
Abstract
In the field of wastewater treatment, the regulation of free radical and non-radical routes has been one of the major challenges. This study investigates the regulation of radical and non-radical oxidation pathways in the peroxymonosulfate (PMS) oxidation system by controlling the calcination temperature of carbon materials and constructing bimetallic single-atom catalysts (NC-FeMn(TA)). Density functional theory calculations and experimental tests indicate that increasing the pyridinic nitrogen content and incorporating single metal atoms in nitrogen-doped carbon materials result in a predominantly non-radical oxidation process. In contrast, enhancing the content of graphitic and pyrrolic nitrogen species and introducing bimetallic catalytic centers promote a radical oxidation pathway. The NC-FeMn(TA)/PMS system demonstrates high oxidation performance over a broad pH range, exhibiting significant interference resistance and stability, with 100% degradation of target pollutants after 22 cycles and complete removal of emerging pollutants (including pharmaceuticals and personal care products, endocrine disrupting chemicals, dyes and chemical materials) within 5 min. This system’s remarkable performance suggests broad application potential in water pollution control field.
Similar content being viewed by others


Introduction
Peroxysulfate-based advanced oxidation processes (PS-AOPs) have attracted significant attention for their effectiveness in treating emerging organic contaminants, due to their wide applicability, rapid degradation rates, and high selectivity for aromatic compounds1. In the domain of environmental catalysis, single-atom catalysts (SACs) adeptly merge the high efficiency of homogeneous catalysts with the robustness of heterogeneous catalysts2. SACs demonstrate significantly enhanced catalytic performance in PS-AOPs, attributed to their well-defined active sites, exceptional atomic utilization efficiency, and distinctive unsaturated coordination environments3. SACs feature simpler and more well-defined geometric configurations compared to their bulk counterparts, facilitating the investigation of charge transfer mechanisms at single-atom centers under different coordination environments during reaction processes4. The distinctive Metal-Nitrogen doped-Carbon (M-N-C) structure in SACs not only provides highly active atomically dispersed metal sites and defect carbon reaction sites for peroxymonosulfate (PMS) activation but also affects the local electronic configuration of the catalyst by modifying the M-N coordination structure5,6,7, thereby regulating the electron transfer process between the catalyst and PMS. The coordination number of the single-atom catalytic center is a crucial factor influencing its catalytic activity and PMS activation pathway. Investigating the regulatory mechanisms of SACs in PS-AOPs can aid in understanding the relationship between the coordination environment of single atoms and the activation pathways of PMS. It provides technical support for enhancing the performance of PS-AOPs systems and for the custom design of catalysts to meet various treatment requirements.
In the activation process of PMS, two oxidation pathways are discernible: radical and non-radical8. Systems dominated by radical species show high chemical oxygen demand (COD) removal efficiency in real wastewater treatment, while systems dominated by non-radical species significantly enhance the biodegradability of wastewater. For instance, a system with a high proportion of radical pathways (81.33%) exhibited superior oxidation capability, achieving an ideal COD removal rate of 75.16% for real soil wastewater within 36 h9. Thus, to address the varying treatment requirements of different water bodies, efficient water treatment can be achieved by adjusting the balance between radical and non-radical species in the oxidation pathways9. These pathways are influenced by the nature of the catalytic site atoms and their electronic configurations10. In PS-AOPs, the regulation of radical and non-radical pathways remains a challenging area of current research. PMS can function as an electron acceptor, being cleaved to generate highly reactive radical species, or as an electron donor, being oxidized to form SO5·−, which then reacts with water to produce mildly reactive non-radical species (1O2)11,12. By manipulating the metal-nitrogen coordination number and the electronic density of the central metal atom, along with the electronic distribution of the edge carbon atoms, PMS could function as both an electron acceptor and donor throughout the reaction, thus controlling its activation and oxidation pathways13. Moreover, spatial confinement strategies can enhance the catalytic activity by concentrating isolated reaction centers at corners and edge positions. On the other hand, the adsorption mode and cleavage manner of the peroxy bond (O-O) in PMS can also significantly influence its activation pathway14. Increasing the PMS dosage promotes radical generation in the Co-N4-C/PMS system, with PMS adsorption on Co sites significantly stretching the O-O bond to produce radicals5. Moreover, the O-O bond in PMS more readily generates hydroxyl radicals (·OH) and sulfate radicals (SO4·−) through electron transfer from the Fe-graphite N sites15. Conversely, when the PMS activation sites shift to Fe-pyridinic N and Fe-pyrrolic N sites, a transition in active species from radicals to 1O2 is noted16,17. The activity of single-atom catalysts largely hinges on the type and loading of metals on the support. Traditional single catalytic active centers are unable to facilitate reactions based on multi-site adsorption. The pathways for generating active species in PMS advanced oxidation systems activated by bimetallic single-atom catalysts remain ambiguous.
Herein, a series of high-loading Fe-Mn bimetallic single-atom catalysts (NC-FeMn(TA)) were developed to elucidate the mechanism of regulating radical and non-radical actions during the activation of PMS by bimetallic single-atoms. By modifying the support structure and adjusting the Fe/Mn ratio, the proportion of radical and non-radical species was regulated, metal loading was increased, and high pollutant degradation efficiency was achieved. Additionally, the differing reaction pathways of iron-manganese bimetallic single-atom catalysts were systematically examined by integrating experimental results with density functional theory (DFT) theoretical calculations. The influence of defect structures on the types of nitrogen species and the valence state of the iron catalytic center was investigated, revealing alterations in the pathways for active species generation. This study clarified the adsorption sites of PMS on bimetallic single-atom catalysts and elucidated the mechanism by which bimetallic species promote PMS radical activation pathways, thereby enabling the regulation of both radical and non-radical pathways.
Results
Characterizations of NC-FeMn(TA)
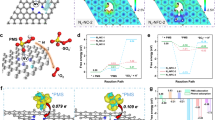
The NC-FeMnx(TAy) was synthesized by coordinating trivalent iron and divalent manganese during the self-assembly process of tannic acid and melamine, followed by pyrolysis (Supplementary Fig. 1). Materials with different Fe/Mn ratios were synthesized by varying the amount of Mn precursor, while the Fe content was fixed at 0.4 mmol unless otherwise stated. The parameter x represents the Fe/Mn molar ratio, where x = 0.1 corresponds to Fe:Mn = 4:1, x = 0.2 to 4:2, and so forth. The variable y represents the pyrolysis temperatures, which were set at 750, 800, 850, and 900 °C. The TEM image of NC-FeMn0.3(TA800) reveals its structure as an ultrathin nanosheet (Supplementary Fig. 2). Notably, no metal clusters were observed on the surface of the nitrogen-doped carbon nanosheets. The C and N elements mapping diagram aligns seamlessly with the material’s morphology, and Fe and Mn elements are evenly distributed on the surface of the carbon material (Fig. 1a and Supplementary Fig. 3). This evidence substantiates the successful preparation of nitrogen-doped carbon material modified by Fe and Mn bimetals. Furthermore, high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) of NC-FeMn(TA) clearly illustrates the uniform anchoring of Fe and Mn on the material’s surface (Fig. 1b), with most metal atoms existing as single atom. And the distances between adjacent Fe and Mn atoms are about 2.30–2.50 Å (Fig. 1c). Additionally, HAADF-STEM images of NC-FeMn0.3(TA750), NC-FeMn0.1(TA800) and NC-FeMn0.4(TA800) (Supplementary Fig. 4) also indicate that different iron-manganese ratios influence the loading of metal single atoms and the number of Fe-Mn atom pairs, but neither the iron-manganese ratio nor the calcination temperature affects the existence form of metal single atoms.

a Elemental mapping of NC-FeMn(TA). b Aberration-corrected HAADF-STEM image of NC-FeMn0.3(TA) (Single atoms are marked by yellow circles; Bimetallic single atoms are marked by boxes). c Bond length of adjacent atoms. d FT-EXAFS spectra at the Fe K-edge of NC-FeMn(TA) and their reference samples. e FT-EXAFS spectra at the Mn K-edge of NC-FeMn(TA) and their reference samples. f 57Fe Mössbauer spectrum of pristine NC-FeMn(TA) measured at room temperature. g Wavelet transform of NC-FeMn(TA) and reference samples.
X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectral analysis techniques were employed to investigate the chemical states and coordination environments of Mn and Fe species in NC-FeMn0.3(TA800). The iron absorption threshold of NC-FeMn(TA) falls between FeO and Fe2O3 (Supplementary Fig. 5a), while the manganese absorption threshold lies between MnO and Mn2O3 (Supplementary Fig. 5b). This positioning indicates that both Fe and Mn species in NC-FeMn0.3(TA800) predominantly exist in valence states ranging from +2 to +318. Examination of the Fourier-transformed extended X-ray absorption fine structure spectrum (FT-EXAFS) at the Fe K edge in NC-FeMn(TA) revealed a prominent peak at ≈1.41 Å (Fig. 1d). This peak resembles the main peak found in FePc and is typically associated with the Fe-N coordination. Moreover, a secondary small peak at ≈2.28 Å was distinctly observed in NC-FeMn0.3(TA800), which is comparable to the main peak of Fe-Fe at ≈2.19 Å in the first shell of iron foil (Fig. 1d and Supplementary Fig. 5c), indicating the presence of Fe-Mn bonds within the material19. As depicted in Fig. 1f, the Mössbauer spectrum of pristine sample (NC-FeMn0.3(TA800)) can be deconvoluted into three doublets, corresponding to distinct spectral components (Supplementary Table 1). Doublet D1 is attributed to high-spin FeIIIN3, doublet D2 to intermediate-spin FeIIN3, and doublet D3 to high-spin FeIIN320. The absence of characteristic sextets and singlets associated with α-Fe and γ-Fe indicates the lack of zero-valent Fe crystalline phases, confirming that the iron in NC-FeMn0.3(TA800) exists as single atoms. To characterize the peaks mentioned above, wavelet transform (WT) EXAFS analysis was conducted on both NC-FeMn0.3(TA800) and standard samples, as shown in Fig. 1g. The wavelet transform (WT) contour plot in NC-FeMn0.3(TA800) displays an intensity maximum at k ≈ 3.7 Å−1, mirroring the pattern seen in FePc and reinforcing the distinctive feature of isolated Fe species in the material21. Nonlinear EXAFS fitting analysis was performed on NC-FeMn0.3(TA800). According to the Fe K-edge EXAFS fitting results of NC-FeMn0.3(TA800), the peak observed at 2.01 Å in NC-FeMn0.3(TA800) can be attributed to the Fe-N3 coordination bond. The FT-EXAFS spectrum at the Mn K-edge of NC-FeMn0.3(TA800) displays a main peak at around 1.526 Å (Fig. 1e), assigned to the Mn-N coordination. Moreover, a secondary minor peak in NC-FeMn0.3(TA800) was also distinctly observed at approximately 2.31 Å, which is comparable to the main peak of Mn-Mn at ≈2.21 Å in the first shell of manganese foil, further confirming the presence of Fe-Mn bonds (Fig. 1e and Supplementary Fig. 5d)22. According to the Mn K-edge EXAFS fitting results of NC-FeMn0.3(TA800), the Mn species exhibit three neighboring N atoms at 2.15 Å, indicative of a Mn-N3 coordination structure. Overall, the analysis results corroborate the predominant structure in NC-FeMn0.3(TA800) as N3-Fe-Mn-N3. To verify whether only specific iron-manganese ratios can form such a coordination structure, we also performed XANES and EXAFS analyses on NC-FeMn0.1(TA800) and NC-FeMn0.4(TA800). As shown in Supplementary Figs. 6 and 7, these two catalysts also exhibit the same coordination structure as NC-FeMn0.3(TA800), indicating that the N3-Fe-Mn-N3 structure is universally present in NC-FeMn(TA).
Path control strategy and involved oxidation mechanism
The radical and non-radical pathways in a bimetallic single-atom catalyst/PMS oxidation system can be modulated in two approaches (Fig. 2a): 1) By constructing bimetallic single-atom catalysts and adjusting the iron/manganese ratio, the adsorption model of PMS on the catalyst surface can be altered, leading to the elongation of the O-O bond in PMS. This elongation promotes the generation of radicals, thereby increasing the contribution of the radical pathway. This method allows for the adjustment of the radical contribution rate from 1.82% to 27.70%. 2) By controlling the calcination temperature of bimetallic single-atom catalysts, the type of nitrogen species coordinated with the metal can be modified. Higher calcination temperatures reduce the content of pyridinic nitrogen while increasing the content of graphitic and pyrrolic nitrogen. This adjustment enhances the catalyst’s ability to acquire electrons from PMS, promoting radical generation and enabling the regulation of PMS activation pathways. This approach allows the radical contribution rate to be tuned within a range of 5.96% to 69.00%.

a Schematic illustration of radical and non-radical pathway regulation in PMS oxidation systems by tuning Fe/Mn ratio and calcination temperature in bimetallic single-atom catalysts. b Degradation of BPA in NC-FeMnx(TA800)/PMS systems (inset: Degradation kinetics in NC-FeMnx(TA800)/PMS systems; Conditions: [BPA]0 = 20 mg L−1, [PMS]0 = 0.24 g L−1, [Cat.] = 0.16 g L−1, pHini = 7, x = 0.1, 0.2, 0.3, 0.4). c Contribution of active species during the activation of PMS for BPA degradation in NC-FeMnx(TA800) (x = 0.1, 0.2, 0.3, 0.4) with different iron-to-manganese ratios. d Iron and manganese content on NC-FeMn(TA800) before reaction and iron dissolution rate after reaction. e Adsorption energy of PMS adsorbed on different sites. f EPR of NC-FeMn(TA)/PMS systems. g57Fe Mössbauer spectrum of used NC-FeMn0.3(TA800) measured at room temperature.
In comparison to bimetallic single-atom catalysts, materials containing only a single type of metal demonstrate significantly lower performance in activating PMS for BPA degradation (Fig. 2b). The oxidation kinetic constants for BPA by catalysts containing only iron or manganese are 1.24 min−1 and 1.13 min−1, respectively (insert of Fig. 2b). Furthermore, under conditions of similar Fe content, the kinetic constants of NC-FeMn0.3(TA800) are more than twice those of NC-Fe(TA800) (3.427 min−1 and 1.693 min−1, respectively), while the kinetic constant of NC(TA800) is only 0.005 min−1 (Supplementary Fig. 8). Unlike the effect of calcination temperature, variations in the Fe/Mn ratio had negligible influence on the coordination environment of nitrogen species or the oxidation states of Fe and Mn. Consequently, the proportion of radical and non-radical pathways in the oxidation system could only be adjusted within a narrow range. This is attributed to the fact that single-metal catalysts predominantly follow non-radical pathways, which has a slower oxidation rate, whereas bimetallic single-atom catalysts engage both radical and non-radical pathways (Fig. 2c). In the single-metal systems, the non-radical pathway dominates, accounting for nearly 100% of the overall reaction mechanism (Fig. 2c and Supplementary Table 2). As the Mn content in NC-FeMnx(TA800) increases, the non-radical contribution in the NC-FeMnx(TA800)/PMS oxidation system gradually rises (Fig. 2c). When the Fe/Mn ratio is 4:3, the bimetallic single-atom catalyst achieves its highest radical contribution, reaching 27.70% (Fig. 2c and Supplementary Table 2), and the oxidation rate constant for BPA degradation reaches up to 3.59 min−1 (insert of Fig. 2b). This enhanced activity is attributed to the introduction of manganese, which facilitates the formation of Fe-Mn bonds. The incorporation of bimetallic single atoms significantly enhances the loading of single atoms in nitrogen-doped carbon materials. As shown in Fig. 2d, introducing manganese increases iron loading, with peak values reaching 4.80 wt% for iron and 5.10 wt% for manganese. However, excessive manganese reduces the iron and manganese loadings, due to impeded stable coordination. Additionally, manganese reduces iron leaching from 1.28% in single-atom iron catalysts to 0.14% in bimetallic catalysts, indicating improved stability. However, when the Fe/Mn ratio reaches 4:4, iron leaching increases due to unstable bond formation.
Notably, the adsorption mode of PMS shifts from monodentate to a bidentate adsorption (Supplementary Fig. 9), with the adsorption energy significantly decreasing from −2.878 eV (Fe-N3(Pyridine)) and −2.715 eV (Fe-N(graphite)N2(Pyrrole)) to −6.644 eV (Fig. 2e). The bimetallic single-atom catalyst also exhibits a higher adsorption capacity for the target pollutant (BPA) compared to the monometallic single-atom catalyst (Supplementary Fig. 10), indicating a greater affinity and more efficient charge transfer capability. Notably, NC-FeMn0.3(TA800) shows a smaller specific surface area but a relatively larger average pore diameter compared to NC-Fe(TA800), indicating that it does not possess a textural advantage in terms of surface area (Supplementary Fig. 11). Furthermore, the isoelectric points of NC-Fe(TA800) and NC-FeMn0.3(TA800) were determined to be 5.51 and 6.11, respectively, with NC-FeMn0.3(TA800) maintaining a consistently higher zeta potential across the pH range of 5–10 (Supplementary Fig. 12). Considering that BPA mainly exists in a neutral molecular form at pH values below its pKa (10.29 ± 0.1)23, the stronger surface potential of NC-FeMn0.3(TA800) in the pH 7–9 range is more conducive to BPA adsorption via enhanced electrostatic and van der Waals interactions. These results suggest that the superior adsorption capacity of NC-FeMn0.3(TA800) originates not from surface area advantages, but from its more favorable surface charge environment enabled by bimetallic coordination. The primary reactive species generated during PMS activation by NC is singlet oxygen (Supplementary Fig. 13)24. Compared to monometallic single-atom catalysts, bimetallic single-atom catalysts activate PMS to produce a greater amount of highly oxidative reactive species, including hydroxyl radicals, sulfate radicals, and high-valent iron species (Fig. 2f). This can be attributed to the elongation of the O–O bond in PMS induced by bidentate adsorption, which facilitates the generation of radicals. Compared to the bond lengths of PMS adsorbed on Fe–N3(pyridine) (1.471 Å) and Fe–N(graphitic)N2(pyrrole) (1.483 Å), the O–O bond length in bidentately adsorbed PMS is significantly extended to 2.717 Å. However, further increases in manganese content led to a decrease in degradation efficiency. This decline may result from the excessive overlap of iron and manganese atoms, which reduces the number of active sites and impedes Fe-Mn bond formation, leading to performance degradation and metal atom loss.
In the NC-FeMn(TA)/PMS system, radical generation occurs through two distinct pathways: 1) The surface of the material, abundant in C=O groups, interacts with PMS to form -C=O-H-O-OSO3, which promotes the cleavage of the O-O bond, thereby generating SO4•−. Simultaneously, the oxygen-containing functional groups are oxidized to a metastable [-C=O]+ state and subsequently reduced back to -C=O by another PMS molecule (Eqs. (1) and (2))25. 2) PMS is adsorbed onto Fe and Mn atoms through dual-site adsorption, which elongates the O-O bond, making it more susceptible to cleavage and resulting in radical formation. The NC-FeMn(TA)/PMS system can selectively convert methyl phenyl sulfoxide (PMSO) to methyl phenyl sulfone (PMSO2) (Supplementary Fig. 14). This suggests that, in addition to singlet oxygen, the NC-FeMn(TA)/PMS system produces high-valent iron species, which are non-radical species capable of degrading target pollutants. In the NC-FeMn(TA)/PMS system, Fe2+ coordinates with the oxygen atom in PMS, forming the [FeIIOOSO3]+ complex (Eq. (3)). The primary active species, FeIV = O, is generated through the cleavage of the O-O bond within the [FeIIOOSO3]+ complex (Eq. (4)).
After reaction, the relative proportion of high-spin FeIIIN3 increased significantly (from 50.5% to 69.7%), while the relative proportion of high-spin FeIIN3 decreased (from 29.8% to 15.5%) (Fig. 2g and Supplementary Table 1). Initially, PMS adsorbs onto FeIIN3, forming the unstable complex [FeIIOOSO3]+ (Eq. (3)). The primary active species, FeIV = O, is generated through the heterolytic cleavage of the O-O bond within the [FeIIOOSO3]+ complex (Eq. (4)). After oxidizing BPA, these active high-valent iron species are converted to high-spin FeIIIN3 (Eq. (5)).
During the activation of PMS, the electrons on the catalyst can be effectively excited through covalent bonds in the conjugated system of the sp3 hybridized carbon and adjacent sp2 hybridized carbon, producing a more intensive electron cloud and simultaneously transferring electrons to the PMS molecule to promote the formation of singlet oxygen. Specifically, PMS first adsorbs on the active sites of NC-FeMn (TA), which promotes electron transfer that was originally enriched on the catalyst surface. This results in the elongation of the O-O bond and activation of PMS (*SO52− and *HSO5−), which then contacts each other on the catalyst surface to be converted into 1O2 (Eqs. (6) and (7))26.
Optimal iron/manganese ratio in bimetallic single-atom catalysts not only increases single-atom loading but also improves the catalyst’s adsorption of the target pollutant and PMS, enhances charge transfer, and enables fine-tuning of the radical pathway, thereby achieving superior performance.
In addition to constructing bimetallic single-atom catalysts and adjusting their Fe/Mn ratio, the type of N atoms coordinating with single atoms can also significantly influence the ratio of radical to non-radical contributions in the NC-FeMn(TA)/PMS system. Specifically, the strong electron-donating ability of pyridinic nitrogen can facilitate the reduction of Fe3+ to Fe2+ in single-atom catalysts27. And the generation of high-valent iron species in the PMS oxidation system predominantly originates from the Fe2+ within the catalyst28. Therefore, by altering the content of pyridinic nitrogen in single-atom modified nitrogen-doped carbon materials, it is possible to influence the Fe2+ content within the catalyst, thereby regulating the level of Fe(IV) formation. Higher the pyrolysis temperature significant enhances the defect density in NC-FeMn0.1(TAy), promoting the conversion of edge nitrogen configurations (pyridinic (398.05–398.24 eV) and pyrrolic nitrogen (400.01–400.03 eV)) to graphitic nitrogen (400.8–401.04 eV), thereby reducing the content of pyridinic nitrogen (Fig. 3a, c)29,30. In addition, the calcination temperature is negatively correlated with the content of Fe2+ (2p1/2, 723.01–723.52 eV; 2p3/2, 709.2–709.78 eV) and positively correlated with the content of Fe3+ (2p1/2, 725.66–726.68 eV; 2p3/2, 712.55–713.54 eV) in NC-FeMn0.1(TAy) materials (Fig. 3b, c)31,32. It can lead to a reduction in the production of high-valent iron species during PMS activation. Carbonization at temperatures above 850 °C leads to the appearance of zero-valent (704.99 eV) iron on the material surface (Supplementary Fig. 15), indicating that high temperatures cause the breaking of some Fe-N bonds and increase the disorderly movement of iron atoms, resulting in the aggregation of iron atoms into clusters. However, calcination at 850 °C with a reduced dosage of the iron precursor can also prevent the formation of zero-valent iron (Supplementary Figs. 16, 17)33. This indicates that minimizing the concentration of iron atoms around Fe under high-temperature conditions can prevent their aggregation into nanoclusters. The ID/IG ratio in Raman spectra can effectively reflect the defect level and graphitization degree of materials34. By comparing the ID/IG values of NC-FeMn0.1(TAy) (y = 750, 800, 850, 900) (Supplementary Fig. 18), it can be observed that as the annealing temperature increases, the ID/IG ratio gradually increases, indicating an increase in the generation of vacancies and edge defects. Additionally, reducing the Fe precursor content from 0.4 mmol to 0.3 mmol results in an ID/IG ratio for NC-Fe0.3Mn0.1 (TA850) that is lower than that of NC-Fe0.4Mn0.1 (TA850) (Supplementary Fig. 19), indicating that decreasing the Fe precursor content also reduces the defect level and graphitization degree of the material.

XPS spectra of (a) N 1s and (b) Fe 2p in NC-FeMn0.1(TA) at different calcination temperatures. c The percentage of ferrous iron and pyridinic nitrogen in of NC-FeMn0.1(TAy) (y = 750, 800, 850, 900). d Degradation of BPA in NC-FeMn0.1(TAy)/PMS systems (inset: Degradation kinetics in NC-FeMnx(TA800)/PMS systems; The kinetic data in the inset were fitted using a pseudo-first-order model; Conditions: [BPA]0 = 20 mg L−1, [PMS]0 = 0.24 g L−1, [Cat.] = 0.16 g L−1, pHini = 7, y = 750, 800, 850, 900). e The contribution of active species during the degradation of BPA in the NC-FeMn0.1(TAy)/PMS system (y = 750, 800, 850, 900). Gibbs free energy diagram of f Fe-N3(Pyridine) and g Fe-N(graphite)N2(Pyrrole) in PMS activation and corresponding intermediates adsorption models in DFT calculation. h The charge difference of the system after PMS is adsorbed on different materials.
The impact of calcination temperature on the BPA degradation performance is depicted in Fig. 3d. The degradation rate constants for the materials calcined at 750 °C, 800 °C, 850 °C, and 900 °C are 1.318 min−1, 1.562 min−1, 4.108 min-1, and 10.8 min−1, respectively (insert of Fig. 3d). It is evident that the degradation rate of BPA increases with rising temperature. This improvement is attributed to the greater contribution of radical pathways at elevated carbonization temperature. The variation in calcination temperature not only influences the coordination environment of nitrogen species bonded to metal atoms but also alters the valence states of the metals, thereby significantly affecting the relative contributions of radical and non-radical pathways in the catalytic system. Specifically, when the calcination temperature is ≥850 °C, the degradation of pollutants is primarily driven by radical-mediated pathways. Conversely, at temperatures ≤800 °C, non-radical mechanisms dominate the oxidation process. As the carbonization temperature increases, the proportion of graphitic nitrogen species in the catalyst also rises, facilitating the generation of radicals with faster oxidation rates. Consequently, the kinetic constant for BPA oxidation reaches its maximum at a calcination temperature of 900 °C. As shown in Fig. 3e, Supplementary Fig. 20 and Supplementary Table 3, the NC-FeMn0.1(TAy)/PMS (y < 850 °C) system predominantly comprises active species consisting of high-valent iron and singlet oxygen, suggesting a predominance of non-radical pathways. Additionally, heightened temperatures induce chemical bond cleavage and rearrangement of carbon chains within the carbon material. Moreover, thermal annealing results in the removal of nitrogen, thereby creating additional vacancies, edge defects, and topological defects. The structural features such as edge defects, curvature, and vacancies can generate unstable σ bonds, facilitating electron transfer from the carbon material to PMS by releasing π electrons from edge carbons35. Consequently, this process generates SO4•− and ·OH radicals, resulting in a decrease in the proportion of non-radical pathways to 31%, coupled with an increase in radical pathways (Fig. 3e and Supplementary Table 3). SO4•− and ·OH radicals play a dominant role in phenolic degradation, owing to their high redox potentials that enable swift pollutant degradation. Based on the electron cloud density of PMS adsorbed on various site types, Fe-N3(Pyridine) atoms are more prone to electron loss and tend to form high-valent iron species during PMS activation. Conversely, Fe-N(graphite)N2(Pyrrole) sites are more likely to gain electrons, facilitating electron transfer from PMS and thereby promoting radical formation (Fig. 3h). Figure 3f, g illustrates the Gibbs free energy of various active species generated by different Fe sites during PMS activation. The formation of FeIV = O has a lower Gibbs free energy when Fe is coordinated with pyridinic nitrogen36. Conversely, the formation of *SO5 has a lower Gibbs free energy for Fe-N(graphite)N2(Pyrrole), suggesting that Fe coordinated with non-pyridinic nitrogen is more conducive to the formation of SO5•ˉ(Fig. 3g)37. DFT calculations have demonstrated that the regulation of both radical and non-radical species in the PMS oxidation system can be effectively achieved by modifying the types of nitrogen coordination species in nitrogen-doped carbon materials and by constructing bimetallic catalytic sites (Fig. 3h). Thus, by modulating the carbonization temperature and fabricating bimetallic single-atom catalysts while adjusting the relative ratio of the bimetallic constituents, precise control over the balance between radical and non-radical pathways within the single-atom catalyst/PMS system can be achieved.
Application potential and environmental impact
Oxidation systems dominated by radical pathways are highly susceptible to pH fluctuations, whereas those dominated by non-radical pathways can effectively resist the inhibitory effects of strong alkaline conditions. For example, the monometallic single-atom catalyst (NC-Fe(TA))/PMS system, which is dominated by non-radical pathways, maintains a relatively high oxidation rate even at a pH of 11 (Fig. 4a), and this high oxidation rate does not result from alkali activation (Supplementary Fig. 21). In contrast, a strong alkaline environment significantly inhibits the degradation efficiency of BPA in a radical-dominated system, such as NC-FeMn0.1(TA850)/PMS. By adjusting the ratio of radical to non-radical contributions, it is possible to achieve optimal conditions for both degradation rate and resistance to pH interference. For instance, NC-FeMn0.3(TA800)/PMS can achieve complete degradation of BPA within 5 min across a pH range from 1 to 11, with a kinetic constant greater than 1.11 min−1 (Supplementary Fig. 22). The inhibition of the oxidation system’s performance under strongly alkaline conditions can be attributed to several factors. The dissociation constant (pKa) of HSO5− is 9.423, with H2SO5 predominating under highly acidic conditions and SO52− under alkaline conditions38. The isoelectric point of NC-FeMn(TA) is 6.11, resulting in an increasingly negative Zeta potential as the pH rises (Supplementary Fig. 23). The negative charge on the material’s surface causes electrostatic repulsion with HSO5− or SO52−, inhibiting their adsorption and activation, thereby impeding the generation of active species and slowing down BPA degradation. Additionally, BPA’s pKa is 10.29 ± 0.139. Thus, in acidic conditions (pH below the pKa), BPA is predominantly neutral, and the surface of the material is positively charged, which facilitates the adsorption and activation of target molecules. Overall, the NC-FeMn(TA)/PMS system demonstrates effective oxidation across a wide pH range, overcoming the limitations of traditional Fenton processes.

a pH interference resistance across systems with varying free radical proportions (Conditions: [BPA]0 = 20 mg L−1, [PMS] = 0.24 g L−1, [Cat.] = 0.16 g L−1). b 28 cycles sustained degradation of BPA by NC-FeMn0.3(TA800)/PMS system and NC-Mn0.3(TA800)/PMS system (Conditions: [PMS] = 0.24 g L−1, [Cat.] = 0.16 g L−1). c Effects of different anions on BPA degradation in the NC-FeMn0.3(TA800)/PMS system (Conditions: [BPA]0 = 20 mg L−1, [PMS] = 0.24 g L−1, [Cat.] = 0.16 g L−1, pHini = 7, Cl− = 135.3 mg L−1, Br− = 1.0 mM, HCO3− = 272 mg L−1, NO3− = 13.6 mg L−1, SO42ˉ = 124.0 mg L−1, H2PO4− = 5.0 mg L−1 and HA = 10.0 mM). d Comparison of k-value of BPA degradation in different PMS-AOP systems. e Elimination efficiency and corresponding kobs for different types of pollutants in the NC-FeMn0.3(TA800)/PMS system. f NC-FeMn0.3(TA800)/PMS and NC-Fe(TA800)/PMS system for actual wastewater biochemical performance, COD and BOD removal (Conditions: [PMS] = 2.4 g L−1, [Cat.] = 1.6 g L−1). g COD removal of industrial sewage in NC-FeMn0.3(TA800)/PMS system (Conditions: [PMS]0 = 2.4 g L−1, [Cat.] = 1.6 g L−1). h BPA removal performance of NC-FeMn0.3(TA800)/PVDF/PMS system and NC-FeMn0.3(TA800)/PVDF system as a function of permeate solution at a pressure of 2 bar (Conditions: [PMS]0 = 40 mg L−1, [BPA]0 = 1.75 × 10−5 M). i Image of NC-FeMn0.3(TA800)/PVDF membrane and cross-flow filtration equipment. j FESEM images of the cross-sectional view of NC-FeMn0.3(TA800)/PVDF membrane and its elemental mapping images.
Thanks to the stability and activity of the bimetallic single-atom catalyst, NC-FeMn0.3(TA800) can still achieve 100% removal of the target pollutant even after 22 cycles, compared to NC(TA) and NC-Mn(TA) (Fig. 4b). In comparison, NC-Mn0.3(TA800) can achieve 100% BPA degradation only within the first 8 cycles and becomes nearly inactive after 12 cycles, primarily because of manganese atom leaching. These findings further illustrate that the synergistic interaction between iron and manganese effectively mitigates metal leaching, thus ensuring prolonged decontamination efficacy (Supplementary Table 4). Additionally, FITR and XPS analyses before and after the NC-FeMn0.3(TA800) reaction also indicate that the material exhibits excellent stability (Supplementary Figs. 24 and 25). FTIR spectra show that the characteristic peaks of the catalyst after the reaction are essentially consistent with those before the reaction, indicating that the material structure has not undergone significant changes before and after the reaction. The N 1 s energy spectra of the catalyst before and after the reaction (Supplementary Fig. 25a, d) show a slight decrease in the pyridine nitrogen content (from 42.73% to 39.53%). The Fe 2p energy spectra before and after the reaction (Supplementary Fig. 25b, e) show changes in the Fe2+ content (from 47.88% to 44.59%), which can be attributed to the partial participation of Fe2+ in the formation of Fe(IV) = O and the slight leaching of iron. The organic carbon transfer process (OCTP) can be used to describe the accumulation of organic pollutant reaction intermediates on the catalyst surface during a multiphase Fenton-like reaction. The proportion of organic pollutants accumulated on the catalyst surface via OCTP can be expressed by the Eq. (8)40:
The COD content of NC and BPA before and after the reaction increased from 134 mg L−1 to 188.5 mg L−1 and decreased from 263.5 mg L−1 to 153 mg L−1, respectively. Therefore, the OCTP of this reaction process can be calculated as 49.32% (Supplementary Fig. 26). Since increasing the OCTP enhances the removal of pollutants in various advanced oxidation processes, while decreasing the OCTP reduces the accumulation of reaction intermediates on the catalyst surface, thereby improving catalyst stability. The low OCTP of the NC-FeMn0.3(TA800)/PMS system indicates that even at high BPA concentrations (100 mg L−1), reaction intermediates do not easily aggregate on the catalyst surface, demonstrating the catalyst’s excellent stability.
Notably, in the presence of various inorganic anions and humic acid (HA), the NC-FeMn0.3(TA800) catalyst maintains a 100% BPA degradation rate (Fig. 4c), underscoring the NC-FeMn0.3(TA800)/PMS system’s robustness and practical applicability. However, when coexist with Cl−, HCO3−, NO3−, or H2PO4−, the BPA oxidation kinetics constant decreases, indicating that these anions compete for and consume ·OH and SO4•− radicals. Furthermore, according to the Nernst equation (Eq. (9))41, the reduction potential of sulfate radicals is lowered in the presence of SO42ˉ.
Interestingly, Br− enhances the BPA degradation rate (from 4.11 min−1 to 4.82 min−1), likely due to the rapid formation of active bromine species (BrO·, Br·, Br2·–, HOBr) that contribute to the degradation process42. In the NC-FeMn0.3(TA800)/PMS system, the addition of humic acid (HA) led to an increase in the BPA degradation rate by 0.449 min−1. This enhancement is likely attributed to HA acting as an electron donor, reducing Fe3+ to Fe2+, which subsequently participates in the activation of PMS. To further assess the system’s resistance to interference, the degradation of BPA was tested under various water matrixes. As illustrated in Supplementary Fig. 27, the NC-FeMn0.3(TA800)/PMS system achieved a 100% removal efficiency of BPA within 5 min in both Qingchun river water and tap water matrices. The degradation rate constants were 3.54 min−1 for Qingchun river water and 3.45 min−1 for tap water, slightly lower than the 3.59 min−1 observed in deionized water, likely due to the presence of organic matter and inorganic ions in the river and tap water (Supplementary Table 5). Moreover, three-dimensional fluorescence analysis of BPA-containing water before and after the reaction further corroborated these findings (Supplementary Fig. 28). The intensity of the characteristic fluorescence peaks of BPA in deionized water and river water (230/310 nm and 275/310 nm) significantly decreased, indicating effective degradation of BPA in these water matrixes. This demonstrates that the NC-FeMn0.3(TA800)/PMS system effectively resists interference from coexisting substances in river water and industrial wastewater.
The activated PMS performance of the NC-FeMn0.3(TA800) catalyst surpasses that of a variety of carbon materials, metal oxides, metal-carbon composites, single-atom catalysts, and bimetallic single-atom catalysts (Supplementary Table 6 and Fig. 4e). The degradation performance of the NC-FeMn0.3(TA800)/PMS system was assessed using a variety of typical organic pollutants as model molecules, encompassing pharmaceuticals, endocrine-disrupting chemicals, dyes, and chemical intermediates (Fig. 4d, Supplementary Tables 7, 8). The findings reveal that the NC-FeMn0.3(TA800)/PMS system exhibits rapid degradation of pollutants with low ionization potentials (e.g., PCMX, aniline, TC, phenol), attributed to the Fe(IV) species in the system preferentially attacking electron-donating groups (EDG) (e.g., hydroxyl, amino, alkoxy, and phenol ester groups)43,44. In contrast, while the degradation rate constants for pollutants with electron-withdrawing groups (e.g., NOF, BPS) are below 1.0 min−1. Notably, the NC-FeMn0.3(TA800)/PMS system still achieves a 100% removal efficiency for high ionization potential pollutants within 5 min45. This efficacy is due to the involvement of both non-radical and radical species in the NC-FeMn0.3(TA800)/PMS system, with radicals indiscriminately targeting pollutants with either electron-withdrawing groups (EWG) or EDG. Furthermore, the NC-FeMn0.3(TA800)/PMS system effectively decolorizes typical dyes by eliminating their chromophoric functional groups46. The NC-FeMn0.3(TA800)/PMS system exhibits a wide pH applicability, exceptional cyclic stability, and robust resistance to interference. Its effectiveness in degrading various typical organic pollutants underscores its promising potential for practical applications.
In oxidation processes, non-radical species possess lower oxidative strength than radicals. Nonetheless, their longer lifespans and higher selectivity make them highly effective in enhancing wastewater biodegradability9. Non-radical pathways can thus be utilized to improve the biodegradability of high COD wastewater. Conversely, in practical wastewater treatment, as COD levels decrease, achieving further reductions becomes progressively challenging. This is especially evident when attempting to lower COD from 200 mg L−1 to 50 mg L−1, a process that proves to be particularly difficult. Radicals, due to their potent oxidative capabilities, can effectively degrade low concentration COD wastewater that non-radical species cannot efficiently address. Therefore, by modulating the proportion of radical and non-radical pathways within the system, the treatment approach can be tailored to meet the specific requirements of different wastewater types. The wastewater treatment capability of the NC-Fe0.3(TA800)/PMS system, which exhibits a nearly 100% non-radical contribution ratio, was assessed using industrial wastewater with extremely low biodegradability (BOD/COD ratio of only 0.033). Following treatment with the NC-Fe0.3(TA800)/PMS system, the BOD/COD ratio increased to 0.336, indicating an over tenfold improvement in biodegradability relative to the influent (Fig. 4f). Furthermore, compared to the system involving both radical and non-radical pathways (NC-FeMn0.3(TA800)/PMS), the NC-Fe (TA800)/PMS system demonstrated a twofold enhancement in improving wastewater biodegradability. As illustrated in Fig. 4g, the FeMn0.3(TA800)/PMS system exhibits substantial treatment efficacy for both high and low concentration COD wastewater. After 24 h of treatment, the COD of high concentration wastewater decreased from 899 mg L−1 to 116 mg L−1, reflecting a reduction of 783 mg L−1. For the more difficult low concentration wastewater, the COD decreased from 110 mg L−1 to 35 mg L−1, achieving a reduction of 75 mg L−1. These final COD levels are significantly low, meeting the Class I-A discharge standards for municipal wastewater treatment plants (COD < 50 mg L−1), highlighting the system’s substantial potential for the advanced treatment of real wastewater47.
The NC-FeMn0.3(TA800) catalyst was immobilized onto commercially available PVDF membranes, and its continuous pollutant removal efficacy was evaluated using a flow-through configuration (Fig. 4i). The resulting NC-FeMn0.3(TA800)/PVDF membrane, with a diameter of 3.1 cm and a thickness of 420 μm, featured a porous surface and interconnected stacking layers, with Fe and Mn elements uniformly distributed throughout the nitrogen-doped carbon material structure (Fig. 4j). Incorporation of graphene and octyltrimethoxysilane enhanced the membrane’s mechanical strength, adhesion, and wettability, reducing its susceptibility to damage. After treating 2.5 L of influent (Fig. 4h), the flow-through catalytic system maintained a 100% BPA removal efficiency and exhibited a high-water flux (~7500 L m−2 h−1 bar−1). Conversely, the system lacking PMS addition demonstrated less than 20% removal efficiency, highlighting the reliance of the system on the activation of PMS by single-atom iron and manganese species to produce active species for BPA degradation, as mere adsorption by the material alone proved inadequate for BPA removal48. The NC-FeMn0.3(TA800)/PMS demonstrates outstanding adaptability, durability, and degradation efficiency in complex wastewater environments, underscoring its substantial potential for real-world applications.
Discussion
This study elucidates the regulation of radical and non-radical oxidation pathways in the PMS oxidation system by constructing a bimetallic single-atom catalyst (NC-FeMn(TA)) and adjusting its iron-to-manganese ratio and calcination temperature. By increasing the pyridinic nitrogen content and incorporating single metal atoms into nitrogen-doped carbon materials, a predominantly non-radical oxidation process is realized. In contrast, augmenting the graphitic and pyrrolic nitrogen species and introducing bimetallic catalytic centers favor a radical oxidation pathway. The bimetallic catalyst enhances the bidentate adsorption of PMS on Fe and Mn, facilitating PMS adsorption and O-O bond elongation, thereby generating radicals. By adjusting the radical and non-radical contributions in the NC-FeMn(TA)/PMS system, high oxidation performance, significant resistance to interference, and stability were achieved across a wide pH range, maintaining 100% degradation of the target pollutant after 22 cycles. Additionally, it achieves complete removal of high ionization potential pollutants within 5 min. Remarkably, NC-FeMn(TA)/PMS significantly enhances the biodegradability of poorly biodegradable wastewater. This system’s outstanding performance indicates extensive application potential in environmental fields. The findings offer theoretical insights into the radical and non-radical generation pathways in PMS activation by Fe/Mn bimetallic single-atom catalysts.
Methods
Preparation of nitrogen-doped carbon-iron-manganese (tannic acid) NC-FeMn(TA)
Dissolve 1 g of tannic acid in 100 mL of an ethanol-water solution (VH2O: VEtOH = 1:1) and stir for 10 min. Gradually add 2 mL of a mixed solution of FeCl3·6H2O (0.108 g, 0.4 mmol) and MnSO4 (0.0453 g, 0.3 mmol) to this solution. Then, introduce 10 g of melamine and continue stirring at room temperature for 30 min. Transfer the resulting mixture to an oil bath at 80 °C for 24 h to fully evaporate the solvent. Grind the resulting solid into a powder. Calcine the obtained purple powder in a tube furnace at 800 °C under a nitrogen atmosphere for 2 h (with a heating rate of 3 °C min−1) to produce the sample, labeled as NC-FeMn(TA) (Supplementary Fig. 1). Ultimately, a 0.065 g sample can be obtained.
Pollutant degradation experiments
In a 50 mL solution of the pollutant at a specified concentration, add the catalyst at the designated concentration and ultrasonicate for 5 min. Adjust the pH using HCl (0.1 M) and NaOH (0.1 M). After stirring for a set period, add PMS at the specified concentration and initiate timing. At fixed reaction intervals, withdraw 0.65 mL of the solution and quench the reaction with an equal volume of MeOH. Filter the samples using a 0.22 μm hydrophilic PTFE syringe filter to remove any solid catalysts. Subsequently, measure the real-time concentration of the target pollutant using HPLC or a UV-Vis spectrophotometer (for dye samples). The HPLC conditions are as follows: injection volume of 1 μL, flow rate of 1.0 mL min−1, and pollutant concentration determined using a reverse-phase C-18 column (4.6 × 250 mm, 5 μm, Agilent). Each data point represents the mean of duplicate measurements, and error bars indicate the standard deviation. The operation details, detection wavelengths and mobile phase of different organic pollutants were listed in Supplementary Tables 7 and 8.
Quenching experiments
Quenching experiments typically involve introducing a specific concentration of a quencher into a mixture of catalyst and pollutant, followed by the addition of an oxidant to initiate the reaction. Methanol is employed as a quencher for SO4·− and ·OH radicals, tert-butanol (TBA) for ·OH radicals, superoxide dismutase for O2·− radicals, furfuryl alcohol (FFA) for 1O2, and dimethyl sulfoxide (DMSO) for detecting high-valent species in the reaction. Additionally, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) is utilized as a spin-trapping agent in radical capture experiments. DMPO in aqueous solution serves as a spin trap for SO4·− and ·OH radicals, DMPO in methanol solution for O2·− radicals, and 4-amino-2,2,6,6-tetramethylpiperidine (TEMP) for 1O2.
Active species capture experiments
The catalyst is dispersed in an aqueous solution and ultrasonicated for uniform dispersion. A specific amount of PMS is then added, and the mixture is continuously stirred. After 30 s, an excess of the spin-trapping agent is introduced, and the reaction is allowed to proceed for an additional 2 min. The paramagnetic signals of the mixture are then detected using electron paramagnetic resonance (EPR) spectroscopy. The EPR settings include a central magnetic field range of 3400.0 to 3500.0 G, a scan width of 100.0 G, and three scans.
The percentage contributions of radical and non-radical pathways can be calculated from the results of various radical capture experiments using Eqs. (10) and (11)49.
where N0 is the total removal efficiency without any quencher, and N1 is the BPA degradation efficiency when methanol is used as the quencher.
As shown in Fig. 2f and Supplementary Figs. 13, 14, 20, quenching and EPR experiments indicate that Fe(IV) = O is the primary active species in the NC-FeMnx(TAy)/PMS system, while SO4·−, ·OH, and 1O2 act as secondary reactive species. Given methanol’s ability to quench ·OH and SO4·− (k·OH = (1.2–2.8) ×109 M−1 s−1, kSO4·− = (0.78–1.0) ×109 M−1 s−1), and the kinetic constants are the results of linear fitting of the degradation efficiency, the contribution of ROS to BPA degradation can be further assessed by comparing kinetic constants before and after methanol addition. For instance, in the NC-FeMn0.1(TA800)/PMS system (Fig. 2c), the rate constants decrease from 1.56 min−1 to 1.4 min−1 upon methanol addition, indicating radical and non-radical contributions of 10.2% and 89.8%, respectively. The remaining data on the radical and non-radical contribution ratios in the NC-FeMn0.1(TAy)/PMS system and the NC-FeMnx(TA800)/PMS system are shown in Supplementary Tables 2 and 3.
Assembly of the membrane device
A specific amount of catalyst is added to a beaker, followed by the addition of an appropriate quantity of graphene solution, octyltrimethoxysilane, and deionized water. The mixture is then dispersed using ultrasonication for thirty min. Subsequently, the dispersed solution is deposited onto commercially available PVDF membranes using a filtration method. Finally, the membranes are placed in a cross-flow filtration device to complete the assembly of the membrane device.
Data availability
All data are available in the main text or in the Supplementary information and in the source data files. Source data are provided in this paper. Source data are provided with this paper.
References
Gao, X., Yang, Z., Zhang, W. & Pan, B. Carbon redirection via tunable Fenton-like reactions under nanoconfinement toward sustainable water treatment. Nat. Commun. 15, 2808 (2024).
Cao, J. et al. Gambling of homogeneous and heterogeneous Fenton in wastewater treatment. Cell Rep. Phys. Sci. 5, 101966 (2024).
Chen, R., Chen, S., Wang, L. & Wang, D. Nanoscale metal particle modified single-atom catalyst: synthesis, characterization, and application. Adv. Mater.36, 2304713 (2024).
Chen, Y. et al. Isolating single and few atoms for enhanced catalysis. Adv. Mater.34, 2201796 (2022).
Yang, M. et al. Unveiling the origins of selective oxidation in single-atom catalysis via Co–N4–C intensified radical and nonradical pathways. Environ. Sci. Technol. 56, 11635–11645 (2022).
Liang, X. et al. Coordination number dependent catalytic activity of single-atom cobalt catalysts for fenton-like reaction. Adv. Funct. Mater. 32, 2203001 (2022).
Gu, C.-H. et al. Upcycling waste sewage sludge into superior single-atom Fenton-like catalyst for sustainable water purification. Nat. Water 2, 649–662 (2024).
Li, Y., Zhou, Y. & Zhou, Y. Ultrasonic enhancement of persulfate oxidation system governs emerging pollutants decontamination. Green. Energy Environ. 9, 1666–1678 (2024).
Liu, X., Yan, X., Liu, W., Yan, Q. & Xing, M. Switching of radical and nonradical pathways through the surface defects of Fe3O4/MoOxSy in a Fenton-like reaction. Sci. Bull. 68, 603–612 (2023).
Ren, W. et al. Activation of peroxydisulfate on carbon nanotubes: electron-transfer mechanism. Environ. Sci. Technol. 53, 14595–14603 (2019).
Huang, Y.-F. & Huang, Y.-H. Behavioral evidence of the dominant radicals and intermediates involved in Bisphenol A degradation using an efficient Co2+/PMS oxidation process. J. Hazard. Mater. 167, 418–426 (2009).
Gao, Y., Chen, Z., Zhu, Y., Li, T. & Hu, C. New insights into the generation of singlet oxygen in the metal-free peroxymonosulfate activation process: important role of electron-deficient carbon atoms. Environ. Sci. Technol. 54, 1232–1241 (2020).
Cheng, J. et al. Directing the persulfate activation reaction pathway by control of Fe-Nx/C single-atom catalyst coordination. Chem. Eng. J.481, 148603 (2024).
Song, X. et al. Unraveling the discriminative mechanisms for peroxy activation via atomically dispersed Fe-N5 sites for tunable water decontamination. Appl. Catal. B Environ. 340, 123240 (2024).
Zhang, L. et al. Mechanical insight into direct singlet oxygen generation pathway: pivotal role of FeN4 sites and selective organic contaminants removal. Appl. Catal. B Environ. 339, 123130 (2023).
Zhang, X. et al. Fe(II) and Pyridinic N complex sites synergy to activate PMS for specific generation of 1O2 to degrade antibiotics with high efficiency. Sci. Total Environ. 892, 164067 (2023).
Zheng, J. et al. Efficient activation of peroxymonosulfate by Fe single-atom: The key role of Fe-pyrrolic nitrogen coordination in generating singlet oxygen and high-valent Fe species. J. Hazard. Mater. 462, 132753 (2024).
Cui, J. et al. Efficient single-atom Fe-catalyzed Fenton-like reaction involving peroxymonosulfate for BPA degradation by high-valent Fe(IV)=O. ACS EST Water 2, 2698–2705 (2022).
Zhang, S. et al. Fe/Cu diatomic catalysts for electrochemical nitrate reduction to ammonia. Nat. Commun. 14, 3634 (2023).
Li, X. et al. Identification of the electronic and structural dynamics of catalytic centers in single-Fe-atom material. Chem 6, 3440–3454 (2020).
Fu, H. et al. Axial coordination tuning Fe single-atom catalysts for boosting H2O2 activation. Appl. Catal. B Environ. 321, 122012 (2023).
Luo, G. et al. Regulating the double-site Mn2-N6 electronic structure by manganese clusters for enhanced oxygen reduction. Appl. Catal. B Environ. 350, 123939 (2024).
Berhane, T. M., Levy, J., Krekeler, M. P. S. & Danielson, N. D. Adsorption of bisphenol A and ciprofloxacin by palygorskite-montmorillonite: Effect of granule size, solution chemistry and temperature. Appl. Clay Sci. 132-133, 518–527 (2016).
Zhou, Y. et al. Efficient photocatalytic NADH regeneration with Rh-loaded Z-scheme mediator-free system. Chin. Chem. Lett. 35, 108690 (2024).
Duan, X., Sun, H. & Wang, S. Metal-free carbocatalysis in advanced oxidation reactions. Acc. Chem. Res. 51, 678–687 (2018).
Wei, J. et al. Highly dispersed Fe7S8 anchored on sp2/sp3 hybridized carbon boosting peroxymonosulfate activation for enhanced EOCs elimination though singlet oxygendominated nonradical pathway. J. Hazard. Mater. 461, 132607 (2024).
Li, B., Sun, X. & Su, D. Calibration of the basic strength of the nitrogen groups on the nanostructured carbon materials. Phys. Chem. Chem. Phys. 17, 6691–6694 (2015).
Wang, Z. et al. Further understanding the involvement of Fe(IV) in peroxydisulfate and peroxymonosulfate activation by Fe(II) for oxidative water treatment. Chem. Eng. J.371, 842–847 (2019).
Dong, C. et al. Boosting bi-directional redox of sulfur with dual metal single atom pairs in carbon spheres toward high-rate and long-cycling lithium–sulfur battery. Adv. Energy Mate. 13, 2301505 (2023).
Zhao, L. et al. Cascade anchoring strategy for general mass production of high-loading single-atomic metal-nitrogen catalysts. Nat. Commun. 10, 1278 (2019).
Ma, C., Zhang, H., Kong, W., Shen, B. & Lyu, H. Bimetallic atomic Fe–Mn metal-nitrogen active sites for synergistic enhancement of CO2 electroreduction efficiency. Environ. Funct. Mater. 1, 284–297 (2022).
Zhou, Y., Zhou, L., Zhou, Y., Xing, M. & Zhang, J. Z-scheme photo-Fenton system for efficiency synchronous oxidation of organic contaminants and reduction of metal ions. Appl. Catal. B-Environ. 279, 119365 (2020).
Zhang, S.-H. et al. Mechanism investigation of anoxic Cr(VI) removal by nano zero-valent iron based on XPS analysis in time scale. Chem. Eng. J.335, 945–953 (2018).
Lv, W. et al. A polymer tethering strategy to achieve high metal loading on catalysts for Fenton reactions. Nat. Commun. 14, 7841 (2023).
Ouyang, D. et al. Activation mechanism of peroxymonosulfate by biochar for catalytic degradation of 1,4-dioxane: important role of biochar defect structures. Chem. Eng. J.370, 614–624 (2019).
Wang, Z. et al. High-valent iron-oxo species mediated cyclic oxidation through single-atom Fe-N6 sites with high peroxymonosulfate utilization rate. Appl. Catal. B-Environ. 305, 121049 (2022).
Das, T. N. Reactivity and role of SO5•- radical in aqueous medium chain oxidation of sulfite to sulfate and atmospheric sulfuric acid generation. J. Phys. Chem. A 105, 9142–9155 (2001).
Duan, X., Sun, H., Wang, Y., Kang, J. & Wang, S. N-doping-induced nonradical reaction on single-walled carbon nanotubes for catalytic phenol oxidation. ACS Catal. 5, 553–559 (2015).
Chu, Y. et al. Efficient destruction of emerging contaminants in water by UV/S(IV) process with natural reoxygenation: effect of pH on reactive species. Water Res. 198, 117143 (2021).
Xing, M. et al. Organic carbon transfer process in advanced oxidation systems for water clean-up. Nat. Water 3, 334–344 (2025).
Wang, J. & Wang, S. Effect of inorganic anions on the performance of advanced oxidation processes for degradation of organic contaminants. Chem. Eng. J.411, 128392 (2021).
Xue, Y. et al. Role of halide ions on organic pollutants degradation by peroxygens-based advanced oxidation processes: a critical review. Chem. Eng. J. (Lausanne) 433, 134546 (2022).
Yan, Y. et al. Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes. Environ. Sci. Technol. 57, 12153–12179 (2023).
Zhou, Y., He, J., Li, X., Lu, J. & Zhou, Y. Efficient removal of roxarsone and emerging organic contaminants by a solar light-driven in-situ Fenton system. Chem. Eng. J.435, 132434 (2022).
Zhou, Z. et al. Activatable singlet oxygen generation from lipid hydroperoxide nanoparticles for cancer therapy. Angew. Chem. Int. Ed. 56, 6492–6496 (2017).
Liu, B. et al. Peroxymonosulfate activation by cobalt(II) for degradation of organic contaminants via high-valent cobalt-oxo and radical species. J. Hazard. Mater. 416, 125679 (2021).
Bao, Y. et al. Generating high-valent iron-oxo ≡FeIV=O complexes in neutral microenvironments through peroxymonosulfate activation by Zn−Fe layered double hydroxides. Angew. Chem. Int. Ed. 61, e202209542 (2022).
Chu, C. et al. Cobalt single atoms on tetrapyridomacrocyclic support for efficient peroxymonosulfate activation. Environ. Sci. Technol. 55, 1242–1250 (2021).
Li, F. et al. Ce-doped CuCoO2 delafossite with switchable PMS activation pathway for tetracycline degradation. Chem. Eng. J.481, 148633 (2024).
Acknowledgements
This work was supported by National Natural Science Foundation of China (No.22376065, 22325602, 22176060), and the Program of Shanghai Academic/Technology Research Leader (23XD1421000). Project supported by Shanghai Municipal Science and Technology Major Project (Grant No.2018SHZDZX03) and the Program of Introducing Talents of Discipline to Universities (B16017). Science and Technology Commission of Shanghai Municipality (20DZ2250400, 22ZR1418600). Authors thank Research Center of Analysis and Test of East China University of Science and Technology for the help on the characterization.
Author information
Authors and Affiliations
Contributions
Yi Zhou and Mingyang Xing designed research. Yi Zhou, Wenxuan Guo and Ming Gao performed research. Yanpan Li, Ming Gao, Xuning Li, Wenyuan Liu, Zhuan Chen Xiaohui Zhang and Yanbo Zhou contributed new reagents/analytic tools. Yi Zhou, Wenxuan Guo, Ming Gao and Xuning Li analyzed data. Yi Zhou and Mingyang Xing wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, Y., Guo, W., Li, Y. et al. Insights into free radical and non-radical routes regulation for water cleanup. Nat Commun 16, 7753 (2025). https://doi.org/10.1038/s41467-025-63235-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63235-2
