
Abstract
Branched PFOA and PFOS have shorter half-lives, lower toxicity, and weaker serum protein binding than linear ones, offering better environmental and health safety. Yet methods to access such branched motifs remain under developed. We now introduce a one-step dehydroxy-perfluoro-tert-butylation of alcohols, in which perfluoro-tert-butyl phenyl sulfone serves both to activate the C–O bond and to deliver the perfluoro-tert-butyl group. Mechanistic studies—including DFT calculations—reveal that perfluoro-tert-butyl phenyl sulfone first generates perfluoroisobutylene in situ to effect C–O bond cleavage, after which a catalytic iodide engages in the C–C bond-forming event. Remarkably, the isolated perfluoro-tert-butyl anion salt, characterized by single-crystal X-ray diffraction, displays significant negative hyperconjugation, as evidenced by the elongated C–F bonds. This operationally simple protocol tolerates a wide array of functional groups and complex substrates, providing rapid access to branched perfluoroalkyl scaffolds with broad implications for drug discovery and advanced materials.
Similar content being viewed by others
Introduction
Fluorinated compounds, renowned for their distinct physicochemical properties, have found widespread utility in agrochemicals, pharmaceuticals, and materials science1,2,3. Among synthetic fluorinated compounds, poly- and perfluoroalkyl substances (PFASs)—including perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA)—are prevalent in industrial and commercial applications4,5. Their strong C–F covalent bonds render PFASs environmentally persistent and bioaccumulative, leading to their detection in humans and wildlife worldwide6,7,8. Nevertheless, branched isomers of PFOA and PFOS exhibit shorter elimination half-lives9,10, reduced toxicity11,12, and weaker binding to total serum proteins13,14,15 compared to their linear counterparts. This distinction underscores the potential of branched fluorinated structures to mitigate environmental and health risks. Consequently, there remains an increasing urgency to develop innovative synthetic methods for creating novel branched fluorinated compounds. In particular, as a “fat” analogue of the widely used trifluoromethyl (CF3) group, the perfluoro-tert-butyl [-C(CF3)3, PFtB] group emerges prominently as one of the most sterically hindered and electron-withdrawing functional groups.
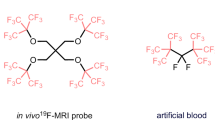
Molecules featuring the PFtB group have been utilized in the design of 19F MRI molecular probes16,17,18,19,20,21. Furthermore, our research has unveiled a high-resolution 19F-labeled probe showcasing the high importance of PFtB group (Fig. 1a)22. To synthesize more structurally diverse molecules containing PFtB group, two new reagents for perfluoro-tert-butylation, namely 1,1-dibromo-2,2-bis(trifluoromethyl)ethylene (DBBF)23 and perfluoro-tert-butyl phenyl sulfone (PFtBS)22 were developed. Mechanistically, these reagents efficiently generate the PFtB carbanion [(CF3)3C−] in the presence of fluoride salts, enabling nucleophilic perfluoro-tert-butylation under mild conditions. The PFtB carbanion was initially characterized using 19F nuclear magnetic resonance spectroscopy (NMR) in 197524,25. In 2021, we also detected the same 19F NMR signal for the PFtB carbanion employing DBBF as the precursor23. Further structural insights into the PFtB anion could be gleaned through X-ray diffraction (XRD) analysis. In 1986, Dixon and coworkers presented a calculated structure of the PFtB anion to illustrate the negative hyperconjugation effect26. Additionally, in 1988, Farnham and coworkers obtained XRD data for 1,3-bis(trifluoromethyl)−2,2,3,4,4-pentafluorocyclobutanide, offering compelling evidence for fluorine negative hyperconjugation27. However, to date, the XRD single-crystal structure of the PFtB anion has never been reported.

a Applications of perfluoro-tert-butylated molecules. b Dual roles of dehydroxy-functionalization reagents. c Dehydroxy-functionalization reagents: state of the art. d Our design.
While alcohols are ubiquitous structural motifs in bioactive molecules and serve as privileged synthons for molecular diversification via strategic C(sp³)–O bond functionalization28,29,30, their direct utilization is hindered by the high bond dissociation energy of C(sp3)–O linkages (ca. 91–105 kcal/mol). This thermodynamic barrier necessitates pre-activation of alcohols into reactive intermediates (e.g., halides, sulfonates), significantly complicating synthetic sequences. To circumvent the inherent low reactivity of alcoholic C(sp3)–O bonds, significant efforts have been devoted to developing activation strategies. Notably, emerging in situ activation strategies utilizing dehydroxy-functionalization reagents offer significant synthetic advantages by bypassing conventional pre-activation steps, thereby enhancing overall efficiency. The key innovation lies in the bifunctional nature of this type of reagents, which could generate both an electrophile (E; to activate alcohol substrate) and a nucleophile (Nu; to form C–Nu bond) via a synergic activation–substitution process. This dual reactivity enables a direct single-step conversion from R–OH to R–Nu (Fig. 1b). This approach has been exemplified by dehydroxy-trifluoromethylthiolation of alcohols with silver(I) trifluoromethanethiolate (AgSCF3)31 and dehydroxytrifluoromethoxylation of alcohols with trifluoromethyl arylsulfonate (TFMS)32. Despite the advancement of these methods, the direct construction of C–C bonds via a single reagent-enabled synergic dehydroxy-functionalization of alcohols remains a significant challenge (Fig. 1c)33,34,35,36,37,38,39. In this context, we envisioned that the above-mentioned PFtBS could serve as dehydroxy-fluoroalkylation reagent, since the reaction between PFtBS and CsF gives benzenesulfonyl fluoride (PhSO2F) and PFtB carbanion [(CF3)3C−]22, and PhSO2F is an activator (E) for alcohol and (CF3)3C− is a good nucleophile (Nu) (see Fig. 1b). In this work, we report the PFtBS reagent-enabled dehydroxy-perfluoro-tert-butylation protocol that enables a wide array of alcohols to efficiently undergo this transformation to form a C–C bond (Fig. 1d). This versatile method proves particularly valuable for late-stage functionalization of complex natural alcohols. Interestingly, through our detailed mechanistic study, we surprisingly discovered that the alcohol substrate is not activated by the in situ formed benzenesulfonyl fluoride.
Results and discussion
Screening of the reaction conditions
Initially selected as a key reagent, PFtBS (1) was synthesized using an optimized method from our previous report (for details, see Supplementary section 2 for details)22. A π-activated alcohol 4-fluorophenylmethanol (2’) was then chosen as the model substrate to react with PFtBS in the presence of fluoride. The initial reaction conditions were established using 2.0 equivalents of PFtBS, 3.0 equivalents of cesium fluoride (CsF) as PFtBS activator and base, and NMP as the solvent at the concentration of 0.1 M, resulting in a 45% yield of product 3’ (Table 1, entry 1). Inspired by Qing’s work31, tetrabutylammonium iodide (TBAI, 0.1 equiv) was introduced as an additive, which significantly improved the yield to 77% (Table 1, entry 2). Replacing TBAI with potassium iodide (KI, 0.1 equiv) led to a slight enhancement, achieving a yield of 81% (Table 1, entry 3). Solvent screening identified N-methylpyrrolidone (NMP) as the optimal one (Table 1, entries 3–5). Other fluoride sources (such as TMAF, KF and KHF2) showed inferior performance compared to CsF (Table 1, entries 6–8). This difference may be attributed to the different countercations exerting distinct stabilization effects on the PFtB anion. Further optimization by lowering the reaction temperature to 0 °C enhanced the product yield (Table 1, entries 9–10). Through systematic parameter refinement (see Supplementary Table S1-2 for details), optimal conditions were established by using 3.0 equivalents of CsF, 2.0 equivalents of 1, and 0.1 equivalents of KI in NMP under N2 at 0 °C, achieving 89% yield of 3’ (determined by 19F NMR).

Furthermore, initial attempts to extend the reaction conditions to unactivated alcohols proved unsuccessful. Consequently, 3-phenylpropan-1-ol was chosen as the model substrate for further optimization. After systematic screening, the optimal conditions were identified as employing 3.0 equivalents of CsF, 2.5 equivalents of 1, and 0.5 equivalents of KI in DMF under an N2 atmosphere at 80 °C, affording the desired product in 70% yield (see Supplementary Table S3-4 for details).
Mechanistic investigation
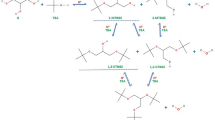
To elucidate the mechanism of dehydroxy-perfluoro-tert-butylation with PFtBS, systematic mechanistic studies were conducted. Initial attempts to detect alkyl phenylsulfonate or the in situ generated benzenesulfonic anion (PhSO3−) via mass spectroscopy (MS) were unsuccessful, which makes our hypothesized benzenesulfonic ester-mediated pathway (as outlined in Fig. 1d) unlikely. To confirm this result, 4-fluorobenzenesulfonyl fluoride (4) was introduced to activate the alcohol substrate, generating alkyl 4-fluorophenylsulfonate and the corresponding 4-fluorobenzenesulfonate anion. These species were specifically chosen for their distinct 19F NMR signals, enabling real-time monitoring of the reaction (Fig. 2a). Remarkably, 19F NMR analysis revealed that no consumption of 4 or the in situ formed benzenesulfonyl fluoride throughout the reaction (Fig. 2a, entries B–E). Furthermore, the addition of 4 showed negligible yield improvement when employing substoichiometric amounts of reagent 1 (Fig. 2a, entries C vs E), while much better yields of 3’ were obtained when 2 equivalents of 1 was employed. These findings conclusively demonstrate that the alcohol activation does not proceed via benzenesulfonyl fluoride intermediate. The energy profile for PFIB generation via β-elimination of perfluoro-tert-butyl anion was calculated (Fig. 2b), revealing an energy barrier of +14.2 kcal/mol and a thermodynamic loss of +15.5 kcal/mol. To further support this conclusion, we in situ prepared PFtB anion from 1,1-dibromo-2,2-bis(trifluoromethyl)ethylene (DBBF) and CsF23, and subsquently added substrate 2’. It was found that the desired product 3’ was formed in 39% yield (Fig. 2c), which suggests that the PFtB anion serves both as nucleophilic perfluoro-tert-butylating agent and as an activator of alcohol substrate.

a 19F NMR analysis of dehydroxy-perfluoro-tert-butylation. b β-Elimination of perfluoro-tert-butyl anion. c Dehydroxy-perfluoro-tert-butylation of alcohol using DBBF. d Proposed pathways for dehydroxy-perfluoro-tert-butylation.
Notably, three 19F NMR peaks [δ 4.17–3.88 (m, 1 F), −48.75 – −48.97 (m, 3 F), −50.11 – −50.32 (m, 3 F) in NMP, referenced against PhOCF3 as internal standard] were consistently observed in all cases as shown in Fig. 2a. These signals correspond to the reported 19F NMR chemical shifts of perfluoro-2-methyl-1-propenol-1-ate anion (PFMPA)40. Building the equilibrium of the PFtB anion with perfluoroisobutylene (PFIB) and fluoride ion24, we propose an PFIB-mediated alcohol activation pathway as outlined in Fig. 2d. The alcohol reacts with PFIB to form an alkyl perfluoroisobutenyl ether intermediates. Subsequently, the activated ester undergoes nucleophilic substitution with nucleophiles (PFtB anion or I−), ultimately yielding the desired perfluoro-tert-butylated products. This proposed mechanism is also supported by Knunyants’ report that alkyl perfluoroisobutenyl ethers, derived from alcohols and perfluoroisobutylene (PFIB), can act as effective alkylating agents41.
To further elucidate the mechanism of the deoxy-perfluoro-tert-butyalation and the structural characteristics of the PFtB anion, we attempted to prepare the single crystal of PFtB anion salts with cesium acting as the counter cation. Based on our previous observation of “CsC(CF3)3” by 19F NMR spectroscopy23, after a simple screen of crown ethers, we successfully isolated the complex {[(cis-syn-cis-DCy-18-C-6)Cs]+C(CF3)3−}2 (5) as a white crystalline solid through THF/hexane recrystallization under inert atmosphere. Single-crystal XRD analysis unambiguously confirmed the structure of 5 (Fig. 3a). The Cs−F bond length in anion salt 5 is between 3.230(4) Å (Cs1−F7) and 3.565(4) Å (Cs1−F5), which is longer than the sum of the covalent radii of cesium atom and fluorine atom (2.96 Å)42,43, but substantially shorter than the sum of the van der Waals radii (4.78 Å), revealing a characteristic Cs-F interaction. The crystal structure of 5 showed a nearly planar configuration of the PFtB anion44, as evidenced by a C21-C22-C23-C24 torsion angle/tetrahedron volume of −19.168°/0.215. This planarity is significantly greater than that observed in compound 3z [C-C-C-C torsion angle/tetrahedron = −37.161°/0.496, see Fig. 4]. The conformation of trifluoromethyl groups in 3z was staggered due to steric hindrance. However, in PFtB salt 5, the two trifluoromethyl groups (C22 and C24) exhibited an eclipsed conformation. This eclipsed arrangement was attributed to the interaction between the lone pair electrons on C21 and antibonding orbitals of C22−F6 and C24−F5 σ-bonds. This suggests a strong negative hyperconjugation effect45 between the carbanion center and these two C−F bonds. Moreover, the C22−F6 bond [1.360(6) Å] and C24−F5 bond [1.374(5) Å] are significantly elongated compared to other C−F bonds in PFtB anion salt 5 [1.291(6) Å to 1.345(5) Å] and compound 3z [1.325(2) Å to 1.342(2) Å]. The structure of PFtB anion salt 5 also reveals that overlap of the carbanion’s electron density with the C–F σ* orbitals weakens these two C−F bonds, and the counteraction effect of cesium promotes the β-elimination of PFtB anion.

a PFtB anion salt 5. b PFMPA anion salt 6. With thermal ellipsoids at the 30% probability level. The hydrogen atoms are omitted for clarity. Selected bond lengths of 5 (Å): C22−F6 1.360(6), Cs1−F6 3.470(6), C24−F5 1.374(5), Cs1−F5 3.565(4), Cs1−F7 3.230(4). Selected bond lengths of 6 (Å): Cs1−O7 2.994, Cs1−F5 3.663, C23−F5 1.335(7) Estimated standard deviations are in parentheses.

Unless mentioned otherwise, substrate 2 (1.0 equiv), PFtBS (1, 2.0 equiv), CsF (3.0 equiv), KI (0.1 equiv) and NMP (0.1 M) were added to a sealed tube. The mixture was stirred at 0 °C for 24 h. Yields referred to isolated products. a1 (4.0 equiv) and CsF (6.0 equiv) were used. b(4-(bromomethyl)phenyl)methanol was used as substrate. 1 (3.0 equiv) and CsF (4.0 equiv) was used, KI was not used. c1 (2.5 equiv) and KI (0.5 equiv) were used, DMF (0.1 M) instead of NMP (0.1 M), reactions were stirred at 80 °C for 24 h. dYields were determined by 19F NMR with PhOCF3 as an internal standard.
Unexpectedly, the addition of both CsF and potassium bifluoride (KHF2) was found to significantly accelerate PFtB anion hydrolysis (a plausible mechanism of PFtB anion hydrolysis was shown in Supplementary information Figure S9.). The 19F NMR analysis revealed concurrent formation of both perfluoro-2-methyl-1-propenol-1-ate anion (PFMPA) and HC(CF3)3. High-quality single crystals of PFMPA salt 6 were obtained through recrystallization from a THF/hexane mixed solvent system under inert atmosphere (Fig. 3b). XRD analysis unambiguously confirmed the structure of PFMPA salt 6, with no significant elongation observed in the C23−F5 [1.335(7) Å] and C23−F6 [1.337(9) Å] bonds. Additionally, the four carbon atoms of the anion adopting complete planarity (C21-C22-C23-C24 torsion angle/tetrahedron volume = −0.250/0.003). This near-perfect planar geometry provides definitive evidence for sp2 hybridization at the central carbon, conclusively establishing the enolate anion character of this species. Particularly, the chemical shift of perfluoro-2-methyl-1-propenol-1-ates from our 19F NMR studies was identical to the 19F NMR spectroscopy of 6.
Building upon these insights, density functional theory (DFT) calculations were performed for the dehydroxy-perfluoro-tert-butylation of benzyl alcohol to elucidate the reaction mechanism (Fig. 5). For computational simplicity, counterion effects were not considered in these calculations. To elucidate the alcohol activation

Method: M06-2X/6-311 + G(2 d,p)-SDD(I)-SMD(DMF)//M06-2X/6-31 + G(d,p)-LanL2DZ(I)-SMD(DMF). The bond lengths are in angstroms (Å).
mechanism, two potential alcohol-activation pathways were both calculated. Our calculations revealed that while the formation of alkyl sulfonate (sfx-2) is thermodynamically favored, the generation of benzyl perfluoroisobutenyl ether (deoxy-2) occurs through a kinetically facile and barrierless process. These computational results, together with our experimental observations (Fig. 2), provide compelling evidence that the dehydroxy-perfluoro-tert-butylation proceeds via in situ generated perfluoroisobutene (PFIB)-mediated alcohol activation. Upon completing the potential energy surface calculation for the dehydroxy-tert-butylation of the benzyl alcohol anion, the results revealed that: (1) perfluoroisobutylene (PFIB) generation (Fig. 2b) exhibits a lower energy barrier compared to benzyl alcohol anion condensation with phenyl sulfonyl fluoride (ΔΔG≠ = -2.1 kcal/mol), which indicates that PFIB-mediated alcohol activation is the more favorable pathway; (2) the in situ generated PFIB remains trapped in the system due to the highly exergonic and barrierless condensation with the alcohol nucleophile; (3) the resulting perfluoroisobutenyloxy group serves as a good leaving group that can be readily displaced by incoming nucleophiles; and (4) the inclusion of catalytic amount of iodide substantially reduces the activation barrier for the nucleophilic perfluoro-tert-butylation step from +21.1 kcal/mol to +17.6 kcal/mol (Fig. 5). These computational results provide a comprehensive insight into the reaction’s energy profile and the crucial role of iodide ion in facilitating the transformation. Additionally, PFIB, generating via β-elimination of the PFtB anion, exhibits electrophilicity during condensation with alcohol anion, while the PFtB anion demonstrates nucleophilicity in the perfluoro-tert-butylation process. To our best knowledge, such type of dual roles (as both nucleophile and electrophilic activator) of a carbanion in organic synthesis has never been reported.
Substrate scope
With the optimized conditions established, the scope of the dehydroxy-perfluoro-tert-butylation reaction was explored using a diverse range of π-activated alcohols (Fig. 4). Substrates bearing 4-phenylbenzyl or 2-naphthyl groups delivered excellent yields of 90% and 99% for 3b and 3c, respectively, whereas the sterically hindered 2,6-dimethylbenzyl derivative gave a reduced yield of 70% (3a). Activated alcohols featuring aromatic substituents, including vinyl, ethynyl, halogens (F, Cl, Br, and I), ether, nitro, sulfonyl, carbonyl, nitrile, ester, and amide groups, were all compatible, yielding products in moderate to excellent yields (3d–3t, 41–91%). Furthermore, pharmacologically relevant heterocycles such as triazole (3 u, 81%) pyridine (3 v, 57%), quinoline (3w, 75%), oxadiazole (3x, 50%), benzofuran (3 y, 64%), benzothiophene (3aa, 63%), and N-methylindole (3ab, 56%) were successfully transformed into their perfluoro-tert-butylated analogs. The protocol also accommodated common ligand motifs, including triphenylphosphine (3z, 58%) and ferrocenyl groups (3ac, 89%). The bis-dehydroxy-perfluoro-tert-butylated product 3ae can be obtained from diphenyl diol in synthetically acceptable yield (16%). Notably, when (4-(bromomethyl)phenyl)methanol was treated with 3 equivalents of 1 under identical conditions, the di-tert-butylated product 3af was isolated in 92% yield without requiring iodide additive. This result suggests that the in situ generated bromide anion can effectively replace iodide additives in this transformation. The protocol’s versatility was further demonstrated through the synthesis of protected perfluoro-tert-butylated dipeptide 3ad (77% yield) and the successful transformation of allylic alcohols 2ah-2ai to their perfluoro-tert-butylated analogs 3ah-3ai (72–75% yields).
Subsequently, we investigated the applicability of this transformation to unactivated alcohols. Gratifyingly, the method demonstrated both robustness and generality, affording products 3ai-3ap in 57–89% yield while tolerating diverse functional groups including amine (3al, 57%) and amide (3am, 76%). The synthetic utility of this dehydroxy-perfluoro-tert-butylation protocol was further highlighted through successful late-stage functionalization of pharmaceutically relevant molecules. Specifically, we achieved efficient modification of oxaprozin (3aq, 84%), perphenazine (3ar, 64%), and estrone (3as, 77%) derivatives, delivering the corresponding perfluoro-tert-butylated derivatives in good to excellent yields.
Discussion
In summary, we have established a straightforward dehydroxy-perfluoro-tert-butylation of alcohols using our perfluoro-tert-butyl phenyl sulfone (PFtBS) reagent. Comprehensive mechanistic investigations, supported by both experimental and computation studies, reveal that alcohol activation proceeds through in situ generated perfluoroisobutylene (PFIB). The XRD result of the perfluoro-tert-butyl (PFtB) anion is reported to elucidate the formation of PFIB via the β-elimination of the PFtB anion. The PFtB anion in salt 5 exhibited significant negative hyperconjugation, as evidenced by the elongation of two C–F bonds. Notably, the PFtB anion demonstrates dual roles in the reaction, serving as both a nucleophile and a latent electrophilic activator. This rare duality in carbanion reactivity has been structurally validated via the crystallographic evidence of the PFtB anion and perfluoro-2-methyl-1-propenol-1-ate (PFMPA) anion. Meanwhile, it has been found that a catalytic amount of iodide ion plays a critical role by lowering the activation barrier of the nucleophilic perfluoro-tert-butylation step. The protocol streamlines the synthesis of perfluoro-tert-butylated derivatives from alcohols, demonstrating broad substrate compatibility.
Methods
General procedure for dehydroxy-perfluoro-tert-butylation of π-activated alcohols
To an oven-dried sealed tube were added PFtBS (144.0 mg, 0.4 mmol, 2.0 equiv), CsF (91.2 mg, 0.6 mmol, 3.0 equiv) and KI (3.3 mg, 0.02 mmol, 0.1 equiv) in glove box. Then in fume hood, NMP (2 mL) and π-activated alcohol (0.2 mmol, 1.0 equiv) were successively added under N2 atmosphere. The reaction mixture was stirred at 0 °C for 24 h. After the reaction was completed, the reaction mixture was quenched with 20 mL saturated NH4Cl aqueous solution, extracted with ethyl ether (20 mL × 3). The combined organic layer was washed with brine (30 mL), dried over Na2SO4, and concentrated under vacuum. The residue was purified by column chromatography on silica gel to afford the desired product.
General procedure for dehydroxy-perfluoro-tert-butylation of unactivated alcohols
To an oven-dried sealed tube were added PFtBS (180.0 mg, 0.5 mmol, 2.5 equiv), CsF (91.2 mg, 0.6 mmol, 3.0 equiv) and KI (16.6 mg, 0.1 mmol, 0.5 equiv) in glove box. Then in fume hood, DMF (2 mL) and alkyl alcohol (0.2 mmol, 1.0 equiv) were successively added under N2 atmosphere. The reaction mixture was stirred at 80 °C for 24 h. After the reaction was completed, the reaction mixture was quenched with 20 mL saturated NH4Cl aqueous solution, extracted with ethyl ether (20 mL × 3). The combined organic layer was washed with brine (30 mL), dried over Na2SO4, and then concentrated under vacuum. The residue was purified by column chromatography on silica gel to afford the desired product.
Data availability
All data that support the findings of this study are available within the paper and its supplementary information files, and also available from the corresponding author upon request. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2018169 (3z), 2018189 (5) and 2018191 (6). Source data are provided with this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk. Source data are provided with this paper.
References
Müller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886 (2007).
O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 37, 308–319 (2008).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Washburn, S. T. et al. Exposure Assessment and Risk Characterization for Perfluorooctanoate in Selected Consumer Articles. Environ. Sci. Technol. 39, 3904–3910 (2005).
Kissa, E. Fluorinated Surfactants and Repellents. (Marcel Dekker, New York; 2001).
Chang, E. T. et al. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit. Rev. Toxicol. 44, 1–81 (2014).
Kannan, K. et al. Perfluorooctanesulfonate and Related Fluorochemicals in Human Blood from Several Countries. Environ. Sci. Technol. 38, 4489–4495 (2004).
Giesy, J. P. & Kannan, K. Global Distribution of Perfluorooctane Sulfonate in Wildlife. Environ. Sci. Technol. 35, 1339–1342 (2001).
Ross, M. S., Wong, C. S. & Martin, J. W. Isomer-Specific Biotransformation of Perfluorooctane Sulfonamide in Sprague–Dawley Rats. Environ. Sci. Technol. 46, 3196–3203 (2012).
De Silva, A. O., Tseng, P. J. & Mabury, S. A. Toxicokinetics of perfluorocarboxylate isomers in rainbow trout. Environ. Toxicol. Chem. 28, 330–337 (2009).
O’Brien, J. M. et al. Technical-grade perfluorooctane sulfonate alters the expression of more transcripts in cultured chicken embryonic hepatocytes than linear perfluorooctane sulfonate. Environ. Toxicol. Chem. 30, 2846–2859 (2011).
Loveless, S. E. et al. Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (APFO). Toxicology 220, 203–217 (2006).
Beesoon, S. & Martin, J. W. Isomer-Specific Binding Affinity of Perfluorooctanesulfonate (PFOS) and Perfluorooctanoate (PFOA) to Serum Proteins. Environ. Sci. Technol. 49, 5722–5731 (2015).
Benskin, J. P. et al. Disposition of perfluorinated acid isomers in sprague-dawley rats; Part 1: Single dose. Environ. Toxicol. Chem. 28, 542–554 (2009).
Gao, Y. et al. Differential Accumulation and Elimination Behavior of Perfluoroalkyl Acid Isomers in Occupational Workers in a Manufactory in China. Environ. Sci. Technol. 49, 6953–6962 (2015).
Tirotta, I. et al. A Superfluorinated Molecular Probe for Highly Sensitive in Vivo19F-MRI. J. Am. Chem. Soc. 136, 8524–8527 (2014).
Jiang, Z.-X., Liu, X., Jeong, E.-K. & Yu, Y. B. Symmetry-Guided Design and Fluorous Synthesis of a Stable and Rapidly Excreted Imaging Tracer for 19FMRI. Angew. Chem. Int. Ed. 48, 4755–4758 (2009).
Peng, Q. et al. Paramagnetic nanoemulsions with unified signals for sensitive 19F MRI cell tracking. Chem. Commun. 54, 6000–6003 (2018).
Akazawa, K. et al. Perfluorocarbon-Based 19F MRI Nanoprobes for In Vivo Multicolor Imaging. Angew. Chem. Int. Ed. 57, 16742–16747 (2018).
Zhang, H. et al. Fluorinated cryptophane-A and porphyrin-based theranostics for multimodal imaging-guided photodynamic therapy. Chem. Commun. 56, 3617–3620 (2020).
Zhang, R. et al. Selective radical-type perfluoro-tert-butylation of unsaturated compounds with a stable and scalable reagent. Nat. Commun. 16, 4458 (2025). A novel radical perfluoro-tert-butylation reagent has been introduced recently. For more details, see.
Wei, Z. et al. Regioselective Aromatic Perfluoro-tert-butylation Using Perfluoro-tert-butyl Phenyl Sulfone and Arynes. J. Am. Chem. Soc. 144, 22281–22288 (2022).
Wang, Q. et al. Fluorination Triggers Fluoroalkylation: Nucleophilic Perfluoro-tert-butylation with 1,1-Dibromo-2,2-bis(trifluoromethyl)ethylene (DBBF) and CsF. Angew. Chem. Int. Ed. 60, 27318–27323 (2021).
Dyatkin, B. L., Delyagina, N. I. & Sergey, R. S. The Perfluoro-t-butyl Anion in the Synthesis of Organofluorine Compounds. Russ. Chem. Rev. 45, 607 (1976).
Delyagina, N. I. Thesis on observing the generation of perfluoro-tert-butyl anion via 19F NMR. Candidate’s thesis, USSR Academy of Sciences (1975).
Dixon, D. A., Fukunaga, T. & Smart, B. E. Structures and stabilities of fluorinated carbanions. Evidence for anionic hyperconjugation. J. Am. Chem. Soc. 108, 4027–4031 (1986).
Farnham, W. B., Dixon, D. A. & Calabrese, J. C. Molecular and electronic structure of an isolated perfluorocarbanion. Crystal structure of tris(dimethylamino)sulfonium 1,3-bis(trifluoromethyl)-2,2,3,4,4-pentafluorocyclobutanide. J. Am. Chem. Soc. 110, 2607–2611 (1988).
Henkel, T., Brunne, R. M., Müller, H. & Reichel, F. Statistical Investigation into the Structural Complementarity of Natural Products and Synthetic Compounds. Angew. Chem. Int. Ed. 38, 643–647 (1999).
Ertl, P. An algorithm to identify functional groups in organic molecules. J. Cheminform. 9, 36 (2017).
Ertl, P. & Schuhmann, T. A Systematic Cheminformatics Analysis of Functional Groups Occurring in Natural Products. J. Nat. Prod. 82, 1258–1263 (2019).
Liu, J.-B., Xu, X.-H., Chen, Z.-H. & Qing, F.-L. Direct Dehydroxytrifluoromethylthiolation of Alcohols Using Silver(I) Trifluoromethanethiolate and Tetra-n-butylammonium Iodide. Angew. Chem. Int. Ed. 54, 897–900 (2015).
Jiang, X., Deng, Z. & Tang, P. Direct Dehydroxytrifluoromethoxylation of Alcohols. Angew. Chem. Int. Ed. 57, 292–295 (2018).
He, B.-Q. et al. Alcohol activation by benzodithiolylium for deoxygenative alkylation driven by photocatalytic energy transfer. Angew. Chem. Int. Ed. e202423795
Pang, X. & Shu, X.-Z. Reductive deoxygenative functionalization of alcohols by first-row transition metal catalysis. Chin. J. Chem. 41, 1637–1652 (2023).
Cai, Q., McWhinnie, I. M., Dow, N. W., Chan, A. Y. & MacMillan, D. W. C. Engaging alkenes in metallaphotoredox: a triple catalytic, radical sorting approach to olefin-alcohol cross-coupling. J. Am. Chem. Soc. 146, 12300–12309 (2024).
Chen, R. et al. Alcohol-alcohol cross-coupling enabled by SH2 radical sorting. Science 383, 1350–1357 (2024).
Cook, A. & Newman, S. G. Alcohols as substrates in transition-metal-catalyzed arylation, alkylation, and related reactions. Chem. Rev. 124, 6078–6144 (2024).
Xu, W., Fan, C., Hu, X. & Xu, T. Deoxygenative transformation of alcohols via phosphoranyl radical from exogenous radical addition. Angew. Chem. Int. Ed. 63, e202401575 (2024).
Großkopf, J., Gopatta, C., Martin, R. T., Haseloer, A. & MacMillan, D. W. C. Generalizing arene C−H alkylations by radical−radical cross-coupling. Nature (2025).
Volkonskii, A. Y. & Rokhlin, E. M. Mechanism of the cleavage of mixed anhydrides of α-hydrohexafluoroisobutyric acid by triethylamine. Russ. Chem. Bull. 31, 1205–1210 (1982).
Snegirev, V. F., Makarov, K. N. & Knunyants, I. L. Alkylating properties of fluorine-containing vinylic ethers. J. Fluor. Chem. 17, 441–445 (1981).
Pyykkö, P. & Atsumi, M. Molecular single-bond covalent radii for elements 1–118. Chem. – A Eur. J. 15, 186–197 (2009).
Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: a summary. J. Phys. Chem. A 119, 2326–2337 (2015).
The inversion barriers of CF3- anion and PFtB anion were calculated to illustrate the structural difference of these two anions. For details, see Supplementary information.
Alabugin, I. V., Gilmore, K. M. & Peterson, P. W. Hyperconjugation. WIREs Comput. Mol. Sci. 1, 109–141 (2011).
Acknowledgements
Financial support of this work by the National Natural Science Foundation of China (22261132514, J.H. and 22271299, C.N.) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0590000, J.H.) is gratefully acknowledged. K. Zhu thanks Syngenta for a PhD scholarship. We thank Dr. Xuebing Leng for his assistance in the XRD measurement and data analysis carried out at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. We thank Dr Lingling Li from instrumental analysis center of Shanghai Jiao Tong University for the single crystal analysis. We thank Dr. Yinlin Chen (Stockholm University) for his advice on the XRD data analysis. We also thank Mr. Zhenxing Li (Nanjing University) for his advice on the DFT calculation. We acknowledge Prof. Xiaosong Xue for his insight on DFT calculation.
Author information
Authors and Affiliations
Contributions
J.H. conceived the concept. K.Z., Q.L. and P.X. performed the experiments. K.Z. conducted the isolation of anion salts. K.Z. and Z.N. performed the DFT calculations. K.Z., Q.L., P.X. and S.S. contributed to the analysis and interpretation of the data. K.Z., M.H., C.N. and J.H. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, K., Luo, Q., Xu, P. et al. Dehydroxy-Perfluoro-tert-butylation of alcohols with PhSO2C(CF3)3 and insights into the structure of the [C(CF3)3]− anion. Nat Commun 16, 7969 (2025). https://doi.org/10.1038/s41467-025-63180-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63180-0
