
Abstract
The production of light olefins from polyethylene (PE) has significant industrial potential. Zeolites have been widely used in petroleum refining for their ability to cleave C–C/C–H bonds and facilitate light olefins selectivity, thanks to their adjustable acidity and pore structure. However, the interaction between zeolites and conventional hydrocarbons or polymer reactants is quite different, a distinction frequently overlooked but has great influence on their reaction. Based on this, we describe a PE self-confined cracking mechanism that can produce C3-C8 olefins with exceptional yields, surpassing 70% under mild conditions (300 °C). Interestingly, the product distribution is only dependent on the degree of self-confinement and melt mass-flow rate (MFR) of PE, regardless of the porous structure, metal content, and internal acid properties of zeolite. Most importantly, this process allows for flexible tandem catalytic reforming to yield more than 67% C2–C4 light olefins and 23% separable BTX, demonstrating great potential to promote chemical recycling of waste polyolefin plastics.
Similar content being viewed by others

Introduction
The rapid growth of polyolefin (PO) plastic waste represents a significant economic and environmental challenge1,2,3,4. While energy recovery and mechanical recycling have been widely practiced, chemical recycling has gained increasing attention in recent research as it offers a promising solution by reducing CO2 emissions and providing the potential to upcycle polyolefins into valuable products such as plasticizers, detergents, and functional materials5,6,7,8,9,10. However, with global PO waste production reaching ~53 million tons (Mt) in 2022, and the market demand for these byproducts (~16 Mt in 2022) falling short of addressing the entire waste polyolefin stream, the issue remains largely unresolved11,12. Light olefins, which are considered a cornerstone of modern chemistry, are produced and consumed in ever-increasing volumes (~170 Mt in 2022)13. Directly converting waste PE into light olefins, rather than relying on fossil-based feedstocks, not only mitigates the plastic waste crisis but also optimizes the utilization of hydrocarbon-rich waste plastics, contributing to a truly circular economy14,15,16.
Current technologies for converting waste PE to light olefins predominantly rely on high-temperature pyrolysis due to the endothermic nature of C–C bond cleavage17. However, conventional pyrolysis processes produce significant quantities of gasoline and diesel fractions and require temperatures exceeding 800 °C to achieve a product distribution dominated by light olefins, thereby exacerbating energy consumption and CO2 emissions18. Catalytic cracking, which lowers the required temperature and enhances olefin selectivity, has been shown in recent studies to convert PE to a broadly distributed range of C3-C7 iso-olefins under milder conditions (280 °C), although the isolation and utilization of these mixed olefins remain challenging19,20. Recent advancements in tandem catalytic strategies for PO depolymerization have significantly improved the selectivity of light olefin products, exemplified by the selective conversion of PE and polypropylene (PP) with C2H4 to C3H6 and i-C4H8 through dehydrogenation, olefin isomerization, and olefin metathesis21,22,23. However, this process consumes large amounts of C2H4 and metathesis catalysts are environmentally sensitive and easy poisoned by impurities of real-life plastics, hindering their widespread use larger scale.
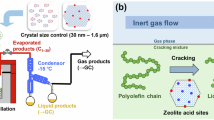
Enlightened by petroleum cracking and refining processes, PE cracking tandem reforming shows promise for the production of light olefins and BTX (benzene, toluene, and xylene). This approach is particularly attractive for industrial applications, as it typically employs inexpensive zeolite catalysts and does not require co-feeds (e.g., H2, C2H4)3. While petroleum refining technologies can be adapted for polyolefin upgrading due to their similar atomic compositions, significant differences must be addressed. In the conventional fluid catalytic cracking (FCC) process24, a gas-solid reaction system is employed, with zeolite channel constraints playing a key role in product distribution (Fig. 1a)25,26. However, a crucial distinction is that polyolefins do not fully vaporize during the reaction, unlike petroleum feedstocks. Instead, a molten layer often forms on the zeolite surface due to Van der Waals forces and capillary effects (Fig. 1b). Recent studies have highlighted the potential impact of this molten layer, which constrains the interface between the zeolite and substrate27,28. This phenomenon is unique to polymer chemical recycling, yet its influence on the reaction remains insufficiently explored.

a Gas-solid phase catalytic cracking of heavy oil: The diffusion of substrate and product molecules is relatively free and limited only by the structure of the catalyst. b Polyolefin catalytic cracking system: The catalyst is encapsulated by a PE molten layer, and diffusion of products will be constrained by PE molten layer.
Herein, we elucidate the role of this self-confined space in PE cracking through experiments and molecular dynamic simulations. Due to this self-confinement, PE directly breaks down into light olefin products without a stepwise cracking path and prevents the conversion of olefin intermediates into aromatics. Interestingly, the distribution of products is determined solely by the degree of self-confinement and the melt mass-flow rate (MFR) of polyethylene (PE), irrespective of the zeolite’s porous structure, metal content, or internal acid properties. Most importantly, this process facilitates flexible tandem catalytic reforming without any co-feeds, yielding over 67% C2-C4 olefins and 23% separable BTX (benzene, toluene, xylene), establishing an efficient chemical recycling pathway for waste polyolefins.
Results
PE self-confined catalytic cracking behaviors and their affecting factors
We began our studies by exploring the differences in catalytic cracking between PE and conventional light hydrocarbon. As illustrated in Fig. 2a, the yield of C1-C12 fractions increased significantly with chain length, from ~18% (C18) to ~90% (C1000). More interestingly, an increase in the chain length of the substrate resulted in a shift in product distributions towards smaller carbon numbers, while significantly improving the selectivity for olefin products. PE (C1000) stood out by producing the highest amount of C3-C8 olefins at 66 wt.%, indicating its potential as a feedstock for direct conversion to light olefins. The reaction temperature is crucial for the high yield of olefin products for PE cracking over zeolites. At higher temperatures, the yield of olefins decreased significantly while the yields of aromatics and paraffins increased (Fig. 2b and Supplementary Fig. 5). We have also ruled out the possible contribution from direct pyrolysis of PE at 300 °C, and the high yield of olefin products can be confidently assigned to catalytic cracking on zeolites. (Supplementary Fig. 6).

a Cracking product distribution of reactant with different chain lengths (C18 and C30 represent conventional FCC feedstocks, and C160 and C1000 represent PE). Reaction conditions: 250 mg of commercial ZSM-5 zeolite, 500 mg of reactants, 100% Ar feed gas at a flow rate of 40 mL/min, 300 °C for 2 hours. Error bars represent standard deviation (SD). b Yields of different fractions products of pyrolysis and catalytic cracking of PE (C1000) at different temperatures (300, 400, 500 °C) for 2 h. c Yields of different hydrocarbon fractions vary with C/P ratio from 5 to 0.2 during PE cracking at 300 °C for 2 h. Error bars represent standard deviation (SD). d Adsorption of C3H6 on mixed samples of commercial ZSM-5 and PE with different mass ratios (∞, 2, 1, 0.5, 0) at 200 °C, with 4.48 mmol of C3H6 injected per pulse. Mixtures were hold at 200 °C under Ar flow to ensure complete melt mixing. e Yields of paraffins, olefins, and aromatics after varying times of PE cracking under different C/P of 1 (top) and 0.5 (bottom) at 300 °C. Error bars represent standard deviation (SD). f Schematic of PE self-confined cracking process.
Next, we explored the effect of the mass ratio of catalyst to PE (C/P) under 300 °C. Figure 2c and Supplementary Fig. 7 illustrate that as the C/P ratio decreased, the yields of paraffins and total aromatics in the products significantly decreased, while the yields of olefins significantly increased. In the reaction network of hydrocarbon catalytic cracking29, olefin intermediates need to be adsorbed again onto the Brønsted acid sites (BAS) for aromatization30. Consequently, a low C/P ratio may hinder the re-contact between olefin intermediates and some BAS, thereby preventing aromatization. To investigate the accessibility of these BAS during PE cracking, chemisorption experiments using C3H6 as a probe molecule are conducted. As shown in Fig. 2d, the pure ZSM-5 catalyst exhibited the highest cumulative amount of C3H6 adsorption, indicating the greatest accessibility of active sites to C3H6, while PE hardly adsorbed any C3H6. As the C/P ratio decreased from 2 to 0.5, the adsorption capacity of C3H6 gradually decreased. It indicates that mixing PE and zeolite catalyst at temperatures sufficient for the molten layer formation hindered the accessibility of acid sites. N2 physisorption results for mixtures of molten PE and catalysts with different mass ratios further evidence that the micropores are more challenging to access as the PE layer around the outer surface of the zeolite thickened (Supplementary Fig. 8 and Supplementary Table. 2).
Considering that the cleavage of PE is accompanied by decreases in both molecular weight and mass, we monitored the product evolution during the reaction. As shown in Fig. 2e, the product distribution during the entire cracking process appears to be divided into 3 stages: 1) In the first few minutes, only light alkane products were detected, with almost no olefins (Supplementary Figs. 9, 10). This is in agreement with the non-classical alkane cracking mechanism with the five-coordinated carbonium ions as a transition state followed by the formation of alkanes (in gas phase) and carbenium ions (absorbed on BAS)31. 2) As the concentration of carbenium ions increases, C3+ olefins are observed as the primary product, possibly due to the β-cleavage in the classical pathway where carbenium ion is the critical reaction intermediate32,33. 3) Then, aromatic fractions start to appear in the product mainly because of a secondary reaction involving light olefins. This includes hydrogen transfer from light olefins to aromatics and alkanes30. Over time, the selectivity of these reactions gradually increases until the polyethylene is fully consumed. Notably, the aromatics generation stage exhibits a significant time lag when the C/P shifts from 1 to 0.5 (Fig. 2e, Supplementary Figs. 11 & 12).
Based on the above findings, we propose a scenario where the PE molecules encapsulate the catalyst (Fig. 2f). Here, we tend to avoid using the word “cover” as it might be confused with the general concept of “coverage” in catalysis. When such encapsulation is sufficient under a low C/P ratio, the highly constrained space will limit the diffusion of any hydrocarbon molecules no matter how the catalytic reaction proceeds. The only exception is light hydrocarbons with small enough carbon numbers. In addition, not only does the molten-layer encapsulation hinder the product diffusion, but it also limits it to the secondary reactions. These findings suggest the presence of the polymer molten-layer encapsulation that can be considered as a spatial-limiting area and serves as the basis of self-confined effects of PE catalytic cracking over zeolite.
Distinct features of PE self-confined cracking
The above-stated encapsulation hinders the accessibility of active sites. From this point of view, it is naturally speculated that the reactants/products hardly diffuse into the zeolitic bulk so that acid sites at the outer surface would perform superior to those in the bulk. To investigate this, we prepared ZSM-5@SiO2 catalysts with passivated outer surface acid sites via chemical liquid deposition (Supplementary Fig. 13)34. As shown in Fig. 3a, PE is hardly converted on ZSM-5@SiO2, indicating that the self-confined cracking of PE occurs at the outer surface acid sites of the catalyst. However, in the C30 cracking experiments, we found that passivating the acid sites on the outer surface resulted in only a slight decrease in conversion, indicating that the catalytic cracking of low-carbon hydrocarbons occurs mainly within the zeolite pores. Thus, the structure-performance relationships of PE self-confined cracking differ from those observed in the catalytic cracking of classical low-carbon hydrocarbons.

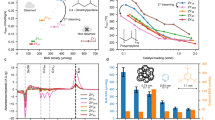
a Yields of different fractions of products on ZSM-5 (with external acid sites) and ZSM-5@SiO2 (without external acid sites). Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg catalyst, 500 mg HDPE or C30, 100% Ar gas, 40 mL/min flow rate, 2 h. b Yields of different fractions of products on various modified ZSM-5 zeolites. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg catalyst, 500 mg HDPE, 2 h. c Distribution of PE catalytic cracking products of zeolites with three different topologies at C/P = 2 and 0.5. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg catalyst, 500 mg HDPE, 2 h. d PE conversion and olefin selectivity for all catalysts. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg catalyst, 500 mg HDPE, 2 h. e Yields of different fractions of products of PE with different MFRs and structures. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg commercial ZSM-5, 500 mg PE, 2 h. f olefin yield and selectivity of PE with different MFRs and structures. Curves were plotted based on visual trends. g Reaction rates of PE with different MFRs and structures. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg commercial ZSM-5, 500 mg PEs with different MFRs, 0–20 min.
For the cracking of light hydrocarbons, optimizing diffusion within the zeolite crystal or reducing acid density can improve the selectivity for light olefins25. Hence, we tested PE-self-confined cracking over a series of modified ZSM-5 zeolites with varying acid densities, crystal sizes, and mesoporous structures (Supplementary Figs. 14–17 and Supplementary Tables. 2 & 3). Interestingly, all the catalysts show very similar product distributions with light olefin yield over 65 wt.% (Fig. 3b and Supplementary Fig. 18). In addition, for light hydrocarbon cracking, the incorporation of metal ions is effective in yield enhancement and product distribution adjustment35. For PE cracking, although the metal modification reduced the reaction rate (Supplementary Fig. 19), which was associated with a decrease in the Brønsted acid density36, the effect on the distribution of the final product was negligible as the reaction is only dependent on the acid properties at the outer surface, regardless of the porous structure, metal content, acid properties inside the micropores, etc.
Additionally, we investigated the cleavage of PE on zeolites with varying topologies (Supplementary Fig. 20 and Supplementary Table. 2). As illustrated in Fig. 3c and Supplementary Fig. 21, under C/P = 2, both alkanes and aromatics generated on ZSM-5, USY, and Beta zeolite increased markedly compared to the yields at C/P = 0.5, whereas the olefin yields decreased substantially. These results suggest that the enhancement of the self-confined effect of PE inhibits hydrogen transfer side reactions, regardless of the zeolite catalyst used. However, expanding the size of the zeolite pores led to a notable decrease in olefin yields due to the larger pore size accommodating bimolecular hydrogen transfer reactions37. This pore-size effect might not be as impactful for the primary cracking reaction, but it becomes more apparent for secondary reactions when encapsulation is less significant. As shown in Fig. 3d, we further conducted PE cracking experiments using various solid acid catalysts without micropores (Supplementary Fig. 22). The olefin selectivity in the PE cracking products exceeded that of the zeolite-based catalysts. However, a significant decrease in conversion over the same period was observed due to the lower acid density of these non-microporous catalysts (Supplementary Fig. 23).
Based on above results, the physicochemical properties of the catalyst exert minimal influence on olefin yield when the PE molten layer fully encapsulates the catalyst. In this scenario, the primary determinant of olefin yield is the in-situ melting of PE and its encapsulation of the catalyst, which we hypothesize is linked to the melt flow rate (MFR) of the polymer reactant. To investigate this further, we conducted catalytic cracking experiments on high-density polyethylene (HDPE), low-density polyethylene (LDPE), and linear low-density polyethylene (LLDPE) with varying MFRs at 300 °C (see Supplementary Table. 4). As illustrated in Fig. 3e and Supplementary Fig. 24, all PE feedstocks exhibit similar olefin yield when MFR is ≥ 1, where the PE has thoroughly melted and coated the catalyst (Supplementary Fig. 25). However, when the MFR falls significantly below 1, there is a marked decrease in olefin yield, accompanied by a substantial increase in aromatic selectivity. This trend can be attributed to the insufficient encapsulation of the catalyst by PE at low MFRs.
Moreover, Fig. 3f demonstrates that when MFR is ≥ 1, both olefin yield and selectivity remain stable within a defined range, showing no further changes with increasing MFR. In contrast, when MFR is <1, an increase in MFR leads to a gradual rise in olefin yield, while selectivity remains relatively constant. Subsequent evaluations of the cracking reaction rates for these PE feedstocks (see Fig. 3g and Supplementary Fig. 26) reveal a rapid increase in reaction rate as MFR rises from 0.04 to 1, which can be ascribed to improved contact between PE and acid sites. Beyond an MFR of 1, the reaction rate increases at a slower pace with further MFR increments. Additionally, the chain structure of PE does not significantly impact the reaction rate within the examined MFR range. These results underscore the critical role of MFR in optimizing olefin production from various PE feedstocks, and in situ melting of PE and complete encapsulation of the catalyst are a prerequisite for high olefin yields.
Insights from molecular dynamics into PE self-confined catalytic cracking
To further elucidate the scenario of the aforementioned encapsulation, molecular dynamics (MD) simulations were applied to model the interactions between the substrates, olefin products, and the zeolite catalyst (Supplementary Fig. 27 and Supplementary Note. 2). As illustrated in Supplementary Figs. 28, 29, compared with light hydrocarbons, macromolecules ( > C100) form a denser molten layer that encapsulates zeolite, and few molecules permeate the zeolite channels. This is due to the strong van der Waals force between the long-chain paraffin and the zeolite framework and the high affinity among macromolecule chains38,39,40.
Then, we investigated the diffusion behavior of C6H12 (model molecule to represent the olefin intermediates) in different molten layers created by paraffins of varying chain lengths (see Supplementary Movie 1 for a detailed illustration of the diffusion process)41,42,43. The spatial restriction imposed by the paraffins was assessed by evaluating hindrance to product diffusion, quantified by mean square displacement (MSD) and diffusion coefficient D. As shown in Fig. 4a, after simulations over the same time period, C6H12 traveled the greatest distance in the C20 melt, whereas the diffusion distance decreases as the carbon chain length increases. This observation is further supported by the MSD curves (Fig. 4b), where hexene’s diffusion coefficient showed a marked reduction in melts of paraffin chains longer than C100. Moreover, in a PE molten layer environment, only products with chain lengths shorter than C12 can effectively diffuse through the layer, while longer-chain molecules experience substantial spatial hindrances (Supplementary Fig. 30). This spatial confinement effect correlates with the experimental observation that PE cracking products are predominantly distributed around C4–C5. These results indicate that the polymer substrate exerts a substantial confinement effect on the intermediate products during PE cracking, resulting in the direct formation of light olefins on zeolite surface.

a Structural models for C6H12 diffusion in molten layers of different paraffins (C20, C100, C500, and C1000, in gray) after 200-ps simulations. b MSD curves depicting C6H12 diffusion in various hydrocarbon molten environments within 200 ps. Error bars represent standard deviation (SD). c Structural model illustrating the diffusion of C3H6 towards a bare zeolite slab after 100-ps simulations. d Final structure in the diffusion simulation of C3H6 towards zeolite encapsulated by PE molten layer, with a ratio of 4.84 mg PE/m² ZSM-5. e z-MSD curves of C3H6 diffusion towards zeolite encapsulated by varying amounts of PE molten layers within 100 ps. Error bars represent standard deviation (SD).
We next modeled the space confinement effects of the PE molten layer on the BAS of zeolite (Supplementary Fig. 31). As shown in Fig. 4c, in the absence of a PE molten layer, C3H6 can transfer freely between zeolite particles, easily traveling through the pores, which are the primary sites where olefins undergo side reactions to form C7-8 aromatics. When the PE molten layer encapsulates the zeolite, no C3H6 is visibly in contact with the zeolite surface. The diffusion distance of C3H6 gradually decreases as the PE molten layer thickens (Fig. 4d and Supplementary Fig. 32). To exclude the influence of the carbon chain length of the diffusing molecule, we also simulated the diffusion of C6H12 toward zeolite, and similar results were obtained (Supplementary Fig. 33). Figure 4e presents the mean square z-displacement (z-MSD) along the b-axis direction of the zeolite for cracking systems of different P/C ratios. During the initial stage of diffusion, the z-MSD of C3H6 increases over time, with the slopes of the curves positively correlated to P/C ratios, implying a difference in initial diffusion rates. The correlation between diffusion rate and P/C ratio can be attributed to the increase in density of adsorbed PE with respect to the P/C ratio (Supplementary Fig. 34), resulting from the long-range PE-PE and PE-zeolite attractions44,45. Additionally, the diffusion distance decreases in the thicker PE molten layer. Besides, simulations of longer times were also conducted, showing the extent of diffusion reached around 100 ps (Supplementary Fig. 35). These results suggest that raising the P/C ratio leads to a thickened PE molten layer, which enhances the confinement effect, thereby preventing olefin intermediates from re-exposure to the active sites for side reactions. This aligns with previous experimental findings on the inhibition of aromatic formation.
Practical applications of PE self-confined cracking
To assess the applicability of self-confined cracking in polymers, we conducted cracking experiments on several common PE plastic commodities. Figure 5a depicts several types of PE waste, including PE agricultural sheets, PE bags, and bottles. The cracking of these waste PEs resulted in the isolation of more than 60% of C3-C8 light olefins (Supplementary Fig. 36). Notably, the cracking experiments on agricultural sheets and packaging bags achieved C3-C8 olefin yields of 68 wt.% and 71.9 wt.%, respectively—the highest yields obtained under similar conditions to date. Interestingly, the optimal reaction temperature varied slightly with the molecular weight of the waste polyethylene. This variation is attributed to the low MFR of this plastic and the different additives used during its synthesis, which can hinder the formation of the PE molten layer (Supplementary Fig. 37). Furthermore, over four consecutive cycles, the olefin yield showed only a slight decrease, and the regenerated catalyst fully restored both the activity and selectivity observed with the initial fresh catalyst (Supplementary Fig. 38). Remarkably, the scaled-up experiment conducted at the 5 g level still achieved an olefin yield exceeding 65% (Supplementary Fig. 39), demonstrating the outstanding industrial application potential of this self-confined cracking strategy.

a Yields of various product fractions after cracking different types of real-life PE. Reaction conditions: 167 mg of hierarchical ZSM-5, 500 mg of PE, 100% Ar feed gas at a flow rate of 40 mL/min, 330–350 °C for 4 hours. Note: agricultural sheets and bags were cracked at 330 °C, while closures and bottles were cracked at 350 °C. b Changes in olefin selectivity (the products in the gas pocket) as the reaction proceeded. Reaction conditions: 300 °C, 100% Ar gas, 40 mL/min flow rate, 250 mg catalyst, 500 mg HDPE, 2 hours. Error bars represent standard deviation (SD). c Analysis of products from PE self-confined cracking tandem catalytic reforming. Left: product yield from PE self-confined cracking. Medium: selectivity of C2–C4 light olefins, BTX, and all other products (excluding solid residues) after catalytic reforming. Right: product yield after catalytic reforming. PE self-confined cracking reaction conditions: 500 mg of HDPE, 167 mg of Nano ZSM-5, 300 °C, 40 mL/min Ar, 4 h. Catalytic reforming conditions: 25 mg of reforming catalysts, 600 °C.
On the other hand, continuous high-yield light olefin production was achieved when we kept feeding the PE substrate. In contrast to a decrease of olefin selectivity after 60 minutes without adding more PE, when we added more PE to the reactor after 1 h, the olefin selectivity remained stable, as high as 80% (Fig. 5b). Thus, collecting olefinic fractions during the self-confined cracking process of PE is analogous to distillation in traditional brewing; olefin-rich fractions should be collected only during the phase where the self-confined effect is active. Since catalytic cracking in a refinery typically involves continuous feedstock supply, we hypothesize that by carefully controlling the PE feed rate to match the PE reaction rate, we can maintain the PE’s self-confinement effect and ensure sustained high olefinic selectivity over an extended period. This suggests that the self-confined cracking of PE holds great potential for industrial applications.
Finally, we performed catalytic reforming of the products from PE self-confined cracking (Supplementary Fig. 40) using two practical catalysts (Cat-1 and Cat-2), acquired from industrial production lines of SINOPEC Catalyst Co., Ltd. As illustrated in Fig. 5c, both catalysts produced approximately 65 wt.% C2-C4 light olefins. More importantly, this process offers the flexibility to adjust the output based on capacity requirements, yielding either primarily a mixture of C3H6 and C4H8 (Cat-1) or a combination of C2H4, C3H6, and separable BTX (benzene, toluene, xylene) fractions (Cat-2). Remarkably, over 90 wt.% of C2-C4 light olefins and separable BTX were produced through the PE self-confined cracking process combined with tandem reforming reactions. In contrast, without a self-confined cracking unit, PE yields only about ~ 34% C2-C4 light olefins over Cat-2 under reforming conditions, excluding the potential of product high-yield C2-C4 light olefins from PE directly (see Supplementary Fig. 41). Additionally, when C/P = 2, this tandem process yields only ~40% C2-C4 light olefins, illustrated maintaining PE self-confined cracking is essential for the efficient production of C2-C4 light olefins in this tandem process (see Supplementary Fig. 42). Moreover, when evaluating various reported pathways for converting PE to light olefins, as summarized in Supplementary Table. 5, our self-confined cracking tandem reforming approach yields the highest profit margin, achieving ~ $248 per ton of waste polyethylene. These findings underscore the efficacy of this tandem strategy and highlight its crucial role in developing a chemical recycling framework for waste polyolefin plastics.
Discussion
The self-confined cracking process of PE, influenced by the reaction conditions and the melt flow rate (MFR) of the polymer reactant, exhibits remarkable selectivity for C3-C8 olefins. This method produces over 67% C2-C4 light olefins and 23% separable BTX when integrated with tandem catalytic reforming, effectively establishing a closed-loop recycling system for waste polyolefin plastics. A defining characteristic of this process is the self-confinement of PE, which enhances the concept of spatial constraints in catalytic reactions of polymeric reactants. Unlike petroleum reforming, the distribution of products in the catalytic cracking of PE is determined more by the degree of self-confinement rather than contact time. Achieving a balance in these spatio-temporal effects is essential for the efficient operation of a sustainable refinery capable of adapting to diverse feedstocks.
Methods
Synthesis of hollow ZSM-5 zeolite
Hollow ZSM-5 zeolite was synthesized with alkali etching method46. Typical procedure was as follow: 0.36 g of NaOH and 30 g of water were mixed and stirred at room temperature for 5 min. Then, 3 g of N-Z5 was added and stirred for another 30 min. Then, the mixture was transferred into an autoclave and thermally treated at 170 °C for 2 days. The solid product was collected by centrifuging, washed with deionized water until the rinse liquids reached a pH of 7–8, then heated in a convection oven at 120 °C overnight, and calcining at 550 °C in air for 6 h to remove the organic template. After calcining, the solid product was treated with 1.0 mol/L NH4Cl solution at 80 °C for 10 h and then calcined at 550 °C for 6 h in an airflow.
Synthesis of hierarchical ZSM-5 zeolite
Hierarchical ZSM-5 zeolite was synthesized with reference to Qin et al.47. First, aluminum isopropoxide was dissolved in an aqueous solution of tetrapropylammonium hydroxide and potassium hydroxide to get a clear solution. Another solution was prepared by mixing colloidal silica with a proper amount of distilled water and added slowly into the template-aluminum mixed solution under stirring. The resultant mixture was stirred for another 60 min. Then the synthesis 2 gel was loaded to a 100 mL Teflon-lined stainless-steel autoclave. The sealed autoclave was transferred into a preheated air-circulated oven and heated at 170 °C under static conditions. After 48 h the autoclave was removed from the oven and quenched immediately with cool tap water. Solid material was recovered by vacuum filtration and subsequently washed with distilled water until the pH of the wash was ~7.0. The solid cake was then recovered and dried at 100 °C overnight, and the solid product was calcined at 550 °C for 6 h. After calcining, the solid product was treated with 1.0 mol/L NH4Cl solution at 80 °C for 10 h and then calcined at 550 °C for 6 h in an airflow.
Synthesis of Na+ ion-changed ZSM-5 zeolite
2 g of dry ZSM-5 zeolite was dispersed in 20 mL of deionized water and stirred well. After that, the weighed NaCl was added, and the solid was collected by centrifugation after stirring at 90 °C for 6 h. Finally, the dried solid powder was calcined in air at 550 °C for 4 hours to obtain the Na-ion-exchanged ZSM-5 catalyst. The samples were named as x% Na/ZSM-5, where x stand for the amount of Na+ load.
Synthesis of metal (Fe, Mn, and Ni) loaded ZSM-5 zeolite
Metal loading on ZSM-5 zeolites is accomplished by impregnation. A typical synthesis procedure is as follows: 2 g of dry ZSM-5 zeolite and 3 g of deionized water were added to a 10 ml beaker and stirred thoroughly. After that, the weighed metal salt was added and stirred at room temperature for 6 h and then transferred to an oven for drying. The dried sample was calcined in air at 550 °C for 4 h to obtain the metal-loaded ZSM-5 catalyst. The samples were named as x%y/ZSM-5, where x represents the metal loading and y represents the metal species.
Synthesis of F/Al2O3 catalyst
2 g calcined γ-Al2O3 was combined with an aqueous solution (2.0 mL, roughly corresponding to the pore volume) of NH4F (5 wt.% of Al2O3). The mixture was dried in air in an oven at 80 °C for 2 h, followed by ramping the temperature to 550 °C at a rate of 2 °C min-1 and holding for 5 h.
Synthesis of sulfated activated carbon (SO4 -/AC)
In a round-bottomed flask, 2 g of vacuum-dried activated carbon was mixed with 50 mL of 6 mol/L sulfuric acid and refluxed while stirring overnight at 90 °C. After cooling, the excess sulfuric acid solution was removed by centrifugation, and the sample was dried and calcined at 600 °C for 4 h under a nitrogen atmosphere to obtain the SO4-/AC catalyst.
Synthesis of H3PO4/SiO2
2 g of dried silica 3 g of phosphoric acid (85%) was added dropwise, and the mixture was ground well in a mortar and pestle, after which it was transferred to a crucible. The H3PO4/SiO2 catalyst was obtained after calcination at 400 °C for 6 h in a muffle furnace.
Characterizations
X-ray diffraction (XRD) spectra of the catalysts were collected on a Shimadzu XRD 6100 with Cu curved monochromator and using Cu-Kα-radiation (λ = 1.5406 Å). Sample scanning 2θ from 5-50 degrees. Nitrogen sorption isotherms were measured at 77 K using a JBK-200W to calculate the micropore volume, external, and specific surface area. The Brunauer-Emmett-Teller (BET) surface areas were calculated from the adsorption data. Surface areas were calculated using the BET equation. Micropore volume and external surface area were calculated using the t-plot method with the Jura-Harkins equation for determining the thickness of the adsorbed layer. The samples were previously outgassed at 350 °C for 12 h under vacuum before the measurements. The size and morphology of zeolite were determined by transmission electron microscopy (TEM) which performed on a JEOL-2000 electron microscope with an acceleration voltage of 200 kV. Temperature-programmed desorption of ammonia (NH3-TPD) was carried out on ICHEM 700 Pulse Chemisorption System. The weighed sample (50 mg) was pretreated at 650 °C for 1 h under He, then cooled to 30 °C. The NH3-Ar (10 vol% NH3) gas was introduced at 80 °C for 1 h to ensure the saturation adsorption of NH3. The sample was then purged with He for 1 h until the signal returned to the baseline as monitored by a thermal conductivity detector (TCD). The desorption curve of NH3 was acquired by heating the sample from 80 to 650 °C at a rate of 10 °C/min under Ar with the flow rate of 30 mL/min. The internal and external acid characterization of the catalyst was determined by 2,6-2-tertbutyl-pyridine IR (py-IR). Each sample was pressed into a self-supported wafer (ca. 10 mg, diameter: 13 mm). The wafers were first pretreated at 400 °C for 1 h under vacuum. After the temperature dropped to 150 °C, a spectrum was collected as the background. Subsequently, the samples were exposed to 2,6-2-tertbutyl-pyridine vapor for 5 min at room temperature and outgassed at 300 °C for 30 min. When the temperature dropped to 150 °C, py-IR spectra were recorded.
General procedure for the catalytic cracking of PE and heavy oil
The experiments on catalytic cracking of PE and heavy oil were conducted using a self-built fixed-bed reactor (Supplementary Fig. 1), the fixed-bed reactor is composed of three sections: a carrier gas supply section located at the front end of the reaction tube, an infrared heating tube furnace for heating the reaction tube, and a product collection section located at the back end of the reaction tube. In a typical procedure, the catalyst powder and substrate powder were mixed in a certain mass ratio and packed into the middle of the quartz reaction tube (φ 10*1 mm), and the quartz cotton (10 μm) was used to fill the two ends of the catalyst bed. The reaction tube was then purged with argon atmosphere (40 mL/min) for 30 min to replace the air. The catalyst bed was heated to the reaction temperature with a ramp rate of 10 °C/s, and then maintained at 300 °C for 2 h. During the reaction, the products generated by PE cracking are carried by a carrier gas (Ar), and a cold trap (cooled with dry ice) and gas bag were used to collect the liquid and gaseous products, respectively. After reaction, when the reaction tubes and cold trap were returned to room temperature (20 °C), they were washed with dichloromethane (DCM) to collect all liquid products. The solid residue insoluble in DCM in the reaction tube is in two main parts: the wax product condensed on the reactor wall and the catalyst bed after the reaction. The wax product was dissolved in hot hexane, and then the remaining catalyst was collected and stored separately.
Catalytic cracking of commodity PE
In this paper, four commercial PEs were selected for catalytic cracking experiments. These commodity plastics are agricultural sheets, plastic bags, plastic bottles, and bottle cap. These materials were washed using deionized water and dried, and subsequently crushed into 40–80 mesh pellets at dry ice temperature. Then, 163 mg of hierarchical catalyst powder and 500 mg PE powder were mixed and packed into the middle of the quartz reaction tube. The reaction time was 4 h, and the reaction temperature was selected between 300-350 °C.
Catalyst stability testing
In the continuously running PE cracking experiments, the catalyst was removed from the reaction tube after reaction. Then without any treatment, it was mixed with the next batch of PE feedstock and continued the cracking experiment. After four single runs, the catalyst was removed and then calcined in flowing air at 800 °C for 2 h. The regenerated catalyst will be reused in PE cracking experiments.
Evaluation of reaction rates
For the evaluation of reaction rates, the typical procedure is as follows: 250 mg of commercial ZSM-5 catalyst powder and 500 mg PE powder were mixed and packed into the middle of the quartz reaction tube and purged with argon atmosphere (40 mL/min) for 30 minutes to replace the air. After that, the catalyst bed was heated to 300 °C at a ramp rate of 10 °C per second and then held for 5, 10, 15, and 20 min. Then, the heating was stopped, and the upper lid of the heater was opened to allow the reaction tube to cool down rapidly and quench the reaction.
Continuous feeding experiment
In order to verify the potential of the PE self-limiting domain cracking strategy for industrial applications, we made up the HDPE feedstock halfway through the reaction, which was carried out as follows: when the PE cracking reaction was carried out for 60 min, the heating was stopped and the upper lid of the heater was opened to allow the reaction tube to cool down rapidly and quench the reaction. After that, 0.5 g of HDPE was added to the reaction tube and mixed well with the residual solids in the reaction tube. The reaction tube was purged using argon at room temperature for 60 min, after which the temperature was slowly increased to 200 °C under the protection of the argon gas stream and maintained for 1 h to ensure that the added HDPE could be completely melted and mixed well. Afterwards, the temperature was raised to 300 °C to continue the reaction, and the gaseous products were collected and analyzed.
Tandem cracking reforming experiment
The experimental setup for PE self-confined cracking tandem catalytic reforming is shown in Supplementary Fig. 38. In a typical procedure, 0.5 g of HDPE and 0.167 g of Nano ZSM-5 were mixed well and loaded into a reaction tube, and 0.025 g of reforming catalyst was added to the U-shaped quartz tube. Connect the tubing of the tandem reactor, ensure that the gas tightness is intact, and insulate the transfer line at 160 °C. The reformed bed was heated to 600 °C under argon purge to activate the catalyst. After that, the PE cracking reactor was heated up to 300 °C and kept for 4 h. After reaction, when the reaction tubes and cold trap were returned to room temperature (20 °C), they were washed with dichloromethane (DCM) to collect all liquid products, and the gas product was collected in gas bag.
Propene adsorption experiment
The propylene pulse adsorption experiments with catalyst and PE mixtures were carried out on a ICHEM 700 Pulse Chemisorption System. Typical experimental operation was as follows: firstly, the mixed catalyst and PE powder were loaded into U-shaped sample tubes and then purged in Ar gas stream (30 mL/min) for 1 h. Then, in order to melt and adsorb the PE onto the zeolite, the reaction tube was heated to 200 °C under argon atmosphere and kept for 2 h, during which the signal from the TCD detector proved that no cracking of the PE occurred. Subsequently, 200 μL of propylene was injected into the U-tube every 3 min until saturation of propylene adsorption.
N2 adsorption experiment for Cat/PE
Typical experimental operation of the N2 adsorption experiments with catalyst and PE mixtures was as follow. First, ZSM-5 was kept under vacuum at 350°C for 6 h for degassing, and after the sample tube was cooled to room temperature, weighed HDPE powder was quickly added to the sample tube and mixed well. The mixture was then kept under vacuum at 200 °C for 2 h to ensure that the PE was completely melted and the catalyst was encapsulated. Then, the nitrogen sorption isotherms were measured at 77 K using a JBK-200W to calculate the micropore volume, external and specific surface area.
TGA with FTIR analysis
The TGA-FTIR analysis was performed using simultaneous TGA (TGA STA8000, PerkinElmer) coupled with FTIR spectrometer (PerkinElmer). Approximately, 10 mg of the ZSM-5 zeolite and 10 mg of was mixed and then heated under N2 gas flow rate of 30 mL/min from 30 °C to 300 °C and then held for 1 h at the heating rate of 20 °C/min. The evolved gases were transported to the FTIR spectrometer through a connecting line (heated at 280 °C to prevent the evolved gases from condensing). The FTIR spectra were recorded at a resolution of 4.0 cm−1 from 4000 to 550 cm−1, and 16 scans were conducted for each spectrum. The thermogravimetric (TG) curve and the FTIR spectra were recorded. The chemicals from decomposed samples were analyzed by FTIR.
Product analysis
The methodologies for product analysis are given in the Supplementary Note 1.
Molecular dynamics (MD) simulations
Details on molecular dynamics simulations are given in the Supplementary Note 2.
Data availability
All data are available within the article and its Supplementary Information files and from the corresponding authors upon request. Source data are provided in this paper. Source data are provided with this paper.
References
Thiounn, T. & Smith, R. C. Advances and approaches for chemical recycling of plastic waste. J. Polym. Sci. 58, 1347–1364 (2020).
Gan, L. et al. Beyond conventional degradation: catalytic solutions for polyolefin upcycling. CCS Chem. 6, 313–333 (2024).
Dong, Z. et al. Understanding the structure–activity relationships in catalytic conversion of polyolefin plastics by zeolite-based catalysts: a critical review. ACS Catal. 12, 14882–14901 (2022).
Sun, J. et al. Catalytic upcycling of polyolefins. Chem. Rev. 124, 9457–9579 (2024).
Li, H. et al. Hydroformylation of pyrolysis oils to aldehydes and alcohols from polyolefin waste. Science 381, 660–666 (2023).
Neidhart, E. K. et al. C–H functionalization of polyolefins to access reprocessable polyolefin thermosets. J. Am. Chem. Soc. 145, 27450–27458 (2023).
Fazekas, T. J. et al. Diversification of aliphatic C–H bonds in small molecules and polyolefins through radical chain transfer. Sciece 375, 545–550 (2022).
Zhao, B. et al. Catalytic Conversion of mixed polyolefins under mild atmospheric pressure. Innovation 5, 100586 (2024).
Vialon, T. et al. Upcycling polyolefin blends into high-performance materials by exploiting azidotriazine chemistry using reactive extrusion. J. Am. Chem. Soc. 146, 2673–2684 (2024).
Sun, J. et al. Bifunctional tandem catalytic upcycling of polyethylene to surfactant-range alkylaromatics. Chem. 9, 2318–2336 (2023).
The Future of Surfactants to 2022 https://www.smithers.com/services/market-reports/materials/the-future-of-surfactants-to-2022.
Circular Economy for Plastics–A European Analysis https://plasticseurope.org/knowledge-hub/the-circular-economy-for-plastics-a-european-analysis-2024/.
Feng S. Global Olefins and Polyolefins Outlook https://commodityinsights.spglobal.com/.
Zou, L. et al. Chemical recycling of polyolefins: a closed-loop cycle of waste to olefins. Natl. Sci. Rev. 10, nwad207 (2023).
Abbas-Abadi, M. S. et al. Challenges and opportunities of light olefin production via thermal and catalytic pyrolysis of end-of-life polyolefins: towards full recyclability. Prog. Energy Combust. Sci. 96, 101046 (2023).
Chernyak, S. A., Corda, M., Dath, J. P., Ordomsky, V. V. & Khodakov, A. Y. Light olefin synthesis from a diversity of renewable and fossil feedstocks: state-of the-art and outlook. Chem. Soc. Rev. 51, 7994–8044 (2022).
Zhang, F. et al. From trash to treasure: chemical recycling and upcycling of commodity plastic waste to fuels, high-valued chemicals and advanced materials. J. Energy Chem. 69, 369–388 (2022).
Fu, Z., Hua, F., Yang, S., Wang, H. & Cheng, Y. Evolution of light olefins during the pyrolysis of polyethylene in a two-stage process. J. Anal. Appl. Pyrol. 169, 105877 (2023).
Duan, J. et al. Coking-resistant polyethylene upcycling modulated by zeolite micropore diffusion. J. Am. Chem. Soc. 144, 14269–14277 (2022).
Feng, J. et al. Micelles cascade assembly to tandem porous catalyst for waste plastics upcycling. Angew. Chem. Int. Ed. 63, e202405252 (2024).
Conk, R. J. et al. Polyolefin waste to light olefins with ethylene and base-metal heterogeneous catalysts. Science 385, 1322–1327 (2024).
Wang, N. M. et al. Chemical Recycling of Polyethylene by Tandem Catalytic Conversion to Propylene. J. Am. Chem. Soc. 144, 18526–18531 (2022).
Conk, R. J. et al. Catalytic deconstruction of waste polyethylene with ethylene to form propylene. Science 377, 1561–1566 (2022).
Du, Y. et al. Revisiting a large-scale FCC riser reactor with a particle-scale model. Chem. Eng. Sci. 249, 117300 (2022).
Wang, Y., Yokoi, T. & Tatsumi, T. Selective production of light olefins over zeolite catalysts: impacts of topology, acidity, and particle size. Micropor. Mesopor. Mater. 358, 112353 (2023).
Blay, V. et al. Engineering zeolites for catalytic cracking to light olefins. ACS Catal. 7, 6542–6566 (2017).
Rejman, S. et al. Transport limitations in polyolefin cracking at the single catalyst particle level. Chem. Sci. 14, 10068–10080 (2023).
Jaydev, S. D., Martín, A. J., Garcia, D., Chikri, K. & Pérez-Ramírez, J. Assessment of transport phenomena in catalyst effectiveness for chemical polyolefin recycling. Nat. Chem. Eng. 1, 565–575 (2024).
Corma, A. & Orchillés, A. V. Current views on the mechanism of catalytic cracking. Micropor. Mesopor. Mater. 35, 21–30 (2000).
Müller, S. et al. Hydrogen transfer pathways during zeolite catalyzed methanol conversion to hydrocarbons. J. Am. Chem. Soc. 138, 15994–16003 (2016).
Kotrel, S., Knözinger, H. & Gates, B. C. The Haag–Dessau mechanism of protolytic cracking of alkanes. Micropor. Mesopor. Mater. 35, 11–20 (2000).
Hiraoka, K. & Kebarle, P. Stabilities and energetics of pentacoordinated carbonium ions. The isomeric protonated ethane ions and some higher analogs: protonated propane and protonated butane. J. Am. Chem. Soc. 98, 6119–6125 (1976).
Thomas, C. L. Chemistry of cracking catalysts. Ind. Eng. Chem. 41, 2564–2573 (1949).
Miyake, K. et al. Improving hydrothermal stability of acid sites in MFI type aluminosilicate zeolite (ZSM-5) by coating MFI type all silica zeolite (Silicalite-1) shell layer. Micropor. Mesopor. Mater. 288, 109523 (2019).
Wang, L. et al. Mechanistic origin of transition metal modification on ZSM-5 zeolite for the ethylene yield enhancement from the primary products of n-octane cracking. J. Catal. 416, 387–397 (2022).
Rejman, S. et al. External acidity as performance descriptor in polyolefin cracking using zeolite-based materials. Nat. Commun. 16, 2980 (2025).
Manos, G., Garforth, A. & Dwyer, J. Catalytic degradation of high-density polyethylene on an ultrastable-y zeolite. nature of initial polymer reactions, pattern of formation of gas and liquid products, and temperature effects. Ind. Eng. Chem. Res. 39, 1203–1208 (2000).
del Campo, P., Martínez, C. & Corma, A. Activation and conversion of alkanes in the confined space of zeolite-type materials. Chem. Soc. Rev. 50, 8511–8595 (2021).
Corma, A. & Ortega, F. J. Influence of adsorption parameters on catalytic cracking and catalyst decay. J. Catal. 233, 257–265 (2005).
Sławek, A. et al. Adsorption of alkanes in zeolites LTA and FAU: Quasi-equilibrated thermodesorption supported by molecular simulations. J. Phys. Chem. C. 123, 29665–29678 (2019).
Yuan, J. et al. Hyperloop-like diffusion of long-chain molecules under confinement. Nat. Commun. 14, 1735 (2023).
Liu, Z. et al. Asymmetric rotations slow down diffusion under confinement. Nat. Commun. 16, 2018 (2025).
Liu, Z. et al. Molecular self-gating inside a zeolite catalyst. J. Am. Chem. Soc. 147, 6126–6136 (2025).
Oseli, A., Vesel, A., Žagar, E. & Perše, L. S. Mechanisms of single-walled carbon nanotube network formation and its configuration in polymer-based nanocomposites. Macromolecules 54, 3334–3346 (2021).
Doruker, P. & Mattice, W. L. Simulation of polyethylene thin films on a high coordination lattice. Macromolecules 31, 1418–1426 (1998).
Groen, J. C. et al. Creation of hollow zeolite architectures by controlled desilication of Al-zoned ZSM-5 crystals. J. Am. Chem. Soc. 127, 10792–10793 (2005).
Qin, Z. et al. The mosaic structure of zeolite crystals. Angew. Chem. Int. Ed. 55, 15049–15052 (2016).
Acknowledgements
This work was financially supported by the National Key R&D Program of China (2021YFA1501700, Z.F.), the National Science Foundation of China (22272114, Z.F), and the Funding for Hundred Talent Program of Sichuan University (20822041E4079, Z.F). This work has also been supported by WeiFang Benzo Chemical Co., Ltd. (No. 24H1191, Z.F) and SINOPEC Research Institute of Petroleum Processing Co., Ltd. via collaborative project No. 36800000-24-ZC0607-0175.
Author information
Authors and Affiliations
Contributions
F.Z., B.P., X.L., M.L., X.W., M.W., and D.M. conceived the project and designed the experiments. Z.D. performed experiments. N.X. and T.L. performed the Molecular dynamics (MD) simulations. C.L., W.C., Z.Q., and R.Z. contributed to the synthesis of catalysts. Z.D., N.X., B.P., and F.Z. wrote the paper. All authors contribute to the discussion and revision of the paper.
Corresponding authors
Ethics declarations
Competing interests
Z.D. and F.Z. are among the inventors on a patent application partially based on this work. All other authors declare that they have no competing interests.
Peer review
Peer review information
Nature Communications thanks Wei Li and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, Z., Peng, B., Xiao, N. et al. Efficient conversion of polyethylene to light olefins by self-confined cracking and reforming. Nat Commun 16, 7964 (2025). https://doi.org/10.1038/s41467-025-63116-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63116-8
This article is cited by
-
Acid sites in hydrogen-involved polyolefin upcycling: mechanistic insights and design principles
Science China Chemistry (2026)
