
Abstract
Conventional genome editing tools rely on DNA double-strand breaks (DSBs) and host recombination proteins to achieve large insertions, resulting in heterogeneous mixtures of undesirable outcomes. We recently leveraged a type I-F CRISPR-associated transposase, PseCAST, for DSB-free DNA integration in human cells, albeit at low efficiencies; multiple lines of evidence suggest DNA binding may be a bottleneck for higher efficiencies. Here we report structural determinants of DNA recognition by the PseCAST QCascade complex using single-particle cryogenic electron microscopy (cryoEM), revealing subtype-specific interactions and RNA-DNA heteroduplex features. By combining structural data, library screens, and rationally engineered mutants, we uncover variants with increased integration efficiencies and modified PAM stringencies. We further leverage transpososome structural predictions to build hybrid CASTs that combine orthogonal DNA binding and integration modules. Our work provides unique structural insights into type I-F CASTs and showcases diverse strategies to investigate and engineer RNA-guided transposase architectures for human genome editing applications.
Similar content being viewed by others

Introduction
Canonical CRISPR-Cas systems that have been leveraged for programmable gene editing, such as Cas9 nucleases, cause targeted DNA double-strand breaks (DSBs) that provoke the cell to activate DNA repair mechanisms1,2. Non-homologous end joining (NHEJ) is the most efficient repair pathway in human cells, which leads to indel mutations, and although homology-directed repair (HDR) offers the ability to generate precise modifications or insertions, it is inefficient in most cell types, inaccessible in non-dividing cells, and requires large homology arms for each new insertion site3,4. Furthermore, HDR efficiencies decrease drastically with insertion size, and aberrant editing pathways that occur at non-negligible frequencies can cause large chromosomal truncations and/or rearrangements5,6,7,8,9,10. Second generation editors, including base and prime editors, employ nickase-variant Cas proteins to bypass DSB intermediates, but indel byproducts still arise and edits are generally restricted to single-base pair (bp) changes or small insertions (<50 bp)11,12,13,14, thus failing to address the need for large DNA insertion technology. CRISPR-associated transposases (CASTs), on the other hand, leverage a CRISPR-associated DNA targeting module and a transposase effector module that allow for highly specific and programmable insertions, which are both DSB-free and multi-kilobases in size15,16,17.
To date, four CAST subtypes have been characterized in bacteria: type I-B, I-D, I-F, and V-K15,16,18,19. These subtypes encode unique architectures for both the targeting and integration steps of the transposition pathway: type I CASTs rely on TnsABC proteins for integration and a multi-subunit complex for DNA targeting that includes TniQ and Cascade components (TniQ-Cascade, hereafter simply QCascade), with Cascade itself comprising 3–5 unique protein components in varying oligomeric states20,21,22; whereas type V-K CASTs rely on only TnsBC for integration16,23,24 and a simpler Cas12k-TniQ-S15 co-complex for DNA targeting25. Individual homologs within each of these CAST subtypes also vary in sequence identity26,27, subunit composition and fusion connectivity18,24,28, DNA targeting modules, crRNA guide sequence18,26,29, and host factor requirements17,25,30, thus representing a diverse pool of potential starting points for tool development. Although type V-K CASTs are more compact systems (~5 kb versus ~8 kb coding size), they exhibit multiple undesirable biochemical properties in heterologous cellular contexts assays—including reduced specificity31,32,33, low overall editing efficiencies16,31, and poor product purity24,34,35—that would necessitate extensive optimization for potential research and therapeutic applications. In contrast, type I-F CASTs exhibit highly specific and homogeneous integration products, with demonstrably greater efficiencies than types I-B, I-D, and V-K in E. coli15,16,17,18,19,24.
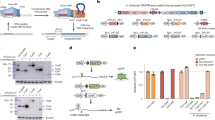
CAST systems have been the focus of extensive structural efforts using cryoEM in recent years. The type V-K ShCAST system from Scytonema hoffmannii has been systematically investigated25,36,37,38,39,40, with a recent report of the holo transpososome architecture that revealed intricacies of the megadalton complex containing Cas12k, TniQ, TnsB, TnsC, single-guide RNA, partial donor and target DNA substrates, and the bacterial host factor S1539. Structural studies of type I-B and I-F CASTs have largely focused on the QCascade DNA targeting module and the accessory TnsC ATPase20,21,41,42,43,44, with no structures of the endonuclease-transposase TnsAB module described to date. Intriguingly, QCascade structures exhibit distinct conformations across different systems: type I-B CASTs feature a single TniQ monomer that recruits TnsC to the Cascade-bound target DNA21, whereas type I-F CASTs feature a TniQ homodimer that is stably associated with Cascade20. Thus far, two I-F CAST systems from subtypes I-F3a and I-F3b have been structurally characterized—VchCAST (Tn6677) and AsaCAST (Tn6900), respectively—both of which are only distantly related to a type I-F CAST from the Pseudoalteromonas Tn7016 transposon (PseCAST), a system that we recently exploited for targeted DNA integration in human cells17.
The PseCAST RNA-guided transposase was identified as a lead candidate for human genome engineering applications through a systematic screen of diverse type I-F CAST systems (Fig. 1a)17. Although our first study reported editing activities that reached single-digit efficiencies at genomic target sites in the presence of a ClpX host factor, representing a > 100-fold improvement over our original candidate, VchCAST, these efficiencies remain limiting for downstream applications. We hypothesized that identifying bottlenecks in the system would inform more targeted rational engineering, developed several assays to investigate intermediate events and overall integration efficiencies in human cells17, and then applied these assays to VchCAST and PseCAST, the only type I-F CASTs shown to successfully perform RNA-guided integration in human cells. Intriguingly, while PseCAST promotes comparatively robust DNA integration, it exhibits markedly weaker DNA binding activity relative to VchCAST. We therefore hypothesize that, alongside parallel efforts to engineer and evolve hyperactive transposase variants, the PseCAST QCascade module represents a promising focus area to improve DNA targeting and thus editing efficiencies.

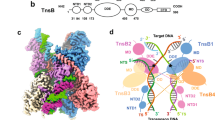
a Phylogenetic tree of type I-F CRISPR-associated transposons (CASTs), based on previous work in the lab26. Systems with previously solved QCascade structures are marked with red arrows, while PseCAST is marked with a green arrow. Phylogenetic clades are colored. b Experimental design to investigate both DNA binding and overall integration activities for CAST systems in human cells17. DNA binding is extrapolated from two different transcriptional activation assays, one in which VP64 is fused to Cas7 and one in which VP64 is fused to TnsC. Overall integration efficiencies are measured via amplicon sequencing. c Comparison of VchCAST and PseCAST across different assays in human cells. Although PseCAST exhibits consistently weak transcriptional activation compared to VchCAST, its absolute DNA integration activity at genomic target sites is approximately two orders of magnitude greater. DNA integration data is adapted from a previous publication17. Data are shown as mean for n = 2 biologically independent samples for Cas7 and TnsC activation. Data are shown as mean ± s.d. for n = 3 biologically independent samples for DNA integration. Source data are provided as a Source Data file. d Operonic architecture of PseCAST components from the PseCAST transposon, with genes encoding the QCascade complex labeled accordingly. e Left, dominant reference-free 2D cryoEM class averages. Right, cryoEM densities with colored map regions corresponding to Cas8 (blue), Cas7 monomers 1–6 (light blue), Cas6 (purple), TniQ monomers 1–2 (orange, yellow), crRNA (gray), and target DNA (red) indicated. f Refined model for the Cas8 ɑ-helical domain and its positioning relative to the TniQ dimer interface.
Towards that goal, here we report the cryoEM structure of PseCAST QCascade and the effect of targeted mutations in the PAM- and crRNA-interacting regions on DNA integration. Separately, we leverage AlphaFold-Multimer to predict protein-protein interactions within the TnsABC co-complex, inspiring the rational design of chimeric CAST systems that enable divergent DNA targeting and DNA integration modules to be combined into a single functional system. Collectively, this work establishes multiple biochemically- and structurally-guided approaches to engineer CAST systems for improved editing efficiencies in human cells.
Results
CryoEM structure of PseCAST QCascade complex
We previously demonstrated that VchCAST and PseCAST, two distantly related type I-F CASTs17,26, exhibit distinct DNA binding and integration efficiencies (Fig. 1a–c). Given our previous mechanistic and structural studies of the QCascade complex from VchCAST20,41, we hypothesized that structure-guided engineering of the PseCAST QCascade complex might reveal novel interactions and open a path to improve overall integration efficiencies. We therefore purified recombinant PseQCascade after carefully optimizing the expression vector design (Supplementary Fig. 1) and set out to determine the cryoEM structure.
We incubated the purified PseQCascade complex, which is expected to comprise a 1:6:1:2:1 stoichiometry of Cas8:Cas7:Cas6:TniQ:crRNA components (Fig. 1d), with a double-stranded DNA (dsDNA) substrate containing a 32-bp target sequence and 5′-CC-3′ PAM, and then subjected the sample to electron microscopy. Preliminary cryoEM experiments revealed a homogeneous behavior with multiple views and no apparent aggregation or disassembly (Supplementary Fig. 2a), and the overall architecture was consistent with other type I-F QCascade complexes, comprising six Cas7 monomers (named hereafter Cas7.1 to Cas7.6) that form a pseudo-helical assembly coating the crRNA molecule (Fig. 1e). The Cas8 protein contains two domains: a bulky domain that interacts with Cas7.1 and binds the crRNA 5′ end and PAM sequence, and a second ɑ-helical domain that exhibited a dynamic behavior (Fig. 1f). Towards the crRNA 3′ end (hereafter PAM-distal region), the RNA hairpin is stabilized by Cas6, which also binds the TniQ dimer. Preliminary maps exhibited greater mobility for the TniQ dimer compared to other QCascade components (Supplementary Fig. 2b,c). The quality of the maps approaching the TniQ dimer region degrades rapidly, contrasting the excellent map quality for the PAM-adjacent region (Supplementary Fig. 2d). Multibody approaches in Relion4 improved the overall resolution, with approximately 2.6 Å and 3.0 Å resolution estimates in the PAM-proximal and PAM-distal regions, respectively (“Methods”).
To further characterize the dynamics of the system and confirm the existence of novel interactions, we complemented our multibody analysis in Relion4 with cryoDRGN45, a machine-learning approach for cryoEM analysis (Supplementary Fig. 3). CryoDRGN revealed multiple populations of the complex, with the TniQ dimer populating a wide range of positions relative to the rest of the complex that pivot around Cas6 and Cas7.6. The dimer adopts an ‘open’ conformation that lacks any direct interactions with Cas8, as well as multiple intermediate, ‘closed’ conformations that approach the tip of the Cas8 ɑ-helical domain (Supplementary Fig. 3b). In a recent structure of a homologous QCascade complex bound to target DNA, the Cas8 ɑ-helical domain exhibits a different conformation, almost perpendicular to the inner face of the TniQ dimer and aligned with the bulky domain of Cas822; we were unable to identify such conformations within our dataset. Both the TniQ dimer and the Cas8 ɑ-helical domains remain in parallel configurations, with only marginal contacts at the periphery of the complex. Despite the apparent flexibility in this interaction (Supplementary movie 1 and 2), the Cas8 ɑ-helical domain is likely essential for RNA-guided DNA integration activity and/or QCascade complex formation, as revealed by the complete loss of human cell activity and partial loss of QCascade complex formation in E. coli, when we replaced the domain with a flexible glycine-serine linker (Supplementary Fig. 4).
Stabilizing protein-RNA and protein-protein interactions
The overall architecture of the TniQ dimer is similar to the VchCAST QCascade dimer20, with an antiparallel head-to-tail configuration, forming a compact unit that laterally approaches the interface formed by Cas6 and Cas7.6 (Fig. 2a). The C-terminal domain of one TniQ monomer interacts with Cas6, and the N-terminal domain of the other TniQ monomer interacts with Cas7.6. At the core of this four-fold interface, the crRNA appears to play a critical role, with residues 40–45 establishing multiple RNA-protein stacking interactions (Fig. 2b, c).

a Overall view of the cryoEM reconstruction of the PseCAST QCascade complex. b Magnified view of the dashed region in (a), highlighting the cryoEM density (colored and semi-transparent) for interactions between the indicated crRNA nucleotides and protein subunits. c Magnified view of the dashed regions in (b), highlighting interactions between the crRNA and Cas6 (left), TniQ.1 (middle), and both TniQ.2 and Cas7.6 (right). Key interacting residues are labeled. d Normalized RNA-guided DNA integration efficiency at the genomic AAVS1 locus in HEK293T cells, as measured by amplicon sequencing. The indicated alanine mutations were designed to perturb specific RNA-protein interactions highlighted in (c), and were compared to WT. NT, non-targeting crRNA. Data are shown as mean ± s.d. for n = 3 biologically independent samples. Source data are provided as a Source Data file. e Comparison of the crRNA conformation within the PAM-distal region, adjacent to the site of RNA hairpin stabilization by Cas6, for VchCAST (PDB: 6PIJ) and PseCAST (this study). The region around nucleotide G41 exhibits a distinct configuration for PseCAST, likely affecting the behavior of the adjacent TniQ dimer.
We hypothesized that crRNA interactions with Cas6, Cas7.1, TniQ.1, and TniQ.2 are crucial for robust QCascade complex formation, and that disrupting them would prevent transposase recruitment and abolish integration activity. We therefore introduced alanine point mutations to disrupt nucleobase-side chain stacking interactions and investigated the resulting effects in human genomic DNA integration assays. Alanine substitutions to Cas6 and TniQ residues contacting the crRNA were well tolerated, whereas a Cas7 R143A mutation (Cas7R143A) abolished integration activity (Fig. 2d). To investigate whether the loss of integration activity corresponded to defects in QCascade stability, we individually expressed and purified QCascade variants using a histidine-tagged TniQ subunit and performed gel filtration analyses to compare QCascade intactness. This revealed a complete loss of QCascade complex formation (Supplementary Fig. 5a), suggesting that this residue is critical to mediate Cas7 assembly onto the crRNA. The crRNA trajectory in the hinge region between Cas7.6 and Cas6 differs in PseCAST and VchCAST (Fig. 2e), and PseCAST crRNA residue G41 seems to play a key role as an interaction ‘hub,’ establishing coincident contact with TniQ.1, TniQ.2, and Cas7.6 by adopting a unique, extruded conformation.
We next explored protein-protein interactions that we similarly hypothesized would contribute to QCascade function, in part by playing a role in downstream transposase recruitment to the target site. The first of these interactions involved a hydrophobic patch on Cas6 cradling hydrophobic residues in the loop connecting TniQ.1 α-helices W262–K275 and F312–S327 (Fig. 3a, b), which is conserved across homologous QCascade complexes, with minor variations. Specifically, a hydrophobic residue in the TniQ.1 connecting loop (I282 in PseCAST, V270 in VchCAST) inserts deeply into the Cas6 hydrophobic patch to anchor the TniQ monomer to the Cascade module (Fig. 3c). The cradle structure of this interaction potentially acts as a pivot point, facilitating dynamic TniQ movement. Disruption of these hydrophobic interactions via introduction of charged arginine residues in either TniQ or Cas6 led to a marked reduction in genomic integration efficiencies (Fig. 3d). The other TniQ monomer (TniQ.2) interacts electrostatically with Cas7.6 via α-helix Y33–L47 and adjacent residues (Fig. 3e). Alanine mutations within the α-helix Y33–L47 led to a reduction in genomic integration efficiencies, while mutations along the adjacent interactions (residues K68, H69, and N70) led to a complete loss of activity (Fig. 3). Protein purification and gel filtration experiments suggest that these adjacent interactions are critical to stabilize QCascade formation (Supplementary Fig. 5b). Given the multimeric assembly of Cas7 monomers along the crRNA, loop regions observed to interact with TniQ.2 may have pleiotropic functions, possibly participating in Cas7 monomer-monomer interactions (Supplementary Fig. 6). With the goal of selectively perturbing Cas7.6-TniQ.2 interactions to investigate its importance, we avoided mutagenizing residues that might affect the Cas7 monomer-monomer contacts and thus focused on loops A and B (Supplementary Fig. 6b); mutations within Cas7 had surprisingly little-to-no impact on overall DNA integration activity (Fig. 3f).

a Overall view of the PseCAST QCascade complex, oriented to highlight the TniQ dimer (dark/light orange). b Magnified view of the region indicated in (a), showing how TniQ.1 (dark orange) interacts with a hydrophobic cavity on Cas6. The two visual renderings are colored either by Cas6 surface (purple, top) or hydrophobicity (bottom). c Comparison of the hydrophobic interactions between TniQ.1 and Cas6 in PseCAST (left) and VchCAST (right, PDB: 6PIJ), with residues labeled. d Normalized RNA-guided DNA integration efficiency at the genomic AAVS1 locus in HEK293T cells, as measured by amplicon sequencing. The indicated arginine point mutations were designed to perturb TniQ.1-Cas6 hydrophobic interactions. NT, non-targeting crRNA. Source data are provided as a Source Data file. e Magnified views of hydrogen bonding (top) and electrostatic (bottom) interactions between Cas7.6 (blue) and TniQ.2 helix (yellow). f Normalized RNA-guided DNA integration efficiency at the genomic AAVS1 locus in HEK293T cells, as measured by amplicon sequencing. Alanine mutations perturbing Cas7.6-TniQ interactions are generally tolerated. Source data are provided as a Source Data file. Data in (d, f) are shown as mean ± s.d. for n = 3 biologically independent samples.
Protein engineering modulates PAM stringency and improves DNA integration
In comparison to other type I-F CASTs, PseCAST exhibits a remarkably flexible PAM preference, with almost no sequence preference at both the –1 and –2 positions in E. coli transposition assays26; this property may lead to a dramatic increase in the effective search space for the 32-bp guide. Inspired by previous work investigating CRISPR-Cas9 activity and PAM search space46, we hypothesized that inefficient DNA targeting due to a flexible PAM preference may represent a rate-limiting step in RNA-guided DNA integration, especially within the cellular milieu of human cells, whose genome is ~1000× larger than E. coli. We therefore set out to specifically engineer QCascade variants that might exhibit altered PAM specificity and thus direct altered DNA integration efficiencies.
After leveraging the excellent quality of our cryoEM map in the area surrounding Cas8, we identified two hydrophobic alanine residues at the center of the PAM-interacting region. In contrast, systems with stricter PAM preferences—VchCAST, AsaCAST, and PaeCascade from a Pseudomonas aeruginosa type I-F1 CRISPR-Cas system26,47—feature polar residues at the equivalent positions, which allow for hydrogen bonding with specific PAM nucleotides (Fig. 4a, b, Supplementary Fig. 7a). Based on these observations, we reasoned that mutating A243 and A244 to residues with greater hydrogen bonding potential might improve PAM stringency, reduce the effective search space, and result in more efficient DNA targeting. We also chose to mutagenize residues 125–127, as this region also interacts with the PAM (Fig. 4b, Supplementary Fig. 7a). We analyzed the sequence conservation at these PAM-interacting regions and compared PseCAST to other Cascade homologs that have previously exhibited either robust DNA integration activity or stringent PAM preferences (Supplementary Fig. 7b, c). Collectively, we designed fifteen Cas8 variants with PAM-interacting mutations, varying from single point mutations at A243 or A244 to larger mutations in which the entire PAM-interacting region was grafted from a type I-F Cascade homolog.

a Top, overall view of the PseCAST QCascade complex, oriented to highlight the target DNA recognition. Bottom, Magnified view of the experimental cryoEM density map around Cas7.1 and Cas7.2, showing interactions with the crRNA (gray) and DNA target strand (TS, red). NTS, DNA non-target strand. b Magnified views of the PAM binding pocket, with Cas8 and DNA shown in blue and red, respectively. Residues A243 and A244 lack any base-specific, hydrogen-bonding interactions with the DNA. c Normalized DNA integration efficiency at the genomic AAVS1 locus in HEK293T cells for the indicated Cas8 mutants (top), plotted above the WebLogo for PAM preferences in the –1 and -2 positions (bottom) derived from integration into pTarget. (For additional PAM specificity data, see Supplementary Fig. 7e.) Integration efficiency data are shown as mean ± s.d. for n = 3 biologically independent samples. Source data are provided as a Source Data file. d Overlay of the refined atomic model and cryoEM density (semi-transparent) for the seed region of QCascade bound to the DNA target strand. e Schematic representation showing angles for the first five RNA-DNA base pairs (BP 1–5) within the R-loop. f View of the RNA-DNA heteroduplex at right, highlighting the unfavorable base-pairing surrounding flipped out nucleobases within the first 18 base pairs of the R-loop. g Magnified view of the RNA-DNA heteroduplex segments aligned at the flipped out base pair, revealing consistent unfavorable angles at the adjacent base pairs. h Normalized RNA-guided DNA integration efficiency at the genomic AAVS1 locus in HEK293T cells for the indicated Cas7 mutations, as measured by amplicon sequencing. Data are shown as mean ± s.d. for n = 3 biologically independent samples. Source data are provided as a Source Data file.
We quantified changes in PAM preference by performing an episomal PAM library screen in HEK293T cells, in which a target plasmid (pTarget) contained an AAVS1 target site directly downstream of a randomized 4-bp PAM library (Supplementary Fig. 7d). After transiently transfecting cells with pTarget, pDonor, and all the necessary protein-RNA expression vectors, we isolated plasmid DNA, sequenced the PAM motifs from all successful integration products, and constructed a consensus motif for each Cas8 variant; in parallel, we also quantified absolute genomic integration efficiencies at the AAVS1 site, which contains a 5′-CC-3′ PAM (Fig. 4c). The results revealed that certain mutations led to improvements in integration efficiencies by as much as 3.5-fold, but without a clear correlation between PAM stringency and overall genomic integration activity (Fig. 4c). For example, the variant with the greatest improvement in integration activity, Cas8R241K,A244S, actually exhibited a reduced PAM preference, compared to the stronger preference for cytidine in the -2 position with WT Cas8 (Fig. 4c, Supplementary Fig. 7e). Interestingly, Cas8A243Q,A244N exhibited decreased PAM preference, whereas when we grafted the entire PAM region from a type I-F1 system (241RPAAV245 > KPQNI), the resulting mutant restored a strong preference for cytidine at the -1. Mutations within the upstream PAM-interacting region (residues 125–127) showed moderate improvements on integration activity, with either unchanged or moderately reduced PAM stringency (Fig. 4c). A Cas8R241A mutant with disrupted ‘R-wedge,’ which normally forms stacking interactions with the -1 PAM position to help unwind dsDNA48,49, unexpectedly exhibited both WT integration efficiencies and PAM stringency (Fig. 4c).
Together, mutational profiling of the PAM-interacting region revealed key residues whose mutation improved integration efficiencies, but the combination of PAM specificity and integration activity results failed to support the hypothesis that PAM promiscuity is a key bottleneck towards achieving higher efficiency PseCAST integration activity in human cells (Fig. 4c, Supplementary Fig. 7e). This suggests that there may be a more complex network of interactions that dictate substrate preference, DNA unwinding, and transposase recruitment, though future studies will be needed to better inform further rational engineering efforts.
We also focused on PAM-proximal interactions with the upstream double-stranded DNA region as another potential point of engineering and optimization. Previous work on canonical type I-F1 defense systems revealed key interactions between dsDNA and the N-terminal region of Cas848,49,50, with a positively charged vise domain undergoing a conformational change to ‘clamp’ onto the PAM-adjacent sequence in a non-specific fashion. When comparing PseCas8 (from type I-F3 PseCAST) to PaeCas8 (from type I-F1 PaeCascade; Supplementary Fig. 8a), we observed a markedly different conformation of the N-terminus, with the vise domain absent. Given this potential deficiency, we hypothesized that substituting the PaeCas8 vise domain in PseCas8 could improve DNA binding affinity and thus CAST activity. However, a thorough screening of chimeric Cas8 constructs for human cell integration activity revealed a clear intolerance of PseCas8 to sequence perturbations in this region (Supplementary Fig. 8b). We pursued additional synthetic strategies to improve DNA binding of PseQCascade by fusing a variety of DNA-binding domains to the PseCas8 N-terminus of PseCas8 (Supplementary Fig. 8c), inspired by engineering strategies previously applied to polymerases51,52, reverse transcriptases53, and ligases54. However, these fusions exhibited no improvement relative to WT, and in some cases reduced overall genomic integration efficiencies (Supplementary Fig. 8c). Collectively, these experiments suggest that either the DNA binding affinity of PseCas8 is not a critical bottleneck in the overall transposition pathway, or that the tested variants fail to improve upon the WT activity in this regard.
Unfavorable nucleobase positioning along the RNA-DNA heteroduplex
Cascade complexes bind the target DNA by forming a discontinuous RNA-DNA heteroduplex in 6-bp segments48,55, and we could clearly resolve RNA-DNA base pairs for the first 4 segments engaged by Cas7 monomers within the PseQCascade complex. However, the remaining two segments featured weaker RNA density, with only density for the DNA phosphate backbone visible at lower thresholds. To assist visualization of the entire R-loop, we included in the final model atomic position corresponding to the DNA phosphate backbone but excluding atomic positions corresponding to the nucleotide bases. Density for the RNA-DNA heteroduplex across the first 3 segments (crRNA residues 9 to 26) was exceptionally good, with clear separation within base pairs and features compatible with a local resolution beyond 3 Å. We were therefore able to accurately model RNA-DNA interactions to a high level of confidence in these regions of the map. The resulting view revealed peculiarities in the base-pair geometry, with acute divergence from ideal values in some base pairs. The third and fourth base pair within each segment exhibited severe deviation from ideal planarity values (buckling), while the first and fifth base pair exhibited exacerbated propeller twist deviations. Only the second base pair across distinct segments exhibited geometric and hydrogen-bonding distance values closer to energetically favored conditions (Fig. 4d–g).
Type I-F Cascade complexes bind the target DNA, such that the two-stranded β-sheet ‘finger’ motif of each Cas7 monomer engages the crRNA to flip out every sixth nucleotide of the 32-nt spacer, thereby preventing RNA-DNA basepairing20,48. We hypothesized that finger motif residues involved in this nucleotide dislocation might promote the consistent distortion of adjacent base pairs, and to explore this effect, we introduced Cas7 mutations intended to relax this distortion, hoping to promote energetically favorable hydrogen-bonding geometries and stabilize the RNA-DNA heteroduplex. Taking advantage of the high local resolution around this region, we identified numerous bulky hydrophobic residues—including I69, L70, and L224—that were not highly conserved across nearby homologs (Supplementary Fig. 9a–c) and subjected them to site-directed mutagenesis.
After generating the desired Cas7 mutations, we performed genomic DNA integration experiments in HEK293T cells at the AAVS1 locus (Fig. 4h). Intriguingly, the Cas7 heteroduplex-interacting residues, though not highly conserved, appeared to have low tolerance for mutations. While Cas7L224F and multiple valine mutations exhibited near-WT integration efficiencies, all other mutations, including Cas7I69P, resulted in detrimental impacts on DNA integration (Fig. 4h). L70H, which would theoretically recapitulate a stacking interaction observed in our previous VchCAST structure20, completely abolished integration activity (Fig. 4h). This unexpected loss of integration activity across multiple variants inspired us to investigate the stability of QCascade; although we observed a range of behaviors, most variants were unable to form QCascade complexes at similar efficiencies to WT (Supplementary Fig. 9d). The Cas7I69A mutant, which exhibited no detectable genomic integration activity (Fig. 4h), demonstrated the greatest ability to form QCascade complexes, albeit markedly less efficiently than WT, suggesting that this mutation specifically destabilizes DNA-binding. Together, the intolerance to perturbations in the Cas7 finger domain suggests these residues help stabilize QCascade complex and R-loop formation.
Structure-based engineering of chimeric CAST systems
Rational engineering of PseQCascade yielded only moderate improvements in integration activity, suggesting a non-trivial path forward to overcome the apparently weak DNA binding activity in human cells17. Although recent studies shed light on the kinetics of Cascade target search and recognition56,57, the intermediate steps of Cascade complex formation, TniQ-Cascade association, and 3D-diffusion remain poorly understood, particularly in human cells. PseCAST was originally identified through a homolog screen that investigated both overall integration activity and several subunit-specific properties: crRNA processing, TnsB-donor DNA interactions, and targeted transcriptional activation17. Through this screening process, VchCAST (Tn6677) and PseCAST (Tn7016) were the only two systems that yielded detectable DNA integration in human cells, despite exhibiting distinct subunit-specific activities. Based on these results, we hypothesized that natural CAST systems may be unlikely to possess optimal human cell properties across all recombinant components, and we therefore set out to design chimeric CAST systems that would enable ‘crosstalk’ between otherwise orthogonal components. Our specific goal was to combine highly active DNA targeting and DNA integration machineries derived from divergent CASTs (Fig. 5a).

a Schematic showing the approach to generate a chimeric CAST system by combining optimal DNA targeting and DNA integration machineries from distinct CAST systems. b AlphaFold-generated structure prediction of the TnsABC co-complex from PseCAST. The C-terminal ‘hook’ region of TnsB that putatively interacts with TnsC is marked. c Visualization of select TnsB graft points within the predicted PseTnsABC structure. Residues where Pse-Vch chimerism was introduced are colored in blue, and the three top performing graft points (V585, S589, Q594; PseTnsB numbering) from panel (e) are labeled. d Experimental workflow to test chimeric TnsAB constructs for RNA-guided DNA integration activity. E. coli BL21(DE3) cells containing a pEffector encoding VchQCascade and VchTnsC were transformed with a plasmid encoding a mini-transposon (mini-Tn) and TnsAB, with TnsAB derived from either VchCAST, PseCAST, or a chimeric combination thereof. Integration efficiency was measured by qPCR (bottom). e E. coli DNA integration efficiencies for each tested TnsAB chimera. The amino acid listed represents the position at which the reading frame was grafted from PseTnsB (red) to VchTnsB (blue). “Custom” denotes a variant in which multiple different VchTnsB sequences were substituted (see Supplementary Data 3 for details). Source data are provided as a Source Data file. Data are shown as mean for n = 2 biologically independent samples.
To identify robust DNA targeting homologs, we tested DNA binding activity across 20 type I-F CASTs via transcriptional repression in E. coli41,58 (Supplementary Fig. 10a). Surprisingly, QCascade complexes from only two systems—VchCAST and Tn7005—exhibited RFP repression under the tested conditions, with only weak activity from PseCAST and Tn7000 (Supplementary Fig. 10b). Yet when we tested the overall DNA integration activity of VchCAST and PseCAST at the exact same sites used for transcriptional repression in E. coli, we again observed greater integration activity for PseCAST, mirroring our results in human cells17 (Supplementary Fig. 10c). This reinforced the conclusion that the weak DNA targeting activity of PseCAST may impose a lower ceiling on achievable DNA integration efficiencies in diverse cell types, despite having co-evolved with a highly active transposition (TnsABC) module.
We sought to address this potential bottleneck by combining the TnsABC machinery from PseCAST with the QCascade machinery from VchCAST. We previously demonstrated that intrinsic CAST modularity precludes simply mixing and matching components from evolutionary diverse systems26, but we were emboldened to attempt a more nuanced approach by taking advantage of recent high-resolution structures21,39, predicted structures via structural alignments41, and AlphaFold-multimer59 predicted structures. (Fig. 5b, Supplementary Fig. 11). In particular, a model for the putative TnsABC co-complex from PseCAST featured the expected heptameric arrangement of TnsC, similar to our empirical structures for VchCAST41, while also revealing predicted interactions between PseTnsC and the C-terminus of PseTnsB that were reminiscent of the TnsB ‘hook’ described for type V-K ShCAST37,39,40 (Fig. 5b, Supplementary Fig. 11a). This model, in conjunction with experimentally determined type V-K structures and biochemical studies of Tn7 60, led us to speculate that the C-terminal tail of TnsB functions as a key mediator of TnsC interactions, and that the specificity of CAST transpososome assembly would be dictated in part by cognate TnsB-TnsC interactions. Importantly, we hypothesized that reengineering this interaction would enable the TnsAB and donor DNA components from one CAST system to be combined with the QCascade and TnsC components from an orthogonal system.
To test this hypothesis, we designed 16 chimeric TnsAB constructs in which different lengths of the PseTnsB C-terminus were substituted with corresponding residues from the VchTnsB C-terminus (Fig. 5c). These variants were then screened for RNA-guided DNA integration activity in E. coli, in conjunction with VchQCascade and VchTnsC, but with a pDonor containing transposon ends compatible with PseTnsB (Fig. 5d). As expected, given our previous work26, WT PseTnsAB, lacking any chimeric substitutions, showed undetectable activity when combined with VchCAST DNA targeting machinery (Fig. 5e). Remarkably, however, several chimeric TnsAB designs were able to robustly rescue activity, showing up to ~10% integration efficiencies (Fig. 5e). These designs, which only reprogrammed 20 – 29 amino acids in the C-terminus of PseTnsAB, exhibited graft points between the Pse and VchTnsB sequence in an unstructured region that links the “hook” region of the C-terminus to the remainder of the protein sequence (Fig. 5c); furthermore, when comparing this region to solved type V-K complexes, it is located in a similar region as the 52-residue long “flexible linker” that was unresolved in type V-K CAST structure39,40. Analyzing the primary sequence of TnsB from both Vch and Pse, we observed a clear lack of conservation within C-terminal disordered regions61 (Supplementary Fig. 12a). We concluded that substitutions in this C-terminal region minimize disruptions to the overall protein fold, while nonetheless providing a chimeric hook that is compatible for cognate interactions with VchTnsC. Next, we investigated whether the best-performing chimeras were active for genomic DNA integration in human cells. At our current limits of detection with NGS (.005–.01%17), we were unable to detect targeted DNA integration with either WT VchCAST or best chimeric designs (Supplementary Fig. 12b), nor was activity detected for orthogonal combinations of PseTnsAB with VchQCascade-TnsC. Nevertheless, we envision that the future combination of chimeric designs with evolved CAST components would provide more optimal starting points for human cell editing experiments.
We next set out to test the reciprocity of these chimeric designs by pairing PseQCascade-TnsC with similar chimeric VchTnsAB variants; we were also able to detect integration activity with the converse combination (Supplementary Fig. 12c). Furthermore, when we applied these chimeric designs to a broader range of homologous TnsAB variants and their cognate mini-Tn donor substrates, we also observed integration activity for chimeric designs derived from additional transposon variants, denoted Tn7005 and Tn701526. Intriguingly, TnsAB chimeras derived from Tn7010 and Tn7011 showed no evidence of activity (Supplementary Fig. 12d), suggesting that some CASTs may require targeted screening to identify tolerable chimeric graft points. Next, we explored whether this engineering approach could also generate compatible chimeras between divergent CRISPR-associated transposons, candidate type I-F (VchCAST) and type V-K (ShCAST) systems, each of which comprise distinct transposase architectures and likely arose from unique domestication events23. TnsB variants derived from ShCAST exhibited low but detectable levels of activity (Supplementary Fig. 12e), and when we investigated the transposon insertion orientation preference for type I/V CAST chimeras, we observed that chimeras in which the TnsB was derived from ShCAST exhibited a T-LR insertion preference, as typically observed in previous ShCAST studies16,35, while type I-F CASTs exhibit a T-RL preference15,26 (Supplementary Fig. 12f). Furthermore, insertion profiles for all three chimeric designs exhibited an unchanged preference of 49–50 bp downstream of the 3’ edge of the target site (Supplementary Fig. 12g), similar to cognate type I-F15,26 CAST systems, suggesting that the TnsC footprint along DNA is the key determinant in the insertional regiospecificity.
Together, these results reveal that rational, structure-guided engineering of diverse CAST systems can overcome their intrinsic orthogonality, enabling diverse genome editing designs.
Discussion
The unexpected paradox of poor DNA binding and strong overall integration activity of PseCAST (Fig. 1b,c, Supplementary Fig. 10), inspired us to determine cryoEM structures of PseQCascade and pursue rational engineering methods to improve DNA targeting. Given the unique phenomenon among CAST systems to harbor ‘homing’ crRNAs that target conserved, often essential, genes within the host genome18,26,28,29, CAST-derived CRISPR modules may have been naturally selected for weak DNA binding relative to their defense-associated CRISPR-Cas counterparts, thereby reducing transcriptional repression of these essential genes. This possibility underscores the need to develop a comprehensive understanding of all molecular requirements and intermediate steps within the CAST transposition pathway.
The structure of PseQCascade resembles previously determined DNA-bound type I-F CAST structures20,22, but several knowledge gaps still limit a complete understanding of the mechanistic requirements for RNA-guided transposition. First, the functional relevance of the Cas8 helical bundle remains uncertain. When comparing between three distinct, DNA-bound QCascade structures20,22, three different conformational states of the helical bundle have been observed: a state in which the domain is unresolved, suggesting a conformationally dynamic mode related to the open versus closed state of the overall QCascade complex20; a state in which the domain is resolved, with close contact to the PAM-distal DNA22; and a state in which the helical bundle is resolved but does not contact TniQ or the PAM-distal DNA (Fig. 1e, f). Observation of PAM-distal contacts led to speculation that this conformation is required for recruitment of downstream transposase proteins22. Our deletion experiments suggest that the helical bundle is crucial for overall DNA integration to occur (Supplementary Fig. 4), but the decreased stability of the QCascade complex after deletion of the Cas8 helical bundle limits our ability to draw confident conclusions about putative Cas8-TnsC interactions (Supplementary Fig. 4c). Another area that will require future study is the manner in which the QCascade complex binds TnsC, since these interactions have not yet been captured for a type I-F CAST system. Mutations in Cas7 that theoretically disrupt Cas7.6 interactions with TniQ.2 appear to be tolerated (Fig. 3e, f); although unexpected, this lends credence to the possibility that only one of the two TniQ monomers present in type I-F CAST complexes interacts with TnsC, which is supported by similar CAST structures from type I-B and type V-K systems in which only one TniQ is present with TnsC at the target site (Supplementary Fig. 11)21,25,39. Further in vitro biochemical studies, combined with structural insights into the holo transpososome, will be necessary to shed light on these mechanistic aspects, including the extent to which the Cas8 helical bundle may regulate TnsC recruitment, and thus the targeting discrimination between on- and off-target sites during CAST transposition41.
Beyond defining structural requirements for transposition, our QCascade structure revealed potential targets for rational engineering, most notably within the PAM-interacting regions of Cas8. The presence of alanine residues at this interface, rather than polar residues, differentiates PseCAST from homologous type I-F CAST systems (Supplementary Fig. 7a). Interestingly, one of these homologous systems — VchCAST — exhibited higher DNA binding activity than PseCAST in both human cells and E. coli (Fig. 1c, Supplementary Fig. 10), leading us to hypothesize that reinstating polar residues might stabilize DNA-protein interactions, thereby increasing DNA binding activity and integration efficiency. Mutation of even one of these alanine residues yielded QCascade variants with integration efficiencies 2- to 3-fold above wild-type, but unexpectedly, these changes did not accompany concomitant increases in PAM stringency (Fig. 4c), suggesting that polar residues may stabilize DNA binding in a sequence non-specific fashion. This inconsistency between overall integration efficiency and PAM stringency suggests a complex, poorly understood relationship between DNA binding and integration activity within type I-F CASTs. Furthermore, our episomal PAM screen in human cells revealed a wild-type ‘CN’ preference that had not previously been observed in E. coli, and we hypothesize that this difference may result from the larger DNA search space in the human cell milieu. Although it is possible that the episomal PAM screen does not reflect the genomic PAM preference, we suspect that the episomal PAM assay better controls for confounding variables related to chromatin landscape and other context-dependent effects.
The quality of our cryoEM maps also provided a detailed view of RNA-DNA base-pairing interactions, enabling visualization of energetically unfavorable nucleobase positioning along the heteroduplex (Fig. 4d–g). Close analysis of the surrounding Cas7 residues implicated several hydrophobic side chains in enforcing this positioning (Supplementary Fig. 9), and we therefore introduced mutations with less bulky side chains to potentially stabilize heteroduplex formation. Interestingly, however, most Cas7 variants completely abolished integration activity (Fig. 4h) and marked destabilization of the QCascade complex (Supplementary Fig. 9d). These data suggest that the nucleobase positioning and unusual stacking interactions may be required for stable Cas7 assembly onto the larger Cascade complex. Further in vitro and biochemical work to dissect the mechanistic requirements of Cas7 polymerization within both CASTs and canonical CRISPR systems will improve future rational engineering for improved DNA targeting.
Alongside our efforts at engineering specific PseCAST components for DNA integration activity improvements, we considered a parallel path that would instead leverage pre-existing components from homologous CAST systems. Our previous experiments revealed the orthogonal properties of diverse type I-F CAST systems, which precluded simple mixing-and-matching of homologous components into single systems26. We hypothesized that a more nuanced, structure-guided approach could reveal unique opportunities for the construction of synthetic chimeric designs that would retain key protein-protein interactions necessary for transposition. To this end, we leveraged AlphaFold59 to generate predicted structures of TnsA-TnsB interacting with a heptameric TnsC ring (Fig. 5b), and based on the resemblance to previously determined type V-K transpososome structures (Supplementary Fig. 11a)39,40, we envisioned that reprogramming the TnsB C-terminus could uncover functional chimeric CASTs. This hypothesis was borne out with data demonstrating that chimeric CASTs, in which the DNA targeting module of VchCAST was combined with the DNA integration module of PseCAST, functioned for RNA-guided DNA integration (Fig. 5). When testing these initial chimeric variants in human cells, we were unable to detect genomic integration activity (Supplementary Fig. 12b). While this is not unexpected considering these variants exhibit ~10% activity relative to wild type in E. coli, this underscores the value in developing/combining diverse engineering and evolution methods to improve CAST enzymes, including homolog screening17,26,35, rational engineering (this study), and directed evolution62, as no single method may prove self-sufficient.
We further extended these chimeric designs to a variety of type I-F systems and demonstrated the first example of coordinated activity between type I-F and type V-K machineries (Supplementary Fig. 12). Several chimeric designs, however, did not show detectable activity in E. coli (Tn7010 and Tn7011, Supplementary Fig. 12d). We believe this is due to limited homology of these systems with both VchCAST and PseCAST26, and that these systems may require slightly different graft points. However, given our ability to generate chimeras between VchCAST and multiple type I-F variants, as well as across type I-F and V-K systems, we believe that TnsB chimeras are a generalizable opportunity to increase CAST modularity. Based on these results, we expect that further modifications will enable additional chimeric starting points for future engineering, such as at the TniQ-TnsC interface (Supplementary Fig. 11b, c).
The ability to coordinate targeted integration with transposase proteins derived from unique families23 opens the door to diverse chimeric CAST designs that can sample combinatorial sequence spaces unexplored by evolution. With growing evidence that additional CAST subtypes can be leveraged for genome editing applications in human cells63,64,65,66, the ability to exchange modules with ease may be key for future CAST engineering efforts. Collectively, our work showcases diverse, structure-guided approaches to understand and improve CAST function, and opens the door to a far greater combinatorial space for leveraging CASTs systems as genome editing tools.
Methods
Protein purification
The TniQ-Cascade complex from PseCAST (PseQCascade) was overexpressed and purified as previously described20, with the following modifications. After initial expression testing showed low expression of PseCAST components, all proteins were codon optimized and placed downstream of consensus RBS sequences, and TniQ contained an N-terminal 10xHis-TEV tag. The minimal CRISPR array was encode upstream of cas7 and contained a 32 bp spacer targeting the AAVS1 locus (see Supplementary Data 1 for detailed plasmid sequences). After overnight expression at 0.5 mM IPTG, cell pellets were resuspended in QCascade lysis buffer (50 mM Tris-Cl, pH 7.5, 700 mM NaCl, 0.5 mM PMSF, EDTA-free Protease Inhibitor Cocktail tablets (Roche), 1 mM dithiothreitol (DTT), 5% glycerol) and lysed by sonication. Lysates were clarified by centrifugation at 15,000 x g for 30 min at 4 °C. Initial purification was performed by immobilized metal-ion affinity chromatography with NiNTA Agarose (Qiagen) using NiNTA wash buffer (50 mM Tris-Cl, pH 7.5, 700 mM NaCl, 10 mM imidazole, 1 mM DTT, 5% glycerol) and NiNTA elution buffer (50 mM Tris-Cl pH 7.5, 700 mM NaCl, 300 mM imidazole, 1 mM DTT, 5% glycerol). The sample was further purified by size exclusion chromatography over a Superose 6 Increase 10/300 column (GE Healthcare) equilibrated with QCascade storage buffer (20 mM Tris-Cl, pH 7.5, 700 mM NaCl, 1 mM DTT, 5% glycerol). Fractions were pooled, concentrated, snap frozen in liquid nitrogen, and stored at −80 °C. TEV cleavage was not performed.
Plasmid construction
Bacterial expression plasmids for PseQCascade were codon-optimized for E. coli and synthesized by GenScript. For human cell transfections, genetic components encoding PseCAST proteins were codon-optimized for human cells, synthesized by GenScript, and cloned into pcDNA3.1 expression vectors. All CAST constructs were cloned into plasmids using a combination of restriction digestion, ligation, Gibson assembly, and Golden Gate assembly. All PCR fragments for cloning were generated in-house using Q5 DNA Polymerase (New England Biolabs (NEB)) and gel purified using Qiagen Gel Extraction.
To clone the 4 N PAM library used for HEK293T cell episomal integration assays, two overlapping oligos containing ‘NNNN’ were phosphorylated with T4 PNK (NEB) and hybridized at 95 °C for 2 min before cooling to room temperature. The resulting oligoduplex was ligated into a target plasmid vector predigested with BsmBI (55 °C for 2 h) using T4 DNA ligase (NEB). Cloning reactions were transformed into chemically competent NEB Turbo E. coli, plated on agar plates with the appropriate antibiotic to grow overnight, and inoculated in 5 µL LB media and antibiotic for approximately 7 h. Colony counting was then performed to ensure sufficient library diversity. Plasmids were then purified using Qiagen Miniprep columns verified by a combination of Sanger sequencing (Azenta/Genewiz) and whole-plasmid nanopore sequencing (Plasmidsaurus), and ultimately characterized by high-throughput sequencing (Illumina).
CryoEM structure determination
Purified PseQCascade was serially diluted in a modified buffer (20 mM Tris-Cl, pH 7.5, 200 mM NaCl, 1 mM DTT) for initial imaging experiments. Target DNA (NTS: 5ʹ-TTCATCAAGCCATTGGACCGCCACAGTGGGGCCACTAGGGACAGGATTGGTGACCTTCGCCTTGACGGCCAAAA-3ʹ, TS: 5ʹ-TTTTGGCCGTCAAGGCGAAGCTGAAAAGCAATGAAGCCAA AGCGTCCTGTAAGGCGGTCCAATGGCTTGATGAA-3ʹ) was duplexed by mixing the NTS and TS in equimolar concentrations, heated to 95 ˚ C, and then cooled to room temperature. 50 µM aliquots were then snap frozen. Purified PseQCascade aliquots were incubated with a 5X molar excess of target DNA for 10 min at room temperature with a total reaction volume of 50 µL. The complex (2–4 µM range) was initially imaged in a Talos L120C (Thermo Fisher) electron microscope equipped with a LaB6 electron source and a Ceta-M camera. Negative staining experiments were carried out using uranyl-formate solution at 0.75% (w/v) in water. CF-400 (EMS) continuous carbon grids were activated for 30 s using a Ar/O2 gas mix plasma at 25 W using a Solarus2 plasma cleaner (Gatan). Immediately after plasma activation, 3 µL of the PseQCascade/DNA complex at concentrations of 1, 2 and 4 µM were applied to the activated grids. After 1 min incubation, the excess solution was gently blotted away, and 3 µL of 0.75% uranyl-formate solution was added for an additional 1 min incubation. Excess staining solution was blotted away and the grids were left on the bench drying for 5 min. Grid screening revealed well stained, homogeneous, and dispersed particles with a circular shape compatible in dimensions and shape with the estimated molecular size of the complex, as well as showing similarities with previously reported images of other Cascade complexes (Supplementary Fig. 2a).
We chose the 1 µM concentration grid for manual collection of 10 negative staining images (pixel size 2.5 Å/pixel, 1 s exposure, –2 to –3 µm defocus) for exploratory class-2D analysis in Relion467. The resulting negative staining C2D averages confirmed the homogeneity of the sample and its potential for high-resolution (Supplementary Fig. 2a, left). Next, we explored the behavior of the complex under cryogenic conditions using the negative stain conditions as a reference starting point. We vitrified UltraAu foil 1.2/1.3 ‘Gold’ grids68 (Quantifoil) using a VitroBot Mark IV (Thermo Fisher) set up to 100% humidity and 4 °C. The sample concentration was in the 2–4 µM range. Grids were plasma cleaned with the same protocol described for the negative staining grids, and after application of 3 µL solution, the grids were blotted and plunged frozen in liquid ethane. Vitrobot settings were: blot force –5, drain and waiting time 0 with blotting times variating between 2.5 and 3.5 s. Following these parameters, we froze 8 grids, 4 grids at 2 µM concentration and 4 grids at 4 µM concentration. 2 grids, one at 2 µM and another at 4 µM concentration were transferred to a cooled 910 side entry holder (Gatan) for screening under cryogenic conditions in the same Talos L120C microscope used for negative staining using similar imaging conditions. Both grids showed good ice distribution, with the 2 µM grid showing better particle distribution and contrast in ice. Using SerialEM69, we collected 10 images with similar settings as in negative staining experiments for exploratory reference-free C2D analysis in Relion4 under cryogenic conditions (Supplementary Fig. 2a, middle). The resulting C2D averages were promising, with distinctive and multiple views of the complex. The grid was recovered and stored for high resolution data collection in a Titan Krios G3i electron microscope equipped with a BioQuantum/K3 energy filter and direct detection.
High resolution data was collected at high magnification with 2x hardware binning in the K3 detector (0.6485 Å/pixel size after binning) at a fluence of ~20e–/pixel/s and 1 s exposure time for a total dose of ~50 e–/Å2. Defocus range was adjusted to vary between -0.8 to -2 µm, and the total number of K3 fractions was adjusted to 50. 24 h collection on the recovered grid yielded ~22,000 images which were on-the-fly motion corrected in Relion4 with ctf estimation in ctffind470. Image processing was integrally done in Relion 4 and cryoDRGN45. First, we manually selected 100 images for Laplacian picking, which yielded ~4000 particles that were normalized and extracted with 8 times binning. Fast C2D analysis using the VDAM algorithm generated C2D averages in multiple orientations that were selected and used as training set for Topaz, used through the Relion wrapper. Using the optimized trained model from Topaz71, the full dataset of ~22,000 images yielded ~1.5 million particles that after two C2D steps using T parameters of 3 and then 6 was reduced to ~667,000 particles. ArnA contamination accounted for the bulk of the eliminated particles. Next, we refined the reduced dataset using a filtered map of VchQCascade as reference. We did not perform alignments with this initial classification (K20, tau fudge T = 6).
We identified multiple classes with damaged or poorly aligned particles, a class without the TniQ dimer, and a dominating class with better features. A re-extraction step was then performed with the recenter option activated and at 4x binning (2.594 Å/pixel). After selection of 2D class averages showing secondary structure features, an ab-initio 3D model was reconstructed using the Stochastic Gradient Descent (SGD)72 algorithm with all selected particles from the class 2D job (K4, tau fudge T = 3). A second 3D refinement produced a consensus refinement in the 5 Å range that upon inspection showed clear secondary features and substantial heterogeneity at the PAM distal region hosting the TniQ dimer. A soft-mask (10 pixel extension, 8 pixel soft edge and initial threshold of 0.002) was used for 3D classification without alignment using 20 classes and T parameters 3, 6 and 8. A minor population (~8% of the particles) of Cascade without TniQ was identified and removed from the dataset, together with poorly aligned or damaged particles, reducing the total dataset to ~128,000 particles. Re-refinement of this dataset after re-extraction to binning 2 ( ~ 1.2 Å/pixel) produced a sub-3Å map, but exacerbated heterogeneity of the TniQ dimer region was evident.
Using focused classification of this region of the map produced multiple classes without clear discrete states, suggesting continuous heterogeneity. Before applying a multibody approach, we re-refined the ~128,000 particle dataset after refining the ctf parameters (defocus values per particle and astigmatism per micrograph) followed by Bayesian particle polishing for signal decay and local particle movement correction. We defined via soft masking (6 pixel mask extension, 6 pixel soft edge decay, initial threshold 0.002) three rigid body groups: the first body included Cas8, and the first Cas7 monomer (Cas7.1), the second body contained Cas7 monomers 2 to 5, and the third body included the TniQ dimer, Cas6, Cas7.6, and the crRNA 3′-proximal hairpin. Residual rotation priors were defined to 10 degrees with translation offset of 2 pixels. We designed two wide masks: one (body 1) covering the best part of the map and including Cas8, the first five Cas7 proteins, and surrounding densities including the corresponding sections of the crRNA-DNA heteroduplex; and a second soft mask (body 2) covering Cas7.6, Cas6, and the TniQ dimer. Multibody refinement produced maps with exceptional quality for each body, with clear sub 3 Å features for the Cas8 and the Cas7 regions. The maps for the PAM-distal body, including the TniQ dimer, improved substantially, but residual heterogeneity remained, especially at the distal end of the TniQ dimer.
We used ModelAngelo73 for initial model building using the improved maps from the multibody analysis. With default options and sequence information from the cloned constructs, ModelAngelo correctly built approximately 90% of the residues. Manual inspection of the built model corrected limited errors and completed areas where the resolution did not allow accurate placement of side chains. The built models were refined against the multibody maps independently, first with phenix refine (secondary structure restrain activated) and then with Refmac5, adjusting the experimental/ideal geometry weights manually to avoid overfitting. CryoDRGN analysis was performed with the final set of ~128,000 particles used for multibody analysis in Relion. This set of particles was re-extracted to a box size of 128 pixels and an initial training in 1 dimension (Zdim=1) was performed. After assessing the homogeneity of this set of particles, 3 different training were performed with 2, 4 and 8 dimensions (Zdim=2, 4 and 8). Principal component analysis (PCA), UMAP, and K-means clustering dimensionality reduction techniques were used to explore the derived latent spaces, producing similar results irrespective of the Zdim used. We perform a final training with particle re-extracted to 256 pixels size and Zdim 2 and 8. Exploration of the latent space derived from these training revealed multiple conformations of the TniQ dimer, as shown in Supplementary Fig. 3.
Mammalian cell culture and transfections
HEK293T cells (ATCC CRL-3216) used in this study are a gift from Dr. Alejandro Chavez. Cells were routinely tested for mycoplasma and cultured at 37 °C and 5% CO2 and maintained in DMEM media with 10% FBS and 100 U/mL of penicillin and streptomycin (Thermo Fisher Scientific). 24 h before transfection, a 48-well plate was coated with poly-D-lysine (Thermo Fisher Scientific) and seeded with 10,000 cells per well. Cells were transfected with DNA mixtures and 1 μL of Lipofectamine 2000 (Thermo Fisher Scientific) per the manufacturer’s instructions. Transcriptional activation and integration assays were performed as previously described17. For plasmid-based PAM library assays, cells were co-transfected with the following PseCAST CAST plasmids: 200 ng pTnsAB, 50 ng pTnsC, 75 ng pQCascade, 100 ng pCRISPR (crRNA), 200 ng pDonor, and 100 ng pTarget (4 N PAM library). Cells were harvested 4 days after transfection using previously described methods17. Flow cytometry was performed as previously described17 using a NovoCyte Quanteon Flow Cytometer, and a figure exemplifying the gating strategy is shown in Supplementary Fig. 14.
Analysis of HEK293T integration assays
Genomic integration assays were analyzed as previously described17. In brief, 5 µL of genomic lysate (10% of total lysate volume) was used for 2 rounds of PCR. In the first PCR, a forward primer was used that anneals to the AAVS1 locus, and a reverse primer was used that anneals to both the AAVS1 locus and a primer binding site in the donor DNA (see Supplementary Data 4 for oligonucleotide sequences). These oligos included 5′ overhangs encoding read 1 and read 2 Illumina adapters. In the second PCR, ‘universal’ primers were used, which anneal to the read 1 and read 2 sequences and append unique index sequences and the remaining Illumina adapter sequences for next generation sequencing. Samples were then pooled, gel purified, and sequenced on a NextSeq 500/550 with at least 75 cycles in read 1. The relative abundance of reads that contain a PseCAST transposon end sequence (representing an integration read) vs. downstream AAVS1 sequence (unintegrated read) was calculated.
For the episomal PAM library assay, samples were prepared as above except a different forward oligo was used that anneals directly upstream of the degenerate PAM library in PCR 1, such that we would capture both the PAM sequence and the presence of the transposon end sequence with the forward read (see Supplementary Data 4 for oligonucleotide sequences). PCR 1 cycles were reduced to 15 cycles. After Illumina sequencing, reads were filtered to have a transposon end sequence, thus representing a PAM library member which was successfully targeted by PseCAST for DNA integration. The input library was sequenced as well, to calculate enrichment and depletion scores. Library members were then ranked by their enrichment values (proportion of output library / proportion of input library). The top 10% of library members were used to generate a consensus WebLogo (Version 2.8.2, 2005-09-08, weblogo.berkeley.edu) for the PAM preference of each Cas8 variant. All library members and their associated enrichment values were used to generate PAM wheels using Krona74.
E. coli repression and integration assays
E. coli transcriptional repression assays were performed as previously described41,58, with some minor modifications. In brief, an E. coli strain expressing mRFP from the chromosome, a gift from L. S. Qi, was transformed with pQCascade. We initially attempted to use pQCascade plasmids with a strong J23119 promoter, but due to toxicity associated with strong PseQCascade expression, we switched to a weaker J23101 promoter for all pQCascade constructs. We designed crRNA sequences to target the template strand of mRFP proximal to the 5′ end of the coding region (60 bp downstream of the mRFP start codon). Two replicates were performed for each unique transformation, and relative mRFP repression was analyzed as previously described41.
Integration assays were performed as previously described15,41, with the following modifications. Although J23101 promoters were used for QCascade, J23119 promoters were still used for constitutive expression of all TnsABC cassettes, as there was no observed toxicity. In brief, TnsABC expression vectors harboring donor DNA (pDonor-TnsABC) encoded a tnsA-tnsB-tnsC operon downstream of a strong constitutive promoter (J23119), as well as a mini-transposon donor DNA of 0.9 and 1.2 kb in length for VchCAST and PseCAST, respectively, all on a pUC19 backbone. Strains harboring medium-strength J23101 promoter-controlled pQCascade constructs were first made chemically competent, followed by duplicate transformations with pDonor-TnsABC and lysate generation for qPCR after an 18 h incubation at 37 °C. Lysates were analyzed via qPCR, as previously performed15,41. Amplicon sequencing of integration products shown in Supplementary Fig. 12g were analyzed as previously performed15,26. In brief, genomic lysates were diluted 100-fold, followed by two rounds of PCR to amplify T-LR integration products and append Illumina sequencing barcodes, and sequenced on an Aviti sequencer. Oligos used to amplify the integration products are listed in Supplementary Data 4.
Statistics and reproducibility
Integration efficiencies are presented normalized to the wild-type condition shown in each graph. The mean value and all biological replicates are plotted, and when appropriate (n > 3), ± one standard deviation is plotted as well. Sample sizes are indicated in figure legends. No statistical method was used to predetermine sample size, and no data were excluded from the analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cryo-EM maps and models have been deposited in the Electron Microscopy Data Bank with accession code EMD-51543. The coordinates for the atomic model have been deposited in the Protein Data Bank (PDB) with accession code 9GS9. Source data for protein gels are included as Supplementary Fig. 13. Amplicon sequencing of PAM libraries have been deposited to the National Center for Biotechnology Information’s Sequence Read Archive under BioProject Accession PRJNA1161457. Source data are provided as a Source Data file. Source data are provided with this paper.
References
Branzei, D. & Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 9, 297–308 (2008).
Heyer, W.-D., Ehmsen, K. T. & Liu, J. RegulAtion Of Homologous Recombination In Eukaryotes. Annu. Rev. Genet. 44, 113–139 (2010).
Pawelczak, K. S., Gavande, N. S., VanderVere-Carozza, P. S. & Turchi, J. J. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem. Biol. 13, 389–396 (2018).
Kanca, O. et al. An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. eLife 8, e51539 (2019).
Zuccaro, M. V. et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell 183, 1–15 (2020).
Adikusuma, F. et al. Large deletions induced by Cas9 cleavage. Nature 560, E8–E9 (2018).
Kosicki, M., Tomberg, K. & Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 36, 765–771 (2018).
Leibowitz, M. L. et al. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 53, 895–905 (2021).
Nahmad, A. D. et al. Frequent aneuploidy in primary human T cells after CRISPR–Cas9 cleavage. Nat. Biotechnol. 40, 1807–1813 (2022).
Tsuchida, C. A. et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell 186, 4567–4582.e20 (2023).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Kim, Y. B. et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 35, 371–376 (2017).
Gaudelli, N. M. et al. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Klompe, S. E., Vo, P. L. H., Halpin-Healy, T. S. & Sternberg, S. H. Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225 (2019).
Strecker, J. et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 364, 48–53 (2019).
Lampe, G. D. et al. Targeted DNA integration in human cells without double-strand breaks using CRISPR-associated transposases. Nat. Biotechnol. 42, 87–98 (2023).
Saito, M. et al. Dual modes of CRISPR-associated transposon homing. Cell 184, 2441–2453.e18 (2021).
Hsieh, S. & Peters, J. E. Discovery and characterization of novel type I-D CRISPR-guided transposons identified among diverse Tn7-like elements in cyanobacteria. Nucleic Acids Res. 51, 765–782 (2023).
Halpin-Healy, T. S., Klompe, S. E., Sternberg, S. H. & Fernández, I. S. Structural basis of DNA targeting by a transposon-encoded CRISPR–Cas system. Nature 577, 271–274 (2020).
Wang, S., Gabel, C., Siddique, R., Klose, T. & Chang, L. Molecular mechanism for Tn7-like transposon recruitment by a type I-B CRISPR effector. Cell 186, 4204–4215.e19 (2023).
Park, J. U. et al. Multiple adaptations underly co-option of a CRISPR surveillance complex for RNA-guided DNA transposition. Mol. Cell 83, 1827–1838.e6 (2023).
Faure, G. et al. CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nat. Rev. Microbiol. 17, 513–525 (2019).
Vo, P. L. H., Acree, C., Smith, M. L. & Sternberg, S. H. Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA 12, 1–17 (2021).
Schmitz, M., Querques, I., Oberli, S., Chanez, C. & Jinek, M. Structural basis for the assembly of the type V CRISPR-associated transposon complex. Cell 185, 4999–5010.e17 (2022).
Klompe, S. E. et al. Evolutionary and mechanistic diversity of Type I-F CRISPR-associated transposons. Mol. Cell 82, 616–628.e5 (2022).
Roberts, A., Nethery, M. A. & Barrangou, R. Functional characterization of diverse type I-F CRISPR-associated transposons. Nucleic Acids Res. 50, 11670–11681 (2022).
Rybarski, J. R., Hu, K., Hill, A. M., Wilke, C. O. & Finkelstein, I. J. Metagenomic discovery of CRISPR-associated transposons. Proc. Natl. Acad. Sci. USA. 118, e2112279118 (2021).
Petassi, M. T., Hsieh, S. & Peters, J. E. Guide RNA Categorization Enables Target Site Choice in Tn7-CRISPR-Cas Transposons. Cell 183, 1757–1771.e18 (2020).
Walker, M. W. G., Klompe, S. E., Zhang, D. J. & Sternberg, S. H. Novel molecular requirements for CRISPR RNA-guided transposition. Nucleic Acids Res. 51, 4519–4535 (2023).
Vo, P. L. H. et al. CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat. Biotechnol. 39, 480–489 (2021).
Rubin, B. E. et al. Species- and site-specific genome editing in complex bacterial communities. Nat. Microbiol. 7, 34–47 (2022).
George, J. T. et al. Mechanism of target site selection by type V-K CRISPR-associated transposases. Science 382, eadj8543 (2023).
Strecker, J., Ladha, A., Makarova, K. S., Koonin, E. V. & Zhang, F. Response to Comment on “RNA-guided DNA insertion with CRISPR-associated transposases”. Science 368, 1–2 (2020).
Tou, C. J., Orr, B. & Kleinstiver, B. P. Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nat. Biotechnol. https://doi.org/10.1038/s41587-022-01574-x (2023)
Park, J. U. et al. Structural basis for target site selection in RNA-guided DNA transposition systems. Science 373, 768–774 (2021).
Park, J. U., Tsai, A. W. L., Chen, T. H., Peters, J. E. & Kellogg, E. H. Mechanistic details of CRISPR-associated transposon recruitment and integration revealed by cryo-EM. Proc. Natl. Acad. Sci. USA 119, 1–9 (2022).
Querques, I., Schmitz, M., Oberli, S., Chanez, C. & Jinek, M. Target site selection and remodelling by type V CRISPR-transposon systems. Nature https://doi.org/10.1038/s41586-021-04030-z (2021)
Park, J. U. et al. Structures of the holo CRISPR RNA-guided transposon integration complex. Nature 613, 775–782 (2023).
Tenjo-Castaño, F. et al. Conformational landscape of the type V-K CRISPR-associated transposon integration assembly. Mol. Cell 84, 2353–2367.e5 (2024).
Hoffmann, F. T. et al. Selective TnsC recruitment enhances the fidelity of RNA-guided transposition. Nature 609, 384–393 (2022).
Jia, N., Xie, W., de la Cruz, M. J., Eng, E. T. & Patel, D. J. Structure–function insights into the initial step of DNA integration by a CRISPR–Cas–Transposon complex. Cell Res. 30, 182–184 (2020).
Wang, B., Xu, W. & Yang, H. Structural basis of a Tn7-like transposase recruitment and DNA loading to CRISPR-Cas surveillance complex. Cell Res. 30, 185–187 (2020).
Li, Z., Zhang, H., Xiao, R. & Chang, L. Cryo-EM structure of a type I-F CRISPR RNA guided surveillance complex bound to transposition protein TniQ. Cell Res. 30, 179–181 (2020).
Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 18, 176–185 (2021).
Moreb, E. A., Hutmacher, M. & Lynch, M. D. CRISPR-Cas ‘non-Target’ Sites Inhibit On-Target Cutting Rates. CRISPR J. 3, 550–561 (2020).
Tuminauskaite, D. et al. DNA interference is controlled by R-loop length in a type I-F1 CRISPR-Cas system. BMC Biol. 18, 1–16 (2020).
Rollins, M. C. F. et al. Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol. Cell 74, 132–142.e5 (2019).
Guo, T. W. et al. Cryo-EM structures reveal mechanism and inhibition of DNA targeting by a CRISPR-Cas surveillance complex. Cell 171, 414–426.e12 (2017).
Chowdhury, S. et al. Structure reveals mechanisms of viral suppressors that intercept a CRISPR RNA-guided surveillance complex. Cell 169, 47–57.e11 (2017).
Wang, Y. et al. A novel strategy to engineer DNA polymerases for enhanced processivity and improved performance in vitro. Nucleic Acids Res. 32, 1197–1207 (2004).
de Vega, M., Lázaro, J. M., Mencía, M., Blanco, L. & Salas, M. Improvement of φ29 DNA polymerase amplification performance by fusion of DNA binding motifs. Proc. Natl. Acad. Sci. USA 107, 16506–16511 (2010).
Oscorbin, I. P., Wong, P. F., Boyarskikh, U. A., Khrapov, E. A. & Filipenko, M. L. The attachment of a DNA-binding Sso7d-like protein improves processivity and resistance to inhibitors of M-MuLV reverse transcriptase. FEBS Lett. 594, 4338–4356 (2020).
Tong, C. L., Kanwar, N., Morrone, D. J. & Seelig, B. Nature-inspired engineering of an artificial ligase enzyme by domain fusion. Nucleic Acids Res. 50, 11175–11185 (2022).
Jackson, R. N. et al. Structural biology. Crystal structure of the CRISPR RNA-guided surveillance complex from Escherichia coli. Science 345, 1473–1479 (2014).
Xue, C., Zhu, Y., Zhang, X., Shin, Y. K. & Sashital, D. G. Real-Time Observation of Target Search by the CRISPR Surveillance Complex Cascade. Cell Rep. 21, 3717–3727 (2017).
Aldag, P. et al. Dynamic interplay between target search and recognition for a Type I CRISPR-Cas system. Nat. Commun. 14, 3654 (2023).
Qi, L. S. et al. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Skelding, Z., Sarnovsky, R. & Craig, N. L. Formation of a nucleoprotein complex containing Tn7 and its target DNA regulates transposition initiation. EMBO J. 21, 3494–3504 (2002).
Blum, M. et al. InterPro: the protein sequence classification resource in 2025. Nucleic Acids Res. 53, D444–D456 (2025).
Witte, I. P. et al. Programmable gene insertion in human cells with a laboratory-evolved CRISPR-associated transposase. Science 388, eadt5199 (2025).
Zhang, F., Saito, M. & Faure, G. Type I-B CRISPR-Associated Transposase Systems. (2024).
Metagenomi Technologies, LLC. Form S-1 Registration Statement. https://www.sec.gov/Archives/edgar/data/1785279/000119312524003477/d425213ds1.htm.
Strecker, J., Zhang, F. & Ladha, A. CRISPR-associated transposase systems and methods of use thereof. International patent WO2021257997A2 (2021).
Liu, J. et al. Integration of therapeutic cargo into the human genome with programmable type V-K CAST. Nat. Commun. 16, 2427 (2025).
Zivanov, J. et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. eLife 11, e83724 (2022).
Russo, C. J. & Passmore, L. A. Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380 (2014).
Schorb, M., Haberbosch, I., Hagen, W. J. H., Schwab, Y. & Mastronarde, D. N. Software tools for automated transmission electron microscopy. Nat. Methods 16, 471–477 (2019).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Bepler, T. et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Jamali, K. et al. Automated model building and protein identification in cryo-EM maps. Nature https://doi.org/10.1038/s41586-024-07215-4 (2024)
Ondov, B. D., Bergman, N. H. & Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12, 385 (2011).
Acknowledgements
We thank Z. Akhtar for laboratory support, R.T.K. for assistance in mammalian assay design and cloning, L. F. Landweber for qPCR instrument access, the Columbia Stem Cell Initiative Flow Cytometry Core, the JP Sulzberger Columbia Genome Center for NGS support, and the CryoEM facility of the St. Jude Children’s Research Hospital in Memphis, Tennessee, USA where the cryoEM high resolution data was collected. Some of this work was performed at the National Center for CryoEM Access and Training (NCCAT) and the Simons Electron Microscopy Center located at the New York Structural Biology Center is supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and NY State Assembly. S.H.S. was supported by NIH grant DP2HG011650, a Pew Biomedical Scholarship, a Sloan Research Fellowship, an Irma T. Hirschl Career Scientist Award, and a generous startup package from the Columbia University Irving Medical Center Dean’s Office and the Vagelos Precision Medicine Fund.
Author information
Authors and Affiliations
Contributions
G.D.L., A.R.L., and S.H.S. conceived of and designed the project. G.D.L. purified PseQCascade. G.D.L. and A.R.L. performed all cellular experiments and cellular experimental analyses, with the exception of E. coli repression and integration assays, which were performed by D.J.Z and A.R.L. I.S.F. collected cryoEM data and performed structure determination. G.D.L., A.R.L., I.S.F., and S.H.S. discussed the data and wrote the manuscript, with input from D.J.Z.
Corresponding authors
Ethics declarations
Competing interests
Columbia University has filed a patent application related to this work. G.D.L., A.R.L., D.J.Z., and S.H.S. are inventors on other patents and patent applications related to CRISPR–Cas systems and uses thereof (application numbers US20240279629, US20250163410, WO2020181264, WO2022261122, WO2022266492, WO2023245010, WO2024124048, WO2025029727, WO2025085782, WO2025085787). S.H.S. is a co-founder of and scientific advisor to Dahlia Biosciences, a scientific advisor to Prime Medicine and CrisprBits and an equity holder in Dahlia Biosciences and CrisprBits. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lampe, G.D., Liang, A.R., Zhang, D.J. et al. Structure-guided engineering of type I-F CASTs for targeted gene insertion in human cells. Nat Commun 16, 7891 (2025). https://doi.org/10.1038/s41467-025-63164-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63164-0
