
Abstract
Pulmonary large cell neuroendocrine carcinoma (LCNEC) is a rare, aggressive lung tumor marked by significant molecular heterogeneity. In a study of 590 patients across two independent cohorts, we observe comparable overall survival across treatment regimens (chemotherapy, chemoimmunotherapy, immunotherapy) without unexpected adverse events. Genomic analysis identifies distinct non-small cell lung cancer-like (NSCLC-like, KEAP1, KRAS, STK11 mutations) and SCLC-like (RB1, TP53 mutations) LCNEC subtypes, with 80% aligning with SCLC transcriptional profiles. Serial sampling reveals stable mutational but shifting transcriptomic landscapes over time. Here we show, elevated FGL-1 (a LAG-3 ligand) and SPINK1 expression in NSCLC-like LCNECs, and higher levels of DLL3 in SCLC-like LCNECs. Immunofluorescence confirms FGL-1 expression in NSCLC-like LCNECs, and H&E slide analyses indicates fewer tumor-infiltrating lymphocytes in LCNECs versus other lung cancers. These findings highlight LCNEC’s distinct immunogenomic profile, supporting future investigations into LAG-3, SPINK1, and DLL3-targeted therapies.
Similar content being viewed by others

Introduction
Under the 2015 World Health Organization (WHO) guidelines, pulmonary large cell neuroendocrine carcinoma (LCNEC) is classified as a high-grade neuroendocrine tumor1. For patients with advanced LCNEC, median survival is typically between 7 and 12 months2. However, optimal systemic treatment strategies for this aggressive disease remain undefined due to limited data. Compounding the challenge is the scarcity of clinical studies and the relative rarity of LCNEC, which accounts for only 3% of all lung carcinomas3. At the core of this issue lies the unresolved biological relationship between LCNEC and other lung neoplasms. Gene expression and limited genomic studies have produced inconsistent findings on the connection between LCNEC and small cell lung cancer (SCLC), with certain reports indicating highly similar biology4 while others have suggested distinct gene expression and mutational profiles5,6. Additionally, molecular alterations typical of adenocarcinoma, such as EGFR mutations7,8, ALK rearrangements9, and KRAS mutations10, have been identified in LCNEC without adenocarcinoma components, sharply contrasting with classic de novo SC.
Previous integrative genomic and transcriptomic analyses of 75 LCNECs delineated two distinct molecular subtypes—Type I, characterized by co-occurring TP53 and STK11/KEAP1 alterations, and Type II, defined by bi-allelic inactivation of TP53 and RB111. Despite overlapping genomic landscapes, these subtypes demonstrated divergent transcriptional programs: Type I LCNECs display a neuroendocrine-enriched phenotype marked by ASCL1 and DLL3 expression with attenuated NOTCH signaling, whereas Type II LCNECs demonstrate diminished neuroendocrine differentiation, heightened NOTCH pathway activity, and enrichment of immune-related signatures. This discordance between mutational architecture and transcriptional identity underscores the biological heterogeneity of LCNEC and challenges reductionist models that rely solely on genomic alterations for subtype classification.
Recent genomic analyses have indicated that LCNEC can be divided into non-small cell lung cancer (NSCLC)-like (characterized by lack of RB1 genomic alterations and presence of mutations in the KRAS, STK11, and KEAP1 genes) and SCLC-like genomic subtypes (characterized by concurrent TP53 and RB1 mutations or loss)3,11,12,13. Unfortunately, patients with advanced LCNEC consistently exhibit poor outcomes regardless of the molecular subtype, underscoring the urgent need for new treatment paradigms14.
Immune checkpoint inhibitors (ICIs) have markedly revolutionized the treatment landscape for various cancers, including both NSCLC and SCLC15,16,17,18,19,20,21,22,23,24,25,26,27. However, the clinical efficacy data of ICIs in advanced LCNEC predominantly stems from case reports and small retrospective studies23,28,29,30,31. A recent analysis of 125 patients with advanced LCNEC suggested a potential survival benefit from immunotherapy-based regimens32. However, all patients received ICIs after front-line therapy—a treatment sequence no longer standard in NSCLC and SCLC. Prospective evaluation of ICIs in LCNEC is in its infancy, with only a small number of patients enrolled across several ongoing clinical trials (NCT03352934, NCT03190213, NCT03136055, NCT03290079, NCT0372836133, NCT0283401), and biomarker data remain sparse. Given the paucity of effective systemic therapies for LCNEC, there is an urgent need for strategies to improve outcomes. In this study, we analyze two independent cohorts comprising 590 patients with advanced LCNEC to define survival outcomes by front-line treatment regimen, including those incorporating ICIs. Through integrative analyses—spanning targeted and whole-exome sequencing (WES), digital pathology with machine learning, and whole-transcriptome sequencing (WTS)—we identify therapeutic targets and molecular vulnerabilities, informing future clinical trial development.
Results
Characteristics of clinical cohorts
Cohort 1 consisted of 217 patients with LCNEC treated with first-line systemic treatments. Cohort 2 comprised 373 patients diagnosed with LCNEC, of whom a subset had available data on first-line systemic treatment (n = 146; Table 1, Supplementary Fig. 1, Supplementary Data 2 and 3). Median age was 66 years (range: 18–88) and 67 (range: 38–89) for Cohorts 1 and 2, respectively (Table 1, Supplementary Data 2 and 3). The median follow-up time for Cohorts 1 and 2 was 48.6 months (95% CI: 38–62) and 29.5 months (95% CI: 25.3–36.7), respectively. The majority of patients identified as white in both cohorts (Cohort 1: n = 168, 81%, Cohort 2: n = 238, 64%; Table 1). For patients with available systemic treatment data, treatment regimens included chemotherapy (n = 121 (56%) for Cohort 1, n = 46 (32%) for Cohort 2), chemoimmunotherapy (n = 82 (38%) for Cohort 1, n = 88 (60%) for Cohort 2), and immunotherapy (n = 14 (6.4%) for Cohort 1, n = 12 (8.2%) for Cohort 2). There were no differences in baseline characteristics across the 3 systemic treatments (Table 1).
Survival outcomes to first-line systemic therapy
There was no significant difference in median OS across the 3 treatment groups in both cohorts (Fig. 1A, B). In Cohort 1, median OS was 15 months (95% CI: 8.1–17.4) in the chemotherapy group, 12 months (95% CI: 7.4–18.3) in the chemoimmunotherapy group, and 13.6 months (95% CI: 6.8–25.2) in the immunotherapy group. In Cohort 2, median OS was 14.9 months (95% CI: 9.3–26.1) in the chemotherapy group, 17.6 months (95% CI: 13.2–21.2) in the chemoimmunotherapy group, and 21.7 months (95% CI: 6.0–NR) in the immunotherapy group. To evaluate the potential influence of treatment year on clinical outcomes, we first performed an analysis of OS within the chemotherapy-treated cohort. The analysis revealed no significant difference in OS between patients treated prior to January 1, 2019 (n = 71), and those treated thereafter (n = 48; p = 0.57). Subsequently, we compared OS among patients treated with chemotherapy alone (n = 32) versus immunotherapy alone (n = 8) versus those treated with chemoimmunotherapy (n = 74) after March 1st, 2019, and similarly observed no significant difference (p = 0.3). Among patients who received chemotherapy as first-line systemic treatment, 61 went on to receive a subsequent line of therapy (28 non-ICI-based, 33 ICI-based). Within this group, there was no significant difference between patients who received subsequent ICI-based therapy and those who received non-ICI-based therapy (p = 0.2). In Cohort 2, there was no significant difference in OS between patients with NSCLC-like LCNECs who received NSCLC-based chemotherapy regimens and those with SCLC-like LCNECs treated with SCLC-based chemotherapy regimens (HR = 1.20, 95% CI: 0.59–2.31, p = 0.65, Supplementary Fig. 2). In Cohort 1, this analysis was limited by small sample size (n = 5 per group), and thus underpowered to detect meaningful differences.

Kaplan–Meier analysis of A overall survival (OS) in Cohort 1, B OS in Cohort 2, and C real-world progression-free survival (rwPFS) in Cohort 1, comparing patients with pulmonary large cell neuroendocrine carcinoma treated with chemotherapy (n = 119 for Cohort 1, n = 47 for Cohort 2), chemoimmunotherapy (n = 81 for Cohort 1, n = 99 for Cohort 2), or immunotherapy (n = 14 for Cohort 1, n = 12 for Cohort 2). Survival distributions were compared using a two-sided log-rank test. D Tornado plot depicting treatment-related adverse events for patients treated with any first-line systemic therapy in Cohort 1 (n = 216). Any grade (right) and ≥grade 3 (left). HR hazard ratio, ref reference. Statistical significance is defined as p < 0.05.
In the ICI-treated group from Cohort 2, six patients exhibited a real-world overall survival (rwOS) exceeding 20 months. Among these, 33% (2 out of 6) demonstrated high TMB, and 50% (3 out of 6) were positive for programmed death-ligand 1 (PD-L1) expression. In patients receiving ICI-based therapies, GSEA revealed a significant enrichment of pro-inflammatory immune pathways in those with a rwOS exceeding 20 months compared to those with an rwOS of less than 20 months (Supplementary Fig. 3). Expanding the biomarker analysis to include the chemoimmunotherapy group in Cohort 2, where the sample size permitted more robust comparisons, the median rwOS was not significantly different between TMB-high (>19) versus TMB-low tumors (≤19; p = 0.7, Supplementary Fig. 4A). Furthermore, within the chemoimmunotherapy group, rwOS did not significantly differ based on PD-L1 status (p = 0.5, Supplementary Fig. 4B).
Among the 216 evaluable patients in Cohort 1, median rwPFS was 5.1 months (95% CI: 3.4–5.5) in the chemotherapy group, 5.4 months (95% CI: 4.4–6.1) in the chemoimmunotherapy group, and 3.9 months in the immunotherapy group (95% CI: 2–6.5). After adjusting for ECOG, M stage, sex, and age, the chemotherapy group had a statistically significantly lower rwPFS compared to the chemoimmunotherapy group (p = 0.03; HR: 1.43 [95% CI: 1.04–1.99]). In contrast, the immunotherapy group did not show a significant difference in rwPFS (HR: 1.3 [95% CI: 0.69–2.58]) (Fig. 1C). In Cohort 2, rwPFS was not available, so ToT was used as a surrogate endpoint. Median ToT was 2.4 months (95% CI: 2.1–3.6) in the chemotherapy group, 7.5 months (95% CI: 5.2–10.4) in the chemoimmunotherapy group, and 6.3 months (95% CI: 1.3–18.0) in the immunotherapy group. Patients treated with chemotherapy had significantly worse ToT compared to those receiving chemoimmunotherapy (HR: 1.44, p = 0.05, Supplementary Fig. 5).
Toxicity profiles in Cohort 1
Overall, 112 (52%) patients developed treatment-related adverse events (trAE) of any grade (Fig. 1D) with similar frequencies across treatment groups (chemotherapy: n = 61, 50%; chemoimmunotherapy: n = 45, 55%; immunotherapy: n = 6, 43%). Grade ≥ 3 trAE occurred in 22% (95% CI: 16–31), 26% (95% CI: 17–36), and 0% (95% CI: 0–22) patients in the chemotherapy, chemoimmunotherapy, and immunotherapy groups, respectively (Supplementary Fig. 6). Toxicity led to discontinuation of systemic treatment in 10% (95% CI: 5.8–17), 15% (95% CI: 8.6– 24), and 14% (95% CI: 2.5–40) patients in the chemotherapy, chemoimmunotherapy, and immunotherapy groups, respectively (Supplementary Data 4).
Genomic map and clinical outcomes of LCNEC molecular subtypes
Prior genomic mapping of LCNEC has delineated these tumors into SCLC-like and NSCLC-like categories11. Utilizing a similar stratification approach, we classified 217 tumors in Cohort 1 into SCLC-like (characterized by concurrent TP53 and RB1 mutations) and NSCLC-like (characterized by mutations in either STK11, KRAS, or KEAP1 and wild-type RB1 status). Tumors that did not conform to either of these subtypes were designated as unclassified. In Cohort 1, 85 patients had genomic data that allowed molecular classification. Of these, 25 (29%) were classified as NSCLC-like, 19 (22%) were SCLC-like, and 41 (48%) were unclassified (Fig. 2A, Supplementary Fig. 1, Supplementary Data 5). The remainder of tumors (n = 132) did not have full mutation profiling of the genes of interest (KEAP1, KRAS, STK11, TP53, and RB1) and thus were labeled unknown. In Cohort 2, 89 (23.9%) tumors were genomically NSCLC-like, 136 (36.5%)were SCLC-like, and 148 (39.7%) were unclassified (Fig. 2B). In addition to the previously mentioned genes, commonly altered genes included other drivers such as SMARCA4, KMT2D, CDKN2A, PTEN, ARID1A, and NF1 (Fig. 2B). Targetable alterations were detected in 22 of 373 (5.9%) LCNECs and included KRASG12C (n = 13), EGFR activating mutations (n = 5), ERBB2 mutation (n = 1), and fusions (EML4::ALK, n = 3; ETV6::NTRK2, n = 1).

A CoMut plot for 85 patients with LCNEC. For each tumor, from top to bottom, the molecular subtype, sex, age at first-line systemic treatment, first-line systemic treatment, and prevalent molecular alterations. B CoMut plot for 373 patients with LCNEC. For each tumor, from top to bottom, the tumor mutational burden (mutations/Mb), LCNEC molecular subtype, sex, age, and prevalent molecular alterations. C Heatmap depicting the genomic driver and transcriptional profile evolution of two temporally different biopsies from four LCNECs in Cohort 1 and five LCNEC patients in Cohort 2*.IHC-PD-L1 (22c3) positivity ≥1†. TMB-High >19 mutations (muts)/megabase (Mb). D Scatter plot showing the prevalence of genomic alterations and FDA-approved ICI biomarkers prevalence across NSCLC-like (n = 89) and SCLC-like (n = 136) LCNEC in Cohort 2. A two-sided Chi-Square test was employed with statistical significance defined as p < 0.05. E Bar-and-whisker plot comparing FDA-approved ICI biomarkers prevalence across NSCLC-like (n = 89), SCLC-like (n = 136), and unclassified (n = 148) LCNECs in Cohort 2. A two-sided chi-squared test was employed with statistical significance defined as p < 0.05. ****<0.0001 (p value for unclassified vs NSCLC-like comparison: 000013; p value for unclassified vs SCLC-like comparison: 0.00002). 1L First-line, TF Transcription factor, mut mutation, dMMR Mismatch repair deficient, MSI-H microsatellite instability high, TMB Tumor mutational burden, PD-L1 Programmed death-ligand 1.
To refine molecular classification of unclassified LCNECs, we developed a support vector machine (SVM) classifier trained on transcriptomic profiles from NSCLC-like and SCLC-like LCNEC subtypes (see “Methods”). Gene selection was guided by both high inter-sample variance and differential expression (adjusted p < 0.01), yielding 2168 gene transcripts as input features. The model, trained on 80% of labeled samples (n = 174) and validated on the remaining 20% (n = 44), demonstrated high discriminatory performance (AUC = 0.98; accuracy = 90.1%) (Fig. 3A, B). Applying the trained classifier to the 143 previously unclassified tumors, 101 (70.6%) were reclassified as SCLC-like and 42 (29.4%) as NSCLC-like. Dimensionality reduction using UMAP revealed three distinct transcriptomic clusters, with strong concordance between classifier-predicted subtypes and spatial clustering (Fig. 3C, D). Notably, reclassified samples localized proximally to their respective subtype clusters, supporting the biological plausibility of the predictions. With the refined classification, we next evaluated OS and found no significant difference across the four LCNEC subtypes (log-rank P = 0.23, Supplementary Fig. 7).

A Receiver operating characteristic (ROC) curve demonstrating the performance of a support vector machine (SVM) classifier trained to distinguish NSCLC-like from SCLC-like LCNECs based on 2168 transcriptomic features (AUC = 0.98). B Confusion matrix showing classification accuracy within the validation cohort. C Unsupervised UMAP projection of transcriptomic profiles reveals three distinct molecular clusters. D Overlay of classifier-derived labels onto the UMAP demonstrates concordance between predicted subtypes and transcriptomic clustering, enabling reclassification of previously unclassified LCNECs into biologically coherent groups. NSCLC non-small cell lung cancer, Unc Unclassified.
To assess whether LCNECs maintain their genomic subtype over time, we analyzed data in Cohorts 1 and 2 from nine patients with two temporally distinct tumor specimens each. The median time between serial samples was 9.5 months (range 1.6–63 months) in Cohort 1 and 13 months (range 11–15 months) in Cohort 2. Our analysis revealed that the genomic drivers were consistently retained across the specimens, with no acquisition of additional genomic alterations that would reclassify the tumors. In comparison, the transcriptional subtypes exhibited greater fluidity over time, with 4 out of 5 tumor pairs demonstrating a shift in their transcriptional profiles(Fig. 2C).
In comparison to NSCLC-like LCNECs, KMT2D genomic alterations were predominantly observed in SCLC-like LCNECs, whereas SMARCA4 alterations were more prevalent in NSCLC-like LCNECs (Fig. 2D). Tumors with high tumor mutational burden (TMB-high, defined as at least 10 mutations per megabase) were found in 56.3% (n = 49) of NSCLC-like LCNECs and 49.6% (n = 67) of SCLC-like LCNECs. PD-L1 positivity (at least 1%) exhibited similar rates across the three treatment groups. Mismatch repair deficiency, determined by immunohistochemistry, was identified in 2 (1.47%) SCLC-like LCNECs (Fig. 2E) and was absent in both NSCLC-like and unclassified LCNECs. There was no difference in rwPFS and OS outcomes to front-line therapy among NSCLC-like, SCLC-like, and unclassified LCNECs. Mutation analyses of key driver genes, including EGFR, KRAS, KEAP1, RB1, SMARCA4, and STK11, revealed that in Cohort 1, tumors harboring mutations in TP53 or STK11 were significantly associated with inferior OS compared to their wild-type counterparts (Supplementary Fig. 8). In contrast, no other genomic alterations demonstrated a statistically significant association with survival in this cohort. Similarly, in Cohort 2, none of the evaluated genomic alterations were significantly correlated with OS.
LCNEC tumors are enriched for the ASCL1 and YAP1 transcriptomic subtypes
SCLCs have been classified into one of four transcriptional subtypes: ASCL1, NEUROD1, POU2F3, and YAP1 based on transcription factor (TF) expression levels34,35. We leveraged an independent cohort of 1704 SCLC from Caris Life Sciences for comparisons between SCLC and LCNECs (Supplementary Data 6). Of the 1704, 1643 SCLC had WTS data. Hierarchical clustering of 1643 SCLC and 361 LCNECs showed enrichment of ASCL1 in SCLC-like LCNEC compared with both NSCLC-like (36.56% versus 23.81%, p = 0.04) and unclassified (36.56% versus 11.12%, p < 0.001, Fig. 4A). The YAP1 subtype was prevalent in about 26.19% of NSCLC-like LCNECs compared to 14.18% and 31.76% of SCLC-like and unclassified LCNECs, respectively. YAP1 LCNECs were characterized by enriched CD8 infiltration as previously described for YAP1-enriched SCLC tumors36 (Fig. 4B, Supplementary Fig. 9). SCLC-like LCNECs were enriched for STK11 and KEAP1 mutations and had a significantly higher TMB compared to SCLC (Fig. 4C, D). SCLC and SCLC-like LCNEC had significantly higher expression of DLL3 compared to unclassified LCNEC (SCLC vs unclassified LCNEC: median TPM = 8.3 vs 3.9, p < 0.0001; SCLC-like LCNEC vs unclassified LCNEC: median TPM = 6.3 vs 3.9, p < 0.05, Fig. 4E). There was no significant difference in DLL3 expression between NSCLC-like and SCLC-like LCNECs. However, DLL3 expression was significantly higher in SCLC compared to NSCLC-like LCNECs (median TPM = 8.3 vs 5.7, p < 0.05, Fig. 4E).

A Heatmap illustrating hierarchical clustering of SCLC (n = 1643, Caris Life Sciences) and LCNECs (n = 361, Cohort 2) for established SCLC transcriptional subtypes (ASCL1, NEUROD1, POU2F3, and YAP1). B Bar plot showing the distribution of SCLC transcriptional subtypes across LCNECs (n = 361, Cohort 2). The non-parametric two-sided Wilcoxon rank sum test was used with statistical significance defined as p < 0.05. C Comparison between the prevalence of genomic alterations and FDA-approved ICI biomarkers between SCLC (n = 1643, Caris Life Sciences) and SCLC-like LCNEC (n = 136, Cohort 2). A two-sided Chi-Square test was employed with statistical significance defined as p < 0.05. D Bar plot illustrating the prevalence of NSCLC-like genomic drivers and FDA-approved ICI biomarkers between SCLC (n = 1643, Caris Life Sciences) and SCLC-like LCNEC (n = 136, Cohort 2). The non-parametric two-sided Wilcoxon rank sum test was used with statistical significance defined as p < 0.05. The p value for STK11 mutations was 0.003, while p values for both KEAP1 mutations and tumor mutational burden (TMB) were <0.0001. E Comparison of DLL3-transformed gene expression across NSCLC-like LCNEC (n = 84), SCLC-like LCNEC (n = 134), unclassified LCNEC (n = 143), and SCLC (n = 1643). The non-parametric two-sided Wilcoxon rank sum test was used with statistical significance defined as p < 0.05. Dot plots with median values are shown. *<0.05; **<0.01; ****<0.0001. The p value for unclassified versus SCLC-like LCNEC was <0.0001; for unclassified versus NSCLC-like LCNEC, 0.04; and for NSCLC-like versus SCLC-like LCNEC, 0.04. dMMR Mismatch repair deficient, MSI-H microsatellite instability high, TMB Tumor mutational burden, NSCLC non-small cell lung cancer, SCLC small cell lung cancer, LCNEC large cell neuroendocrine carcinoma.
Fibrinogen-like protein 1 (FGL-1) and serine peptidase inhibitor, Kazal type 1 (SPINK1) overexpression in NSCLC-like LCNECs suggest potential therapeutic vulnerabilities
De novo differential gene expression analysis between NSCLC-like and SCLC-like LCNECs in Cohort 2 revealed substantial differences in the expression of 1061 genes (p < 0.05, fold change > 2, Fig. 5A). Among these, FGL-1 and SPINK1 were markedly enriched in NSCLC-like LCNECs relative to SCLC-like LCNECs. This enrichment was characterized by ubiquitous overexpression in NSCLC-like LCNECs, in contrast to the low expression observed in other LCNEC subtypes and SCLC molecular subtypes (Fig. 5B). Notably, SFTPB, a hallmark gene of type II alveolar cells, exhibited elevated expression in both NSCLC-like and unclassified LCNECs, suggesting a potentially distinct cellular origin compared to SCLC-like tumors.

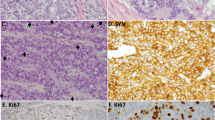
A Volcano plot showing differentially expressed genes between NSCLC-like (n = 89) and SCLC-like (n = 136) LCNECs in Cohort 2. Y-axis displays the −log10 p value derived from a two-sided Kolmogorov–Smirnov test. Genes with a False discovery rate of 5% and an absolute value of the log10 fold change of 0.5. B Heatmap of the top differentially expressed genes identified in (A), applicable to LCNEC and SCLC molecular subtypes. C Comparison of FGL-1 and SPINK1 log-transformed gene expression across LCNEC subtypes: NSCLC-like (n = 19), SCLC-like (n = 16), and unclassified (n = 31) LCNECs, using previously published data from George et al.11. The non-parametric two-sided Wilcoxon rank sum test was used with statistical significance defined as p < 0.05. For FGL-1, the p value for NSCLC-like versus SCLC-like LCNEC was 8.4 × 10−6; for NSCLC-like versus unclassified LCNEC, 0.0003; and for unclassified versus SCLC-like LCNEC, 0.01. For SPINK1, the p value for NSCLC-like versus SCLC-like LCNEC was 1 × 10−6; for NSCLC-like versus unclassified LCNEC, 8.3 × 10−5; and for unclassified versus SCLC-like LCNEC, 0.003. D Comparison of relative FGL-1 protein expression across 54 cell lines from various cancer types, using data from the DepMap dataset. E Box-and-whisker plots comparing median FGL-1 expression across 20 cancer types from Caris Life Sciences (n = 125,632 tumor samples). Dashed lines from top to bottom represent median FGL-1 expression in NSCLC-like, all, and SCLC-like LCNECs, respectively. For the box-and-whisker plots, the center line indicates the median, the bounds of the box represent the 25th and 75th percentiles (interquartile range), and the whiskers extend to the minimum and maximum values. Each point represents an individual patient tumor (biological replicate). F GSEA plots showing pathways enriched in FGL-1 high versus FGL-1 low NSCLC-like LCNECs. G Representative immunofluorescence staining of FGL-1 (green) and DAPI (white) in 2 NSCLC-like LCNECs, 1 SCLC-like LCNEC, 3 NSCLC, and 4 SCLC (H). 20× magnification is shown. The experiment was repeated using independent biological replicates (no technical replicates). Dot plot comparing tumor-infiltrating lymphocyte (TIL) counts among patients with lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), small cell lung cancer (SCLC), and large cell neuroendocrine carcinoma (LCNEC). Median values are shown per group. The non-parametric two-sided Wilcoxon rank sum test was used with statistical significance defined as p < 0.05. TIL tumor-infiltrating lymphocytes, LUSC lung squamous cell carcinoma, LUAD lung adenocarcinoma, NSCLC non-small cell lung cancer, SCLC small cell lung cancer, LCNEC large cell neuroendocrine carcinoma. The p value for SCLC versus NSCLC-like LCNEC was 0.005; for LUAD versus NSCLC-like LCNEC, 0.009; and for LUSC versus NSCLC-like LCNEC, 0.006.
Unsupervised clustering analysis of all LCNECs, irrespective of their mutational status, delineated four distinct clusters (Supplementary Fig. 10A). Using the top differentially expressed genes between the two largest clusters (B and D, Supplementary Fig. 10B), hierarchical clustering of LCNEC samples, irrespective of molecular subtype, showed enrichment of FGL-1 and SPINK1 in cluster A, whereas FGL-1 expression was minimal in the other three LCNEC clusters (Supplementary Fig. 10C).
Given the prior identification of FGL-1 as an MHC II-independent ligand for LAG-337, we conducted further in-depth analysis to further explore this relationship within our dataset. Analysis of TCGA-LUAD data38 indicated that FGL-1 expression was significantly elevated in NSCLC-like LCNEC (n = 6) compared to NSCLC tumors (n = 503, Supplementary Fig. 11). Additionally, RNA expression data from a previously published dataset of 75 LCNECs11 demonstrated significant enrichment of FGL-1 in NSCLC-like LCNECs (n = 19) compared to SCLC-like LCNECs (n = 16) and unclassified LCNECs (n = 31, Fig. 5C).
Examination of the DepMap dataset, encompassing 54 cell lines from various cancer types, revealed the highest protein expression of FGL-1 in the LCNEC cell line NCIH1155 (Fig. 5D, Supplementary Data 7). Furthermore, WTS data from Caris Life Sciences, spanning 125,632 tumor samples across 20 cancer types, indicated that median FGL-1 expression in NSCLC-like LCNECs was the third highest, following intrahepatic cholangiocarcinoma and hepatocellular carcinoma(Fig. 5E). SPINK1 shares 50% sequence homology with epidermal growth factor expression and has been shown to engage both EGFR and MAPK pathways39,40. As these are potentially targetable pathways, we leveraged the study by George et al.11 and showed enrichment of SPINK1 expression in NSCLC-like LCNECs compared to SCLC-like and unclassified LCNECs (Fig. 5C). This observation suggests promising therapeutic strategies targeting NSCLC-like LCNECs through LAG-3 and/or SPINK1 inhibition.
GSEA of Hallmark gene sets, a collection of genes curated to provide a comprehensive summary of key cellular pathways and functions41, was performed on FGL-1 high versus low NSCLC-like LCNECs. GSEA revealed, among other pathways, significant enrichment of the KRAS signaling pathway in FGL-1 high NSCLC-like tumors compared to FGL-1 low ones, suggesting a potential cross-talk between KRAS signaling and FGL-1 (Fig. 5F). FGL-1 immunofluorescence staining was positive in 1 out of 2 (50%) NSCLC-like LCNEC, 0 out of 1 (0%) SCLC-like, 3 out of 3 (100%) NSCLC, and 0 out of 4 (0%) SCLC, respectively (Fig. 5G).
Depletion of tumor-infiltrating lymphocytes in LCNECs compared to other lung cancer cohorts
Clinical evidence suggests that the blockade of immune checkpoint pathways, such as PD-1, is most efficacious in tumors that have already initiated an endogenous T cell response. However, the observed therapeutic response in certain PD-L1–negative tumors implies that the induction of tumor rejection via PD-1 blockade does not necessarily depend on the preexistence of an immune response, as conventionally indicated by the presence of tumor-infiltrating T cells42. Given the potential for targeting alternative immune pathways through LAG-3 inhibition in NSCLC-like LCNECs, we investigated the level of immune infiltration in LCNEC tumors in comparison to SCLC and NSCLC. Employing computational pathology analysis, we quantified TILs on H&E slides, following the methodology previously established by our group43. Our analysis revealed that LCNECs (n = 16) exhibited significantly lower TIL counts compared to lung adenocarcinomas (n = 353), lung squamous cell carcinomas (n = 63), and SCLC (n = 122) (Fig. 4H, Supplementary Data 8). However, we were underpowered to perform analyses stratified by LCNEC molecular subtypes, as there were 6 NSCLC-like, 4 SCLC-like, and 6 unclassified LCNECs with TIL assessments.
Integrating mutational subtype classification and RNA expression data leads us to propose a model that may be associated with a unique response to therapies and can be prospectively tested in clinical trials (Fig. 6).

The dashed line corresponds to a potential therapeutic target. NSCLC non-small cell lung cancer, SCLC small cell lung cancer.
Discussion
Currently, there is no consensus on the optimal systemic treatment for LCNEC. The advent of immunotherapy has created new treatment paradigms, but comprehensive comparative analyses of first-line treatment regimens in pulmonary LCNEC are limited, particularly due to the scarcity of clinical trial data for this patient population. This gap underscores the importance of real-world studies. Our study represents the most comprehensive characterization of LCNEC to date, encompassing detailed clinical cohorts, tumor DNA sequencing, WTS, and an evaluation of the TME. Our findings reveal comparable efficacy and toxicity among patients treated with chemotherapy, chemoimmunotherapy, and immunotherapy alone. Building on existing LCNEC subtyping research, we identify therapeutic targets that have the potential to expand the treatment landscape for this aggressive malignancy, and we propose a framework to reclassify unclassified LCNECs.
Recent studies in the post-front-line setting indicate that immunotherapy-based strategies may hold promise for patients with LCNEC. For instance, a retrospective study involving 23 patients treated with immunotherapy in advanced LCNEC reported a median PFS of 4.2 months31. Another study, including 17 patients treated with nivolumab in the second-line setting, reported a median OS of 12.1 months and an overall response rate of 29.4%, with a median PFS of 3.9 months44. Our analysis did not reveal significant differences in OS outcomes across various treatment groups, including immunotherapy-based regimens. There was a statistically significantly lower rwPFS for patients treated with chemotherapy compared to chemoimmunotherapy, although the difference was not clinically significant (median rwPFS difference of 0.3 months). In general, patients exhibited typical poor outcomes regardless of the systemic treatment regimen employed.
Genomic analysis from our study revealed that close to 6% of LCNEC possess targetable genomic alterations amenable to existing FDA-approved therapies for lung cancer, corroborating previous findings, and supporting the use of WES in this patient population at the time of diagnosis7,8. Previous studies have classified LCNEC into genomic subtypes paralleling either SCLC or NSCLC3,11,14. In the vast majority of patients lacking targetable driver mutations, our results demonstrate that current systemic treatments do not significantly enhance clinical outcomes across these genomic subtypes. Notably, our data indicate that patients with NSCLC-like LCNECs exhibit elevated expression of FGL-1 and SPINK1 at the RNA level with variable protein expression of FGL-1, suggesting potential therapeutic benefits from targeting LAG-3 or SPINK1 pathways. This emphasizes the critical need for clinical trials investigating LAG-3 inhibitors or FGL-1 antibody-drug conjugates in this context. Furthermore, SPINK1-positive cancers could potentially benefit from interventions targeting downstream effectors such as the MAPK pathway45,46,47,48. While our study primarily focuses on the molecular and clinical characterization of LCNEC, the functional significance of FGL-1 and SPINK1 remains unresolved. Future in vitro and in vivo studies are warranted to elucidate its role in tumor progression and immune evasion, which may further support its development as a therapeutic target.
SCLC-like and NSCLC-like LCNECs exhibit elevated DLL3 expression, suggesting that DLL3 antibody-drug conjugates or bispecific antibodies, or T cell engagers, may provide a promising therapeutic approach for targeting these tumors in a manner analogous to SCLC49. Ongoing clinical trials (NCT05882058 and NCT05619744) are actively investigating DLL3-targeted therapies in patients with LCNEC. We also utilized digital assessment of TILs to show a significant reduction of TILs in LCNECs compared to other lung cancer types. The low absolute levels of TILs in LCNECs could suggest that these tumors are either altered or cold immune tumors, potentially explaining the modest efficacy of immunotherapy-based approaches observed so far. Overall, these findings underscore the urgent requirement for innovative clinical trials and the exploration of therapeutic strategies to improve outcomes for patients with LCNEC.
A key contribution of our study is the resolution of previously unclassified LCNECs through integrative transcriptomic modeling. Utilizing an SVM classifier trained on NSCLC-like and SCLC-like subtypes, we reclassified the majority of unclassified tumors into biologically coherent groups with high discriminatory performance (AUC = 0.98). This refined molecular taxonomy offers a critical framework for aligning LCNEC subtypes with targeted therapeutic strategies. Nonetheless, prospective validation in independent cohorts is warranted to confirm the robustness and clinical applicability of this reclassification schema.
Recent studies in SCLC have questioned the existence of a YAP1-defined subtype, as immunohistochemical and molecular profiling analyses failed to confirm its distinction within SCLC50,51. However, emerging evidence suggests that YAP1 plays a biologically significant role in pulmonary LCNEC. In our cohort, YAP1 subtypes were found in more than a quarter of NSCLC-like, SCLC-like, and unclassified LCNECs. A recent study also demonstrated that YAP1 expression defines two intrinsic subtypes of LCNEC with distinct molecular characteristics and therapeutic vulnerabilities52. The YAP1-high subtype is associated with a mesenchymal and inflamed phenotype, frequent SMARCA4 and CDKN2A/B genomic alterations, and vulnerability to MEK and AXL-targeted therapies. In contrast, the YAP1-low subtype shares genomic and transcriptomic similarities with SCLC, including RB1 and TP53 co-mutations, a neuroendocrine phenotype, and potential susceptibility to SCLC-directed therapies, such as DLL3 and CD56-targeting CAR T therapies. These findings underscore the biological significance of YAP1 in LCNEC and highlight its potential role in guiding therapeutic strategies. Future research should further investigate whether YAP1 expression influences tumor plasticity, immune microenvironment interactions, and treatment response, particularly in the context of emerging therapies for LCNEC.
Our study has several limitations that warrant consideration. First, the retrospective design inherently introduces biases and limits the ability to draw causal inferences. Second, the clinical data were incomplete, and follow-up intervals were not standardized, potentially introducing variability in the calculation of rwPFS. Moreover, the retrospective nature of the study introduces variability in treatment decisions based on evolving clinical guidelines and physician discretion. While PD-L1 expression and TMB were assessed where available, additional factors such as histologic subtype, prior treatment history, and disease burden also influenced therapy initiation. However, due to the lack of standardized prospective selection criteria, we cannot fully account for all variables that may have guided immunotherapy decisions. Overall, these limitations reflect the inherent heterogeneity of real-world data collection and may affect the robustness of rwPFS estimates. As such, we emphasize the need for prospective studies to validate and build upon our findings, thereby enhancing their translational potential. Third, in Cohort 1, the use of variable targeted sequencing platforms to identify mutations and copy number alterations posed a challenge. Differences in gene composition and baitset coverage across these platforms limited the comprehensiveness of genomic analyses. To overcome this limitation, we included Cohort 2, which underwent systematic and uniform genomic and transcriptomic characterization, thereby providing a more consistent and robust dataset of equivalent size. Fourth, matched germline testing was not uniformly available across sequencing platforms, and this limitation was further compounded by variability in germline filtering algorithms. These factors may influence the interpretation of mutational drivers and TMB estimates. While this may have led to occasional false-positive somatic calls, it reflects current practice across CLIA-certified platforms, which largely rely on tumor-only sequencing and population databases for germline exclusion. Fifth, the study lacked detailed information on the specific biopsy methods used for diagnosing LCNEC. This limitation may impact the interpretation of diagnostic challenges associated with small biopsy specimens; however, all cases were reviewed and confirmed by board-certified thoracic pathologists. Sixth, our study is limited by the under-representation of non-White populations, which reduces the generalizability of our findings and limits the statistical power to identify genomic and survival associations within these subgroups. This highlights the critical need for more inclusive research to ensure findings are applicable across diverse patient populations. Moreover, in Cohort 1, LCNEC diagnoses were made by local pathologists without centralized pathological review, raising the possibility of case overestimation and inadvertent inclusion of tumors with mixed histologic features. However, a validation study conducted by Caris Life Sciences on a subset of samples initially classified as LCNEC revealed that 95% of these cases were confirmed upon central pathological review, supporting the accuracy of the classifications. Additionally, the use of FFPE material introduces the potential for sequencing artifacts, although standardized quality control measures were employed to minimize this risk. Finally, given the rarity of LCNEC, we extended the study period to accumulate a sufficiently large sample size. This approach, while necessary, may have introduced variability in the reliability of estimates when comparing treatment strategies due to temporal trends. To account for this, sensitivity analyses stratified by treatment year were conducted to evaluate potential temporal influences.
Despite these limitations, our analyses consistently revealed similar clinical outcomes across the two distinct cohorts, underscoring the robustness of our findings. The complementary nature of these datasets allowed us to capture a broader spectrum of clinical and molecular characteristics of LCNEC, leveraging the unique strengths of each cohort to provide a more comprehensive understanding of this rare malignancy. By analyzing the cohorts independently for most outcomes, we effectively mitigated the confounding effects of methodological differences, ensuring the integrity of our results. Collectively, the two cohorts represent the most extensive and integrative analysis of LCNEC to date, offering critical insights into its genomic landscapes and clinical behavior, and paving the way for future research and therapeutic innovations.
In conclusion, while the systemic treatment of LCNEC remains an area of unmet clinical need, our study advances the field by offering the most extensive and integrative analysis of this malignancy to date. Through meticulous examination of clinical outcomes, genomic landscapes, and the TME, we illuminate the complexity of LCNEC and highlight critical avenues for therapeutic intervention. Our findings challenge the efficacy of current systemic therapies across LCNEC subtypes, underscoring the urgent need for treatment strategies tailored to the molecular underpinnings of this aggressive cancer. The identification of actionable targets such as FGL-1, SPINK1, and DLL3 opens new frontiers in LCNEC therapy, with ongoing clinical trials poised to transform the treatment landscape. However, the modest responses to immunotherapy observed in our study and the paucity of TILs in LCNEC tumors suggest that future efforts must also focus on overcoming immune evasion mechanisms. To truly shift the paradigm in LCNEC treatment, it will be imperative to conduct robust, prospective clinical trials that not only evaluate the efficacy of emerging therapies but also ensure inclusivity across diverse patient populations.
Methods
Patient cohorts
To provide a broad description of treatment patterns in patients with LCNEC, we gathered data from two large historical cohorts: Cohort 1 is a multicenter study of 217 patients with LCNEC treated with 1st line systemic treatment between 1/2014 and 12/2023. Clinical information was gathered from 26 participating institutions in Belgium, Germany, Italy, Spain, the United Kingdom, and the United States (Supplementary Data 1). This study was conducted in accordance with the principles of the Declaration of Helsinki and received approval from the Yale New Haven Hospital Institutional Review Board (IRB) as well as the IRBs of the respective participating institutions. Although the study relied exclusively on de-identified data, we acknowledge that genetic data, while de-identified, retains inherent identifiability due to its unique nature. In compliance with HIPAA guidelines and considering GDPR classifications of genetic data as personal data, stringent safeguards were implemented to protect patient confidentiality. No direct identifiers were accessible to study investigators, and data were managed within secure, access-controlled environments. Based on the use of de-identified data and the minimal risk posed to participants, written informed consent was waived by the IRBs. For Cohort 1, the pathologic diagnosis of LCNEC was reviewed at the local treating institution and confirmed by pulmonary pathologists according to the 5th edition of the WHO Classification of Lung Tumors53. The diagnosis of pulmonary LCNEC required the presence of neuroendocrine morphology (organoid nesting, palisading, rosettes, or trabeculae) and expression of at least one neuroendocrine marker (chromogranin A, synaptophysin, INSM1, CD56) by immunohistochemistry. High mitotic activity (>10 mitoses per 2 mm²) and/or extensive necrosis were also required for classification. Tumor specimens with mixed histologic components (adenocarcinoma, squamous cell carcinoma, or SCLC) other than LCNEC were excluded to enrich for LCNECs.
Cohort 2 represents a historical cohort collected by Caris Life Sciences (Phoenix, AZ, USA) between 1/2015 and 11/2023. This included 373 patients diagnosed with LCNEC who underwent tissue-based genomic profiling by a commercial laboratory (Caris Life Sciences). The specimens were primarily composed of diagnostic biopsy or surgical tumor samples. Of these, a subset of 146 patients met the inclusion criteria for clinical outcome analyses, consistent with Cohort 1, defined as having advanced LCNEC treated with first-line systemic therapies. This focus was driven by the study’s objective to investigate first-line treatment outcomes in advanced LCNEC—a critical and understudied area in the field. The remaining patients, who either did not have advanced LCNEC or were not treated with first-line systemic therapies, were excluded from the clinical outcome analyses but included in genomic and transcriptomic correlates. This approach ensured alignment with the study’s objectives to investigate the treatment landscape and outcomes for advanced LCNEC. Clinical data were acquired from insurance claims, and the selection of systemic therapies was at the discretion of the treating physician. The sex and age of patients were determined from medical forms. For Cohort 2, pathologic diagnosis was initially confirmed at local institutions and later reviewed centrally at Caris Life Sciences for accuracy in a subset of 142 tumors with a diagnostic accuracy rate of 94.3%. Systemic treatments for both cohorts included chemotherapy alone, chemoimmunotherapy, and immunotherapy alone. An independent cohort of 1704 SCLCs from Caris Life Sciences was utilized for comparison with LCNECs.
Genetic analysis
In Cohort 1, local institutions utilized standard-of-care genomic sequencing platforms to identify mutations and copy number alterations in key oncogenic drivers, including ALK, EGFR, KEAP1, KRAS, MET, RB1, SMARCA4, STK11, and TP53. The use of institution-specific platforms introduced variability in gene coverage and analytical methodologies but reflects the diversity inherent in clinical practice.
In Cohort 2, a more standardized approach was employed. Tumor samples underwent microdissection prior to nucleic acid isolation to enrich for tumor content. Next-generation sequencing (NGS) was then conducted on genomic DNA using either the NextSeq platform (Illumina, Inc., San Diego, CA, USA) for a targeted panel of 592 cancer-relevant genes (n = 84 samples) or the Illumina NovaSeq 6000 platform (Illumina, Inc., San Diego, CA, USA) for WES (n = 289 samples). For NextSeq-sequenced tumors, a custom-designed SureSelect XT assay (Agilent Technologies, Santa Clara, CA, USA) was employed to enrich for the 592 target genes. For NovaSeq-sequenced tumors, a hybrid pull-down panel of baits was used to achieve high coverage and read depth for >700 clinically relevant genes (average 500×), with additional enrichment for >20,000 genes at an average depth of 200x. Genetic variants were detected with >99% confidence and classified by board-certified molecular geneticists using previously established criteria54.
These methodological differences between Cohorts 1 and 2 highlight the real-world heterogeneity in clinical and genomic data acquisition. To ensure scientific rigor, analyses were conducted separately where appropriate, accounting for the inherent differences in data generation and processing between the two cohorts.
Variant assessment
For Cohort 1, variants assumed to be oncogenic or likely oncogenic on OncoKB were considered pathogenic55,56. For Cohort 2, genomic alterations were reviewed by board-certified clinical geneticists according to criteria established by the American College of Medical Genetics and Genomics57.
RNA sequencing
We obtained publicly available RNA WTS data from The Cancer Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) data collection (n = 515 tumors)38. For each specimen, the normalized transcripts-per-million (TPM) counts were calculated, and the data were log2 transformed. Gene set enrichment analysis (GSEA http://software.broadinstitute.org/gsea/index.jsp) was performed using the clusterProfiler package (version 4.12.2) in R (version 4.4.1), with hallmark gene sets from the Molecular Signatures Database (MSigDB v2023.2).
For Cohort 2, RNA WTS was conducted using a hybrid-capture approach from formalin-fixed paraffin-embedded (FFPE) tumor samples (n = 373) with the Agilent SureSelect Human All Exon V7 bait panel (Agilent Technologies; RRID) and the Illumina NovaSeq platform (Illumina, Inc.). Pathology review of FFPE specimens was performed to determine the percent tumor content and tumor size, requiring at least 20% tumor content in the area for microdissection to allow for enrichment and extraction of tumor-specific RNA. Extraction was carried out using a Qiagen RNA FFPE Tissue Extraction Kit, and the RNA quality and quantity were assessed with the Agilent TapeStation. Biotinylated RNA baits were hybridized to the synthesized and purified cDNA targets, followed by a post-capture PCR amplification of the bait-target complexes. The resulting libraries were quantified, normalized, pooled, denatured, diluted, and sequenced. Raw data were demultiplexed using the Illumina DRAGEN FFPE accelerator. Briefly, FASTQ files were aligned with the STAR aligner (Alex Dobin, release 2.7.4a, GitHub, https://github.com/alexdobin/STAR/releases/tag/2.7.4a). A complete 22,948-gene dataset of expression data was generated by Salmon, which offers fast and bias-aware quantification of transcript expression58. BAM files from the STAR aligner (RRID: SCR_004463) were further processed for RNA variants using a proprietary custom detection pipeline. The reference genome used was GRCh37/hg19, and analytical validation of this test showed ≥97% positive percent agreement, ≥99% negative percent agreement, and ≥99% overall percent agreement with a validated comparator method.
Immune cell fractions within the tumor microenvironments (TMEs) were estimated by deconvoluting RNA expression profiles using quanTIseq (RRID:SCR_022993)59. QuanTIseq is a computational tool that quantifies the abundance of ten immune cell populations from WTS. The algorithm is validated against flow cytometry and immunohistochemistry for determining the absolute fractions of myeloid dendritic cells, regulatory T cells (Tregs), CD8+ and CD4+ T cells, natural killer (NK) cells, neutrophils, monocytes, M1 and M2 macrophages, and B cells.
Immunohistochemistry for PD-L1 status and immunofluorescence for FGL-1
For Cohort 1, PD-L1 status was determined using one of the following anti-PD-L1 antibodies: 22c3, 28-8 (Agilent, Dako), and SP263 (Ventana). For Cohort 2, PD-L1 status was determined using the 22c3 anti-PD-L1 antibody (Dako) on FFPE sections. The evaluation involved calculating the percentage of positively stained tumor cells to obtain a tumor proportion score60.
For FGL-1 immunofluorescence, tumor regions from paraffin-embedded sections were delineated by a board-certified pathologist using corresponding hematoxylin and eosin (H&E)-stained slides. Unstained FFPE slides from NSCLC-like LCNEC (n = 2), SCLC-like LCNEC (n = 1), NSCLC (n = 3), and SCLC (n = 4) were immersed in Xylene I/II, absolute ethyl alcohol, 95% and 85% alcohol to deparaffinize the tissue sections. The slides were then subjected to antigen retrieval using Tris-EDTA buffer (pH = 8.0) at 98 °C for 20 min. Slides were blocked with 1% BSA, 4% Horse Serum, 0.4% Triton-X100 in PBS for 30 min, then incubated overnight at 4 °C with an anti-FGL-1 rabbit polyclonal primary antibody (Proteintech, 16000-1-AP) mouse monoclonal primary antibody (Proteintech, 66483-1-Ig) at 1:200. An anti-rabbit corresponding secondary antibody was used at a 1:1000 dilution, for 2 h at room temperature. Sections were then mounted with Fluoroshield histology medium containing DAPI (Sigma, F6057). Confocal imaging was acquired with an LSM880 microscope with Airyscan, and data were analyzed by using ImageJ.
Digital pathology assessment of tumor-infiltrating lymphocytes
For DFCI lung tumor samples, H&E slides were digitized using the Aperio AT at a resolution of 0.49 microns per pixel. The detailed method is reported previously43. Briefly, the images were processed in QuPath (v.4.0) using built-in functions. This involved color deconvolution to estimate stain vectors and normalize the RGB channels for each image. For cell detection, watershed segmentation was employed to identify cells based on size, shape, and the optical density of nuclei in the hematoxylin channel. Additional features were calculated by adding intensity and smoothed object features, computing Haralick texture features, and determining Gaussian-weighted averages per object/cell. A random forest algorithm was used to train an object classifier to identify tumor-infiltrating lymphocytes (TILs), tumor cells, and stromal cells. TILs were defined as mononuclear immune cells, including lymphocytes and plasma cells.
Mismatch repair status
Multiple test platforms were used to determine the MSI or MMR status. These included fragment analysis (MSI Analysis System kit; Promega, Madison, WI, USA), immunohistochemistry staining (MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 antibody; and PMS2, EPR3947 antibody; Ventana Medical Systems, Tucson, AZ, USA), and NGS (examining 7000 target microsatellite loci and comparing them to the reference genome hg19 from the University of California Santa Cruz (UCSC) Genome Browser database). The results from these three platforms were highly concordant. In rare cases of discordant results, the microsatellite stability or MMR status of the tumor was determined in the order of immunohistochemistry, fragment analysis, and NGS61.
Tumor mutational burden
In Cohort 2, tumor mutational burden (TMB) was assessed by counting all nonsynonymous missense, nonsense, in-frame insertion/deletion, and frameshift mutations in each tumor that were not previously identified as germline alterations in dbSNP151, the Genome Aggregation Database (gnomAD), or as benign variants by Caris’s geneticists. TMB-High was defined as having >19 mutations per megabase (muts/Mb), in accordance with the KEYNOTE-158 pembrolizumab trial62.
Statistical analyses
For Cohorts 1 and 2, no statistical method was used to predetermine the sample size. To ensure robust analyses and minimize confounding due to cohort-specific biases, clinical outcomes were analyzed separately for Cohorts 1 and 2. Overall survival (OS) in the ICI cohort was calculated from the time of first anti-PD-1/L1 drug treatment (pembrolizumab, nivolumab, atezolizumab, durvalumab, avelumab, or cemiplimab) to death or last follow-up. OS in the chemotherapy and chemoimmunotherapy cohorts was calculated from the time of the first systemic treatment to death or last follow-up. Real-world progression-free survival (rwPFS) was calculated from the date of initiation of first-line systemic therapy to the date of progression or death. Disease progression was determined based on available clinical records, imaging studies, or treating physician assessments, as documented in patient charts or claims data. Alive patients were censored at the date of last follow-up. Time on treatment (ToT) was calculated from the start date of first-line systemic therapy to the end date. Patients who were still alive and receiving ongoing treatment were censored at the date of their last follow-up. Survival functions were estimated using the Kaplan–Meier method, and survival distributions were compared using a two-sided log-rank test. P values less than 0.05 were considered significant. Multivariable Cox proportional hazards regression models for rwPFS and OS were performed and adjusted for variables selected a priori: Sex, ECOG performance status, age at time of systemic treatment, and M stage (M1a, M1b, M1c). For the analysis of TME biomarkers and GSEA, a false discovery rate of 0.05, determined by the Benjamini–Hochberg procedure, was used to define statistical significance. Median follow-up time was determined by the reverse Kaplan–Meier method. Analyses were conducted using Python 3.12.5 and RStudio 2024.04.2 + 764.pro1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw sequencing data for Figs. 1b, 2b–e, 3, 4 and 5a, b were generated by Caris Life Sciences and are not publicly available due to patient privacy concerns and proprietary restrictions. These data are considered third-party clinical datasets and are owned by Caris Life Sciences. Access is restricted due to legal and privacy protections. Raw data cannot be deposited in a public repository. Aggregated and de-identified data may be made available for academic research purposes upon request. Researchers should contact the corresponding author with a brief description of the data required and the intended use. All requests will be reviewed by the Caris data access team, and a response will be provided within 4 weeks. Once access is granted, data will remain available for the duration of the agreed-upon research project, subject to compliance with Caris Life Sciences’ data use agreement. External datasets used in this study are publicly available11,38. All other data supporting the findings of this study, including source data underlying the figures and tables (excluding the Caris-derived panels), are provided in the accompanying Source data file. Source data are provided with this paper.
References
Travis, W. D. et al. The 2015 World Health Organization Classification of Lung Tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015).
Naidoo, J. et al. Large cell neuroendocrine carcinoma of the lung: clinico-pathologic features, treatment, and outcomes. Clin. Lung Cancer 17, e121–e129 (2016).
Rekhtman, N. et al. Next-generation sequencing of pulmonary large cell neuroendocrine carcinoma reveals small cell carcinoma-like and non-small cell carcinoma-like subsets. Clin. Cancer Res. 22, 3618–3629 (2016).
Jones, M. H. et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet 363, 775–781 (2004).
Bari, M. F. et al. BAI3, CDX2 and VIL1: a panel of three antibodies to distinguish small cell from large cell neuroendocrine lung carcinomas. Histopathology 64, 547–556 (2014).
Simbolo, M. et al. Lung neuroendocrine tumours: deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J. Pathol. 241, 488–500 (2017).
De Pas, T. M. et al. Large-cell neuroendocrine carcinoma of the lung harboring EGFR mutation and responding to gefitinib. J. Clin. Oncol. 29, e819–e822 (2011).
Aroldi, F. et al. Tyrosine kinase inhibitors in EGFR-mutated large-cell neuroendocrine carcinoma of the lung? A case report. Case Rep. Oncol. 7, 478–483 (2014).
Omachi, N. et al. A case of large-cell neuroendocrine carcinoma harboring an EML4-ALK rearrangement with resistance to the ALK inhibitor crizotinib. J. Thorac. Oncol. 9, e40–e42 (2014).
Karlsson, A. et al. Mutational and gene fusion analyses of primary large cell and large cell neuroendocrine lung cancer. Oncotarget 6, 22028–22037 (2015).
George, J. et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 9, 1048 (2018).
Derks, J. L. et al. Molecular subtypes of pulmonary large-cell neuroendocrine carcinoma predict chemotherapy treatment outcome. Clin. Cancer Res. 24, 33–42 (2018).
Miyoshi, T. et al. Genomic profiling of large-cell neuroendocrine carcinoma of the lung. Clin. Cancer Res. 23, 757–765 (2017).
Zhuo, M. et al. The prognostic and therapeutic role of genomic subtyping by sequencing tumor or cell-free DNA in pulmonary large-cell neuroendocrine carcinoma. Clin. Cancer Res. 26, 892–901 (2020).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 20, 1239–1251 (2019).
Marabelle, A. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
Andre, T. et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N. Engl. J. Med. 383, 2207–2218 (2020).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Burtness, B. et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394, 1915–1928 (2019).
Gandhi, L. et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).
Morris, V. K. et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 18, 446–453 (2017).
Wang, V. E. et al. Checkpoint inhibitor is active against large cell neuroendocrine carcinoma with high tumor mutation burden. J. Immunother. Cancer 5, 75 (2017).
Reck, M. et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).
Horn, L. et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 379, 2220–2229 (2018).
Paz-Ares, L. et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer: 3-year overall survival update from CASPIAN. ESMO Open 7, 100408 (2022).
Cheng, Y. et al. Durvalumab after chemoradiotherapy in limited-stage small-cell lung cancer. N. Engl. J. Med. 391, 1313–1327 (2024).
Mauclet, C. et al. Complete tumor response of a locally advanced lung large-cell neuroendocrine carcinoma after palliative thoracic radiotherapy and immunotherapy with nivolumab. Lung Cancer 128, 53–56 (2019).
Zhang, X., Sun, Y., Miao, Y. & Xu, S. Immune checkpoint inhibitor therapy achieved complete response for drug-sensitive EGFR/ALK mutation-negative metastatic pulmonary large-cell neuroendocrine carcinoma with high tumor mutation burden: a case report. Onco Targets Ther. 13, 8245–8250 (2020).
Oda, R. et al. Long-term survivor of pulmonary combined large cell neuroendocrine carcinoma treated with nivolumab. Thorac. Cancer 11, 2036–2039 (2020).
Sherman, S. et al. Efficacy of immune check-point inhibitors (ICPi) in large cell neuroendocrine tumors of lung (LCNEC). Lung Cancer 143, 40–46 (2020).
Dudnik, E. et al. Real-world survival outcomes with immune checkpoint inhibitors in large-cell neuroendocrine tumors of lung. J. Immunother. Cancer 9, https://doi.org/10.1136/jitc-2020-001999 (2021).
Owen, D. H. et al. A phase II clinical trial of nivolumab and temozolomide for neuroendocrine neoplasms. Clin. Cancer Res. 29, 731–741 (2023).
Chen, H. Z. et al. Genomic and transcriptomic characterization of relapsed SCLC through rapid research autopsy. JTO Clin. Res Rep. 2, 100164 (2021).
Rudin, C. M. et al. Author Correction: Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer 19, 415 (2019).
Owonikoko, T. K. et al. YAP1 expression in SCLC defines a distinct subtype with T-cell-inflamed phenotype. J. Thorac. Oncol. 16, 464–476 (2021).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347.e12 (2019).
Cancer Genome Atlas Research Network Author Correction: Comprehensive molecular profiling of lung adenocarcinoma. Nature 559, E12 (2018).
Ateeq, B. et al. Therapeutic targeting of SPINK1-positive prostate cancer. Sci. Transl. Med. 3, 72ra17 (2011).
Chen, F. et al. Targeting SPINK1 in the damaged tumour microenvironment alleviates therapeutic resistance. Nat. Commun. 9, 4315 (2018).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Wei, S. C., Duffy, C. R. & Allison, J. P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018).
Rakaee, M. et al. Association of machine learning-based assessment of tumor-infiltrating lymphocytes on standard histologic images with outcomes of immunotherapy in patients with NSCLC. JAMA Oncol. 9, 51–60 (2023).
Agar, C. et al. Brief report on the efficacy of nivolumab in patients with previously treated advanced large-cell neuroendocrine cancer of the lung. JTO Clin. Res. Rep. 2, 100129 (2021).
Ozaki, N. et al. Serine protease inhibitor Kazal type 1 promotes proliferation of pancreatic cancer cells through the epidermal growth factor receptor. Mol. Cancer Res. 7, 1572–1581 (2009).
Wang, C. et al. Serine protease inhibitor Kazal type 1 promotes epithelial-mesenchymal transition through EGFR signaling pathway in prostate cancer. Prostate 74, 689–701 (2014).
Tiwari, R. et al. Correction: SPINK1 promotes colorectal cancer progression by downregulating Metallothioneins expression. Oncogenesis 10, 16 (2021).
Marchbank, T., Mahmood, A. & Playford, R. J. Pancreatic secretory trypsin inhibitor causes autocrine-mediated migration and invasion in bladder cancer and phosphorylates the EGF receptor, Akt2 and Akt3, and ERK1 and ERK2. Am. J. Physiol. Ren. Physiol. 305, F382–F389 (2013).
Ahn, M. J. et al. Tarlatamab for patients with previously treated small-cell lung cancer. N. Engl. J. Med. 389, 2063–2075 (2023).
Baine, M. K. et al. SCLC subtypes defined by ASCL1, NEUROD1, POU2F3, and YAP1: a comprehensive immunohistochemical and histopathologic characterization. J. Thorac. Oncol. 15, 1823–1835 (2020).
Ng, J. et al. Molecular and pathologic characterization of YAP1-expressing small cell lung cancer cell lines leads to reclassification as SMARCA4-deficient malignancies. Clin. Cancer Res. 30, 1846–1858 (2024).
Stewart, C. A. et al. YAP1 status defines two intrinsic subtypes of LCNEC with distinct molecular features and therapeutic vulnerabilities. Clin. Cancer Res. 30, 4743–4754 (2024).
Rindi, G. et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 33, 115–154 (2022).
Muquith, M. et al. Tissue-specific thresholds of mutation burden associated with anti-PD-1/L1 therapy benefit and prognosis in microsatellite-stable cancers. Nat. Cancer https://doi.org/10.1038/s43018-024-00752-x (2024).
Chakravarty, D. et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017, https://doi.org/10.1200/PO.17.00011 (2017).
Suehnholz, S. P. et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 14, 49–65 (2024).
Richards, S. et al. Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Finotello, F. et al. Correction to: Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 11, 50 (2019).
Kim, S. Y. et al. Characterization of MET Exon 14 skipping alterations (in NSCLC) and identification of potential therapeutic targets using whole transcriptome sequencing. JTO Clin. Res Rep. 3, 100381 (2022).
Wang, J. et al. Mutational analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: a retrospective cohort study. Lancet Oncol. 24, 151–161 (2023).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020).
Acknowledgements
D.J.P. acknowledges grant support from the UKRI IAA Healthy Society Grant Scheme (Round 2023), the Cancer Treatment and Research Trust (CTRT), and infrastructural support by the Imperial Experimental Cancer Medicine Centre and the NIHR Imperial Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.H.N., C.K., A.R.N., A.C.C. Data curation: A.H.N., T.A., E.B.F., H.A., M.R., K.M., S.F.L., Y.T., A.O., F.A.L., T.L., S.R., S.A., J.E.G., J.H., D.K., J.B., M.Z.B., F.A.N., C.G., H.L., F.C., A.C., E.C.M., D.P., M.D., T.J.E.C., J.M.C., J.J.L., A.S.W., D.R.C., A.S., K.P., F.Cro., T.U.M., V.A., M.Ah., K.S., H.Kw., J.Z., D.H.O., M.L. (Mingjia Li), M.N., D.J.P., N.A., K.A., S.P., U.Z., D.M.G., C.L., H.K., J.Lia., A.I.V., T.B., L.M., M.M., P.R., M.E., A.V., A.E., J.N., G.L., P.C.M., H.B., M.L2. (Matthew Lee), L.Y., R.A., H.M., D.J.K., R.S.H., R.A.F., A.R.N., A.C.C. Formal analysis: A.H.N., T.A., H.A. Funding acquisition: none. Investigation: A.H.N., C.K., A.R.N., A.C.C. Methodology: A.H.N., C.K., T.A., A.R.N., A.C.C. Project administration: A.H.N., T.A., A.R.N., A.C.C. Resources: A.H.N., C.K., A.R.N., A.C.C. Supervision: A.H.N., A.R.N., A.C.C. Validation: not applicable. Visualization: A.H.N., T.A., H.A. Writing—original draft: A.H.N., C.K., A.R.N., A.C.C., H.A. Writing—review and editing: All authors listed under data curation.
Corresponding authors
Ethics declarations
Competing interests
A.H.N.: honoraria: the Korean Society of Medical Oncology, TEMPUS, OncLive, Oklahoma University, Targeted Oncology; travel compensation: Korean Society of Medical Oncology, American Association for Cancer Research; consultation fees: Guidepoint Global, Putnam Associates, Capvision; compensation from Outlier.ai to provide feedback on data analysis tools, AI development; Equity in Revolution Medicine, Summit Therapeutics. M.G.E. receives full-time employment, travel/speaking expenses, and stock/stock options from Caris Life Sciences. A.C.C.: advisory boards: AbbVie, Amgen, BI, Merck, Jazz, and Research funding: Zai Labs. D.J.P.: Lecture fees: Bayer Healthcare, AstraZeneca, EISAI, Bristol Myers Squibb, Roche, Ipsen, OncLive; Travel expenses: Bristol Myers Squibb, Roche, Bayer Healthcare; Consulting fees: Mina Therapeutics, Boeringer Ingelheim, Ewopharma, EISAI, Ipsen, Roche, H3B, AstraZeneca, DaVolterra, Starpharma, Boston Scientific, Mursla, Avammune Therapeutics, LiFT Biosciences, Exact Sciences; Research funding (to institution): MSD, BMS, GSK, EISAI. N.A.: No conflicts to declare. P.R. reports travel support from AstraZeneca, MSD, BMS, and Kiowa Kirin outside the submitted work. M.R. received lecture fees from AstraZeneca. F.A.-L.: Research PI (AZ, Alira Health). A.I.V. received consulting honorarium from AstraZeneca, AbbVie, Janssen, Regeneron, Merus, and Novocure. T.A.: Employee of Caris Life Sciences. J.Z. reported the following: Grants/Contracts: AbbVie, AstraZeneca, BeiGene, BridgeBio, Genentech, Hengrui Therapeutics, InnoCare Pharma, Janssen, Kahr Medical, Merck, Mirati Therapeutics, Nilogen, Novartis, Champions Oncology, BMS. Consulting fees: AstraZeneca, Hengrui Therapeutics, Mirati Therapeutics, Novartis, Novocure, Regeneron, Sanofi, and Takeda Oncology. Payment or honoraria for lectures, presentations, speakers, bureaus, manuscript writing, or educational events: AstraZeneca, MJH Life Sciences, Novartis, Regeneron, Sanofi, and Takeda. A.S.W. has performed consulting work for MJH Life Sciences and received speaking fees from The Binaytara Foundation and Janssen. L.M.: Travel support: BMS. J.B.: grants for consultancies/advisory boards: BMS, Roche, AstraZeneca. Speaker fees: AstraZeneca, Lilly, Johnson and Johnson. Travel support: Roche, AstraZeneca, MSD, Johnson and Johnson. Research funding (to institution): SEOM. C.G.: grants for consultancies/advisory boards: MSD, BMS, Oncowissen, AstraZeneca, REGENERON, Roche. Speaker fees: AstraZeneca, Boehringer Ingelheim, Chugai, Pierre-Fabre, MSD, Sanofi/REGENERON. Writing/Editorial activity: BMS, MSD. Travel support: Sanofi/REGENERON, MSD. Research fundings (to institution): BMBF/Deutsche Krebshilfe/Deutsche Forschungsgemeinschaft. D.O.: Honorarium: Chugai. Research funding (to institution): Merck, BMS, Palobiofarma, Genentech, AbbVie, Nuvalent, Onc.AI. J.K.H. received consulting honorarium from Jackson Laboratory for Genomic Medicine and ARUP. H.B.: Research Support (Clinical Trials): BMS, Lilly, Amgen; Advisory Board/Consultant: BMS, Lilly, Genentech, Pfizer, Merck, EMD Serono, Boehringer Ingelheim, AstraZeneca, Novartis, Genmab, Regeneron, BioNTech, Amgen, Axiom, PharmaMar, Takeda, Mirati, Daiichi, Guardant, Natera, Oncocyte, Beigene, iTEO, Jazz, Janssen, Puma, BerGenBio, Bayer, Iobiotech, Grid Therapeutics, RAPT; Data and Safety Monitoring Board: University of Pennsylvania: CAR T Program, Takeda, Incyte, Novartis, Springworks; Scientific Advisory Board: Sonnetbio (Stock Options); Inspirna (formerly Rgenix, Stock Options); Nucleai (stock options); Honoraria: Amgen, Pfizer, Daiichi, Regeneron; Travel: Amgen, BMS, Merck, Lilly, EMD Serono, Genentech, Regeneron, Mirati. M.D.: Advisory boards; Sanofi/Genzyme, Regeneron, Janssen, AstraZeneca, Gilead, Bristol Myer Squibb, Catalyst Pharmaceuticals, Novocure, Guardant Consulting: AbbVie, Janssen, Gilead, Daiichi Sankyo, Bristol Myer Squibb Research: Merck, Genentech, CellSight, Novartis, Varian. A.E.: Employee of Caris Life Sciences. J.J.L. has served as a compensated consultant for Genentech, C4 Therapeutics, Blueprint Medicines, Nuvalent, Bayer, Elevation Oncology, Novartis, Mirati Therapeutics, AnHeart Therapeutics, Takeda, CLaiM Therapeutics, Ellipses, Hyku BioSciences, AstraZeneca, Bristol Myers Squibb, Daiichi Sankyo, Yuhan, Merus, Regeneron, Pfizer, Nuvation Bio, and Turning Point Therapeutics; has received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Bayer, Elevation Oncology, Roche, Linnaeus Therapeutics, Nuvalent, and Novartis; and travel support from Pfizer and Merus. C.K.: Research funding (to institution): AstraZeneca, Novartis, Regeneron, Janssen, Genentech, Lyell, Daiichi Sankyo, Gilead, Macrogenics, Boehringer Ingelheim, Black Diamond Therapeutics. Consulting fees: Arcus, AstraZeneca, Daiichi Sankyo, Eisai, Regeneron, Sanofi, Takeda, J&J, Pinetree, Boehringer Ingelheim, Gencurix. M.N. is on the advisory board for AstraZeneca, Daiichi Sankyo, Takeda, Novartis, EMD Serono, Janssen, Pfizer, Eli Lilly and Company, Bayer, Regeneron, BMS and Genentech; consultant for Caris Life Sciences (virtual tumor board); speaker for Blueprint Medicines, Janssen, Mirati and Takeda; and reports travel support from AnHeart Therapeutics. Reports stock/stock options from MBrace Therapeutics. T.U.M. currently or has previously served on Advisory and/or Data Safety Monitoring Boards for Rockefeller University, Regeneron, AbbVie, Merck, EMD Serono, Storm, Geneos, Bristol-Meyers Squibb, Boehringer Ingelheim, Atara, AstraZeneca, Genentech, Celldex, Chimeric, DrenBio, Glenmark, Simcere, Arrowhead, Surface/Coherus, G1 Therapeutics, NGMbio, DBV Technologies, Arcus, Fate, Ono, Storm, Replimmune, Larkspur, Avammune, and Astellas, and has research grants from the National Institutes of Health (NCI), the Cancer Research Institute, Regeneron, Genentech, Bristol Myers Squibb, Merck, and Boehringer Ingelheim. A.R.N. reports: Funding to Institution for Trials he is PI on: Loxo@Lilly, Surface Oncology, ADC Therapeutics, IGM Biosciences, EMD Serono, Aravive, Nikang Therapeutics, Inspirna, Exelexis, Revolution Medicine, Jacobio, Pionyr, Jazz Pharmaceuticals, NGM Biopharmaceuticals, Immunocore, Phanes Therapeutics, Kymera Therapeutics, Dren Bio, Daichi; Consultant Editor Compensation: JCO Precision Oncology; Travel Compensation from: SITC/ AACR/ Conquer Cancer Foundation/BinayTara Foundation and Foundation Med/ Caris Life Sciences/ ASCO; Advisory Board: Foundation Med, Astellas, NGM biosciences, Natera, Regeneron; Honoraria: BinayTara Foundation, Foundation Med, Medlive; Grant Support: SOWG Hope Foundation. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shigeki Umemura and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nassar, A.H., Kim, C., Adeyelu, T. et al. Integrated molecular and clinical characterization of pulmonary large cell neuroendocrine carcinoma. Nat Commun 16, 7717 (2025). https://doi.org/10.1038/s41467-025-63091-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63091-0
