
Abstract
Electrocatalytic upgrade of CO2 offers a promising approach for recycling of global CO2 emissions, facilitating the achievement of carbon neutrality. Nevertheless, direct utilization of practical dilute CO2 is urgently important yet rather difficult, which is hindered by the balance of reaction kinetics and mass transport of CO2 to the catalytic sites. Herein, we propose coordinating the local environment and active catalyst by constructing covalent organic frameworks (COF) on single-atomic In-doped Cu2O (In1@Cu2O) for a high tolerance of CO2 inlet concentrations (15% to 100%). The optimized amounts of COF functionalized by the trifluoromethyl group act as the local CO2/CO diffusion channels via steric confinement effects and C···F electronic effects. Besides, the formation of key intermediates for C2+ products is greatly facilitated by the promoted COOH adsorption. Hence, a total current of 81.7 A is realized in a 4 × 100 cm2 electrolyzer stack with over 770 mmol/h C2+ products at an inlet of dilute CO2. Such a electrode architecture sheds light on the dilute CO2 electrolysis at the potential industrial scale.
Similar content being viewed by others


Introduction
Recycling CO2 emissions at ambient conditions by electrocatalytic conversion with renewable electricity grants an elegant carbon-neutral route1,2,3. Such electrochemical CO2 reduction reactions (CO2RR) have shown techno-economic viability to synthesize valuable chemicals and feedstocks4. Nevertheless, the imbalance of reaction kinetics and mass transport with an increasing conversion rate and industrial dilute CO2 limits the large-scale applications of this technique. So far, most of the research on CO2RR is focused on pure CO2 gas to benchmark catalyst or device performances5,6,7. However, the general CO2 concentration is only around 70–95% after capture and separation by the membrane plants from flue gas (15–20%)8,9,10. Hence, the direct utilization of impure or dilute CO2 inlets is highly significant.
The intrinsic challenge for continuous electrolysis of dilute CO2 is that the solubility or partial pressure of CO2 becomes too low to be fed sufficiently to the catalytic sites11,12. Therefore, a concerted manner of concentrating CO2 at a local level and efficiently converting CO2 is urgently needed for further practical developments. Particularly, it is crucial to manage both localized reaction environments and catalytic sites for enrichments of local reactants or intermediates and improvements of conversion rates, respectively. Researchers have been investigating the use of covalent organic frameworks as sorption materials for capturing CO2 because of a high affinity for CO213. Meanwhile, COF-based materials have also been extensively studied for CO2RR, either using them as potential catalysts with tuned active sites or introducing them into catalytic layers with functionalized groups14. Attributed to the unique porous configuration and conjugated electronic structure, COF materials can be employed for hydroxide and gaseous feedstock adsorption15 or hydrophilic–hydrophobic nanochannels16 for regulating the local environment.
Herein, we coordinate reaction kinetic promotions and mass transport regulations by constructing localized mass transport channels for the scale-up of the electrolyzer stack with industrial-level electrosynthesis of C2+ products from dilute CO2. As shown in Fig. 1a, through constructing mass transport channels with a COF layer on the single atomic In doped Cu2O (In1@Cu2O), CO2 concentrating and converting processes are coupled together. Hence, the dilute CO2 (70–90%) and even simulated flue gas (15% CO2) are efficiently converted into C2+ products at ambient conditions. In detail, a COF functionalized by the trifluoromethyl group (TfCOF) acts as localized channels for localized CO2/CO mass transport. Besides, active In1@Cu2O is found to strengthen the adsorption of *COOH for the production of CO key intermediate toward C2+ products17. Correspondingly, such TfCOF-In1@Cu2O electrode converts dilute CO2 with a Faradaic efficiency for C2+ products (FEC2+) of 83.5% at Ecell of 3.4 V, presenting only a 3.4% decrease in FEC2+ compared with the performance testing under 100% pure CO2 inlet. Such high selectivity is well maintained for over 96 h with a current density of over 700 mA/cm2. Additionally, a 4 × 100 cm2 electrolyzer stack is assembled to achieve over 770 mmol/h C2+ products with a total current of 81.7 A with a dilute CO2 inlet.

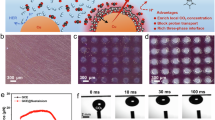
a Schematic of relationships among reaction kinetics, mass transport, and industrial-level electrosynthesis. The atomic schematic is a magnified view of the highlighted region in the green boxes of the catalytic layer to highlight the reactive interface between TfCOF and In1@Cu2O. TfCOF layer on single atomic In1@Cu2O surface acting as localized channels for CO and CO2 diffusion, which synergistically balances the reaction kinetics promotions and facilitates the scale-up of electrolyzer stacks. b Synthetic scheme of TfCOF by an ion-exchange process. Tp is 1,3,5-triformyl phloroglucinol and BTBD is 2,2′ Bis(trifluoromethyl)benzidine. c The schematic structure of TfCOF with the pore size of 18 Å. d WAXS patterns of TfCOF and the carbon paper background. e TEM, HRTEM, STEM, and element mapping images of synthesized TfCOF before the activation step. f HAADF-STEM image of TfCOF-In1@Cu2O. The yellow circles highlight representative single In atoms, which are brighter compared with Cu atoms. g Three-dimensional visualization of region \({{\rm{\alpha }}}\) in (e) to depict single atomic In atoms. h Element mapping images of TfCOF-In1@Cu2O.
Results
Electrode synthesis and in-situ activation
The TfCOF is proposed to tune the mass transport at the local environment near the catalytic sites, which is synthesized from 1,3,5-triformyl phloroglucinol (Tp) and 2,2′-Bis(trifluoromethyl)benzidine (BTBD) with the formation of Enol-imine form and further Keto-enamine form (Fig. 1b). The structures of TfCOF are illustrated as shown in Figs. 1c, S1, and S2a. The pore size is around 18 Å, which is characterized by the wide-angle X-ray scattering (WAXS, Fig. 1d), the nitrogen adsorption/desorption analysis (Fig. S2b, c), and the small-angle X-ray diffraction (XRD) patterns (Fig. S2d). The high crystallinity and the compositions of TfCOF are also observed by the crystalline lattice in transmission electron microscopy (TEM) images and elemental mapping images (Fig. 1e), respectively.
On the other hand, the active site is also delicately designed to promote the reaction kinetics of CO2-to-C2+ products. As reported18, In doping of Cu-In alloy highly suppresses the hydrogen evolution and regulates the electronic structure of Cu for selective synthesis of CO intermediate. Therefore, single atomic In is further introduced to modify the surface Cu2O site. The synthesis process of TfCOF-In1@Cu2O and the structure evolutions are presented in Fig. S3. An epoxide gelation method was firstly employed to prepare In-Cu gel, which consists of Cu2(OH)3Cl and In(OH)3. This In-Cu gel was aged in the oven for 24 h, and then it was mixed with TfCOF to prepare catalyst ink, which was drop-casting on the carbon paper to form the electrode (Fig. S4). Under CO2RR conditions, the synthesized electrode was subjected to in-situ electrochemical activation at a constant potential for 30 min, and then TfCOF-In1@Cu2O electrode was eventually obtained.
It is revealed by scanning electron microscopy (SEM) images that abundant interlaced channels are formed in the as-prepared TfCOF-In1@Cu2O by cross-linking of nanoparticles (Figs. S5 and S6). To further investigate this material, the high-resolution TEM (HRTEM) is examined as depicted in Figs. S7 and S8. It is found that such nanoparticles are quite fine and highly dispersed. Moreover, as shown in the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images, isolated bright spots are identified as representing atomically dispersed In atoms in the Cu2O matrix (Figs. 1f and S9). The lattice fringe with a d-spacing of 0.245 nm corresponds to the Cu2O (111) plane, which is increased to 0.250 nm with the substitution of single atomic In over the Cu2O matrix. This structural distortion directly evidences In incorporation into the bulk lattice19,20. As shown in Fig. 1g, h, the 3D visualization and element mapping images further depict the distribution of single atomic In atoms.
Synchrotron X-ray absorption spectroscopy (XAS) characterizations are further performed to reveal the local electronic structures and coordination environment of Cu and In in TfCOF-In1@Cu2O catalyst21,22. The Cu K-edge of X-ray absorption near-edge structure (XANES) for TfCOF-In1@Cu2O is depicted in Fig. 2a, as well as the spectra of copper foil, Cu2O, and CuO references. The near-edge absorption peak of TfCOF-In1@Cu2O is located between those of Cu foil and Cu2O, indicating the chemical valence of copper species is between 0 and +1, and close to +123,24. As for the catalyst modification, it is well known that monovalent Cu(I)-based catalysts are favorable for C–C bond formation25,26,27, while single-atom alloys are reported to steer the electronic features of host material with the introduction of foreign atoms20.

a XANES spectra of Cu K-edge. EXAFS spectra of b Cu K-edge and c In K-edge, respectively. d Operando WAXS patterns detected during the activation process. Operando WAXS spectra of e In(OH)3 leaching and f Cu2O generation during the activation step. g 3D images of TOF-SIMS elemental depth distribution of Cu and In elements before and after in-situ activation step. The bottom images are the surface distributions of single atomic In formation. Quasi in-situ NAP-XPS spectra of h Cu LMM and i In 3d spectra during the activation process.
With the analyses of the extended X-ray absorption fine structure (EXAFS) for Cu K-edge (Fig. 2b), the peak in R space at 1.46 Å is ascribed to the contribution of Cu-O bonds from Cu2O28,29. The broad peak at 2.35 Å is ascribed to the co-existence of Cu-In and Cu-Cu paths24,30. Meanwhile, XAS curves of In K-edge for TfCOF-In1@Cu2O, indium foil, and In2O3 references are shown in Figs. 2c and S10. Two peaks at 1.64 Å and 2.47 Å are attributed to In-O and In-Cu paths, respectively31. Notably, there is no In-In coordination in TfCOF-In1@Cu2O, revealing that indium is atomically dispersed on Cu2O.
Single atomic In is formed during the in-situ activation step, and such evolution of crystal structures and 3D elemental distribution of TfCOF-In1@Cu2O is captured by operando synchrotron 2D-WAXS measurements as shown in Fig. 2d–g. Before the in-situ activation (0 min), the crystal patterns of Cu2(OH)3Cl (JCPDS#50-1559) and In(OH)3 (JCPDS#16-0161) are presented. Notably, these peaks gradually disappear and the existence of Cu2O (JCPDS#78-2076) can be detected as the activation proceeds24,32. It is worth noting that no In-In peaks are observed for the TfCOF-In1@Cu2O material, which further confirmed that there are no In particles formed. These results are further clearly verified by the integrated XRD curves in Fig. 2e, f. The In(OH)3 (2 theta = 5.04) leaches out rapidly, while Cu2(OH)3Cl (2 theta = 3.65, Fig. S11) is converted to Cu2O (2 theta = 8.27).
Time-of-flight secondary-ion mass spectroscopy (TOF-SIMS) with vacuum interconnection was also performed to analyze the elemental distribution in different depths of the electrode33. After the activation in the glove box, the electrodes were transferred to the test station for TOF-SIMS measurements immediately. The 3D rendering of Cu and In species in the electrode before and after the activation step is depicted in Fig. 2g. In elements are observed to be distributed along different depths of the electrode before the activation step. Then the content of In is greatly reduced after the activation step (Fig. S12), which is consistent with the 2D-WAXS results. These results reveal that a large amount of In atoms leach out during the in-situ activation process. The distribution of other elements for TfCOF-In1@Cu2O catalysts is shown in Figs. S13 and S14. Notably, the uniform distribution of K element in Fig. S11b indicates that the TfCOF-In1@Cu2O catalyst holds porous structures for electrolyte penetrations.
Additionally, quasi in-situ near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) is performed to further elucidate the evolution of electronic structure and chemical states34,35. As shown in Fig. S15, the binding energy of all Cu 2p3/2 spectra for TfCOF-In1@Cu2O catalyst is around 932.7 eV, attributing to Cu0 or Cu+ peaks24,36. The Cu LMM Auger spectra further clarify the major Cu+ species in the material after the activation step (Fig. 2h). Moreover, according to In 3d spectra in Fig. 2i, the In3+ peaks of In-Cu gel are located at 452.5 eV and 444.9 eV, which show a positive shift of about 0.23 eV after the activation step. The result indicates that In atom has a higher valence state than In3+, probably due to re-oxidations after the in-situ activation process.
Localized mass transport channels for dilute CO2 electrolysis
We employ TfCOF-In1@Cu2O for regulating both catalytic sites and localized diffusion channels to simultaneously promote the reaction kinetics and regulate local mass transport for efficient dilute CO2 electrolysis (Fig. 3a). In detail, In1@Cu2O plays the role of accelerating the production of CO key species for C–C coupling via enhancing the adsorption of *COOH intermediate as illustrated in the following Density Functional Theory (DFT) simulations. On the other hand, the TfCOF layer acts as the CO and CO2 diffusion channels and makes it possible for them to contact with electrons from the catalyst and protons from the electrolyte, as illustrated by combined Multiphysics and Molecular Dynamic simulations. It is worth mentioning that the TfCOF layer also avoids the rapid departure of CO species to the electrolyte.

a Schematic diagram of the specialized roles for In1@Cu2O and TfCOF to promote reaction kinetics and regulate mass transport, respectively. Inset in the middle revealing the steric effect for CO2 by calculating CO2 adsorption distribution. b FEs of different products for Cu2O and TfCOF-In1@Cu2O with dilute 80% CO2 inlets. The cell voltage of Ecell is reported without iR correction. The error bars represent the standard deviation from the measurement of three independent electrodes. c FFs toward total carbon products (FECtot) and d C2+ products (FEC2+) for TfCOF-In1@Cu2O in a flow cell with different concentrations of CO2 inlets. The simulated flue gas is 15% CO2 and 85% N2. e Calculated 2D distributions of CO concentration around electrodes with and without TfCOF layer with Multiphysics simulations. f Calculated coverages of adsorbed *CO and CO2 on the catalytic surface. The error bars represent the standard deviation from the measurement of three independent electrodes. Source data are provided as a Source Data file.
With the target of extending the commercial utilization of CO2RR systems, we employ a customized flow cell with a CO2-saturated aqueous inlet to deliver the carbon sources3. The architecture of neutral electrolyte flow-through electrodes not only eliminates the degradation from carbonate salt precipitations and electrolyte leakage but also highly reduces carbon losses (<5%)37 with a cation exchange membrane to avoid the crossover of bicarbonate and carbonate anions3. The catholytes are bubbled by a series of pure and impure CO2 gases: 100%, 90%, 80%, and 70% CO2. These typical concentrations are selected to simulate the CO2 waste gas after the membrane plants from fuel gases9,10. As shown in Fig. S16, FE toward total carbon-containing products (FECtot) shows a sharp decrease from 82.6% to 49.4% at Ecell of 3.4 V on pure Cu2O when changing the inlet from pure CO2 to 80% CO2. It is attributed to that low concentrations of CO2 inlet reduce the partial pressure of CO2 (PCO2) and further decrease the solubility of CO238, which highly reduces the selectivity of CO2RR on pure Cu2O.
Dilute CO2 is concentrated according to the electrostatic interaction by the modifications of TfCOF in the electrode13,14,39. As shown in Fig. S17a, Faradaic efficiency toward total carbon-containing products (FECtot) is promoted to 86.9% using TfCOF-Cu2O and 80% CO2 inlet. We further performed the measurements of electrochemically active surface area (ECSA, Fig. S18) and ECSA-normalized current densities to exclude the porosity effect (Fig. S19). It can be found that which TfCOF-Cu2O electrode still maintains substantially higher C2+ partial current density (5.6 mA cm−2 at 3.4 V) compared to that of Cu2O electrode (2.7 mA cm−2 at 3.4 V). This indicates that the enhanced performance is dominated by the interaction between the TfCOF and the CO2 reactant. Nevertheless, the selectivity of C2+ products is still less than 75%. The possible reason is the production and concentration of CO intermediate adsorbed on the catalysts are too low to proceed for the further C–C coupling on pure Cu2O for impure CO2 electrolysis. Meanwhile, it is found that In1@Cu2O has a higher ability to produce CO than Cu2O (Fig. S20), which confirms the critical role of single atomic In doping for the facilitating of reaction kinetics.
As expected, the selectivity of C2+ products on TfCOF-In1@Cu2O demonstrates strong tolerance on a wide CO2 concentration window, which shows high FECtot and FEC2+ in the flow cell as shown in Fig. 3b. A maximum value of 86.3 ± 2.4% at Ecell of 3.4 V is delivered for pure CO2 electrolysis. The obtained FECtot and FEC2+ using 90% CO2 inlet are almost the same as those using pure inlet at a wide potential window between 2.6 V and 3.6 V (Fig. S18a). Meanwhile, FEC2+ reaches 83.5% and FECtot is 95.5% at Ecell of 3.4 V with 80% CO2 inlet, showing comparable performance with 100% pure CO2 and the performance damping is less than 5%. The high selectivity and activity are attributed to the balance of effective capture of CO2 by TfCOF and faster reaction kinetic by In1@Cu2O.
FECtot drops when the inlet changes to 70% CO2, which is around 85.8% ± 2.2% at Ecell of 3.4 V (Fig. 3c). The 11.7% reduction in FECtot compared with pure inlet is likely due to the limited CO2 supply for the reactions40, which is more evident under larger cell voltages. With the inlet of simulated flue gas of 15% CO2, TfCOF-In1@Cu2O still presents a high conversion ability of CO2, and the FECtot is over 60% at Ecell between 2.8 V and 3.6 V (Figs. 3d and S18b).
To reveal the effect of the TfCOF layer on the localized concentrations and coverages of CO and CO2, Multiphysics simulations coupled with MD calculations are employed with considerations of the mass transport, buffer reactions, and electrochemical reactions in the near-surface region of the catalyst (Fig. 3e, f)41,42. The diffusion coefficients of CO and CO2 in the catholyte with diffusion channels are calculated from Mean Squared Displacement functions from MD calculations (Fig. S21), which are the inlet parameters used for Multiphysics simulations.
It can be seen that the local concentrations of CO and CO2 around the catalyst are much higher after the modification by TfCOF (Figs. 3f and S22). The coverage of adsorbed CO (*CO) varies from 0.06 to 0.28 after adopting TfCOF layer, which shows 4.7 times promotion. This high coverage of *CO provides sufficient *CO for further reduction, which accounts for the higher FEC2+ on TfCOF-In1@Cu2O than that on In1@Cu2O. The addition of such TfCOF also facilitates the coverage of *CO2, swinging from 0.22 to 0.31, which provides more carbon sources for CO2 electrolysis on the catalytic sites43. The simulated results reveal that the existence of TfCOF layer could highly increase the coverage and localized concentration of CO and CO244,45. The enrichment of these species around active sites can realize the further reduction reactions toward valuable C2+ products.
Besides Multiphysics numerical simulation, the calculated CO2 distribution probability using MD simulations is also shown in the middle inset of Fig. 3a. The red dots in the pores of TfCOF indicate the distribution possibilities of CO2 accumulation region. High possibilities occur at the corners of nanopores in TfCOF, which reflect the special steric confinement effect on CO2 by TfCOF46. TfCOF holds a large amount of CO2 with ample adsorption sites (Table S1), ascribing to the high electrostatic interaction of CO2. Such TfCOF additive layer further breaks the solubility limitations from the low pCO2 and promotes the local concentration of CO2 for the reactions, which further improves the tolerance of lower inlet concentrations of CO2.
High yield and stability with impure CO2 inlets
The synthesized TfCOF-In1@Cu2O not only presents a high selectivity for C2+ products but also exhibits improved current densities and yields. As shown in Fig. 4a, the total current density (J) for TfCOF-In1@Cu2O exceeds 1400 mA/cm2 at Ecell of 3.8 V with the inlet of pure CO2 in the flow cell. FECtot is also over 87% at such a high current, which suggests a pronounced catalytic activity and CO2 utilization. With the inlets of dilute 90% and 80% CO2, the total current densities remain high at Ecell of 3.8 V, which are 1355 mA/cm2 and 1245 mA/cm2, respectively. TfCOF-In1@Cu2O also presents a total current density of 200 mA/cm2 at Ecell of 3.4 V for the direct conversion of simulated flue gas with 15% CO2. Meanwhile, the maximum selectivity of C2H4 reaches 72.9 ± 2.2% at Ecell of 3.4 V, with FEC2+ of 83.5% and FECtot of 95.5% when using the 80% CO2 inlet. Likewise, we observe that TfCOF-In1@Cu2O electrode delivers a superior partial current density toward C2+ products (JC2+) of 837 mA/cm2 at 3.4 V with pure CO2 inlet, while it remains 768 mA/cm2 with 80% impure CO2 inlet (Fig. 4b), showing only 9% decrease in the production rate with 20% decrease of CO2 concentration at such a high current density.

a Total current densities (J) of TfCOF-In1@Cu2O with different CO2 concentrations. b Partial current densities toward C2+ products under different potentials and CO2 concentrations. c Yield of CO, CH4, C2H5OH, and C2H4 with the inlet of 80% CO2. The yield for C2H6 is not shown in the figures due to the low selectivity (FE < 3%). d Total current density and partial current density for C2+ and C1 products with different adding amounts of TfCOF. e Stability measurements of TfCOF-In1@Cu2O at Ecell of 3.4 V with inlet of 80% CO2. f Photograph of the electrolyzer stack. g Total current (JTtot) and partial current for C2+ (JTC2+) and C1 (JTC1) products with stack potentials in the electrolyzer stack with inlet of 80% and 100% CO2. The error bars represent the standard deviation from the measurement of three independent electrodes. Source data are provided as a Source Data file.
As depicted in Fig. 4c, we realize a total yield for carbon products of 3.45 mmol/h/cm2 at Ecell of 3.6 V with the impure inlet of 80% CO2, and the C2H4 yield is 2.36 mmol/h/cm2. It is worth mentioning that the separations of C2H4 from CO and CH4 are comparatively mature industrial processes, which makes the TfCOF-In1@Cu2O catalyst a promising material for CO2 electrolysis. The efficient conversion of dilute CO2 is ascribed to the simultaneous steric confinement effect and electronic effects of TfCOF on CO2 and CO as discussed previously. Specifically, dilute CO2 molecules are concentrated by TfCOF with ample adsorption sites47, and the adsorption energies are revealed by the DFT simulations (Figs. S23 and S24).
Different amounts of TfCOF additives are also conducted with weight ratios of 0.1, 0.2, and 0.3 relative to the In-Cu gel (Figs. S25 and S26). The amount of TfCOF is critical to ensure the high yield of the C2+ products and the optimized ratio of TfCOF is around 0.2. The morphologies of different additions of TfCOF remain similar to the fine particles after the in-situ activations (Figs. S27 and S28). The JC2+ of 698 mA/cm2 with 80% dilute CO2 inlet is realized on the catalyst with TfCOF(0.3), showing around 10% reduction compared with that of TfCOF-In1@Cu2O. On the other hand, TfCOF(0.1)-In1@Cu2O shows 18.5% decrease of JC2+ compared with that of TfCOF-In1@Cu2O (Figs. 4d and S29). This could be due to electron conductivity loss at higher TfCOF loadings and reduced CO/CO2 concentrating effects at lower loadings.
The stability of TfCOF-In1@Cu2O is also measured with 80% CO2 inlet. A current density of 760 mA/cm2 is well maintained for 96 h at Ecell of 3.4 V with FECtot over 95% and FEC2+ over 80% during continuous operation (Fig. 4e). The high yield of C2H4 is around 2.1 mmol/h/cm2, which is about 200 mmol after a four-day electrochemical CO2 upgrade with 1 cm2 electrode. It can be found that the morphology (Fig. S30) and electronic structures (Fig. S31) of TfCOF-In1@Cu2O are well-maintained during the 96-h stability testing (Fig. S32). Such high yield and stability performance with 80% CO2 inlet surpasses lots of reports with pure CO2 inlet (Table S2).
The single-pass conversion efficiency consistently surpasses 25% starting from an applied voltage of 3.0 V when using simulated flue gas with 15% CO2 feed concentration (Fig. S33), demonstrating promising prospects for industrial application. As the partial pressure of feed CO2 decreases to 70% and 15%, the total carbon conversion rate exhibits a moderate decline. Consequently, under a feed condition of 15% CO2, the energy conversion efficiency drops to approximately 20% at 3.2 V with a current density of 200 mA/cm2. Both the single-pass conversion efficiency and energy efficiency surpass the reported values under the same conditions (the same current density and CO2 inlet rate), demonstrating significant competitiveness3,48,49.
With the target of potential industrial applications, an electrolyzer stack is further assembled as shown in Fig. 4f, which consistents of four 10 × 10 cm2 modules connected in series3,50,51. The maximum production rate of C2+ products is over 770 mmol/h with an inlet of 80% CO2, corresponding to a total current of 81.7 A (Fig. 4g). With the utilization of pure CO2 inlet, the maximum total current of 95.9 A is achieved with the C2+ production rate of around 800 mmol/h. Therefore, such a dual strategy of mass transport regulations and reaction kinetics promotions is scalable for potential industrial-level applications.
Underling mechanism
To evaluate the catalytic site of a single atomic In dopant combined with monovalent Cu(I) on CO formation, we firstly set up four surface models by the periodic DFT computations: Cu (111), Cu2O(111), In1@Cu (111) and In1@Cu2O (111) as shown in Figs. S34–S37, respectively. The atomic coordinates of the optimized computational models are shown in Supplementary Data 1 file. The reaction energies are depicted in Fig. 5a for the pathways of CO production52,53,54,55. Among the four surface models, In1@Cu2O (111) shows the lowest Gibbs free energy for COOH* formation, indicating the promoting effect from single atomic In doping in Cu2O. The reaction energy for COOH formation and CO desorption steps are all lower on In1@Cu2O (111) facets compared with those on pure Cu2O (111). This indicates that the observed performance improvements stem from the relatively stronger COOH adsorption and weaker CO adsorption by In-induced electronic modulation of Cu2O surface.

a Relative Gibbs free energy (ΔG) profiles of the CO2-to-CO reaction on Cu(111), Cu2O (111), In1@Cu (111), and In1@Cu2O (111) facets. b Crystal orbital Hamiltonian population (COHP) analysis between COOH with In1@Cu2O(111) substrate and Cu(111) substrate. The dashed line in pink and green color indicates the weighted average location of the antibonding level. c Schematic diagram of COHP analysis. DOS is density of states. d C⋯F electronic effect for CO, and the molecular electrostatic potential map of CO interacted with TfCOF. Inset: representative snapshots showing the pure TfCOF and close interactions of CO-TfCOF, respectively. Cyan, red, blue, white, and pink sticks: C, O, N, H, and F atoms. e Radial distribution function (RDF) and coordination numbers (CN) of CO and CO2 with the distance from TfCOF. Inset is the representative snapshot showing close interactions of TfCOF-CO. An electric field of 5 V/nm was added in the Z-direction. f CO and CO2 distributions in the lattice implemented in the MD simulation considering reactive surface (yellow), K+ (blue balls), H2O (red dots), HCO3− (short-stick structures, red, white, cyan: O, H, C) and TfCOF (long-stick structures, blue, white, and pink sticks: N, H, and F). The element color of (e) is the same as (f). g The density distribution of CO along the Z-direction of the modeling lattice. h, i In-situ FTIR spectrum of TfCOF-In1@Cu2O for CO2RR in different wavenumber regions. Source data are provided as a Source Data file.
To better understand the intrinsic mechanism for the enhanced activity for CO formation on In1@Cu2O, the projected density of states of the C atom in the key COOH* adsorbate and corresponding surface Cu atoms in Cu(111), In1@Cu (111), and In1@Cu2O (111) were calculated (Fig. S38). The electron density and the wave function are decomposed to reveal the atomic orbital contributions56,57. The overlaps between the C-2p and Cu-3d states on In1@Cu2O are the largest among all the models, which shows the strong interaction between the C and Cu atoms58.
The crystal orbital Hamiltonian population (COHP) analysis was further performed. The weighted average locations of antibonding levels were also identified in this work. We found that a higher antibonding orbital level ((d-π*)*) relative to the Fermi level was delivered for In1@Cu2O (111) than Cu substrate after the interaction of the frontier orbitals with COOH. Additionally, the calculated -ICOHP values reveal significantly stronger bonding between COOH and the In1@Cu2O (111) substrate (–ICOHP = 2.156, Fig. 5b) compared to that between COOH and Cu surface (–ICOHP = 1.943, Fig. 5b).
We also calculated the C–C coupling step to produce *OCCO intermediate from two *CO (Figs. S39 and S40)48. We found that the reaction energy for *OCCO formation is lower on In1@Cu2O (111) facets compared with that on the pure Cu (111) surface. This indicates that the observed performance improvements stem from both the relatively stronger COOH adsorption and *OCCO intermediate stabilization by In1@Cu2O (111) facets.
Hence, such a strategic combination of foreign In atomic In atoms doped in oxidized monovalent Cu(I) leads to the highest selectivity towards CO by holding specific electronics and features that are rather different from their counterpart metals. The proposed mechanism is also depicted by the schematic of Fig. 5c. Incorporating single atomic In into Cu2O results in a larger energy split of hybrid orbitals when incorporated with the *COOH34,59. A larger –ICOHP results in stronger interactions between substrate and molecules.
TfCOF possesses unique 3D porous structures, which can availably regulate the mass transport and enrichments of CO and CO2 near the catalyst surface. The electrostatic potential of the F atom becomes larger after interacting with CO (Fig. 5d), which is attributed to the electron density transfer from F atom of TfCOF to C atom in CO. Hence, the negative regions in the molecular electrostatic potential maps extend to the oxygen atom of CO. The distance of C and F atoms is found to be 2.25 Å (Figs. 5e and S41). Such C···F interactions make the TfCOF become CO diffusion channels near the catalytic surface. As revealed, the strong electrostatic interaction between CO and TfCOF is further confirmed by the radial distribution function (RDF) and coordination numbers (CN) around TfCOF.
The distributions of CO and CO2 are shown in the lattice for the MD simulation (Figs. 5e, f and S42). The random TfCOF molecules are gathering around 5 nm away from the catalytic surface. This assembled structure is likely causing the existence of multiple layers of CO and CO2 between the TfCOF layer and catalytic surface due to different electrostatic interactions, which results in the analogous normal density distribution of it along the Z-direction (Fig. 5f). The top layers of CO (5–8 nm) are enriched by TfCOF layer ascribed to the stronger van der Waals’ interactions, while the bottom layers of CO with moderate interactions are close to the catalytic sites (around 1 nm in the simulation lattice) and can be released from TfCOF to the catalytic surface for further reduction. This is the proposed mechanism for the simultaneous capturing and releasing of CO and CO2 molecules by TfCOF with the C\(\cdots\)F electronic effect and steric effect, respectively.
Such mass transport regulation effects are further depicted by the experimental confirmations with in-situ electrochemical attenuated total reflection Fourier-transform infrared (ATR-FTIR) spectroscopy to monitor the intermediates of In1@Cu2O/COF at various potentials (Fig. 5h, j)60,61,62. The important intermediates for the generation of C2+ products were observed, including the CO* (at 2065 cm−1)63, and CO bound to the copper surface (at 1967, 1919 cm−1)64. The intensity of these peaks increased with the cathode potentials, indicating the high coverage of *CO species. The enhancement in *CO adsorption promoted the subsequent C−C coupling. Moreover, the characteristic peak at 1541, 1423, and 1396 cm−1 corresponds to COOH* and *COO− (two O coordinated), respectively17,65,66. The *H2O peak is observed at 1649 cm−1 (Fig. 5i)63. The peaks at 1743, 1554, and 1519 cm−1 are attributed to CO32− species67. The pronounced peak at 2344 cm−1 is assigned to the CO2* (Fig. S43), demonstrating the adsorption of CO2 on TfCOF-In1@Cu2O. These findings further demonstrate the effective regulations of CO and CO2 species near the catalytic sites by the TfCOF.
In short summary, the C2+ product formation is enhanced by the combined effect of In doping and TfCOF. Besides, the high tolerance of CO2 concentration during CO2RR is delivered by the TfCOF-mediated confinement effects. In detail, (1) the role of In doping: stronger COOH adsorption and *OCCO intermediate stabilization are obtained by In-induced electronic modulation of Cu2O surface (Fig. 5a–c); (2) the role of TfCOF: the optimized amounts of COF functionalized by the trifluoromethyl group act as the local CO2/CO diffusion channels via steric confinement effects and C···F electronic effects (Fig. 5d–g).
Discussion
Efficient electrolysis of dilute CO2 (70–90% and 15%) toward C2+ products is realized through employing combined TfCOF and single-atom active sites of In1@Cu2O. Supported by the multi-scale simulation and experimental characterizations, TfCOF is proved to be the local diffusion channels of CO2/CO and optimized amounts of TfCOF provide the proper concentrating of these molecules. In1@Cu2O plays the role of promoting the production of CO key intermediate for C–C coupling. Hence, the maximum FEC2+ of 83.5% and FECtot of 95.5% are obtained at Ecell of 3.4 V when using the 80% CO2 inlet. The high C2H4 yield of 2.1 mmol/h/cm2 is delivered for four days with an impure CO2 inlet. Such a dual strategy is further scalable to the electrolyzer stack with a high yield of C2+ products of over 770 mmol/h. The primary contributions of this work are: (1) a high tolerance of CO2 inlet concentrations (15%–100%) is realized; (2) steric confinement effects and C···F electronic effects of the Tf Group are implemented; (3) CO2 concentrating and converting processes are coupled together.
Methods
Chemicals
Copper(II) chloride dihydrate (CuCl2·2H2O, metal basis >99.99%), indium nitrate hydrate (In(NO3)3·5H2O, metal basis >99.99%), ethanol (>99.7%), propylene oxide (>99.5%), and isopropanol (> 99.7%) were purchased from Aladdin. Nafion solution (5 wt.%) was purchased from Macklin. Nafion 211 cation-exchange membranes were obtained from Dupont. All of the chemicals were used without further purification. The ultrapure water was obtained from the Milli-Q system.
Synthetic procedures
Preparation of TfCOF: 1 mmol 1,3,5-triformyl phloroglucinol (Tp, 210 mg) and 1 mmol 2,2′-Bis(trifluoromethyl)benzidine (BTBD, 324 mg) were added to a Pyrex tube containing 10 mL mixed solvent (dioxane/mesitylene = 1:1, volume ratio) with 1 mL acetic acid (6 M), which was then sonicated for 20 min. The mixture was flash-frozen under liquid nitrogen. After that, the mixture was sealed to the oven and dried at 120 °C for 3 days. The resultant precipitate was collected by filtration and washed with acetone, dichloromethane, and tetrahydrofuran, respectively. The precipitates were then heated at 120 °C for 24 h in a vacuum oven to get the material, which was named TfCOF.
Preparation of In-Cu gel: The CuCl2·2H2O (3.0 mmol) and In(NO3)3·5H2O (0.2 mmol) were dissolved in 2 ml isopropanol. Under consistent bath sonication, 2 ml propylene oxide was added to the resulting solution and mixed for 20 s. Then, 0.2 ml H2O was added and sonicated continuously until a homogeneous grey-green gel was formed. The above grey-green gel was aged for 24 h and eventually dried at 60 °C, lasting 24 h in a vacuum oven.
Preparation of TfCOF-In1@Cu2O electrode: First, the above In-Cu gel was formulated into ink solution in the usual way. In a typically prepared procedure of the working electrode, 5 mg of the dried In-Cu gel was mixed with TfCOF (In-Cu gel: TfCOF = 5:1, weight ratio), 1 ml water-isopropanol solution with a volume ratio of 7:3 and 80 μl Nafion solution (5 wt%) to prepare an ink solution. Then, the pristine electrode was obtained by loading the ink solution onto Sigracet 29 BC carbon paper of 1.0 × 1.0 cm and dried in an infrared drying oven. Finally, under CO2RR conditions, the TfCOF-In1@Cu2O electrode was in-situ electrochemically activated in a CO2-saturated 0.5 M KHCO3 electrolyte at a constant potential of −3.14 V vs. RHE for 30 min (E(RHE) = E(SCE) + 0.235 + 0.0591 × pH). TfCOF-In1@Cu2O(0.1) and TfCOF-In1@Cu2O(0.3) electrodes were synthesized as described above, but with different additions of TfCOF (weight ratios of In-Cu gel:TfCOF = 1:0.124, In-Cu gel: TfCOF = 1:0.372, respectively).
Preparation of In1Cu2O/Cu electrode and Cu nanoparticles (Cu NPs): In1Cu2O/Cu electrode and Cu nanoparticles were synthesized in a similar way to that used for the TfCOF-In1@Cu2O catalyst, except that TfCOF powder and In(NO3)3·5H2O were not added.
Electrochemical measurements
Electrochemical CO2 reduction was conducted in a customized flow cell with a Nafion 211 membrane to separate the two compartments, preventing the reoxidation of CO2RR products at the anode3,40. Different inlets of pure and impure CO2 were continuously bubbled into the catholyte bottle to saturate 0.5 M KHCO3 solution (pH value is 7.2 ± 0.1). N2 is used to balance and simulate the impure CO2 from fuel gas9,10. The flow rate of gas was controlled at 60 ml/min by a mass flow meter. The anolyte was also 0.5 M KHCO3 solution. All electrochemical experiments were tested using a Biologic VMP-300 Potentiostat with a current booster. The cell voltage of Ecell is reported without iR correction. The current densities were normalized to the geometric surface area in this work. The partial current densities were equal to the total current densities multiplied by the Faradaic efficiency. Three independent measurements were performed under ambient pressure and room temperature (25 °C).
The electrolyzer stack was assembled by four module units with an electrode area of 10 × 10 cm2. Two 1 L electrolyte bottles were employed for 0.5 M KHCO3 solutions as the anolyte and catholyte. The inlet was separated into four modules in parallel, and the outlets were collected in the bottom flow channels.
Products analysis
The gaseous products were quantified by the online gas chromatograph (GC, GC-2014 Shimadzu) with a packed molecular sieve column. The online GC is equipped with a flame ionization detector (FID 1) for CO and CH4, a flame ionization detector (FID 2) for C2H4, C2H6, and C2H2, as well as a thermal conductivity detector for H2. The flow rates of the gaseous outlet are quantified by an additional flow-meter between the catholyte bottle and online GC. Liquid products in the catholyte were analyzed on a nuclear magnetic resonance spectroscopy (400 MHz 1H liquid NMR spectrometer, Bruker Avance NEO) with the water suppression method. 1 ml of product-containing electrolyte was collected from the cathodic compartment after electrolysis. It was mixed with an internal standard of dimethyl formamide (Sigma-Aldrich, > 99.9%) and D2O (Sigma-Aldrich, 99.9%), and then sealed for NMR measurements.
The Faradaic efficiency (FE) can be calculated as the following equation:
where, \(z\) is the number of electrons exchanged in the reaction, \(n\) is the number of moles of product formed, \(F\) is the Faraday constant, and \(Q\) is the total amount of charges passed through the CO2RR process. The partial current density was then obtained by multiplying the Faradaic efficiency with the average current density.
Characterizations
The XRD was performed to disclose the crystal structure of the as-prepared materials by Bruker/D8 ADVANCE X-ray diffractometer. Scanning electron microscope (SEM, ZEISS Sigma 500) and transmission electron microscopy (TEM, FEI Talos F200X) were applied to observe the morphologies and microstructures of materials and electrodes. The FEI Talos F200X TEM combines high-resolution STEM and TEM imaging with energy-dispersive X-ray spectroscopy (EDS) signal detection and 3D chemical characterization with compositional mapping. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) characterization was carried out on a Titan Cubed Themis G2 300 thermal field emission microscope with a high-tension voltage range of 60–300 kV.
XANES and EXAFS spectra of Cu K-edge and In K-edge were acquired on HXMA and BioXAS beamlines at Canadian Light Source (CLS), respectively. The results were analyzed via Athena and Artemis software. Solid-state 13C NMR experiments were recorded on an AVANCE NEO 600 MHz spectrometer (Bruker BioSpin Corp.) at the resonance frequency of 150.72 MHz for 13 C using the cross-polarization (CP), magic-angle spinning (MAS), and a high-power 1H decoupling. The powder samples were placed in a pencil-type zirconia rotor of 4.0 mm o.d. The spectra were obtained at a spinning speed of 10 kHz (4.2 μs 90° pulses), a 2 ms CP pulse, and a recycle delay of 3 s. The C signal of tetramethylsilane at 0 ppm was used as the reference of 13 C chemical shift.
Time-of-flight secondary ion mass spectroscopy (TOF-SIMS) was employed to study the elemental composition of various catalysts. All depth profiles were acquired in non-interlaced mode, namely, sequential sputtering. A 50 × 80 µm2 area centred on the analysis area was sputtered using a Cs+ ion beam (1 KeV ion energy, ~40 nA measured sample current).
Details of the operando, in-situ, and quasi in-situ measurements
The operando synchrotron two-dimensional wide-angle X-ray scattering (2D-WAXS) images were performed on the High Energy Wiggler Beamline in the Brockhouse Diffraction Sector (BXDS-WHE) at CLS, Canada.
Thermo Fisher Nicolet IS50 spectrometer was employed for in-situ electrochemical attenuated total reflection Fourier-transform infrared (ATR-FTIR) experiments. The Si prism with an incidence of a 60° angle was assembled into the bottom of the three-electrode in-situ electrochemical cell.
Quasi in-situ characterizations were conducted to explore the information on elemental composition and oxidation states, which were collected by a near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) located at the Vacuum Interconnected Nanotech Workstation (Nano-X), Suzhou Institute of Nano-Tech, and Nano-Bionics, Chinese Academy of Sciences.
DFT calculations
We used the Vienna Ab-initio Simulation Package68 with the projector augmented wave method69 for all DFT calculations. The Perdew-Burke-Ernzerhof (PBE) functional70 combined with DFT-D3 correction71 was employed to treat the exchange-functional for describing the weak interactions between atoms. During the calculations, the plane-wave basis had a cut-off energy of 420 eV. The Brillouin zone integration was conducted with a 2 × 2 × 1 Monkhorst-Pack k-points for optimizing the geometry and lattice size. We performed structural relaxation until the electronic structure self-consistency loop reached 1 × 10−4 eV. Partial occupancies were permitted for the Kohn-Sham orbitals by using the Gaussian smearing method and a width of 0.1 eV. The energy change in a geometry optimization was considered convergent when it was less than 0.01 eV Å−1. The implicit dipole and solvent corrections72,73 were also taken into account in this work. For each elementary step, Gibbs reaction free energy ΔG is the difference of free energies between the initial and final states and is defined as follows:
where the reaction energies of reactants calculated by DFT correspond to ΔE, ΔZPE, and ΔS are the zero-point energy and entropy changes due to the reaction. The definitions of adsorption energy (Eads) for the adsorbate are depicted in the reported literature44,72,74.
Molecular dynamics simulations
The all-atom MD simulations were performed using the Gromacs-4.6.7 software package75, with a general AMBER force field76 featuring RESP charges77. The system was in a relaxed liquid configuration at 298 K, and the equilibrium MD simulation ran for a total of 10 ns NPT. We used the relaxed system as a starting configuration and performed energy minimization with a composite protocol of steepest descent, using 100 kJ/mol·nm termination gradients, prior to system relaxation MD. To maintain the equilibrium temperature at 298 K, the Nose´-Hoover thermostat78 was employed, while periodic boundary conditions were enforced on all three dimensions. The Particle Mesh-Ewald method79,80 was utilized to calculate long-range electrostatics with a relative tolerance of 1 × 10−6, and a 1 nm cut-off distance was applied to real-space Ewald interactions and van der Waals interactions. The bond lengths of hydrogen atoms were constrained using the LINCS algorithm81, and we used a leap-frog algorithm82 with a time step of 1 fs. Meanwhile, an electric field of 5 V/nm was added in the Z-direction.
Data availability
All data needed to evaluate the conclusions in the paper are present in the Supplementary Information/Source data file. Source data are provided with this paper.
Code availability
VESTA (https://jp-minerals.org/vesta/en) and Athena software (http://bruceravel.github.io/demeter) are all open-source packages.
References
Wang, G. et al. Electrocatalysis for CO2 conversion: from fundamentals to value-added products. Chem. Soc. Rev. 50, 4993–5061 (2021).
Wen, G. et al. Bridging trans-scale electrode engineering for mass CO2 electrolysis. JACS Au 3, 2046–2061 (2023).
Wen, G. et al. Continuous CO2 electrolysis using a CO2 exsolution-induced flow cell. Nat. Energy 7, 978–988 (2022).
Sheng, X. et al. Engineering the NiNC catalyst microenvironment enabling CO2 electroreduction with nearly 100% CO selectivity in acid. Adv. Mater. 34, e2201295 (2022).
Zhang, Z. et al. Membrane electrode assembly for electrocatalytic CO2 reduction: principle and application. Angew. Chem. Int. Ed. 62, e202302789 (2023).
Li, H. et al. Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over Ni-N-C catalyst in a membrane electrode assembly electrolyzer. Energy Environ. Sci. 16, 1502–1510 (2023).
Wen, G. et al. Engineering electrochemical surface for efficient carbon dioxide upgrade. Adv. Energy Mater. 12, 2103289 (2021).
Wu, H. et al. Membrane technology for CO2 capture: from pilot-scale investigation of two-stage plant to actual system design. J. Membr. Sci. 624, 119137 (2021).
Shi, Z. et al. Robust metal-triazolate frameworks for CO2 capture from flue gas. J. Am. Chem. Soc. 142, 2750–2754 (2020).
Zhang, Z. et al. Porous metal electrodes enable efficient electrolysis of carbon capture solutions. Energy Environ. Sci. 15, 705–713 (2022).
Lee, G. et al. Electrochemical upgrade of CO2 from amine capture solution. Nat. Energy 6, 46–53 (2020).
Ye, K. et al. Resolving local reaction environment toward an optimized CO2-to-CO conversion performance. Energy Environ. Sci. 15, 749–759 (2022).
Sullivan, I. et al. Coupling electrochemical CO2 conversion with CO2 capture. Nat. Catal. 4, 952–958 (2021).
Liu, H. et al. Covalent organic frameworks linked by amine bonding for concerted electrochemical reduction of CO2. Chem 4, 1696–1709 (2018).
Ozden, A. et al. Energy- and carbon-efficient CO2/CO electrolysis to multicarbon products via asymmetric ion migration–adsorption. Nat. Energy 8, 179–190 (2023).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Deng, B. et al. The crystal plane is not the key factor for CO2 -to-methane electrosynthesis on reconstructed Cu2O microparticles. Angew. Chem. Int. Ed. 61, e202114080 (2022).
Kibria, M. G. et al. Electrochemical CO2 reduction into chemical feedstocks: from mechanistic electrocatalysis models to system design. Adv. Mater. 31, e1807166 (2019).
Li, X. et al. Molecule confined isolated metal sites enable the electrocatalytic synthesis of hydrogen peroxide. Adv. Mater. 34, e2104891 (2022).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386–1393 (2021).
Wen, G. et al. Ternary Sn-Ti-O electrocatalyst boosts the stability and energy efficiency of CO2 reduction. Angew. Chem. Int. Ed. 59, 12860–12867 (2020).
Yuan, L.-J. et al. Strategies and mechanism for enhancing intrinsic activity of metal–nitrogen–carbon catalysts in electrocatalytic reactions. Renewables 1, 514–540 (2023).
Zhao, Y. et al. Industrial-current-density CO2-to-C2+ electroreduction by anti-swelling anion-exchange ionomer-modified oxide-derived Cu nanosheets. J. Am. Chem. Soc. 144, 10446–10454 (2022).
Jung, H. et al. Electrochemical fragmentation of Cu2O nanoparticles enhancing selective C-C coupling from CO2 reduction reaction. J. Am. Chem. Soc. 141, 4624–4633 (2019).
Zhou, Y. et al. Stabilization of Cu+ via strong electronic interaction for selective and stable CO2 electroreduction. Angew. Chem. Int. Ed. 61, e202205832 (2022).
Zhang, T. et al. Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes. Nat. Catal. 5, 202–211 (2022).
Zhao, Y. et al. Elucidating electrochemical CO2 reduction reaction processes on Cu(hkl) single-crystal surfaces by in situ Raman spectroscopy. Energy Environ. Sci. 15, 3968–3977 (2022).
Kim, Y. et al. Time-resolved observation of C–C coupling intermediates on Cu electrodes for selective electrochemical CO2 reduction. Energy Environ. Sci. 13, 4301–4311 (2020).
Li, P. et al. In situ dual doping for constructing efficient CO2-to-methanol electrocatalysts. Nat. Commun. 13, 1965 (2022).
Shi, H. et al. Atomically dispersed indium-copper dual-metal active sites promoting C-C coupling for CO2 photoreduction to ethanol. Angew. Chem. Int. Ed. 61, e202208904 (2022).
Tsoukalou, A. et al. Hydrogen dissociation sites on indium-based ZrO2-supported catalysts for hydrogenation of CO2 to methanol. Catal. Today 387, 38–46 (2022).
Lum, Y. & Ager, J. W. Stability of residual oxides in oxide-derived copper catalysts for electrochemical CO2 reduction investigated with 18O labeling. Angew. Chem. Int. Ed. 57, 551–554 (2018).
Zhang, X. et al. Regulating ethane and ethylene synthesis by proton corridor microenvironment for CO2 electrolysis. J. Energy Chem. 87, 368–377 (2023).
Ren, B. et al. Dual-scale integration design of Sn-ZnO catalyst toward efficient and stable CO2 electroreduction. Adv. Mater. 34, e2204637 (2022).
Zhang, W. et al. Atypical oxygen-bearing copper boosts ethylene selectivity toward electrocatalytic CO2 reduction. J. Am. Chem. Soc. 142, 11417–11427 (2020).
Yang, P.-P. et al. Protecting copper oxidation state via intermediate confinement for selective CO2 electroreduction to C2+ fuels. J. Am. Chem. Soc. 142, 6400–6408 (2020).
Wen, G. et al. Cu-in dual sites with sulfur defects toward superior ethanol electrosynthesis from CO2 electrolysis. Adv. Mater. 36, e2310822 (2024).
Takahashi, T. et al. Global air-sea flux of CO2: an estimate based on measurements of sea-air pCO2 difference. Proc. Natl. Acad. Sci. USA 94, 8292–8299 (1997).
Zhu, H. J. et al. Efficient electron transmission in covalent organic framework nanosheets for highly active electrocatalytic carbon dioxide reduction. Nat. Commun. 11, 497 (2020).
Ren, B. et al. Nano-crumples induced Sn-Bi bimetallic interface pattern with moderate electron bank for highly efficient CO2 electroreduction. Nat. Commun. 13, 2486 (2022).
Singh, M. R. et al. Mechanistic insights into electrochemical reduction of CO2 over Ag using density functional theory and transport models. Proc. Natl. Acad. Sci. USA 114, E8812–E8821 (2017).
Weng, L. C., Bell, A. T. & Weber, A. Z. Modeling gas-diffusion electrodes for CO2 reduction. Phys. Chem. Chem. Phys. 20, 16973–16984 (2018).
Akinoglu, E. M. et al. Vertically aligned multiwalled carbon nanotube/Cu Catalysts for CO2 electroreduction. ACS Appl. Nano Mater. 5, 10399–10408 (2022).
Ren, B. et al. New mechanistic insights into CO2 reduction in solid oxide electrolysis cell through a multi-scale modelling approach. J. Power Sources 490, 229488 (2021).
Ren, B., Croiset, E. & Ricardez–Sandoval, L. A theoretical study on CO2 electrolysis through synergistic manipulation of Ni/Mn doping and oxygen vacancies in La(Sr)FeO3. J. Catal. 383, 273–282 (2020).
Hou, B. et al. Confinement-driven enantioselectivity in 3D porous chiral covalent organic frameworks. Angew. Chem. Int. Ed. 60, 6086–6093 (2021).
Kang, C. et al. Insertion of CO2 in metal ion-doped two-dimensional covalent organic frameworks. Proc. Natl. Acad. Sci. USA 120, e2217081120 (2023).
Xie, Y. et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat. Catal. 5, 564–570 (2022).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Feng, F. et al. Two-dimensional noble metal-based intermetallics for electrocatalysis. Energy Mater Devices 1, 9370008 (2023).
Chang, J. & Yang, Y. Advancements in seawater electrolysis: progressing from fundamental research to applied electrolyzer application. Renewables 1, 415–454 (2023).
Cai, Y. et al. Insights on forming N,O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 12, 586 (2021).
Jia, Y. et al. Probing the roles of indium oxides on copper catalysts for enhanced selectivity during CO2-to-CO electrochemical reduction. Nano Lett. 23, 2262–2268 (2023).
Shi, R. et al. Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces. Nat. Commun. 11, 3028 (2020).
Wen, G. et al. Constructing a localized buffer interlayer to elevate high-rate CO2-to-C2+ electrosynthesis. J. Am. Chem. Soc. 147, 18110–18121 (2025).
Wu, B. et al. Compression stress-induced internal magnetic field in bulky TiO2 photoanodes for enhancing charge-carrier dynamics. JACS Au 3, 592–602 (2023).
Wang, X. et al. Deciphering electrochemical interactions in metal–polymer catalysts for CO2 reduction. Energy Environ. Sci. 16, 4388–4403 (2023).
Wen, G. et al. Orbital interactions in Bi-Sn bimetallic electrocatalysts for highly selective electrochemical CO2 reduction toward formate production. Adv. Energy Mater. 8, 1802427 (2018).
Gu, M. W. et al. Tuning surface d bands with bimetallic electrodes to facilitate electron transport across molecular junctions. Nat. Mater. 20, 658–664 (2021).
Chen, C. et al. Coupling N2 and CO2 in H2O to synthesize urea under ambient conditions. Nat. Chem. 12, 717–724 (2020).
Raciti, D., Livi, K. J. & Wang, C. Highly dense Cu nanowires for low-overpotential CO2 reduction. Nano Lett. 15, 6829–6835 (2015).
Liu, Y. et al. Atomically inner tandem catalysts for electrochemical reduction of carbon dioxide. Energy Environ. Sci. 16, 5185–5195 (2023).
Zhang, Z.-Y. et al. Cu-Zn-based alloy/oxide interfaces for enhanced electroreduction of CO2 to C2+ products. J. Energy Chem. 83, 90–97 (2023).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Shangguan, W. et al. Molecular-level insight into photocatalytic CO2 reduction with H2O over Au nanoparticles by interband transitions. Nat. Commun. 13, 3894 (2022).
Zhang, H. et al. Mechanistic insights into CO2 conversion chemistry of copper bis-(terpyridine) molecular electrocatalyst using accessible operando spectrochemistry. Nat. Commun. 13, 6029 (2022).
Wang, J. et al. Atomically dispersed metal–nitrogen–carbon catalysts with d-orbital electronic configuration-dependent selectivity for electrochemical CO2-to-CO reduction. ACS Catal. 13, 2374–2385 (2023).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Ren, B. et al. First-principles based microkinetic modeling of CO2 reduction at the Ni/SDC cathode of a solid oxide electrolysis cell. J. Phys. Chem. C. 122, 21151–21161 (2018).
Han, N. et al. Ultrathin bismuth nanosheets from in situ topotactic transformation for selective electrocatalytic CO2 reduction to formate. Nat. Commun. 9, 1320 (2018).
Ren, B. et al. A density functional theory study on the carbon chain growth of ethanol formation on Cu-Co (111) and (211) surfaces. Appl. Surf. Sci. 412, 374–384 (2017).
Hess, B. et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Wang, J. et al. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Bayly, C. I. et al. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97, 10269–10280 (1993).
Berendsen, H. J. C. et al. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Astrakas, L. G., Gousias, C. & Tzaphlidou, M. Structural destabilization of chignolin under the influence of oscillating electric fields. J. Appl. Phys. 111, 074702 (2012).
Hess, B. et al. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Van Gunsteren, W. F. & Berendsen, H. J. C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1, 173–185 (2007).
Acknowledgements
Thanks for the support of the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0600100, Z. C.), and Energy Revolution S&T Program of Yulin Innovation Institute of Clean Energy (YIICE E411060316, Z. C.). The authors appreciate Dr. Graham King and Dr. Al Rahemtulla for the synchrotron WAXS on BXDS-WHE beamline at the Canadian Light Source (CLS). The authors thank Dr. Ning Chen, Dr. Roman Chernikov, and Dr. Emilio Heredia for XAS characterizations on HXMA and BioXAS beamlines at CLS. The authors also acknowledge the technical support for quasi in-situ NAP-XPS characterizations from Nano-X from Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences (SINANO). This work is financially supported by the National Natural Science Foundation of China (22472049, G.W.; 22402230, B.R.) and the Provincial Natural Science Foundation of Hunan (2025JJ20013, G.W.; 2025JJ40015, B.R.). The authors gratefully acknowledge the computational resources provided by the National Supercomputer Center in Guangzhou.
Author information
Authors and Affiliations
Contributions
Z.C. and B.R. devised the concept and wrote the manuscript. B.R. and X.Z. contributed to electrochemical experiments and material characterizations. B.R. and L.Y. synthesized catalysts. Z.C., B.R., and G.W. conceived and coordinated the project. S.D., H.X., Y.L., L.T., and X.D. participated in the discussion and analysis of the results. G.W., X.W., and Z.C. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yang-Gang Wang and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, B., Zhang, X., Yang, L. et al. Localized mass transport channels for electro-upgrade of dilute CO2 toward high-yield C2+ products. Nat Commun 16, 8383 (2025). https://doi.org/10.1038/s41467-025-63178-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63178-8
