO2 + CaCO3 was only 0.046 h−1, much lower than that of the commercial PtSn/γ-Al2O3 (0.121 h−1) and K- CrOx/γ-Al2O3 (0.331 h−1) under the same conditions (Supplementary Fig. 14). In addition, the PtSn/SiO2 + CaCO3 tandem catalytic system offers significant advantages in terms of energy consumption and net CO2 emissions compared to the commercial PtSn/γ-Al2O3 catalytic system, with energy savings of about 8.3% and CO2 emission reductions of 60.6% for ethylene production per unit (Supplementary Fig. 15). This suggests that the catalytic performance and economic potential of our proposed tandem catalytic process has a significant advantage over commercial catalytic systems under near-realistic and challenging operating conditions. We also evaluated the cyclic stability of the tandem catalyst for CO2-ODHE under various feed conditions (high ethane/CO2 ratios, 75% high concentration of ethane). After several regeneration cycles, the catalytic performance was basically restored (Fig. 1g). To verify the potential of the tandem catalytic system for industrial scale-up, we appropriately increased the reactor scale (total flow rate: 400 mL/min) and tested its catalytic performance (Supplementary Fig. 16). The ethane conversion, ethylene selectivity, and stability were almost identical to the results obtained in the laboratory-scale reactor.
Structural characterization of tandem catalysts
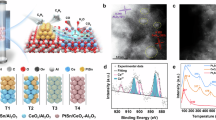
XRD and high-resolution transmission electron microscope (HRTEM) of PtSn/SiO2 showed that the PtSn nanoparticles were uniformly dispersed on the amorphous SiO2 support (Supplementary Figs. 17 and 18). For the tandem catalyst, high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDX) images discerned independent nano-CaCO3 (ca. 52 nm) and highly dispersed PtSn nanoparticles (ca. 1.9 nm) on SiO2 with nanoscale intimacy (Fig. 2b–g and Supplementary Figs. 19–23), as described in Fig. 2a. The shortest distance between PtSn nanoparticles and CaCO3 was less than 1 nm in the analysis of the HRTEM image (Fig. 2h and Supplementary Fig. 23). Pt 4f X-ray photoelectron spectroscopy (XPS) and Pt L3-edge X-ray absorption near-edge structure (XANES) spectra verified the electron transfer from Sn to Pt in the PtSn/SiO2 and tandem catalyst, possibly due to the alloying effect of PtSn (Fig. 2i and Supplementary Fig. 24)37,38, as evidenced by the Pt-Sn bond formation determined by Pt L3-edge extended X-ray absorption fine structure (EXAFS) spectra and wavelet transform (Fig. 2j and Supplementary Fig. 25)37,39. Importantly, the chemical states of Pt and Sn would not be subject to physical integration with CaCO3, as sustained by XPS and Pt L3-edge XANES spectra (Supplementary Figs. 24–27 and Supplementary Table 6). In addition, a J factor was defined as the ratio of CNPt-Sn/ (CNPt-Sn + CNPt-Pt) versus the theoretical value of thermally stable phase (Supplementary Table 7)30,40. As JPt3Sn was close to 1, Pt3Sn was affirmed as the main phase in PtSn/SiO2 and the tandem catalyst, consistent with the results of HRTEM. We also conducted in situ ambient pressure XPS to further investigate the valence states of Pt and Sn during the CO2-ODHE (Supplementary Fig. 28). The difference in the binding energy shifts of Pt 4f in Pt/SiO2 and PtSn/SiO2 after introducing the reaction gas indicates that the PtSn alloy effect weakens the interaction between Pt and H*20.

a Schematic spatial distribution of PtSn/SiO2 + CaCO3. b–h HAADF-STEM images (b, c), EDX-mappings (d–g) and HRTEM image (h) of PtSn/SiO2 + CaCO3. The inset in (c) shows the size distribution of Pt3Sn nanoparticles. The inset in (h) shows Inverse Fast Fourier Transform (IFFT) processing of single Pt3Sn nanoparticle from the selected square area. i, j Normalized Pt L3-edge XANES spectra (i) and k2-weighted Pt L3-edge EXAFS spectra (j) of PtSn/SiO2, PtSn/SiO2 + CaCO3 and reference samples.
To investigate the cause of catalyst deactivation, we conducted HRTEM, HAADF-STEM, EDS mapping, solid-state NMR, and Raman spectroscopy on both the spent and regenerated catalysts. The XRD, HAADF-STEM, HRTEM, and EDS mapping results indicated that the structure of the catalyst and the size of PtSn particles remained unchanged after 10 h of reaction and subsequent regeneration (Supplementary Figs. 29–32). However, Raman spectroscopy revealed the presence of defective carbon (D band: 1326 cm−1) and graphitic carbon (G band: 1594 cm−1) on spent catalysts (Fig. 3a)29. Notably, individual PtSn/SiO2 and the tandem catalyst showed similar defective and graphitic carbon contents and ID/IG, indicating that the introduction of CaCO3 does not affect the coke-induced deactivation of PtSn/SiO2. Compared to the commercial PtSn/γ-Al2O3 system, the tandem catalytic system exhibited lower coke content, which accounts for the lower kd observed. Solid-state NMR analysis showed abundant saturated aliphatic carbon species on the spent tandem catalyst (Supplementary Fig. 31), suggesting that coke formation may be associated with ethylene polymerization products at high temperature41. Additionally, O2-TPO analysis of spent catalysts after different reaction times revealed that coke accumulation increased with reaction time, although the deposition rate gradually slowed (Supplementary Fig. 33). The accumulation of the coke may cover the active sites and lead to catalyst deactivation19,42. However, after the regeneration, the disappearance of graphite carbon and defective carbon on the catalyst indicated that the coke was completely removed, thereby restoring the catalytic activity.

a Raman spectra of PtSn/γ-Al2O3, PtSn/SiO2, PtSn/SiO2 + CaCO3 catalysts after 10 h of reaction and PtSn/SiO2 + CaCO3 after regeneration. b, c Catalytic performance of tandem catalysts comprising PtSn/SiO2 and CaCO3 with different integration manners in EDH or CO2-ODHE. b Ethane conversion, CO2 conversion, and ethylene selectivity. c Ethylene yield, H2 yield, and carbon balance in CxHy. Reaction conditions: T = 600 °C, WHSV = 8.14 h−1, total gas flow rate of 20 mL/min. d, e Effect of the ethane (d) and CO2 (e) partial pressure on the ethane conversion over different catalysts. Reaction conditions: T = 600 °C. f Arrhenius plots of different catalytic systems.
Importance of CaCO3 integrated with PtSn/SiO2
Temperature-programmed surface reaction (TPSR) of C2H6/CO2 (Ar) showed that the introduction of CaCO3 significantly increased the yield of ethylene by PtSn/SiO2, accompanied by noticeable consumption of CO2 and decreased production of H2. This suggests that CaCO3 refines the process of RWGS in CO2-ODHE (Supplementary Fig. 34). For the tandem catalyst, H2 and CO2 conversion of 36.0% and 35.4%, respectively, approached the thermodynamic equilibrium value of RWGS (38.3%) at 600 °C (Supplementary Fig. 35), higher than those of PtSn/SiO2 without CaCO3 (28.7% H2 and 28.2% CO2 conversion). These results are mutually corroborated with the improved performance in the CO2-ODHE mediated by the integration of CaCO3.
For manipulating the distance between dehydrogenation sites and the CaCO3 hydrogen acceptor, we coupled PtSn/SiO2 with CaCO3 in three tandem modes denoted as M1, M2, and M3 (Fig. 3b, c)13. Based on Monte Carlo estimations, the distances between the two components in M1, M2, and M3 are approximately 2.4 mm, 472.8, and 43.6 μm, respectively (Supplementary Fig. 36). A strong correlation was found between the distance of PtSn/SiO2 and CaCO3 and the catalytic performance. As the distance between PtSn/SiO2 and CaCO3 decreases, the ethane reaction rate, ethylene selectivity, and CO2 conversion significantly increase (M3 > M2 > M1), accompanied by lower H2 yield (Supplementary Fig. 37). According to the concept of “the closer, the better” for tandem sites43, the shorter distance between reactive centers is conducive to the migration of intermediates or active species. Therefore, we ascribe this proximity-dependent phenomenon to the intensified intermediate species spillover between PtSn/SiO2 and CaCO3 at the nanoscale.
We conducted kinetic experiments to further investigate the crucial role of CaCO3 in the CO2-ODHE. At 600 °C, by varying the partial pressure of C2H6 or CO2, we determined the reaction orders of ethane and CO2 over PtSn/SiO2 and the tandem catalyst, as shown in Fig. 3d, e. The reaction orders of ethane were significantly higher than those of CO2 (0.84 > 0.16, 0.75 > 0.33). The ethane reaction rate increased with increasing CO2 partial pressure, which can be attributed to the consumption of H* by CO2 via the RWGS reaction, thereby promoting ethane conversion Notably, the reaction order of CO2 on the tandem catalyst (nCO2 = 0.16) was lower than that on PtSn/SiO2 (nCO2 = 0.33), suggesting that the incorporation of CaCO3 reduces the dependence of ethane conversion on CO2 partial pressure. In addition, we tested the apparent activation energies (Ea) of ethane over the tandem catalyst and PtSn/SiO2 (in the presence and absence of CO2) (Fig. 3f). For PtSn/SiO2, the introduction of CO2 significantly lowered the Ea from 127.2 kJ/mol (without CO2) to 111.2 kJ/mol (with CO2). After the tandem integration of CaCO3 and PtSn/SiO2, the Ea was further reduced to 84.8 kJ/mol, indicating that CaCO3 can activate CO2 to further consume H* and thereby improve ethylene production. The kinetic analysis of propane revealed similar trends (Supplementary Fig. 38), confirming the mechanistic consistency of the tandem catalyst in promoting alkane conversion.
Mechanistic studies of hydrogen spillover-mediated EDH and RWGS
The smooth migration of intermediates or H* species between reactive sites in closer proximity over the nanoscale tandem catalyst is of paramount importance to accelerate tandem catalytic coupling processes13,44,45. For EDH, the kinetic analysis indicated that promptly removing H2 or H* from the dehydrogenation sites can enhance the reaction rate of EDH to ethylene (Supplementary kinetic analysis). We performed H2-TPR and NH3 temperature-programmed reduction (NH3-TPR) to validate the spillover performance of hydrogen intermediates on the tandem catalyst (Fig. 4a and Supplementary Fig. 39). The temperature of reduction peak over CaCO3 decreased from 726 °C to 671 °C when coupling with PtSn/SiO2 (Fig. 4a). As shown by mass spectrometry, hydrogenation products formed more easily at a lower temperature on the tandem catalyst compared with individual CaCO3.

a H2-TPR profiles of PtSn/SiO2 + CaCO3 and CaCO3. b, c In situ XRD patterns of PtSn/SiO2 + CaCO3 (b) and CaCO3 (c) under 10 vol.% H2/Ar at 600 °C. d XRD patterns of PtSn/SiO2 + CaCO3 after different reaction states. e 13CO2/H2-TPSR of PtSn/SiO2 + CaCO3. f Schematic of tandem coupling of EDH and RWGS over PtSn/SiO2 + CaCO3. g–i In situ DRIFTs of PtSn/SiO2 + CaCO3 (g), PtSn/SiO2 (h), and relative content of Si-OH (i) under 10 vol.% C2H6 at 600 °C. The inset schematic in (i) depicts the evolution processes of Si-OH over PtSn/SiO2 + CaCO3.
H2-TPR for tandem catalyst with different tandem modes also reflects that the distance between two components is strongly interrelated to hydrogen spillover (Supplementary Fig. 40). Specifically, as the distance between the components decreases, the reduction temperature of CaCO3 shifts earlier, which is attributed to the enhanced hydrogen spillover from PtSn/SiO2 to CaCO3. For tandem catalyst, the H2 signal increased slowly initially, while CO and H2O signals exhibited a rapid rise, suggesting a higher rate of CaCO3 hydrogenation in the presence of PtSn/SiO2 (Supplementary Fig. 41). In situ XRD under H2/Ar at 600 °C (Fig. 4b, c) and ex situ Raman Spectroscopy (Supplementary Fig. 42) demonstrated that CaCO3 in the tandem catalyst was completely reduced into CaO at 600 °C within approximately 60 min in contrast to the poor reducibility of individual CaCO3. These results hint that the occurrence of hydrogen spillover induced by nanoscale contact between two components in the tandem catalyst should result in the acceleration of the CaCO3 hydrogenation rate.
To deeply delve into the role of CaCO3 in the hydrogen spillover process, we switched the reaction atmospheres and observed the changes of it during CO2-ODHE (Fig. 4d). XRD revealed that CaCO3 was first reduced to CaO via H2/Ar pretreatment. When switching to C2H6/CO2, CaO rapidly underwent carbonation back to CaCO3, corresponding to the induction period in the former 30-min reaction of CO2-ODHE (Supplementary Fig. 10). This suggests that the supplement of CO2 is faster than consumption, leading to the existence of CaCO3 under the reaction condition. Subsequently, on exposure to C2H6/Ar, the CaO phase reappears. Isotopic 13CO2 and H2 TPSR (13CO2-H2/TPSR) on the tandem catalyst indicated that the RWGS reaction dominated below 485 °C with 13CO as the main products (Fig. 4e). In the temperature range of 485–650 °C, 12CO signal increased continuously while 13CO signal remained almost unchanged, illuminating that H* preferably reacts with the carbonates in CaCO3 rather than 13CO2 molecules. Therefore, we conclude that the active carbonates in CaCO3 are responsible for driving hydrogen spillover and rapidly consuming H* to reinforce the equilibrium shift of CO2-ODHE. Subsequently, the reduced CaCO3 captures CO2 molecules to accomplish the regeneration cycle of carbonates, as illustrated in Fig. 4f.
In situ diffuse reflectance infrared Fourier transform (DRIFT) experiments further unravel the hydrogen spillover mediated by carbonates in CaCO3 during CO2-ODHE (Fig. 4g, h). After feeding ethane at 600 °C, the signals at 3728–3734 cm−1 assigned to the Si−OH groups46,47 were observed in both the tandem catalyst and individual PtSn/SiO2. The relative content of Si−OH species over individual PtSn/SiO2 rapidly increased to 87.8% of the infrared signal during the steady state within 15 min (Fig. 4i). In contrast, the Si-OH species on the tandem catalyst increased slowly and accounted for only 16.8% of the integral intensity of the steady state, pointing to the fact that the adjacent CaCO3 with nanoscale proximity triggers H* migration from the Pt3Sn sites toward carbonates producing CO (2110 cm−1 and 2175 cm−1)48, thus expediting the removal of H*. After the 15-min period, the continuous consumption of carbonates without replenishment leads to weakened impetus for H* spillover, accounting for the rapid increase of Si-OH content.
Theoretical study
Density functional theory (DFT) calculations were carried out on the surface of CaCO3 (010) and Pt3Sn (100) (Supplementary Fig. 43). Figure 5a presents a uniform charge distribution in CO2 molecule, with both C = O bond lengths of 1.18 Å. Comparably, the carbonate of CaCO3 due to uneven charge distribution was comprised of three distinct C−O linkages with relatively elongated bond lengths of 1.27, 1.31, and 1.32 Å, enabling the easier activation of C−O bonds per se. Potential density of states (PDOS) analysis (Fig. 5b) demonstrates a more dispersive electron distribution in carbonates of CaCO3, confirming the weaker strength of C−O bonds in carbonates compared with CO2 molecules. Collectively, we infer that carbonates within CaCO3 are more accessible to undergo the RWGS reaction compared with CO2 molecules. Notably, the hydrogen adsorption on O-CaCO3 (−1.12 eV) was considerably stronger than those on Ca-CaCO3 (−0.26 eV), C-CaCO3 (−0.62 eV), Pt–Pt3Sn (−0.79 eV), and Sn–Pt3Sn (–0.71 eV) (Fig. 5c and Supplementary Fig. 44), illustrating that the hydrogen spillover from Pt3Sn sites to O-CaCO3 is thermodynamically favorable.

a Microstructure and charge density difference between carbonates in CaCO3 and CO2 molecules. b PDOS for carbonates in CaCO3 and CO2 molecules. c Hydrogen adsorption energy on the O-CaCO3, Ca-CaCO3, C-CaCO3, Pt-Pt3Sn, and Sn-Pt3Sn. d Calculated relative energy diagrams of EDH on Pt3Sn clusters. e Calculated relative energy diagrams of the reaction between H* and CO2 molecules on Pt3Sn. f Calculated relative energy diagrams of the reaction between H* and carbonates in CaCO3 and CO2 molecules replenishment of vacancies. Insets in (d–f) are the corresponding structures along the reaction paths. IS indicates the initial state, and TS indicates the transition state. Color scheme: red, gray, white, green, purple, and blue balls stand for O, C, H, Ca, Pt, and Sn atoms, respectively.
The molecular-level details of EDH and RWGS processes were elaborated through the calculation of relative energy. For the EDH process, ethane is first adsorbed onto the Pt3Sn site and then dehydrogenated to form ethylene and hydrogen (Fig. 5d and Supplementary Figs. 45 and 46). The highest energy barrier in this reaction corresponds to the first C-H bond cleavage of ethane (TS1 in Fig. 5d, E0 = 1.67 eV), which aligns with the kinetic observation that the reaction order of ethane is relatively high. In addition, the energy barrier for hydrogen formation is 1.56 eV (TS3 in Fig. 5d, E1). In the reaction pathway of RWGS involving CO2 molecules on the Pt3Sn surface, the formation of COOH* from H* and CO2* is the rate-determining step (TS1 in Fig. 5e, E2 = 1.47 eV), with a lower energy barrier than hydrogen formation in the EDH process, indicating CO2 is kinetically viable to consume H*. In contrast, the RWGS process steered by CaCO3 involving H* and carbonates requires a much lower energy barrier (TS2 in Fig. 5f, E3 = 1.38 eV, E3 < E2 < E1). This is consistent with the kinetic results showing that the Ea of the tandem catalyst is significantly lower than that of the individual PtSn/SiO2 catalyst (84.8 kJ/mol <111.2 kJ/mol). Therefore, H* is energetically favored to undergo RWGS with the carbonates in CaCO3, which is consistent with the results of 13CO2-H2/TPSR. In this process, H* adsorption steps are exothermic, suggesting the feasibility of hydrogen spillover from Pt3Sn sites to CaCO3. Based on the above calculation results, we propose a reaction mechanism of the CO2-ODHE process. Pt3Sn serves as the dehydrogenation site to convert ethane into ethylene and H*. Subsequently, H* spills over to the nanoscale-adjacent CaCO3 and reacts with carbonates via RWGS to produce H2O and CO. Finally, CO2 replenishes the vacancies in CaCO3 to complete the cycle.
Related tandem catalysts
To assess the generality of hydrogen spillover effects over carbonate-mediated tandem catalytic systems, we utilized other alkaline earth metal carbonates (such as MgCO3, SrCO3, and BaCO3) to replace CaCO3 as hydrogen acceptors in the tandem catalyst. As expected, these carbonates consumed hydrogen to varying degrees, thereby increasing the ethylene single-pass yield. All tandem catalysts showed enhanced ethylene yield that exceeded the non-oxidative EDH process equilibrium value (22.5%), with selectivity greater than 93% (Supplementary Figs. 47 and 48). These results support the universal principle of carbonate-induced hydrogen spillover in heterogeneous catalysis.
Discussion
We describe a CaCO3-mediated strategy for tandem catalytic process of EDH and RWGS to tackle the imposing constraint on the selectivity and yield of ethylene in CO2-ODHE. PtSn/SiO2 coupled with nanoscale-adjacent CaCO3 achieved 31.9% ethylene single-pass yield with 96.7% ethylene selectivity under industrially relevant conditions, exceeding the equilibrium yield of the non-oxidative EDH process and outperforming commercial CrOx- and Pt-based catalyst systems. Moreover, the tandem catalytic system showed lower energy consumption and net CO2 emissions compared to commercial PtSn/γ-Al2O3 catalytic EDH system. The comprehensive studies elucidate that PtSn/SiO2 catalyzes EDH to produce ethylene and H*, while the intrinsic carbonates in CaCO3 induce H* migration and thus shift the CO2-ODHE toward high-efficiency ethylene production. Simultaneously, the carbonates in CaCO3 could be reversibly replenished by CO2 molecules to complete the cycle. This tandem catalytic system also works efficaciously for CO2-ODHP to produce propylene. Our study offers an inspiration for catalyst design with application in the reactions involving hydrogen transfer catalysis and processes.
Methods
Preparation of tandem catalysts
The tandem catalysts comprising PtSn/SiO2 and nano-CaCO3 (average particle size of 50 nm, Macklin) were prepared by physical mixing. The actual contents of Pt and Sn in PtSn/SiO2 were determined by inductively coupled plasma mass spectrometry to be 0.98 wt% and 0.59 wt%, respectively. The control of the spatial distribution distance between PtSn/SiO2 and CaCO3 were prepared by three different tandem modes. M1: initially, PtSn/SiO2 and nano-CaCO3 were individually pressed, crushed, and sieved to granules between 20 and 40 mesh, respectively. Subsequently, PtSn/SiO2 granules were placed on the top layer, CaCO3 granules were placed on the bottom layer, and a quartz wool layer approximately 2 mm thick was used to separate the two layers. M2: the equal mass sieved PtSn/SiO2 (20–40 mesh) and nano-CaCO3 (20–40 mesh) were placed into a 10 mL centrifuge tube, followed by shaking to ensure homogeneous mixing. M3: equal masses of PtSn/SiO2 and nano-CaCO3 powders were ground together in the agate mortar for 10 min and then placed into a 10 mL centrifuge tube, followed by shaking to achieve uniform mixing. Subsequently, the obtained mixtures were pressed, crushed, and sieved to granules between 20 and 40 mesh. Tandem catalysts were mixed using the M3 method unless otherwise specified.
Preparation of commercial catalyst analogues
Common commercial catalyst analogues in EDH (PDH) system are PtSn/γ-Al2O3 and K-CrOX/Al2O3. In a typical synthesis of PtSn/γ-Al2O39,38, H2PtCl6 (Macklin) and SnCl4·5H2O (Meryer) were added to a 0.1 M HCl solution and ultrasonically mixed for 30 min in which the final molar concentration of Pt and Sn ions were 102 mM and 510 mM. Then, 1 mL of the above solution was diluted to 40 mL of deionized water, which was added to 2 g of γ-Al2O3 and stirred at room temperature for 12 h. The theoretical loading of Pt was controlled at 1%. For synthesis of K-CrOx/Al2O317,47, 1.58 g of γ-Al2O3 support was dissolved in 40 mL of deionized water, followed by the addition of 3.08 g of Cr(NO3)3·9H2O (Meryer) and 0.05 g of KNO3 (Meryer), and stirred at room temperature for 12 h. The theoretical loadings of Cr and K were controlled at 20% and 1%, respectively. The above solution was dried overnight at 373 K. Finally, the obtained solid was calcined in air at 550 °C for 3 h.
Catalytic performance tests
The EDH and CO2-ODHE catalytic performance were conducted in a continuous flow fixed-bed reactor at atmospheric pressure13. Typically, 0.1 g of PtSn/SiO2 and 0.1 g of nano-CaCO3 (or other mixtures) composing the tandem catalyst (20–40 mesh) were placed in a quartz tube (inner diameter 8 mm), with quartz wool filled both above and below the tandem catalyst. The temperature of the catalyst bed was controlled by a K-type thermocouple. The catalyst was pretreated in 10 vol.% H2/Ar (30 mL/min) at 600 °C for 2 h, followed by Ar (50 mL/min) purging for 10 min. Next, the specified reaction conditions (C2H6: CO2 = 1:1, total flow rate of 20 mL/min) was switched into the reactor and stabilized for 30 min. The effluent from the reactor outlet was thoroughly mixed with pure N2 (5 mL/min) before entering the gas chromatograph (GC) for analysis. For the tests at different temperatures, the gas composition and catalyst loading were consistent with the above conditions, except that the reaction temperature was decreased to the desired value (450–650 °C) and data points were collected after stabilization for 30 min. For tests with different ethane volume fractions, the flow rates of C2H6 and CO2 were adjusted to 10, 8, 6, 4, and 2 mL/min, respectively, while Ar was used as a balance gas with flow rates of 0, 4, 8, 12, and 16 mL/min, maintaining a total flow rate of 20 mL/min. For the tests on individual PtSn/SiO2, SiO2 was used instead of CaCO3 to mix with PtSn/SiO2. For EDH tests, Ar was used instead of CO2 to mix with C2H6 (C2H6: Ar = 1:1). For RWGS tests, H2 was used instead of C2H6 to mix with CO2 (H2: CO2 = 1:1).
The products were analyzed by an online GC (F70, Fuli Instruments) equipped with Pora PLOT Q column (CH4, C2H4, C2H6, C3H6, and C3H8), another Pora PLOT Q column (CO and CO2), and MS 5 A Plot column (H2 and N2). N2 was used the internal standard for GC quantification of all gases. The C2H6 conversion (Eq. (1)), CO2 conversion (Eq. (2)), C2H4 yield (Eq. (3)), H2 yield (Eq. (4)), C2H4 selectivity in CxHy (Eq. (5)), total C2H4 selectivity (Eq. (6)), carbon balance in CxHy (Eq. (7)), and total carbon balance (Eq. (8)) were defined as follows13,21:
where \({{F}_{x}}^{{{{\rm{in}}}}}\) and \({{F}_{x}}^{{{{\rm{out}}}}}\) represent the inlet and outlet flow rates of given species (mL min−1), respectively. The weight hourly space velocity (WHSV, Eq. (9)), gaseous hourly space velocity (Eq. (10)) and C2H4 STY (Eq. (11)) were calculated as follows:
where \({\rho }_{{{{{\rm{C}}}}}_{2}{{{{\rm{H}}}}}_{6}}\) is the density of ethane (1.356 g/L), \({m}_{{{{\rm{PtSn}}}}/{{{\rm{Si}}}}{{{{\rm{O}}}}}_{2}}\) is the mass of PtSn/SiO2 (g), mmetal is the mass of active metal in the catalyst (g), Vm is the molar volume of gas (24.5 L mol−1). The calculation equations related to the RWGS, PDH, and CO2-ODHP were defined similarly to the CO2-ODHE.
The deactivation constant (kd) of the catalyst was defined using a first-order deactivation model17, based on the following Eq. (12):
Where Xfinal and Xinitial represent the conversion measured at the initial and final periods of the experiment, t represents the reaction time (h), kd is the deactivation rate constant (h−1).
The kinetic experiments were conducted with the conversion of ethane and CO2 controlled to be below 15% (well below the thermodynamic equilibrium at 600 °C), thus eliminating the effects of heat and mass transfer on the kinetic testing. The tandem catalyst loading was 20 mg (10 mg PtSn/SiO2 + 10 mg CaCO3). The ethane reaction order was tested by controlling the CO2 flow rate at 10 mL/min, while the ethane flow rate (10.0, 9.6, 9.0, 8.6, 8.0 mL/min) was adjusted using Ar as the balance gas (total flow rate of 20 mL/min). The CO2 reaction order was tested by controlling the ethane flow rate at 10 mL/min, while the CO2 flow rate (10, 8, 6, 4, 2 mL/min) was adjusted using Ar as the balance gas (total flow rate of 20 mL/min). Arrhenius plots were tested in different temperature (570, 580, 590, 600 °C) under conditions where both ethane and CO2 flow rates were 10 mL/min (total flow rate of 20 mL/min). The apparent activation energy (Ea) was calculated using the following Eq. (13):
where k is the rate constant, A is the pre-exponential factor, and T is the temperature (K). Ea was obtained by fitting the values of ln(r) at different temperatures.
Catalyst regeneration
For the catalyst regeneration process, the reaction temperature was lowered to 500 °C, and the reactor was purged with Ar (50 mL/min) for 10 min to ensure the complete removal of ethane and CO2. Then, 1 vol.% O2/Ar (30 mL/min) was introduced for 30 min, followed by purging with Ar (50 mL/min) for 10 min to ensure the complete removal of O2. Next, the gas was switched to 10 vol.% H2/Ar (30 mL/min) as the temperature increased to 600 °C and maintained for 2 h. Finally, purge with Ar (50 mL/min) for 10 min to ensure the complete removal of H2.
In situ XRD
In situ X-ray diffraction (In situ XRD) experiments were conducted on a 45 kV, 220 mA, and 9 kW Rigaku Smartlab using Cu Kα radiation filtered with graphite (λ = 1.5406 Å) and equipped with a high temperature cell. The powder samples of CaCO3 + SiO2 and PtSn/SiO2 + CaCO3 were separately loaded into high temperature cell vessels, and then purged with 10 vol.% H2/Ar (20 mL/min) for 10 min. Firstly, an XRD pattern was recorded at room temperature. Then, the sample was heated at a rate of 20 °C/min under 10 vol.% H2/Ar (20 mL/min) to 600 °C and maintained for 2 h. The XRD patterns were recorded immediately upon reaching 600 °C with 2θ range of 25–75° at a scan speed of 5°/min and at intervals of 15 min during the isothermal process. The in situ XRD experiments during the reaction process were conducted similarly. After treatment under 10 vol.% H2/Ar (20 mL/min) for 2 h, the gas was switched to the reaction atmosphere (50 vol.% C2H6/CO2), and XRD patterns were recorded.
XAS
X-ray absorption spectroscopy (XAS) spectra obtained at the BL14W beamline of the Shanghai Synchrotron Radiation Facility (SSRF). The storage rings at SSRF operated at 3.5 GeV with a stable current of 200 mA. Data was collected using a Si (111) double-crystal monochromator. The spectra of Pt L3-edge and Sn K-edge were collected in transmission mode and fluorescence mode by Lytle detector, respectively at room temperature.
H2-TPR and H2-IMS
H2 temperature-programmed reduction (H2-TPR) and H2 isothermal mass spectrometry (H2-IMS) experiments were conducted on a chemical adsorption instrument (Micromeritics AutoChem II 2920) equipped with a mass spectrometer (HRP-20 EGA) at atmospheric pressure. The samples (0.1 g) of PtSn/SiO2 + CaCO3 or SiO2 + CaCO3 (mass ratio of 1:1) were separately loaded into U-shaped quartz tubes. For H2-TPR testing process, the sample was pretreated with pure Ar (30 mL/min) at 150 °C for 1 h and cooled to 50 °C, followed by ramping the temperature at a rate of 10 °C/min from 50 °C to 850 °C in 10 vol.% H2/Ar (30 mL/min). For H2-IMS testing process, the sample was heated from room temperature to 600 °C at a ramp rate of 10 °C /min in pure Ar (30 mL/min) and stabilized for 10 min, then the gas was switched to 10 vol.% H2/Ar (30 mL/min) and maintained at 600 °C for an additional 2 h. The exhaust gases were analyzed for signals of H2 (m/z = 2), CO (m/z = 28), CO2 (m/z = 44), and H2O (m/z = 18) via mass spectrometer. Other temperature-programmed experiment details were showed in the supporting information.
In situ DRIFTS
In situ diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy was conducted on a Thermo Scientific Nicolet iS50 spectrometer, equipped with a high temperature cell featuring ZnSe windows and a liquid nitrogen-cooled detector. The catalyst powder was loaded into the center of the high temperature cell, and surface was leveled to minimize interference from reflections. The sample was initially heated to 600 °C in pure Ar (30 mL/min) flow and held for 0.5 h. Subsequently, background spectra were recorded under steady state conditions, followed the 10 vol.% C2H6/Ar (20 mL/min) was introduced into the high temperature reaction cell at 600 °C, with spectra recorded at intervals of one minute during this process.
DFT calculations
All DFT calculations were carried out utilizing the Materials Studio and the ultrasoft method. Details were shown in the supplementary.
Data availability
All data generated in this study are provided in the Article and Supplementary Information. The other data that support the findings of this study are available from the corresponding author upon request. Source data are provided with this paper.
References
Ro, I. et al. Bifunctional hydroformylation on heterogeneous Rh-WOx pair site catalysts. Nature 609, 287–292 (2022).
Froese, R. D. et al. A commercially viable solution process to control long-chain branching in polyethylene. Science 383, 1223–1228 (2024).
Pan, X., Jiao, F., Miao, D. & Bao, X. Oxide-zeolite-based composite catalyst concept that enables syngas chemistry beyond Fischer-Tropsch synthesis. Chem. Rev. 121, 6588–6609 (2021).
Arora, S. S., Nieskens, D. L. S., Malek, A. & Bhan, A. Lifetime improvement in methanol-to-olefins catalysis over chabazite materials by high-pressure H2 co-feeds. Nat. Catal. 1, 666–672 (2018).
Najari, S. et al. Oxidative dehydrogenation of ethane: catalytic and mechanistic aspects and future trends. Chem. Soc. Rev. 50, 4564–4605 (2021).
Dai, Y. et al. Recent progress in heterogeneous metal and metal oxide catalysts for direct dehydrogenation of ethane and propane. Chem. Soc. Rev. 50, 5590–5630 (2021).
Xie, Z. & Chen, J. G. Bimetallic-derived catalytic structures for CO2-assisted ethane activation. Acc. Chem. Res. 56, 2447–2458 (2023).
Qin, X. et al. Domino catalysis for selective dehydrogenation of ethane with shifted thermodynamic equilibrium. Joule 7, 753–764 (2023).
Liu, L. et al. Rivet of cobalt in siliceous zeolite for catalytic ethane dehydrogenation. Chem 9, 637–649 (2023).
Maeno, Z. et al. Isolated indium hydrides in CHA zeolites: speciation and catalysis for nonoxidative dehydrogenation of ethane. J. Am. Chem. Soc. 142, 4820–4832 (2020).
Zeng, L. et al. Stable anchoring of single rhodium atoms by indium in zeolite alkane dehydrogenation catalysts. Science 383, 998–1004 (2024).
Almallahi, R., Wortman, J. & Linic, S. Overcoming limitations in propane dehydrogenation by codesigning catalyst-membrane systems. Science 383, 1325–1331 (2024).
Wang, W. et al. Tandem propane dehydrogenation and surface oxidation catalysts for selective propylene synthesis. Science 381, 886–890 (2023).
Jiang, X. et al. Oxidative dehydrogenation of propane to propylene with soft oxidants via heterogeneous catalysis. ACS Catal. 11, 2182–2234 (2021).
Yan, H. et al. Tandem In2O3-Pt/Al2O3 catalyst for coupling of propane dehydrogenation to selective H2 combustion. Science 371, 1257–1260 (2021).
Wang, C. et al. Main-group catalysts with atomically dispersed in sites for highly efficient oxidative dehydrogenation. J. Am. Chem. Soc. 144, 16855–16865 (2022).
Chen, S. et al. Defective TiOx overlayers catalyze propane dehydrogenation promoted by base metals. Science 385, 295–300 (2024).
Liu, J. et al. Highly-dispersed zinc species on zeolites for the continuous and selective dehydrogenation of ethane with CO2 as a soft oxidant. ACS Catal. 11, 2819–2830 (2021).
Liu, J. et al. Influence of the zeolite surface properties and potassium modification on the Zn-catalyzed CO2-assisted oxidative dehydrogenation of ethane. Appl. Catal. B 304, 120947 (2022).
Yang, J. et al. Atomically synergistic Zn-Cr catalyst for iso-stoichiometric co-conversion of ethane and CO2 to ethylene and CO. Nat. Commun. 15, 911 (2024).
Xing, F., Nakaya, Y., Yasumura, S., Shimizu, K. -i & Furukawa, S. Ternary platinum-cobalt-indium nanoalloy on ceria as a highly efficient catalyst for the oxidative dehydrogenation of propane using CO2. Nat. Catal. 5, 55–65 (2022).
Yan, B. et al. Active sites for tandem reactions of CO2 reduction and ethane dehydrogenation. Proc. Natl. Acad. Sci. USA 115, 8278–8283 (2018).
Biswas, A. N., Xie, Z. & Chen, J. G. Can CO2-assisted alkane dehydrogenation lead to negative CO2 emissions? Joule 6, 269–273 (2022).
Gomez, E., Yan, B., Kattel, S. & Chen, J. G. Carbon dioxide reduction in tandem with light-alkane dehydrogenation. Nat. Rev. Chem. 3, 638–649 (2019).
Jiang, X. et al. CO2-assisted oxidative dehydrogenation of propane over VOx/In2O3 catalysts: interplay between redox property and acid–base interactions. ACS Catal. 12, 11239–11252 (2022).
Kang, L. et al. Surface structure-dependent mechanistic modulation of the selective oxidative dehydrogenation of ethane with CO2 over iron oxide catalysts. ACS Catal. 15, 5378–5390 (2025).
Xie, Z., Wang, X., Chen, X., Liu, P. & Chen, J. G. General descriptors for CO2-assisted selective C-H/C-C bond scission in ethane. J. Am. Chem. Soc. 144, 4186–4195 (2022).
Zheng, Y. et al. Evolution of Co species in CO2-assisted ethane dehydrogenation: competing cleavage of C-H and C-C Bonds. ACS Catal. 14, 4749–4759 (2024).
Zheng, Y. et al. Effect of CO2 co-feeding on the stabilization of atomically dispersed iron species over MgAl2O4 during ethane dehydrogenation reactions. ACS Catal. 13, 11153–11163 (2023).
Zhai, P. et al. CO2-mediated oxidative dehydrogenation of propane enabled by Pt-based bimetallic catalysts. Chem 9, 3268–3285 (2023).
Buelens, L. C., Galvita, V. V., Poelman, H., Detavernier, C. & Marin, G. B. Super-dry reforming of methane intensifies CO2 utilization via Le Chatelier’s principle. Science 354, 449–452 (2016).
Baamran, K., Lawson, S., Rownaghi, A. A. & Rezaei, F. Reactive capture and conversion of CO2 into hydrogen over bifunctional structured Ce1–xCoxNiO3/Ca perovskite-type oxide monoliths. J. Am. Chem. Soc. Au 4, 101–115 (2024).
Al-Mamoori, A., Lawson, S., Rownaghi, A. A. & Rezaei, F. Oxidative dehydrogenation of ethane to ethylene in an integrated CO2 capture-utilization process. Appl. Catal. B 278, 119329 (2020).
Han, R. et al. Progress in reducing calcination reaction temperature of calcium-looping CO2 capture technology: a critical review. Chem. Eng. J. 450, 137952 (2022).
Sun, S. et al. Potassium-promoted limestone for preferential direct hydrogenation of carbonates in Integrated CO2 Capture and Utilization. J. Am. Chem. Soc. Au 4, 72–79 (2024).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Liu, L. et al. Structural modulation and direct measurement of subnanometric bimetallic PtSn clusters confined in zeolites. Nat. Catal. 3, 628–638 (2020).
Motagamwala, A. H., Almallahi, R., Wortman, J., Igenegbai, V. O. & Linic, S. Stable and selective catalysts for propane dehydrogenation operating at thermodynamic limit. Science 373, 217–222 (2021).
Liu, L. et al. Regioselective generation and reactivity control of subnanometric platinum clusters in zeolites for high-temperature catalysis. Nat. Mater. 18, 866–873 (2019).
Frenkel, A. I. Applications of extended X-ray absorption fine-structure spectroscopy to studies of bimetallic nanoparticle catalysts. Chem. Soc. Rev. 41, 8163–8178 (2012).
Jianhao, J. et al. Coke deposition mechanisms of propane dehydrogenation on different sites of Al2O3 supported PtSn catalysts. Chem. Synth. 5, 19 (2025).
Zhou, H. et al. Cobaltosilicate zeolite beyond platinum catalysts for propane dehydrogenation. Nat. Catal. 8, 357–367 (2025).
Weisz, P. B. & Swegler, E. W. Stepwise reaction on separate catalytic centers: isomerization of saturated hydrocarbons. Science 126, 31–32 (1957).
Dong, C. et al. Disentangling local interfacial confinement and remote spillover effects in oxide–oxide interactions. J. Am. Chem. Soc. 145, 17056–17065 (2023).
Garg, S., Xie, Z. & Chen, J. G. Tandem reactors and reactions for CO2 conversion. Nat. Chem. Eng. 1, 139–148 (2024).
Tuo, J. et al. Promoting syngas to olefins with isolated internal silanols-enriched Al-IDM-1 aluminosilicate nanosheets. Angew. Chem. Int. Ed. 62, e202313785 (2023).
Zhao, D. et al. In situ formation of ZnOx species for efficient propane dehydrogenation. Nature 599, 234–238 (2021).
Gu, H. et al. Adjacent single-atom irons boosting molecular oxygen activation on MnO2. Nat. Commun. 12, 5422 (2021).
Acknowledgements
We acknowledge the National Key R&D Program of China (2023YFA1507800), the National Natural Science Foundation of China (22121004, 22361142706, and 22376077) and the State Key Laboratory of Catalysis (2024SKL-A-015) for financial support. This work is supported by the XPLORER PRIZE.
Author information
Authors and Affiliations
Contributions
X.L. and J.G. conceived and supervised the research. Z.W., X.L., S.C., and J.G. designed the studies, analyzed the data, and wrote the paper. Z.W. performed catalyst preparation, catalytic performance testing, and most of the characterizations. Y.L. and Z.Z. performed DFT calculations and valuable discussions. S.C. and J.Z. participated in data analysis and assisted with chemisorption characterization. W.Z. assisted with catalytic performance testing. All authors contributed to the discussion of the research and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Z., Liu, Y., Chen, S. et al. Ethane dehydrogenation over CaCO3-mediated tandem catalysts. Nat Commun 16, 7722 (2025). https://doi.org/10.1038/s41467-025-63063-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63063-4
