
Abstract
Membrane scaffold protein-based nanodiscs have facilitated unprecedented structural and biophysical analysis of membrane proteins in a near-native lipid environment. However, successful reconstitution of membrane proteins in nanodiscs requires prior solubilization and purification in detergents, which may impact their physiological structure and function. Furthermore, the detergent-mediated reconstitution of nanodiscs is unlikely to recapitulate the precise composition or asymmetry of native membranes. To circumvent this fundamental limitation of traditional nanodisc technology, we herein describe the development of membrane-solubilizing peptides to directly extract membrane proteins from native cell membranes into nanoscale discoids. By systematically protein engineering and screening, we create a class of chemically modified Apolipoprotein-A1 mimetic peptides to enable the formation of detergent-free nanodiscs with high efficiency. Nanodiscs generated with these engineered membrane scaffold peptides are suitable for obtaining high-resolution structures using single-particle cryo-EM with native lipids. To further highlight the versatility of our approach, we directly extract a sampling of membrane signaling proteins with their surrounding native membranes for biochemical and biophysical interrogations.
Similar content being viewed by others

Introduction
The invention of nanodiscs (NDs) by the Sligar group twenty years ago revolutionized all aspects of membrane biology1,2. By engineering amphipathic membrane scaffold proteins (MSPs) from Apolipoprotein-A1 (AopA-I), membrane protein complexes can be embedded in a lipidic environment for in-depth biophysical characterizations that are usually challenging in traditional membrane mimetic systems. Although the ND technology has emerged as a powerful tool for various applications, its assembly principle is rather straightforward3. The hydrophobic face of MSPs encircles a nanoscale patch of lipid bilayers inside NDs for the reconstitution of membrane proteins, whereas the outside hydrophilic face ensures water solubility of the assembled discoidal particles. Since the first generation of MSPs reported by Bayburt et al., the simple yet elegant framework of NDs inspired extensive efforts to further expand the structure and function of this highly useful platform using distinct designs of protein and polymer scaffolds4,5,6,7,8,9,10,11,12. These studies have culminated in an increasingly versatile ND toolkit for reconstituting diverse lipids and membrane proteins of interest. In addition, the rigid structure of NDs with diameters ranging from 5–50 nanometers has enormous therapeutic potential as a delivery vehicle for small molecules, vaccines, and immunotherapeutic drugs13,14,15,16,17,18,19,20.
Despite the tremendous progress in the development and application of ND technology over the past decade, many membrane protein complexes remain recalcitrant to this approach. The main difficulty is that current ND reconstitutions rely on the self-assembly of lipids, MSPs, and membrane proteins of interest upon the removal of detergents21. A prerequisite of this assembly process is to obtain stable membrane proteins in detergent micelles. Unfortunately, this prerequisite is a notoriously unmet challenge for membrane protein complexes that often unfold or fall apart once extracted from lipid bilayers using detergents. To address this problem, chemical crosslinkers are employed to increase the stability and homogeneity of membrane protein complexes for structural studies22,23. However, these crosslinked protein complexes are no longer suitable for other biochemical and functional interrogations because their dynamics are markedly distorted. Therefore, a pressing need exists to bypass the limitation of detergent-mediated reconstitution of NDs.
On this front, amphipathic polymers have been developed for detergent-free reconstitution of membrane proteins into native NDs7. In recent years, these NDs have been widely utilized to elucidate the function of biological membranes in regulating the conformational state and stoichiometry of membrane proteins in diverse transmembrane signaling pathways24,25,26. However, the sensitivity of original polymer NDs for bivalent cations and the altered packing of extracted native lipids makes it challenging for functional characterizations of many membrane proteins12,27,28,29. Although recent improvements in charge-free, electroneutral polymers have greatly mitigated these issues30, lipids undergo rapid exchange between these NDs31, indicating that polymer-based approaches may need additional optimization to resemble the properties of lipid bilayers. In addition, it is known that screening a large library of different polymers for ND reconstitution is often required to overcome some unforeseeable difficulties27.
In this work, we propose to develop a different and orthogonal approach for detergent-free ND reconstitution through the engineering of membrane scaffold peptides. The design of this approach is based on previous studies of membrane disruption by antimicrobial and ApoA-I mimetic peptides13,14,32,33,34,35,36,37. Through extensive rational design and screening of these amphipathic peptides, we generate a library of membrane scaffold peptides capable of directly extracting membrane protein complexes from native cell membranes into NDs without the need for detergents. The resulting NDs can maintain integral membrane proteins in functional states and provide an appropriate lipidic environment for the characterization of peripheral membrane proteins. Moreover, our approach allows for the preservation of membrane protein complexes that are usually disrupted by detergents, thereby enabling biophysical interrogations of detergent-sensitive membrane proteins unattainable in previous studies. Finally, we demonstrate that our detergent-free native ND technology using re-engineered membrane scaffold peptides is compatible with a wide range of transporters, receptors, and ion channels in bacterial and eukaryotic cells.
Results
Screening membrane scaffold peptides for detergent-free ND reconstitution
It is well established that amphipathic peptides can drastically remodel membranes and transform small unilamellar vesicles into discoidal ND structures without detergents38. By virtue of this advantage, these peptides are invaluable reagents for therapeutic development against bacterial resistance, cancer vaccines, and viral infections14,39,40. Inspired by these findings, we set out to characterize the ability of these peptides to extract a bacterial prototype ATP-binding cassette transporter MalFGK2 into NDs from proteoliposomes (Fig. 1a). MalFGK2 is composed of two transmembrane proteins (MalF and MalG) and two copies of the ATPase component (MalK) that powers the uptake of maltose into bacteria in the presence of the maltose binding protein (MBP)41. Previous studies have shown that the basal ATPase activity of MalFGK2 in proteoliposomes is low and tightly coupled to the transport of maltose42,43. In contrast, the detergent-solubilized MalFGK2 complex is over 100-fold more active and is uncoupled from maltose and MBP. Thus, the ATPase activity of MalFGK2 is highly sensitive to the lipid environment and is a useful feature in evaluating the performance of membrane mimetic reconstitution systems.

a Illustration of the peptide screening strategy. Potent scaffold peptides will insert themselves into lipid bilayers of proteoliposomes and extract the maltose transporter MalFGK2 into NDs. Inset, surface charges of 18 A as an exemplary amphipathic scaffold peptide. The structural model of 18 A is generated by Alphafold2. MalF, blue; MalG, orange; MalK, yellow. b Representative blue-native gel analysis of detergent-free ND reconstitution. MalFGK2 proteoliposomes prepared using PC lipids were incubated with the indicated peptide scaffolds (33, 100 and 300 µM) in the absence of detergents and then analyzed by native electrophoresis. In control experiments, the same proteoliposomes were analyzed alone (lane 1) or treated with DDM (lane 2). Similar results were obtained with three independent experiments. c SEC profiles of detergent-free MalFGK2 NDs formed from proteoliposomes (pL) using 18 A, in comparison with control experiments of pL alone. Samples were fractionated on a Superdex S200 3.2/300 column. d ATPase activities of MalFGK2 in detergent micelles, pL, and 18A-enclosed NDs. Data are shown as mean ± s.d., n = 3 independent experiments. e Negative stain EM micrograph of MalFGK2 NDs encased by 18 A. Red circles highlight the monomer NDs. Scale bar, 30 nm. Similar results were obtained with three independent experiments.
We began our study with a panel of amphipathic peptides ranging from ApoA-I mimetic peptides to antimicrobial peptides (Fig. 1b, Supplementary Fig. 1a and Table 1). Formation of MalFGK2 NDs from direct incubation of proteoliposomes with peptides was characterized using blue-native electrophoresis (native PAGE) (Fig. 1b and Supplementary Fig. 1a). Once extracted from lipid bilayers into micelles or incorporated into NDs, MalFGK2 migrated as a single membrane protein complex with a molecular weight of ~ 200 kDa, as observed with the detergent DDM. This simple readout enabled us to rapidly screen more than a dozen amphipathic peptides for detergent-free reconstitution of MalFGK2 into NDs. The results showed that several ApoA-I mimetic peptides (e.g., 18 A36, 22A14, 4F34, and NSP35) were able to transform MalFGK2 proteoliposomes into NDs as compared to other amphipathic or antibacterial peptides. However, these NDs contained a mixture of monomer, dimer, and different oligomers of MalFGK2 since multiple bands of protein complexes with higher molecular weights were observed on the native PAGE (Fig. 1b).
Because 18 A is the most effective for the extraction of MalFGK2 monomer, we then attempted to purify MalFGK2 NDs formed by 18 A (18A-MalFGK2 NDs) through size-exclusion chromatography (SEC) (Fig. 1c). As expected, soluble particles were isolated after incubating MalFGK2 proteoliposomes with 18 A. In control experiments, we only observed large, insoluble proteoliposomes in the absence of peptides. Quantitative analysis also confirmed the presence of ~ 100 copies of lipids per ND (Supplementary Fig. 1b), consistent with the idea that 18 A directly transforms proteoliposomes into discoidal particles. Moreover, MalFGK2 encased in 18A NDs is functional as its ATPase activity remains coupled to maltose and MBP (Fig. 1d), in line with previous studies obtained using proteoliposomes43. In contrast, MalFGK2 was inactive and not responding to maltose and MBP after extraction into NDs using amphipathic polymers (Supplementary Fig. 1c, d). The lack of detectable ATPase activities of MalFGK2 in polymer NDs is probably due to the sensitivity of these polymers to bivalent cations29, causing the aggregation of the transporter in ATP hydrolysis buffer containing high concentrations of MgCl2 (Supplementary Fig. 1e). In addition, other unknown problems might exist with polymer-based scaffolds for extracting MalFGK2 from membranes. We also characterized new versions of charge-free, electroneutral polymers that were more resistant to divalent cations (Supplementary Fig. 1f). However, the MalFGK2 transporter in these polymers was still much less active as compared to proteoliposomes or NDs formed with 18 A.
We further characterized the 18A-MalFGK2 NDs using negative-stain electron microscopy (EM). Although most purified particles from SEC are indeed showing diameters of 10–20 nanometers (Fig. 1e), they are quite poly-disperse, consistent with the data from native PAGE analyses (Fig. 1b). In addition, the structural feature of the MalFGK2 transporter is difficult to identify from these raw micrographs, suggesting that 18 A needs further engineering to improve its performance in ND reconstitution for biophysical studies of membrane proteins. For simplicity, thereafter, we named these peptides DeFrMSPs for the reconstitution of detergent-free NDs through amphipathic membrane scaffold peptides.
Potentiation of DeFrMSPs for ND reconstitution through fatty acid modifications
Next, we sought to improve the efficacy of 18 A for the reconstitution of native NDs. The key lies in the ability of the peptide to insert itself into lipid bilayers and stably enclose membranes, which can be potentiated by N- and C-terminal modifications (Supplementary Fig. 2a and Table 1). First, we attempted to fuse 18 A with antimicrobial peptides to enhance its membrane insertion activities. Second, we set out to functionalize 18 A peptides with distinct chemical groups, such as amide, acetyl, and fatty acids. To assay the efficiency of native ND reconstitution, the same MaFGK2 proteoliposomes were incubated with an increasing amount of these modified peptides and analyzed by native electrophoresis. The results showed that fatty acid modifications of 18 A profoundly enhanced the monodispersity of MalFGK2 NDs (Fig. 2a and Supplementary Fig. 2a), migrating as a single band on non-denaturing native gels. Among all the fatty acid modifications, hexanoic acid functionalized 18 A (Hex18A) resulted in the highest efficiency and was selected for further characterization.

a Blue native gel analysis of detergent-free ND reconstitution. MalFGK2 proteoliposomes prepared using PC lipids were incubated with the indicated peptide scaffolds (33, 100 and 300 µM) modified with fatty acids in the absence of detergents and then analyzed by native electrophoresis. Similar results were obtained with three independent experiments. b SEC profiles of detergent-free MalFGK2 NDs formed using Hex18A. Samples were fractionated on a Superdex S200 3.2/300 column. c Representative negative stain EM micrograph of MalFGK2 NDs encased by Hex18A. Scale bar, 30 nm. MalFGK2 NDs were highlighted in red circles. Similar results were obtained with three independent experiments. d, e Single particle cryoEM analysis of MalFGK2 in Hex18A NDs. d 2D class average. e 3D reconstructed model overlaid with density map (white).
We purified MalFGK2 NDs enclosed by Hex18A from SEC and determined their ATPase activities (Fig. 2b and Supplementary Fig. 2b). Again, the transporter is fully functional and stimulated by maltose and MBP. In addition, lipids were coeluted with these NDs (Supplementary Fig. 2c), as expected for the conversion of MalFGK2 proteoliposomes into ~ 10–15 nm discoidal particles. We then analyzed these samples by negative stain EM (Fig. 2c and Supplementary Fig. 2d). In contrast to the results obtained with 18 A, NDs formed by Hex18A were much more monodisperse, with clearly appreciable features of the cytosolic ATPase subunit of MalFGK2. The diameters of these MalFGK2 NDs formed with Hex18A averaged at ~ 12 nm, which would provide a ~ 2.5 nm layer of PC lipids around the transporter.
The vastly improved homogeneity of the Hex18A MalFGK2 NDs suggested that these samples are suitable for structural studies using single particle analysis by cryogenic electron microscopy (cryoEM). Indeed, the structure of the MalFGK2 complex in Hex18A NDs was solved in the closed state at 3.3 Å in the absence of ATP (Fig. 2d and e, Supplementary Fig. 3, and Table 2), and is overall in agreement with previous crystal structures of the transporter stabilized with truncations in the N-terminus of MalF or arrested by the glucose enzyme EIIA44,45. Together, we concluded that Hex18A can directly extract MalFGK2 from membranes into DeFrMSP-enclosed, detergent-free NDs (DeFrNDs) for biochemical and structural characterization.
DeFrNDs enclose stable and functional membranes
Although the above results obtained with MalFGK2 as a proof-of-principle study are encouraging, the properties of membranes in DeFrNDs remain unclear. In many cases, previous studies have shown that the stability and function of membrane mimetics greatly affect the conformational state of membrane proteins46,47,48. Furthermore, the lipids in polymer NDs rapidly diffuse from one ND to another by collisional transfer, unlike lipids in vesicles or traditional NDs formed by protein scaffolds31. The rapid lipid exchange between polymer nanodiscs suggests they may possess a more dynamic nature, which could limit their ability to emulate the native organization of cellular membranes. Therefore, it is critical to assess the membrane property of DeFrNDs.
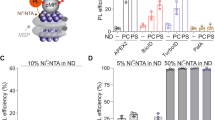
First, we determined if DeFrMSP can encase reconstituted lipid bilayers into monodisperse NDs. To this end, protein-free liposomes were prepared with various synthetic lipids or membrane extracts. These samples were then incubated with increasing amounts of Hex18A followed by fractionation on SEC (Supplementary Fig. 4a–f). At the optimized Hex18A/lipid ratio, well-defined ~ 15 nm NDs were formed with all the lipid mixtures tested in these experiments. Interestingly, we also observed the formation of 30–50 nm NDs at lower Hex18A/lipid ratios, indicating that the diameter of DeFrNDs is flexible and potentially programmable. We employed a fluorescent probe, Laurdan, to assay the lipid packing properties in DeFrNDs and found similar levels of minor alternations as the most recent version of charge-free, electroneutral polymers, which are much better than the original SMA200 polymer30 (Supplementary Fig. 4g, h).
Second, we employed a classic FRET-based membrane fusion assay to determine if the lipids in DeFrNDs are stably embedded or readily exchanging among individual NDs49. In these experiments (Fig. 3a), donor NDs or liposomes were prepared with a FRET pair (NBD-PE and Rho-PE) and then incubated with acceptor liposomes that did not harbor fluorescent lipids50,51. If the lipids in DeFrNDs are unstable, the FRET pair, upon incubation with acceptor liposomes, will be diluted and separated from each other, increasing NBD fluorescence. However, we only observed a negligible fluorescence increase after incubating protein-free donor DeFrNDs with acceptor liposomes (Fig. 3b), similar to the negative control conditions using protein-free donor liposomes. In positive control experiments, we employed the classic membrane fusion machinery, soluble N-ethylmaleimide-sensitive-factor attachment protein receptors (SNAREs). Specifically, the cognate vesicle (v) and targeted-membrane (t) SNAREs were reconstituted into donor NDs and acceptor liposomes, respectively. Membrane fusion between NDs and liposomes mediated by the SNARE complex resulted in the massive dilution of the FRET pair and the dequenching of NBD fluorescence. Furthermore, we also assayed the lipid exchange between DeFrNDs using the same FRET reporter (Supplementary Fig. 4i). Interestingly, we did not observe significant levels of lipid exchange as reported in SMA polymer NDs31. Thus, lipids are stably enclosed in DeFrNDs for the characterization of membrane dynamics and do not freely diffuse between NDs unless fusogenic proteins are present.

a Illustration of the membrane fusion assay. Donor NDs harboring NBD-PE and Rho-PE are incubated with acceptor liposomes. Fusion mediated by SNARE proteins will result in robust dequenching of NBD fluorescence. b Fusion activities of DeFrNDs. Fusion assays were carried out using protein-free (pf) or v-SNARE (v) liposomes and NDs (donor, 0.5 µM) with liposomes (acceptor, 5 µM) that either contained t-SNARE (t) or were pf. Fusion assays were performed in the presence of Ca2+ and the soluble fragment of Syt1. Data are shown as mean ± s.d., n = 3 independent experiments. c Characterization of syt1-lipid interactions using DeFrNDs. Illustration (top) and quantification (bottom) of the syt1 membrane binding and penetration assay. The soluble fragment of syt1 (C2AB, 10 nM) was labeled with NBD and incubated with NDs (5 µM) encasing the indicated lipids (ND-PS/PC and ND-PC) in the presence or absence of Ca2+. Fluorescence of C2AB-NBD was quantified before (F0) and after (F1) the addition of NDs. Data are shown as mean ± s.d., n = 3 independent experiments. d Asymmetric membranes trapped in DeFrNDs as demonstrated by SecA-lipid interactions. SecA (1 µM) was incubated with DeFrNDs (2 µM) formed with the indicated lipid composition. Samples were then analyzed by clear native electrophoresis. 1, SecA alone; 2, SecA with NDs prepared with only PG lipids (ND-PG); 3, SecA with ND prepared with symmetric PG and PC lipids (sym ND-PG/PC); 4, SecA with NDs prepared with asymmetric PG and PC lipids (asym ND-PG/PC); 5, SecA with NDs prepared with only PC lipids (ND-PC). The bands corresponding to the SecA monomer in complex with one or two sides of NDs were highlighted in red boxes. With symmetric NDs, we also observed large SecA-ND oligomers formed probably because of the self-association of SecA. Similar results were obtained with three independent experiments.
Finally, we assessed if the DeFrNDs can functionally capture protein-lipid interactions. For this purpose, NDs were prepared with different lipid mixtures to characterize membrane binding and remodeling by synaptotagmin-1 (syt1), the Ca2+ sensor responsible for synaptic transmission52,53,54. Using a well-established membrane penetration assay55, syt1-lipid interactions will cause a drastic fluorescence increase only in the presence of Ca2+ and anionic lipids, as we found using DeFrNDs (Fig. 3c). Consistently, minor increases were observed in the absence of Ca2+ or with NDs harboring solely neutral lipids. Together, we conclude that DeFrNDs can encase stable and functional lipid bilayers for biophysical characterizations of membrane biology.
Exploring DeFrNDs to reconstitute membrane asymmetry
One distinct feature of native membranes is lipid asymmetry that is critical for numerous cellular signaling pathways56,57. However, it is challenging to reconstitute this lipid asymmetry in traditional NDs because detergent-mediated reconstitution will disrupt the structural organization of membranes. We suspect that our peptide-mediated formation of native NDs might help address this dilemma by obviating the need for detergent solubilization.
We first formed asymmetric PC/PG liposomes using a previously described pH-driven method58 and incubated these liposomes with DeFrMSPs to form NDs. If membrane asymmetry is preserved in DeFrNDs, they should contain mainly PG lipids on one side and largely PC lipids on the other side. We then used SecA, which has a high affinity for PG lipids, to assess the asymmetry of PG and PC lipids in DeFrNDs (Fig. 3d). SecA is a ~ 100 kDa protein and forms a dimer in solution, which migrated at the position of the 240 kDa protein marker on clear native gels. Interestingly, it dissociates into monomers upon binding to negatively charged lipids59. By incubating SecA with DeFrNDs harboring only PG or symmetric PG/PC lipids (Lane 2 and 3), we thus readily observed two additional bands showing up on native gel electrophoresis. The upper band of ~ 350 kDa was probably the complex of two SecA monomers bound to each side of NDs. The lower band of ~ 200 kDa corresponded to one monomeric SecA with NDs, which migrated below the SecA dimer band most likely because of the negative charge of PG lipids. Other gel bands above the 480 kDa protein marker could be different forms of high-order SecA-ND complexes. These observations were further validated using crosslinking experiments (Supplementary Fig. 5a). SecA alone was readily crosslinked into a dimer, which was not formed with DeFrNDs harboring negatively charged lipids. In addition, incubation of pre-crosslinked SecA dimer with even saturated concentrations of DeFrNDs did not generate the lower band of the monomeric SecA-ND complex on native gels (Supplementary Fig. 5b). As a control experiment, DeFrNDs prepared with only PC lipids (lane 5) had little impact on the migration of the protein on the native gel compared to SecA alone because its affinity for PC lipids was considerably weaker than PG (lane 1)59. With asymmetric membranes (lane 4), the lower band of the ND-SecA monomer complex was much more enriched, indicating that membrane asymmetry was perhaps moderately reconstituted in DeFrNDs. The distribution of monomeric SecA binding to one or two sides of NDs was different by titrating ND and SecA concentrations (Supplementary Fig. 5c, d). At lower ND or saturated SecA concentrations, we observed a significant increase of two monomeric SecA proteins bound to both sides of asymmetric NDs, indicating that these NDs were not 100% asymmetrical with PG lipids located on only one leaflet. This problem could arise at the stage of generating asymmetric liposomes or as a result of ND formation using DeFrMSPs.
We also assessed the stability of the asymmetric NDs using the SecA-ND binding assay. The results showed that the enclosed membranes in NDs were relatively stable for a week when kept on ice (Supplementary Fig. 5e). However, our data do not exclude that lipids might still flip in NDs. It is probably just not sufficient to the level that can significantly disturb the interaction of SecA with lipids.
Encouraged by the above SecA-based experiments, we further examined if DeFrNDs could maintain the structural organization of native cell membranes. For this purpose, we focused on ADAM10, an abundant protease expressed in many cells that is activated by PS externalization60 (Supplementary Fig. 5f–i). DeFrNDs were prepared from HEK293 cells to assay ADAM10 activities. The results showed that ADAM10 activities in DeFrNDs and crude membranes were quite similar. In comparison, the activity of ADAM10 was significantly increased if we prepared vesicles from crude membranes using detergent-mediated reconstitution or if we generated DeFrNDs from these reconstituted vesicles, probably because the detergent completely disrupted native cell membranes and caused the redistribution of PS lipids between the two leaflets of reconstituted vesicles. Despite these promising results of recapitulating functional protein-lipid interactions in the context of native cell membranes, we noted that our data were not direct evidence to demonstrate the reconstitution of membrane asymmetry in DeFrNDs and would need more comprehensive interrogations in future studies.
Reconstitution of bacterial membrane protein complexes into DeFrNDs
In contrast to detergent-mediated reconstitution, the main advantage of DeFrNDs is the potential to stabilize membrane protein complexes with native lipids (Fig. 4a). To test this idea, we incubated crude membranes from E.coli expressing MalFGK2 with Hex18A and performed affinity purification. As expected, we can directly isolate MalFGK2 in Hex18A-encased native NDs (Fig. 4b and Supplementary Fig. 6a). However, we noticed that Hex-18A was much less efficient than the detergent DDM to extract MalFGK2, albeit significantly higher than 18 A or negative control experiments. To further enhance the performance of Hex18A, we set out to optimize the peptide sequence (Supplementary Table 1). The amphipathic peptide 18A was designed to combine positively charged residues with hydrophobic ones37. The main membrane-penetrating residue of 18A is Phe. However, it is known that Trp is more potent to destabilize lipid bilayers. Therefore, we gradually replaced Phe with Trp and assessed their efficacies in extracting MalFGK2 into NDs from proteoliposomes. These peptides are all modified with the hexanoic acid (Hex) group at their N-termini and are designated as F1W, F2W, F3W, and F4W. In addition, we slightly increased the amphipathic repeat of 18A to 20 or 22 amino acids (20B and 22B) to expand the surface area of these peptides for engaging lipids. As compared to Hex18A, several of these redesigned peptides showed improvement in extracting MalFGK2 into NDs from proteoliposomes (Supplementary Fig. 6b), with 20B exhibiting the highest efficacy. We then placed Trp substitutions in 20B and assessed their performance for the reconstitution of MalFGK2 NDs from crude E.coli membranes. Consistent with the results obtained using proteoliposomes, Trp substituted 20B peptides were indeed more effective than the original Hex18A, giving rise to a ~ 2.5-fold increase in the yield of native NDs (Fig. 4b). Because Hex20B1WA showed the highest reconstitution efficiency, we purified native NDs formed with this peptide for further characterizations. These MalFGK2 NDs encased by Hex20B1WA were quite homogenous as shown by SEC, negative stain EM and dynamic light scattering (DLS) analyses (Fig. 4c and Supplementary Fig. 6c–e) and contained native bacterial lipids (Supplementary Fig. 6f). The associated lipids with MalFGK2 were mainly PE and PG. The absence of other bacterial lipids, such as cardiolipin, indicated that the transporter was surrounded by a specific local lipid environment. Inspired by these results, we also characterized the structure of MalFGK2 isolated in native DeFrNDs by single particle cryoEM in the presence of ATP and vanadate (Fig. 4d and e, Supplementary Figs. 7, 8 and Table 3), which has not been resolved in previous studies. Interestingly, we identified both the inward- and outward-facing conformational states of MalFGK2 in this condition, supporting that the transporter is in a metastable state and needs its cofactor MalE for the structural transition to the fully activated outward-facing configuration44. Thus, DeFrNDs can directly isolate membrane proteins from native cell membranes for structure determination by single particle cryoEM.

a Illustration of the detergent-free, native ND reconstitution procedure. Native NDs were formed by one-step incubation of crude cell membranes with the designed DeFrMSPs in the absence of detergents. b ND formation efficiencies of MalFGK2 with different designs of DeFrMSPs. DDM and PBS buffer were used as positive and negative controls for data normalization. Top: representative gels of extracted MalFGK2 in native NDs. Bottom, ND formation efficiencies of MalFGK2 were quantified based on the density of the MalF and MalK bands normalized to that of DDM-extracted samples. Data are shown as mean ± s.d., n = 3 independent experiments. c Negative stain EM micrograph of MalFGK2 NDs encased by Hex20B1WA. Native MalFGK2 NDs were highlighted in red circles. Scale bar, 30 nm. Similar results were obtained with three independent experiments. d, e Single particle cryoEM analysis of MalFGK2 native NDs during ATP hydrolysis. d, 2D classes; (e) local resolution maps of the inward- and outward-facing states of MalFGK2 obtained in the presence of ATP (2 mM), MgCl2 (5 mM), and Vanadate (0.2 mM). f Blue native gel analysis of isolated MalFGK2 mutants in DDM or Hex20B1WA NDs. The red arrow indicates the position of the intact MalFGK2 complex. Most of these mutant complexes were unstable once extracted by DDM from cell membranes and thus dissociated into individual subunits. In contrast, many of them can be stably extracted into native NDs by Hex20B1WA. Similar results were obtained with three independent experiments.
Since our DeFrNDs can retain native lipids, they might also be able to trap challenging membrane protein complexes that are otherwise disrupted by detergents. To test this idea, we assayed the performance of Hex20B1WA to isolate MalFGK2 mutants that fall apart once extracted out of membranes by detergents61. These mutants behave very differently from the wild-type protein and are useful tools to advance our understanding of ABC transporters in general62. However, in-depth dissections of these mutants are unattainable because they are not stable in detergents. As shown by native electrophoresis, several MalFGK2 mutants were dissociated at various degrees after affinity purification in DDM (Fig. 4f, left). In contrast, Hex20B1WA directly extracted many of these MalFGK2 mutants into NDs with high efficiency and good stability for biochemical characterizations (Fig. 4f, right). We found that the basal ATPase activities of these mutants were much higher than the wild-type MalFGK2 transporter (Supplementary Fig. 6g), indicating escalated alternations in their conformational dynamics for MBP-independent maltose translocation.
Reconstitution of eukaryotic membrane proteins into DeFrNDs
To further assess the utility of DeFrNDs for biophysical characterizations of membrane biology, we first explored if they are appropriate tools for reconstituting a human GPCR, the metabotropic glutamate receptor 7 (mGluR7). We expressed the receptor from HEK293 cells and assayed if we could extract it from crude cell membranes into native NDs using DeFrMSPs (Fig. 5a and Supplementary Fig. 9a). We genetically fused mGluR7 with eGFP, allowing for sensitive fluorescent measurements to determine the efficiency of ND formation. We then incubated our collection of DeFrMSPs with mGluR7-eGFP membranes and quantified the efficiency of NDs formation by measuring the fluorescence of eGFP in the solubilized fractions (Supplementary Fig. 9a). The formed native NDs were then purified and analyzed using denaturing and native electrophoresis followed by in-gel fluorescence imaging (Fig. 5b and Supplementary Fig. 9b). We found that Hex20B1WA was the most effective to extract mGluR7 into soluble and homogeneous NDs as a single band of the reconstituted complex was observed on native PAGE (Fig. 5b). We purified mGluR7 native NDs formed by Hex20B1WA through affinity purification and SEC (Supplementary Fig. 9b, c). Using negative stain EM and DLS measurements, we found that mGluR7 native NDs are indeed quite monodisperse (Supplementary Figs. 6d, e and 9d). Moreover, they are fully functional to bind with high affinity to the extracellular domain of the trans-synaptic binding partners of mGluR763, the extracellular leucine-rich repeat and fibronectin type III domain-containing protein (ELFN-1), as shown by native electrophoresis (Fig. 5b, c).

a Illustration of the mGluR7-ELFN complex at neuronal synapses. mGluR7 is a prominent metabotropic glutamate receptor that modulates synaptic transmission. Recent studies demonstrate that its function is also regulated by trans-synaptic interactions with ELFN-162. b Representative clear native gel of mGluR7 in Hex20B1WA NDs binding to ELFN-1. Native NDs harboring mGLuR7 were incubated with increasing concentrations of the extracellular fragment of ELFN-1. Samples were analyzed by native electrophoresis and in-gel fluorescence imaging. c Quantification of mGluR7 interaction with ELFN-1 from native gel analysis as described in (b). The decreased densities of the mGluR7 band normalized to the protein alone (lane 1) was used to calculate the relative binding to ELFN-1 at various concentrations. The binding data of mGluR7 with ELFN-1 was then fitted with the Hill equation (Kd = 8 ± 1 µM; n = 2.6). Data are shown as mean ± s.d., n = 3 independent experiments.
In addition to mGluR7, we also tested the performance of our engineered DeFrMSPs for native ND reconstitution of the HCN1 channel (Fig. 6). Consistently, we can extract HCN1 into NDs from crude HEK293 membranes (Supplementary Fig. 10a), as characterized by SEC, negative stain EM and DLS (Supplementary Figs. 6d, e and 10b, c). Interestingly, Hex18A was the best scaffold for the extraction of HCN1, indicating that its local lipid environment might be different from mGluR7. The SEC profile of HCN native NDs showed more oligomerized proteins than mGluR7, perhaps because the fused GFP on this tetrameric channel tends to self-associate into high-order oligomers in the absence of detergents. Using a powerful single-molecule fluorescence imaging assay in zero-mode waveguides64, we looked at the binding activity of fcAMP to HCN1 channels (Fig. 6a, b). The results showed that the HCN1 tetramer is functional in DeFrNDs, retaining the ability to bind fcAMP (Fig. 6c, d). The overall binding activity and affinity of HCN1-cAMP interactions in native NDs were very different from data obtained in detergent micelles, supporting the role of lipids in regulating the property of this ion channel. In contrast, we could not efficiently purify HCN1 native NDs using amphipathic polymers (Supplementary Fig. 10d), let alone maintain their activities. We noted that the presented model in Fig. 6b is a simplified illustration of cAMP binding to HCN1. The underlying molecular mechanism is a subject of debate and will require more extensive characterization to generate in-depth insights into the interaction of cAMP with HCN1.

a Illustration of the single molecule fluorescence ligand binding assay to probe the interaction of fcAMP with HCN1. b fcAMP binds to HCN1 in native NDs with higher affinity than HCN1 in detergent micelles. Top, independent sequential ligand binding model for fcAMP binding, white boxes indicate the unbound state (U) and black boxes (B) indicate the bound state. “L” represents the ligand, and “K” represents the transition rate constant. Bottom, predicted binding curves for HCN1 in detergent (red) and HCN1 in native NDs (black) were generated by calculating the bound probability at various concentrations by using the Adair equation (see “Methods”). The binding probability of the first ligand was obtained from experimental data, and the binding probabilities of the 2nd, 3rd, and 4th ligands were estimated, assuming that these binding events occur independently. c Representative fluorescence-time traces overlaid with idealized fit showing fcAMP (50 nM) binding to HCN1 in detergent micelles (left) or native ND (right). d Representative fluorescence-time traces displayed as heat maps show multiple traces of fcAMP (50 nM) binding to HCN1 in detergent micelles (upper) or native ND (bottom). White spaces indicate an unbound state of the channel, whereas the color coding indicates duration and transition to various ligation states - 1 bound (purple), 2 bound (blue), 3 bound (green) or 4 bound (red). Across all concentrations, for the detergent sample, total N = 386 molecules, total time – 35 h, total events- 35661. For HCN1 in native NDs, total N = 119 molecules, total time − 9.9 h, total events -13143.
Moreover, we performed thin-layer chromatography (TLC) experiments to analyze the extracted lipids in native NDs (Supplementary Fig. 10e, f). Interestingly, we found that mGluR7 was associated with more PE lipids than HCN1. This difference might suggest the role of lipids in regulating the function of membrane proteins. Further, these results demonstrate that DeFrMSP NDs can isolate surrounding native lipids with eukaryotic membrane proteins. Hence, we analyzed the lipids extracted by DeFrNDs. By performing lipidomic analysis, we found that DeFrNDs did not show much preference for extracted lipids as compared to crude membranes (Supplementary Fig. 11), suggesting that they can serve as useful tools to characterize the impact of native membranes on different kinds of membrane proteins. However, quantitative lipidomic characterization is needed in future studies, especially for purified DeFrNDs harboring the membrane protein of interest.
Finally, we assessed whether DeFrNDs are useful for isolating proteins embedded in the ER and secretory vesicles. As expected, Sec61β from ER microsomes and VAMP2 from dense-core vesicles were well extracted by DeFrMSPs (Supplementary Fig. 12). Thus, our current test cases encompass a wide range of protein families and support that DeFrNDs are versatile tools for different kinds of membrane proteins resided in various cellular membranes.
Discussion
In this manuscript, we engineered membrane scaffold peptides for detergent-free reconstitution of membrane proteins into NDs from native membranes. Our DeFrND approach bypasses the limitation of detergent-mediated reconstitution used in traditional methods (Supplementary Fig. 13), thereby enabling structural and functional studies of membrane protein-lipid complexes previously unattainable. In addition, the membranes in DeFrNDs can more closely mimic native lipid environments for biophysical dissections of both integral and peripheral membrane proteins involved in various transmembrane signaling pathways. We anticipate that our approach will nicely complement current ND platforms and serve as a useful tool for both basic and translational research of membrane proteins.
While previous studies on ApoA1 mimetic peptides have established that some of them can solubilize synthetic membranes with defined lipid compositions33,34,38,65, our engineered DeFrMSP peptides can extract membrane protein-bearing discoids with high efficiency from native membranes. The reconstituted signaling membrane proteins (channels, receptors, and transporters) are never exposed to detergents throughout the reconstitution process, and the resultant NDs maintain their structure, function and single molecule dynamics (Figs. 2–6). In addition, DeFrMSPs are compatible with various membrane proteins and vastly simplify the workflow for the reconstitution of NDs compared to previous methods. The extraction activity of DeFrMSPs mostly depends on two parameters: insertion into membranes and stabilization of discoidal ND structures. Specifically, these peptides first need to penetrate lipid bilayers and then stably encircle a rigid patch of membranes into the ND framework. Antimicrobial peptides can readily insert themselves into membranes but cannot form stable discoidal structures with lipids. Hence, they are unable to extract membrane proteins into NDs as compared to ApoA-I mimetic peptides such as 18 A. In contrast, longer amphipathic peptides (e.g., NSP and NSPr) can form stable NDs35. However, they are much less effective in penetrating lipid bilayers to allow for detergent-free ND reconstitution (Fig. 2a and Supplementary Figs. 1a and 2a), probably due to the increased sizes. Consistently, it is known that traditional membrane scaffold proteins derived from ApoA-I are completely unable to disrupt membranes, most likely because of their over 10-fold larger molecular weights. Therefore, the successful development of DeFrMSPs is realized by optimizing membrane insertion and ND stability through extensive screening and rational design.
Further, DeFrMSPs greatly complement native ND reconstitution systems. Current native NDs are formed using different kinds of polymers that often require careful optimization7,27, such as the concentration of divalent ions and pH in the reconstitution buffer. In addition, it is difficult to predict the compatibility of these polymers for a specific membrane protein, because their efficiencies for forming native NDs vary significantly from case to case. The compatibility of DeFrNDs with bivalent cations makes it a great complementary and orthogonal system to current detergent-free reconstitution systems based on polymers. For example, DeFrNDs are suitable for membrane proteins that are not active in polymer-based NDs (Figs. 1–3 and Supplementary Fig. 1). However, we also noted that the current polymer-based system provides more options and versatility than DeFrND. Despite rapid lipid exchange between polymer NDs, they are stable and can withstand multiple free-thaw cycles66.
Moreover, DeFrMSPs will extend the potential of NDs for fundamental research of membrane biology. From our assessment of DeFrNDs with different transporters, receptors and ion channels, we found that the size of DeFrNDs adapted to the embedded transmembrane domain (Supplementary Fig. 6d). In addition, the size of DeFrNDs can be expanded to 30–50 nm when generated from protein-free liposomes (Supplementary Fig. 4a–c), indicating that DeFrNDs probably can accommodate much larger membrane protein complexes than the ones assayed in the current study. Another advantage of DeFrND is the potential to reconstitute asymmetric membranes with synthetic and native lipids. By virtue of this advantage, further improvement of DeFrND might enable a variety of biophysical approaches to investigate how lipid asymmetry regulates the conformational dynamics of membrane protein complexes as observed in cells. In conjunction with single-particle cryoEM, we expect that the DeFrND technology will help to unravel the molecular mechanism underlying the regulation of myriad transmembrane signaling pathways by native cell membranes. Based on our cryoEM analysis, DeFrNDs have not exhibited preferred orientation as sometimes found in other membrane mimetic systems67. This advantage will provide a useful alternative option for structural characterizations of membrane proteins in a near native lipid environment using single-particle cryoEM.
DeFrMSPs will also benefit therapeutic developments based on ApoA-I mimetic peptides. NDs formed with ApoA-I mimetic peptides are potent vehicles for vaccination14,16,17,18, because of their nanoscale sizes, enhanced tissue penetration, and low immunogenicity. As such, these nanomaterials can avidly engage with antigen-presenting cells and reprogram the immune system to control cancer and viral infections. We expect that the enhanced stability and efficiency of detergent-free ND reconstitution by DeFrMSPs will further promote therapeutic applications in these areas. Another potential application is to facilitate the delivery of therapeutic protein complexes (e.g., genome-editing enzymes)68,69. In conjunction with cationic cell-penetrating peptides, several amphipathic peptides were recently engineered to bind protein therapeutics and boost their entry into cells through unconventional clathrin-independent endocytosis and macropinocytosis. The delivery efficiency is determined by the membrane-penetrating activities of these amphipathic peptides. Since we found that DeFrMSPs also could spontaneously insert themselves into cell membranes, we expect that fine-tuning of our peptide designs will generate robust platforms for delivering therapeutic proteins.
A few potential issues will limit the utility of DeFrNDs. As synthesis of amphipathic peptides is challenging, we currently have to purchase them from outside vendors. The cost of DeFrMSPs is thus higher than traditional detergents and amphipathic polymers. Further, the additional aromatic amino acids in DeFrMSPs will interfere with spectroscopic experiments and protein quantification assays.
Together, our study establishes a new approach for native ND reconstitution through the development of membrane scaffold peptides, DeFrMSPs. The application of this powerful approach will elucidate how the structural organization of native membranes governs the dynamics and function of membrane protein complexes, thereby shedding light on new ideas to steer membrane biology for therapeutic developments against human diseases. Further improvement and optimization are critical to expand the utility of DeFrMSPs. One of the main challenges is the lower extraction efficiency of DeFrMSPs than detergents. This problem will need refinement of DeFrMSP designs to increase their solubilities and efficacies in extracting membrane proteins from native biological membranes. More design strategies can be learned from the characterization of novel amphipathic peptides and proteins70. Furthermore, we expect that machine learning and computational protein design approaches will greatly help our efforts in improving the performance of DeFrMSPs, as evidenced by the discovery of new potent antimicrobial peptides71.
Methods
Chemicals and Reagents
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-PE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl (Rho-PE), 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (PS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (PE) and 1,2-dioleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (PG) were obtained from Avanti Polar Lipids. Nitrilotriacetic acid (Ni2+-NTA)-chelating Sepharose, Superdex 200 3.2/300 GL, Superdex 200 10/300 GL and Superose 6 increase 10/300 GL were purchased from GE Healthcare. 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate) (Dil), N,N’-Dimethyl-N-(Iodoacetyl)-N’-(7-Nitrobenz-2-Oxa-1,3-Diazol-4-yl)Ethylenediamine (IANBD amide) and Oregon GreenTM 488 (OG) maleimide were obtained from ThermoFisher. All other chemicals were acquired from Sigma.
Plasmids
pTrc-MalFGK2 was a gift from Dr. Franck Duong61. pEThis-vamp2 was a gift from Dr. James Rothman49. pGEX-syt1 and pDuetRSF t-dimer were a gift from Dr. Edwin Chapman51,55. All other constructs in this work were made using the In-Fusion® HD Cloning Kit (Takara Bio USA).
Proteins and peptides
Protein expression and purification for syt1, MBP, SecA, MalFGK2 and SNAREs were performed as described previously42,50,51,72,73,74. Briefly, plasmids were transformed into BL21 cells that were grown in LB supplemented with Km (50 mg/ml) or Amp (100 mg/ml) to OD600 ~ 0.7. Protein expression was induced with 0.2 mM IPTG at 16 °C, overnight (O/N). Bacteria were harvested by centrifugation at 3428 x g for 20 min, resuspended in Buffer A (50 mM Tris-HCl (pH 8),100 mM NaCl, 5% glycerol, 2 mM β-mercaptoethanol), and lysed using a Branson cell disrupter. Cell lysates were clarified by centrifugation at 8184 x g for 45 min. For syt1, MBP and SecA, the supernatants were loaded onto a 1 ml NTA column (GE Healthcare), followed by two times wash using buffer B (50 mM Tris-HCl (pH 8), 20 mM Imidazole, 400 mM NaCl, 5% glycerol, 2 mM β-mercaptoethanol). Proteins were eluted in buffer C (50 mM Tris-HCl (pH 8), 500 mM Imidazole, 400 mM NaCl, 5% glycerol, 2 mM β-mercaptoethanol), desalted in buffer A using PD MiDiTrap G-25 (GE Healthcare) for syt1 and MBP, and stored at − 80 °C. SecA was further purified by SEC using a Superdex 200 10/300 column in 50 mM Tris-HCl (pH 8), 100 mM NaCl, 5% glycerol, 1 mM DTT. Soluble SecA dimer fractions were pooled together and concentrated to ~ 2 mg/ml and stored at − 80 °C. For MalFGK2 and SNAREs, membrane fractions were resuspended in Buffer A and solubilized with 1% DDM (4 °C, O/N). Solubilized membrane extracts were isolated by centrifugation at 112,000 × g for 1 h. Supernatants were loaded onto a 1 ml NTA column (GE Healthcare), followed by two times wash using buffer B supplemented with 0.02% DDM. Proteins were eluted in buffer C plus 0.02% DDM, desalted in buffer A supplemented with 0.02% DDM using PD MiDiTrap G-25 (GE Healthcare) and stored at − 80 °C.
Ecto-ELFN1-Fc (residues 1–399) were expressed as previously described63. An eGFP fusion was added to the C-terminus of mGluR7 and expressed as before63. Briefly, mGluR7 and Ecto-ELFN1-Fc constructs were inserted into the modified pEG BacMam vector75. The bacmid was then transfected into Sf9 cells (ThermoFisher Scientific, 11496015) to produce baculovirus. The supernatant of the third generation of virus production (P3) was then harvested and added to HEK293S GnTi- cells (ATCC, CRL-3022) grown at 37 °C with 8% CO2 at a confluency of 2.0-3.0 × 106 cells/mL in FreeStyle 293 expression media. After 18 h, sodium butyrate (Sigma-Aldrich, B5887) was added to 10 mM. The cells were then incubated for a further 48 h and pelleted by centrifugation. For the purification of ELFN-1, the supernatant of cell lysates clarified by centrifugation (8184 x g, 1 h) was then affinity purified using Pierce™ anti-DYKDDDDK affinity resin (ThermoFisher Scientific, A36801) and eluted with 3 x Flag peptide (GenScript, RP21087), concentrated using an Amicon™ Ultra-4 centrifugal filter (Sigma-Aldrich, UFC805024), analyzed by SDS-PAGE, aliquoted and flash frozen.
Expression of HCN1 tagged with eGFP and Single-molecule fluorescence imaging were performed as described previously64.
All peptides were custom-synthesized by GenScript with 95% purity. LC-MS/MS data of the final optimized peptide designs were provided in Supplementary Fig. 14. Peptides were stored at − 20 °C and dissolved at 100 mg/ml in DMF before use.
Purification of dense-core vesicles
PC12 cells were cultured in Dulbecco’s modified Eagle’s medium, high glucose (Gibco) supplemented with 10% horse serum (CellGro), and 10% calf serum (Fe + ) (Hyclone), with 1% penicillin/streptomycin mix. Cell media was changed every 2 to 3 days and split when cells reached ~ 90% confluency by incubating for 5 min in Hank’s balanced salt solution, resuspended by adding fresh medium, and replating them at a 6:1 ratio. Dense core vesicles were then purified from 15 10 cm plates (90% confluent) by scraping them into phosphate-buffered saline (PBS) solution. Cells were pelleted by centrifugation at 1000 x g for 5 min, resuspended, and washed once by repeating the centrifugation step in homogenization media (0.26 M sucrose, 5 mM MOPS, and 0.2 mM EDTA). Cell pellet was resuspended in 3 mL of homogenization medium containing protease inhibitor (Roche Diagnostics), the cells were then physically lysed by passing cells through a ball bearing homogenizer 10 times, which contained a 0.6367 cm bore and a 0.6340 cm diameter ball. Nuclei and large debris were removed by centrifugation in a fixed-angle microcentrifuge at 1000 x g for 10 min at 4 °C. The post-nuclear supernatant was collected, and mitochondria were removed by centrifugation at 8000 x g for 15 min at 4 °C. The post-mitochondria supernatant was then collected and adjusted to 5 mM EDTA and incubated on ice for 10 min to assist with removing ribosomes from the endoplasmic reticulum to reduce contamination in the dense core vesicle fraction. A working solution of 50% OptiPrep (iodixanol) was made by mixing five volumes of 60% OptiPrep with one volume 0.26 M sucrose, 30 mM MOPS, and 1 mM EDTA. This working solution was mixed with homogenization media to prepare solutions of 30% iodixanol and 14.5% iodixanol for a discontinuous gradient. A SW55 tube was prepared by laying 3.8 mL of 14.5% iodixanol on top of 0.5 mL of 30% iodixanol. Then 1.2 mL of the post-mitochondrial supernatant was layered on top of this iodixanol gradient. The sample was then centrifuged at 190,000 x g for 5 h. A clear white band at the interface between the 30% iodixanol and 14.5% iodixanol was collected as the dense core vesicle sample. The dense core vesicles sample was then extensively dialyzed in a cassette with 10,000 kDa molecular weight cutoff (24 h with 3 buffer changes of 5 L each) into 120 mM potassium glutamate, 20 mM potassium acetate, and 20 mM HEPES, pH 7.4. After dialysis, sucrose was spiked in to make a final concentration of 10% sucrose and then the sample was flash frozen and stored at − 80 °C.
Fluorescent labeling of proteins
Purified syt1 were desalted using Zeba Spin columns (Thermo Fisher) in buffer D (50 mM Tris-HCl, pH 8, 100 mM NaCl, 5% glycerol) and labeled with a 3-fold excess of IANBD amide or OG maleimide in the presence of TCEP (0.2 mM) at room temp for 2 h. Free dyes were removed by passing through Zeba Spin columns in buffer A.
Preparation for proteoliposomes and liposomes
Lipids were dried under a gentle stream of nitrogen and further with vacuum for 2 h followed by rehydration in reconstitution buffer (25 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM DTT). Samples were then extruded through 200 nm filter for liposome preparation. To monitor the binding of Syt1 to protein-free NDs via fluorescence spectrometry, protein-free liposomes were made using 70% PC and 30% PS. To study the interaction of SecA with lipids, protein-free liposomes were made using 100% PC, 100% PG, or 50% PC with 50% PG. We induced the formation of asymmetric liposomes with PC/PG lipids by generating a transmembrane pH gradient as described previously58. For proteoliposome reconstitution, purified proteins (MalFGK2 and SNAREs) and lipids were incubated at a ratio of 1:1000 in reconstitution buffer plus 1% OG on ice for 30 min. MalFGK2 proteoliposomes were prepared with 100% PC lipids. For membrane fusion and lipid exchange assays, donor v-SNARE and protein-free vesicles were prepared with 12% PE, 40% PS, 45% PC, 1.5% NBD-PE and 1.5% Rho-PE, whereas t-SNARE and protein-free vesicles were prepared with 15% PE, 25% PS and 60% PC. Detergents were removed by the addition of Bio-beads (1/3 volume) and gentle shaking (O/N). Finally, proteoliposomes were purified by flotation in an Accudenze step gradient as described previously50.
Nanodisc reconstitution
For initial screening with MalFGK2 proteoliposomes, DeFrMSPs (100 mg/ml in DMF) diluted to 25 mg/ml in PBS were added at the indicated concentrations at 4 °C (O/N), followed by blue native electrophoresis. For biochemical and structural characterizations, MalFGK2 NDs were formed with DeFrMSP at 0.2 mg/ml and fractionated by SEC on a Superdex 200 3.2/300 column in buffer A. Samples were concentrated to 1-2 mg/ml and stored at − 80 °C. Protein-free and v-SNARE NDs were prepared by incubating the respective liposomes with DeFrMSPs at a ratio of 1/30 and then purified using a Superose 6 10/300 column in buffer A.
To directly reconstitute membrane proteins into DeFrMSP native nanodiscs, we first lysed cells using a cell disrupter (Fisher) and then isolated crude membranes by ultracentrifugation (184,000 × g, 1 h). These membrane preparations (~ 10 mg/ml) were resuspended in buffer A and then incubated with DeFrMSPs at 1 mg/ml (4 °C, O/N). Extracted samples were clarified by ultracentrifugation (112,000 × g, 1 h), and supernatants were subjected to affinity purification. MalFGK2 NDs were purified using Ni2+-NTA beads as described above. mGluR7 NDs were purified using Ni2+-NTA and anti-DYKDDDDK (Fisher) affinity resins as ELFN-1. HCN1 NDs were purified using Streptactin affinity resin (IBA Life Sciences) and eluted in buffer D (50 mM Tris-HCl (pH 8), 300 mM NaCl, 10% glycerol, 2 mM DTT) plus 5 mM desthiobiotin. Finally, MalFGK2 and mGluR7 NDs were further fractionated on a Superdex S200 3.2/300 column in buffer A, whereas HCN1 NDs were in buffer D.
We assayed the extraction of Sec61β into DeFrNDs from Canine pancreatic ER microsomes, which were a generous gift from Dr. Andrey Karamyshev at Texas Tech University Health Sciences Center. ER microsomes were incubated with DeFrMSPs as for crude membranes described above. ER Solubilized microsomes were then spun down for 10 min at 12,000 x g at 4°C in a Sorvall Legend X1R tabletop centrifuge. The pellet was discarded, and the supernatant was subjected to western blot analysis using a Sec61β antibody (Novus Biologicals, NBP2-13290, 1/1000 dilution) and a goat anti-rabbit secondary antibody (ThermoFisher, A32735, 1/10000 dilution).
To evaluate the extraction of VAMP2 into DeFrNDs from dense-core vesicles, samples were incubated with DeFrMSPs. Extracted samples were clarified by centrifugation at 112,000 x g at 4 °C for 1 h and then analyzed by western blot using a VAMP2 antibody (Synaptic Systems, 104211, 1/2000 dilution) and a secondary mouse IgG Fc binding protein (Santa Cruz Biotechnology, sc545209, 1/5000 dilution).
To generate polymer NDs, MalFGK2 proteoliposomes, protein-free liposomes or HCN1 crude membranes were incubated (4 °C, O/N) with the indicated polymers at a final concentration of 1% as recommended by the vendor (Curi Bio). Samples were clarified by ultracentrifugation (112,000 × g, 1 h). For proteoliposomes and protein-free liposomes, extracted NDs were then purified by SEC using Superdex 200 10/300 in buffer A. For extracted HCN1 NDs from crude membranes, samples were subjected to affinity purification using Streptactin affinity resin as described above for DeFrNDs.
Negative stain electron microscopy
Formvar/carbon-coated copper grids (01754-F, Ted Pella, Inc.) were glow-discharged (15 mA, 25 secs) using PELCO easiGlowTM (Ted Pella, Inc). NDs (10 µg/ml) were applied onto the grids for 30 secs, followed by staining with 0.75% uranyl formate for 1 min. Images were collected using a ThermoFisher Science Tecnai G2 TEM (100 kV) equipped with a Veleta CCD camera (Olympus). All TEM data were analyzed using Fiji to determine ND sizes.
CryoEM data collection
For structure determination of MalFGK2 at the apo state in Supplementary Fig. 3, 3 µl purified NDs at 2 mg/ml was applied to glow discharged 300 mesh UltraAufoil gold R1.2/1.3 (quantifoil) and vitrified using a Vitrobot Mark IV (Thermo Fisher Scientific/FEI) at 10 °C and 100% humidity with a blot time of 4 s, and a blot force of 0. To solve the structure of MalFGK2 with native lipids at ATP hydrolysis condition reported in Supplementary Fig. 7, purified NDs at 2 mg/ml were incubated with ATP (2 mM) and vanadate (0.2 mM) for 10 min at 37 °C in Buffer A supplemented with MgCl2 (5 mM). Samples were then used to prepare grids following the same procedure described above for the transporter at the apo state. Cryo-EM data for single particle analysis were collected at New York Structural Biology Center on a 300 kV Titan Krios electron microscope (Thermo Fisher Scientific/FEI) with a Gatan K3-Bioquantum direct electron detector (Gatan, Inc.) and energy filter, using leginon software76. Movies for MalFGK2 NDs in the apo state were captured at a magnification of 81,000 x (pixel size of 1.058 Å). A total of 12522 movies in the defocus range of − 0.8 to − 2.2 μm were recorded with a total accumulated dose of 55.80 e-/Å. Movies for native MalFGK2 NDs in the presence of ATP, MgCl2 and vanadate were captured at a magnification of 105,000 x super-resolution mode (pixel size of 0.413 Å). A total of 22799 movies in the defocus range of − 0.8 to − 2.2 μm were recorded with a total accumulated dose of 52.78 e-/Å.
Image processing, 3D reconstruction, modeling, and refinement
All Data processing was performed using cryoSPARC v4.4.177. Raw movies were aligned using patch motion correction, and the micrograph contrast transfer function (CTF) parameters were estimated via patch CTF estimation. Micrographs were picked using a blob picker, and an initial particle set was chosen through 2D classification. Selected 2D classes were used for template particle picking and extracting particles for data processing.
For MalFGK2 NDs in the apo state, 5,805,546 particles were extracted from template picking, followed by multiple rounds of 2D-classifications to remove junk particles. 1,662,509 particles were further subjected to ab initio reconstruction to generate the initial maps. Ab-initio jobs were run with four classes, and the top three classes (highlighted in the box of Supplementary Fig. 3) were used for three rounds of heterogeneous refinement to remove junk particles. Finally, 452,428 particles in the best class were used for local CTF and non-uniform refinement, resulting in the final map of 3.3 Å based on the 0.143 Fourier Shell Correlation (FSC) criterion. Details of the Cryo-EM classification and processing are shown in Supplementary Fig. 3.
For MalFGK2 NDs prepared with ATP, MgCl2 and vanadate, 724,346,435 particles were extracted from template picking, followed by multiple rounds of 2D-classifications to remove junk particles. 694,732 particles were subjected to Ab initio reconstruction, and the top five classes (highlighted in the box of Supplementary Fig. 7) were used for three rounds of heterogeneous refinement to remove the junk particles and separate the particles into the inward-facing state with 163,557 particles and the outward-facing state with 246,297 particles for further data processing. Another three rounds of heterogeneous refinement were carried out to remove junk particles for both inward- and outward-facing states. Finally, 130,237 particles in the inward-facing state and 125,454 particles in the outward-facing state were used for local CTF and non-uniform refinement, achieving 3.5 Å and 3.4 Å resolution for the two respective states, based on the 0.143 FSC criterion. Details of the Cryo-EM classification and processing are shown in Supplementary Fig. 7. In the density maps shown in Fig. 4 and Supplementary Figs. 3 and 7, we have adjusted the threshold in the volume viewer of Chimera to filter the low-resolution density of lipid nanodiscs.
Initial models were built starting from PDB structures of 3fh6 and 3rlf. These models were rigid body docked into the density and adjusted in Chimera78 and coot79. Loops were manually fit into the density map using coot. Subsequently, the real space refinement was performed, with remaining manual adjustments were performed in Coot. Models were validated using Molprobity in Phenix80. A summary of model refinement statistics is provided in Supplementary Table 2 and 3.
Lipidomics
NDs or crude membranes were subjected to lipid extraction by MTBE (methyl tert-butyl ether) by tip sonication 3 times, vortex mixing 3 times, and the upper phase containing the lipids was collected and dried by SpeedVac with no heat. Dried samples were reconstituted with 50 μL of 50% EtOH and used for the LC-MS/MS using Vanquish Horizon UHPLC (Thermo Scientific) and Orbitrap Tribrid ID-X (Thermo Scientific) mass spectrometry. Database search was carried out by Compound Discoverer 3.3 SP2.
Fusion assays
v-SNARE vesicles harboring the FRET reporter (NBD-/Rho-PE lipids) and t-SNARE vesicles were prepared as described previously50,51. Fusion assays were performed by incubation of v-SNARE NDs or vesicles (0.5 µM) with t-SNARE vesicles (5 µM) in reconstitution buffer (20 mM Tris-HCl, pH 7.5, 100 mM NaCl) at 37 °C for 30 min. For lipid exchange assays, NDs (0.5 µM) were prepared from protein-free liposomes bearing the same FRET reporter and incubated with another set of NDs (5 µM) prepared from protein-free liposomes using PC/PE/PS lipids as described above. NBD fluorescence was monitored using a Synergy H1M plate reader with excitation at 460 nm and emission at 530 nm. After each run, the percentages of membrane fusion or lipid exchange were calculated by normalization of data to the maximal fluorescence signal increase after the addition of 0.5 % DDM to each reaction.
Fluorescence spectroscopy
The NBD-labeled soluble fragment of syt1 (C2AB, 10 nM) was incubated with the indicated NDs (5 µM). The fluorescence spectrum of samples was collected on a Synergy H1M plate reader with excitation at 460 nm and emission from 500 to 650 nm.
Laurdan (D250, ThermoFisher, 2 µM) was incubated with liposomes or the indicated nanodiscs at a ratio of 1:100. Fluorescence spectrum was collected on the Synergy H1M plate reader with excitation at 380 nm and emission from 410 to 540 nm. Laurdan GP values were quantified as described previously28.
Single molecule cAMP binding measurements
Single-molecule cAMP binding measurements were done using zero-mode waveguides fabricated as described previously64. Imaging was done on a custom micromirror TIRF setup (Mad City Labs) equipped with a NA 60X oil immersion objective lens (Olympus). Micromirror TIRF excitation field generated either with a 488 nm or 561 nm laser (OBIS, coherent) to excite the GFP and fcAMP, respectively. The subsequent fluorescence emission was recorded on a 512 × 512 EMCCD camera (Andor iXON Ultra) at a frame rate of 10 Hz. Data collection was done using Micromanager 2.0. The emission was passed filtered through a dichroic filter (T565lpxr Chroma) and then a band-pass filter (Chroma ET550/25 nm) for GFP and a band-pass filter (Semrock bright line 593/40 nm) for fcAMP were in the corresponding emission pathways. The binding data were recorded from each molecule for a minimum of 240 s with 100 msec exposure time. Single-molecule data was analyzed as described in the previous work64. Briefly, single-molecule traces were extracted and analyzed using the DISC software for the colocalization and image projection to obtain time-dependent fluorescence intensity changes. The traces were then idealized using the DISC software81. The equilibrium constant for intrinsic binding affinity, K, was calculated from the dwell times corresponding to the singly-bound state as described previously using QUB software64. Assuming that the binding of each of the four ligands binds to the channel independently, the binding curve was generated by calculating microscopic rate constants k1 = K/4, k2 = 2/3(K), k3 = 3/2(K), and k4 = 4 K and substituting them in Adair’s equation82.
ADAM10 reconstitution and biochemical assays
Crude cell membranes were prepared from HEK293S GnTi- cells (ATCC, CRL-3022) grown to a confluency of 2.0-3.0 × 106 cells/mL and converted to DeFrNDs using Hex18A (1 mg/ml, 4 °C, O/N) as described above for mGluR7. Samples were then clarified by ultracentrifugation (112,000 x g, 1 h) and fractionated on a Superdex 200 10/300 column in buffer A. Soluble fractions containing ADAM10 DeFrNDs were identified by western blot using an anti-ADAM10 antibody (Abcam, ab1997) and concentrated to ~ 0.5 mg/ml for biochemical assays. For detergent-mediated reconstitution of ADAM10, cell membranes were solubilized with 1% OG (4° C, O/N) and then allowed to reform vesicles by the addition of Bio-beads (1/3 volume) and purified by flotation in an Accudenze step gradient. The orientation of ADAM10 in reconstituted vesicles was assayed by Trypsin digestion in the absence or presence of Triton X-100 (1%) for 1 h on ice, followed by SDS-PAGE and western blot using the anti-ADAM10 antibody (1/1000 dilution) from Abcam (ab124695). Activities of ADAM10 in DeFrNDs or vesicles formed by detergent-mediated reconstitution were determined using an ADAM10 assay kit (BPS Bioscience) in comparison with crude membranes. Data were normalized based on the amount of ADAM10 levels measured by western blot.
Other methods
TLC was carried out using silica gel plates (Sigma) and stained with 5% primuline83. Crosslink experiments were performed using BS3 (10 µM) in PBS buffer at room temperature for 10 min and quenched using 50 mM Tris-HCl (pH 8). For SecA-ND binding assays, crosslinked SecA were further purified using Zeba spin desalting columns and then incubated with NDs. SDS-PAGE and Native PAGE electrophoresis were performed using 12% or 4–15% TGX protein gels (Bio-Rad). ATPase assays and quantification of phospholipids were performed as described previously42,43,73. Source Data are available as a source data file.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The coordinate and cryo-EM maps of MalFGK2 NDs have been deposited at the PDB and Electron Microscopy Data Bank under accession codes 9BCR (EMD-44435), 9NQJ (EMD-49659), and 9NXC (EMD-49901) for the apo, outward- and inward-facing states, respectively. All other data supporting the findings of this study are available within this manuscript. Source data are provided in this paper.
References
Bayburt, T. H., Grinkova, Y. V. & Sligar, S. G. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2, 853–856 (2002).
Denisov, I. G. & Sligar, S. G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 23, 481–486 (2016).
Sligar, S. G. & Denisov, I. G. Nanodiscs: A toolkit for membrane protein science. Protein Sci. 2, 297–315 (2020).
Frauenfeld, J. et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat. Methods 13, 345–351 (2016).
Nasr, M. L. et al. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nat. Methods 14, 49–52 (2017).
Autzen, H. E., Julius, D. & Cheng, Y. Membrane mimetic systems in CryoEM: keeping membrane proteins in their native environment. Curr. Opin. Struct. Biol. 58, 259–268 (2019).
Brown, C. J., Trieber, C. & Overduin, M. Structural biology of endogenous membrane protein assemblies in native nanodiscs. Curr. Opin. Struct. Biol. 69, 70–77 (2021).
Bao, H. Developing nanodisc-ID for label-free characterizations of membrane proteins. Commun. Biol. 4, 514 (2021).
Zhang, S. et al. One-step construction of circularized nanodiscs using SpyCatcher-SpyTag. Nat. Commun. 12, 5451 (2021).
Ren, Q., Zhang, S. & Bao, H. Circularized fluorescent nanodiscs for probing protein-lipid interactions. Commun. Biol. 5, 507 (2022).
Brady, N. G., Workman, C. E., Cawthon, B., Bruce, B. D. & Long, B. K. Protein extraction efficiency and selectivity of esterified styrene-maleic acid copolymers in thylakoid membranes. Biomacromolecules 22, 2544–2553 (2021).
Ravula, T. & Ramamoorthy, A. Synthesis, characterization, and nanodisc formation of non-ionic polymers. Angew. Chem. Int. Ed. Engl. 60, 16885–16888 (2021).
Kuai, R., Li, D., Chen, Y. E., Moon, J. J. & Schwendeman, A. High-density lipoproteins: Nature’s multifunctional nanoparticles. ACS Nano 10, 3015–3041 (2016).
Kuai, R., Ochyl, L. J., Bahjat, K. S., Schwendeman, A. & Moon, J. J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017).
Nam, J. et al. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 4, 398–414 (2019).
Noh, I. et al. Cellular nanodiscs made from bacterial outer membrane as a platform for antibacterial vaccination. ACS Nano 17, 1120–1127 (2022).
Guo, Z. et al. Cancer cell membrane nanodiscs for antitumor vaccination. Nano Lett. 23, 7941–7949 (2023).
Sun, L. et al. Synthesis of erythrocyte nanodiscs for bacterial toxin neutralization. Angew. Chem. Int. Ed. Engl. 62, e202301566 (2023).
Mulder, W. J. M., Ochando, J., Joosten, L. A. B., Fayad, Z. A. & Netea, M. G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 18, 553–566 (2019).
Priem, B. et al. Trained immunity-promoting nanobiologic therapy suppresses tumor growth and potentiates checkpoint inhibition. Cell 183, 786–801 (2020).
Denisov, I. G., Grinkova, Y. V., Lazarides, A. A. & Sligar, S. G. Directed self-assembly of monodisperse phospholipid bilayer nanodiscs with controlled size. J. Am. Chem. Soc. 126, 3477–3487 (2004).
Dong, D. et al. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature 573, 546–552 (2019).
Dong, Y. et al. Structural principles of B cell antigen receptor assembly. Nature 612, 156–161 (2022).
Yu, J. et al. Mechanism of gating and partial agonist action in the glycine receptor. Cell 184, 957–968 (2021).
Walker, G. et al. Oligomeric organization of membrane proteins from native membranes at nanoscale spatial and single-molecule resolution. Nat. Nanotechnol. 19, 85–94 (2024).
Dadsena, S. et al. Lipid unsaturation promotes BAX and BAK pore activity during apoptosis. Nat. Commun. 15, 4700 (2024).
Qiu, W. et al. Structure and activity of lipid bilayer within a membrane-protein transporter. Proc. Natl. Acad. Sci. USA 115, 12985–12990 (2018).
Real Hernandez, L. M. & Levental, I. Lipid packing is disrupted in copolymeric nanodiscs compared with intact membranes. Biophys. J. 122, 2256–2266 (2023).
Ravula, T., Hardin, N. Z. & Ramamoorthy, A. Polymer nanodiscs: Advantages and limitations. Chem. Phys. Lipids 219, 45–49 (2019).
Glueck, D. et al. Electroneutral polymer nanodiscs enable interference-free probing of membrane proteins in a lipid-bilayer environment. Small 18, e2202492 (2022).
Cuevas Arenas, R. et al. Fast collisional lipid transfer among polymer-bounded nanodiscs. Sci. Rep. 7, 45875 (2017).
Avrahami, D. & Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J. Biol. Chem. 279, 12277–12285 (2004).
Miyazaki, M., Tajima, Y., Handa, T. & Nakano, M. Static and dynamic characterization of nanodiscs with apolipoprotein A-I and its model peptide. J. Phys. Chem. 114, 12376–12382 (2010).
Kariyazono, H. et al. Formation of stable nanodiscs by bihelical apolipoprotein A-I mimetic peptide. J. Pept. Sci. 22, 116–122 (2016).
Carlson, M. L. et al. The Peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. ELife 7, e34085 (2018).
Kanellis, P. et al. Studies of synthetic peptide analogs of the amphipathic helix. Effect of charged amino acid residue topography on lipid affinity. J. Biol. Chem. 255, 11464–11472 (1980).
Segrest, J. P., De Loof, H., Dohlman, J. G., Brouillette, C. G. & Anantharamaiah, G. M. Amphipathic helix motif: classes and properties. Proteins 8, 103–117 (1990).
Bechinger, B. & Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta 1758, 1529–1539 (2006).
Hancock, R. E., Haney, E. F. & Gill, E. E. The immunology of host defence peptides: beyond antimicrobial activity. Nat. Rev. Immunol. 16, 321–334 (2016).
Jackman, J. A. et al. Therapeutic treatment of Zika virus infection using a brain-penetrating antiviral peptide. Nat. Mater. 17, 971–977 (2018).
Chen, J. Molecular mechanism of the Escherichia coli maltose transporter. Curr. Opin. Struct. Biol. 23, 492–498 (2013).
Bao, H. & Duong, F. Discovery of an auto-regulation mechanism for the maltose ABC transporter MalFGK2. PloS ONE 7, e34836 (2012).
Bao, H., Dalal, K., Wang, V., Rouiller, I. & Duong, F. The maltose ABC transporter: action of membrane lipids on the transporter stability, coupling and ATPase activity. Biochim. Biophys. Acta 1828, 1723–1730 (2013).
Khare, D., Oldham, M. L., Orelle, C., Davidson, A. L. & Chen, J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell 33, 528–536 (2009).
Chen, S., Oldham, M. L., Davidson, A. L. & Chen, J. Carbon catabolite repression of the maltose transporter revealed by X-ray crystallography. Nature 499, 364–368 (2013).
Zhang, Y. et al. Visualization of the mechanosensitive ion channel MscS under membrane tension. Nature 590, 509–514 (2021).
Notti, R. Q. & Walz, T. Native-like environments afford novel mechanistic insights into membrane proteins. Trends Biochem. Sci. 47, 561–569 (2022).
Petroff, J. T. et al. Open-channel structure of a pentameric ligand-gated ion channel reveals a mechanism of leaflet-specific phospholipid modulation. Nat. Commun. 13, 7017 (2022).
Weber, T. et al. SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772 (1998).
Bao, H. et al. Exocytotic fusion pores are composed of both lipids and proteins. Nat. Struct. Mol. Biol. 23, 67–73 (2016).
Bao, H. et al. Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263 (2018).
Geppert, M. et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727 (1994).
Jahn, R. & Sudhof, T. C. Synaptic Vesicles and Exocytosis. Annu. Rev. Neurosci. 17, 219–246 (1994).
Tucker, W. C., Weber, T. & Chapman, E. R. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304, 435–438 (2004).
Bai, H. et al. Different states of synaptotagmin regulate evoked versus spontaneous release. Nat. Commun. 7, 10971 (2016).
Lorent, J. H. et al. Plasma membranes are asymmetric in lipid unsaturation, packing and protein shape. Nat. Chem. Biol. 16, 644–652 (2020).
Doktorova, M., Symons, J. L. & Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 16, 1321–1330 (2020).
Hope, M. J., Redelmeier, T. E., Wong, K. F., Rodrigueza, W. & Cullis, P. R. Phospholipid asymmetry in large unilamellar vesicles induced by transmembrane pH gradients. Biochemistry 28, 4181–4187 (1989).
Or, E., Navon, A. & Rapoport, T. Dissociation of the dimeric SecA ATPase during protein translocation across the bacterial membrane. EMBO J. 21, 4470–4479 (2002).
Drexhage, L. Z. et al. Apoptosis-mediated ADAM10 activation removes a mucin barrier promoting T cell efferocytosis. Nat. Commun. 15, 541 (2024).
Bao, H. & Duong, F. Nucleotide-free MalK drives the transition of the maltose transporter to the inward-facing conformation. J. Biol. Chem. 289, 9844–9851 (2014).
Covitz, K. M. et al. Mutations that alter the transmembrane signalling pathway in an ATP binding cassette (ABC) transporter. EMBO J. 13, 1752–1759 (1994).
Dunn, H. A., Patil, D. N., Cao, Y., Orlandi, C. & Martemyanov, K. A. Synaptic adhesion protein ELFN1 is a selective allosteric modulator of group III metabotropic glutamate receptors in trans. Proc. Natl. Acad. Sci. USA 115, 5022–5027 (2018).
White, D. S. et al. cAMP binding to closed pacemaker ion channels is non-cooperative. Nature 595, 606–610 (2021).
Ikeda, K. & Nakano, M. Self-reproduction of nanoparticles through synergistic self-assembly. Langmuir 31, 17–21 (2015).
Stroud, Z., Hall, S. C. L. & Dafforn, T. R. Purification of membrane proteins free from conventional detergents: SMA, new polymers, new opportunities and new insights. Methods 147, 106–117 (2018).
Kampjut, D., Steiner, J. & Sazanov, L. A. Cryo-EM grid optimization for membrane proteins. iScience 24, 102139 (2021).
Zhang, Z. et al. Efficient engineering of human and mouse primary cells using peptide-assisted genome editing. Nat. Biotechnol. 42, 305–315 (2023).
Foss, D. V. et al. Peptide-mediated delivery of CRISPR enzymes for the efficient editing of primary human lymphocytes. Nat. Biomed. Eng. 7, 647–660 (2023).
Gaudet, R. G. et al. A human apolipoprotein L with detergent-like activity kills intracellular pathogens. Science 373, https://doi.org/10.1126/science.abf8113 (2021).
Huang, J. et al. Identification of potent antimicrobial peptides via a machine-learning pipeline that mines the entire space of peptide sequences. Nat. Biomed. Eng. 7, 797–810 (2023).
Gold, V. A. et al. The action of cardiolipin on the bacterial translocon. Proc. Natl Acad. Sci. USA 107, 10044–10049 (2010).
Bao, H. & Duong, F. ATP Alone Triggers the Outward Facing Conformation of the Maltose ATP-binding Cassette Transporter. Proc. Natl. Acad. Sci. USA 288, 3439–3448 (2013).
Chicka, M. C. & Chapman, E. R. Concurrent binding of complexin and synaptotagmin to liposome-embedded SNARE complexes. Biochemistry 48, 657–659 (2009).
Goehring, A. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585 (2014).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
White, D. S., Goldschen-Ohm, M. P., Goldsmith, R. H. & Chanda, B. Top-down machine learning approach for high-throughput single-molecule analysis. ELife 9, e53357 (2020).
Adair, G. S. The hemoglobin system. VI. The oxygen dissociation curve of hemoglobin. J. Biol. Chem. 63, 529–545 (1925).
Bao, H., Dalal, K., Cytrynbaum, E. & Duong, F. Sequential action of MalE and Maltose allows coupling ATP hydrolysis to translocation in the MalFGK2 transporter. J. Biol. Chem. 290, 25452–25460 (2015).
Acknowledgements
Mass Spectrometry analyses were performed by the Mass Spectrometry Technology Access Center at the McDonnell Genome Institute (MTAC@MGI) at Washington University School of Medicine, supported by the Diabetes Research Center/NIH grant P30 DK020579, Institute of Clinical and Translational Sciences/NCATS CTSA award UL1 TR002345, and Siteman Cancer Center/NCI CCSG grant P30 CA091842. We thank Drs. Michael Purge and David Cooper at the University of Virginia and Drs. Naomi Kamasawa and Debby Guerrero-Given from the imaging center at the Max Planck Florida Institute for Neuroscience for assistance in negative stain EM. CryoEM data were collected at the National Center for CryoEM Access and Training (NCCAT) and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the NIH Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539), NIGMS (R24 GM154192), and by grants from the Simons Foundation (SF349247) and NY State Assembly. We thank Dr. Susovan Roy Chowdhury at WUSTL and Dr. Scott T. Retterer from Oak Ridge National Laboratory for generating ZMWs used in this study. We would also like to thank Dr. Franck Duong for the plasmid pTRC-MalFGK2, Dr. James Rothman for the plasmid pEThis-Vamp2, and Dr. Edwin Chapman for the plasmid pGEX4T-syt1-C2AB and pDuetRSF t-SNARE dimer. J.S. was partially supported by Hankuk University of Foreign Studies Research Fund. This work was supported by NIH (DP2GM140920 R21AG078699, and R35GM156801 to H.B.; EY034339 and NS128039 to K.M.; NS124758 to W.G.L.; R35NS116850 to B.C.; P01GM072694 to L.K.T). In addition, this research was conducted while H.B. was a Hevolution/AFAR New Investigator Awardee in Aging Biology and Geroscience Research.
Author information
Authors and Affiliations
Contributions
H.B. conceived the project and wrote the manuscript with input from all authors. Q.R., S.Z. (Shanwen Zhang), and H.B. performed protein purification, biochemical reconstitutions, and characterizations of NDs by gel electrophoresis, SEC and ATPase assays. J.W., Q.R., and H.B. performed single-particle cryoEM analysis of NDs. J.S. and Q.R. performed negative stain EM analysis of NDs. H.C. contributed to the purification of SecA. V.I., S.R.C., and B.C. provided cells expressing HCN1 and carried out single-molecule imaging experiments and data analysis, and lipidomic experiments and analysis. A.J.B.K., V.K., and L.K.T. provided dense core vesicles. S.Z. (Sara Zdancewicz) and A.J. performed the extraction of Sec61β from ER microsomes. W.G.L. and K.M. purified ELFN-1 proteins and provided cells expressing mGluR7. L.H. and I. L. contributed to TLC experiments.
Corresponding authors
Ethics declarations
Competing interests
Huan Bao has filed a patent application through the University of Florida related to the DeFrMSPs described in this work (International Patent Application No. PCT/US25/10215). All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jennifer Cash and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, Q., Wang, J., Idikuda, V. et al. DeFrND: detergent-free reconstitution into native nanodiscs with designer membrane scaffold peptides. Nat Commun 16, 7973 (2025). https://doi.org/10.1038/s41467-025-63275-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63275-8
