
Abstract
Optimizing the catalyst-electrolyte interface structure is crucial for enhancing the performance of electrochemical alkaline hydrogen evolution reaction. Traditional approaches typically focus on regulating the thermodynamic barriers of adsorption and desorption for reactants, intermediates, and ions at active sites on the solid electrode surface. However, the structure of the electrical double layer influences the concentration of intermediates, adsorption energy, and surface reaction kinetics. Here, we dynamically construct a dense epitaxial hydroxide layer on nickel molybdate, forming an effective protective barrier to prevent molybdenum leaching and enhance material stability. This optimization enhances local electric field increasing the concentration of hydrated potassium ions within the outer Helmholtz plane. As a result, the interfacial hydrogen-bond network improves, water availability on the catalyst surface increases, and reaction kinetics accelerate. The optimized material operates stably for 1400 h at a current density of 0.45 A cm−2 in an industrial alkaline electrolyzer. Our dual-optimization strategy of dynamically constructing an epitaxial catalytic layer offers valuable insights for developing stable, high-current-density electrocatalytic materials.
Similar content being viewed by others


Introduction
The alkaline hydrogen evolution reaction (HER) holds notable potential for large-scale industrial hydrogen production. However, its efficiency is hindered by sluggish reaction kinetics, particularly in the initial water dissociation step (Volmer process, H2O ⇋ H + OH) and the subsequent OH* desorption step (OH* + e− ⇋ OH−), both of which introduce high energy barriers that limit overall alkaline water electrolysis performance1,2,3. Traditional approaches focus on optimizing the chemical and electronic structures of electrocatalytic materials to reduce the water dissociation activation energy and improve the stability of the catalyst surface4,5,6. Notably, the electrical double layer (EDL) structure at the catalyst-electrolyte interface, influenced by material properties and electrolyte characteristics, contributes to the regulation of adsorption behavior, interfacial electric fields, and reactant concentration7,8,9. This enhances the availability of reactants at the interface, accelerates the transfer of reaction intermediates, and reduces the energy barriers for adsorbed reactants and intermediates. These findings provide valuable insights for designing efficient and stable alkaline electrolysis systems by achieving simultaneous optimization of the catalyst surface and electrolyte interface.
According to Koper’s hemolytic and heterolytic models10,11, the proton-supplying ability of the electrolyte, specifically the concentration of reactants near the surface active sites, is a key descriptor in determining the overall catalytic activity for HER. During electrocatalytic reactions, the adsorption of various species on the electrode surface (including reactants, products, intermediates, and specifically adsorbed ions) plays a crucial role in governing the reaction’s progress12,13,14,15. Traditionally, optimizing alkaline HER active sites has focused on regulating the adsorption and desorption barriers of H2O/OH at the surface or within the inner Helmholtz plane3,16,17,18. However, changes in the concentration, arrangement, and configuration of solvent molecules and cations in the outer Helmholtz plane (OHP) within the EDL structure can also influence the amount and adsorption energy of reaction intermediates at active sites19,20,21,22,23. These interfacial changes further impact proton-coupled electron transfer kinetics between interfacial solvent molecules and surface intermediates24,25,26. Therefore, mass transfer and local microenvironment regulation near the catalytic sites requires focusing on the OHP to optimize the electric field, ion distribution, and hydrogen bond network structure at the catalyst-electrolyte interface, ultimately enhancing the overall performance of alkaline HER.
In this work, we revealed the epitaxial growth of a catalytic layer on nickel molybdate, utilizing it as a platform to investigate the local electrochemical microenvironment regulation mechanisms at the catalyst-electrolyte interface. The localized enhancement of the electric field around the dendritic epitaxial hydroxide layer increased the concentration of hydrated potassium ions within the OHP, facilitating the rearrangement of the surface hydrogen bond network and improving HER kinetics. Additionally, the dynamically structured epitaxial layer exhibited a denser structure, effectively suppressing molybdenum dissolution and providing enhanced stability against corrosive degradation during HER. Ultimately, the material achieved continuous operation for 1400 h at a current density of 0.45 A cm−2 in an industrial alkaline water electrolyzer with a large electrode assembly. This study demonstrates a dynamic construction strategy for optimizing epitaxial layers at the catalyst-electrolyte interface, offering a perspective for the design of high-performance electrocatalysts.
Results
Synthesis and characterizations
Nickel molybdate constructed with epitaxial layer (e-NiMoO4) is commonly fabricated using a two-step approach involving hydrothermal processing followed by electrochemical synthesis (Figure S1a). In order to establish a robust three-dimensional substrate model, initial synthesis focused on producing NiMoO4 precursor microrods (Fig. 1a, and Supplementary Fig. 1, 2). In the subsequent cathodic electrochemical synthesis, tailored KOH electrolytes comprising nickel chloride as an additional nickel source and sodium citrate as chelating agent27. This approach aims to effectively anchor the epitaxial hydroxide layer on the surface of the precursor microrods. Subsequent morphological and structural analyses confirm surface modifications while maintaining the fundamental stability of the material (Supplementary Fig. 3). Concurrently, the originally smooth surface of the precursor microrods transforms into a rough texture. The dynamically constructed layer also has densely packed nanodendrites smaller than 10 nm in size (Fig. 1b and Supplementary Fig. 4a–d). Dendrites on the epitaxial layer show lattice fringes corresponding to the (100) facet of nickel hydroxide (Supplementary Fig. 4e–f). SEM elemental mapping confirms the uniform distribution of Ni, Mo, and O (Supplementary Fig. 5a). Additionally, STEM mapping at various magnifications, including aberration-corrected imaging, clearly illustrates the epitaxial relationship between the NiMoO4 core and the Ni(OH)2 shell (Supplementary Fig. 5b, c). X-ray photoelectron spectroscopy (XPS) was conducted to explore the bonding states of Ni and Mo (Supplementary Fig. S6). High-resolution XPS spectra of Ni 2p and Mo 3 d reveal minimal changes in their main peak separations. Following the formation of e-NiMoO4, Mo 3 d shifts to lower binding energies with the emergence of low valence Ni peak in Ni 2p spectrum, demonstrating the reduction and reconstruction of e-NiMoO4 surface28. The X-ray absorption near-edge structure (XANES) and corresponding extended X-ray absorption fine-structure (EXAFS) spectra of NiMoO4 and e-NiMoO4 at the Ni K-edge have been investigated (Fig. 1c and Supplementary Table 1). Ni K-edge XANES spectra indicate a lower oxidation state of Ni in e-NiMoO4 compared to NiMoO4 (Fig. 1d). Corresponding EXAFS spectra show Ni-O coordination peaks at 1.54 Å for NiMoO4 and 1.52 Å for e-NiMoO4 (Fig. 1e)29. During electrochemical synthesis process, the fundamental Ni-O-Ni/Mo bonds remain unaffected by surface dissolution of molybdenum. The slight enhancement of the Ni-OH peak at 2.74 Å demonstrates the successful growth of the epitaxial hydroxides layer (Supplementary Fig. 7). Further analysis of the wavelet transform of the Ni K-edge EXAFS oscillations confirms that this reduction in oxidation state is due to the presence of nickel hydroxides with trace amounts of nickel, which are also similar to XPS results (Fig. 1f). Meanwhile, the bonding coordination environment of Mo has been further analyzed (Supplementary Fig. 8). The Mo coordination environment of e-NiMoO4 is similar to that of NiMoO4, with a slight increase in the Mo-O bond length. These findings indicate that Mo dissolution causes minor structural loosening while preserving the overall integrity of the Mo-containing framework, highlighting the stability and structural flexibility of the epitaxially grown Ni(OH)2 dendritic layer.

a, b TEM image of NiMoO4 (a) and e-NiMoO4 (b) with schematic illustration of lattice structure. c, Ni K-edge XANES spectra of e-NiMoO4 and the control samples. d The oxidation state of Ni in e-NiMoO4. e Fourier transformed magnitudes of Ni K-edge EXAFS spectra. f WT contour plots of the EXAFS signal for e-NiMoO4 and the control samples.
Electrochemcial HER activity and stability
Following HER standard measurements, the HER activities of the e-NiMoO4 catalyst and control samples are evaluated in 1.0 M KOH electrolyte. The electrochemical synthesis was optimized by adjusting the cathodic potential, synthesis duration, and electrochemical parameters for different electrode sizes to obtain the optimal e-NiMoO4 with varying sizes (Supplementary Fig. 9–14). Prior to evaluating the activity of all synthesized samples, the performance of commercial Pt/C, Raney Ni and control samples has been initially assessed (Fig. 2a). The control sample NiMoO4 exhibits HER activity with an overpotential (η10) of 238 mV and Tafel slope of 125.1 mV dec−1, suggesting the Volmer reaction as the rate-determining step (Fig. 2b). NiMoO4 may exhibit moderate hydrogen adsorption but inadequate hydroxyl adsorption30,31. The HER activity of e-NiMoO4 is further optimized via electrochemical synthesis (Supplementary Fig. 15–17). The optimized catalyst demonstrates enhanced alkaline HER activity, featuring a low Tafel slope of 45.7 mV dec−1 and η10 of 32 mV, comparable to that of Pt/C and Raney Ni (Fig. 2b). In particular, the e-NiMoO4 hybrid catalyst achieves low η100 and η200 at 170 mV and 251 mV, respectively. The variation in Tafel slope indicates a transition in the alkaline HER mechanism. For NiMoO4, the high Tafel slope (~120 mV/dec) aligns with the Volmer–Heyrovsky pathway, where the rate-determining step (RDS) is the dissociation of H2O (Volmer step: H2O + e− → H* + OH−), followed by electrochemical desorption (Heyrovsky step: H* + H2O + e− → H2 + OH−)4. The NiMoO4 surface exhibits strong H* adsorption but poor H2O activation. In contrast, e-NiMoO4 displays a lower Tafel slope (~40 mV/dec), indicating a shift to the Volmer–Tafel pathway, where the RDS transitions to the chemical recombination of adsorbed H*. This transformation is attributed to the epitaxial Ni(OH)2/NiMoO4 interface, which optimizes H* adsorption energy and facilitates rapid H* coupling. Furthermore, the optimized e-NiMoO4 demonstrates a substantial increase in electrochemical active surface area (ECSA) and high Cdl, nearly five times that of NiMoO4, indicating an enhancement in active site density (Supplementary Figs. 18, 19). When considering ECSA-normalized HER activity, e-NiMoO4 shows specific activity compared to NiMoO4 (Supplementary Fig. 20). The constructed nickel hydroxides layer on the surface of NiMoO4 fundamentally improved the intrinsic HER activity of the material32. This improvement suggests that the enhanced HER kinetics and enlarged surface area contribute to the performance of the hybrid catalyst. These synergistic improvements distinguish it among a series of non-noble metal materials (Supplementary Fig. 21).

a Polarization curves without iR corrections of e-NiMoO4 and control samples (scan rate 5 mV s−1). b Corresponding Tafel analysis of (a). c Chronopotentiometry durability test of e-NiMoO4. d Ni K-edge XANES spectra of NiMoO4 and e-NiMoO4 before and after stability testing. e The corresponding Fourier transformed magnitudes of Ni K-edge EXAFS spectra. f Bode phase plots of NiMoO4 and e-NiMoO4 at different operating potentials. g Nyquist diagrams of NiMoO4 and e-NiMoO4 with corresponding Rct at different operating potentials.
While achieving high HER activity is crucial, ensuring robust stability is paramount for practical applications. The optimized e-NiMoO4 shows stable operation for over 160 h at currents close to 1 A cm−2 (Fig. 2c). Following stability testing, e-NiMoO4 retains good HER activity (Supplementary Figs. 22, 23). TEM analysis revealed that a thick layer of Ni(OH)2 nanosheets formed on the NiMoO4 surface, whereas the Ni(OH)2 dendritic layer on e-NiMoO4 remained largely intact, with only minor particle aggregation (Supplementary Figs. 24, 25). Subsequent ICP analysis confirmed the dissolution of Ni and Mo in the NiMoO4 system (Ni: 0.12 ppm, Mo: 7.08 ppm), while their dissolution in the e-NiMoO4 system was suppressed (Ni: 0.09 ppm, Mo: 0.85 ppm) (Supplementary Table 2). XPS analysis after the reaction reveals that e-NiMoO4 exhibits stability compared to NiMoO4 (Supplementary Fig. 26). Considering the impact of surface molybdenum dissolution in the comparative sample NiMoO4, we investigated the Ni K-edge XAFS before and after the reaction. The Ni K-edge XANES spectrum suggests that in e-NiMoO4, although the oxidation state of nickel slightly shifts, overall stability remains preserved (Fig. 2d). In contrast, NiMoO4 exhibits a notable decrease in nickel oxidation state, possibly attributed to molybdenum dissolution33,34. The corresponding EXAFS spectrum of e-NiMoO4 displays nearly identical peak shapes before and after the reaction (Fig. 2e and Supplementary Fig. 27). The coordination structure of NiMoO4 undergoes changes due to extensive molybdenum dissolution. Based on the above structural characterization, it can be inferred that NiMoO4 and e-NiMoO4 exhibit distinct catalytic behaviors during the reaction process. Operando EIS has been conducted to elucidate the electrode kinetics and interfacial dynamics between the electrode and electrolyte. The Bode plot of NiMoO4 indicates a marked decrease in phase angle at low frequencies under open circuit voltage conditions compared to e-NiMoO4 (Fig. 2f). This indicates that NiMoO4 undergoes electrochemically induced structural evolution before the HER process35. Notably, e-NiMoO4 exhibits a distinct frequency response, with a smaller corresponding voltage at the same frequency. Additionally, the Nyquist plot of e-NiMoO4 displays a smaller semicircle and lower charge transfer resistance (Rct), indicating enhanced charge transfer kinetics. (Fig. 2g and Supplementary Fig. 28). The observed electrode stability and efficient charge transfer capability can be attributed to the successful construction of the epitaxial hydroxide layer on the surface of nickel molybdate.
In-situ XAFS during HER and structure stability analyses
To explore the structure stability of e-NiMoO4, in-situ EXAFS analysis revealed the structural dynamics associated with HER catalytic behavior. To gain deeper insights into the electronic structure changes of NiMoO4 and e-NiMoO4, potential-dependent in-situ XAFS experiments have been conducted. Both NiMoO4 and e-NiMoO4 exhibit highly similar patterns in the Ni K-edge XANES spectra at different potentials (Fig. 3a, f and Supplementary Table 3-4). As the applied potential increases, the peak position of NiMoO4 shifts to lower energies, indicating reduction in the oxidation state of nickel during the HER process (Fig. 3b)36. Fourier transform of the Ni K-edge EXAFS spectra reveal a primary coordination peak corresponding to Ni-O coordination, fitted at 1.54 Å (Fig. 3c and Supplementary Figs. 29–31). At different potentials, the peak positions of Ni-O and Ni-Ni coordination peaks of NiMoO4 show negligible changes, while the intensity of the Ni-Ni coordination peak gradually increases. Quantitative EXAFS fitting is also performed to obtain coordination information. At open circuit potential (OCP), the coordination number of Ni is 6.1 (Fig. 3d). With increasing applied bias, the coordination number rapidly decreases due to the influence of oxidation states and structural changes. Simultaneously, molybdenum dissolution induces rapid and irreversible overall structural changes in the material, thereby impacting its stability (Fig. 3e). Interestingly, in the e-NiMoO4 system, the oxidation state of Ni oscillates within a small range during bias application (Fig. 3f). Moreover, oscillations in Ni-O bond lengths were observed with negligible changes (Fig. 3g and Supplementary Figs. 32, 33). The corresponding coordination numbers also exhibit random oscillations (Fig. 3h). This phenomenon is attributed to structural relaxation induced by molybdenum dissolution under bias, where epitaxial hydroxide layer provides some protection against the structural collapse triggered by molybdenum dissolution (Fig. 3i). Confident in stabilizing the coordination number of Ni, we have mitigated structural changes in NiMoO4. These observations align with our expectation that dynamic construction of epitaxial hydroxide layer on NiMoO4 during electrochemical synthesis will influence subsequent HER processes (Fig. 3j).

a, f In situ XANES spectra recorded at the Ni k-edge of NiMoO4 (a) and e-NiMoO4 (f) at different operating potentials from ocp to -0.5 V vs. RHE during the HER process. b, h The corresponding enlarged area of in situ XANES spectra of (a, f). c, g Fourier transformed magnitudes of Ni k-edge of NiMoO4 (c) and e-NiMoO4 (g) EXAFS spectra under various potentials. d, i The coordination number of Ni extracted from EXAFS fitting results of NiMoO4 (d) and e-NiMoO4 (i). e, j Scheme of stability mechanism of NiMoO4 (e) and e-NiMoO4 (j).
DFT investigations and MD simulations
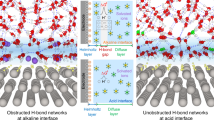
To further elucidate the activity mechanism on e-NiMoO4, density functional theory (DFT) calculations have been conducted. Aqueous decomposition free energy calculations are firstly performed to assess the influence of epitaxial hydroxide layer on NiMoO4 (Supplementary Fig. S34, Table 5, 6). Hybridization are observed among the Ni 3 d, Mo 4 d, and O 2p orbitals, with the centers of the Mo 4 d and O 2p orbitals shifting downward relative to the Fermi level (Fig. 4a, b and Supplementary Table 7). Low-coordination surface oxygen exhibit minimal contribution to the electronic states near the Fermi level (Supplementary Fig. S35). Through strong bonding interactions with adjacent Ni and Mo atoms, these oxygen atoms stabilize the epitaxial interface. Introducing epitaxial hydroxide layer further lowers the d-band center in e-NiMoO4, thereby enhancing the binding affinity of HER intermediates37. This suggests that epitaxial hydroxide layer can tailor the surface reactivity of NiMoO4 and reduce the strength of H adsorption. Furthermore, five key stages are considered for the theoretical exploration of the HER reaction pathway, including initial water adsorption and dissociation to form H* intermediates (Volmer step) and hydrogen generation (Tafel step or Heyrovsky step)38. Due to the high bond energy between hydrogen and nickel sites, and the substantial water dissociation barrier of NiMoO4, the epitaxial hydroxide layer in e-NiMoO4 can serve as active sites for hydroxyl adsorption, effectively lowering the following reaction barrier (Fig. 4c and Supplementary Fig. 36, 37). This synergy highlights how epitaxial hydroxide layer interacting with NiMoO4 enhance both the thermodynamics and kinetics of the HER reaction. Alternatively, we have simulated the electric field intensity distribution at the surface to accurately capture differences in the alkaline interface double-layer structure (Fig. 4d)39. Considering that the nanodendrite structure could influence the effect of the electric field on ion concentration, we used finite element numerical methods to study the surface electric field and ion distribution in the epitaxial dendritic layer. Unlike the uniform current distribution on the smooth NiMoO4 surface, the current on the e-NiMoO4 dendritic layer is mainly concentrated at the top regions of the nanodendrites and at the Ni(OH)2-NiMoO4 interface (Fig. 4e). Additionally, charge accumulation was observed at the top regions of the dendritic epitaxial layer, along with a localized increase in the concentration of hydrated potassium ions at the interface (Supplementary Fig. 38). Compared to NiMoO4, the density peak in the e-NiMoO4 system is higher and sharper. Structural simulations further confirmed that solvent-induced polarization charges promote the distribution of surface-hydrated potassium ions. On the e-NiMoO4 surface, hydrated potassium ions are primarily distributed at a distance of around 25 Å from the surface, which is lower than in the NiMoO4 system (Fig. 4f and Supplementary Fig. 39). To further investigate the influence of K+ ions on the reaction pathway of active sites, we analyzed their effect on e-NiMoO4 (Supplementary Fig. 40, 41). On the e-NiMoO4 surface, the dissociation barrier of H2O is 0.69 eV, primarily due to the slow cleavage of the O-H bond (Supplementary Fig. 42)15,40. At Ni sites adjacent to K+, the free energy barrier for the conversion of H2O to OH-H reduction occurs because K+ reduces the localization of charge density on H*, thereby facilitating faster OH*-H* decoupling. Additionally, K+ lowers the Volmer barrier by rebalancing the equilibrium between H* adsorption and desorption. At Ni sites adjacent to K+, the transition state energy barrier is the lowest (0.70 eV), with a final state energy of -0.22 eV, indicating that K+ further reduces the HER energy barrier at these sites (Supplementary Fig. 43-46, Table 8). Moreover, MD simulations confirmed that the localized enhancement of the electric field around the dendritic layer increases the concentration of hydrated potassium ions within the OHP, disrupting the rigid hydrogen bond network at the interface (Fig. 4g–i and Supplementary Fig. S47, 48). This disruption enhances the availability of H2O/OH* on the catalyst surface, thereby improving reaction kinetics41,42.

a, b Calculated projected density of states (PDOS) of NiMoO4 (a) and e-NiMoO4 (b). c Free energy profile for the alkaline HER process on NiMoO4 and e-NiMoO4. d Local electric field around NiMoO4 and the epitaxial catalytic layer. e Computed potential, current density, and K+ concentration near the surface of NiMoO4 and e-NiMoO4 electrodes via COMSOL simulations. f Distance distribution of potassium ion on the surface of NiMoO4and e-NiMoO4. g–i The distribution of K+ ions and water molecules in the NiMoO4 (g), Ni(OH)2 (h) and e-NiMoO4 models (i).
Industrial device performance
Benefiting from the enhanced HER activity and stability, we have further explored its capability for overall water splitting. Compared to e-NiMoO4 and RuO2, NiMoO4 exhibits the earliest OER onset and the fastest growth of the current with the applied potential (Supplementary Fig. 49a). NiMoO4 achieves smallest overpotentials (428 mV for 200 mA cm−2 and 480 mV for 300 mA cm−2), which are lower than those of e-NiMoO4 (508 and 635 mV), and RuO2 (496 and 610 mV), respectively. Due to the outstanding HER performance of e-NiMoO4 and OER performance of NiMoO4 under 1 M KOH conditions, it is integrated as the anode and cathode to construct an alkaline water electrolysis cell for overall water splitting. A noble metal electrode Pt/C | |RuO2 is used as a comparative system. NiMoO4 | |e-NiMoO4 (+,−) achieves a current density of 400 mA cm−2 at only 2.11 V (Supplementary Fig. 49b). At 50 mA cm−2, the driving voltage of NiMoO4 | |e-NiMoO4 (+,−) is much lower than (−)Pt/C | |RuO2 (+). Moreover, NiMoO4 | |e-NiMoO4 (+,−) exhibits comparable stability over 7 days at high catalytic current density for overall water splitting (≈500 mA cm−2), along with good activity reproducibility (Supplementary Fig. 49c). The ultimate challenge for alkaline HER catalysts lies in their performance in industrial-scale systems. The alkaline electrolyzer is structured in a series-connected monopolar filter-press configuration, consisting of multiple electrolysis chambers (Fig. 5a, b). Each chamber comprises an anode electrode plate, anode catalyst (for oxygen evolution), membrane, sealing ring, cathode catalyst (for hydrogen evolution), and cathode electrode plate43. We have assembled an alkaline electrolyzer cell with e-NiMoO4 serving as both the anode and cathode. At different cell potentials, the current density of the electrolyzer cell employing e-NiMoO4 electrodes is higher than that of the Raney Ni electrode electrolyzer cell. Furthermore, increasing the temperature from 30 °C to 50 °C leads to a gradual increase in current density. The current-voltage characteristic curves (without iR compensation) exhibit peak activity at an operating temperature of 50 °C, achieving a driving voltage of only 2.14 V at 0.4 A cm−2. This is lower than the 2.66 V required for commercial Raney nickel (Fig. 5c). At a consistent current density of 450 mA cm−2, continuous operation for 1400 h demonstrates the lowest operating voltage with no increase, highlighting comparable stability of e-NiMoO4 in industrial electrolyzers and its promising potential for industrial applications (Fig. 5d). After stability testing at high current densities, the microrods structure of e-NiMoO4 remains largely intact. While there were instances of surface aggregation and collapse, the material retained good structural stability overall, confirming its robustness (Supplementary Fig. 50).

a Photograph of an industrial electrolyzer device. b Enlarged view of the industrial electrolyzer. c Current-voltage polarizations of the industrial electrolyzer using e-NiMoO4 and commercial Raney Ni at different operating temperatures. d The cell voltage of the electrolyzer held at 450 mA cm−2 at 50 oC and 2 MPa with 30 wt% KOH solution supply.
Discussion
In summary, we have developed a highly active and stable HER catalyst capable of maintaining stability for at least 1400 h under a 0.45 A cm−2 water-splitting current in an alkaline electrolyzer cell. The durable epitaxial hydroxide layer constructed on the surface of NiMoO4 synergistically enhances active hydroxyl adsorption, thereby reducing the hydrogen evolution reaction barrier. Moreover, the locally enhanced electric field on the dendritic epitaxial catalytic layer increases the concentration of hydrated potassium ions, optimizing the rigid hydrogen-bond network at the interface and further accelerating HER reaction kinetics. Our discoveries provide an effective strategy to optimize the electrochemical reaction kinetics by controlling the electrode-electrolyte interface microenvironment, thereby enhancing the HER performance of industrial electrolyzer electrodes.
Methods
Chemicals
The following chemicals were purchased and used as-received without further purification. Nickel nitrate hexahydrate (≥99.9%, Sigma Aldrich), ammonium molybdate (≥99%, Sigma Aldrich), nickel chloride (≥98%, Sigma Aldrich), Potassium hydroxide (≥98%, Sigma Aldrich), sodium citrate (≥99%, Sigma Aldrich), 20 wt% Pt/C (Sigma Aldrich), Raney nickel and nickel foam (CeTech Co., Ltd).
Catalyst preparation
In a typical procedure for synthesizing the NiMoO4 precursor, nickel nitrate hexahydrate (0.8 mmol), ammonium molybdate (0.2 mmol), and nickel foam (1.5 cm × 1.5 cm) were dissolved in 50 mL of deionized water. After vigorous ultrasound for 1 h, the homogeneous suspension with nickel foam was evaporated in an oven at 160 °C for 6 h. For the synthesis of larger-sized materials, the corresponding amounts of raw materials were adjusted: nickel nitrate hexahydrate (1150 mg), ammonium molybdate (1000 mg), and nickel foam (7.0 cm × 20.0 cm) in 300 mL of water, using a 500 mL oven. Electrochemical synthesis of NiMoO4 with an epitaxial catalytic layer (e-NiMoO4) was achieved using a standard three-electrode system on a CHI 760E electrochemistry workstation in a mixed solution of nickel chloride (0.04 M), KOH (1.0 M), and sodium citrate (0.06 M). The NiMoO4 precursor served as the working electrode, a Ag/AgCl electrode as the reference electrode, and platinum as the counter electrode. For the electrochemical synthesis of large-scale electrodes, a DC power supply (Maisheng KKS605N, 0 ~ 60 V, 0 ~ 5 A) equipped with a peristaltic pump-driven circulation system was employed under ambient temperature and pressure. The current was maintained at 200 mA/cm², and the synthesis duration was kept consistent with that of smaller electrodes. Further optimization involved applying negative voltage and adjusting the electrochemical synthesis time; the resulting samples were cleaned and dried in a vacuum oven overnight at 80 °C.
Characterizations
Materials characterizations
Powder X-ray diffraction (XRD) patterns of the materials were recorded on a Rigaku Ultima IV diffractometer (Japan) with Cu Kα radiation (λ = 0.15406 nm), scanning 2θ from 10° to 80° with a step size of 0.04° to analyze crystalline structures. Surface chemical composition and elemental valence states were determined using an X-ray photoelectron spectrometer (AXIS SUPRA, Kratos) equipped with a monochromatic Al Kα source and a charge neutralizer. All binding energies were calibrated to the C 1 s peak at 284.6 eV of adventitious carbon. Scanning electron microscopy (SEM) images were acquired using a Hitachi Regulus 8100 microscope to observe morphological features. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained on a JEOL JEM-2100Plus microscope operating at 200 kV. Samples were prepared by ultrasonic dispersion in solution, dropping onto copper grids with polyvinyl formal support films, and air-drying. Atomic-scale phase distribution was characterized via high-resolution aberration-corrected scanning transmission electron microscopy (AC-HADDF-STEM, FEI Themis Z) at 60 kV and 60 pA.
X-ray absorption fine structure experiments
X-ray absorption fine structure (XAFS) experiments, including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), were performed at the Shanghai Synchrotron Radiation Facility (BSRF) with a Si (111) monochromator. The storage ring operated at 8.0 GeV with an average current of 99.5 mA. Data reduction and analysis were conducted using Athena and Artemis software (Demeter package), with FEFF6 for EXAFS fitting. Energy calibration was performed using a standard Ni foil, Ni(OH)2 and NiOOH. Pre-edge background was subtracted linearly, edge jumps were normalized, and k3-weighted χ(k) data were Fourier-transformed with a Hanning window (Δk = 1.0). EXAFS fitting in R-space used a previously determined amplitude reduction factor (S02 = 0.85) from Ni foil fitting.
In situ XAFS experiments
In situ XAFS measurements were carried out in a custom cell with 1.0 M KOH electrolyte. Catalyst inks (5 mg/mL) were loaded onto 1 × 1 cm2 carbon paper (1 mg cm−2) and sealed with Kapton film. Potentials ranging from −0.10 to −0.50 V vs. RHE were applied, and spectra were collected in triplicate using quick-scanning XAFS for signal reliability, with absorption edge position (E0) calibrated against Ni foil.
Electrochemical measurements
The electrochemical performance of samples for the HER was measured using a standard three-electrode system on CHI 760E electrochemistry workstation in 1.0 M KOH electrolyte (All electrolytes were prepared immediately before use). The as-synthesized electrodes, a graphite rod and a Hg/HgO electrode were used as the working, counter and reference electrodes, respectively. Ti meshes loaded with catalysts served as the working electrode. Nernst equation (ERHE = EHg/HgO + 0.0591 × pH + 0.098) was applied to record all potentials vs a reversible hydrogen electrode (RHE). In this work, all potentials were reported vs a reversible hydrogen electrode (RHE). Electrochemical surface area (ECSA) was calculated according to the equation ECSA=Cdl/Cs. The double-layer capacitance (Cdl) was evaluated on the basis of cyclic voltammetry (CV) curves within non-faradic potential window of 0.1 to 0.2 V, with various scan rates range from 5 to 30 mV s−1, and the Cs is 40 μF cm−2. It is noted that all the linear sweep voltammetry (LSV) curves without iR corrections at a scan rate of 5 mV s−1 was performed to evaluate the electrochemical performance of catalysts towards the HER. The Tafel slope was extracted by the Tafel equation, η = b log j + a, where η stands for the overpotential for the HER, b denotes the Tafel slope, j denotes the current density.
Industrial measurements
The industrial testing system was purchased from Cockerill Jingli Hydrogen, China. The industrial electrolyzer was operated using 30 wt% KOH solution. The e-NiMoO4 disc electrodes with diameter of 6 cm were integrated as both anode and cathode, respectively. NiMoO4 and commercial Raney Ni acts the control electrodes. The current density of water splitting was simultaneously monitored at different temperatures. The stability of the industrial electrolyzer was evaluated by measuring chronopotentiometry at 0.45 A cm−2 and the temperature of 50 °C.
DFT computational details
All the calculations were performed using the Vienna Ab initio Simulation Package (VASP) (Supplementary Data 1). The electron-ion interaction was described by the projector augmented-plane-wave (PAW) method, and exchange-correlation effects were treated with the generalized gradient approximation (GGA) using the Perdew-Burke-Ernzerhof (PBE) functional44. The van der Waals interactions were corrected via the Grimme DFT-D3 model45. NiMoO4 (110) and Ni(OH)2 (001) were selected as model surfaces, with lattice parameters: a = 15.3 Å, b = 12.9 Å, c = 30 Å for NiMoO4 (110); a = 13.5 Å, b = 12.2 Å, c = 30 Å for Ni(OH)2 (001). A plane-wave cutoff energy of 450 eV was used, and the Brillouin zone was sampled with a 2 × 2 × 1 Gamma-centered k-point mesh46. Structural relaxations converged when residual forces on atoms were <0.01 eV/Å, with an energy convergence threshold of 10-5 eV per cell. A 15 Å vacuum layer was added perpendicular to the surface to avoid periodic image interactions.
Free energy of hydroxyl adsorption on are defined as:
where Gsur-OH, GOH, and Gsur are the free energies of species adsorbed, hydroxyl species and clean surfaces, respectively. At equilibrium potential of HER:
Thus,
TS values are from previous report:
Solvent-induced polarization charges were computed using the PWmat code, adopting computational parameters consistent with the aforementioned setup. Atomic structures were visualized using the VESTA software. For alkaline HER, four key steps were considered in the theoretical analysis: water adsorption, water activation, formation of H* intermediates, and H2 generation.
The Gibbs free energy (G) of each intermediate or product is given by
Here, ΔE represents the reaction energy directly derived from DFT calculations; ΔEZPE (zero-point energy correction) and ΔS (entropy correction) were obtained from vibrational frequency analyses; T was set to 298.15 K in this work47.
MD simulation details
Atomistic molecular dynamics (MD) simulations were carried out using the GROMACS package (version 2020.6) under cubic periodic boundary conditions, with the CHARMM 36 general force field and TIPS3P water model adopted for parameterization (Supplementary Data 1). The equations of motion for all atoms were integrated via the classic Verlet leapfrog algorithm, with a time step of 1.0 fs. Short-range van der Waals and real-space electrostatic interactions were truncated at a radius of 1.6 nm, while long-range electrostatic interactions in reciprocal space were handled using the particle-mesh Ewald (PME) summation method, with an interpolation order of 4 and a Fourier grid spacing of 0.16 nm. Periodic boundary conditions were applied in all three directions, and the Newtonian equations of motion were integrated using the leapfrog algorithm.
The simulation system was constructed by randomly introducing 167 K+ and OH- ions into a box (≈5.4 × 5.4 × 9 nm3) containing either a NiMoO4 surface or a Ni(OH)2 layer-supported NiMoO4 surface. Water molecules were then added to solvate the system. Following energy minimization over thousands of steps, semi-isotropic NPT MD simulations (with fixed surface side lengths) were performed to equilibrate the system. Subsequent production runs were conducted under the canonical ensemble (NVT) for 20 ns, with the temperature maintained at 298 K using the Nose-Hoover thermostat. Non-bonded interactions were truncated at 1.2 nm, and long-range electrostatic interactions were computed via the PME method with a Fourier spacing of 0.1 nm. All covalent bonds involving hydrogen atoms were constrained using the LINCS algorithm.
COMSOL simulation details
Using the COMSOL Multiphysics finite element solver (https://www.comsol.com/), we simulated the free electron density on the electrode, as well as the electric field and potassium ion density near the electrode. The Electric Currents module was employed to solve for the free electron density on the electrode under a specific electrode bias potential48,49. The electric field E was calculated as the negative gradient of the potential V, given by E = − ∇V.
f
The diffusion layer consists of potassium ions that freely diffuse within the electrolyte. A concentration gradient is formed toward and away from the electrode surface through the tracking of charged particles.
Combine the Electrostatics and Diluted Species Transport modules to solve for the potassium ion density in the electric double layer50,51.
Data availability
All the data that support the findings of this study are available within the paper and its Supplementary Information files. Source data are provided with this paper.
References
Huang H. et al. Manipulation of oxidation states on phase boundary via surface layer modification for enhanced alkaline hydrogen electrocatalysis. Adv. Mater. 36, 2405128 (2024).
Fairhurst, A. R., Snyder, J., Wang, C., Strmcnik, D. & Stamenkovic, V. R. Electrocatalysis: from planar surfaces to nanostructured interfaces. Chem. Rev. 125, 1332–1419 (2025).
Chen, C. et al. Local reaction environment in electrocatalysis. Chem. Soc. Rev. 53, 2022–2055 (2024).
Fu, H. Q. et al. Hydrogen spillover-bridged volmer/tafel processes enabling ampere-level current density alkaline hydrogen evolution reaction under low overpotential. J. Am. Chem. Soc. 144, 6028–6039 (2022).
Quan, L., Jiang, H., Mei, G., Sun, Y. & You, B. Bifunctional electrocatalysts for overall and hybrid water splitting. Chem. Rev. 124, 3694–3812 (2024).
Tüysüz H. et al. Alkaline water electrolysis for green hydrogen production. Acc. Chem. Res., 57, 558–567 (2024).
Goyal, A., Louisia, S., Moerland, P. & Koper, M. T. M. Cooperative effect of cations and catalyst structure in tuning alkaline hydrogen evolution on Pt electrodes. J. Am. Chem. Soc. 146, 7305–7312 (2024).
Chang B. et al. Dynamic redox induced localized charge accumulation accelerating proton exchange membrane electrolysis. Adv. Mater. 37, e2405447 (2025).
Schott, C. M. et al. How to assess and predict electrical double layer properties. Implications for electrocatalysis. Chem. Rev. 124, 12391–12462 (2024).
McCrum, I. T. & Koper, M. T. M. The role of adsorbed hydroxide in hydrogen evolution reaction kinetics on modified platinum. Nat. Energy 5, 891–899 (2020).
Fernández-Vidal, J. & Koper, M. T. M. Effect of a physisorbed tetrabutylammonium cation film on alkaline hydrogen evolution reaction on Pt single-crystal electrodes. ACS Catal. 14, 8130–8137 (2024).
Shah, A. H. et al. The role of alkali metal cations and platinum-surface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 5, 923–933 (2022).
Chang, B. et al. Electronic perturbation of isolated fe coordination structure for enhanced nitrogen fixation. ACS Nano 18, 288–298 (2023).
Electrochemistey C. S. o The top ten scientific questions in electrochemistry. J. Electrochem. 30, 2024121 (2024).
Cui, W. G. et al. Insights into the pH effect on hydrogen electrocatalysis. Chem. Soc. Rev. 53, 10253–10311 (2024).
Xie, L. et al. Flexible tungsten disulfide superstructure engineering for efficient alkaline hydrogen evolution in anion exchange membrane water electrolysers. Nat. Commun. 15, 5702 (2024).
Levell, Z. et al. Emerging atomistic modeling methods for heterogeneous electrocatalysis. Chem. Rev. 124, 8620–8656 (2024).
Demnitz, M. et al. Effect of iron addition to the electrolyte on alkaline water electrolysis performance. iScience 27, 108695 (2024).
Li, P., Jiao, Y., Huang, J. & Chen, S. Electric double layer effects in electrocatalysis: Insights from ab initio simulation and hierarchical continuum modeling. JACS Au 3, 2640–2659 (2023).
Khani, H., Puente Santiago, A. R. & He, T. An interfacial view of cation effects on electrocatalysis systems. Angew. Chem. Int. Ed. 135, e202306103 (2023).
Zhu, X., Huang, J. & Eikerling, M. Hierarchical modeling of the local reaction environment in electrocatalysis. Acc. Chem. Res. 57, 2080–2092 (2024).
Ringe, S. Cation effects on electrocatalytic reduction processes at the example of the hydrogen evolution reaction. Curr. Opin. Electrochem. 39, 101268 (2023).
Munz, M., Poon, J., Frandsen, W., Cuenya, B. R. & Kley, C. S. Nanoscale electron transfer variations at electrocatalyst-electrolyte interfaces resolved by in situ conductive atomic force microscopy. J. Am. Chem. Soc. 145, 5242–5251 (2023).
Chen C., Zheng Y., Qiao S.-Z. Understanding the local environment in electrocatalysis. Nat. Sci. Rev., 11, nwae250 (2024).
Qin, X., Hansen, H. A., Honkala, K. & Melander, M. M. Cation-induced changes in the inner- and outer-sphere mechanisms of electrocatalytic CO2 reduction. Nat. Commun. 14, 7607 (2023).
Ovalle, V. J., Hsu, Y.-S., Agrawal, N., Janik, M. J. & Waegele, M. M. Correlating hydration free energy and specific adsorption of alkali metal cations during CO2 electroreduction on Au. Nat. Catal. 5, 624–632 (2022).
Li, X. & Binnemans, K. Oxidative dissolution of metals in organic solvents. Chem. Rev. 121, 4506–4530 (2021).
Lee, K. et al. Modulation of molybdenum oxidation state via catalytic-oxidation. Appl. Surf. Sci. 615, 156330 (2023).
Fu, G. et al. Coordination effect-promoted durable Ni(OH)2 for energy-saving hydrogen evolution from water/methanol co-electrocatalysis. Nanomicro Lett. 14, 200 (2022).
Chen, Y. et al. Room-temperature sulfur doped NiMoO4 with enhanced conductivity and catalytic activity for efficient hydrogen evolution reaction in alkaline media. J. Colloid Interface Sci. 664, 469–477 (2024).
Ren, J.-T. et al. Synergistic activation of crystalline Ni2P and amorphous NiMoOx for efficient water splitting at high current densities. ACS Catal. 13, 9792–9805 (2023).
Ze, H. et al. In situ probing the structure change and interaction of interfacial water and hydroxyl intermediates on Ni(OH)2 surface over water splitting. J. Am. Chem. Soc. 146, 12538–12546 (2024).
Li, G. L., Qiao, X. Y., Miao, Y. Y., Wang, T. Y. & Deng, F. Synergistic effect of n-nimoo4/ni heterogeneous interface with oxygen vacancies in N-NiMoO4/Ni/CNTs for superior overall water splitting. Small 19, e2207196 (2023).
An, L. et al. Epitaxial heterogeneous interfaces on N-NiMoO4/NiS2 nanowires/nanosheets to boost hydrogen and oxygen production for overall water splitting. Adv. Funct. Mater. 29, 1805298 (2019).
Qi, Y. et al. Insights into the activity of nickel boride/nickel heterostructures for efficient methanol electrooxidation. Nat. Commun. 13, 4602 (2022).
Dixon, D., Mangold, S., Knapp, M., Ehrenberg, H. & Bhaskar, A. Direct observation of reductive coupling mechanism between oxygen and iron/nickel in cobalt-free Li-rich cathode material: An in operando X-ray absorption spectroscopy study. Adv. Energy Mater. 11, 2100479 (2021).
Ren, J. T. et al. Interface metal oxides regulating electronic state around nickel species for efficient alkaline hydrogen electrocatalysis. Small 19, e2206196 (2023).
Lao, M. et al. From fundamentals and theories to heterostructured electrocatalyst design: an in-depth understanding of alkaline hydrogen evolution reaction. Nano Energy 98, 107231 (2022).
Li, P. et al. Revealing the role of double-layer microenvironments in pH-dependent oxygen reduction activity over metal-nitrogen-carbon catalysts. Nat. Commun. 14, 6936 (2023).
Yang, Y. et al. Electrocatalysis in alkaline media and alkaline membrane-based energy technologies. Chem. Rev. 122, 6117–6321 (2022).
Li, P. et al. Hydrogen bond network connectivity in the electric double layer dominates the kinetic pH effect in hydrogen electrocatalysis on Pt. Nat. Catal. 5, 900–911 (2022).
Chen, X. H. et al. Enhancing neutral hydrogen production by disrupting the rigid hydrogen bond network on Ru nanoclusters through Nb2O5-mediated water reorientation. Energy Environ. Sci. 17, 5091–5101 (2024).
Zhang, B. et al. High-valence metals improve oxygen evolution reaction performance by modulating 3d metal oxidation cycle energetics. Nat. Catal. 3, 985–992 (2020).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Norskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Gomez-Tames, J., Gonzalez, J. & Yu, W. Influence of different geometric representations of the volume conductor on nerve activation during electrical stimulation. Comput. Math. Methods Med. 2014, 489240 (2014).
Joucla, S., Gliere, A. & Yvert, B. Current approaches to model extracellular electrical neural microstimulation. Front. Comput. Neurosci. 8, 13 (2014).
Banerjee, T., Ghoshal, S. & Bhattacharya, B. B. Comsol-based design and validation of dilution algorithm with continuous-flow lab-on-chip. INAE Lett. 2, 55–63 (2017).
Adam, T. & Hashim, U. Comsol multiphysics simulation in biomedical engineering. Adv. Mater. Res. 832, 511–516 (2013).
Acknowledgements
This work received financial support from King Abdullah University of Science and Technology (KAUST) and Center of Excellence for Renewable Energy and Storage Technologies under award number 5937, National Natural Science Foundation of China (52202366), Natural Science Foundation of Shandong Province (2025HWYQ-050, ZR2021QE011, ZR2021JQ15), Taishan Scholar Project of Shandong Province (tstp20240515, tsqn202312217), and Innovative Team Project of Jinan (2021GXRC019). For computer time, this research used Shaheen III and Ibex managed by the KAUST Supercomputing Core Laboratory under project K10175.
Author information
Authors and Affiliations
Contributions
B. C., W. Z. and H. Z. constructed and planned the whole project. B. C. and J. H. carried out the synthesis of the samples and electrochemical experiments. X. L., M. K. and R. A. carried out industrial testing. S. Z. carried out the XANES and EXAFS characterizations. M. H. and Y. L. performed the HRTEM and high-resolution HAADF STEM imaging under the guidance of H. A. and H. Z. Y. R. helped to discuss the reaction mechanism and improve the manuscript. D. W. performed COMSOL simulation and analysis. W. L., R. H., Y. H. and H. L. performed the theoretical computations and participated in various aspects of the experiments and discussions. B. C., X. L. and S. Z. wrote the paper. All authors discussed the results and commented on the manuscript
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yao Zheng, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chang, B., Liu, X., Zuo, S. et al. Dynamic construction of a durable epitaxial catalytic layer for industrial alkaline water splitting. Nat Commun 16, 7959 (2025). https://doi.org/10.1038/s41467-025-63361-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63361-x
