
Abstract
Despite the efficacy of chimeric antigen receptor (CAR)-T cells in selected hematological malignancies, further improvement on CAR-T designs is still desirable. We hypothesize that modifying the CAR structure to enhance immunological synapse (IS) stabilization and CAR target-binding may be a feasible strategy. Here we show that the membrane protein, CD99, is critical for IS formation in T cells by mediating actin-microtubule interaction. CD99 deficiency abolishes IS formation and prevents effective in vivo T cell immunity. Mechanistically, CD99 interacts with microtubules and actins through the transmembrane and cytoplasmic domains, respectively, with which myosin and IQGAP1 interact. As such, incorporating the transmembrane and juxtamembrane domains of CD99 into the CAR structure enhances IS formation and improves the therapeutic efficacy of human CAR-T cells against lymphoma in immune-deficient mice. Our data thus suggest that CD99-mediated IS stabilization may help improve CAR design and efficacy.
Similar content being viewed by others


Introduction
Chimeric antigen receptor (CAR) is a recombinant receptor in which an extracellular tumor-targeting antibody moiety is linked to the intracellular signaling domain of the T cell receptor1. When a CAR gene is introduced into T cells (CAR-T cells) from cancer patients, these T cells can recognize the tumor antigen and be activated via the CAR to kill the tumor cells. Although CAR-T cell therapy has shown remarkable efficacy in some hematological malignancies, its efficacy in general needs to be significantly improved2. For this purpose, attempts have been made to modify the CAR structure. Most efforts to date have focused on the addition of one or more signaling domains from costimulatory molecules, such as CD28, 41BB, and OX40, to the intracellular region of the CAR3. However, increasing the number of costimulatory domains incorporated into a single CAR molecule may lead to excessive activation of CAR-T cells, resulting in adverse side effects, such as cytokine release syndrome4,5. Therefore, a different approach may be required to further improve the CAR structure, such as strengthening the immunological synapse (IS) between CAR-T cells and the target tumor cells. This could be achieved by incorporating the domains from a key surface receptor responsible for IS formation into CAR. However, our current understanding of the molecular mechanism of IS formation and the receptors responsible is very limited.
The IS is a specialized structure formed at the site of contact of T cells with antigen-presenting cells (APCs) or target cells, and is known to enhance T cell activation and effector function6,7,8. The mature IS consists of three radially symmetric substructures, known as the central (c-), peripheral (p-), and distal (d-) supramolecular activation clusters (SMACs)9. In the cytosol, the actin cytoskeleton forms distinct networks: a hypodense network in the cSMAC, a lamella-like arc network in the pSMAC, and a lamellipodia-like branched network in the dSMAC10. Microtubules are arrayed with radial symmetry, while the microtubule-organizing center (MTOC) is located immediately beneath the plasma membrane in the cSMAC11. The unique shape of the IS cytoskeleton network is formed by reorganization of the actin and microtubule networks after TCR signaling. TCR signaling triggers actin polymerization at the T-APC contact site, promoting lamellipodia extension toward the APC12,13. Actin is then depolymerized at the center of the contact site, followed by its retrograde flow from the lamellipodia to the central area along with maturation of the IS structure. TCR signaling also induces microtubule polymerization/elongation, which occurs in parallel with lamellipodia extension14. Subsequently, the MTOC translocates to the synapse side from the distal region (uropod) of T cells15,16. Importantly, these actin and microtubule reorganization processes are tightly intertwined, probably through the physical interaction of actin and microtubule filaments16,17,18,19. However, the mechanisms underlying the actin-microtubule physical interaction, and the existence of a membrane protein that orchestrates actin and microtubule reorganization are unclear.
CD99 is a transmembrane protein that is expressed at basal levels on various types of cells, but is upregulated on activated T cells and some tumor cells20. Although CD99 was initially identified as a cell adhesion molecule21, its involvement in transendothelial migration of leukocytes and tumor cells has been demonstrated22,23,24,25. Roles for CD99 in the generation of costimulatory effects on T cells26,27, intracellular molecular trafficking in T cells28,29,30, and recruitment of CD99 into IS during T cell activation31 have also been reported. Mechanistically, all of these functions are known to be associated with actin polymerization/reorganization28,29,32. We also reported previously that the absence of the intact cytoplasmic domain of CD99 impairs molecular trafficking33, suggesting the molecular interaction of this domain with cytoskeletal components. However, the molecular mechanisms remain unclear.
Here, based on the possibility of CD99-cytoskeleton interaction and the importance of actin polymerization in IS formation, we investigate the hypothesis that CD99 may also play a role in cytoskeleton reorganization to achieve IS formation. Our results demonstrate that CD99 ensures the interaction between actin and microtubules, which is crucial for coordinated cytoskeletal reorganization. CAR-T cells containing CD99 functional domains in the CAR structure exhibit enhanced IS formation and functional efficacy. Thus, this study identifies CD99 as a key membrane protein for IS formation and proposes a novel IS-stabilizing CAR design that could be generally applicable to enhance CAR-T cell therapy.
Results
Requirement of CD99 for IS formation and actin reorganization
To test the aforementioned hypothesis, we first examined the impact of the absence of CD99 on IS formation by comparing T-APC conjugate formation by CD99-deficient (Cd99−/−) and wild type (WT) murine T cells in vitro. When cells from a WT CD8 cytotoxic T lymphocyte line specific for a minor histocompatibility antigen (MiHA) H6034,35,36 (WT H60-CTL) were incubated with H60-peptide-presenting dendritic cells (H60-DCs), T-APC conjugates were formed, with binding of about three T cells on average per DC and > 25% of the H60-DCs being occupied by three or more WT H60-CTLs per DC (Fig. 1a). In contrast, Cd99−/− H60-CTLs did not form such conjugates with H60-DCs, suggesting that CD99 is required for T-APC conjugate formation. Similar results were obtained with naïve TCR-transgenic J1537,38 or OT-1 T cells specific for H60 or ovalbumin (OVA) complexed with H-2Kb, respectively (Supplementary Fig. 1a, b), implying that the CD99 dependency of T-APC conjugate formation is independent of antigen-experience and the antigen-specificity of T cells. In subsequent experiments, Cd99−/− and WT H60-CTLs (hereafter, Cd99−/−-cells and WT-cells, respectively) were used representatively. Cd99−/−- and WT-cells exhibited comparable cell sizes and expressed similar levels of TCR and LFA on their cell surfaces prior to TCR-stimulation (Supplementary Fig. 1c). After T cell stimulation with APCs, however, the polarization of polymerized actin, TCR, and LFA-1 to the contact sites with cognate H60-DCs, indicators of mature IS formation in WT-cells, were not observed in Cd99−/−-cells, in fluorescence staining experiments (Fig. 1b, Supplementary Fig. 1d).

a Fluorescence microscopy images of wild type (WT) or Cd99−/− H60-CTLs (green) coincubated with cognate H60-DCs or control OVA-DCs (red). Numbers of CTLs in contact with one DC (n = 263, 369, 386, and 192 conjugates, from left to right) and the proportion of DCs in contact with more than three CTLs (n = 5 fields per group) are plotted. b Confocal images of phalloidin-stained WT or Cd99−/− H60-CTLs, and control OVA-DC or cognate H60-DCs (cognate DC) after 15 min of coincubation. Polarized CTLs were quantified by identifying cells with higher F-actin intensity at the IS compared to the rear of the CTL (n = 6, 12, 5, and 12 fields, from left to right). Representative single Z-section images of the synapse are shown. Each data point in the graph indicates percentage of polarized cells per field. c–e Longitudinal analysis of actin reorganization in anti-CD3-stimulated WT- and Cd99−/−-cells (see Supplementary Movie 1). Time-lapse images of F-actin (Lifeact-mCherry) against black or differential interference contrast (DIC) image background (c). Time in minutes:seconds. Plots of cellular fluorescence intensity expressed in arbitrary units (arb.units) (n = 6 cells per group) (d). Plots of the synapse area (a, white dotted line on cell image) and lamellipodia width (b, black line) at different time points, and distance of the actin-microcluster (MC) from the plasma membrane (c, yellow double-headed arrow) measured at 15–20 min (n = 9 cells per group) (e). f Quantification of nuclear translocation of NFAT (n = 9, 35, 28, 40, 8, 40, 33, and 35 cells, from left to right) and NFκB (n = 23, 24, 35, 38, 24, 20, 37, and 35 cells, from left to right), and phosphorylation of ERK (n = 28, 48, 46, 43, 38, 43, 39, and 34 cells, from left to right) at the indicated time points (see Supplementary Fig. 1e–g). All representative images were obtained from at least 5 randomly selected fields from each group. Data (a, b, d–f) are presented as mean ± SEM from at least three independent experiments. Statistical significance was assessed using unpaired two-tailed t-test (a, b, d, f), and two-way repeated measures ANOVA with Sidak post-hoc test (d, e). Exact p-values are presented in the figure. Source data are provided as a Source Data file.
The effects of CD99 on IS-forming actin dynamics were then examined in a longitudinal real-time actin-tracing experiment using Lifeact-mCherry39-expressing retrovirus-transduced Cd99−/−-cells and WT-cells that were activated on anti-CD3 antibody-coated plates. Confocal live-cell imaging of WT-cells showed that the lamellipodial actin ring expanded in the early phase (3:40 image in Fig. 1c), followed by the establishment of a radially symmetric actin network, typical of IS, with centripetal actin retrograde flow (6:20 and 14:20 images in Fig. 1c) in 10–15 min post-stimulation (Supplementary Movie 1). In Cd99−/−-cells, however, despite initial actin polymerization, the typical IS actin network was not established within the ~30-min observation period, with detection of actin-free cell cortex area (Fig. 1c, Supplementary Movie 1). Both the basal and maximal intensities of filamentous actin (F-actin; polymerized) were significantly lower in CD3-stimulated Cd99−/−-cells, with significantly smaller synapse size and lamellipodia width, compared to the WT counterparts (Fig. 1d, e). In addition, retrograde actin flow in Cd99−/−-cells was not centripetal (after about 10:00 min in Supplementary Movie 1), so that dense actin-microclusters were rarely found in the central area of the cells (10:15 and 18:30 images for Cd99−/−-cells in Fig. 1c, e). In line with these observations, although the TCR signal could be delivered by anti-CD3 stimulation in Cd99−/−-cells, as demonstrated by nuclear translocation of NFAT and NFκB, and Erk phosphorylation in confocal microscopy of fixed cells, the degree of activation was far less than that in WT cells (Fig. 1f, Supplementary Fig. 1e–g). Taken together, these findings indicated that CD99 is required for the progression and amplification of TCR signaling-initiated actin dynamics leading to mature IS formation in T cells.
Requirement of CD99 for the physical interaction between actin and microtubules, and for microtubule reorganization
As actin flows toward the centrally localized MTOC in IS formation10, we assumed that the atypical localization of actin-microculsters in Cd99−/−-cells reflected inappropriate microtubule reorganization. Therefore, we stained Cd99−/−-cells, stimulated with plate-bound anti-CD3 antibody or H60-DCs, with anti-α-tubulin antibody to visualize the microtubules. As expected, Cd99−/−-cells had fewer and less intensely stained perpendicularly growing microtubules (pMTs) compared to WT-cells, and showed impaired MTOC-translocation (Fig. 2a, supplementary Fig. 2a).

a Confocal images of WT- and Cd99−/−-cells stained with anti-α-tubulin antibody at 15 min after anti-CD3-stimulation. Plots showing the number of perpendicularly growing microtubules (pMT) in the cell body (CB) and lamellipodia (LP) (n = 12 cells per group), and cellular microtubule (MT) fluorescence intensity (n = 24 cells per group). b, c Longitudinal analysis of microtubule reorganization in Lifeact+ WT- and Cd99−/−-cells (see Supplementary Movies 2, 3). Time-lapse images of microtubules overlaid on DIC (b) and F-actin (c) images. Actin-microtubule colocalization shown in white. Time in minutes:seconds. Plots of microtubule staining (SiR-tubulin+) intensities and pMT numbers (n = 5 cells per group) (b), and actin-microtubule colocalization according to Manders’ coefficient of microtubules colocalized with F-actin (n = 3 cells per group) (c). d Confocal images of F-actin (red) and microtubules (green), and their colocalization (yellow) in WT- and Cd99−/−-cells stained with phalloidin and anti-α-tubulin antibody at 15 min after CD3-stimulation. Plot of actin-microtubule colocalization (n = 12 cells per group). e Immunoblot of actin in coprecipitates of α-tubulin from stimulated WT- or Cd99−/−-cells. Data a–e are representative of at least three independent experiments. Data a–d are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test (a, b, d), and two-way repeated measures ANOVA with Sidak post-hoc test (b, c). Exact p-values are presented in the figure. Source data are provided as a Source Data file.
Then, actin-microtubule interactions were examined in detail in a real-time microtubule-tracing experiment using Lifeact-transduced Cd99−/−- and WT-cells stained with a tubulin-visualizing agent SiR-tubulin40. Upon plate-bound anti-CD3 stimulation, the microtubules of WT-cells were newly polymerized, elongated, and tethered to the cell cortex, concomitant with lamellipodial extension (Fig. 2b, Supplementary Fig. 2b, Supplementary Movie 2). In Cd99−/−-cells, however, there was little new microtubule outgrowth, and the microtubules were also untethered. Additionally, in WT-cells, shortening (contraction) or curling (bending) of pMTs near the cell cortex was seen, as noted previously16, followed by the generation of numerous and intensely staining pMTs and horizontally growing microtubules (hMTs), with an IS-typical microtubule network formed within 10–15 min. In contrast, in Cd99−/−-cells, the new microtubules quickly fragmented and/or depolymerized, so that their staining intensity was low and their numbers declined over time (Fig. 2b, Supplementary Fig. 2b, Supplementary Movie 2). Therefore, CD99 is also crucial for appropriate microtubule dynamics following TCR-stimulation.
Importantly, simultaneous visualization of actin (Lifeact+) and microtubule (SiR-tubulin+) dynamics revealed that the two cytoskeletal components rarely interacted with each other in Cd99−/−-cells, resulting in significantly less colocalization (indicated by a lower Manders’ coefficient of microtubules with F-actin) in the cell body (cytoplasmic core plus lamella) and lamellipodia than seen in WT-cells (Fig. 2c, Supplementary Fig. 2c, Supplementary Movie 3). Staining of the fixed cells with phalloidin and anti-α-tubulin antibody, in the absence of Lifeact and SiR-tubulin, reproduced colocalization of actin and microtubules, particularly in the peri-MTOC, lamella, and cortex regions of WT-cells, but not Cd99−/−-cells (Fig. 2d). Structured illumination microscopy (SIM) further confirmed the presence of reduced levels of actin-microtubule colocalization and intensity of F-actin and microtubules in Cd99−/−-cells compared to WT-cells in both the synapse plane and z-projection images (Supplementary Fig. 2d, Supplementary Movie 4). Interestingly, treatment of WT-cells with SMIFH2 or CK666, inhibitors of formin- or Arp2/3-mediated actin polymerization, respectively10, did not fully recapitulate the low levels of actin-microtubule colocalization and intensity of F-actin and microtubules observed in Cd99-/--cells, despite inducing an overall reduction in the aforementioned parameters (Supplementary Fig. 2e). Furthermore, even when treated with phorbol 12-myristate 13-acetate and the ionophore ionomycin (PMA/Iono), agents that deliver artificial TCR signaling without TCR engagement and promote actin polymerization41,42, Cd99−/−-cells still exhibited lower microtubule and actin-microtubule colocalization levels compared to the WT-counterparts (Supplementary Fig. 2f), confirming that CD99 not only affects actin polymerization but also mediates proper microtubule reorganization and actin-microtubule colocalization. Consistently, the coimmunoprecipitation of actin and tubulin observed in WT-cells was not seen in Cd99−/−-cells (Fig. 2e). Collectively, these results demonstrated that CD99 is critical for the physical interaction of actin and microtubules during IS formation, suggesting the importance of CD99 function-mediated actin-microtubule interaction for appropriate and coordinated reorganization of the two cytoskeletal components.
Importance of the cytoplasmic and transmembrane domains for CD99 function
Next, we analyzed the intracellular localization of CD99 to investigate how it functions in actin-microtubule interaction. Cd99−/−-cells were transduced such that they expressed myc-tagged recombinant CD99-YFP fusion protein (rWT-CD99) as a reporter protein (Fig. 3a). Flow cytometric analysis confirmed expression of rWT-CD99 on the surface of stably transduced cells (rWT-cells) and showed comparable cell size and surface levels of TCR and LFA-1 between rWT-cells and mock-transduced cells (Supplementary Fig. 3a). Fixed-cell analysis of the rWT-cells showed that IS formation was successfully restored (as determined by the actin/microtubule colocalization index, actin ring width, pMT number, and MTOC-translocation), in contrast to mock-transduced Cd99−/−-cells (Fig. 3b, Supplementary Fig. 3b, c). Live-cell imaging confirmed the proper dynamics and interaction of actin and microtubules in rWT-cells (Fig. 3c, Supplementary Fig. 3d, Supplementary Movie 5). Analysis of rWT-CD99 revealed its molecular localization in the cell body (but not in the MTOC area) and lamellipodia (Fig. 3c, Supplementary Movie 6), and suggested pSMAC and dSMAC localization in the IS. The pSMAC localization of CD99 was confirmed by positive costaining of endogenous CD99 with LFA-1, but not with the TCR, on the surface of WT-cell ISs (Supplementary Fig. 3e). Furthermore, rWT-CD99 was markedly enriched in the peri-MTOC, lamella, and cortical regions (Supplementary Fig. 3f), i.e., major sites of colocalization between actin and microtubules (Fig. 2d), in rWT-cells. Therefore, three-way colocalization of rWT-CD99, actin, and microtubules in these regions was seen frequently (Fig. 3d), suggesting that CD99 can interact simultaneously with these cytoskeletal proteins.

a Schematic diagram showing the rWT and mutant CD99s, and mock-YFP. L leader sequence, Myc Myc epitope tag, Ext extracellular domain, TM transmembrane domain, Cyt cytosolic domain. b Plots of actin-microtubule colocalization (n = 6 cells per group), lamellipodia actin width (n = 58, 71, 70, and 71 cells, from left to right), and the number of pMTs (n = 41 cells per group). Related images are shown in Supplementary Fig. 3b. c–g Longitudinal analysis of cytoskeleton reorganization and the localization of recombinant CD99 (see Supplementary Movies 5–7). Time-lapse confocal images of microtubules (cyan), F-actin (red), recombinant CD99-YFP (orange-yellow) in rWT, Cyt-Mut, or TM-Mut cells (c). Zoomed images of the peri-MTOC (PM) and lamella-lamellipodia (LLP) regions of the rWT-cells shown in (c, d). Molecular colocalization shown in white. Plots of F-actin and microtubule intensities (n = 5 cells per group) (e), actin-microtubule colocalization in the PM and LLP regions designated in (c, f), and colocalization of F-actin or microtubules with recombinant CD99 with in the cell body (CB) and lamellipodia (LP) at 11 min (rWT-cells) or 14 min (mutant-cells) after stimulation (g). At least 9 randomly selected regions from 5 cells of each group were analyzed from quantitative colocalization of two proteins (f, g). h Localization and domain-mediated interaction with the cytoskeletons of rWT-CD99, Cyt-Mut-CD99, and TM-Mut-CD99. Data b–g are representative of at least three independent experiments. Data b, e–g are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test (b, f, g), and two-way repeated measures ANOVA with Sidak post-hoc test (e). Exact p-values are presented in the figure. Source data are provided as a Source Data file.
Insight into the mechanism of action of CD99 was obtained in studies using two mutants, Cyt-Mut-CD99 and TM-Mut-CD99, in which either the cytoplasmic or transmembrane domain was replaced with the corresponding nonfunctional domain from human CD4 (Fig. 3a). When the two mutants were introduced into Cd99−/−-cells (Cyt-Mut-cells or TM-Mut-cells), no substantial differences in cell size and surface levels of TCR and LFA-1 were observed between the two mutant-cells and rWT-cells in the unstimulated state (Supplementary Fig. 3a). However, upon stimulation, the cytoskeleton reorganization for IS formation, defined according to the aforementioned criteria, was largely unrestored in both mutant-cells, as determined in both fixed (Fig. 3b, Supplementary Fig. 3b, c) and live (Fig. 3c, e, f, Supplementary Movie 6) cells. In addition, both mutants showed significantly less colocalization with actin and microtubules compared to rWT-CD99 (Fig. 3g), indicating that both domains are required for the function of CD99. However, the two mutants exhibited distinct defects. While Cyt-Mut-CD99 did not localize at the cell cortex, TM-Mut-CD99 was rarely detected at the cell body (Fig. 3c, Supplementary Movie 6). This was confirmed by flow cytometric analysis which showed a defect in cell-surface expression of Cyt-Mut-CD99, consistent with the defect in molecular trafficking to the plasma membrane of the mutant33, while TM-Mut-CD99 showed increased cell-surface expression (Supplementary Fig. 3a). Furthermore, Cyt-Mut-CD99 showed drastic reduction of colocalization with actin, whereas colocalization of TM-Mut-CD99 with microtubules was greatly reduced (Fig. 3c, g, Supplementary Movie 7). These differences in cellular localization and impaired colocalization with either actin or microtubules between the two mutants were not due to aberrant functioning of the substituted human CD4 domains, since expression in Cd99−/−-cells of alternative mutants containing human CD19 domains, designated CythCD19-Mut and TMhCD19-Mut, resulted in the same effects (Supplementary Fig. 3g). These observations indicated that the cytoplasmic and transmembrane domains make different contribution to the function of CD99.
A detailed analysis of cytoskeleton dynamics revealed significantly fewer pMTs in Cyt-Mut-cells, with defective growth of peri-MTOC microtubules and untethering of microtubules from actin (Fig. 3c, f, Supplementary Fig. 3h, Supplementary Movie 6). The lack of actin-microtubule interaction required for lamellipodial extension resulted in highly unstable actin dynamics and an abnormal predominance of actin-free cortex (Fig. 3c, Supplementary Fig. 3h). Full lamellipodia extension was transient and followed by rapid retraction (Supplementary Fig. 3i). These observations were consistent with a previous report that actin-microtubule linkage enables coordinated microtubule growth and actin filament elongation43. Therefore, CD99 was suggested to use its cytoplasmic domain to support interdependent actin and microtubule growth probably by actin-guided microtubule growth and microtubule-induced actin polymerization, thus enabling lamellipodial extension.
In TM-Mut-cells, actin polymerization persisted near the cell cortex, where TM-Mut-CD99 resides, albeit expressed at low levels (Fig. 3c and Supplementary Movie 7). Additionally, there were a greater number of long pMTs than in Cyt-Mut-cells, although the overall number of pMTs was lower than in rWT-cells (Fig. 3b, c, e). A low level of actin-microtubule colocalization, as detected in the peri-MTOC area (Fig. 3b, c, f), may facilitate pMT generation. This supports a role for the cytoplasmic domain (which is intact in TM-Mut-CD99) in pMT generation and actin polymerization in lamellipodia. However, tethering of pMTs to actin at the cell cortex was absent, and pMTs in the cortical region were broken and decayed over time, so that intense staining of pMTs and hMTs was not observed (Fig. 3c, Supplementary Fig. 3j). Due to this lack of pMT-actin interaction, periodic retraction of the lamellipodia was reduced, and the actin accumulation necessary to form an actin-arc network near the lamella was hindered (Fig. 3c, Supplementary Fig. 3i). These results indicated that the transmembrane domain of CD99 is required for stable tethering between actin and preformed pMTs near the cell cortex and for the retraction of actin and extended lamellipodia.
In summary, our results indicated that in the cell body, the cytoplasmic domain of CD99 is required for pMT formation and F-actin extension, while the transmembrane domain is for microtubule retention (Fig. 3h). In lamellipodia, the cytoplasmic domain of CD99 is needed for actin polymerization and lamellipodial extension, while the transmembrane domain is for pMT-tethering to cortical actin, which allows lamellipodia retraction.
Proteomic identification of IQGAP1 and myosin IIA as domain-mediated interaction partners of CD99
The molecular mechanism of action of CD99 was further investigated by proteomics analysis using coprecipitates of rWT-CD99. As expected, cytoskeletal proteins, including various components of the actomyosin (Actbl2, Iqgap1, Coro1a, Actc1, Myh9, Myl6), microtubule (Tuba1, Tuba4a, Tubb4b, Tubb5), and molecular trafficking (Fmnl, Rac2, and Rab27) systems, were identified as a major subset of the CD99 interactome (Fig. 4a, Supplementary Fig. 4a, b, Supplementary Data 1–4).

a Venn diagram of representative CD99 interactors in the actomyosin and microtubule systems shown in Supplementary Fig. 4a, b, and Supplementary Data 1. b, c Levels of cytoskeletal components in the Cyt-Mut-CD99 or TM-Mut-CD99 interactome, compared to the levels in the rWT interactome. Plots of the log2 fold change (FC) in the values of the majority of the interactors (except for myosin and IQGAP1), myosin (MyoII) proteins, and IQGAP1, as detected in the Cyt-Mut-CD99 and TM-Mut-CD99 interactomes relative to the rWT interactome (n = 2 samples per group) (b). Schematic illustration of the binding of MyoII and IQGAP1 to different recombinant CD99 molecules (c), based on the log2FC values in Supplementary Fig. 4d. d, e Immunoblots of MyoIIA or IQGAP1 in coimmunoprecipitates of rWT-CD99, Cyt-Mut-CD99, and TM-Mut-CD99 (using anti-myc antibody) from the corresponding cells (d), and MyoIIA, IQGAP1, actin, and α-tubulin in coimmunoprecipitates of CD99 (using anti-CD99 antibody) from WT-cells after anti-CD3-stimulation (e). f–h Colocalization analysis in rWT and mutant cells. Images of two-way (green) and three-way (white) molecular colocalization (based on Supplementary Fig. 4e) (f). Image of three-way colocalization (rWT-CD99, IQGAP1, and MyoIIA) overlaid on DIC image of rWT-cells, and plot of three-way colocalization signal intensities measured along the line (cyan) across the MTOC (red asterisk) (g). Plots of two-way colocalizations (n = 9 cells per group) (h). i, j Interaction between IQGAP1 and MyoIIA in in WT- or Cd99-/--cells. Confocal images of colocalization (white) between IQGAP1 and MyoIIA on DIC image background of WT- and Cd99−/−-cells. Plots of the IQGAP1 and MyoIIA signal intensities measured along the MTOC-crossing line, and the colocalization (n = 10 cells per group) (i). Immunoblot of MyoIIA in coprecipitates of IQGAP1 (j). k, l Interaction of microtubules with IQGAP1 or MyoIIA in WT- or Cd99−/−-cells. Confocal images of microtubules with IQGAP1 or MyoIIA, and colocalization (yellow). Plots of colocalization of microtubules with IQGAP1 or MyoIIA (n = 9 cells per group) (k). Immunoblot of actin, MyoIIA, and IQGAP1 in coprecipitates of α-tubulin (l). The tubulin and actin immunoblots were reused from Fig. 2e. m CD99 domain-mediated interaction of IQGAP1, MyoIIA, actin, and microtubules. Data (a, b, d–l) are representative of two (a, b, j) or three (d–i, k, l) independent experiments. Data (h, i, k) are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test. Exact p-values are presented in the figure. Source data are provided as a Source Data file.
Next, we assessed the domain-specific interactions of CD99 with cytoskeletal components by comparing coimmunoprecipitates of Cyt-Mut-CD99 and TM-Mut-CD99 with those of rWT-CD99. Principal component analysis (PCA) of the interactome data revealed clear clustering of replicates and distinct separation between the three recombinant protein samples (Supplementary Fig. 4c). In general, cytoskeletal interactors were present at lower levels in the coprecipitates of both mutants than in those of rWT-CD99, with less binding to Cyt-Mut-CD99 than to TM-Mut-CD99 (Fig. 4b, Supplementary Fig. 4d, Supplementary Data 1, 5–7). Exceptionally, however, myosin (MyoII) proteins (Myh9 heavy (MyoIIA) and Myl6 light chains) showed less binding to TM-Mut-CD99 than Cyt-Mut-CD99, indicating that myosin proteins preferentially bind to the transmembrane rather than the cytoplasmic domain of CD99 (Fig. 4b, c). In contrast, IQGAP1 showed less binding to Cyt-Mut-CD99 than TM-Mut-CD99, although binding to both mutants was relatively strong. Therefore, IQGAP1 was suggested to have greater affinity for the cytoplasmic than the transmembrane domain of CD99. This pattern was confirmed by immunoblotting of each coimmunoprecipitate (Fig. 4d). IQGAP1 is a candidate molecule that mediates actin-microtubule linkage with domains capable of binding to F-actin and microtubules44. The actin motor protein MyoIIA has been reported to play a role in microtubule reorganization45. Therefore, IQGAP1 and MyoIIA were inferred to be cytoplasmic and transmembrane domain-mediated principal interactors involved in CD99-associated actin-microtubule interaction. Indeed, both MyoIIA and IQGAP1, as well as actin and α-tubulin, coprecipitated with CD99 in WT-cells (Fig. 4e), suggesting that these five proteins form a multiprotein complex.
CD99 facilitates interactions between IQGAP1, myosin IIA, and the cytoskeleton
To gain further insight into the above biochemical interactions, rWT-cells and mutant-cells were stained for the localization of IQGAP1 and MyoIIA, and examined by confocal microscopy. In rWT-cells, IQGAP1 and MyoIIA were enriched in the lamella, as well as in peri-MTOC and cortical regions, of the IS (Supplementary Fig. 4d), as reported previously46,47. Additionally, rWT-CD99 colocalized with these two molecules in these regions (Fig. 4f, g). In contrast, the colocalization of Cyt-Mut-CD99 with IQGAP1 was greatly reduced, as was that of TM-Mut-CD99 with MyoIIA (Fig. 4f, h, Supplementary Fig. 4e), consistent with the biochemical data. Notably, the colocalization of MyoIIA and IQGAP1, as well as the three-way colocalization of MyoIIA, IQGAP1, and rWT-CD99, was significantly reduced in Cyt-Mut- and TM-Mut-cells, compared to rWT-cells (Fig. 4f, h, Supplementary Fig. 4e). The requirement of CD99 in the MyoIIA-IQGAP1 interaction was further supported by the reduced colocalization and coprecipitation of these two molecules in Cd99−/−-cells (Fig. 4i, j, Supplementary Fig. 4f).
As CD99 mediates both actin-microtubule and MyoIIA-IQGAP1 interactions, we sought to determine whether CD99 is necessary for the interaction of the actin modulators MyoIIA and IQGAP1 with microtubules. The colocalization of microtubules with IQGAP1 or MyoIIA was significantly reduced in Cd99−/−-cells compared to WT-cells, such that neither MyoIIA nor IQGAP1 coprecipitated with α-tubulin (Fig. 4k, l). These results suggest a role for CD99 as a hub protein that coordinates actin-microtubule crosstalk by binding to IQGAP1 and MyoIIA and modulating their interaction (Fig. 4m).
Ineffective T cell activation and in vivo immunity in the absence of CD99
Given the significance of IS for T cell activation and effector function7, the effects of CD99 deficiency on the efficiency of T cell activation were investigated. After stimulation with anti-CD3 and CD28 antibodies, polyclonal T cells, both CD4 and CD8 T cells, from Cd99−/− mice were capable of dividing, but showed delayed cell division (CFSE-dilution) and activation (upregulation of CD69, CD25, and CD44), compared to the WT counterparts (Fig. 5a, b, Supplementary Fig. 5a, b). This was also the case in antigen-specific T cells, i.e., Cd99−/− OT1 T cells stimulated with OVA-DCs (Supplementary Fig. 5c). Consistently, TCR signaling events, including the phosphorylation of ZAP70 and Erk, nuclear translocation of NFAT and NFκB, and sustained Akt activation, were all reduced in Cd99−/− T cells (Supplementary Fig. 5d–f), recapitulating the biochemical features observed in Cd99−/− CTL cells (Fig. 1f, Supplementary Fig. 1e–g). Furthermore, Cd99−/− CD8 T cells exhibited lower expression levels of FasL and granzyme B compared to the WT-counterparts (Supplementary Fig. 5g).

a, b Comparison of in vitro activation of CD8+ T cells from WT and Cd99−/− mice. Flow cytometry profiles of CFSE dilution (n = 5 mice per group) (a) and the expression of CD69, CD25, and CD44 (n = 5 mice per group) (b) by anti-CD3/CD28 stimulated WT and Cd99−/− T cells. Plots of the proportions (%) of T cells in each diluted CFSE peak (a) and marker-positive cells (b). Data a, b are pooled from two independent experiments (n = 2–3 mice/group/experiment). c In vivo activation of WT and Cd99−/− J15 CD8+ T cells. Flow cytometry profiles of female Thy1.2+ WT or Cd99−/− J15 CD8+ T cells in the spleens of adoptive hosts (Thy1.1+). The numbers of CD8+Thy1.2+ cells in the spleen on days 3 and 7 post-immunization are plotted (n = 7 mice per group) using data pooled from three independent experiments (n = 2–3 mice/group/experiment). d Comparison of allograft-rejection between female WT and Cd99−/− B6 recipients. Representative images of male BALB.B tail-skin (red arrow) rejected (dotted line) or surviving (solid line), and syngeneic B6 tail-skin (black arrow) on the tail of a WT or Cd99−/− B6 recipient on day 14 post-transplantation. Kaplan-Meier survival plot of the grafts, with median survival time (MST) shown in parentheses. Flow cytometric profiles of H60-tetramer- or H4-tetramer-binding CD11a+ CD8 T cells in blood on day 12 post-transplantation. Plots of the percentage of tetramer-binding cells in blood CD8 T cells. Data represent two independent experiments (n = 9 mice/group/experiment). e B16F10-OVA tumor growth in B6 mice to which were transferred naive CD45.1+ WT or Cd99−/− OT-I CD8 T cells. Tumor sizes and percentages of OVA-tetramer-binding CD45.1+ cells in blood CD8 T cells are plotted. Representative flow cytometric profiles of OVA-tetramer-binding CD45.1+ CD8 T cells on day 10 post-transfer. Data represent two independent experiments (n = 5 mice/group/experiment). Data a–e are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test (a–e), and log-rank (Mantel-Cox) test (d graft survival). Exact p-values are presented in the figure. Source data are provided as a Source Data file.
Whether the activation of Cd99−/− T cells was also delayed in vivo was investigated by the adoptive transfer of WT or Cd99−/− H60-specific J15 (Thy1.2+) T cells into B6 mice, which were then injected intraperitoneally with H60-expressing splenocytes from H60-congenic mice38. In response to H60-challenge, the expansion of Cd99−/−J15 T cells in the spleen and the migration of these cells to the antigen-bearing site (the peritoneum) in the host were slower, compared to the WT counterparts (Fig. 5c, Supplementary Fig. 5h). Based on this inefficient reactivity of Cd99−/− T cells for MiHA H60, whether CD99 deficiency in T cells would delay the rejection of MiHA-mismatched allografts was investigated using a well-established B6 anti-BALB.B skin-allograft model48,49. When BALB.B tail-skin was transplanted onto WT or Cd99−/− B6 recipients, Cd99−/− recipients showed a significant delay in allograft rejection, with the T cells specific for MiHAs (H60 and H4) showing lower peak levels in the blood compared to WT recipients (Fig. 5d).
The physiological relevance of CD99 for T cell activation was further investigated in an antitumor T cell therapy model. WT OVA-specific OT-1 (CD45.1+) T cells, when adoptively transferred, reduced tumor growth in OVA-expressing B16 melanoma-bearing mice, which was not the case with Cd99−/− OT-1 T cells (Fig. 5e). Consistently, Cd99−/− OT-1 T cells were detected at lower levels in host blood compared to WT OT-1 cells. Collectively, these results suggest that CD99 deficiency in T cells limits the efficiency of T cell immunity in vivo, probably through defective CD99-mediated actin-microtubule crosstalk. However, as CD99 has been known to be also important for transendothelial migration of leukocytes50, it should be noted that defective migration of T cells in the absence of CD99 cannot be formally excluded.
Transmembrane and juxtamembrane domains are minimal essential domains of mouse and human CD99
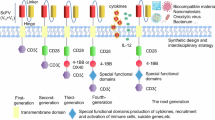
Having confirmed a critical role of CD99 in IS formation and T cell function, we investigated whether incorporation of the CD99 transmembrane and cytoplasmic domains into a tumor-targeting CAR would enhance the antitumor efficacy of CAR-T cells by potentiating IS formation. To minimize the CD99 components to be incorporated into the CAR, additional domain studies were performed. First, two additional transmembrane domain mutants, TM-SR (small-portion restored) and TM-LR (large-portion restored), were generated, in which small and large portions of the human CD4 transmembrane domain of TM-Mut-CD99 were restored to mouse CD99 sequences, respectively (Fig. 6a). In transduced Cd99−/− cells, greater inward localization of mutant-CD99 from the cell cortex was achieved with TM-LR than TM-SR (Fig. 6b, Supplementary Fig. 6a). Concomitantly, hMT generation and actin-microtubule colocalization were gradually restored in the order TM-Mut < TM-SR < TM-LR. However, complete restoration was achieved only in rWT-cells, indicating that the intact transmembrane domain is essential for CD99 function.

a Schematics of the TM-SR and TM-LR mutant CD99. b Confocal images of recombinant CD99-YFP, F-actin, and microtubules, including their colocalization (white). Plots of microtubule colocalization with actin in MTOC-proximal (n = 8 cells per group) and -distal (n = 10 cells per group) regions in transduced Cd99−/−-cells after 15 min of anti-CD3 stimulation. c Alignment of CD99 amino acid sequences from different organisms using Clustal Omega75. Identical sequences are highlighted in red; fully conserved sequences are marked with an asterisk, strongly conserved sequences with a colon, and weakly conserved sequences with a period. Jux, juxtamembrane region. d Functional restoration by TJ-CD99 introduced into Cd99−/−-cells. Schematic of TJ-CD99. Confocal images of recombinant CD99-YFP, F-actin, microtubules, including their colocalization (white) in transduced Cd99−/−-cells. pMT numbers (n = 16 cells per group) and microtubule colocalization with actin (n = 8 cells per group) are plotted. e Cytoskeletal derangements in CD99 knocked-down (KD-HuCD99) Jurkat T cells. Schematic of the DNA construct for human CD99 (sh1-HuCD99) knockdown. Confocal images of actin or microtubules (cyan) in transfected (mCherry-positive) Jurkat T cells after 15 min of anti-CD3 stimulation. Plots of F-actin (n = 12, 15 cells) and microtubule (n = 20, 11 cells) intensities. f Restored IS-cytoskeletal network following HuTJ-CD99 expression in KD-HuCD99-Jurkat cells. Schematic of HuTJ-CD99, mock, and KD (sh1-HuCD99) plasmids. Ms murine CD99, Hu human CD99. Confocal images of HuTJ-CD99 or mock expression (YFP, green) with F-actin or microtubules (red and cyan), and their colocalization (yellow) in KD-HuCD99-Jurkat cells. Plots of lamellipodia (LP) width (n = 21, 25 cells) and pMT numbers (n = 10 cells per group). Data represent three (b, d) or two (e, f) independent experiments. Data b, d–f are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test. Exact p-values are presented in the figure. Source data are provided as a Source Data file.
A comparison of CD99 amino acid sequences from different species showed that the transmembrane and cytoplasmic domains, especially the transmembrane-juxtaposing sequences (hereafter referred to as Juxta, i.e., the juxtamembrane region), are highly conserved (Fig. 6c). Therefore, we generated a mutant (TJ-CD99) that retained the transmembrane and Juxta (TJ) regions but with deletion of the rest of the cytoplasmic domain (Fig. 6d). When introduced into Cd99−/− cells, TJ-CD99 fully restored the colocalization and reorganization of actin and microtubules, as well as molecular localization, similar to rWT-CD99 (Fig. 6d, Supplementary Fig. 6b). Thus, the TJ region of CD99 is essential for IS formation.
Considering application to human CAR-T-cell generation, we then investigated whether the mouse CD99 findings could be recapitulated in human T cells. Human CD99 exists as two isoforms resulting from alternative splicing, one with a short cytoplasmic domain and the other with a long cytoplasmic domain, but both isoforms share the transmembrane and juxtamembrane regions21, enabling the evaluation of a single human TJ mutant. First, the function of human CD99 was tested using human Jurkat T cells. After transfection of an anti-CD99 short hairpin RNA (shRNA) plasmid selected for the effective knockdown (KD) of human CD99 (Supplementary Fig. 6c–e), the CD99 knocked-down (KD-HuCD99) Jurkat T cells exhibited defective formation of the actin and microtubule networks of the IS, in contrast to control cells transfected with scramble-shRNA (Fig. 6e). Furthermore, when the human TJ-mutant of CD99 (HuTJ-CD99-YFP) was expressed in KD-HuCD99-Jurkat cells, both actin and microtubules were properly reorganized (Fig. 6f, Supplementary Fig. 6f). HuTJ-CD99-YFP molecules localized to the cell body and cell cortex, where they were colocalized with actin and microtubules. Next, we extended our analysis to human primary T cells, utilizing alloreactive T cells generated through a mixed leukocyte reaction (MLR) of peripheral blood mononuclear cells (PBMCs) from allogeneic individuals. These alloreactive T cells were transfected with CD99 single guide RNA (sgRNA) to induce CD99 gene deletion, generating sgCD99-T cells. Upon TCR-stimulation, sgCD99-T cells also exhibited defects in actin and microtubule network formation of IS, including the polarization of F-actin and MTOC toward the coincubated target allogeneic cells (Supplementary Fig. 6g, h). Furthermore, sgCD99-T cells showed reduced biochemical and phenotypic readouts of TCR-signaling events, including nuclear translocation of NFAT and NFκB, phosphorylation of ERK, sustained activation of AKT, and upregulation of activation markers, compared to sgControl-T cells (Supplementary Fig. 6i-k). Altogether, these results provide further evidence for the conserved function of CD99 and the essentiality of the TJ region in human T cells.
Potentiated IS formation and antitumor effects via CD99 domain-incorporated CAR design
Finally, we evaluated whether the function of human CAR-T cells bearing the CD99-TJ region would be enhanced compared to conventional CAR-T cells. In the conventional CAR structure, such as the BBz-CAR targeting human CD19-expressing cells51, the antibody moiety is linked to the signaling cytoplasmic domains of 41BB and CD3 zeta chain via the extracellular and transmembrane domains of CD8 (Fig. 7a). These CD8-derived domains serve only as a physical linker rather than a functional entity. Therefore, we designed a new BBz variant-CAR, named HuTJ-BBz-CAR, incorporating the functional extracellular and TJ domains of human CD99 instead of the CD8-derived linker domains (Fig. 7a). CAR-T cells expressing BBz-CAR or HuTJ-BBz-CAR, with comparable CAR expression levels, were utilized in subsequent experiments (Fig. 7b). IS formation and effector function were then compared between HuTJ-BBz- and conventional BBz-CAR-T cells at multiple time points after coincubation with target (CD19+) Raji human lymphoma cells. Consistent with normal IS formation between T cells and APCs, cell-cell conjugate formation with the target cells was seen more frequently, and actin and MTOC were more polarized to the target cells in BBz-CAR-T cells than in untransduced (UnTD) control T cells (Fig. 7b, c, Supplementary Fig. 7a). Notably, HuTJ-BBz-CAR-T cells exhibited more efficient cell-cell conjugate formation, and earlier completion of F-actin polarization and MTOC-translocation than BBz-CAR-T cells (Fig. 7b, c, Supplementary Fig. 7a). HuTJ-BBz-CAR-T cells, both the CD4 and CD8 fractions, upregulated CD69, CD25, and CD44 to greater extents than BBz-CAR-T cells after the coculture with Raji tumor cells (Fig. 7d, supplementary Fig. 7b, c). The enhanced activation of HuTJ-BBz-CAR-T cells relative to BBz-CAR-T cells was maintained even after the deletion of the T cell’s endogenous CD99 gene (Supplementary Fig. 7d). Notably, CD99 deletion did not significantly impact the activation levels of either HuTJ-BBz-CAR or BBz-CAR-T cells, suggesting that CAR-signaling does not strictly depend on the function of endogenous CD99. Then, when subjected to long-term tumor-killing assay, HuTJ-BBz-CAR-T cells expanded greatly to significantly higher levels and exerted superior antitumor effects in comparison with BBz-CAR-T cells (Fig. 7e, Supplementary Fig. 7e). Regarding safety, although there has been some controversy in the literature regarding the oncogenic and oncosuppressive roles of CD9925, in the absence of tumor target cells and cytokine supplementation during long-term culture, both HuTJ-BBz- and BBz-CAR-T cells, as well as UnTD-T cells, exhibited a continuous decline in cell numbers, suggesting their minimal oncogenic potential (Supplementary Fig. 7f). Collectively, these results indicated that the incorporated CD99-derived domains were functional in CAR-T cells, mediating potentiated IS formation, activation, and tumor-killing efficiency of HuTJ-BBz-CAR-T cells.

a Schematic of BBz-CAR and HuTJ-BBz-CAR. b Histogram showing CAR expression on untransduced (UnTD) human T, BBz-CAR-T, or HuTJ-BBz-CAR-T cells. Percentage and mean fluorescence intensity (MFI) values are included. Plots of conjugate formation of UnTD human T, BBz-CAR-T, or HuTJ-BBz-CAR-T cells with Raji tumor cells at different time points of coincubation. In b from left to right, n (fields of coverslips) = 5, 7, and 7. Data are pooled from two independent experiments. c IS formation by CAR-T cells. Z-stacked confocal images of F-actin (red) and microtubules (green), and their colocalization (yellow) in UnTD T, BBz-CAR-T, or HuTJ-BBz-CAR-T cells interacting with CMTMR-labeled Raji cells (blue) after staining with phalloidin and anti-tubulin-α antibody. Arrows indicate polymerized actin enriched near the contact areas (yellow arrows) and the MTOC (green arrows) in T and CAR-T cells. F-actin polarization (Polr) and MTOC translocation (Traslc) are denoted on the image of HuTJ-BBz-CAR-T cells, and plotted (n = 12 conjugates per group). Data represent three independent experiments. d Plots of MFI values of CD69, CD25, and CD44 expressed on UnTD T, BBz-CAR-T, or HuTJ-BBz-CAR-T cells after coculture with Raji cells (n = 4 replicates per group). e In vitro tumor killing assay. Numbers of CAR-T and residual tumor cells were quantified during coculture (n = 3 replicates per group). Data represent two (b, c, e) or three (d) independent experiments. Data b–e are shown as mean ± SEM. Statistical significance was assessed using unpaired two-tailed t-test. Exact p-values are presented in the figure. Source data are provided as a Source Data file.
CD99 domain-incorporated CAR enhances CAR-T cell efficacy in vivo
The in vivo antitumor efficacies of the two lines of human CAR-T cells were compared in Raji tumor-bearing immunodeficient NSG mice. Tumor burden was monitored by implantation of luciferase-expressing Raji cells. After infusion of BBz-CAR-T cells (5 × 106 cells), tumor growth was significantly suppressed by day 7 post-infusion (Fig. 8a). However, tumors re-grew and eventually killed all the mice (Fig. 8a, b). Notably, the infusion of HuTJ-BBz-CAR-T cells resulted in earlier tumor eradication and a much better survival rate for the mice than was the case with BBz-CAR-T cells (Fig. 8a, b). At the lower dose of CAR-T cells (1 × 106 cells), with which BBz-CAR-T cells could not control tumor growth, HuTJ-BBz-CAR-T cells still completely eliminated tumors, indicating more than five-fold enhanced efficacy (Fig. 8c, d). HuTJ-BBz-CAR-T cells expanded with much faster kinetics in the peripheral blood than BBz-CAR-T cells, for both CD4 and CD8 fractions, in that HuTJ-BBz-CAR-T cells peaked on day 7, whereas BBz-CAR-T cells began to show major expansion on day 14 (Fig. 8e). Therefore, efficient expansion and tumor-killing activity of HuTJ-BBz-CAR-T cells were observed in vivo as well as in vitro. Altogether, the incorporation of CD99 TJ domains into the CAR improved CAR-T functionality, suggesting that CD99 functional domain-mediated IS stabilization enhances CAR-T cell functionality.

a, b Experimental scheme and longitudinal bioluminescence images of tumor cells after injection of high dose (5 × 106) CAR-T cells (a), and plots of bioluminescence intensities and Kaplan-Meier survival curves of hosts, with MSTs shown in parentheses. ud, undetermined (b). c, d Bioluminescence images of tumor cells after injection of low dose (1 × 106) CAR-T cells (c) and plot of bioluminescence intensities of tumor hosts (d). e Plots of BBz-CAR-T or HuTJ-BBz-CAR-T cell numbers in host peripheral blood after infusion. Data are representative of more than three (a) and two (c–e) independent experiments (n = 5 mice/group/experiment). Data b, d, e are shown as mean ± SEM. Statistical significance was assessed using two-way repeated measures ANOVA with Sidak post-hoc test (b, d), log-rank (Mantel-Cox) test (b) and unpaired two-tailed t-test (e). Exact p-values are presented in the figure. Source data are provided as a Source Data file.
Discussion
This study demonstrated that CD99 mediates the physical interaction between actin and microtubules and that this interaction is critical for mature IS formation and effective T cell immunity, leading to the novel design of an IS-stabilizing CAR structure.
CD99 contributes to several biological processes and inflammatory responses, such as T cell adhesion21,52,53; activation and differentiation26,27; intracellular membrane protein trafficking in T, B, and epithelial cells28,29,30,33,54; and lymphocyte diapedesis into inflamed vascular endothelium32,50. Nevertheless, the uncertainty surrounding its molecular function and mechanism of action has hindered the acknowledgement of CD99 in the immunological field. Actin reorganization has been considered a molecular target of CD99 action, as the aforementioned phenomena are related to the actin cytoskeleton, and stimulation of CD99 by an agonistic antibody has been shown to induce action polymerization28,29. Therefore, the requirement of CD99 for proper actin reorganization during IS formation is unsurprising. However, the contribution of CD99 to microtubule dynamics is a novel and completely unexpected finding. Furthermore, the observed physical and functional dissociation of actin and microtubules in the absence of CD99 strongly suggests that CD99 is the long-sought master-controller membrane protein that orchestrates cytoskeleton reorganization during IS formation - a protein that has been proposed but remained unidentified55,56. Regarding human CD99, the alternatively spliced isoforms can exhibit divergent functions, depending on the cell type21, with both isoform being recruited to the IS in activated T cells31. Given that both isoforms share functional domains (TJ regions), we propose that the core function of CD99—namely, its role in controlling the reorganization and cross-talk between actin and microtubules—would be maintained, irrespective of the cooperative or counteractive relationships these isoforms may exhibit within the IS.
We identified the domains of CD99 responsible for the actin-microtubule linkage and their interactors associated with the linkage: the transmembrane domain for microtubule interaction via MyoII and the cytoplasmic domain for actin interaction via IQGAP1. IQGAP1 is a candidate mediator of the actin-microtubule linkage11,44. Our identification of IQGAP1 as a binding partner of the cytoplasmic domain of CD99 is consistent with the above consensus. However, despite its ability to interact with IQGAP1, TM-Mut-CD99 did not fully restore the actin-microtubule linkage, indicating the involvement of additional factors such as MyoII. MyoII is an actin motor protein known to be primarily involved in lamellar actin-arc contraction in IS45. However, the failure of TM-Mut-CD99 to interact with MyoIIA and restore pMT-actin linkage particularly at the cell cortex suggests that MyoII also plays an important role in actin-microtubule linkage at the cell cortex. This is supported by the results of general cell biology studies indicating that MyoII forms a contractile ring complex with actin and microtubules at the cell cortex when a cell divides, and generates the contraction force needed to close the ring during cytokinesis57,58. Additionally, recruitment of IQGAP1, formin, lipids, and a membrane protein to the ring complex has been reported59,60,61. Therefore, CD99 is likely to be “the” membrane protein component of the ring complex. We have also reported defective cytokinesis in some human CD99-downregulated tumor cells62. Taken together, these observations suggest that CD99-MyoIIA-IQGAP1 constitutes the general machinery for the physical and functional coupling of actin and microtubules in many cellular responses.
During IS formation, the actin-microtubule linkage at the peri-MTOC and cell cortex has been proposed to provide the traction force driving translocation of the MTOC to the synapse site11,55. However, the results of our cytoskeleton dynamics analyses suggest that the actin-microtubule physical interaction has important functional roles beyond simple mechanical traction. At the peri-MTOC, the CD99-mediated interaction enables concurrent pMT generation/growth and actin ring expansion, as inferred from the defects in Cyt-Mut-cells. At the cell cortex, the linkage mediates microtubule contraction, leading to lamellipodial retraction, and the associated retrograde actin flow and then lamellar actin-arc formation, as inferred from the defects in TM-Mut-CD99 cells, which to our knowledge has not been reported previously. Furthermore, at the lamella, we propose that depolymerization of microtubule tips may lead to actomyosin contraction via linked F-actin, enabling inward flow of actin-microclusters across the lamella10,45,63. Similar control of actomyosin contractility by microtubule depolymerization has been demonstrated in cell migration64. Collectively, our findings highlight the significance of CD99-mediated actin-microtubule physical interaction in IS structure formation, providing mechanistic insight into the process of IS formation.
Incorporation of the functional domains of CD99 into a conventional BBz-CAR structure significantly improved the efficacy of CAR-T cells, potentiating IS formation by CAR-T cells. This validated the functionality of CD99 domains in the context of CAR-IS, underscoring the significance of IS formation as a mechanistic component of CAR-T cell functionality. Previous studies have demonstrated a positive correlation between IS quality and CAR-T cell performance in vivo65, and proposed the need for an IS-stabilization strategy to enhance CAR-T cell functionality66,67. However, there have been few reports on such strategies. Our identification of the molecular function and mechanism of action of CD99 represents a key advance enabling the development of IS-potentiating CAR-T cells.
Despite recent advances in CAR design, most CARs use the transmembrane domain as the physical linker between the extracellular antibody moiety and the intracellular signaling domains, with only the cytoplasmic domains being modified. The improvement of the efficacy of CAR-T cells by our novel CAR design, involving replacement of the CD8-linker domains with CD99 transmembrane and juxtamembrane domains, provides a new concept that the transmembrane domain can be transformed to boost CAR-T-cell functionality. This also has the advantage of making more space in the cytoplasmic domain available for further modification. It has been shown that CAR-T cells form nonclassical IS with indistinguishable pSMAC structure, rapidly recruiting lytic granules to the IS and quickly detaching from target cells68,69. Considering the pSMAC-localization of CD99, we propose that the improved functionality of HuTJ-BBz-CAR T cells is a result of the gain-of-pSMAC-localizing CD99 functional domains, which would promote the establishment of classical and stable IS by strengthening actin-microtubule crosstalk in CAR-T cells. This is supported by enhanced activation marker upregulation and proliferation of HuTJ-BBz-CAR T cells.
In summary, we propose CD99 as a novel master regulator of IS stabilization via the actin-microtubule linkage, which could be applied in T-cell-mediated therapeutics.
Methods
Mice
C57BL/6 J (000664; B6), B6.PL-Thy1a/CyJ (000406; Thy1.1+), B6.SJL-PtprcaPepcb/BoyJ (002014; CD45.1+), C.B10-H2b/LiMcdJ (001952; BALB.B), C57BL/6-Tg(TcraTcrb)1100Mjb/J (003831; OT-1) and NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (005557; NSG) mice were purchased from the Jackson Laboratory. H60 congenic mouse strain (B6.CH60; a kind gift from Dr. Derry Roopenian at the Jackson Laboratory, Maine, USA). J15 TCR transgenic mouse strain was developed in our laboratory37,38. Cd99−/− mouse strain (B6.Cd99Gt(pU-21T)44Imeg; CARD ID #718)33 were obtained from the Center for Animal Resources and Development (CARD) of Kumamoto University. All mice were housed in a specific pathogen-free animal facility at the Seoul National University College of Medicine and maintained in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC). The experimental use of animals was approved by the IACUC (SNU-221207-9-2, SNU-221207-5-1, SNU-221207-4-3, SNU-220704-9). Experimental and control group mice at the age of 8-12-week-old were housed separately in individually ventilated cage racks in the same room. Euthanasia was performed by CO2 inhalation.
Cell culture
Cells were cultured with DMEM (for mouse cells) or RPMI-1640 (for human cells) media (Gibco) supplemented with 10% FBS (Hyclone), HEPES, sodium pyruvate, L-arginine, L-glutamine, 2-mercaptoethanol, non-essential amino, gentamycin, streptomycin, and penicillin under an atmosphere of 5% CO2 at 37 °C.
Cell line establishment and cell engineering
H60-specific WT or Cd99−/− CD8 CTL lines were established from WT or Cd99−/− B6 mice immunized intraperitoneally with splenocytes from B6.CH60 mice, and maintained via weekly re-stimulation with irradiated B6.CH60 splenocytes48. For introduction of recombinant CD99 molecules, early (<10) passage Cd99−/− CTL lines were incubated with filtered retroviral supernatants harvested from Plat-E cells (Cell Biolabs #RV-101) transiently transfected with pMSCV-puro vector (Addgene #68469) containing Lifeact-mCherry, rCD99WT-YFP, Cyt-Mut-CD99-YFP, TM-Mut-CD99-YFP, CythCD19-Mut-CD99-YFP, TMhCD19-Mut-CD99-YFP, TM-SR-CD99-YFP, TM-LR-CD99-YFP, or TJ-CD99-YFP gene, or an empty vector (mock expression), using the calcium phosphate transfection or polyethylenimine (PEI) method. After 2 additional days of culture in fresh medium, the transduced CTL cells were re-stimulated for passage in culture medium containing puromycin (Sigma-Aldrich) and selected for mCherry+ or YFP+ cells using the FACS Aria II (BD Biosciences). DC2.4 cells70 transduced to express H60 (LTFNYRNL) or OVA (SIINFEKL), yielding H60-DC and OVA-DC, respectively, were cultured in the presence of puromycin. The B16F10-OVA cell line was generated by transduction of B16F10 murine melanoma cells (ATCC) with the lentivirus harboring the OVA-expressing puromycin-resistance cassette (pLEF-puro-OVA). The transduced cells were selected with puromycin and isolated as clones. The luciferase-GFP-expressing Raji cell line (Raji-Luc) were generated by spin infection with lentivirus (pLEF-LucGFP) harboring the luciferase-P2A-EGFP expression cassette. The GFP-high populations were sorted using the FACS Aria II.
In Jurkat cells, for knockdown of human CD99, anti-CD99 shRNA- or scramble shRNA-expressing plasmid (pSicoR-mCh; Addgene #21907) was transfected into the cells by electroporation using a Nucleofector2b device (Lonza). To express HuTJ-CD99, YFP- or HuTJ-YFP-expressing plasmid (pcDNA3.1; Thermo Fisher Scientific #V79020) was co-transfected with anti-CD99 shRNA-expressing plasmid (see Supplementary Table 1) using the method described above. Live cells were isolated 48 h post-transfection by Ficoll-gradient centrifugation (GE Healthcare #17-1440-02), stimulated on anti-human-CD3 antibody (OKT3; BioXcell #BE0001-2; 10 μg/ml)-coated coverslips, and observed using confocal microscopy.
For human primary T cell experiments, human peripheral blood mononuclear cells (PBMCs) from healthy donor leukapheresis samples were subjected to mixed lymphocyte reaction (MLR) with irradiated (20 Gy) human PBMCs from allogeneic leukapheresis samples to induce alloreactive T cell activation. To delete the CD99 gene, T cells were electroporated on day 4 of MLR with Cas9 ribonucleoprotein containing sgRNA targeting CD99 (cggcgaccagaacacccagc, synthesized by IDT) using the Neon Transfection System (Thermo Fisher Scientific). A scrambled sgRNA (sgControl; #1072544, IDT) was used as a control. The transfected T cells were rested in complete medium for another 6 days and restimulated with irradiated allogeneic PBMCs for 10 days for further expansion. The CD99-deleted alloreactive T cells were then sorted using the SH800S cell sorter (Sony Biotechnology) and further expanded by 1-2 rounds of allogeneic PBMC stimulation at 10-day intervals before use.
Cell-cell conjugate formation assay
Live B6 WT and Cd99−/− CTLs purified by Ficoll-gradient separation at day 3 after re-stimulation for passage were stained with Cell Tracker green CMFDA (5-chloromethylfluorescein diacetate; Thermo Fisher Scientific #C7025) according to the manufacturer’s directions. CMFDA-labeled T cells (1 × 105) and H60-DC or OVA-DC cells (2 × 104) labeled with Cell Tracker orange CMTMR (5-(and-6)-(((4-chloromethyl)benzoyl)amino) tetramethylrhodamine; Thermo Fisher Scientific #C2927) were co-incubated on poly-L-lysine (Sigma-Aldrich #P4832)-coated coverslips at 37°C and examined by fluorescence microscopy. T cells from J15 and OT-1 mice on a WT or Cd99−/− B6 background were purified by negative magnetic-activated cell sorting (MACS; Miltenyi Biotec) and then co-incubated with H60-DCs or OVA-DCs, as described above. After incubation, the cells were fixed with 4% paraformaldehyde for 15 min, washed, and viewed under a microscope (IX71; Olympus).
Confocal and structured illumination microscopy and confocal time-lapse live imaging
To visualize the IS, WT and Cd99−/− CTLs were pre-stained with fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated anti-CD99 (EJ2; Dinona; 1:200) together with FITC-, or PE-conjugated anti-TCRβ (H57-597; Thermo Fisher Scientific #11-5961-81; 1:500) or anti-LFA1 (2D7; BD Biosciences #553120; 1:500) antibody for 1 h at 4 °C. H60-DCs were stained with Alexa 647-conjugated anti-ICAM1 antibody (YN1/1.7.4; Thermo Fisher Scientific #12-0541-81; 1:500). Pre-stained T and H60-DC cells were co-incubated on the same poly-L-lysine coated coverslips; after fixation and washing, they were viewed under a FluoView 1000 confocal microscope (Olympus). For the imaging of fixed cells, T cells (1 × 105) loaded onto anti-CD3 antibody (10 μg/ml; 145-2C11, BD Biosciences #553058)-coated coverslips or plated together with H60-DCs on poly-L-lysine coated coverslips were incubated for 7, 15, 30 min and fixed with 4% paraformaldehyde. For chemical treatment, T cells pretreated with the actin polymerization inhibitors CK666 or SMIFH2 were stimulated on an anti-CD3-coated plate for 15 min before fixation. In another experiment, T cells were treated with PMA (20ng/ml; Sigma-Aldrich #P8139) and Ionomycin (10 μM; Sigma-Aldrich #I0634) for 15min and fixed. The fixed cells were permeabilized with 0.25% Triton in phosphate-buffered saline (PBS) for 10 min and blocked with 10% bovine serum albumin (BSA; GeorgiaChem #BS1005) for 1 h at room temperature. Then, cells were stained with anti-myosin IIA (Cell Signaling Technology #3403; 1:200), anti-IQGAP1 (C-9; Santa Cruz Biotechnology #sc-376021; 1:200), anti-NFAT1 (D43B1; Cell Signaling #5861; 1:200), anti-NFκB p65 (D14E12; Cell Signaling #8242; 1:200), phosphor-Erk1/2 (D13.14.4E; Cell Signaling #4370; 1:200), or anti-α-Tubulin (DM1A; Millipore #05-829; 1:500) antibody (see Supplementary Table 2), washed, and stained with Alexa 647-conjugated phalloidin (Thermo Fisher Scientific #A22287; 1:1000), fluorescence-conjugated secondary antibodies (Thermo Fisher Scientific), and 4’,6-diamidino-2-phenylindole (DAPI; 0.5 μg/ml; Sigma-Aldrich #D9542). For Jurkat cell imaging, cells were stimulated for 15 min on cover slip coated with anti-human CD3 antibody (10 μg/ml; OKT3, BioXcell), and for human primary T cell analysis, cells were either stimulated on the coverslips or co-incubated with irradiated allogeneic PBMCs for 30 min prior to imaging. To perform super-resolution 3D structured illumination microscopy (SIM), the N-SIM system (Nikon) was used. 3D-SIM images of the microtubule and actin cytoskeleton were acquired with a 100×/1.49 CFI Apochromat TIRF oil-immersion objective, using excitation wavelengths of 488 nm and 561 nm. Z-stacks were acquired at 120 nm intervals to scan the entire cell. For live-cell imaging, the sorted cells stably transduced with Lifeact-mCherry for F-actin visualization were isolated by Ficoll separation and incubated for 1 h with 1 μM of SiR-Tubulin dye (Spirochrome AG #CY-SC002) containing 10 μM of the efflux pump inhibitor verapamil (Spirochrome AG #CY-SC002). After the CTLs were loaded onto an anti-CD3 antibody-coated coverslip, time-lapse images at 100× were collected every 20 s for 30–35 min using FluoView 3000 confocal microscope (Olympus).
Image analysis
Single-plane or z-stacked images were generated from raw microscopy data by exporting with FIJI ImageJ (NIH) or rendering with IMARIS (Bitplane). To remove non-specific background fluorescence, unstained control images were acquired using the same imaging settings, and their fluorescence intensities were subtracted. For fluorescence intensity measurements, a single cell was selected as the region of interest (ROI) by automatically defining the cell boundary on the thresholded F-actin image, and cell size (area) was also measured from the same image. Specific parts of the cell were selected using the rectangular selection tool. Distance and thickness measurements were performed using the line tool. Intensity was quantified based on these defined areas. For quantification of NFAT or NFκB nuclear translocation, the nucleus region was defined as the ROI by thresholding the DAPI channel, and fluorescence intensity of NFAT or NFκB was measured within this ROI. Colocalization analysis was conducted by calculating the Manders’ coefficient M1 using IMARIS71 and Pearson’s coefficient using ImageJ and the JACoP plug-in. All time-lapse images were analyzed using IMARIS. To analyze F-actin retraction dynamics, time-lapse images were aligned and straightened using ImageJ software, and the Multi Kymograph plug-in was used to generate kymographs. In most cases, the bottom layer along with one or two (when necessary) upper layer images were presented as a synapse plane image and used for analysis, unless otherwise specified in the figures and figure legends.
In vitro T cell stimulation, co-immunoprecipitation, and immunoblotting
Lymph node or splenic T cells (2 × 106/well) from WT or Cd99−/− mice were resuspended in pre-warmed serum-free DMEM, and stimulated with anti-CD3 (10 μg/ml) and anti-CD28 (0.5 μg/ml; 37.51, BD Biosciences #556620) antibodies at 37 °C for 0, 2, 5, 15, or 30 min before adding cold PBS containing 1 mM sodium orthovanadate. After centrifugation, cell pellet was resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 10% Glycerol, 1% NP-40, 1 mM Na3VO4, 2 mM NaF, Protease inhibitor cocktail) and incubated on ice for 20 min cell lysis. Cell lysates were subjected to immunoblotting using anti-Zap70, anti-pZap70, anti-ERK1/2, and anti-pERK1/2 (Cell Signaling Technology) antibodies, and horseradish peroxidase-conjugated anti-rabbit (Santa Cruz Biotechnology) or anti-mouse (Jackson ImmunoResearch Labs) secondary antibody. For the co-immunoprecipitation assays, lysates were precleared for 1 h with protein-G sepharose beads (BioVision #6511-5) and then incubated with anti-α-tubulin (DM1A), anti-CD99 (EJ2), anti-myc (9E10; Santa Cruz Biotechnology #sc-40), or anti-YFP (3E6; Thermo Fisher Scientific #A11120) antibody. The antibody-bound proteins were pulled down using protein-G sepharose beads and eluted after boiling in sample buffer. Immunoblotting was performed as described above.
Proteomics analysis for CD99-interactome identification
Samples co-immunoprecipitated with anti-YFP antibody, or isotype IgG were fractionated on a 4–12% Bis-Tris gel (Thermo Fisher Scientific) and stained with Coomassie Brilliant Blue (Sigma-Aldrich). The entire gel lane was cut into 7–10 pieces and subjected to in-gel tryptic digestion following the general protocol. The extracted peptides were resuspended in 0.1% formic acid (FA, solvent A) and analyzed on a liquid chromatography-tandem mass spectrometry (LC-MS/MS) system integrated with Dionex 3000 ultra-high-performance liquid chromatograph and Q-Exactive Plus mass spectrometer. Peptides were loaded onto a C18 trap column (Acclaim PepMap100, 75 μm × 2 cm, 100 Å; Thermo Fisher Scientific) and separated on a C18 analytical column (PepMap RSLC, 75 μm × 50 cm, 100 Å; Thermo Fisher Scientific) using a 125-min linear gradient containing 5–35% solvent B (0.1% FA in acetonitrile) at a flow rate of 300 nL/min. The spray voltage of the column was set to 2.2 kV, and the heated capillary to 250 °C. The normalized collision energy was 27 eV. Q-Exactive was run in data-dependent acquisition mode with an MS survey scan followed by 10 MS/MS scans of the most abundant ions. The full MS scan range was 400– 1400 m/z and dynamic exclusion was applied for 20 s.
To assess the interactomes of CD99 and its mutants, the amounts of precipitated recombinant proteins were compared by a test run on SDS-PAGE followed by immunoblotting. After normalization of the levels of precipitated recombinant proteins, the samples were subjected to interactome studies. For quantitative comparison of interactome components among the three recombinant proteins (rWT, Cyt-Mut, and TM-Mut), the levels of proteins in the rWT interactome were used as a standard. For data analysis, differential expression analysis was performed using the edgeR package (v3.36.0)72. Raw spectral count data were normalized using the voom transformation from the limma package, which involved log-transforming the counts and applying precision weights to account for the mean-variance relationship. A linear model was fitted to the data, with contrasts specified to identify differentially expressed proteins. Proteins differentially expressed between WT-YFP and WT-Control with a false discovery rate (FDR) < 0.05 and a log2 fold change > 0 were considered CD99 interactome proteins. These proteins were further classified into functional subsets based on Gene Ontology (GO) pathway terms obtained from the Molecular Signatures Database (MsigDB). Proteins associated with the actin cytoskeleton (GO:0015629), along with the proteins Myl6, Actbl2, Actc1, and Iqgap1, were classified into the actin system group. Proteins associated with microtubule (GO:0005874) and the microtubule cytoskeleton (GO:0015630) were classified into tubulin system group. Proteins associated with vesicles (GO:0031982), cytoplasmic vesicles (GO:0031410), or secretory granules (GO:0030141) were classified into the molecular trafficking system group. Proteins associated with the myosin complex (GO:0016459), along with Myl12a, were classified into the myosin system group. Proteins associated with the cytoskeleton (GO:0005856), along with Rac2, Capza2, Rab1b, Rab27a, and Fmnl1, were classified into other cytoskeleton group. Proteins associated with the ribosome (GO:0005840), along with Arf4, Arf5, Rpn1, and Rps27, were classified into the ribosome group. Proteins associated with TCR signaling pathway (GO:0050852), along with Ptprc, Zap70, Lck, CD247, and CD3e, were classified into the TCR signaling group. Proteins associated with ATP metabolic process (GO:0046034) were classified into the mitochondria group. Interaction pairs from the STRING database (v11.0)73 were used to construct a protein-protein interaction (PPI) network of CD99-interactome proteins, by selecting high-quality pairs with a combined score > 700. The network was visualized using Cytoscape software (v3.9.1; Cytoscape Team)74. PCA was performed on the normalized proteomics data using Partek Flow software (Partek Inc.). Data were normalized using log2-scale transformation, and the first three principal components were visualized in 3D plots to assess sample clustering and variation across experimental groups. To compare the interactomes of CD99 mutants, log2 fold changes of the cytoplasmic mutant (Cyt-Mut) and transmembrane mutant (TM-Mut) relative to wild-type (rWT) were calculated from their average transformed spectral counts. These values were then visualized as a heatmap.
Flow cytometry
For FACS staining, non-specific binding was blocked with an anti-Fc receptor antibody (2.4G2; Tonbo Bioscience #70-0161-M001) prior to staining with the directly labeled monoclonal antibodies indicated in the figures and text. Dead cells were excluded by DAPI staining. The following antibodies (see Supplementary Table 2) were used to stain cell surface molecules. PE/Cy7 anti‑mCD8α (53‑6.7; BD Biosciences #552877; 1:2000), FITC anti‑mTCRβ (H57‑594; Thermo Fisher Scientific #11‑5961‑81; 1:1000), FITC anti‑mCD11a (2D7; BD Biosciences #553120; 1:1000), APC anti‑mCD25 (PC61.5; Thermo Fisher Scientific #17‑0251‑81; 1:2000), APC/Cy7 anti‑m/hCD44 (IM7; BioLegend #103028; 1:2000), eFluor450 anti‑mCD45.1 (A20; Thermo Fisher Scientific #48‑0453‑82; 1:500), PE anti‑mCD69 (H1.2F3; BioLegend #104508; 1:2000), BV605 anti‑mCD90.2 (30‑H12; BioLegend #105343; 1:4000), PE-conjugated H60/Kb-tetramer, H4/Kb-tetramer or OVA/Kb-tetramer, PE anti‑mCD99 (R&D Systems #FAB3905P; 1:500), APC anti‑mCD178 (MFL3; BioLegend #106609; 1:500), BV421 anti‑hCD3 (UCHT1; BioLegend #300434; 1:500), AlexaFluor647 anti‑hCD99 (DN16; Novus Biologicals #NB100‑64930AF647; 1:1000), BV510 anti‑hCD8 (SK1; BioLegend #344732; 1:500), APC/Cy7 anti‑hCD4 (RPA‑T4; BD Biosciences #557871; 1:500), BV605 anti‑hCD69 (FN50; BD Biosciences #562989; 1:250), PE anti‑hCD25 (M‑A251; BioLegend #985802; 1:1000), biotinylated anti‑FMC63 (Y45; Acro Biosystems #FM3‑BY54; 1:50), PE/TexasRed streptavidin (BD Pharmingen #551487; 1:2000), PE streptavidin (BD Pharmingen #554061; 1:8000). Cell proliferation were detected by labeling T cells (5 × 105) from WT and Cd99−/− mice with 5 μM CFSE (carboxyfluorescein diacetate succinimidyl ester; Thermo Fisher Scientific #C34554) and stimulating them with anti-CD3 (1 µg/ml) and anti-CD28 (0.5 µg/ml) antibodies. On days 2 and 3 after the stimulation, cell proliferation was assessed by flow cytometric analysis of CFSE dye dilution. For intracellular staining, cells were treated with PMA (20 ng/ml), Ionomycin (10 μM), and Brefeldin A (1×; Thermo Fisher Scientific) 4 h before harvest, then fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton-containing buffer for granzyme B (NGZB; eBioscience #12-8898-82; 1:4000) staining. Additionally, cells were fixed with 1.5% paraformaldehyde and permeabilized with methanol to stain for phospho-Akt (J1-223.371; BD Biosciences #558275; 10 µl/sample). Fluorescence intensity was assessed with a BD LSRII (BD Biosciences) or Northern Lights (Cytek Biosciences) and analyzed using FlowJo software (TreeStar).
Adoptive T cell transfer into hosts for immunization or into tumor bearing mice
CD8+ T cells from female WT or Cd99−/− J15 mice were purified from the spleen and lymph nodes by negative MACS selection. After labelling with CFSE, cells were adoptively transferred (1 × 106) 1 day before immunization with a single-cell suspension of splenocytes (2 × 107 in 300 μL of PBS) from male B6.CH60 mice. In tumor regression experiments, naïve T cells (5 × 105) from female CD45.1+ WT or Cd99−/− OT-I mice were transferred into a female host implanted with B16F10-OVA (3 × 105) at the flank. Tumor volume was measured every third day using an electronic caliper. On days 7 and 10 after OT-I cell transfer, peripheral blood was collected by eye bleeding for flow cytometric analysis. Tumor growth was monitored every 2–3 days using a digital caliper, and volume was calculated using the formula (length × width2)/2. Mice were euthanized if tumors exceeded 1500 mm3, showed ulceration, or caused distress, in accordance with IACUC guidelines. Euthanasia was performed by CO2 inhalation.
Skin transplantation
Allogeneic tail skin transplantation was performed49. WT or Cd99−/− recipient mice (female, 8–12 weeks) were grafted with three pieces of full-thickness tail skin (1 × 1 × 0.5 cm3) derived from male BALB.B mice (male, 8–12 weeks), and one piece of syngeneic WT or Cd99−/− mice, respectively. Graft survival was evaluated by daily visual inspection. Rejection was defined as necrosis of 75% of the transplanted skin surface.
Generation of CAR-T cells
The ORF sequence of the conventional CAR harboring CD8 extracellular and transmembrane domain (BBz-CAR) was acquired from a patent (US_2013_0287748_A1). HuTJ-BBz-CAR ORF was generated by replacing CD8 parts of BBz-CAR with human CD99 extracellular, transmembrane and juxtamembrane domain (amino acid 88–158, UniProt #P14209-1). The CAR ORF cDNAs were synthesized by Integrated DNA Technologies (IDT) and cloned into MSGV retroviral vector (Addgene #64269). Retroviruses for human T cell transduction were produced by establishing amphitropic retrovirus-producing stable cell lines from PG13 retroviral packaging cells (ATCC). For this purpose, retroviral plasmids were transfected into Phoenix ECO cells (ATCC) using Lipofectamine 3000 (Thermo Fisher Scientific) to produce ecotropic retroviruses. The retroviral supernatant was harvested 48 h after transfection and spin-infected into PG13 cells (derived from a mouse fibroblast) for stable transduction of the retroviral cDNA. Retrovirus-containing supernatants from these PG13 cultures were harvested and concentrated 5–10-fold using a centrifugal filter device (Amicon Ultra,100 kDa cut-off; Millipore) and used for human T cell transduction. For the generation of CAR-T cells, human peripheral blood was obtained from healthy volunteers who provided written informed consent according to a protocol approved by the Institutional Review Board of Seoul National University Hospital (IRB No. H-2008-114-1150). PBMCs prepared by Ficoll-gradient centrifugation (841×g, 20 min, RT) were activated for 48 h with plate-bound anti-CD3 (10 μg/ml; OKT3, BioXcell) and soluble anti-CD28 (2 μg/ml; CD28.2, BD Biosciences) antibodies. The concentrated retroviral solutions were attached to a 24-well retronectin-coated plate (20 μg/ml, TaKaRa) by centrifugation for 2 h at 2000×g. After removal of the viral solution, the activated T cells were added to the plate, followed by 10-min centrifugation at 1000×g and further incubation for 48 h in medium containing human IL-2 (300 IU/ml Proleukin; Novartis). The T cells were then washed and expanded in fresh medium containing human IL-2 (200 IU/ml) for 3–6 days before being used as CAR-T cells. To generate endogenous CD99 gene-deleted HuTJ-BBz- or BBz-CAR-T cells, T cells stimulated with anti-CD3 and CD28 for 48 h were electroporated with Cas9 ribonucleoprotein containing sgRNA targeting CD99 (cggcgaccagaacacccagc, synthesized by IDT) or a scramble-sgRNA control (sgControl; #1072544, IDT) using the Neon Transfection System and transduced with virus containing either HuTJ-BBz-CAR or BBz-CAR.
Evaluation of CAR-T cell activity in vitro and in vivo
To visualize cell-conjugate formation, 2 × 105 CAR-T cells were co-incubated with 1 × 105 CMTMR-labeled Raji B cells on poly-L-lysine-coated coverslips. After 15, 30 or 60 min, the cells were washed, fixed, and permeabilized, and then stained with phalloidin and anti-α-tubulin (DM1A). For flow cytometric analysis of CAR-T cell activation, 1 × 105 CAR-T cells were co-incubated with 2 × 104 Raji B cells in round-bottom 96 well plates for three days. To evaluate tumor killing efficacy of CAR-T cells in vitro, untransduced T or CAR-T cells (4000 cells as CAR+/well) were co-cultured with Raji tumor cells (16,000 cells/well) at an E:T ratio of 1:4 in 96 well plates in the absence of exogenous cytokines. On the day of analysis, dead cells were excluded by staining with 7-aminoactinomycin D (7-AAD) (BioLegend #420404, 5 μl/sample) and counted number of live tumor cells (7-AAD-CD3-) and CAR-T cells (7-AAD−CD3+) using 123 count eBead (containing about 10,000 beads; Thermo Fisher Scientific #01-1234-42, 10 μl/sample). To exclude the tumorigenic potential of CAR-T cells, HuTJ-BBz- and BBz-CAR-T cells were suspended in 500 μl of 10% RPMI-1640 medium (5 × 105 cells/sample) and cultured without additional CAR stimulation or cytokines. Untransduced-T cells and the Jurkat cell line were used as controls. Cell viability and the number of viable cells were determined at each time point. 7-AAD-negative cells were gated as viable cells, and cell numbers were counted using a bead-based flow cytometry method. For Jurkat cells, cells were split by 1/5 or 1/10 every 3–4 days to prevent excessive cell growth, so the dilution ratio was taken into account when calculating the number of viable cells.
CAR-T cell activity in vivo was evaluated by intravenously injecting NSG mice (female and male, 6–8 weeks) with Raji-Luc tumor cells (5 × 105/mouse). After 7 days, BBz-CAR-T cells or HuTJ-BBz-CAR-T cells (5 × 106/mouse) were infused intravenously. Tumor engraftment and growth were measured by bioluminescence imaging (IVIS Spectrum; PerkinElmer) after intraperitoneal injection of D-luciferin (2 mg; Promega #P1043). The tumor burden was quantified as total flux (photons per s) using Living Image software (PerkinElmer). To estimate CAR-T cell numbers in blood, 10 μl 123 count eBeads were added to each RBC-lysed and FACS-stained sample tube. After 2000 beads were acquired by flow cytometer, number of human cells in the blood was calculated by the following formula: blood cell count (cells/μl) = (blood cell count × eBead volume)/(eBead count × blood volume) × eBead concentration.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software version 9 (GraphPad Software Inc.), with statistical tests detailed in the figure legends. Unless otherwise stated, quantitative data are expressed as means ± SEM from data obtained from at least two independent experiments. Statistical significance was determined by one-way ANOVA with the Dunnett’s post-hoc test, two-way repeated measures ANOVA with Sidak post-hoc test, two-tailed unpaired Student’s t-test, or a log-rank (Mantel-Cox) test. P values lower than 0.05 were considered to be significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD039955. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this Article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018).
Labanieh, L. & Mackall, C. L. CAR immune cells: design principles, resistance and the next generation. Nature 614, 635–648 (2023).
Dotti, G., Gottschalk, S., Savoldo, B. & Brenner, M. K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 257, 107–126 (2014).
Morris, E. C., Neelapu, S. S., Giavridis, T. & Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 22, 85–96 (2022).
Morgan, R. A. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010).
Grakoui, A. et al. The immunological synapse: a molecular machine controlling T cell activation. Science 285, 221–227 (1999).
Dustin, M. L., Chakraborty, A. K. & Shaw, A. S. Understanding the structure and function of the immunological synapse. Cold Spring Harb. Perspect. Biol. 2, a002311 (2010).
Demetriou, P. et al. A dynamic CD2-rich compartment at the outer edge of the immunological synapse boosts and integrates signals. Nat. Immunol. 21, 1232–1243 (2020).
Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 (1998).
Hammer, J. A., Wang, J. C., Saeed, M. & Pedrosa, A. T. Origin, Organization, dynamics, and function of actin and actomyosin networks at the T cell immunological synapse. Annu. Rev. Immunol. 37, 201–224 (2019).
Kopf, A. & Kiermaier, E. Dynamic microtubule arrays in leukocytes and their role in cell migration and immune synapse formation. Front Cell Dev. Biol. 9, 635511 (2021).
Ritter, A. T. et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immunity 42, 864–876 (2015).
de la Roche, M., Asano, Y. & Griffiths, G. M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 16, 421–432 (2016).
Martin-Cofreces, N. B. et al. End-binding protein 1 controls signal propagation from the T cell receptor. EMBO J. 31, 4140–4152 (2012).
Kupfer, A. & Dennert, G. Reorientation of the microtubule-organizing center and the Golgi apparatus in cloned cytotoxic lymphocytes triggered by binding to lysable target cells. J. Immunol. 133, 2762–2766 (1984).
Kuhn, J. R. & Poenie, M. Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity 16, 111–121 (2002).
Stinchcombe, J. C., Majorovits, E., Bossi, G., Fuller, S. & Griffiths, G. M. Centrosome polarization delivers secretory granules to the immunological synapse. Nature 443, 462–465 (2006).
Laan, L. et al. Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell 148, 502–514 (2012).
Yi, J. et al. Centrosome repositioning in T cells is biphasic and driven by microtubule end-on capture-shrinkage. J. Cell Biol. 202, 779–792 (2013).
Pasello, M., Manara, M. C. & Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal 12, 55–68 (2018).
Hahn, J. H. et al. CD99 (MIC2) regulates the LFA-1/ICAM-1-mediated adhesion of lymphocytes, and its gene encodes both positive and negative regulators of cellular adhesion. J. Immunol. 159, 2250–2258 (1997).
Schenkel, A. R., Mamdouh, Z., Chen, X., Liebman, R. M. & Muller, W. A. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 3, 143–150 (2002).
Lou, O., Alcaide, P., Luscinskas, F. W. & Muller, W. A. CD99 is a key mediator of the transendothelial migration of neutrophils. J. Immunol. 178, 1136–1143 (2007).
Winger, R. C. et al. Cutting Edge: CD99 Is a Novel Therapeutic Target for Control of T Cell-Mediated Central Nervous System Autoimmune Disease. J. Immunol. 196, 1443–1448 (2016).
Manara, M. C., Pasello, M. & Scotlandi, K. CD99: a cell surface protein with an oncojanus role in tumors. Genes (Basel) 9, 159 (2018).
Waclavicek, M. et al. CD99 engagement on human peripheral blood T cells results in TCR/CD3-dependent cellular activation and allows for Th1-restricted cytokine production. J. Immunol. 161, 4671–4678 (1998).
Oh, K. I. et al. CD99 activates T cells via a costimulatory function that promotes raft association of TCR complex and tyrosine phosphorylation of TCR zeta. Exp. Mol. Med. 39, 176–184 (2007).
Choi, E. Y. et al. Engagement of CD99 induces up-regulation of TCR and MHC class I and II molecules on the surface of human thymocytes. J. Immunol. 161, 749–754 (1998).
Yoon, S. S. et al. Engagement of CD99 triggers the exocytic transport of ganglioside GM1 and the reorganization of actin cytoskeleton. FEBS Lett. 540, 217–222 (2003).
Bremond, A. et al. Regulation of HLA class I surface expression requires CD99 and p230/golgin-245 interaction. Blood 113, 347–357 (2009).
Pata, S. et al. Association of CD99 short and long forms with MHC class I, MHC class II and tetraspanin CD81 and recruitment into immunological synapses. BMC Res. Notes 4, 293 (2011).
Watson, R. L. et al. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 212, 1021–1041 (2015).
Nam, G. et al. Interaction of CD99 with its paralog CD99L2 positively regulates CD99L2 trafficking to cell surfaces. J. Immunol. 191, 5730–5742 (2013).
Roopenian, D., Choi, E. Y. & Brown, A. The immunogenomics of minor histocompatibility antigens. Immunol. Rev. 190, 86–94 (2002).
Choi, E. Y. et al. Immunodominance of H60 is caused by an abnormally high precursor T cell pool directed against its unique minor histocompatibility antigen peptide. Immunity 17, 593–603 (2002).
Choi, E. Y., Choi, K., Nam, G., Kim, W. & Chung, M. H60: a unique murine hematopoietic cell-restricted minor histocompatibility antigen for graft-versus-leukemia effect. Front. Immunol. 11, 1163 (2020).
Ju, J. M. et al. Escape from thymic deletion and anti-leukemic effects of T cells specific for hematopoietic cell-restricted antigen. Nat. Commun. 9, 225 (2018).
Kim, J. et al. Memory programming in CD8(+) T-cell differentiation is intrinsic and is not determined by CD4 help. Nat. Commun. 6, 7994 (2015).
Riedl, J. et al. Lifeact mice for studying F-actin dynamics. Nat. Methods 7, 168–169 (2010).
Lukinavicius, G. et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 11, 731–733 (2014).
Macian, F. et al. Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731 (2002).
Keller, H. U., Niggli, V. & Zimmermann, A. Diacylglycerols and PMA induce actin polymerization and distinct shape changes in lymphocytes: relation to fluid pinocytosis and locomotion. J. Cell Sci. 93, 457–465 (1989).
Preciado Lopez, M. et al. Actin-microtubule coordination at growing microtubule ends. Nat. Commun. 5, 4778 (2014).
Fukata, M. et al. Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109, 873–885 (2002).
Liu, X., Kapoor, T. M., Chen, J. K. & Huse, M. Diacylglycerol promotes centrosome polarization in T cells via reciprocal localization of dynein and myosin II. Proc. Natl Acad. Sci. USA 110, 11976–11981 (2013).
Wang, J. C. et al. The Rap1-cofilin-1 pathway coordinates actin reorganization and MTOC polarization at the B cell immune synapse. J. Cell Sci. 130, 1094–1109 (2017).
Kumari, S. et al. Cytoskeletal tension actively sustains the migratory T-cell synaptic contact. EMBO J. 39, e102783 (2020).
Choi, E. Y. et al. Quantitative analysis of the immune response to mouse non-MHC transplantation antigens in vivo: the H60 histocompatibility antigen dominates over all others. J. Immunol. 166, 4370–4379 (2001).
Yoo, K. I. et al. Subdominant H60 antigen-specific CD8 T-cell response precedes dominant H4 antigen-specific response during the initial phase of allogenic skin graft rejection. Exp. Mol. Med 47, e140 (2015).
Dufour, E. M., Deroche, A., Bae, Y. & Muller, W. A. CD99 is essential for leukocyte diapedesis in vivo. Cell Commun. Adhes. 15, 351–363 (2008).
Milone, M. C. et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464 (2009).
Bernard, G. et al. The E2 molecule (CD99) specifically triggers homotypic aggregation of CD4+ CD8+ thymocytes. J. Immunol. 154, 26–32 (1995).
Bernard, G. et al. CD99 (E2) up-regulates alpha4beta1-dependent T cell adhesion to inflamed vascular endothelium under flow conditions. Eur. J. Immunol. 30, 3061–3065 (2000).
Sohn, H. W. et al. CD99 regulates the transport of MHC class I molecules from the Golgi complex to the cell surface. J. Immunol. 166, 787–794 (2001).
Martin-Cofreces, N. B. & Sanchez-Madrid, F. Sailing to and docking at the immune synapse: role of tubulin dynamics and molecular motors. Front Immunol. 9, 1174 (2018).
Mastrogiovanni, M., Juzans, M., Alcover, A. & Di Bartolo, V. Coordinating cytoskeleton and molecular traffic in T cell migration, activation, and effector functions. Front. Cell Dev. Biol. 8, 591348 (2020).
Chanet, S., Sharan, R., Khan, Z. & Martin, A. C. Myosin 2-induced mitotic rounding enables columnar epithelial cells to interpret cortical spindle positioning cues. Curr. Biol. 27, 3350–3358 e3353 (2017).
Verma, V., Mogilner, A. & Maresca, T. J. Classical and emerging regulatory mechanisms of cytokinesis in animal cells. Biology 8, 55 (2019).
Hedman, A. C., Smith, J. M. & Sacks, D. B. The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16, 427–446 (2015).
Laplante, C., Huang, F., Tebbs, I. R., Bewersdorf, J. & Pollard, T. D. Molecular organization of cytokinesis nodes and contractile rings by super-resolution fluorescence microscopy of live fission yeast. Proc. Natl Acad. Sci. USA 113, E5876–E5885 (2016).
Sundvold, H. et al. Arv1 promotes cell division by recruiting IQGAP1 and myosin to the cleavage furrow. Cell Cycle 15, 628–643 (2016).
Kim, S. H. et al. Generation of cells with Hodgkin’s and Reed-Sternberg phenotype through downregulation of CD99 (Mic2). Blood 92, 4287–4295 (1998).
Hammer, J. A. 3rd & Burkhardt, J. K. Controversy and consensus regarding myosin II function at the immunological synapse. Curr. Opin. Immunol. 25, 300–306 (2013).
Kopf, A. et al. Microtubules control cellular shape and coherence in amoeboid migrating cells. J. Cell Biol. 219, e201907154 (2020).
Xiong, W. et al. Immunological synapse predicts effectiveness of chimeric antigen receptor cells. Mol. Ther. 26, 963–975 (2018).
Liu, D. et al. The role of immunological synapse in predicting the efficacy of chimeric antigen receptor (CAR) immunotherapy. Cell Commun. Signal 18, 134 (2020).
Gao, Y., Wang, Y., Luo, F. & Chu, Y. Optimization of T cell redirecting strategies: obtaining inspirations from natural process of T cell activation. Front Immunol. 12, 664329 (2021).
Davenport, A. J. et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl Acad. Sci. USA 115, E2068–E2076 (2018).
Mukherjee, M., Mace, E. M., Carisey, A. F., Ahmed, N. & Orange, J. S. Quantitative imaging approaches to study the CAR immunological synapse. Mol. Ther. 25, 1757–1768 (2017).
Shen, Z., Reznikoff, G., Dranoff, G. & Rock, K. L. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J. Immunol. 158, 2723–2730 (1997).
Manders, E. M., Stap, J., Brakenhoff, G. J., van Driel, R. & Aten, J. A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 103, 857–862 (1992).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Sievers, F. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 (2011).
Acknowledgements
We thank professor Sang Hee Shim (Korea University, Seoul, Korea) for technical advice on the visualization of tubulin dynamics and Min Jae Kim (Ticaros, Seoul, Korea) for technical assistance. This work was supported by the National Research Foundation (NRF) grants (RS-2024-00405619, EYC; RS-2023-00260454, KC; RS-2024-00400897, KC; 2021R1A4A1029097, EYC and KC) funded by Ministry of Science and ICT, the Korea Drug Development Fund (KDDF) grant (RS-2024-00337759, KC) funded by the Ministry of Science and ICT, Ministry of Trade, Industry, and Energy, and Ministry of Health and Welfare, and KIST intramural grant (2E33701, HR), Republic of Korea.
Author information
Authors and Affiliations
Contributions
Conceptualization: G.N., K.C., and E.Y.C. Investigation: G.N., H.R.Y., H.B.P., H.C., J.H.K., J.E.L., B.K.C., Y.L., S.L., H.L., and E.M.H. Data Curation: H.J., B.K.C., and E.C.Y.; Formal Analysis: S.K., J.Y.A. and W.J.S. Resources: H.R. and J.C. Funding Acquisition: K.C. and E.Y.C. Project Administration: E.Y.C. Supervision: K.C. and E.Y.C. Writing-original draft: G.N., K.C., and E.Y.C. Writing-review & editing: G.N., K.C., and E.Y.C.
Corresponding authors
Ethics declarations
Competing interests
G.N., H.R.Y., H.B.P., J.E.L., K.C., and E.Y.C. are inventors on a filed patent related to the CAR-T cells. K.C. and E.Y.C. are scientific co-founders and shareholders of Ticaros. H.R.Y., H.B.P., and J.E.L. are employees of Ticaros currently. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Claire Hivroz, Michael Kalos, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nam, G., Yeon, H.R., Park, H.B. et al. CD99-mediated immunological synapse formation potentiates CAR-T cell function. Nat Commun 16, 7987 (2025). https://doi.org/10.1038/s41467-025-63184-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63184-w
