
Abstract
Reverse transcriptases (RTs) have well-established roles in the replication and spread of retroviruses and retrotransposons. However, recent evidence suggests that RTs have been conscripted by cells for diverse roles in antiviral defence. Here we determine structures of a type I-A retron, which explain how RNA, DNA, RT, HNH-nuclease and four molecules of a structure maintenance of chromosome (SMC)-family ATPase assemble into a 364 kDa complex that provides phage defence. We show that phage-encoded nucleases trigger degradation of the retron-associated DNA, leading to activation of the HNH nuclease. The HNH nuclease cleaves tRNASer, stalling protein synthesis and arresting viral replication. Taken together, these data reveal diverse and paradoxical roles for RTs in the perpetuation and elimination of genetic parasites.
Similar content being viewed by others


Introduction
Reverse Transcriptases (RTs) were originally discovered in Rous Sarcoma and murine leukemia viruses1,2. Over the next several decades, research on RTs suggested that these enzymes are primarily involved in the replication of selfish genetic elements, and occasionally domesticated for cellular functions (e.g., telomerase and spliceosome)3,4. Initially, RTs were thought to be restricted to the eukaryotic domain of life5,6,7, but in 1989, three independent groups discovered RTs in bacteria that make multicopy single-stranded DNA (msDNA) using an RNA template8,9,10. These systems were called retrons6, but the biological function of these systems remained enigmatic for almost 30 years11. Recently, these systems have been shown to provide bacteria with defence against phage infection12,13,14.
Retrons are diverse (i.e., 13 types and several subtypes), but all retrons consist of an RT with two distinguishing sequence motifs (NAXXH and VTG), and a non-coding RNA (ncRNA) containing an inverted repeat and an invariant guanosine that acts as a primer for branched DNA synthesis5,15,16. Most retrons are associated with putative effectors, that are predicted to interact with cell membranes, cleave DNA, or degrade proteins13,15,17,18. To date, the only available retron structure is that of the type II-A3 (Eco1/Ec86) system19,20,21. These structures reveal an oligomeric assembly of retrons that sequester an NAD+-hydrolyzing toxin, which is activated either by phage-induced methylation of msDNA or, in the case of the type II-A1 retron, by msDNA degradation mediated by the prophage-encoded exonuclease VIII, recE17,19,21.
With the growing appreciation for the functional diversity of prokaryotic RTs in antiphage defence13,22,23, we were particularly intrigued by a class of RTs that are uniquely associated with Structural Maintenance of Chromosomes (SMC) ATPases15. SMC ATPases are conserved across all domains of life, perform critical functions in organizing higher-order chromosome structures, and facilitate DNA repair24,25. Intriguingly, SMC ATPases play critical roles in antiviral immunity in both eukaryotic (e.g., human Smc5/6 and Rad50)26,27,28 and prokaryotic organisms (e.g., PARIS29,30,31, Wadjet32,33,34, Lamassu35,36,37, and Gabija37,38). While RTs that synthesize extrachromosomal DNA and ATPases that recognize foreign DNA are independently recognized as components of antiviral defence, their unique association in type I-A retrons—sometimes called type II Septu39—raises fundamental questions about their functional synergy.
Here we clarify the functional interplay between RTs and SMC-family ATPases in type I-A retrons. We determine three different structures, which explain how RT-generated extrachromosomal msDNA serves as the structural core of a 364 kDa complex composed of the RT, a portion of the non-coding RNA, msDNA, two ATPase homodimers, and an HNH nuclease. Upon infection, a phage encoded nuclease cleaves the msDNA, potentially triggering disassembly of the complex leading to activation of the HNH nuclease. The activated HNH selectively degrades cellular tRNASer, arresting translation and stalling phage replication. Collectively, the structures, structure-guided mutants, phage challenge assays, and biochemical experiments presented here reveal how RT-generated extrachromosomal DNA—once viewed as a hallmark of selfish elements—acts as a crucial host factor that relies on SMC ATPases for antiviral defence.
Results
The type I-A retron system protects bacteria against phages
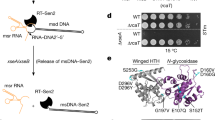
The type I-A retron system consists of a ncRNA, RT, msDNA, ATPase, and HNH nuclease (Fig. 1a, Supplementary Fig. 1a, b)15. Here we focus on a type I-A retron we identified in the FORC 82 strain of Escherichia coli. To determine if the E. coli FORC 82 retron provides phage defence, we cloned the operon along with ~550 bases of the upstream DNA, which is expected to encode the ncRNA (Fig. 1a)15. The retron-containing plasmid or an empty vector control was transformed into E. coli MG1655, and plaque assays were performed using T3, T4, T5, and T7 phages. While the retron conferred defence against T3 and T5, no immunity was observed against T4 or T7 (Fig. 1b, Supplementary Fig. 1c). Notably, the level of defense was stronger against T3 than T5, suggesting potential differences in how these phages activate or evade retron-mediated immunity.

a Schematic of the type I-A retron operon. The retron encodes an RT, ncRNA, ATPase and HNH nuclease. Both the ATPase and HNH nuclease feature C-terminal extensions. b Phage challenge assay demonstrate that the type I-A retron from E. coli protects against phages T3 and T5. The empty vector (EV) control exhibits no defence. Data shown is representative of n = 2 replicates. c A covariance model for ncRNA from type I-A retrons generated from 70 sequences. d Size exclusion chromatography (SEC) of the affinity-purified type I-A retron reveals a monodispersed peak with an estimated molecular weight of 350 kDa. The chromatogram is representative of n = 3 biological replicates. e SDS-PAGE analysis of the main SEC peak reveals proteins corresponding to the ATPase, RT, and HNH. Nucleic acids (ncRNA and msDNA) associated with the retron complex treated with either DNase or RNase and resolved using a 14% urea-PAGE. Data is representative of n = 3 biological replicates. f Reconstructed density maps of the type I-A retron. HNH is bound asymmetrically in an up or down orientation to one of the ATPase homodimers.
To assess the role of each protein component in phage immunity, we introduced active site mutations in the ATPase (D387A), HNH nuclease (H71A), or the RT (YADD to YAAA). Mutations in the ATPase or HNH active sites eliminate retron-based phage defence, while attempts to clone mutations in the RT active site (YADD to YAAA) consistently failed, suggesting that these mutants are toxic (Supplementary Fig. 1d, Supplementary Data 1). Persistent attempts to clone the RT mutant resulted in the emergence of compensatory mutations in the ATPase (Q454K) or the HNH (G44S) and the ATPase (V73A) that rendered the defence system inactive (Supplementary Fig. 1e, Supplementary Data 1). Attempts to revert the ATPase Q454K mutation yielded 13 clones, all of which carried the intended reversion (K454Q) but also contained frameshift mutations within the ATPase, indicating continued selective pressure to mitigate toxicity. These results demonstrate that all protein components of the E. coli FORC82 retron system are indispensable for defence, consistent with observations reported for retron Ec7812,13.
Given the involvement of multiple proteins, DNA, and RNA in the type I-A retron defence system, we hypothesized that the retron forms a multicomponent complex capable of recognizing phages and conferring immunity. To purify the complex, we first identified the 5’ and 3’ boundaries of the ncRNA by developing a covariation model built by aligning regions upstream of type I-A retron RTs (Fig. 1c). The model reveals a long stem-loop structure consistent with previous observations15. The predicted non-coding RNA and each of the protein-coding genes were cloned into an expression vector with affinity tags on either the C-terminus of the HNH nuclease (HNH-Strep), the N-terminus of the RT (Strep-RT), or the N-terminus of the ATPase (Strep-ATPase). Placement of the affinity tags was informed using structural predictions, which indicated that the N-terminus of the HNH nuclease would be buried, while the C-termini of both the RT and ATPase were avoided due to predicted interactions with adjacent proteins or the ncRNA. While the affinity-tagged proteins preserve retron-mediated immunity, only the tagged ATPase effectively pulls down all components of the retron (Supplementary Fig. 1f, g). The affinity-purified complex elutes from size-exclusion chromatography (SEC) as a monodispersed peak with an estimated molecular weight of 350 kDa. SDS-PAGE and mass spectrometry were used to identify all three protein components (i.e., ATPases, RT, and HNH), while denaturing urea-PAGE and nucleases assays were used to identify the ~90 nucleotide msDNA, and a ~ 40 nucleotide ncRNA (Fig. 1d, e, Supplementary Data 2). Our findings suggest that all components of the type I-A retron assemble into a multicomponent complex that is necessary for phage defence.
The purified complex was used for structure determination using cryo-EM (Fig. 1f, Supplementary Fig. 2, Supplementary Table 1). 2D classification revealed that a subset of particles contained an extra lobe of asymmetric density. This was reflected in initial 3D reconstructions which contained weak density for the additional domain. Focused 3D classification performed using a soft mask over the area of weak density produced three distinct structures with overall resolutions of 3.1 Å (Fig. 1f, Supplementary Fig. 3). Preliminary models of the complex were built by docking AlphaFold340 predicted structures into the maps. This process revealed that the RT and a portion of the ncRNA comprise the head of the assembly, while the msDNA protrudes from the RT like a harpoon that extends all the way through the center of the complex. The HNH nuclease is asymmetrically bound to one of the two ATPase homodimers, in either an up or down configuration. Comparison between the HNH-up, HNH-down, and no-HNH structures revealed minimal structural differences between the shared subunits. The RMSD of each of the shared subunits in each of the three structures was less than 0.7 Å. Notably, density for the C-terminal region of the ATPase—which interacts with the HNH subunit—was only visible in the HNH-bound structures.
Extrachromosomal DNA synthesized by RT is a scaffold for the SMC ATPase
To understand how reverse transcription leads to formation of the retron, we compared an AlphaFold3-predicted structure of the ncRNA-RT complex to the experimentally determined structure of the retron complex (Fig. 2a, b). In contrast to the experimentally determined structure (i.e., post-assembly), the predicted structure is likely to represent an early intermediate. In both the predicted and experimentally determined structures, stem-loop 1 (SL-1) forms a molecular handle that interacts with the thumb domain of the RT and likely remains anchored to the RT41,42, while the RNA template is reeled through the active site during reverse transcription (Fig. 2a–d, Supplementary Fig. 4a, c).

a The ncRNA sequence of retron I-A from E. coli is mapped onto the covariance model shown in Fig. 1c. Conserved stem-loop motifs (SL-1, SL-2, and SL-3), the clamp (CL) and termination sequence (TS) are highlighted. b AlphaFold3-predicted structure of the ncRNA-RT complex highlight features of the ncRNA identified in (a). c The ncRNA-RT structure prediction reveals that the RT thumb domain is trapped between SL-1 and SL-3, while SL-3 is positioned between the thumb and palm domains of the RT. The magnified region highlights the conserved VTG motif, which is predicted to make contacts with the DNA backbone, positioning the priming guanosine (G8, yellow) and adenosine 7 (A7, purple) near the active site (YADD, pink). d Like the predicted structure of ncRNA-RT prior to msDNA synthesis (panel c), the experimentally determined structure of retron I-A reveals interactions between the RT thumb domain and SL-1. The duplex formed by the TS and the 3′ end of the msDNA is positioned between the thumb and palm domains, occupying the site previously occupied by SL-3 in panel c. The magnified region highlights the 3′ end of the cDNA contacting the YADD active site. e The IS1 flexible loop of the SMC ATPase contains a series of basic residues that interact with the msDNA ‘harpoon’. f The RT makes extensive interaction with one of ATPases (cyan). g–i Coiled-coil domains in IS2 of the ATPase form distinct structural features on either side of the complex. g On one side, the coiled-coil domains from opposing ATPases bear hug the DNA harpoon. i On the opposite side, the IS2 coiled-coils form dorsal fin-like feature.
In the predicted ncRNA-RT structure, the 5′ and 3′ ends of the ncRNA form a conserved stem-loop (SL-3) that is positioned within a cleft of the RT formed by the thumb and palm domains (Fig. 2a–c)43. The prediction places the branching guanosine of the ncRNA, which serves as the primer for reverse transcription, near the VTG motif and the YADD active site (Fig. 2c)15,16. Reeling of the RNA template into the RT active site is anticipated to require unzipping the predicted stem of the ncRNA, and displacement of SL-3.
The clamp (CL) is a highly conserved feature of the ncRNA (Fig. 2a). This sequence is predicted to interact with the palm domain of the RT, prior to reverse transcription, while in the experimentally determined structure, the clamp is a linchpin that simultaneous contacts the 3′-end of the cDNA, the ATPase, and key domains of the RT, including the finger, palm, and VTG motif (Supplementary Fig. 4d, e). Collectively, these structural comparisons suggest that during reverse transcription, the 3′ end of the ncRNA is reeled through the active site and into the cleft formed by the thumb and palm domain (initially occupied by SL-3) while the SL-1 handle remains bound by the RT. Reeling of the template positions the 3′ end of the msDNA and the ncRNA termination sequence (TS) in close proximity, forming an RNA-DNA hybrid at the site previously occupied by SL-3 (Supplementary Fig. 4d, e).
The ATPase homodimers form an extensive network of electrostatic interactions with the msDNA (Fig. 2e, Supplementary Fig. 5a). Notably, a cluster of basic residues within insertion sequence 1 (IS1)—a conserved feature of SMC ATPases—is oriented towards the msDNA-binding cavity (Fig. 2e)25. However, IS1 is dynamic and density for IS1 is weak (Movie 1). While single-point mutants (K100A, K103A, K109A, K141A, N151A, N152A, K196A, or K201A) at residues involved in msDNA binding had no detectable impact on phage defense (Supplementary Fig. 5b), a single construct with multiple charge-swap mutations in IS1 were unclonable (R195E, K196E, R197E, K198E, K201E, and K203E), suggesting toxicity (Fig. 2e). Similarly, structure-guided mutations that disrupt the interface between the RT and ATPase (RT residues: S217R, N219R and E220R) were also unclonable (Fig. 2f). In contrast, these mutants were successfully cloned in constructs with limited retron expression. Together, these findings underscore the critical role of msDNA and RT in orchestrating ATPase assembly, which is essential for the functional integrity of the retron.
SMC ATPases typically dimerize in the presence of ATP and play a central role in forming chromosomal structures that mediate DNA repair24,25. Insertion sequence 2 (IS2) is a conserved feature of SMC ATPases that forms long coiled-coil domains, which wrap around genomic DNA25,44. In retrons, the ATPase dimers are offset by ~75° relative to each other and coiled-coils from two adjacent ATPases encircle the extrachromosomal msDNA in an ATPase-mediated bear hug on one side of the complex and a dorsal fin shaped structure on the other (Fig. 2g–i). While the IS2-mediated bear hug obscures access to the msDNA, 3D variability analysis reveals a subset of particles with an alternate conformation, where the IS2 arms are disordered and the msDNA is exposed (Movie 1). Collectively, this reveals a conserved role for IS2 in binding chromosomal DNA during repair or msDNA during retron assembly.
ATPase homodimers form a claw that grasps the HNH nuclease
The HNH nuclease is anchored to the retron complex via the C-terminal extension of the ATPase, which forms a triple helix (Figs. 1a, 3a, b, c, Supplementary Fig. 1b). The C-terminal triple helix from adjacent subunits form a claw-like structure that grips the C-terminal region of the HNH nuclease and structure guided mutations in ATPase (D522R, L523R, V526R, F529A, and K530D) result in a complete loss of defence (Fig. 3a, b, d). The nuclease is asymmetrically recruited to one side of the complex (i.e., one pair of C-terminal extensions) in either an up or down orientation (Fig. 1f). Exclusion of the HNH from the opposing homodimer is partially explained by steric hindrance from the RT. In addition, three of the four ATPase nucleotide-binding domains (NBDs) include clear density for ATP, while the fourth NBD (NBD4) is reordered in a way that is incompatible with ATP binding (Supplementary Fig. 6c, d). This arrangement and exclusion of ATP from this NBD is consistent between the HNH-bound and HNH-free structures, demonstrating that the unique pose of NBD4 occurs during assembly rather than during HNH binding. Structural comparison with the type-I Septu ATPase (PDB: 8EEA) reveals a similar pattern of ATP exclusion, suggesting a conserved function in complex assembly, as opposed to HNH binding (Supplementary Fig. 6e)45.

a Reconstructed density for the C-terminal ATPase domain interacting with the HNH nuclease with ATPases shown in blues and HNH shown in orange. b Key residues that mediate interactions between the ATPase and nuclease are highlighted. c Q454 of the ATPase maintains structural integrity of the claw by stabilizing helices from the two oppositely oriented ATPase monomers. d Phage challenge assay performed using a C-terminal mutant of the ATPase (D522R, F529A, K530E, L523R, V526R) or ATPase mutant Q454K. Data shown is representative of n = 2 replicates.
While mutations in the RT active site result in toxicity, a spontaneous mutation in the ATPase (Q454K) renders the RT active site mutants non-toxic. Q454 is conserved across type I-A retron ATPases, and the structure explains a role for this residue in stabilizing the claw-like structure (Fig. 3c). Although Q454 does not directly interact with the HNH nuclease, the Q to K mutation leads to a complete loss of defence, highlighting the importance of the claw-like structure in recruitment of the HNH (Fig. 3d).
The msDNA is a phage sensor
Building on our structural understanding, we hypothesized that retron activation is triggered by specific components of the infecting phage. To test this hypothesis, we screened for phage mutants that escape retron-mediated defence. Cells expressing the retron defence system were infected with phages T3 and T5. Three T3 escape mutants were isolated, but no escapers were recovered for T5 despite repeated attempts (Supplementary Fig. 7a). Whole-genome sequencing of the T3 escape mutants revealed mutations in five phage genes (i.e., serine/threonine kinase, an exonuclease, a small hypothetical protein, a tail protein, and a tail fiber), and the insertion of three guanosines between two genes (YP_009792963.1 and YP_009792964.1) (Fig. 4a). While serine/threonine kinase was toxic by itself, phage tail fiber, phage tail protein, and the exonuclease were expressed either alone or in retron-containing cells. Co-expression of the exonuclease with the retron severely impaired bacterial growth, whereas no growth defect was observed when the exonuclease was expressed alone or with a retron containing an inactive HNH nuclease (H71A) (Fig. 4b, c). These results indicate that T3 exonuclease triggers retron-mediated immunity.

a Genome sequencing of three T3 escape mutants reveals the locations of mutations (red lines). Missense or frameshift mutations observed in serine/threonine kinase (YP_009792931.1), exonuclease (YP_009792953.1), tail protein (YP_009792962.1) and tail fiber protein (YP_009792968.1) are indicated. Arrow indicates frameshift mutation in a small hypothetical protein (YP_009792955.1) and the asterisk marks intergenic mutation between genes YP_009792963.1 and YP_009792964.1. b Dilution series of E. coli cells shows the effects of expressing phage genes alone or in combination with the retron. c Dilution series of E. coli cells expressing retron mutants reveals that mutations in the HNH nuclease (control) or the phage exonuclease, both abolish cell toxicity. d Urea-PAGE analysis confirms the presence or absence of msDNA in samples co-expressing WT or mutant exonuclease with the retron. e Dilution series of E. coli cells expressing exonuclease from phages T7, T4, or T5, with WT or mutant (H71A) retron. Data shown in panel (b, c, e) are representative of n = 2 biological replicates.
An AlphaFold3 predicted structure of the T3 exonuclease reveals that one of the escaper mutations (W48R) is on a β-hairpin ~34 Å from the exonuclease active site (Supplementary Fig. 7d). Co-expression of the T3 W48R exonuclease mutant with the retron did not result in cell growth arrest, suggesting that retron activation is dependent on specific structural features beyond the active site (Fig. 4c). Mapping the remaining escaper mutations on the exonuclease structure revealed that none of the mutations occurred at the active site (Supplementary Fig. 7d). This suggests that the exonuclease activity is not required for retron activation, or this activity is essential for phage replication and mutations that disrupt its function are not tolerated. To determine whether the exonuclease directly degrades msDNA, acts indirectly through other cellular nucleases, or has no impact on msDNA integrity, we mutated the exonuclease active site (D162K) and co-expressed the mutant or the wild-type (WT) exonuclease in retron containing cells. Co-expression of the T3 exonuclease active site mutant (D162K) or T3 phage escaper mutant (W48R) with retron results in no growth arrest and no degradation of the msDNA, while the WT exonuclease results in both growth arrest and msDNA degradation (Fig. 4c, d, Supplementary Fig. 7b, c). Collectively, these data demonstrates that the T3 exonuclease triggers retron activation, that residues outside of the active site are necessary for retron-mediated recognition of the trigger, and that the nuclease active site is necessary for degrading the msDNA.
To better understand the mechanism of retron activation by the T3 exonuclease, we identified and compared related nucleases using a combination of AlphaFold340 and FoldSeek46 (Supplementary Fig. 7d, e). T3 exonuclease is structurally similar to the T4, T5, and T7 flap nucleases that have been proposed to remove Okazaki fragments (Supplementary Fig. 7e)47,48,49,50. The T3, T5 (D15), and T7 nucleases trigger growth arrest in retron-expressing cells, whereas the T4-encoded nuclease had no detectable effect on cell growth (Fig. 4c, e). Consistent with this observation, expression of nucleases from T3, T5, and T7 led to msDNA cleavage, but the nuclease from T4 fails to cleave msDNA (Supplementary Fig. 7f). While growth inhibition by T3 and T5 nucleases in retron-expressing cells aligns with the observed immunity against these phages, T7 phage evades retron-mediated defence, despite its nuclease triggering degradation of the msDNA. This suggests that msDNA cleavage occurs at reduced levels or slower rates during a T7 infection as compared to the overexpression system, or that T7 encodes an anti-retron mechanism that occurs downstream of msDNA degradation. While more work remains to be done regarding the mechanism of retron evasion by T7, our results suggest that retron-mediated protection generally correlates with nuclease-mediated cleavage of msDNA.
Retron activation triggers translation arrest
The type I Septu defence system, composed of an ATPase and an HNH nuclease, degrades viral DNA during infection45. Growth arrest following type I-A retron activation, along with structural similarity between the retron and Septu HNH nucleases, led us to hypothesize that the retron would rely on a similar mechanism (Supplementary Fig. 6e). To test this hypothesis, we repeated experiments done for Septu by inducing E. coli cells expressing either the WT or an HNH-dead mutant (H71A) retron along with the T3 exonuclease and analyzed the cellular DNA using fluorescence microscopy (Fig. 5a). Rather than DNA degradation, we observed nucleoid compaction and propidium iodide exclusion (Fig. 5a, Supplementary Fig. 8). In bacteria, transcription, translation, and membrane insertion of proteins are tightly coupled through a process known as transertion, and disruption of translation has been shown to induce DNA compaction51,52,53. Collectively, these data suggest that retron-mediated growth arrest is due to translational inhibition.

a Fluorescence microscopy images for WT or HNH-inactive (H71A) retron in the presence of T3 exonuclease. Cell membrane is labeled with WGR Oregon (violet). DNA is labeled with DAPI (blue). Arrows indicate cells with DNA compacted away from the cell membrane. Images representative of n = 2 independent biological replicates. b Volcano plot comparing tRNA abundance between cells expressing WT retron and T3 exonuclease versus those expressing an HNH-inactive retron and exonuclease. The p value is calculated using a two-sided negative binomial test. Data shown is representative of n = 3 independent biological replicates. c WT T5 sequence with the deleted region in T5MSU highlighted in cyan. The location of the phage tRNASer are indicated. d Proposed model for antiphage defence by the ATPase-associated RT. The RT reverse-transcribes a ncRNA into an extrachromosomal DNA that recruits SMC ATPases. The HNH toxin is recruited. Upon infection, phage flap nuclease degrades the msDNA, potentially releasing the HNH, which depletes E. coli tRNASer. The flap nuclease from T7 degrades msDNA, but T7 is resistant to type I-A retron mediated defense, suggesting a virally encoded anti-retron.
During the course of this work, Azam et al. reported that expression of a related ATPase and HNH nuclease isolated from the type I-A retron (Ec78) from strain 102598 of E. coli, led to the degradation of bacterial tRNATyr (Supplementary Fig. 1)54. Additionally, they showed that T5 phage encodes a tRNATyr resistant to cleavage by the retron system. Inspired by these findings and results from our microscopy, we performed tRNA sequencing of cells expressing the T3 exonuclease with either the WT retron or HNH-inactive (H71A) control. This analysis reveals a significant depletion of all four serine tRNAs encoded in E. coli MG1655 (Fig. 5b, Supplementary Fig. 9a, b). AlphaFold3 prediction of two HNH nucleases with tRNASer-GGA yielded a low-confidence model of an HNH dimer, in which dimerization is mediated by the C-terminal extension that otherwise facilitates interaction with the ATPase (Fig. 1a, Supplementary Fig. 9c, d).
The T5 phage genome also includes tRNAs for serine, which would be anticipated to complement tRNAs depleted by the activated retron. To verify the T5 genome, we sequenced and compared the T5 strain we were using to the WT T5 genome in NCBI (T5WT). The T5 strain used in these experiments (designated T5MSU) includes a 8.6 kb deletion within the tRNA-rich genomic region (Fig. 5c)55. This deletion eliminates 10 of the 25 tRNAs including a tRNASer containing a GCT anticodon (tRNASer-GCT), which decodes a highly represented codon in both the T5 and E. coli genomes (Supplementary Fig. 9e). The deletion of T5 encoded tRNAs may occur spontaneously during serial passage of the virus in the absence of selective pressures (e.g., immune systems). To investigate whether phage-supplemented tRNAs contribute to evasion of retron immunity, we cloned and expressed tRNASer-GCT in cells infected with T5MSU, which is missing tRNASer-GCT (Supplementary Fig. 10a, b). Surprisingly, supplementation did not confer detectable evasion of retron-mediated phage defense. Northern blot analysis of total RNA from cells co-expressing T5 tRNASer-GCT, the wild-type retron, and the T3 exonuclease revealed a distinct cleavage band for both E. coli and T5 tRNASer-GCT (Supplementary Fig. 10c). In contrast, this cleavage band was absent in cells expressing the HNH-inactive retron. We next asked whether the previously identified retron inhibitor gene (ORF75), in combination with phage-encoded tRNAs, is necessary for phage evasion of the retron54. To address this, we obtained a T5 phage isolate from ATCC (T5ATCC) and performed whole-genome sequencing (Supplementary Fig. 10a, b). The T5ATCC genome includes the full complement of phage tRNAs (including tRNASer) and ORF75. However, phage challenge assays revealed that T5ATCC does not evade FORC 82 retron immunity. Thus, in contrast to retron Ec78 described by Azam et al.54, neither T5-encoded tRNAs nor ORF75 contribute to evasion of FORC 82 retron immunity. Instead, our findings demonstrate that the FORC 82 retron can cleave host and phage-encoded tRNASer.
Finally, we investigated how viral tRNAs differ from E. coli tRNAs that enable retron-mediated elimination by retron Ec78 yet remain ineffective against retron FORC-82. We aligned E. coli tRNATyr and tRNASer sequences with their counterparts encoded in the WT T5 genome (Supplementary Fig. 11). Notably, most differences between the phage and E. coli tRNAs cluster in or around the D-loop, a region recognized by aminoacyl-tRNA synthetases56. To determine if D-loop variation in tRNAs is a more general theme, we aligned E. coli tRNAs to each of the corresponding tRNAs in T5. All 23 tRNAs in the T5 genome contain D-loop mutations relative to analogous isoaccepters in E. coli. However, mutations were not restricted to the D-loop, which is consistent with a wider spectrum of nucleases that target tRNA degradation and modification29,31,57,58.
Discussion
The competing selfish interest of viruses and their hosts results in a biological tit-for-tat that necessitates perpetual innovation to avoid extinction59,60,61. As part of this process, genes with one function are often conscripted, repurposed, or swapped between the competing interests in ways that (temporarily) enhance fitness62,63. Here we determine structures, perform structure-guided mutations, phage challenge assays, and biochemical experiments that collectively explain how bacteria and archaea use RTs to generate extrachromosomal DNA, that serves as a scaffold to recruit SMC ATPases and HNH nucleases for antiviral defence. The experimentally determined structure and structure predictions add new insight to the process of reverse transcription, and explain how the RT links the ncRNA and msDNA to a pair of SMC ATPase homodimers (Fig. 5d). Insertion sequences 1 and 2 (IS1 and IS2), both characteristic features of SMC ATPases, play distinct structural roles: IS1 interacts with the msDNA, while IS2 stabilizes the ATPase homodimer through bear hug and dorsal fin features. Further, the C-terminal extensions from two opposing ATPase subunits form a claw-like structure that engages the HNH nuclease, whereas the corresponding regions in the other two subunits are disordered (Fig. 1). The mechanism by which only a single HNH is recruited per retron, as well as the functional implications of the up, down, or no HNH configurations, remains unclear.
The C-terminal claw of the ATPase and the HNH are structurally similar to that previously reported for Septu (RMSD = 1.1 Å across 56 atom pairs, and 2.7 Å across all 84 pairs) (Supplementary Fig. 6a,b)45. However, Septu systems do not involve RTs, the HNH is reported to be a DNase rather than an RNase, and the viral activator is unknown45. The HNH structure reported here is nearly identical to the HNH from Septu (RMSD = 1.1 Å across 101 pruned atom pairs), so it remains unclear how subtle structural differences result in distinct biological outcomes. Nevertheless, the ability of retron-associated HNHs to cleave tRNAs makes them unique among HNH family nucleases that are frequently associated with DNA cleavage64.
Phage-encoded nucleases trigger msDNA degradation, but the mechanism of degradation remains unclear. According to the structure, the msDNA and/or ncRNA are vulnerable to nuclease activity at the head (SL1, SL2 and/or SL3), the body (shaft of the msDNA), or the poorly ordered tip of the msDNA, which includes 12 unmodeled bases that extend beyond the ATPases. Initially we suspected the tip, because it is most exposed and because the covariance model predicts high variability in this region, which could reflect a mechanism for identifying different phage triggers. However, truncation of the loop had no impact on defence (Supplementary Fig. 7g), suggesting that the tip of the harpoon may not be directly involved in trigger detection. Multiple efforts to use AlphaFold3 to predict a binding site for the trigger never resulted in a high confidence prediction, but most of the models positioned the T3 exonuclease near the head of the complex. Verification of these models will require further structural studies.
The ability of T7 phage to infect cells expressing the FORC 82 retron is intriguing, given that its flap nuclease is structurally similar (RMSD = 0.455 Å across 300 pruned atom pairs) and shares 92% amino acid identity with the T3 nuclease. While overexpression of either nuclease results in msDNA degradation and toxicity in retron-expressing cells, only T3 phage is neutralized by the immune system, suggesting that T7 may encode an anti-retron gene (Fig. 5d). One candidate is the T7 serine/threonine kinase, which was recently shown to phosphorylate and inhibit the toxin RcaT in the type II retron-Eco965. While it is currently unknown whether T7 kinase could inhibit nucleic acid targeting components of retron I-A, T7 kinase shares 46% amino acid identity with a serine/threonine kinase which showed unique mutations in the T3 escaper screen (Fig. 4a). Notably, a missense mutation in the T3 escaper kinase deletes the predicted DNA-binding C-terminal domain, although whether this mutation confers gain-of-function activity in phosphorylating retron components remains unclear. Further investigation is required to determine whether kinase-mediated phosphorylation helps T7 evade immunity by retron type I-A.
Recently, Azam et al. reported that co-expressing the ATPase and HNH nuclease from a type I-A retron in E. coli (Ec78) depletes cellular tRNATyr54, whereas we reveal that the retron from E. coli FORC 82 reduces total serine tRNA levels by half. This divergence in tRNA specificity suggests that different retron systems have evolved distinct substrate preferences. This model parallels what we and others observed for the TOPRIM nuclease in the PARIS defence system, where distinct tRNAs are cleaved by TOPRIM nuclease from related PARIS systems29,58. However, unlike the Ec78 retron and PARIS, in which virally encoded tRNAs can antagonize immune activity, the tRNASer from T5 does not appear to evade cleavage by the FORC 82 retron. This suggests that bacteria continually evolve HNH variants to counteract tRNA-encoding phages, or that retron-mediated tRNA-cleavage is only the first step of a more complex pathway leading to growth arrest. Together, our findings underscore the paradoxical role of RTs in both viral propagation and antiviral defense, highlight the functional plasticity of SMC ATPases across processes like DNA repair and immunity, and expand our understanding of how tRNAs—either resistant or susceptible to HNH cleavage—are shaped by the selective pressures of genetic conflict.
Method
Statistics & reproducibility
Phage challenge assays and bacterial growth assays were performed in two or three independent biological replicates, as shown in the main and Supplementary Fig. panels and detailed in the source data file. Replicate data and uncropped images of gels and plates are provided in the source data file. Gel images showing msDNA cleavage was performed in two or three independent biological replicates (Fig. 4d, Supplementary Fig. 7f; see Source Data File). Fluorescence microscopy was performed in two independent biological replicates and is presented in Fig. 5a, Supplementary Fig. 8, and the source data file. Differential tRNA abundance was calculated using DESeq2, which employs a two-sided negative binomial model to estimate log2 fold changes and to determine statistical significance (p-values).
Plasmid construction
Phage challenge, toxicity assays, tRNA sequencing and msDNA cleavage analysis were performed in E. coli MG1655, with the WT retron operon from E. coli FORC 82 cloned into a pACYCDuet-1 backbone with native promoters. Overexpression and large-scale protein pull-down experiments were performed using T7 promoters in a pRSF backbone, with an N-terminal Strep-tag on the ATPase. The phage triggers were cloned into pCDFDuet-1. Protein expression and pull-down assays were conducted in E. coli BL21 AI cells. The retron operon from E. coli FORC 82 was synthesized as a gene fragment by GenScript. The triggers were amplified and cloned from respective phages.
All primers used for cloning were obtained from Eurofins Genomics. Cloning was performed in either DH5α or NEB Turbo competent E. coli cells, and plasmids were extracted using the Zymo ZR Plasmid Miniprep Kit. The sequence of each cloned construct was verified by whole plasmid sequencing using Oxford Nanopore (Plasmidsaurus).
E. coli cultures were grown on LB agar plates or in LB broth. Constructs with the pACYCDuet-1 backbone were maintained with 25 μg/mL chloramphenicol, pRSF constructs with 50 μg/mL kanamycin, and pCDFDuet-1 constructs with 100 μg/mL Streptomycin unless otherwise specified. All plasmids used in this study are provided in Supplementary Data 1.
Phage challenge assay
Plaque assays were performed using high phage titers (108 PFU/mL) of T3, T4, T5, and T7. E. coli MG1655 cells transformed with plasmids for the WT retron, empty vector (pACYCDuet-1), or mutant retrons were grown to an OD600 of 0.3–0.4 in LB media supplemented with chloramphenicol (34 µg/mL). The cells were added to 0.6% agarose in LB (top layer) and the suspension was poured onto plates containing 3.2% LB agar (bottom agar). Both the top layer and the bottom agar were supplemented with chloramphenicol and 2 mM CaCl2. Once the top layer solidified, 2.5 µL of 10-fold serial dilutions of phage stocks were spotted on the lawn of E. coli cells. The plates were then incubated overnight at 37 °C.
Although wild-type T3 is unable to infect E. coli K-12 strains as it requires a truncated R1-type LPS core66,67, we consistently observe infection of MG1655 using a strain of T3 with mutations in the tail fiber gene. MG1655 was acquired from E. coli Genetic Resource Center (https://ecgrc.net). While the precise mechanism of infection remains unclear, these observations likely reflect strain-specific differences in T3 or MG1655.
Covariance model and sequence comparison for ncRNA and tRNA
Retron type I-A containing genomes15 were downloaded from NCBI (n = 70). 1 kb upstream of each RTs was extracted using a custom Python script and aligned using MAFFTv7.52068 with the following parameters --maxiterate 1000 --localpair. Poorly aligned regions at both ends of the alignment were trimmed, and a well-aligned ~250 bp region corresponding to the conserved ncRNA element was extracted. This region was then realigned with mLocARNA v2.0.069 using --stockholm --concensus-structure alifold. The resulting alignment was used to generate a covariance model that was visualized with R2R v1.0.770.
E. coli MG1655 and WT T5 genomes were downloaded from NCBI database. tRNAscan-SE web server71 was used to predict T5 tRNAs. mLocARNA v2.0.069 was used to align tRNA sequences to generate sequence alignments and covariance model.
Recombinant expression and purification of the retron complex
The N-terminally Strep-tagged ATPase along with the other type I-A retron components, were co-expressed using an inducible T7 promoter in E. coli BL21 AI cells. Cultures were grown in LB media containing 50 μg/mL kanamycin at 37 °C until OD600 ~ 0.5, then induced with 0.2 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 0.1% arabinose and grown for 16 h at 22 °C. Cells were harvested by centrifugation at 3000 × g for 10 min at 4 °C, then lysed by sonication in lysis buffer (20 mM Tris pH 7.5, 200 mM KCl, 2 mM MgCl2, 1 mM DTT). The lysate was clarified by centrifuging at 10,000 × g for 25 min, and the supernatant was passed through a 5 mL StrepTactin column (IBA Lifescence). After washing with the 20-column volumes of the lysis buffer, bound proteins were eluted using lysis buffer supplemented with 2.5 mM desthiobiotin. The retron was further purified using a Superdex 200 10/300 column equilibrated with 20 mM Tris pH 7.5, 200 mM KCl, 2 mM MgCl2, 1 mM DTT, 5% glycerol. Fractions containing the purified retron components were analyzed by 12% SDS-PAGE to resolve protein components, while 15% urea-PAGE was used to visualize ncRNA and msDNA. The fraction containing the retron complex was further concentrated and used for vitrification.
SDS-PAGE bands corresponding to each protein (n = 1) were sent to the IDeA National Resource for Quantitative Proteomics for mass spectrometry analysis. Mass spectrometry analysis for bands corresponding to ATPase, RT and HNH are shown in Supplementary Data 2.
Cryo-EM sample preparation
C-flat copper grids with a mesh size of 300 and R1.2/1.3 hole spacing were glow discharged using a Pelco EsiGlow at 15 mA for 45 seconds. 3 µL of 4 µM retron was applied to prepared grids in a Vitrobot Mk IV (ThermoFischer) set to 100% humidity and 4 °C. Grids were subjected to double-sided blotting with a force of 5 for 5 s before plunge-freezing in liquid ethane. Clipped grids were loaded into autogrid boxes and stored in liquid nitrogen before imaging.
Cryo-EM data collection
Data were collected on a Talos Arctica (Thermo Fischer) equipped with a Gatan K3 direct electron detector at Montana State University’s Cryo-EM core facility using SerialEM72 under the control of SmartScope73 for automated data collection. 15,594 micrographs were collected with a pixel size of 0.9061 Å and apparent magnification of 45,000X. The total dose per exposure was calculated as 56.69 electrons with 37 subframes per exposure (1.53el/px/frame).
Cryo-EM data processing
After filtering micrographs by CTF-fit <8 Å resolution and total motion <100 px, 13,695 exposures were subjected to blob picking in cryoSPARC74 Live (60–200 Å diameter). 1,576,065 blobs were extracted using a box size of 512px (binx2) and sorted by 2D classification to yield a stack of 358,381 selected particles. From these particles, a de novo template volume was generated which contained 245,457 particles and refined to 3.7 Å. Using this volume as a template for particle picking, 7,373,097 particles were extracted with a box size of 512 px (binx4) and subjected to 2D classification with a 200 Å mask. 1,321,836 particles were selected and fed into a 3-class ab initio reconstruction. One class was identified containing 1,244,961 particles that appeared to correspond to the retron complex but exhibited compositional heterogeneity in regions of density corresponding to the HNH. Using a soft-padded mask directed towards the HNH, focused 3D classification yielded three distinct particle classes that varied based on the orientation (up or down), or absence of the HNH subunit. Un-binned particles were then re-extracted with a box size of 480px for non-uniform refinement. After reference-based motion correction, we carried out a final round of 2D classification to remove remaining junk. Final reconstructions were obtained from non-uniform refinements with several options enabled including minimizing over per particle scale, correcting for per-particle defocus and estimating the exposure group parameters Cs, Anisotropic Magnification, and tetrafoil. Phenix’s75 half-map based sharpening was used to aid in model building.
Model building
An initial model was established by docking AlphaFold3 predicted structures into the density40. After rigid body fitting the individual chains using the fit in map command in ChimeraX74, each chain was equilibrated in ISOLDE76 to improve the initial model. For each density map, the corresponding maps and models were provided to Phenix75 for real-space refinement. Per residue CC scores were used to guide the trimming of flexible loops and regions of poor density. Sidechains not visible in the map were removed. Maps were sharpened using Phenix’s implementation of anisotropic half-map sharpening to aid in modeling difficult regions. Additionally, 3DVA volumes were used to inform the position of the ATPase coiled-coil domains77. While these features were not included in the mask during the final iterations of high-resolution refinement, they are clearly visible in lower-resolution refinements of the same particle stacks used to generate the maps submitted to EMDB (Supplementary Table 1). As such, we opted to include the coiled coil domain in the PDBs, which helps to explain how access to the msDNA is gated by ATPase subunits, as well as highlight the role of the coiled coil in the tetrameric assembly of ATPases.
Phage escaper screening and genome sequencing
Phage escaper assay was performed as described in Avigail et al.18. Briefly, E. coli MG1655 cells with or without the retron were grown in LB media at 37 °C to mid-log phase (OD600 ~ 0.4) in the presence of 25 µg/mL chloramphenicol and 2 mM CaCl2. In four separate tubes having 2 mL of LB and 2 mM CaCl2, bacterial cultures were added to obtain the following percentages of defence-containing cells: 0%, 1%, 10%, or 100%. Cultures were inoculated with 20 µL of T3 or T5 phage stock and incubated overnight at 37 °C with shaking. The tubes were centrifuged at 5000 × g for 10 min, and 20 µL of the clarified supernatants were transferred into fresh cultures prepared under the same conditions. This passaging step was repeated for four consecutive rounds. Supernatants from the fourth passage were filter-sterilized (0.22 µm), serially diluted, and plated for plaque assays on defence-containing cells. Phage preparations that formed plaques were re-tested, and three individual plaques were picked for T3. No escapers could be isolated for T5. Each T3 plaque was used to inoculate 2 mL of mid-log defence-containing cultures (25 µg/mL chloramphenicol), which were grown overnight at 37 °C with shaking and then centrifuged at 5000 × g for 10 min at 4 °C. The resulting supernatants were filter-sterilized (0.22 µm) and centrifuged at 113,000 × g for 2 h at 4 °C. Pellets were gently resuspended in 300 µL of SM buffer, and final phage stocks were stored at 4 °C.
Phage genomic DNA was extracted following previously described protocols78. Isolated phage stocks were incubated with DNase I and RNase A for 1 h to eliminate contaminating E. coli DNA. The nucleases were then inactivated by adding EDTA and heating at 65 °C. Phage capsids were digested with proteinase K in the presence of SDS, and the resulting phage genomic DNA was purified by phenol–chloroform extraction. The purified DNA was sequenced using the Native Barcoding Kit V14 (Oxford Nanopore Technologies) on a GridION sequencer. Raw Nanopore reads were concatenated and assembled de novo using the Flye package79, and the assembled genome was compared to the T3 sequence used in the lab.
Bacterial growth assay
Chemically competent E. coli cells containing either WT or HNH-inactive retron (pACYCDuet-1 backbone) was transformed with WT or mutant exonuclease (pCDFDuet-1 backbone) and plated on LB agar having 2% glucose and grown at 37 °C in the presence of appropriate antibiotics (chloramphenicol and streptomycin). Individual colonies were grown overnight in the presence of 2% glucose and antibiotics and 10-fold serial dilution of the liquid culture was spotted in LB agar plates containing either 2% glucose or 0.2 mM IPTG in the presence of antibiotics. The plates were incubated for 16 h at 37 °C and imaged.
msDNA cleavage analysis
Chemically competent E. coli cells containing the WT retron were transformed with WT or mutant T3 exonuclease and cultured overnight in LB media supplemented with 2% glucose and antibiotics (chloramphenicol and streptomycin). The overnight cultures were then used to inoculate a secondary culture and grown to OD600 of ~0.4 in the presence of antibiotics. IPTG was added to induce expression of the exonuclease. DNA was extracted using the Zymo ZR Plasmid Miniprep kit and DNA shorter than 500 bps were resolved by 15% urea-PAGE.
Fluorescence microscopy
Bacterial cells carrying either WT or HNH-inactive retron in the pACYCDuet-1 backbone were transformed with T3 exonuclease and grown in LB medium supplemented with the appropriate antibiotics (chloramphenicol and streptomycin) at 37 °C until reaching an OD600 of 0.5–0.6. Cultures were induced with 0.2 mM IPTG for 2 h at 37 °C with shaking. Following induction, cells were harvested by centrifugation and washed with 1× PBS. Cells were simultaneously incubated with 5 µg/mL Wheat Germ Agglutinin (WGA) Oregon Green (Invitrogen, W7024) and 10 µg/mL propidium iodide for 15 min at RT followed by two washes with 1× PBS. Cells were then incubated with 50 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) for 10 minutes in the dark at room temperature. After staining, cells were transferred to Poly-L-lysine-treated coverslips and allowed to adhere for 5 min before imaging. Imaging was performed using an inverted spinning-disc confocal microscope built by 3i. The system included a Zeis 63×1.4 NA Plan-Apochromat objective, Yokogawa CSU-W1 SORA confocal head, 2.8× magnification changer, 100 mW lasers (40% power: 405 nm, 488 nm, 561 nm), and a Hamamatsu Orca-Fusion BT camera (2x binning to create 74 nm pixels). A piezo stage was used to acquire 10 z planes/sample with a step size of 0.27 µm. Acquired images were analyzed using Fiji80. For presentation, only one z plane was used.
tRNA sequencing and analysis
Chemically competent E. coli cells containing either the WT retron or the HNH-inactive (H71A) retron were transformed with T3 exonuclease and grown overnight in the presence of 2% glucose and antibiotics (chloramphenicol and streptomycin). Secondary cultures were inoculated from the overnight cultures, grown to an OD600 of 0.4–0.5 in the presence of antibiotics, and induced with IPTG for 15 min. Total RNA, including small RNAs, was extracted using the Zymo Quick-RNA Miniprep Kit.
tRNA sequencing (MSR-seq) was performed by MesoRNA (Chicago, IL, USA) as per previously reported protocol81. Total RNA (1 µg) was subjected to deacylation by oxidation with sodium periodate (NaIO4), followed by quenching with ribose and treatment with sodium tetraborate for β-elimination. The deacylated RNA was then end-repaired using T4 polynucleotide kinase (PNK; New England Biolabs, NEB) and ligated to biotinylated capture hairpin oligos (CHO) containing sample-specific barcodes using T4 RNA Ligase I (NEB). After ligation, all barcoded samples were pooled. The pooled samples were captured on streptavidin-coated MyOne C1 Dynabeads (Thermo Fisher Scientific) via the biotinylated CHOs. Following capture, the samples were dephosphorylated with Quick CIP (Roche) and reverse-transcribed overnight using SuperScript IV Master Mix (Thermo Fisher Scientific). RNA templates were subsequently degraded with RNase H (NEB), and cDNA subjected to periodate oxidation. After quenching periodate with ribose, the single-stranded cDNAs were ligated to the second ligation oligo and then released from the beads by incubating at 95 °C. For each sample, 100 ng of purified cDNA was PCR-amplified for 12 cycles (10 s at 98 °C, 15 s at 55 °C, 20 s at 72 °C). The amplified products were purified using the DNA Clean & Concentrator kit (Zymo Research) and resolved on a 10% non-denaturing TBE polyacrylamide gel with appropriate dsDNA size markers. Bands corresponding to the final library size were gel-extracted and ethanol-precipitated. The resulting libraries were sequenced on a NovaSeq X Plus platform using a 1.5B flow cell (Illumina). Raw reads were demultiplexed and processed by MesoRNA to generate tRNA count tables. Reads were demultiplexed and trimmed in accordance with the MSR-seq library preparation protocol81. Quality control was performed with FastQC82. Reads were aligned to tRNA sequences in GtRNAdb83 using bowtie284 to generate tRNA count tables.
tRNA sequencing counts were normalized and subjected to differential expression analysis using the DESeq2 package85 in R with custom scripts. Fold changes in tRNA abundance were calculated by DESeq2, which employs a negative binomial distribution model to estimate dispersion and determine statistical significance (p-value).
Northern blot analysis
Chemically competent E. coli cells containing either the WT retron or the HNH-inactive (H71A) retron, with or without T5 tRNASer-GCT, were transformed with T3 nuclease and grown overnight in LB medium supplemented with 2% glucose and appropriate antibiotics (chloramphenicol, streptomycin, and kanamycin). Secondary cultures were inoculated from the overnight cultures, grown to an OD600 of 0.4–0.5 in the presence of antibiotics, and T3 exonuclease expression was induced with IPTG for 60 minutes. Total RNA, including small RNAs, was extracted using the Monarch Total RNA Miniprep Kit according to the manufacturer’s instructions and samples were resolved on a 10% denaturing urea-PAGE gel run at 180 V for 90 minutes. Following electrophoresis, the gel was washed with 1× TBE buffer for 5 minutes and transferred to an Amersham Hybond-N nylon membrane at 30 V for 2 hours. The membrane was then UV-crosslinked using a transilluminator at maximum power for 5 minutes. For hybridization, the membrane was pre-washed with 30 mL of pre-hybridization buffer (6× SSC containing 0.5% SDS) for 10 minutes, then incubated overnight at 45 °C with 20 nM northern probes specific for 5S rRNA (FAM-labeled), E. coli tRNASer-GCT (ID700-labeled), and T5 tRNASer-GCT (ID800-labeled) in 30 mL of pre-hybridization buffer (Supplementary Data 3). After hybridization, the membrane was washed twice with wash buffer (2× SSC, 0.1% SDS) for 10 minutes each and imaged using an Amersham Typhoon Scanner.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The sequencing data for T3 and T5 strains used in this study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under BioProject ID PRJNA1222438. This BioProject also contains the tRNA sequencing data and whole-genome sequencing data for the T3 phage escaper mutants. The EM maps of the type I-A retron complex and associated models used in this study have been deposited in Electron Microscopy Data Bank (EMDB) under accession code EMD-49053, EMD-49055 and EMD-49056. The experimentally determined structure of the retron IA complex used in this study have been deposited in Protein Databank (PDB) under accession code 9N69, 9N6B, 9N6C. The raw micrograph images used in this study have been deposited in the EMPIAR database under accession code EMPIAR-12550. The mass spectrometry proteomics data used in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository under accession code PXD061597. All plasmids data used in the study are provided in Supplementary Data 1. Sequences for recombinantly expressed proteins and mass spectrometry data used in the study are provided in Supplementary Data 2. DNA oligos used in the study are provided in Supplementary Data 3. All uncropped gel and plate images, and replicate data used in the study have been made available in the Source data are provided with this paper.
Code availability
Custom R script for tRNA sequencing analysis have been made available in GitHub (https://github.com/WiedenheftLab/2025_George_Burman_et_al) and in Zenodo (https://doi.org/10.5281/zenodo.15839760).
References
Temin, H. M. & Mizutani, S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226, 1211–1213 (1970).
Baltimore, D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature 226, 1209–1211 (1970).
Cech, T. R. The generality of self-splicing RNA: relationship to nuclear mRNA splicing. Cell 44, 207–210 (1986).
Gonzalez-Delgado, A., Mestre, M. R., Martinez-Abarca, F. & Toro, N. Prokaryotic reverse transcriptases: from retroelements to specialized defense systems. FEMS Microbiol Rev 45 https://doi.org/10.1093/femsre/fuab025 (2021).
Lim, D. & Maas, W. K. Reverse transcriptase in bacteria. Mol. Microbiol 3, 1141–1144 (1989).
Temin, H. M. Reverse transcriptases. Retrons in bacteria. Nature 339, 254–255 (1989).
Coffin, J. M. & Fan, H. The Discovery of Reverse Transcriptase. Annu Rev. Virol. 3, 29–51 (2016).
Lampson, B. C., Inouye, M. & Inouye, S. Reverse transcriptase with concomitant ribonuclease H activity in the cell-free synthesis of branched RNA-linked msDNA of Myxococcus xanthus. Cell 56, 701–707 (1989).
Lampson, B. C. et al. Reverse transcriptase in a clinical strain of Escherichia coli: production of branched RNA-linked msDNA. Science 243, 1033–1038 (1989).
Lim, D. & Maas, W. K. Reverse transcriptase-dependent synthesis of a covalently linked, branched DNA-RNA compound in E. coli B. Cell 56, 891–904 (1989).
Zimmerly, S. & Wu, L. An Unexplored Diversity of Reverse Transcriptases in Bacteria. Microbiol Spectr 3, MDNA3-0058-2014 https://doi.org/10.1128/microbiolspec.MDNA3-0058-2014 (2015).
Gao, L. et al. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084 (2020).
Millman, A. et al. Bacterial Retrons Function In Anti-Phage Defense. Cell 183, 1551–1561 e1512 (2020).
Bobonis, J. et al. Bacterial retrons encode tripartite toxin/antitoxin systems. bioRxiv, 2020.2006.2022.160168 https://doi.org/10.1101/2020.06.22.160168 (2020).
Mestre, M. R., Gonzalez-Delgado, A., Gutierrez-Rus, L. I., Martinez-Abarca, F. & Toro, N. Systematic prediction of genes functionally associated with bacterial retrons and classification of the encoded tripartite systems. Nucleic Acids Res. 48, 12632–12647 (2020).
Simon, A. J., Ellington, A. D. & Finkelstein, I. J. Retrons and their applications in genome engineering. Nucleic Acids Res 47, 11007–11019 (2019).
Bobonis, J. et al. Bacterial retrons encode phage-defending tripartite toxin-antitoxin systems. Nature 609, 144–150 (2022).
Stokar-Avihail, A. et al. Discovery of phage determinants that confer sensitivity to bacterial immune systems. Cell 186, 1863–1876 e1816 (2023).
Carabias, A. et al. Retron-Eco1 assembles NAD(+)-hydrolyzing filaments that provide immunity against bacteriophages. Mol. Cell 84, 2185–2202.e2112 (2024).
Wang, Y. et al. Cryo-EM structures of Escherichia coli Ec86 retron complexes reveal architecture and defence mechanism. Nat. Microbiol 7, 1480–1489 (2022).
Wang, Y. et al. DNA methylation activates retron Ec86 filaments for antiphage defense. Cell Rep. 43, 114857 (2024).
Tang, S. et al. De novo gene synthesis by an antiviral reverse transcriptase. Science, eadq0876 https://doi.org/10.1126/science.adq0876 (2024).
Wilkinson, M. E., Li, D., Gao, A., Macrae, R. K. & Zhang, F. Phage-triggered reverse transcription assembles a toxic repetitive gene from a noncoding RNA. Science 386, eadq3977 (2024).
Burmann, F. & Lowe, J. Structural biology of SMC complexes across the tree of life. Curr. Opin. Struct. Biol. 80, 102598 (2023).
Krishnan, A., Burroughs, A. M., Iyer, L. M. & Aravind, L. Comprehensive classification of ABC ATPases and their functional radiation in nucleoprotein dynamics and biological conflict systems. Nucleic Acids Res 48, 10045–10075 (2020).
Liu, H. W., Roisne-Hamelin, F. & Gruber, S. SMC-based immunity against extrachromosomal DNA elements. Biochem Soc. Trans. 51, 1571–1583 (2023).
Decorsiere, A. et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389 (2016).
Kondo, T. et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl Acad. Sci. USA 110, 2969–2974 (2013).
Burman, N. et al. A virally encoded tRNA neutralizes the PARIS antiviral defence system. Nature 634, 424–431 (2024).
Rousset, F. et al. Phages and their satellites encode hotspots of antiviral systems. Cell Host Microbe 30, 740–753.e745 (2022).
Deep, A., Liang, Q., Enustun, E., Pogliano, J. & Corbett, K. D. Architecture and activation mechanism of the bacterial PARIS defence system. Nature 634, 432–439 (2024).
Deep, A. et al. The SMC-family Wadjet complex protects bacteria from plasmid transformation by recognition and cleavage of closed-circular DNA. Mol. Cell 82, 4145–4159.e4147 (2022).
Pradhan, B. et al. Loop-extrusion-mediated plasmid DNA cleavage by the bacterial SMC Wadjet complex. Mol. Cell 85, 107–116 e105 (2025).
Roisne-Hamelin, F., Liu, H. W., Taschner, M., Li, Y. & Gruber, S. Structural basis for plasmid restriction by SMC JET nuclease. Mol. Cell 84, 883–896.e887 (2024).
Millman, A. et al. An expanded arsenal of immune systems that protect bacteria from phages. Cell host microbe 30, 1556–1569 e1555 (2022).
Cury, J. et al. Conservation of antiviral systems across domains of life reveals immune genes in humans. Cell Host Microbe 32, 1594–1607.e1595 (2024).
Doron, S. et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, https://doi.org/10.1126/science.aar4120 (2018).
Antine, S. P. et al. Structural basis of Gabija anti-phage defence and viral immune evasion. Nature 625, 360–365 (2024).
Payne, L. J. et al. Identification and classification of antiviral defence systems in bacteria and archaea with PADLOC reveals new system types. Nucleic Acids Res. 49, 10868–10878 (2021).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Inouye, S., Hsu, M. Y., Xu, A. & Inouye, M. Highly specific recognition of primer RNA structures for 2’-OH priming reaction by bacterial reverse transcriptases. J. Biol. Chem. 274, 31236–31244 (1999).
Shimamoto, T., Hsu, M. Y., Inouye, S. & Inouye, M. Reverse transcriptases from bacterial retrons require specific secondary structures at the 5’-end of the template for the cDNA priming reaction. J. Biol. Chem. 268, 2684–2692 (1993).
Hsu, M. Y., Inouye, S. & Inouye, M. Structural requirements of the RNA precursor for the biosynthesis of the branched RNA-linked multicopy single-stranded DNA of Myxococcus xanthus. J. Biol. Chem. 264, 6214–6219 (1989).
Liu, Y. et al. ATP-dependent DNA binding, unwinding, and resection by the Mre11/Rad50 complex. EMBO J. 35, 743–758 (2016).
Li, Y. et al. PtuA and PtuB assemble into an inflammasome-like oligomer for anti-phage defense. Nat. Struct. Mol. Biol. 31, 413–423 (2024).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246 (2024).
Devos, J. M., Tomanicek, S. J., Jones, C. E., Nossal, N. G. & Mueser, T. C. Crystal structure of bacteriophage T4 5’ nuclease in complex with a branched DNA reveals how flap endonuclease-1 family nucleases bind their substrates. J. Biol. Chem. 282, 31713–31724 (2007).
AlMalki, F. A. et al. Direct observation of DNA threading in flap endonuclease complexes. Nat. Struct. Mol. Biol. 23, 640–646 (2016).
Mitsunobu, H., Zhu, B., Lee, S. J., Tabor, S. & Richardson, C. C. Flap endonuclease activity of gene 6 exonuclease of bacteriophage T7. J. Biol. Chem. 289, 5860–5875 (2014).
Mitsunobu, H., Zhu, B., Lee, S. J., Tabor, S. & Richardson, C. C. Flap endonuclease of bacteriophage T7: Possible roles in RNA primer removal, recombination and host DNA breakdown. Bacteriophage 4, e28507 (2014).
Bakshi, S., Choi, H., Mondal, J. & Weisshaar, J. C. Time-dependent effects of transcription- and translation-halting drugs on the spatial distributions of the Escherichia coli chromosome and ribosomes. Mol. Microbiol 94, 871–887 (2014).
van Helvoort, J. M., Kool, J. & Woldringh, C. L. Chloramphenicol causes fusion of separated nucleoids in Escherichia coli K-12 cells and filaments. J. Bacteriol. 178, 4289–4293 (1996).
Zimmerman, S. B. Toroidal nucleoids in Escherichia coli exposed to chloramphenicol. J. Struct. Biol. 138, 199–206 (2002).
Azam, A. H. et al. Evasion of antiviral bacterial immunity by phage tRNAs. Nat. Commun. 15, 9586 (2024).
Wang, J. et al. Complete genome sequence of bacteriophage T5. Virology 332, 45–65 (2005).
Shimada, A., Nureki, O., Goto, M., Takahashi, S. & Yokoyama, S. Structural and mutational studies of the recognition of the arginine tRNA-specific major identity element, A20, by arginyl-tRNA synthetase. Proc. Natl Acad. Sci. USA 98, 13537–13542 (2001).
Jain, I. et al. tRNA anticodon cleavage by target-activated CRISPR-Cas13a effector. Sci. Adv. 10, eadl0164 (2024).
Belukhina, S. et al. Specificity and Mechanism of tRNA cleavage by the AriB Toprim nuclease of the PARIS bacterial immune system. bioRxiv, 2025 2002.2004.636281 https://doi.org/10.1101/2025.02.04.636281 (2025).
Koonin, E. V., Makarova, K. S., Wolf, Y. I. & Krupovic, M. Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat. Rev. Genet 21, 119–131 (2020).
Frost, L. S., Leplae, R., Summers, A. O. & Toussaint, A. Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol 3, 722–732 (2005).
Bobay, L. M., Touchon, M. & Rocha, E. P. Pervasive domestication of defective prophages by bacteria. Proc. Natl Acad. Sci. USA 111, 12127–12132 (2014).
Jangam, D., Feschotte, C. & Betran, E. Transposable Element Domestication As an Adaptation to Evolutionary Conflicts. Trends Genet.: TIG 33, 817–831 (2017).
Benler, S. & Koonin, E. V. Recruitment of Mobile Genetic Elements for Diverse Cellular Functions in Prokaryotes. Front Mol. Biosci. 9, 821197 (2022).
Wu, C. C., Lin, J. L. J. & Yuan, H. S. Structures, Mechanisms, and Functions of His-Me Finger Nucleases. Trends Biochem Sci. 45, 935–946 (2020).
Bartolec, T. et al. Pervasive phosphorylation by phage T7 kinase disarms bacterial defenses. bioRxiv, 2024.2012.2020.629319 https://doi.org/10.1101/2024.12.20.629319 (2024).
Maffei, E. et al. Systematic exploration of Escherichia coli phage-host interactions with the BASEL phage collection. PLoS Biol. 19, e3001424 (2021).
Ando, H., Lemire, S., Pires, D. P. & Lu, T. K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 1, 187–196 (2015).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Will, S., Joshi, T., Hofacker, I. L., Stadler, P. F. & Backofen, R. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA 18, 900–914 (2012).
Weinberg, Z. & Breaker, R. R. R2R-software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinforma. 12, 3 (2011).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44, W54–W57 (2016).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Bouvette, J. et al. Automated systematic evaluation of cryo-EM specimens with SmartScope. Elife 11, https://doi.org/10.7554/eLife.80047 (2022).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D. Struct. Biol. 75, 861–877 (2019).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D. Struct. Biol. 74, 519–530 (2018).
Punjani, A. & Fleet, D. J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J Struct Biol 213, 107702 (2021).
Peters, D. L., Harris, G., Davis, C. M., Dennis, J. J. & Chen, W. Bacteriophage Isolation, Purification, and Characterization Techniques Against Ubiquitous Opportunistic Pathogens. Curr. Protoc. 2, e594 (2022).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Watkins, C. P., Zhang, W., Wylder, A. C., Katanski, C. D. & Pan, T. A multiplex platform for small RNA sequencing elucidates multifaceted tRNA stress response and translational regulation. Nat. Commun. 13, 2491 (2022).
S., A. FastQC: a quality control tool for high throughput sequence data., <http://www.bioinformatics.babraham.ac.uk/projects/fastqc> (2010).
Chan, P. P. & Lowe, T. M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37, D93–97 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Pettersen, E. F. et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput Chem. 25, 1605–1612 (2004).
Acknowledgements
We thank members of the B.W. laboratory for feedback and discussions, C. Hophan-Nichols and the cyber security team at Montana State University for computational support, C.M. Lawrence and Ravi Thakkar for maintenance and operation of the cryo-EM Core Facility at Montana State University. The cryo-EM Core Facility at Montana State University is supported by NSF no. 1828765 and the M.J. Murdock Charitable Trust. We thank Sam Mackintosh from IDeA National Resource for quantitative proteomics for mass spectrometry data. tRNA sequencing was performed by MesoRNA (Chicago, IL, USA). We thank Amy Binny Philip for the valuable suggestions on the statistics for tRNA sequencing analysis. Fluorescent microscopy was possible with support from NIH grant (no. R35GM151262) to S.Z.D. B.W. is the Winifred Asbjornson Plant Sciences Endowed Chair. Research in the Wiedenheft laboratory is supported by the National Institutes of Health (no. R35GM134867) and the Montana State University Agricultural Experimental Station (USDA NIFA). A.D. was supported by the National Science Foundation’s Research Experiences for Undergraduates program (Award 2349117 to Montana State University). N.B. is supported by an F31 from National Institutes of Health (NIH) (no. GM153146) and received support from Montana INBRE (no. P20GM103474). Molecular graphics and analyses were performed with UCSF ChimeraX86, developed by the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco, with support from NIH no. R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. The funders had no role in the conceptualization, designing, data collection, analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Project conceptualization was performed by B.W., R.A.W., J.T.G. and N.B. J.T.G, B.W. and N.B. wrote the manuscript with review and input from all authors. Cryo-EM data collection was performed by N.B. Cryo-EM data processing and structural analysis were generated by N.B. and J.T.G. Plaque assays were performed by S.D., H.L. A.D., A.B.G and J.T.G. Cloning was performed by J.T.G., R.A.W., S.D. and Q.M.P. Protein purification and biochemistry was performed by R.A.W. Bacterial toxicity assays were performed by J.T.G and Q.M.P. tRNA sequencing analysis and Northern blot was performed by J.T.G. Microscopy was performed by J.T.G. and S.Z.D. Covariation model for ncRNA and comparison between retron types was performed by M.B. Comparative analysis between E. coli and phage tRNAs were performed by J.T.G and M.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
George, J.T., Burman, N., Wilkinson, R.A. et al. Structural basis of antiphage defence by an ATPase-associated reverse transcriptase. Nat Commun 16, 8459 (2025). https://doi.org/10.1038/s41467-025-63285-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63285-6
