
Abstract
CRISPR-Cas systems revolutionize life science. Metagenomes contain millions of unknown Cas proteins. Traditional mining relies on protein sequence alignments. In this work, we employ an evolutionary scale language model (ESM) to learn the information beyond sequences. Trained with CRISPR-Cas data, ESM accurately identifies Cas proteins without alignment. Limited experimental data restricts feature prediction, but integrating with machine learning enables trans-cleavage activity prediction of uncharacterized Cas12a. We discover 7 undocumented Cas12a subtypes with unique CRISPR loci. Structural analyses reveal 8 subtypes of Cas1, Cas2, and Cas4. Cas12a subtypes display distinct 3D-folds. CryoEM analyses unveil unique RNA interactions with the uncharacterized Cas12a. These proteins show distinct double-strand and single-strand DNA cleavage preferences and broad PAM recognition. Finally, we establish a specific detection strategy for the oncogene SNP without traditional Cas12a PAM. This study highlights the potential of language models in exploring undocumented Cas protein function via gene cluster classification.
Similar content being viewed by others

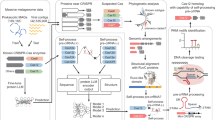

Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute the adaptive immune system in prokaryotes to defend against invasive genetic elements1. The CRISPR-Cas systems keep evolving during natural evolution, currently, two classes have been identified, and new subtypes are emerging2. Class 1 comprises types I, III, and IV, featuring multiple Cas proteins as effector modules. Class 2 comprises types II, V, and VI, mainly using single multidomain proteins, e.g., Cas9, Cas12, and Cas13, as effector modules. Due to its ease of reprogrammability, class 2 Cas proteins have been widely applied in gene editing, nucleic acid detection, imaging, and annotation, which has led to the research revolution from basic science to translational medicine3. Novel CRISPR-Cas discovery is fundamental for the technology iteration. Metagenome provides a precious reservoir for the novel Cas exploration. Recently, the compact gene editor Φ was discovered in the genome of huge phages, which were identified from the metagenomic dataset4,5,6. Nevertheless, millions of unknown Cas proteins, scattered in the metagenomes, are still waiting to be characterized.
Traditional Cas protein mining mainly depends on the primary sequences to predict the protein function and classification2. Sequence similarity-based search could be performed by Basic Local Alignment Search Tool (BLAST) and Hidden Markov Model (HMM)7,8. Based on the sequence similarity search, MacSyFinder and HMMCAS have been developed9,10. But these methods are restrained by the known Cas sequence and are difficult to discover new Cas proteins. Machine learning-based methods predict Cas proteins in a data-driven manner, for example, CASpredict, CASboundary, and CRISPRCasTyper11,12,13. However, the protein function is directly determined by the three-dimensional structure, but not amino acid sequences. Learning the hidden biological information from protein sequences motivates the development of various evolutionary-scale language models14. They can predict secondary structure and tertiary structures15,16. Recently, structure-based protein clustering discovered undocumented clades of deaminase and generated an efficient cytosine base editor, indicating the importance of structural information in protein discovery17. In addition to the three-dimensional structural information, an alternative approach could be the evolutionary-scale language model. The recently developed ESM-2 language model, scaling up to 15 billion trainable parameters, can capture the protein feature at the atomic-resolution level18.
Here, we develop an Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan) strategy based on the ESM language model19. After training with the CRISPR-Cas sequences and their functional annotation, the AIL-Scan can accurately distinguish the different CRISPR-Cas types from the annotated genome sequences. However, only a few Cas proteins are experimentally evaluated. We integrate the ESM and machine learning on small sample size data and develop a trans-cleavage activity prediction model with accuracy. The Cas12a family is taken as an example to explore in the metagenomic database. Different from the classical CRISPR loci of Cas12a, we discovered eight subtypes of the Cas12a family, which are characterized by the unique organization of CRISPR loci and protein sequences. Furthermore, the integrase proteins, i.e., Cas1, Cas2, and Cas4, also have eight subtypes, respectively, according to the structural alignments. The missing integrase proteins result in a decrease in spacer numbers in the CRISPR-loci. In addition, the unreported Cas12a proteins show diverse 3D foldings between subtypes. The CryoEM analyses further discover unique interaction patterns with RNA. Accordingly, these proteins show distinct double-strand and single-strand DNA cleavage preferences and broad PAM recognition, which enables the specific detection of the oncogene single-nucleotide polymorphisms (SNP) without traditional Cas12a PAM and efficient cellular gene editing with minimal off-targets. The study provides new insights into machine learning in the discovery of undocumented functional Cas proteins via gene cluster classification.
Results
Development of an Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan) strategy based on an ESM large language model
We assumed that by embedding the functional feature with protein primary sequences, we could trace the natural evolution rules and identify the CRISPR-Cas proteins in the metagenomics data directly without sequence alignments. To identify the CRISPR-Cas proteins, we developed an Artificial Intelligence-assisted CRISPR-Cas Scan (AIL-Scan) strategy (Fig. 1a). It includes the following steps:
-
1.
CRISPR-Cas training data is created by extracting CRISPR-associated (Cas) proteins from the NCBI database, classifying them by genes, and removing redundant sequences.
-
2.
Supervised fine-tuning of ESM on the CRISPR-Cas training data based on the biological information to predict the Cas protein.
-
3.
Feature analyses of Cas proteins, including cleavage activity, CRISPR-loci type, CRISPR loci-length, direct repeats, spacers, evolutionary analyses, MSA, and structures.

a The ESM language model is trained by Cas proteins, which were collected, classified, and clustered as input sequences. The Cas proteins were embedded and classified with multiple labels. The trans-cleavage activity prediction model was developed based on the ESM and small-scale experimental data of trans-cleavage. The trained model was applied to discover Cas proteins and predict features from the sequences extracted from the metagenome. The protein structures were visualized using Chimera59. The sequence alignment was visualized by Jalview61. b The receiver operating characteristic (ROC) curves and area under the ROC curve (AUC) for 12 Cas proteins and non-Cas proteins. c The test loss and test accuracy curves of AIL-Scan.
We generated our training data using reviewed NCBI gene data. We annotated the Cas1, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9, Cas10, Cas12, and Cas13. Non-Cas proteins were extracted according to the following rules, without the annotation of Cas, and removing the proteins with sequence similarity over 40%. The Cas protein database was separated into a training or validation database using CD-HIT-2D with a 40% identity threshold to remove the redundant sequences and avoid overfitting. We collected 76567 non-redundant positive sequences and 13047 non-Cas proteins, which were deposited in NCBI before July 5, 2023 (Supplementary Fig. 1). The maximal protein length is less than 1764 amino acids. To obtain the best classification, we introduced the “focal loss” in the classification to solve the unbalance of the input data. We obtained the best model during the 13th Epoch of model training and obtained 97.75% accuracy for the ESM 2 model with 650 million (650 M) parameters (Supplementary Fig. 2). Using the 15 billion (15B) parameters model, we achieved the best performance in the 9th Epoch with 98.22% accuracy (Supplementary Fig. 2). This model maintained consistent performance, achieving an accuracy 97.68% on the independent dataset, i.e. TestSet2024, which contains sequences deposited in NCBI from July 6, 2023, to Oct 28, 2024 (Supplementary Tables 1–3). These results indicate a robust generalization of this model. The accuracy and prediction speed of AIL-Scan is comparable to the CRISPRcasIdentifier, which integrates HMMs and machine learning (Table 1 and Supplementary Fig. 3). CASPredict performed with the highest speed among the four software, although its accuracy is lower than the machine learning based software, i.e., AIL-Scan and CRISPRcasIdentifier. However, the NCBI data has been partially annotated by the HMM model, so we turned to validate AIL-Scan’s capability in recognizing “unseen proteins”. We utilized a recent dataset of 3601 Cas12 family protein sequences20, in which 3521 sequences (97.8%) had less than 90% similarity with the training set, meanwhile 3351 sequences (93.1%) had less than 40% similarity with the training set. This test set is named TestSet2025 and is significantly distinct from the training set in sequence space, making it suitable for evaluating generalization ability. AIL-Scan successfully identified 3182 Cas12 proteins, in contrast, the HMM model identified 1240 sequences, demonstrating the strong generalization capabilities of AIL-Scan. Considering the resource consumption, the 650M model is sufficient for the Cas prediction. We used ESM embeddings to reduce dimensionality with t-SNE for 77684 sequences and discovered that ESM can distinguish the differences in various Cas classifications. The ROC curves and AUC indicate the probability that the positive sample’s decision value is greater than the negative sample’s decision value for all the Cas and non-Cas proteins (Fig. 1b). The test loss and test accuracy also indicate that the model generalizes correctly and performs well on unseen data (Fig. 1c). We evaluated the model robustness using the 5-fold cross-validation. The average accuracy is 0.9786 and the standard deviation is 0.0013 (Supplementary Table 4).
We use the Global Microbial Gene Catalog (GMGC) metagenomic database for the Cas protein discovery21. We selected 50,000 bins with high quality from GMGC and extracted 20,000 MAGs, including CRISPR-loci, to test the performance of AIL-Scan. The protein sequences were predicted by Prodigal software22. We collected ca. 20,000,000 protein sequences shorter than 1500 amino acids for prediction. In comparison with the established methods, the AIL-Scan predicts 1379 Cas12a sequences.
Development of a trans-cleavage activity prediction model
The trans-cleavage activity of Cas12a has been used in various applications. Although many CRISPR-Cas12a proteins have been identified, few of them have been tested in the trans-cleavage experiments. Therefore, the main challenge encountered during this study lies in dealing with a small sample size coupled with high-dimensional embeddings, which often leads to convergence issues when employing most models. A total of 69 labeled Cas12a proteins (including three known Cas12a) were included in our analysis (Supplementary Data 1). Their trans-cleavage activities were assessed by the fluorophore-quencher (FQ) reporter assay. The trans-cleavage activity was defined as proteins displaying fluorescence intensity twice that of the negative control. Thirty-three proteins were classified as active in trans-cleavage activity, and the remaining 36 proteins were categorized as inactive. To evaluate the performance of our predictive model, a test set comprising 13 randomly selected proteins (approximately 20% of the sample) was used, while the remaining 56 proteins were employed for training purposes. Initially, we recorded the last embedding layers based on our fine-tuned ESM model for all labeled Cas12a protein sequences. These embeddings (1280 dimensions) were utilized as covariates to predict trans-cleavage activity.
Different forms of decision tree models are evaluated in this task. The results of our study demonstrate that Light Gradient Boosting Machine (LightGBM) achieves the highest accuracy among mainstream machine learning models, with an accuracy rate of 69.2% on the test set trained on embeddings. To address dimensionality-related challenges, principal component analysis (PCA) was employed to extract essential embeddings, with prediction performance evaluated across 2–15 principal components. Alongside PCA, we compared 31 alternative methods, including t-SNE, UMAP, and raw data. Detailed comparisons, training procedures, and results are provided in Table 2, Supplementary Table 5, and the supplementary notes. LightGBM, CatBoost, and RandomForest achieve the accuracy of 92.3% in the test set (12 out of 13 proteins are correctly labeled) with 4, 6, and 8 principal components, respectively. We can see that compared to training models directly with embeddings, extracting essential dimensions with PCA provides higher accuracies in predicting trans-cleavage activity (Supplementary Table 5). However, this model is still limited by the small dataset, more experimental data would improve its prediction accuracy. Additionally, we tested our prediction model on two unreported Cas12a proteins, i.e., the trans-cleavage activity of two Cas12a candidates: ArCas12a_1 (derived from Agathobacter rectale) and LeCas12a_3 (derived from Lachnospira eligens_B). Our model predicted that ArCas12a_1 has trans-cleavage activity but not LeCas12a_3. In the experiment, ArCas12a_1 demonstrated significantly stronger trans-cleavage activity than the negative control, while LeCas12a_3 did not (Supplementary Fig. 4). These experimental outcomes were consistent with our model’s predictions, supporting the generalizability and robustness of the prediction model.
CRISPR-Cas12a loci predicted from the metagenomics
We did further feature analyses of Cas12a candidate proteins. Phylogenetic analysis of Cas12 proteins suggests that the identified Cas12a proteins fall into the Cas12a clade (Fig. 2a). The classical CRISPR-loci, comprising essential elements such as Cas1, Cas2, and Cas4, play a pivotal role in type classification. To delve into these features, we employed AIL-Scan to predict Cas1, Cas2, and Cas4 proteins within the same CRISPR loci adjacent to the Cas12a sequence. Subsequently, we meticulously verified 300 predicted CRISPR loci to gain deeper insights manually. Normally, Cas12a is considered to have a unique CRISPR locus, comprising Cas1, Cas2, and Cas4. Intriguingly, the observed count of Cas1, Cas2, and Cas4 proteins was notably lower than that of Cas12a, suggesting the absence of these small Cas proteins in some Cas12a loci (Fig. 2b, c). Further stratification based on the number of integrase proteins led to the classification of CRISPR loci into eight distinct subtypes. The distribution of integrase proteins across these subtypes exhibited a sparse pattern (Fig. 2d). Notably, subtype VIII lacked any integrase proteins, subtype I encompassed Cas1, Cas2, and Cas4, while subtype VI exclusively featured Cas2. This nuanced classification sheds light on the diversity within CRISPR loci and underscores the intricate variations in the composition of integrase proteins among different subtypes. Our observations may provide unreported perspectives on correlations among different CRISPR-Cas systems and integrase proteins. Remarkably, the analyses using the 1000 predicted CRISPR Cas12a loci without manual verification show a strikingly similar distribution pattern as the result from the 300 manually confirmed ones, indicating this distribution is a universal phenomenon (Supplementary Fig. 5). To provide further insights, we measured the length of CRISPR loci, beginning from the start of the Cas12 protein and concluding at the first spacer. Subtype VIII emerged as the shortest, spanning mere 4200 bp, while subtype I is the longest, extending over 6100 bp. Particularly noteworthy were certain subtype I CRISPR loci exhibiting extraordinary lengths of up to 6700 bp, raising the possibility of harboring enigmatic protein elements (Fig. 2e). Aligned with the integrase variation, the numbers of spacers notably decreased in subtypes IV, VI, and VIII, underscoring the pivotal roles of integrases in spacer capture (Fig. 2f). Despite the divergence in spacer numbers, the stem-loop region corresponding to direct repeat sequences remained conserved (Fig. 2g). This consistent conservation hints at a shared structural element, emphasizing the importance of the stem-loop region in CRISPR loci across different subtypes.

a Phylogenetic tree of Cas12 proteins. The identified Cas12a proteins in this work were highlighted in red in the Cas12a family. b Cas12a subtypes with different combinations of accessory proteins, i.e., Cas4, Cas1, and Cas2. c Statistics of Cas12, Cas1, Cas2, and Cas4 from 300 CRISPR-loci, which were verified manually. The features of the first 1000 CRISPR-loci were analyzed in Supplementary Fig. 5. d Statistics of subtypes in the 300 CRISPR-loci. e Sequence length variation in different subtypes. DNA sequence length was calculated from the start codon of the Cas12a gene to the end of the first repeat. f Statistics of spacers in different subtypes. g Sequence alignment of direct repeats in the 300 CRISPR-loci. The sequence corresponding to the stem loop region of crRNA was highlighted with a gray background. h Distribution of Cas proteins in different subtypes and species. The subtypes were colored in the inner circle. The species were labeled in the outer circle. Error bar indicates mean ± s.e.m. measured from three technical replicates. n = 3. Statistical significance was assessed using one-way ANOVA analysis. The symbol ‘#’ indicated that the metagenomes in the corresponding subtypes did not contain spacer sequences. Source data are provided as a Source Data file.
To explore the distribution of the discovered proteins in the organisms, we constructed a phylogenetic tree using 300 candidate Cas12a proteins, which were manually verified, along with three known Cas12a (LbCas12a, FnCas12a, and AsCas12a). 232 Cas12a proteins from the Lachnospiraceae family cluster into one clade. Within this clade, subclade 1 consisted of 62 subtype I Cas12a proteins, 81 subtype VII Cas12a proteins, and a modest representation of other subtypes. Notably, subtype I and subtype IV emerge as the principal constituents within Subclade 2. Furthermore, Subclade 3 is marked by the exclusive presence of 28 subtype VIII Cas12a proteins originating from the Acutalibacteraceae family. It is worth noting, 94.6% of the identified Cas12a proteins originate from enteric microorganisms (Fig. 2h), which may be due to the ease of recovering high-quality genomes from enteric microorganisms. Additionally, the thermostable YmeCas12a (subtype I) is adjacent to subtype I Cas12a proteins (Supplementary Fig. 6).
Cas integrases in CRISPR loci
New insights highlight the structural diversity and functional roles of Cas integrases in CRISPR loci23,24,25,26,27. Cas1, Cas2, and Cas4 are essential for integrating foreign DNA into bacterial CRISPR systems, which generates bacterial immunity26. AlphaFold228 was applied to predict all protein structures in the eight distinct subtypes, providing insights into their variation, respectively (Fig. 3 and Supplementary Fig. 7). Cas1 proteins, encompassing 92–331 amino acids, are classified into eight types based on structure and sequence (Fig. 3a, b and Supplementary Fig. 7b). Type 8 is the most prevalent Cas1 protein, resembling AfCas1 (PDB: 4N06)29 and its N-terminal and C-terminal domains (NTD, CTD) contain with key catalytic sites in specific helices and loops (Supplementary Fig. 7c). Structural differences across types were analyzed via the Dali server30. The variation in CTD elements does not necessarily hinder foreign DNA acquisition31, emphasizing their structural flexibility. Cas2 proteins, containing 70–146 amino acids, also fall into eight subtypes, with type 8 showing notable structural similarities to E. coli Cas2 (PDB: 5DQT)32 but with unique N-terminal helices (Fig. 3c, d and Supplementary Fig. 7d–f). Other subtypes exhibit varied structural deficiencies, such as missing β-sheets or helices, affecting dimer interfaces and potentially altering DNA binding. This diversity underlines Cas2’s adaptability within Cas1–Cas2 complexes (Supplementary Fig. 7f)33. Cas4 proteins, comprising 79–206 amino acids, exhibit eight types (Fig. 3e, f and Supplementary Fig. 7g, h), with type 8 resembling I-C Cas4 (PDB: 8D3Q)24 but lacking specific helices critical for protospacer cleavage. Structural differences across subtypes, such as missing helices or β-sheets, impact spacer insertion and integration within CRISPR systems (Supplementary Fig. 7i). These findings broaden our understanding of Cas4 structural variations and their functional implications in bacterial immunity. The detailed structural features of integrases are analyzed in the Supplementary Note.

a, c, e The RMSD matrix of Cas1, Cas2, and Cas4 structure models constructed by AlphaFold2. Colors within the heatmap, ranging from dark blue to white, represent the RMSD values ranging from high to low. The protein names were colored based on their structure type classification. The color of each protein name corresponds to the protein structure type displayed in the right panel. b, d, f Typical structure models of Cas1, Cas2, and Cas4, which were classified into different types. Secondary structures were annotated for all protein types. Type 1–7 structures of Cas1, Cas2, and Cas4 were superposed onto each full-length type 8 structure, and secondary structures were labeled. The “αX” in type 1 of (f) indicates that it does not appear in other Cas4 structure types.
Cas12a proteins in the subtypes
The differences in the Cas12a structures are key features of the Cas12a subtypes. We analyzed the motifs of the Cas12a sequences and discovered conserved and distinct motifs in the different subtypes, which are key for the Cas12a functions (Supplementary Fig. 8). The analysis revealed that the catalytic residues within the RuvC and Nuc domains are highly conserved among all subtypes, reflecting their critical roles in enzymatic function. Specifically, the first catalytic aspartate in the triad resides within the conserved motif IGIFRGEERN. The second catalytic glutamate displays subtype-specific distributions, appearing as MED in subtypes I, IV, V, and VI, as M/LEN/D in subtype II, and as MEK/D in subtype VIII. The third catalytic aspartate is consistently located in the motif DADANG, specifically at the second “D”. Additionally, a highly conserved TSKIDP motif was identified across all subtypes, indicating a shared functional mechanism. Other conserved motifs showed variability among subtypes, suggesting distinct sequence characteristics while maintaining overall catalytic and structural integrity. We also built the structure models of 300 Cas12a proteins using AlphaFold2, except for the failed construction, and calculated the root mean square fluctuation (RMSF) for all candidate Cas12a proteins within one subtype (Supplementary Fig. 9). The detailed analyses are appended in the Supplementary Notes. The RMSF reflects the residue-wise structural difference within one subtype. The results suggested that, despite an overall conserved structural architecture, specific regions within the proteins exhibit variability that may reflect structural adaptations specific to each subtype.
Cas12a proteins have distinct cis- and trans-cleavage activities
Cas12a processes the pre-crRNA transcripts into mature crRNA by its endoribonuclease activity. Then the Cas12a–crRNA complex efficiently cis-cleaves a double-stranded DNA (dsDNA), which is initiated by a PAM motif recognition. The cleaved DNA segment that remains bound then induces non-specific degradation of single-strand DNA (ssDNA) (Fig. 4a).

a Scheme of Cas12a activation, cis-, and trans-cleavage. The Cas12a from different subtypes was labeled with different colors. b Binding of Cas12a with crRNAs investigated by electrophoretic mobility shift assay (EMSA). c Binding of Cas12a with DNAs investigated by EMSA. d Scheme of PAM analyses using a double-strand DNA (dsDNA) array. Normalized PAM heatmaps for EvCas12_2 (e), AmCas11a (f), RspCas12a_2 (g), CAGCas12a (h), and RbrCas12a_1 (i). Each heatmap was normalized from 6 genes, including endogenous genes EMX1, DNMT1, and FANCF, 2 sites from eGFP, and 1 site from MERS virus genes. The individual maps were shown in Supplementary Fig. 12. The DNA sequences were listed in Supplementary Table 8. The weblogs of the PAM sequences for each Cas12a variant are shown below the heatmap. Colors within the heatmap range from dark blue to white, illustrating the normalized intensity of each PAM sequence. Source data are provided as a Source Data file.
Therefore, we evaluated the RNA binding efficiency, DNA binding efficiency, cis- and trans-acting DNase activities of sixteen Cas12a proteins from eight subtypes derive from Anaeroglobus micronuciformis (AmCas12a), Eubacterium_G ventriosum (EvCas12a_1 and EvCas12a_2), Erysipelatoclostridium sp. (EspCas12a), Ruminococcus_E sp. (RspCas12a_1 and RspCas12a_2), Agathobacter rectale (ArCas12a), Lachnospira eligens (LeCas12a_1 and LeCas12a_2), UBA3388 sp. (UBACas12a), RC9 sp. (RCCas12a), CAG-127 sp. (CAGCas12a), Ruminococcus_E bromii_B (RbrCas12a_1, RbrCas12a_2, RbrCas12a_3 and RbrCas12a_4) (Fig. 4, Supplementary Fig. 10 and Supplementary Table 6). Remarkably, the direct repeat sequence of these candidate Cas12a proteins is conserved alongside their celebrated counterparts, i.e., LbCas12a (Fig. 2g and Supplementary Fig. 11). Therefore, we chose LbCas12a as the positive control in the following assays, as well as its crRNA scaffold in the screening step. All the Cas12a proteins show RNA and DNA binding ability as expected (Fig. 4b, c, Supplementary Fig. 10c, d, and Supplementary Table 7). However, the DNA binding ability of subtype I and subtype VIII are higher than other Cas12a proteins. According to the inherent trans-DNase activity of Cas12a, as well as the 4 bp PAM length, we developed a simple and efficient PAM detection method. We constructed 6 short dsDNA target arrays by annealing 256 kinds of PAM sequence primer pairs in each well, which target EMX1 site1, DNMT1 site1, FANCF site1, MERS site1, eGFP site1, and eGFP site 3 (Supplementary Table 8). Each dsDNA target was incubated with candidate Cas12a proteins, crRNA and FAM-BHQ reporter to detect fluorescence of each reaction system (Fig. 4d). Using this assay, we determined the PAM preference of EvCas12a_2, AmCas12a, RspCas12a_2, CAGCas12a and RbrCAS12a_1, EcCas12_2, RspCas12a_2, and CAGCas12a recognize T rich PAM, but AmCas12a prefer G-start PAM, RbrCas12a_1 recognize 5-GTV-3 PAM (Fig. 4e–i and Supplementary Figs. 11, 12).
To corroborate the cis-acting DNase activity of candidate Cas12a proteins, we incubated Cas12a proteins with a crRNA and a linearized plasmid dsDNA. All linearized dsDNA were degraded by candidate Cas12a proteins with comparable efficiency to LbCas12a at 37 °C, with the exception of RCCas12a (Fig. 5a and Supplementary Fig 13a). Sanger sequencing of the cleaved DNA ends revealed that AmCas12a introduced INDELs at 18 in NTS and 23 in TS, consistent with other Cas12a orthologs (Supplementary Fig. 13e, f). However, most Cas12a variants exhibited diminished DNase activity, resulting in the production of uncleaved DNA at room temperature (RT), except for subtype VIII Cas12a proteins, which lack integrases. (Fig. 5b and Supplementary Fig. 13b). Subtype II Cas12a variants are slightly less active than LbCas12a in single-strand (ssDNA) degradation, while EspCas12a, EvCas12a_1, EvCas12a_2, and ArCas12a exhibited moderate activity. In contrast, the other Cas12a variants displayed notably lower activity (Fig. 5c and Supplementary Fig. 13c). Most of these Cas12a proteins represent considerable cis cleavage activity but are a bit different in trans-cleavage activity compared to LbCas12a. The ion preference assay reveals that these Cas12a proteins can be activated by Mn2+, similar to the LbCas12a34. Divalent Mg ions prove ineffective in activating the trans ssDNA cleavage activity of low-activity Cas12a variants, and Mn2+ cation emerges as the catalyst for their trans DNase activity. (Fig. 5d and Supplementary Figs. 13d and 14) To investigate the genome-editing ability of candidate Cas12a in eukaryotic cells, we selected 6 target sites with canonical PAM, which can be recognized by all the tested Cas12a (Fig. 5e and Supplementary Table 9). AmCas12a exhibits an average editing efficiency of 49.6% across six sites, with remarkable peaks at sites 3 (85.4%) and 6 (84.9%). In contrast, EvCas12a_2 displays an average editing efficiency of 20.3%, with its highest performance observed at site 1 (25.8%). RspCas12a_2 and RbrCas12a_2, which lack integrase in the loci, yield modest average editing efficiencies of 14.3% and 17.8%, respectively, with notable peaks at site 3 (26.3% and 37.3%, respectively). ArCas12a shows comparable average editing efficiencies with AmCas12a (45.4%), which gets notable peaks at site 3 (75.8%). LeCas12a_1 shows an average editing efficiency of 6.2% and a maximum efficiency of 25.7% at site 2. UBACas12a exhibits nearly negligible editing efficiency, with the highest activity reaching 2.1%. At site 4, CAGCas12a and LeCas12a_2 demonstrate peak genome-editing efficacy, at 81.7% and 73.8%, respectively, with mean editing efficiencies of 28.8% and 26%. AsCpf1 attains an impressive average editing efficiency of 65.5%, with its maximum at site 6 (84.7%). Finally, LbCas12a shows an average editing efficiency of 25.6% and a maximum efficacy of 53.5% at site 6.

a, b Cleavage of dsDNA by Cas12a subtypes at 37 °C (a) and 25 °C (b). c Trans-cleavage of ssDNA by Cas12 subtypes using fluorescence-labeled ssDNA reporter. d Divalent cation ions' preference for the Cas12a variants. Colors within the heatmap, ranging from dark blue to white, indicated the trans-cleavage activity from high to low. Time-course kinetic analyses were analyzed in the Supplementary Fig. 14. e Cellular gene editing efficiency on targeting sites. Two sites were selected from FANCF, EMX1, and DNMT1, respectively. The statistical significance was calculated using the LbCas12a as a reference at each site. The detailed sequences were listed in Supplementary Table 9. Error bar indicates mean ± s.e.m. measured from three technical replicates. n = 3. Statistical significance was assessed using a two-tailed unpaired t-test. Source data are provided as a Source Data file.
The AmCas12a–crRNA binary complex
The protein sequence identity of 16 candidate Cas12a proteins to AsCas12a, FnCas12a, and LbCas12a are low, ranging from 30%-46% (Fig.6a and Supplementary Fig. 15). In the three-dimensional structural landscape, Cas12a proteins within the same subclade exhibit a high degree of structural similarity. However, AmCas12a presents a subtle deviation, distinguishing itself somewhat from its subclade I Cas12a counterparts (Fig. 6d, f and Supplementary Fig. 15).

a Domain organization of the AmCas12a protein. Detailed protein sequences and alignments were supplemented by Supplementary Fig. 19. The REC1, REC2, PI, WED, BH, RuvC, and Nuc domains were highlighted with distinct colors, respectively. b The cartoon representation of the structure of the AmCas12a–crRNA and schematic of the crRNA used for structural analysis. The nucleotides of crRNA are labeled with numbers. c The structure of AmCas12 revealed by cryoEM. (PDB: 8KGF, EMDB: EMD-37219) The structure alignments comparison with known Cas12a and other variants was analyzed in Supplementary Fig. 17. The structural domains were distinguished according to the color codes at the bottom. d The RMSD matrix of Cas12 structure models constructed by AlphaFold2. Colors within the heatmap from dark blue to white represent the RMSD values from high to low. e Interaction network of crRNA with residues in AmCas12a. The detailed interactions of crRNA seed regions with AmCas1a were shown in Supplementary Fig. 18. f The Alphafold2 structure models of Cas12as, which were used in this paper. g Mismatch analyses of AmCas12a. Error bar indicates mean ± s.e.m. measured from three technical replicates. n = 3. Source data are provided as a Source Data file.
To understand the molecular details underlying the RNA binding behavior of AmCas12a, we achieved the cryo-EM map of the crRNA binding complex, which consists of AmCas12a and a 44-nt crRNA, at 2.9 Å resolution (Fig. 6b, c, Supplementary Figs. 16 and 17, and Supplementary Table 10). The AmCas12a–crRNA structure maintains a bilobed architecture (Fig. 6c), similar to other Cas12a structures35,36. Nonetheless, it is noteworthy that the AmCas12a–crRNA complex exhibits a distinct conformation when juxtaposed with its counterparts. Specifically, an observable rotational variance is discernible within the REC domain of AmCas12a when compared to the LbCas12a–crRNA and FnCas12a–crRNA complexes. Relative to LbCas12a and FnCas12a, the REC1 domain of AmCas12a presents a deviation of 7.3° and 9.4°, respectively. Simultaneously, the REC2 domain of AmCas12a manifests a rotational disparity of 4.8° and 6.2°, respectively (Supplementary Fig. 17d, e).
As observed in the LbCas12a and FnCas12a crRNA binary structures, the repeat-derived pseudoknot in the 5’ handle of the crRNA is ordered. However, the crRNA conformation is markedly different from that of the crRNA bound by LbCas12a or FnCas12a. Due to the flexibility of the spacer-derived part of crRNA, it’s almost unclear in the Cas12a–crRNA binary complex35,36. Notably, an extra RNA stem formed by A(1)–A(5) and U(18)–U(22) within the crRNA spacer region makes a part of spacer region including seed sequence well-defined in the central cavity of AmCas12a and adopt an A-form-like helical conformation, but A(−10)–G(−6) and G(6)–A(15) nucleotides of crRNA are unclear (Fig. 6b and Supplementary Fig. 18). To accommodate the double RNA stem substrate, the REC lobe of AmCas12a rotates away from the NUC lobe. Unsurprisingly, the docking of crRNA to Alphafold-generated AmCas12a causes a severe clash in the REC domain (Supplementary Fig. 15c). The attainment of conformational integrity within the extra RNA stem is orchestrated by intricate interplays involving the ribose and phosphate moieties of the crRNA backbone, engaging in multiple interactions with specific residues within the WED, REC1, and RuvC domains of AmCas12a (Fig. 6e). These include residues T19, H751, K522, and H861 from the WED domain, Y50 and R168 from the REC1 domain and Q1003 from the RuvC domain, all of which are conserved with Cas12a orthologs, except Q1003 which form a hydrogen bond with the phosphate of U(18) (Supplementary Fig. 18). Distinct from the FnCas12–crRNA complex, the spacer segment of crRNA major interacts with the WED domain of AmCas12a.
Compared to the LbCas12a–crRNA complex and FnCas12a–crRNA complex, the divalent Mg ions are in the same location (Supplementary Fig. 17a–c). Consistent with a seed sequence-dependent mechanism of DNA targeting and in broad agreement with previous analyses of AsCas12a, LbCas12a activities in vivo, and FnCas12a activities in vitro35,37,38, cleavage of DNA substrates with single-nucleotide mismatches in the seed segment was almost completely impaired, while mismatches in the PAM-distal region of the DNA target were mostly tolerated (Fig. 6g).
Specific detection of single-nucleotide mutation by AmCas12a
Cas12a is a promising tool in the next-generation molecule diagnosis, however, it suffers from the PAM limitation39. The oncogene SNP only has a small sequence window to probe, the traditional PAM, TTTV, could not cover all the SNPs. Therefore, we tested whether the AmCas12a can distinguish the SNPs without a traditional PAM. (Fig. 7a) The oncogene mutants, KRAS c.34 G > T (G12C), did not contain the available TTTV in the adjacent sequences (Fig. 7b). Among the Cas12a proteins that have undergone PAM preference testing, AmCas12a, EvCas12a_2, CAGGCas12a, and RbrCas12a_1 showed potential for recognizing the G12C mutation. The results revealed that AmCas12a exhibited the best performance (Supplementary Fig. 20). We designed the crRNA targeting the SNP (Fig. 7b). According to the fluorescence intensity, we selected the crRNAs inducing the strongest signals, i.e., crRNA 1 for the KRAS mutant (Fig. 7c). The AmCas12a can detect ten copies of the KRAS mutant (Fig. 7d). Furthermore, we diluted the target mutant and evaluated the sensitivity of detection. The AmCas12a can even distinguish 0.1% KRAS mutant in the wild-type gene background, which is more sensitive than the Sanger sequencing (Fig. 7e, f).

a Scheme of single-nucleotide mutant detection by Cas12a. b Synthetic crRNA for single-nucleotide KRAS mutation based on the PAM preference of AmCas12a. The single-nucleotide polymorphism (SNP) site was highlighted in red. c AmCas12a detection of KRAS G12C with various crRNAs and Mn2+. d Detection limit of KRAS mutant using recombinase polymerase amplification (RPA) integrated with Cas12a. The fluorescent images and fluorescence intensity of the 15-min reaction were shown. The copy numbers of the target DNA were shown on the x-axis. e Sensitivity of the AmCas12a detection. KRAS mutant DNA was spiked in the wild type sequences with various ratios, which were shown in the x-axis. f Sanger sequencing results of wild-type KRAS and mutant with different ratios. NC represented the negative control without target DNA. Error bar indicates mean ± s.e.m. measured from three technical replicates. n = 3. Statistical significance was assessed using a two-tailed unpaired t-test. Source data are provided as a Source Data file.
Discussion
CRISPR-Cas system keeps evolving during the arms race between bacteria and phages. The proteins gain or lose function via mutation or domain reorganization, accordingly, the accessory proteins such as integrases Cas1, Cas2, and Cas4 change in the CRISPR loci. Therefore, tracing the Cas protein sequences in the CRISPR loci provides a practical strategy to decipher the evolution of the CRISPR-Cas system. However, this process is complex due to the diversity and the huge data from metagenomics. In this work, the AIL-Scan model leveraged the capability of ESM-2 in residue resolution prediction for the CRISPR-Cas identification. The 15 billion model shows superior prediction capability for all types of CRISPR-Cas in comparison with the 650 million model and other machine learning software. (Table 1) The small Cas proteins, e.g., Cas1, Cas2, Cas3, Cas4, Cas5, and Cas8, are difficult to predict, as shown by the relatively low prediction accuracy (Table 1), because the short sequences provide too limited information to accurately predict. The highly accurate model can extract more precise information from short sequences. When we increase the size of the model, we observe a significant increase in the prediction accuracy for the small Cas proteins. The superior capability of the large model also contributed to the precise prediction of the non-Cas proteins (Table 1). However, considering resource consumption, the 650 M model can practically handle the Cas protein classification.
The interpretability of the large language model is crucial for understanding the principles of prediction and the underlying biological mechanisms. However, the vectors produced by ESM-2 are abstract and highly processed, which limits their interpretability. To address this, we explored the interpretability of the model through the attention mechanisms in ESM. Inspired by studies of protein structure, we successfully extracted attention scores of LbCas12a from the 20 heads of the ESM-2 model and analyzed their correlation with structural domains (Supplementary Table 11). The results showed that attention in the cleavage domains was 2- to 24-fold higher than in the non-cleavage domains across all heads. This indicates that the model primarily focused attention on the cleavage domains of Cas12a, which are critical for protein function. The analyses of AsCas12a and SpCas9 revealed similar results (Supplementary Table 11), indicating that the cleavage domains are the key to distinguishing different Cas proteins. Furthermore, we observed significant correlated attention between cleavage and cleavage domains, which indicates the structural attention between domains in the ESM-2 (Supplementary Fig. 21). These results demonstrated that the ESM-2 model concentrated attention on the cleavage domains, emphasizing their importance in protein function and underscoring the interpretability of the model’s predictions.
However, this model still has certain limitations, including dependence on training data, high computational resource requirements, limited ability to capture complex biophysical properties and rare features (e.g., trans-cleavage activity), and sensitivity to sequence variations in specific protein families. Additionally, the model’s interpretability, efficiency, and the management of imbalanced data still require improvement. Nevertheless, this model has laid an important foundation for advancing protein characterization and structure prediction. Therefore, it is both necessary and reasonable to extend the model’s ability to capture specific sequences based on large models, such as our work on trans-cleavage activity prediction. This model could serve as a platform for further development and extension.
Given the absence of universal cas genes and the frequent modular recombination, CRISPR-Cas classification requires multipronged parameters, including the signature cas genes, sequence similarity between the shared Cas proteins, and the organization of the genes in the CRISPR-Cas loci40,41. The Cas12a proteins are well-characterized Cas proteins, normally containing three integrases, i.e., Cas1, Cas2, and Cas4. Paradoxically, we discovered 7 unreported subtypes with distinct integrase combinations in the metagenomic data (Fig. 2). We aligned the Cas12a protein sequences and integrated them with the information of subtypes and species. Remarkably, even in the same bacterial family, such as Lachnospiraccae and Acutalibacteraccae, the variants are diverse. The Cas12a proteins in the Lachnospireaccae family share high similarities, but the CRISPR-loci are mainly dominated by subtype I (40.7%) and followed by subtype VI (35.9%). Interestingly, most CRISPR-Cas12a proteins in the Acutalibacteraccae family do not contain any integrases, only 11.8% belong to subtype 2. Cas1, Cas2, and Cas4 are located downstream of Cas12 in a tandem pattern. The Cas1–Cas2 complex is necessary for the site-selective CRISPR array expansion during the initial step of bacterial adaptive immunity26. Cas4 is an endonuclease that defines the PAM and assists in the insertion of the spacer into the CRISPR array unidirectionally25,42. Many CRISPR-Cas systems lack Cas4, and some hosts use alternative exonucleases to acquire new CRISPR immune sequences23. Alternatively, parts of hosts encode a solo-Cas4 outside the CRISPR-Cas loci43, but due to the incompleteness of the metagenomics, we could not exclude this possibility in the current study. In the evolution system, loss-of-function in certain components will drive the gain-of-function in other components to keep the robustness of the whole system. Integrase-deficient Cas12a variants can all achieve dsDNA degradation, (Fig. 5a, b and Supplementary Fig. 13a, b). However, it remains unclear whether the absence of either ancillary integrase necessitates alternative genes in the genome, collaboration with integrases from other CRISPR-Cas systems, loss of function in immunological memory acquisition, or gain of function in Cas12a proteins. Notably, the diverse CRISPR-Cas variants within the same family highlight the intricacies of evolution, warranting further studies in the future.
The Cas12 variants showed distinct biochemical properties. They can bind to the crRNA and DNA but with different affinities (Fig. 4b, c). All the Cas12a variants cleave the dsDNA in a temperature-dependent manner. Divalent metal ions play key roles in the cleavage and conformational rearrangements of CRISPR–Cas12a44. Despite Mg2+ ions, Mn2+ is able to activate some Cas12a34,45. EvCas12a_2, AmCas12a, RspCas12a_2, RbrCas12a_1, and CAGCas12a prefer Mn2+ in the ssDNA cleavage. In addition, Co2+ can activate RspCas12a_2 better than Mg2+ (Fig. 5d). The ssDNA cleavage by Cas12a has been successfully applied in nucleic acid detection46,47. Nevertheless, Cas12a proteins are restrained by the PAM sequence48, therefore, Cas12a with different PAM preferences are required. In our work, we have systematically evaluated the PAM preference of the Cas12a proteins (Fig. 4). We found that AmCas12a recognizes a broader PAM. We took the SNP of the oncogene KRAS as the target, which can not be detected by the traditional Cas12a due to the lack of PAM sequences near the mutation site. After optimization, the AmCas12a can specifically distinguish the KRAS G12C mutations, but LbCas12a can not. These findings extend the toolbox of Cas12a detection. Although the Cas proteins demonstrate great potential in biological and medical applications, the potential misuse of CRISPR technology poses significant ethical, security, and ecological challenges. In addition, the risks of off-target effects and unintended genetic changes complicate the ethical considerations, particularly regarding human germline editing. Ensuring proper use of CRISPR requires more precise gene editing tools, stringent regulatory frameworks, and ethical guidelines.
The structure of crRNA is key to the conformational change of Cas12a. The stem-loop region and seed region are stable, but the spacer region in an apo form is largely elusive. Our cryo-EM structure discovered an undocumented folding of the crRNA spacer region, which forms a stem (Fig. 6b). This structure demonstrates the structural tolerance of the AmCas12a for crRNA flexibility. In the crRNA binary complex, Cas12a interacts with the pseudoknot of crRNA with conserved residues. However, due to the extra RNA stem, AmCas12a interacts with the spacer region by the WED domain instead of the REC1 and WED domains.
In summary, we developed an artificial intelligence language model for Cas protein prediction. The increasing parameters in large data models can enhance the accuracy of predicting Cas proteins, especially for short protein sequences. This feature contributes to the undocumented CRISPR loci discovery and analyses of Cas protein evolution. Importantly, some Cas12 proteins have broader PAM recognition patterns and can be developed into efficient genome editors in mammalian cells or specific SNP detection kits. These findings will substantially increase the diversity of CRISPR-Cas12a systems and largely expand the programmable DNA-editing toolbox. This study shows the great potential of the language model with tremendous parameters in protein function exploration. Our study provides new insights into machine learning on the natural evolution of the CRISPR system, and a detailed characterization will discover more valuable gene editing tools.
Methods
Training data for language models
We generated sequences from NCBI databases to train ESM-2 650 million and 15 billion language models. We first used the keyword ‘CRISPR-associated protein’ to download all the gene IDs and then analyzed the annotation ‘gene’ with ‘cas’. We further removed redundant sequences. We finally collected 13047 non-Cas proteins and 76567 Cas protein sequences, including 11248 Cas1, 15148 Cas2, 12309 Cas3, 7708 Cas4, 8656 Cas5, 11330 Cas6, 340 Cas7, 299 Cas8, 6706 Cas9, 2282 Cas10, 334 Cas12, and 207 Cas13. We splited these sequences into two datasets, 80% as training data and 20% as validation data. The preparation details are listed in the Supplementary Notes.
Training ESM language models
We performed fine-tuning on the open-source sequence classifier provided by ESM to adapt it to our application. The model consists of two fully connected layers, which are specifically designed for classification tasks. In the first layer of the model, the fully connected layer applies a linear transformation to the output features of the ESM model, mapping them to the same dimensional space, followed by a hyperbolic tangent (Tanh) activation function to introduce non-linearity. The second fully connected layer then projects the processed features to the dimension of target classes. This model structure effectively combines the excellent feature extraction ability of ESM and the efficient classification performance of fully connected neural networks, achieving effective classification of sequences. This model design maintains the high-dimensional sequence features while effectively learning the classification task.
To further improve training efficiency and model performance, we employ the Accelerate49 and Deepspeed50 training acceleration framework, particularly its ZeRO Stage51 offload feature, to optimize memory utilization and accelerate the training process. AdamW is employed as the optimizer for its weight decay and momentum feature to enhance the training stability and efficiency. Meanwhile, WarmupLR is adopted as the learning rate scheduler, which gradually increases the learning rate in the early stages of training to facilitate model convergence. FocalLoss is used as the training loss function, which adjusts the weights of positive and negative samples to mitigate the class imbalance problem. The details were described in the supplementary methods. The hyperparameter ‘α‘ of FocalLoss is determined by considering the ratio of class sizes in the Cas data. We have tried different learning rates and batch size combinations, i.e., learning rate 0.00001 with batch size 64, learning rate 0.001 with batch size 32, and learning rate 0.001 with batch size 64, separately. The best parameter is a learning rate of 0.00001 with a batch size of 64. We trained multiple epochs and chose the best one for the final prediction (Supplementary Fig. 22). The model architecture and training are implemented in PyTorch, ensuring code modernity and efficiency. The training was conducted on the Zhejiang Lab Alkaid Intelligent Computing Operating System using NVIDIA Volta A100 GPUs for the 15B model and V100 for the 650 M model.
Protein expression and purification
The candidate discovered Cas12a was expressed and purified as previously described52. In brief, the coding sequences of Cas proteins were codon-optimized and synthesized by Tsingke Biotech (China) and then cloned into pET28a (Novagen) with a C-terminal 10× His tag. The pET28a-Cas12a plasmid was transformed into E. coli Rosetta and induced with 0.2 mM IPTG for 16 h at 18 °C before cell harvesting. After cell pellet lysis, the Cas12a protein was purified using a Ni-NTA resin column and a Heparin Sepharose column according to the manufacturer’s instructions (GE Healthcare). Then the purified Cas12a protein was concentrated in storage buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol, 2 mM DTT), quantified using the absorption at 280 nm, and frozen at −80 °C until use.
Nucleic acid preparation
The double-stranded DNA fragment of Cas12a variants was synthesized by Tsingke Biotech (China) and cloned into the pUC57 vector with a T7 primer. The crRNAs were synthesized by GenScript (Nanjing, China), and sequences are listed in Supplementary Table 7.
Cas12a-mediated nucleic acid detection
The detection assays were performed according to previously reported methods with minor modifications52. In a 20 μl detection assay, with 200 ng Cas12a protein, 25 pM ssDNA FQ reporter, 50 nM crRNA, and 10 ng of target dsDNA in a reaction buffer (100 mM NaCl, 50 mM Tris-HCl, 100 µg/mL BSA, pH 7.9) supplied with 10 mM MgCl2 or MnSO4, incubate at 37 °C until detection. A PerkinElmer EnSpire reader with excitation at 485 nm and emission at 520 nm was used for fluorescence detection. For the divalent ion preference screen. The metal ion preference assay was performed as previously described34. In brief, the CRISPR-Cas12 detection assay was supplemented with 10 mM CaCl2, CoCl2, CuSO4, NiSO4, MgSO4, MnSO4, or ZnSO4.
In vitro RNA and DNA binding assays
For RNA binding assays, Cas12a (100 nM) was incubated with Cy3-DNA (10 nM) at room temperature for 10 min in the reaction buffer. The reaction was quenched with glycerol loading buffer (10 mM Tris-HCl,pH 8.0, 10% glycerol). Reaction products were resolved by 12% PAGE and visualized by Typhoon FLA 9500 (GE Health Care).
For DNA binding assays, Cas12a protein was first complexed with crRNA at a 1:2 ratio at room temperature for 10 min in the reaction buffer. Cas12a complex (100 nM) was incubated with annealed FAM-DNA (25 nM) for 10 min at room temperature. The reaction was quenched with glycerol loading buffer (10 mM Tris-HCl,pH 8.0, 10% glycerol). Reaction products were resolved by 12% PAGE and visualized by Typhoon FLA 9500 (GE Health Care).
PAM preference assay
The six short dsDNA target arrays were constructed by annealing 256 types of PAM sequence primer pairs in each well, which target EMX1 site1, DNMT1 site1, FANCF site1, MERS site1, eGFP site1 and eGFP site 3 (Supplementary Table 8), Next, same as nucleic acid detection, Cas12a protein (200 ng), ssDNA FQ reporter (25 pM), crRNA (50 nM) and short target dsDNA (8.5 nM) were mixed in a reaction buffer supplied with 10 mM MgCl2 (EvCas12a_2 and RspCas12a_2) or MnSO4 (AmCas12a, CAGCas12a and RbrCas12a_1), and incubated at 37 °C, ViiA 7 Real-Time PCR system were used for fluorescence tracing. In each detection plate, triple repeats of dsDNA with TTTG PAM are used as the control for intensity normalization.
Phylogenetic analysis
The phylogenetic tree of Fig. 2a was constructed from a dataset of 87 sequences, including 30 Cas9 proteins, 43 Cas12 proteins, and 14 Cas13 proteins. The tree of Fig. 2h was constructed by 300 Cas12a variants, as well as FnCas12a, LbCas12a, and AsCas12a. The sequences were aligned with MAFFT-linsi (v7.480)53. A phylogenetic tree was constructed by FastTree54 with default parameters. The phylogenetic tree is annotated by iTol55.
Targeted deep sequencing
HEK293T cells were from Cell Bank/Stem Cell Bank, Chinese Academy of Sciences, and cultured in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% fetal calf serum (v/v) (Gemini) and 1% penicillin–streptomycin at 37 °C with 5% CO2. For plasmid transfection, cells were in 24-well plates in three biological replicates and transfected with 1.2 μg plasmids (including 900 ng editor and 300 ng sgRNA) per well, when cells reached an approximate 70-90% confluency. Transfections were carried out with the aid of EZ Trans (Life-iLab; Cat. No.: AC04L091) reagent and according to the manufacturer’s protocols. Three days after transfection, cells were harvested for deep sequencing. Target sites were amplified from extracted genomic DNA using Phanta® Max Super-Fidelity DNA Polymerase (Vazyme). PCR products with different barcodes were pooled together for deep sequencing on the Illumina HiSeq X Ten platform (2× 150 PE) by Annoroad Gene Technology (Beijing, China). Different experimental conditions were differentiated by bar codes, and experimental repetitions were included in different pools. Sequencing reads were demultiplexed using AdapterRemoval (version 2.2.2), and the pair-end reads with 11 bp or more alignments were combined into a single consensus read. All processed reads were then mapped to the target sequences using the BWA-MEM algorithm (BWA v0.7.16). Indel frequency was calculated as: the number of indel-containing reads/total mapped reads. The targets and primers used in this study are provided in Supplementary Tables 9 and 12.
Reconstruction of AmCas12a–crRNA complex
AmCas12a was expressed and purified as described above, but further purified by size exclusion column (Superdex 200 Increase 10/300, GE Healthcare) in SEC buffer 1 (10 mM Tris-HCl, pH 7.5, 500 mM NaCl) for complex preparation. The sgRNA was diluted to 100 μM in refolding buffer (50 mM KCl, 5 mM MgCl2) and refolded at 72 °C for 5 min. The AmCas12a–crRNA binary was reconstituted by incubating 25 μM AmCas12a and 30 μM crRNA for 30 min at room temperature in a total volume of 450 μl assembly buffer (10 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM MgCl2). Subsequently, the mixture was purified by size exclusion column in SEC buffer 2 (10 mM Tris-HCl, pH 7.5, 500 mM NaCl, 1 mM MgCl2). The purified aliquots were concentrated to 2 mg/mL, flash frozen, and stocked at −80 °C.
Cryo-EM sample preparation and data collection
Sample vitrification was performed using a Vitrobot Mark IV (Thermo Fisher) operating at 4 °C and 100% humidity. A 4 μl sample was applied to a holey amorphous nickel–titanium alloy foil (ANTA foil 1.2/1.3) that had been glow-discharged for 30 s. The grids were blotted for 4 s at a ‘blot force’ of −2 by standard Vitrobot filter paper (Ted Pella) and were then plunge-frozen in liquid ethane. Cryo-EM data were collected on a Titan Krios electron microscope operated at 300 kV equipped with and Falcon4 direct electron detector with a Quantum energy filter using EPU. Micrographs were recorded in counting mode at a nominal magnification of 165,000×, resulting in a physical pixel size of 0.74 Å per pixel. The defocus was set between −0.6 μm and −1.8 μm. The total exposure time of each movie stack led to a total accumulated dose of 46.73 electrons per Å2, which was fractionated into 32 frames. More parameters for data collection are shown in Supplementary information, Supplementary Table 10.
Image processing and 3D reconstruction
The raw dose-fractionated image stacks were 2× Fourier binned, aligned, dose-weighted, and summed using MotionCor256. CTF-estimation, blob particle picking, 2D reference-free classification, initial model generation, final 3D refinement, and local resolution estimation were performed in cryoSPARC57. The details of data processing were summarized in Supplementary information, Supplementary Fig. 16, and Supplementary Table 10.
Model building and refinement
The initial protein model was generated using AlphaFold2 and manually revised in UCSF-Chimera and Coot28,58,59. The crRNA was manually built in Coot based on the cryo-EM density. The complete model was refined against the EM map by PHENIX in real space with secondary structure and geometry restraints60. The final model was validated in the PHENIX software package. The structural validation details for the final model are summarized in Supplementary information, Supplementary Table 10.
RPA and fluorescence detection
The KRAS gene was cloned into the pcDNA3.1 vector using NheI and KpnI restriction sites, and the construct was subsequently used in RPA as a template. The RPA assay was performed with a GenDx ERA Kit (Suzhou GenDx Biotech, China). According to the instructions in the manual, the 50 µl RPA system contains 2 µl DNA template, 2.5 µl forward primer (10 µM), 2.5 µl reverse primer (10 µM), 10 µl ERA basic buffer, 20 µl reaction buffer, 2 µl activator, and supplementary ddH2O. Three microliters of the RPA reaction product were transferred to the Cas12a reaction. In a 20 μl Cas12a reaction, additional with 100 ng Cas12a protein, 25 pM ssDNA FQ reporter, 50 nM crRNA in a reaction buffer (100 mM NaCl, 50 mM Tris-HCl, 100 µg/mL BSA, pH 7.9) supplied with 2.5 mM MnSO4, incubate at 37 °C until detection. A PerkinElmer EnSpire reader with excitation at 485 nm and emission at 520 nm was used for fluorescence detection.
Statistics and reproducibility
All values in the text and figures are presented as mean ± SEM of independent experiments with given n sizes. For image analysis, images were collected from at least three independent experiments. Graphs were compiled, and statistical analyses were performed with Prism software (GraphPad) and Excel. Statistical significance was evaluated with the two-tailed unpaired t-test when comparing two groups. Differences between more than two samples were calculated using a one-way analysis of variance (ANOVA). Statistical details, including sample sizes (n), are indicated in the figures and legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All source data are provided with this paper in the Source Data file. The deep sequencing data generated in this study have been deposited in the NCBI database under the accession code PRJNA1043844. The structural data generated in this study have been deposited in the RCSB Protein Data Bank under the accession number 8KGF (PDB) and Electron Microscopy Data Bank under the accession number EMD-37219 (EMDB). Source data are provided with this paper.
Code availability
The code was deposited in GitHub (https://github.com/LUCA-BioTech/AIL-scan/) with the (https://doi.org/10.5281/zenodo.15710365). The source code is available under the Apache License 2.0.
References
Hille, F. et al. The biology of CRISPR-Cas: backward and forward. Cell 172, 1239–1259 (2018).
Makarova, K. S. et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83 (2020).
Pickar-Oliver, A. & Gersbach, C. A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507 (2019).
Al-Shayeb, B. et al. Clades of huge phages from across Earth’s ecosystems. Nature 578, 425–431 (2020).
Devoto, A. E. et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 4, 693–700 (2019).
Pausch, P. et al. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science 369, 333–337 (2020).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Eddy, S. R. Profile hidden Markov models. Bioinformatics 14, 755–763 (1998).
Chai, G., Yu, M., Jiang, L., Duan, Y. & Huang, J. HMMCAS: a web tool for the identification and domain annotations of CAS proteins. IEEE/ACM Trans. Comput. Biol. Bioinform 16, 1313–1315 (2019).
Abby, S. S., Néron, B., Ménager, H., Touchon, M. & Rocha, E. P. MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems. PLoS One 9, e110726 (2014).
Padilha, V. A. et al. Casboundary: automated definition of integral Cas cassettes. Bioinformatics 37, 1352–1359 (2021).
Yang, S., Huang, J. & He, B. CASPredict: a web service for identifying Cas proteins. PeerJ 9, e11887 (2021).
Russel, J., Pinilla-Redondo, R., Mayo-Muñoz, D., Shah, S. A. & Sørensen, S. J. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas loci. Crispr j. 3, 462–469 (2020).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl. Acad Sci USA. https://doi.org/10.1073/pnas.2016239118 (2021).
Chowdhury, R. et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 40, 1617–1623 (2022).
Elnaggar, A. et al. ProtTrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127 (2022).
Huang, J. et al. Discovery of deaminase functions by structure-based protein clustering. Cell 186, 3182–3195.e3114 (2023).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Tang, J., Ma, P. & Li, Z. AIL-scan: nature communications. Zenodo https://doi.org/10.5281/zenodo.15710365 (2025).
Tordoff, J. et al. Initial characterization of 12 new subtypes and variants of type V CRISPR systems. Crispr j. 8, 149–154 (2025).
Coelho, L. P. et al. Towards the biogeography of prokaryotic genes. Nature 601, 252–256 (2022).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11, 119 (2010).
Wang, J. Y. et al. Genome expansion by a CRISPR trimmer-integrase. Nature 618, 855–861 (2023).
Dhingra, Y., Suresh, S. K., Juneja, P. & Sashital, D. G. PAM binding ensures orientational integration during Cas4–Cas1–Cas2-mediated CRISPR adaptation. Mol. Cell 82, 4353–4367.e4356 (2022).
Hu, C. et al. Mechanism for Cas4-assisted directional spacer acquisition in CRISPR-Cas. Nature 598, 515–520 (2021).
Wright, A. V. et al. Structures of the CRISPR genome integration complex. Science 357, 1113–1118 (2017).
Xiao, Y., Ng, S., Nam, K. H. & Ke, A. How type II CRISPR-Cas establish immunity through Cas1–Cas2-mediated spacer integration. Nature 550, 137–141 (2017).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Kim, T. Y., Shin, M., Huynh Thi Yen, L. & Kim, J. S. Crystal structure of Cas1 from Archaeoglobus fulgidus and characterization of its nucleolytic activity. Biochem. Biophys. Res. Commun. 441, 720–725 (2013).
Holm, L. Dali server: structural unification of protein families. Nucleic Acids Res. 50, W210–w215 (2022).
Nuñez, J. K. et al. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat. Struct. Mol. Biol. 21, 528–534 (2014).
Wang, J. et al. Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR-Cas systems. Cell 163, 840–853 (2015).
Tang, D. et al. A distinct structure of Cas1–Cas2 complex provides insights into the mechanism for the longer spacer acquisition in Pyrococcus furiosus. Int. J. Biol. Macromol. 183, 379–386 (2021).
Ma, P. et al. MeCas12a, a highly sensitive and specific system for COVID-19 detection. Adv. Sci. https://doi.org/10.1002/advs.202001300 (2020).
Swarts, D. C., van der Oost, J. & Jinek, M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233 (2017).
Dong, D. et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526 (2016).
Kim, D. et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 34, 863–868 (2016).
Kleinstiver, B. P. et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 34, 869–874 (2016).
Kim, H. K. et al. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat. Methods 14, 153–159 (2017).
Makarova, K. S. et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol 13, 722–736 (2015).
Koonin, E. V., Makarova, K. S. & Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78 (2017).
Shiimori, M., Garrett, S. C., Graveley, B. R. & Terns, M. P. Cas4 nucleases define the PAM, length, and orientation of DNA fragments integrated at CRISPR loci. Mol. Cell 70, 814–824.e816 (2018).
Hudaiberdiev, S. et al. Phylogenomics of Cas4 family nucleases. BMC Evol. Biol. 17, 232 (2017).
Son, H. et al. Mg(2+)-dependent conformational rearrangements of CRISPR-Cas12a R-loop complex are mandatory for complete double-stranded DNA cleavage. Proc. Natl. Acad. Sci. USA 118, e2113747118 (2021).
Sundaresan, R., Parameshwaran, H. P., Yogesha, S. D., Keilbarth, M. W. & Rajan, R. RNA-Independent DNA Cleavage Activities of Cas9 and Cas12a. Cell Rep. 21, 3728–3739 (2017).
Li, S. Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, 20 (2018).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Gugger, S. et al. Accelerate: Training and inference at scale made simple, efficient and adaptable. https://github.com/huggingface/accelerate (2022)
Rasley, J., Rajbhandari, S., Ruwase, O. & He, Y. In Proc the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining 3505–3506 (Association for Computing Machinery, Virtual Event, 2020).
Rajbhandari, S., Rasley, J., Ruwase, O. & He, Y. In Proc the International Conference for High Performance Computing, Networking, Storage and Analysis Article 20 (IEEE Press, Atlanta, Georgia, 2020).
Wang, X. et al. CRISPR/Cas12a technology combined with immunochromatographic strips for portable detection of African swine fever virus. Commun. Biol. 3, 62 (2020).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, W293–w296 (2021).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Acknowledgements
We thank the Molecular and Cell Biology Core Facility (MCBCF) and the Molecular Imaging Core Facility (MICF) at the School of Life Science and Technology, ShanghaiTech University, for providing technical support, Shuimu Biosciences for providing technical support on cryo-EM sample evaluation and screening, Dr. Yong-Xiang Gao at the Cryo-EM Center, University of Science and Technology of China, for technical support on Cryo-EM data collection of AmCas12a, the Research Center for Intelligent Computing Platforms at the Zhejiang lab, and Hangzhou LUCA Intelligent Technology for providing the computing resources. The study is supported by the National Key R&D Program of China (2022YFB4501500, 2022YFB4501504 Y.Y.), National Science Foundation of China (22177073 P.M., 32161133022 X.Z.), National Science and Technology Innovation 2030 Major Program (2022ZD0211905 X.Z.), Key R&D Program of Zhejiang (2024C01036 Y.Y.), the Shanghai Science and Technology Committee (23ZR1437600, 24141901302 P.M.), Shanghai Municipal Science and Technology Major Project (23HC1400700 Y.Q.), Key Research Program of Chinese Academy of Sciences (ZDBS-ZRKJZ-TLC008 X.H.), Emergency Key Program of Guangzhou Laboratory (EKPG21-18 X.H.), Key Research Project of Zhejiang Lab (2021PE0AC06 J.T.), Jiangsu Basic Research Center for Synthetic Biology (BK20233003 L.W.), and Shanghai Frontiers Science Center of Degeneration and Regeneration in Skeletal System. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Author information
Authors and Affiliations
Contributions
P.M., X.H., J.T., and X.Z. conceived and designed this project. Y.F., J.S. performed biochemical and cellular experiments. Z.L., Y.L., J.Y., Y.Y., Q.L., and J.T. performed the computational experiments. Y.Q., J.Z., and W.H. contributed to biochemical experiments and analysis. Y.F. and X.Z. performed the structural analyses. J.F.Z. and S.H. performed the bioinformatics analysis. Y.F., P.M., X.H., Q.L., L.W., and C.H. wrote and revised the manuscript with input from the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jin Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, Y., Shi, J., Li, Z. et al. Discovery of CRISPR-Cas12a clades using a large language model. Nat Commun 16, 7877 (2025). https://doi.org/10.1038/s41467-025-63160-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63160-4
