
Abstract
BCL-2 is a central regulator of apoptosis and inhibits cell death by sequestering pro-apoptotic BH3 alpha-helices within a hydrophobic surface groove. While venetoclax, a BH3-mimetic drug, has transformed the treatment of BCL-2–driven malignancies, its efficacy is increasingly limited by acquired resistance mutations that disrupt small-molecule binding yet preserve anti-apoptotic function—reflecting a remarkable structural adaptation. Here, we employ hydrocarbon-stapled alpha-helices derived from the BAD BH3 motif as conformation-sensitive molecular probes to investigate this therapeutic challenge. The stapled peptides not only retain high-affinity binding to all BCL-2 variants but also show enhanced potency to select venetoclax-resistant mutants. Structural analyses, including X-ray crystallography and hydrogen-deuterium exchange mass spectrometry (HDX MS), demonstrate that these stapled helices restore native BH3 engagement by reversing the conformational consequences of resistance mutations. Notably, we identify a serendipitous interaction between the α3–α4 region of BCL-2 and hydrocarbon staple, which further compensates for altered groove conformation and contributes to mutant binding affinity. Together, these findings offer mechanistic insights into BCL-2 drug resistance and reveal a blueprint for designing next-generation inhibitors that overcome this clinically significant barrier to durable treatment responses.
Similar content being viewed by others

Introduction
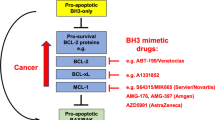
BCL-2 family proteins are central regulators of mitochondrial apoptosis and dictate cellular fate in response to stress through a complex network of protein interactions1. Cancer cells evade apoptosis by overexpressing anti-apoptotic proteins such as BCL-2, BCL-XL, MCL-1, and BFL-1, which sequester pro-apoptotic alpha-helices, called BCL-2 homology 3 (BH3) domains, in a conserved surface groove2. Since the identification of the BH3 “killer” motif of pro-apoptotic members3, a large series of BH3-only proteins have been demonstrated to regulate apoptosis by directly activating the pro-apoptotic executioner proteins BAX and BAK and/or by inhibiting anti-apoptotic proteins, thereby shifting the balance toward cell death4,5. These mechanistic insights have led to two therapeutic strategies: “inhibit the inhibitors” and “activate the activators” to restore apoptosis in cancer6,7,8. Among anti-apoptotic proteins, BCL-2 is the most well-established oncogenic target9,10,11, with its overexpression conferring a survival advantage by neutralizing pro-apoptotic proteins such as BIM, BID, and BAX4. Thus, mimicking BH3 interactions to inhibit BCL-2 and its anti-apoptotic homologs emerged as a BCL-2 family targeting approach to treat human cancer.
Extensive medicinal chemistry efforts toward selective inhibition of anti-apoptotic proteins have led to major pharmacologic breakthroughs. The earliest BH3-mimetic small molecules, such as ABT-7377 and ABT-263 (navitoclax)12, were enabled by the pioneering structure of an anti-apoptotic protein13 and its complex with an alpha-helical BH3 domain2. Such molecules effectively targeted BCL-2 and BCL-XL but were ultimately found to cause dose-limiting thrombocytopenia due to BCL-XL’s physiologic role in maintaining platelet survival14. This challenge spurred the development of venetoclax (ABT-199)—a BCL-2-selective inhibitor, which received FDA approval for the treatment of hematologic malignancies in 201615,16. Venetoclax has demonstrated remarkable efficacy in chronic lymphocytic leukemia, especially in combination with rituximab or obinutuzumab, resulting in durable remissions17,18. Inspired by its success, additional BCL-2 family antagonists targeting MCL-1 (e.g., MIK665, AMG-176, AZD5991) and BCL-XL (e.g., ABT-263, APG-1252, ABBV-155, DT2216) have been advanced to clinical testing with mixed or pending results19,20.
Given its favorable clinical profile, venetoclax use expanded and soon revealed a classic vulnerability of targeted small molecule therapies in cancer—acquired resistance through protein mutagenesis. Several studies identified mutations such as G101V and F104C/L/I, which disrupt venetoclax binding, preserve native BH3 binding, and thus promote resistance21,22,23. Additional mutations, including D103Y/E/V, A113G, R129L, and V156D, as well as in-frame insertions (e.g., R107_R110_dup), have been reported in cases of relapsed leukemia24,25,26,27. Of note, resistance is often polyclonal, with multiple mutations co-occurring, further complicating management24,28. Not surprisingly, venetoclax resistance mutations arise within or around the BH3-binding pocket, where the drug exerts its effect29. Thus, second-generation small molecule BCL-2 inhibitors such as sonrotoclax30 and LP-11831 remain vulnerable to analogous resistance mechanisms. Indeed, the capability of venetoclax-exposed cancer cells to develop mutations adjacent to or within the pocket that block small molecule binding yet preserve native BH3 binding is a striking structural biology feat.
We previously inserted all-hydrocarbon staples into peptide motifs, such as BH3 domains, to restore and fortify their bioactive structure for use as investigative tools and prototype therapeutics32. In the context of targeting the 6-helix fusion bundle of HIV-1, we discovered that pre-folding the bioactive HR2 domain peptide by inserting two staples overcame mutations that thwarted the peptide drug enfuvirtide, which is composed of unmodified, native amino acid sequence33. Here, we reasoned that stapled BH3 peptides could be developed to dissect and target the spectrum of BCL-2 mutants that are otherwise resistant to venetoclax. Early stapled BH3 peptides, derived from BID and BIM, effectively neutralized multiple anti-apoptotic proteins and directly activated BAX and BAK34,35. In contrast to these more promiscuous BH3 domains, others have more discrete binding selectivities. Whereas NOXA BH3 targets MCL-1 and BFL-1, BAD BH3 preferentially engages BCL-2, BCL-XL, and BCL-w5. By generating and screening a stapled peptide library of nature’s diverse BH3 sequences, we determined that the MCL-1 BH3 helix is the only naturally-occurring exclusive MCL-1 inhibitor, and solved the structure of the peptide/protein complex to provide insights into the molecular basis of MCL-1 binding selectivity36. Subsequent work yielded a selective covalent inhibitor of BFL-1 by targeting its unique cysteine within the surface groove using stapled NOXA and BIM BH3 peptides bearing an electrophilic warhead37.
Given the ability of stapled BH3 peptides to recapitulate natural binding selectivities, inform structure-function mechanisms, and deliver precision tools for targeting, we harness peptide stapling to develop a selective BCL-2 inhibitor that effectively targets BCL-2 and its clinical mutants. Complementary structural methods inform both the conformational consequences of BCL-2 mutagenesis and the capacity of a stapled BAD BH3 helix to effectively overcome them.
Results
Identification of a lead stapled peptide for targeting BCL-2
A previous crystal structure of venetoclax bound to the G101V mutant of BCL-2 (aa 1–34, loop residues 35–91 replaced with BCL-XL 29–44, 92–207; hereafter referred to as BCL-2ΔLΔC) showed a conformational change in the canonical groove, where E152 created steric hindrance that blocked the para-chlorine phenol moiety of venetoclax from binding to a subpocket (called P2) of the BH3 groove29 (Fig. 1a). Whereas venetoclax can still engage BCL-2 mutants like G101V, binding activity is impaired by two orders of magnitude (Supplementary Fig. S1a), weakening pro-apoptotic function and thus clinical efficacy. Because such BCL-2 mutants restore apoptotic resistance, we reasoned that the natural BH3 motif of the BCL-2 Associated Agonist of Cell Death (BAD), further reinforced by peptide stapling, might effectively inhibit the range of clinical mutants of BCL-2. Thus, we generated an i, i + 4 staple-scanning library of the BAD BH3 sequence (aa 105–126), whereby all-hydrocarbon staples spanning one turn of the alpha-helix were sequentially installed along the length of the sequence (Fig. 1b, Supplementary Fig. S1b, Supplementary Table 1). Hereafter, the peptides are referred to as BAD Stabilized Alpha-Helices of BCL-2 domains 4.X, or BAD SAHBs 4.X, with “4” representing the staple type and “X” denoting the location of the first (N-terminal) stapling residue of the inserted crosslink, respectively. We first assessed the ability of N-terminally biotinylated BAD SAHBs to bind FLAG-tagged BCL-2 in lysates of transfected HEK 293T cells using streptavidin pull-down and anti-FLAG western analysis. Among the 20 constructs, BAD SAHBs 4.2, 4.6, and 4.9 bound most effectively to FLAG-BCL-2 (Fig. 1c), with their staples clustering at the amphipathic interface and extending into the hydrophobic surface (Fig. 1d).

a Structure of BCL-2ΔLΔC G101V (PDB 6O0K), highlighting the location of the G101V mutation (red) that shifts E152 (orange) and thereby impairs venetoclax engagement of the canonical groove. Mutation of F104 (blue), which lies in the immediate vicinity of G101V, likewise causes venetoclax resistance29. b Staple scanning library of the BAD BH3 sequence, where “X” represents the inserted S-pentenyl alanine residues that form the i, i + 4 staples and amino acid “B” represents norleucine. For the peptide nomenclature, “4” is an abbreviation for the i, i + 4 staple and the number indicates the location of the first (N-terminal) stapling residue. c Comparative streptavidin pull-downs of biotinylated BAD SAHBs incubated with lysate from HEK 293T cells transiently transfected with full-length, FLAG-tagged, WT BCL-2, as evaluated by anti-FLAG western analysis. Asterisks denote the most effective BCL-2-binding BAD SAHBs. The experiment was performed in biological triplicate using independent preparations of lysates and reagents with similar results. d Helical wheel depiction of the BAD BH3 helix, highlighting the location of three lead i, i + 4 staple positions at the amphipathic interface and extending into the hydrophobic groove. Z represents the insertion position of an S-pentenyl alanine residue N- or C-terminal to the native template sequence. e Streptavidin pull-down of biotinylated BAD SAHBs upon exposure to equimolar recombinant BCL-2ΔLΔC, BCL-XLΔC (aa 1–212), and GST-MCL-1ΔNΔC (aa 170–327) proteins (1 µM), as visualized by electrophoresis and silver stain. The experiment was performed in biological triplicate using independent preparations of proteins and reagents with similar results. f Competitive FP of the FITC-BIM BH3 (3 nM) interaction with BCL-2ΔLΔC (8.6 nM) by BAD SAHBs 4.2, 4.6, and 4.9. Data are mean ± S.D. for experiments performed in technical quadruplicate and repeated three times with independent preparations of proteins and reagents with similar results. Source data are provided as a Source Data file.
Since dual BCL-XL/BCL-2 inhibitors like navitoclax can induce thrombocytopenia as a result of BCL-XL targeting14, lead BAD SAHB candidates were further screened for BCL-2 selectivity using a competitive binding assay. Biotinylated constructs were incubated with an equimolar pool of recombinant BCL-2ΔLΔC, BCL-XLΔC (aa 1–212), and GST-MCL-1ΔNΔC (aa 170–327), followed by streptavidin pull-down, electrophoresis, and silver stain. As expected, none of the compounds bound to MCL-1, consistent with the natural selectivity of BAD. Whereas BAD SAHB 4.9 engaged both BCL-2 and BCL-XL, BAD SAHBs 4.2 and 4.6 showed a competitive preference for BCL-2 (Fig. 1e). In a quantitative, competitive fluorescence polarization (FP) assay measuring displacement of a fluorescein isothiocyanate (FITC)-labeled BIM BH3 peptide (unstapled) from recombinant BCL-2ΔLΔC, BAD SAHB 4.2 emerged as the most potent ligand (IC50, 15.7 nM) (Fig. 1f) and was thus selected for further characterization.
BAD SAHB 4.2 maintains inhibitory activity against BCL-2 mutants
BAD SAHB 4.2, the strongest binder of wild-type (WT) BCL-2ΔLΔC, was subjected to direct and competitive FP using BCL-2ΔLΔC G101V and F104L, the earliest venetoclax-resistance mutations21,23. Whereas binding activity was similar for WT and G101V-mutant BCL-2 (direct FP EC50, 6.3 vs. 10.4 nM; competitive FP IC50, 15.7 vs. 17.1 nM), BAD SAHB 4.2 showed somewhat improved affinity for the F104L variant, particularly in the competitive binding assay (direct FP EC50, 6.3 vs. 4.4 nM; competitive FP IC50, 15.7 vs. 3.7 nM) (Fig. 2a, b, Supplementary Fig. S1c). We further derived Kd values from the direct FP data and performed biolayer interferometry measurements, both of which corroborated the EC50 values (Supplementary Figs. S1d, S2a–f). Next, we assessed the capacity of venetoclax to compete with biotinylated-BAD SAHB 4.2 for binding to FLAG-tagged, full-length BCL-2 in lysates from transfected HEK 293T cells, as assessed by streptavidin pulldown and anti-FLAG western blot. Whereas venetoclax effectively displaced BAD SAHB 4.2 from WT BCL-2 in a dose-dependent manner, with near complete dissociation at 1 μM and complete dissociation by 10 μM, its competitive binding efficacy was reduced by more than tenfold for the G101V and F104L mutants (Fig. 2c). To test the functional consequences of these differential binding effects, we used fluorophore-encapsulated liposomes mimicking the mitochondrial outer membrane and measured BAX-mediated membrane poration in response to triggering by the BH3-only protein tBID38. Specifically, we monitored the capacity of WT and the G101V and F104L mutants of BCL-2ΔLΔC to block tBID-triggered BAX activation and the comparative ability of BAD SAHB 4.2 and venetoclax to derepress the activity of these BCL-2 proteins. Whereas BAD SAHB 4.2 similarly restored BAX-mediated poration across BCL-2 variants, venetoclax activity was impaired by G101V or F104L mutagenesis (Fig. 2d, Supplementary Fig. S3). Taken together, these data demonstrate that, in contrast to venetoclax, BAD SAHB 4.2 maintains similar binding and functional activities despite BCL-2 mutagenesis.

a Binding of FITC-BAD SAHB 4.2 (3 nM) to WT, G101V, and F104L BCL-2ΔLΔC proteins, as measured by FP assay. Data are mean ± S.D. for experiments performed in technical quadruplicate and repeated three times with independent preparations of proteins and reagents with similar results. b Binding of Ac-BAD SAHB 4.2 to WT, G101V, and F104L BCL-2ΔLΔC proteins (8.6, 5.8, and 1.6 nM, respectively), as measured by competitive FP assay using FITC-BIM BH3 (3 nM). Data are mean ± S.D. for experiments performed in technical quadruplicate and repeated three times with independent preparations of proteins and reagents with similar results. c Binding of biotin-BAD SAHB 4.2 to FLAG-tagged, full-length WT, G101V, and F104L BCL-2 proteins in the presence or absence of a serial dilution of venetoclax, as assessed by streptavidin pull-down from lysates of transiently-transfected HEK 293T cells and anti-FLAG western analysis. The experiment was repeated three times with independent preparations of lysates and reagents, with similar results. d Effect of BAD SAHB 4.2 vs. venetoclax treatment (3.6 μM) on the capacity of WT, G101V, and F104L BCL-2ΔLΔC proteins (1.8 μM) to suppress tBID-triggered (100 nM), BAX-mediated (300 nM) liposomal release of ANTS/DPX, as measured by AUCCompound/AUCVehicle. Data are mean ± S.D. for experiments performed in technical quadruplicate and repeated three times with independent preparations of proteins and reagents, with similar results. Statistical significance was determined by two-sided, unpaired t test with Welch correction (venetoclax, WT vs G101V, p = 0.0023; venetoclax, WT vs F104L, p = 0.00041). *, p < 0.005; **, p < 0.001; ns no significance. Source data are provided as a Source Data file.
Structural basis for BAD SAHB 4.2 binding to BCL-2 and its G101V mutant
To examine the mechanism by which BAD SAHB 4.2 preserves binding to the G101V mutant of BCL-2, we first solved the crystal structure of WT BCL-2ΔLΔC in complex with BAD SAHB 4.2 (1.73 Å; PDB 9O14) (Fig. 3a, Supplementary Fig. S4a, Supplementary Table 2), as a structure of the BAD BH3 helix bound to BCL-2 was not previously reported. We then solved the structure of BAD SAHB 4.2 bound to BCL-2ΔLΔC G101V (1.99 Å; PDB 9O15) (Fig. 3b, Supplementary Fig. S4b, Supplementary Table 2) and compared the findings. In both complexes, BAD SAHB 4.2 maintains similar peptide backbone alignments within the groove, indicating that, in contrast to venetoclax, G101V mutagenesis does not affect the BAD SAHB 4.2 binding mode. Specifically, the hydrophobic network of the BH3-conserved leucine (L114) of BAD SAHB 4.2 with BCL-2 residues F104, F112, M115, V133, L137, A149, and F153 is preserved (Fig. 3c, d), as are the electrostatic and hydrophilic pairings (BAD SAHB 4.2-BCL-2ΔLΔC) of D119-N143/R146, E120-Y108, R115-E136/D140, and R116-Y108/D111 (Fig. 3e, f). We find that the all-hydrocarbon i, i + 4 staple is directed toward the peptide/protein binding interface and engages a discrete hydrophobic patch formed by residues M115 and L119 of the BCL-2 α3 helix, with V133 (α4) and F153 (α5) further reinforcing the hydrophobic network (Fig. 3c, d). Also of note, BAD SAHB 4.2 contains two C-terminal i, i + 4 phenylalanine residues, F121 and F125, which participate in a network of aromatic contacts with BCL-2 residues Y108, F104, F198, and Y202, demonstrating edge-to-face interactions (T-stacking) that create a stabilized aromatic cage in this region of the binding interface (Fig. 3g, h).

Overall similarity of the BAD SAHB 4.2/BCL-2ΔLΔC WT (a, PDB 9O14) and BAD SAHB 4.2/BCL-2ΔLΔC G101V (b, PDB 9O15) structures, demonstrating the helix-in-groove interaction, with BCL-2 surface residues colored by hydrophobic (yellow), hydrophilic (green), positive charge (blue), and negative charge (red) features. c, d The hydrophobic residues of the BAD SAHB 4.2 binding surface, including the highly conserved leucine (L114), engage with a network of hydrophobic residues of the groove, including F104, F112, M115, V133, L137, A149, and F153. The all-hydrocarbon staple (cyan) binds to a hydrophobic patch composed of M115, L119, V133 and F153. e, f A series of electrostatic and hydrophilic interactions, including (BAD SAHB 4.2-BCL-2ΔLΔC) D119-N143/R146, E120-Y108, R115-E136/D140, and R116-Y108/D111, are also preserved across the two structures. g, h The C-terminal phenylalanine residues of BAD SAHB 4.2 engage a network of aromatic residues, including Y108, F104, F198, and Y202 at the BCL-2 interface.
To interrogate differences in the disposition of residues between BCL-2 proteins bound to venetoclax vs. BAD SAHB 4.2, we generated and analyzed difference distance matrix plots (DDMPs). Comparing the DDMP derived from structures of venetoclax bound to G101V-mutant vs. WT BCL-2ΔLΔC29, we observe a reduction in the distances between discrete residues of helices adjacent to the G101V mutation, specifically α1 and α5, as well as α4 and α6, effectively tightening the protein core through V101’s increased hydrophobic interactions. Distal to the mutation, an apparent allosteric effect increases the distance between the proximal portion of the α1 loop and the α3-α4 hairpin (helix-loop-helix motif) (Fig. 4a, b). Interestingly, these perturbations are not observed in the DDMP for BAD SAHB 4.2 bound to G101V-mutant vs. WT BCL-2ΔLΔC (Fig. 4c), indicating that stapled peptide binding nullifies the conformational consequences of mutagenesis observed for venetoclax interaction. Comparing the relative impact of BAD SAHB 4.2 and venetoclax upon binding to WT BCL-2ΔLΔC, the stapled peptide induces extensive conformational changes throughout the protein, consistent with a larger interface of engagement by the peptide than the small molecule (Fig. 4d). When compared in the context of G101V-mutant BCL-2, the pattern of the changes is largely similar although notably diminished, consistent with the mutant protein exhibiting increased structural rigidity (Fig. 4e). Intriguingly, the regions that maintain a similar extent of conformational difference relate to the α3 helix (aa 109-119), which engages the hydrocarbon staple. Specifically, the distal halves of the α1 and α2 helices, the α6-α7 bend, and a portion of the α5-α6 hairpin demonstrate increased distances from the α3 helix, consistent with the pocket opening up to bind the staple and thus displacing α3 posteriorly from the protein core (Fig. 4e, f). Taken together, these structural data indicate that the more extensive binding interface of BAD SAHB 4.2 as compared to venetoclax (Supplementary Fig. S5), including the added hydrophobic interaction of the hydrocarbon staple itself, effectively overcomes the conformational consequences of BCL-2 mutagenesis that impairs small molecule engagement.

A distance difference matrix plot comparing venetoclax complexes with BCL-2ΔLΔC G101V and WT (PDB 6O0L, 6O0K), with three regions of prominent structural changes boxed in green, purple, and brown (a) and mapped onto the structure of BCL-2ΔLΔC G101V (PDB 9O15; BH3 helix [maroon] shown for reference) using the corresponding colors (b). DDMPs comparing BAD SAHB 4.2 complexes with BCL-2ΔLΔC G101V and WT (PDB 9O15, 9O14) (c), BAD SAHB 4.2 and venetoclax complexes with BCL-2ΔLΔC WT (PDB 9O14, 6O0K) (d), and BAD SAHB 4.2 and venetoclax with BCL-2ΔLΔC G101V (PDB 9O15, 6O0L) (e), with the changes preserved by BAD SAHB 4.2 in the latter boxed in purple, blue, yellow, and orange and mapped onto the structure of BCL-2ΔLΔC G101V (PDB 9O15) with respect to α3 in green (f). Source data are provided as a Source Data file.
Influence of G101V mutagenesis and BAD SAHB 4.2 targeting on protein dynamics
Whereas high-resolution crystal structures provide a snapshot of stable protein conformation, hydrogen-deuterium exchange mass spectrometry (HDX MS) offers alternative and complementary insight by monitoring protein dynamics in real-time. An added advantage of HDX MS in the context of BCL-2 family studies is the ability to investigate the behavior of fully native sequences (Supplementary Fig. S1d), which are often incompatible (such as the native α1-α2 loop of BCL-2) with solving anti-apoptotic protein structures in complex with their BH3 ligands. HDX MS probes protein structure by measuring the deuterium incorporation of backbone amide hydrogens over time. When diluted into deuterium buffer, backbone hydrogens of flexible and/or exposed protein regions rapidly exchange with deuterium, whereas buried domains and/or those regions that contain hydrogen-bonding of backbone amide hydrogens (such as in α-helices) or that are engaged in ligand interactions (such as BH3 binding) demonstrate slowed or suppressed deuterium exchange. Here, when we compare the difference in deuterium exchange of recombinant, WT BCL-2ΔC (aa 1-207) in the presence and absence of BAD SAHB 4.2, a clear pattern emerged, whereby peptides of α2, α3, α4, and α5 that comprise the canonical binding groove are significantly protected from exchange in the presence of bound ligand; in addition, there is allosteric protection of the proximal portion of α6 that abuts α5 at the turn of the α5-α6 hairpin (Fig. 5a, Supplementary Data 1, Supplementary Table 3). Strikingly, the effect of BAD SAHB 4.2 on BCL-2ΔC G101V is essentially identical (if not more protected) (Fig. 5b, Supplementary Data 1, Supplementary Table 3), consistent with preservation of functional binding activity, the crystal structures of the complex, and the derived DDMPs (Figs. 2–4). When comparing the deuterium-exchange profiles of G101V-mutant and WT BCL-2 proteins, those peptides in the vicinity of the valine mutation, including proximal α2 and distal α6 through α8, demonstrate increased protection, consistent with a more rigid and less exposed conformation (Fig. 5c, Supplementary Data 1, Supplementary Table 3). This latter structural distinction in protein dynamics between G101V-mutant and WT BCL-2 is eliminated upon BAD SAHB 4.2 interaction (Fig. 5d, Supplementary Fig. S6a, Supplementary Data 1, Supplementary Table 3).

Deuterium difference plots showing the relative deuterium incorporation of BCL-2ΔC WT/BAD SAHB 4.2 minus BCL-2ΔC WT (a), BCL-2ΔC G101V/BAD SAHB 4.2 minus BCL-2ΔC G101V (b), BCL-2ΔC G101V minus BCL-2ΔC WT (c), and BCL-2ΔC G101V/BAD SAHB 4.2 minus BCL-2ΔC WT/BAD SAHB 4.2 (d), as measured at 10 s, 1 min, and 10 min of deuterium labeling. Regions of protection (green) above the 0.5-Da significance threshold (gray shading) at 1 min of deuterium labeling are mapped onto the structure of BAD SAHB 4.2/BCL-2ΔLΔC WT or G101V (PDB 9O14, 9O15). The protein, stapled peptide, and mutant residue are colored gray, maroon, and red, respectively. Peptides containing the point mutation cannot be compared to WT and are thus represented by bracketed gaps in the plots (c, d). HDX MS experiments were performed twice using independent preparations of BCL-2 proteins and reagents. ND no data. Source data are provided as Supplementary Data 1.
HDX MS reveals the differential impact of BCL-2 mutants on protein conformation
Given the sensitivity of HDX MS in identifying the conformational consequences of a clinically-relevant point mutation in BCL-2, we expanded the analysis to characterize a series of observed mutants (Supplementary Fig. S1d). Comparing the difference in deuterium exchange profile of D103Y-mutant and WT BCL-2ΔC, we observe deprotection of both a portion of the α1-α2 loop and the proximal half of α2 upstream of the point mutation, changes that localize to the vicinity of D103Y in α2 (Fig. 6a, Supplementary Fig. S6b, Supplementary Data 1, Supplementary Table 3). Whereas A113G mutagenesis (located in α3) induces subtle deprotection of α3 through α4 starting just distal to the mutation (Fig. 6b, Supplementary Fig. S6c, Supplementary Data 1, Supplementary Table 3), more prominent changes occur in response to R129L and V156D mutagenesis. For R129L, which is located in α4, there is notable protection of a cluster of peptides in the adjacent regions of α3 and α4, distal α5 and its hairpin loop, and proximal α6; allosteric deprotection occurs within the α1-α2 loop and proximal α2 (Fig. 6c, Supplementary Fig. S6d, Supplementary Data 1, Supplementary Table 3). In contrast, V156D, which is located in the middle of α5, causes circumferential deprotection of α2, the proximal portion of α3, and of α7, which lies in direct proximity to the proximal portion of α2 (Fig. 6d, Supplementary Fig. S6e, Supplementary Data 1, Supplementary Table 3). The latter finding is reminiscent of our previous characterization of BAX α5 core hydrophobic residues, which destabilize the monomeric structure of BAX upon alanine mutagenesis39, with conversion of a hydrophobic to a negatively-charged residue here likely even more consequential. These data collectively highlight that mutagenesis of α2, α3, α4, or α5 residues located in or around the canonical groove causes distinctive patterns of conformational alteration, with each point mutant reported to disrupt venetoclax engagement40.

Deuterium difference plots showing the relative deuterium incorporation of BCL-2ΔC D103Y (a), A113G (b), R129L (c), and V156D (d), minus BCL-2ΔC WT, as measured at 10 s, 1 min, and 10 min of deuterium labeling. Regions of protection (green) and deprotection (orange) outside of the 0.5-Da significance threshold (gray shading) at 1 min of deuterium labeling are mapped onto the structure of BCL-2ΔLΔC WT in complex with venetoclax (PDB 6O0K). The protein is colored gray and mutant sites colored brown, yellow, blue, and purple for D103Y, A133G, R129L, and V156D, respectively. The mapping on BCL-2ΔLΔC A113G is based on the indicated single long peptide above the significance threshold (b). Peptides containing the point mutations cannot be compared to WT and are thus represented by bracketed gaps in the plots (a, c, d). Dotted lines represent a small region lacking sequence coverage (d). HDX MS experiments were performed twice using independent preparations of BCL-2 proteins and reagents. ND no data. Source data are provided as Supplementary Data 1.
BAD SAHB 4.2 reverses the conformational consequences of differential BCL-2 mutagenesis
To determine if BCL-2 targeting by BAD SAHB 4.2 could broadly combat the heterogeneous conformational impacts of individual mutants, HDX MS analysis of each mutant was repeated in the presence or absence of stapled peptide. Strikingly, irrespective of mutation site and resultant structural alteration, BAD SAHB 4.2 binding at the canonical groove was fully preserved (Fig. 7a–e, Supplementary Data 1, Supplementary Table 3), as reflected by the deuterium difference exchange profiles that mirrored the protection of WT BCL-2ΔC (α2, α3, α3-α4 loop, α4-α5 loop, proximal α5, distal α5, α5- α6 loop, proximal α6) upon BAD SAHB 4.2 engagement (Fig. 5a). In calculating the difference in deuterium exchange between BAD SAHB 4.2 when bound to mutant vs. WT BCL-2ΔC proteins, we observed that the D103Y and A113G complexes were essentially indistinguishable from that of WT (Supplementary Fig. S6b, c, S7a, b). For the R129L mutant, BAD SAHB 4.2 interaction reduces the level of protection in the α3-α4 loop and α5-α6 hairpin seen upon comparing the apo proteins (Fig. 6c), although regiospecific changes persist (Supplementary Fig. S6d, S7c). R129 is located in the proximal portion of α4 and, in the crystal structure of the BCL-2ΔLΔC WT/BAD SAHB 4.2 complex, lies adjacent to the all-hydrocarbon staple (Fig. 3a, b). Replacing a positively charged residue with a hydrophobic one appears to enhance and stabilize the local hydrophobic interaction network of the apo protein (e.g., M114, L119, V133, F153 [Fig. 3c]); this effect manifests as persistent protection in the α4 region immediately surrounding the leucine mutation, consistent with enhanced hydrophobic interaction between the staple and the canonical groove (Fig. 6c, Supplementary Fig. S6d, S7c). In contrast, V156D mutagenesis at the α5 core destabilizes apo BCL-2ΔC, resulting in deprotection of α2, proximal α3, and the portion of α7- α8 that abuts α2 (Fig. 6d, Supplementary Fig. S6e). Whereas BAD SAHB 4.2 effectively protects the surface groove of BCL-2ΔC V156D (Fig. 7d), the relative difference in deuterium uptake compared to the other mutants is less negative (e.g., peptide 98–111: D103Y, –5.79; A113G, –5.4; R129L, –5.88, V156D, –4.81). In achieving the surface groove interaction (Fig. 7d), BAD SAHB 4.2 reverses the deprotection seen for the V156D apo protein (Fig. 6d) and, when comparing ligand bound to mutant vs. apo protein, compensatory deprotection of α3, α4, and the α5-α6 hairpin is observed (Supplementary Fig. S6e, S7d).

Deuterium difference plots showing the relative deuterium incorporation of BCL-2ΔC D103Y/BAD SAHB 4.2 (a), BCL-2ΔC A113G/BAD SAHB 4.2 (b), BCL-2ΔC R129L/BAD SAHB 4.2 (c), and BCL-2ΔC V156D/BAD SAHB 4.2 (d), minus the corresponding apo protein mutant, as measured at 10 s, 1 min, and 10 min of deuterium labeling. HDX MS experiments were performed twice using independent preparations of BCL-2 proteins and reagents. e Regions of protection (green) outside of the 0.5-Da significance threshold (gray shading in a–d) across mutants and time points are mapped onto the structure of BCL-2ΔLΔC WT in complex with BAD SAHB 4.2 (PDB 9O14). f Locations of venetoclax resistance mutations within the structure of BCL-2ΔLΔC WT (PDB 9O14). g Competitive FP of the FITC-BIM BH3 (3 nM) interaction with the indicated BCL-2ΔLΔC protein mutants at EC90 (WT, 8.6 nM; G101V, 5.8 nM; D103Y, 21 nM; F104L, 1.6 nM; A113G, 23 nM; R129L, 8.7 nM; V156D, 17 nM) by BAD SAHB 4.2. Data are mean ± S.D. for experiments performed in technical quadruplicate and repeated three times with independent preparations of proteins and reagents with similar results. Source data are provided as a Source Data file and Supplementary Data 1.
In contrast to the reported ~200-fold reduction in venetoclax binding affinity for BCL-2ΔLΔC G101V compared to WT BCL-2ΔLΔC protein, we observe by competitive FP that BAD SAHB 4.2 retains the same binding affinity for WT and G101V-mutant BCL-2ΔLΔC, (competitive FP IC50, 15.7 and 17.1 nM, respectively; Fig. 2b). Across the spectrum of mutations that localize in or around the canonical groove (Fig. 7f), BAD SAHB 4.2 demonstrates between 1.7 and 7.5-fold improvement in competitive binding activity, including for the D103Y, A113G, F104L, and R129L mutants (competitive FP IC50s, 9.1, 6.3, 3.7, and 2.1 nM, respectively) (Fig. 7g). Only the V156D mutant, buried in the core α5 helix, reduced BAD SAHB 4.2 binding activity by 2.5-fold (competitive FP IC50, 39.2 nM), consistent with reduced protection of the canonical groove shown by HDX MS due to disruption of α5 core interactions upon replacement of a hydrophobic residue with a negatively charged one (Fig. 6d).
To further examine the structural basis for the largest enhancement of BAD SAHB 4.2 binding affinity for a clinical mutant, namely R129L, we solved the structure of the BCL-2ΔLΔC R129L/BAD SAHB 4.2 complex (1.73 Å; PDB 9O16) (Fig. 8a, Supplementary Fig. S4c, Supplementary Table 2). Consistent with the increased protection from deuterium exchange observed by HDX MS (Fig. 7c) and enhanced binding affinity measured by FP (Fig. 7g), the crystal structure demonstrates unique interactions between the all-hydrocarbon staple and L129, which extends the hydrophobic groove to the region previously occupied by the positively-charged R129 (Fig. 8b). A DDMP comparing BCL-2ΔLΔC R129L/BAD SAHB 4.2 and BCL-2ΔLΔC WT/BAD SAHB 4.2 highlights that this extension of the hydrophobic groove reduces the distance between the α3 and proximal α4 region and the distal α1 helix (Fig. 8c, d). Evaluating comparative protein/peptide distances for the two complexes by DDMP likewise shows a reduction of the distances between the peptide N-terminus, which bears the staple, to this same region, where a convergence of hydrophobic residues from α3 (M115, L119, and L121), α4 (A126, R129L, F130, and V133), α5 (F153, V156, and M157), and α6 (M116, L169, I173, and W176) forms a network that expands the hydrophobic binding interface and engages the hydrocarbon staple (Fig. 8e). Indeed, superposition of the two complexes reveals that the staple becomes further entrenched in the R129L groove, with associated downward displacement of the peptide backbone (Fig. 8f). Taken together, these data reveal the influence of differential point mutagenesis on the conformational dynamics of BCL-2, and the capacity of a lead BAD SAHB construct to not only target WT BCL-2 and its clinical mutants but also effectively revert the structural consequences of a broad spectrum of mutations. For the R129L mutant in particular, the capacity of all-hydrocarbon stapling to exploit the expanded hydrophobic surface at the canonical groove showcases the utility of SAHBs as tools for both dissecting structure-activity relationships and targeting BCL-2 mutants that confer resistance to small molecule inhibitors.

Structure of the BCL-2ΔLΔC R129L/BAD SAHB 4.2 complex (a, PDB 9O16) demonstrates how replacement of the positively-charged R129 residue with leucine extends the hydrophobic groove, enabling further reinforcement of hydrophobic interactions with the all-hydrocarbon staple (b). DDMP comparing the BCL-2ΔLΔC R129L/BAD SAHB 4.2 and BCL-2ΔLΔC WT/BAD SAHB 4.2 protein complexes (PDB 9O16, 9O14) (c), with prominent differences in intra-protein proximities mapped onto the structure of BCL-2ΔLΔC R129L (d, PDB 9O16). e DDMP comparing the distances between BAD SAHB 4.2 residues and those of BCL-2ΔLΔC in the BCL-2ΔLΔC R129L/BAD SAHB 4.2 and BCL-2ΔLΔC WT/BAD SAHB 4.2 complexes. f Superposition of the two complexes (PDB 9O16, 9O14) highlights the impact of R129L mutagenesis on the position of the staple and associated peptide backbone at the hydrophobic groove. Source data are provided as a Source Data file.
Discussion
The three-decade journey from the discovery of BCL-2 to the development of venetoclax represents a remarkable arc of scientific progress—spanning fundamental mechanistic insights to the surmounting of formidable pharmacological challenges. The realization that BCL-2 exerts its anti-apoptotic function by sequestering BH3 domains within a hydrophobic groove provided the roadmap for drug development2, culminating in small molecule BH3 mimetics, ABT-7377 and ABT-26312. However, dual inhibition of BCL-2 and BCL-XL led to dose-limiting thrombocytopenia in patients14, necessitating the refinement of selectivity in ABT-199 (venetoclax), which specifically targets BCL-2 while sparing BCL-XL16. The clinical success of venetoclax in hematologic malignancies has been tempered by the emergence of resistance mutations, a challenge shared by kinase inhibitors like imatinib (Gleevec)41, with structural alterations at the binding pocket impairing small-molecule binding. The resistance mutations in BCL-2 present a unique structural paradox—they must thwart small-molecule engagement while preserving binding to the native BH3 domains of pro-apoptotic BCL-2 proteins in order to function. Indeed, a point mutation such as G101V accomplishes this by altering the positioning of the adjacent E152 residue, disrupting optimal small molecule engagement of the P2 pocket, while leaving intact the structural framework necessary for BH3 domain interactions29. The ability of cancer cells to evolve structural modifications that selectively disrupt drug binding while maintaining endogenous function underscores the need for alternative therapeutic strategies that can bypass this mechanism of resistance.
Before the advent of the ABT class of BCL-2 inhibitors, we developed stabilized alpha-helices of BCL-2 domains (SAHBs) to interrogate BCL-2 family protein interactions8. Unlike small molecules, stapled BH3 peptides replicate the entirety of the structural and binding features of their native counterparts, making them powerful tools for dissecting selective interactions and apoptotic mechanisms6,34,35,36,37. Here, we leveraged BAD SAHBs in conjunction with biochemical and structural methods to elucidate how clinically-observed venetoclax resistance mutations differentially alter BCL-2 conformational dynamics and how stapled peptides can overcome them. Structural analyses using X-ray crystallography and HDX MS demonstrated that BCL-2 resistance mutations induce both direct and allosteric modifications in distinct subregions of the canonical groove. Despite these alterations, a lead BAD SAHB not only retained but, in most cases, enhanced its binding affinity by engaging a broader interaction surface than venetoclax, including residues of the α3-α4 and α5-α6 helix-loop-helix motifs, effectively counteracting the altered conformational dynamics of BCL-2 mutagenesis.
A recent HDX MS study comparing an apo and venetoclax-bound construct of WT BCL-2ΔLΔC demonstrated protection predominantly in α2, distal α4, and proximal α542. This contrasts with the profile of BAD SAHB 4.2, which protects a broader region (i.e., α2-α6) and to a greater extent than observed for venetoclax. Intriguingly, a reported apo BCL-2ΔLΔC structure42 lacked density in the very α3 sequence engaged by the proximal portion of BAD SAHB 4.2 and which appears folded as an α3 helix in our structures of BAD SAHB 4.2 complexes. Indeed, the strongest BCL-2 interactor—BAD SAHB 4.2—benefits from the serendipitous engagement of its hydrocarbon staple with α3 residues that form a hydrophobic patch, whose interaction surface is further expanded by L129 substitution within α4, making the BCL-2ΔLΔC R129L/BAD SAHB 4.2 interaction the strongest of all. Thus, engagement of the α3-α4 region appears especially important for reinforcing ligand-groove interactions that overcome BCL-2 resistance mutations.
As clinical mutants continue to emerge43, iterative small molecule development to target BCL-2—whether as direct inhibitors or glues to promote its degradation—may continue to face the structural hurdles induced by point mutagenesis. Whereas stapled peptides are not readily thwarted by point mutagenesis owing to a larger, structurally-reinforced interaction surface, their development as therapeutics face a different set of hurdles. Although stapling has largely remedied the proteolytic instability of natural peptides8,33,44, ensuring sufficient intracellular delivery to achieve the requisite potency remains a challenge. The development and testing of a clinical-stage stapled peptide that reactivates p53-mediated apoptosis in cancer by dual targeting of HDM2 and HDMX has yielded learnings that, in time, may prove applicable to advancing stapled BH3 peptides45,46,47. If successfully optimized for delivery, BAD SAHBs could serve as an alternative modality for treating BCL-2-driven cancers, particularly in the setting of venetoclax resistance. Indeed, the evolutionary constraint that BH3-in-groove binding is essential for oncogenic function suggests that mutations capable of evading BAD SAHBs—unlike those conferring venetoclax resistance—would inherently compromise BCL-2’s anti-apoptotic activity. By avoiding mutation-driven escape, BAD SAHBs may offer a uniquely durable strategy to circumvent resistance to BCL-2-targeted therapy.
Methods
Peptide synthesis
Peptides (BAD BH3, aa 105–126: WAAQRYGRELRRBSDEFVDSFK; BIM BH3, aa 146-166: IWIAQELRRIGDEFNAYYARR) were synthesized, derivatized with either biotin, biotin-PEG2, acetyl, or FITC at the N-terminus, purified, and quantified by amino acid analysis as reported in detail48. Briefly, peptides were synthesized by solid phase Fmoc chemistry using rink-amide resin and elongated using hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) as the coupling reagent and N,N-diisopropylethylamine (DIEA) as the base. For stapled peptides, two naturally-occurring amino acids at discrete i, i + 4 positions were replaced with non-natural S-pentenyl alanine pairs followed by olefin metathesis using the Grubb’s first-generation catalyst to form the staple. Peptides were deprotected and cleaved from the resin by a solution of 95:2.5:2.5 (v/v) TFA/TIS/H2O and purified to >95% homogeneity by C18 reverse phase HPLC (Agilent 1260 Infinity II) using an MeCN/H2O gradient containing 0.1% TFA. Peptides were lyophilized and stored at −20 °C until use. Stapled peptide compositions and masses are listed in Supplementary Table 2.
Recombinant protein expression and purification
For biochemical analyses and X-ray crystallography, recombinant human WT BCL-2ΔLΔC containing native residues 1–34, a truncated α1-α2 loop (aa 35–91) containing BCL-XL α1-α2 loop sequences 29–44, and native residues 92–207 (1MAHAGRTGYDNREIVMKYIHYKLSQRGYEWDAGD34-DVEENRTEAPEGTESE-92VVHLTLRQAGDDFSRRYRRDFAEMSSQLHLTPFTARGRFATVVEELFRDGVNWGRIVAFFEFGGVMCVESVNREMSPLVDNIALWMTEYLNRHLHTWIQDNGGWDAFVELYGPSMR207)27 and its point mutants were cloned into the pTYB1 vector (GE Healthcare) and constructs validated by DNA sequencing. Transformed E. coli BL21(DE3) were cultured in Luria Broth (LB) containing ampicillin (0.1 g/L), grown to an OD of 0.6–0.8, and protein expression induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 16 °C. Bacterial pellets were collected by centrifugation and resuspended in lysis buffer (250 mM NaCl, 20 mM Tris HCl, pH 7.2 containing complete protease-inhibitor tablets [Roche, 11873580001, 3 tablets/50 mL]). The cell suspension was lysed using a chilled microfluidizer, centrifuged to remove insoluble cellular debris, applied to chitin resin, and washed with lysis buffer. Chitin-bound BCL-2ΔLΔC was cleaved by overnight incubation with 50 mM dithiothreitol (DTT) in lysis buffer at 4 °C (leaving a residual C-terminal Ala moiety from the chitin binding domain on the cleaved protein), concentrated, and purified by size exclusion chromatography (SEC) using a Superdex S-75 (GE Healthcare) gel filtration system on an AKTA Pure 25 (150 mM KCl, 20 mM HEPES, pH 7.2). Protein purity and identity were verified by Coomassie staining (Supplementary Fig. S1c) and western blot analysis.
Recombinant human GST-MCL-1ΔNΔC (aa 170–327), and BCL-XLΔC (aa 1–212) in pGEX-4T-1 were generated as reported42. Briefly, transformed E. coli BL21(DE3) were cultured in LB as described for BCL-2ΔLΔC and protein expression induced with 1.0 mM IPTG for 4 h at 37 °C. Bacterial pellets were collected by centrifugation and resuspended in lysis buffer (PBS containing 0.1% Triton X-100) containing complete protease-inhibitor tablets (3 tablets/50 mL). The cell suspension was then lysed and centrifuged as described for BCL-2ΔLΔC, applied to glutathione resin and washed with lysis buffer. To generate tagless BCL-XLΔC, glutathione resin-bound protein was incubated overnight at 25 °C with PBS containing thrombin (9 units in 6 mL), leaving a residual N-terminal Gly-Ser moiety on the GST-cleaved protein. To isolate GST-MCL-1, protein was eluted from the glutathione column with elution buffer (50 mM Tris HCl, pH 8.0, 10 mM GSH). Eluted proteins were concentrated and purified by SEC using a Superdex S-75 (GE Healthcare) gel filtration system (150 mM NaCl, 50 mM Tris HCl, pH 7.4). Protein purity and identity were verified by Coomassie staining and western blot analysis.
Recombinant, full-length human BAX (aa 1-192) (pTYB1 vector) was produced as reported38. Transformed E. coli BL21(DE3) were cultured as described for BCL-2ΔLΔC and protein expression induced using 1.0 mM IPTG for 4 h at 37 °C. Bacterial pellets were collected by centrifugation and resuspended, lysed, purified, and verified as described for BCL-2ΔLΔC.
For biolayer interferometry (BLI) and HDX MS experiments, recombinant human BCL-2ΔC1-207 and its mutants, all bearing a native α1-α2 loop, were cloned into the pGEX-4T-1 expression vector (GE Healthcare) and generated, purified, and verified as described for GST-MCL-1ΔNΔC (yielding GST-BCL-2ΔC for BLI) or GST-cleaved BCL-XLΔC (yielding GST-cleaved BCL-2ΔC for HDX MS) (Supplementary Fig. S1d).
Fluorescence polarization binding assays
FP assays were performed as reported49. Briefly, recombinant anti-apoptotic proteins were serially diluted threefold from a starting concentration of 4 μM into assay buffer (5% DMSO, 0.01% Triton X-100 in 1x PBS, pH 7.4) in 96-well plates. FITC-peptide (3 nm) was then added and the mixture incubated in the dark for 30 min at 25 °C. Fluorescence (485 nm excitation, 525 nm emission) was detected on a SpectraMax M5 microplate reader (Molecular Devices) and data subjected to nonlinear regression analysis using Prism software (GraphPad) to calculate EC50s (Y = Bottom + [(Top -Bottom) * XHillslope] / [EC50Hillslope + XHillslope]), and Kds (P = Pf +[(Pb - Pf) * RT] / [Kd + RT]).
For competitive FP, the protein concentration required to achieve the EC90 for FITC-BIM BH3 interaction was first determined by direct FP (BCL-2ΔLΔC WT, 8.6 nM; G101V, 5.8 nM; D103Y, 21 nM; F104L, 1.6 nM; A113G, 23 nM; R129L, 8.7 nM; V156D, 17 nM). N-terminal acetylated BAD SAHBs were serially diluted 3-fold into assay buffer in 96-well plates, followed by addition of FITC-BIM BH3 (3 nM) and BCL-2 ΔLΔC (EC90 concentration). The mixture was incubated in the dark for 30 min at 25 °C. Fluorescence (485 nm excitation, 525 nm emission) was detected on a SpectraMax M5 microplate reader (Molecular Devices) and data subjected to nonlinear regression analysis using Prism software (GraphPad) to calculate IC50s per the equation described above for EC50s.
Streptavidin pull-down of cell lysates
HEK 293T cells (ATCC, CRL-1573) were maintained in DMEM medium supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin/streptomycin, and 2 mM glutamine, and confirmed to be mycoplasma-free using the MycoAlertTM mycoplasma detection kit (Lonza Biologics Inc). Cells at 70% confluence in a T175 Flask were transiently transfected with 3 μg of pCMV-Tag2B plasmid encoding N-terminally FLAG-tagged, full-length BCL-2 protein (aa 1-239) (WT, G101V, or F104L) using Lipofectamine LTX & Plus reagent (ThermoFisher). After 24 h, cells were harvested and lysed in CHAPS buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 1% CHAPS [v/v]). Protein concentration of the soluble fraction was measured by BSA assay following the manufacturer’s protocol (Thermo Scientific). Lysate (300 μg) was incubated with biotinylated peptides (1 μM) in 200 μL volume overnight at 4 °C. Peptide-protein complexes were then captured by high-capacity streptavidin agarose beads (Thermo Scientific) for 2 h at room temperature. Beads were subsequently washed 3x with 200 μL PBS and 3x with 200 μL 1% NP-40 in PBS before eluting in 2x lithium dodecyl sulfate (LDS) containing 300 mM DTT. Samples were subjected to electrophoresis and western blotting using an HRP-conjugated anti-FLAG antibody (1:2000; abcam #ab49763) to evaluate comparative BCL-2 binding activity of BAD SAHBs. For streptavidin pull-downs incorporating venetoclax competition, lysate (300 μg) was incubated with biotinylated BAD SAHB 4.2 (1 μM) in 200 μL volume overnight at 4 °C in the presence (0.1, 1.0, and 10 μM) or absence of venetoclax. N-terminally FLAG-tagged, full-length BCL-2 proteins (WT, G101V, or F104L) bound to biotinylated BAD SAHB 4.2 were captured, washed, and detected as described above.
Competitive streptavidin pull-down of recombinant proteins
Recombinant BCL-2ΔLΔC, BCL-XLΔC, and GST-MCL-1ΔNΔC (1 μM each) were combined and incubated with biotinylated BAD SAHB 4.2 (1 μM) overnight at 4 °C in PBS containing 1 mM DTT. The mixtures were then added to high-capacity streptavidin agarose beads (Thermo Scientific) for 2 h at room temperature, washed 3x with 200 μL PBS and 3x with 200 μL 1% NP-40 in PBS before eluting in 2x LDS containing 300 mM DTT. Samples were then subjected to electrophoresis on a 12% Bis-Tris gel and visualized by silver staining following the manufacturer’s protocol (Pierce).
Biolayer interferometry
BLI binding measurements were performed using an Octet Red384 System (ForteBio Inc.). Streptavidin (SA) sensors were prewetted in PBS and then assay buffer (PBS containing 1% [w/v] BSA) for 2 min prior to use, loaded with 100 nM Biotin-PEG2-BAD SAHB 4.2 for 3 min, and washed with assay buffer for 2 min. The sensors were then transferred into 2.33-fold serial dilutions (starting from 200 nM) of WT or G101V mutant GST-BCL-2ΔC in assay buffer for 15 min (association step), followed by assay buffer alone for 10 min (dissociation step). Because less than 5% dissociation was observed (precluding a kinetic fit), the association endpoints were fit to a concentration-wavelength shift curve (nm, Octet Software, version 9, ForteBio, Inc.) for two biological replicates and dissociation constants (Kds) calculated by Prism software (Graphpad) using the binding saturation equation (one site, specific binding): [Y = Bmax * X / (Kd + X)].
Liposomal release assay
Large unilamellar vesicles (LUVs) were prepared with a lipid composition of 48 mol% phosphatidylcholine, 28 mol% phosphatidylethanolamine, 10 mol% phosphatidylinositol, 10% dioleyl phosphatidyl-serine, and 4 mol% tetraoleoylcardiolipoin (Avanti Polar Lipids). The lipid mixture was prepared from chloroform stocks, dried as a thin film by vacuum (1 mg total lipid), and stored under nitrogen at –80 °C until use. Fluorescent 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS, 6.3 mg) and fluorescence quencher p-xylene-bis-pyridinium bromide (DPX, 19.1 mg) were added to the dried film and rehydrated with 1 mL of liposomal buffer (200 mM KCl, 5 mM MgCl2, 10 mM Hepes, pH 7.0). Following 5 freeze-thaw cycles, the hydrated lipid solution was extruded through a 100-nm membrane (Whatman) using an Avanti mini extruder. LUVs were subsequently isolated from free ANTS/DPX by gravity flow SEC using Sepharose CL-2B resin (Sigma Aldrich) and liposomal buffer. LUVs (5 μL) were mixed with the following reagents singly or in combination as indicated: BAD SAHB (3.6 μM), venetoclax (3.6 μM), BCL-2ΔLΔC (1.8 μM), tBID (100 nM), and BAX (300 nM). To assess BAX-mediated liposomal membrane permeabilization, after adding BAX (F0), the change in ANTS fluorescence (355 nm excitation and 520 nm emission) was monitored at one-minute intervals for 2 h. Triton X-100 was then added to each well at a final concentration of 0.5% (vol/vol) to determine maximal release (F100). Fraction ANTS/DPX release was then calculated as [(F-F0)/(F100-F0)]. BAX reactivation, as measured by AUC, was calculated using the formula [AUCF-AUCF1]/[AUCF100-AUCF1], where F1 represents the suppression of tBID-triggered BAX by BCL-2 in the absence of BAD SAHB or venetoclax.
X-ray crystallography
Recombinant BCL-2ΔLΔC (WT, G101V, or R129L) was preincubated at 1 mg/mL with 3x molar excess of BAD SAHB 4.2 (10 mM DMSO stock, 1.5% DMSO final) for 2 h at 4 ˚C and then concentrated to supersaturated solutions above 15 mg/mL (773 μM). Immediately prior to co-crystallization, samples were centrifuged to remove any aggregates, and concentrates were adjusted to 15 mg/mL. Proteins were mixed with precipitation solutions (200 nL each) and crystallized by hanging-drop vapor diffusion at 20 °C. BCL-2ΔLΔC WT/BAD SAHB 4.2 crystallized in 15% PEG3350 and 150 mM CsCl, whereas BCL-2ΔLΔC G101V/BAD SAHB 4.2 crystallized in 1.4 M ammonium tartrate and 0.1 M Tris HCl, pH 8, and BCL-2ΔLΔC R129L/BAD SAHB 4.2 crystallized in 25% PEG3350, 0.2 M ammonium sulfate, and 0.1 M Tris HCl, pH 8.5. The crystals were transferred into crystallization buffer containing 25% glycerol prior to flash-freezing in liquid nitrogen. Diffraction data were collected at beamline 17-ID-2 (BAD SAHB 4.2 in complex with BCL-2ΔLΔC WT and BCL-2ΔLΔC G101V, 0.9793 Å) and beamline 17-ID-1 (BAD SAHB 4.2 in complex with BCL-2ΔLΔC R129L, 0.9202 Å) of the National Synchrotron Light Source II (Brookhaven National Laboratory), with 0.2˚ oscillation and 0.2 s exposure. Datasets were integrated and scaled using XDS or DIALS packages50. Structures were solved by molecular replacement using the program Phaser (v.2.8.3)51 based on a series of search models. Iterative manual model building and refinement using Phenix (v.1.20.1_4487)52 and Coot (v.0.9.7)53 led to the final models. Ramachandran statistics ([favored (%), allowed (%), outliers (%)]) are [99.32, 0.68, 0.00], [98.88, 1.01, 0.11], and [100.00, 0.00, 0.00] for BAD SAHB 4.2 in complex with WT, G101V, and R129L BCL-2ΔLΔC proteins, respectively.
Distance difference matrix plots
For each crystal structure, we define a distance map as Di,j = | | xi – xj | |, whereby xi, xj are the cartesian coordinates of the alpha carbons from residues i, j and | | xi – xj | | is the Euclidean norm, with each capturing the distance between the subscripted pair of residues. We further quantify relative shifts in structure using the distance difference map (DDM) ΔABi,j = DAi,j – DBi,j, where distance map DA, DB correspond to structures A and B, respectively. Entries of Δ with a positive sign indicates that a pair of residues are further apart in structure A, while a negative sign indicates that a pair of residues are closer together in structure A. All maps were calculated using Python-based analysis tools using the NumPy library54 for numerical analysis and Matplotlib55 for data visualization.
Protein interface analysis
To quantify the interfaces between WT BCL-2ΔLΔC and the BH3-mimetic ligands venetoclax and BAD SAHB 4.2 (PDB IDs 6O0K and 9O14, respectively), we used PDBe PISA (Proteins, Interfaces, Structures, and Assemblies; v1.52, 10/20/2014) to computationally identify interface residues and calculate: (i) the solvent-accessible surface area (ASA) for each ligand, (ii) the interface or buried surface area (Ų) of BCL-2 upon complex formation, and (iii) the relative buried surface area (percentage of total ASA of BCL-2). These parameters were reported as Total Surface Area, Surface Area of BCL-2 Interface, and Relative BCL-2 Coverage, respectively.
Hydrogen deuterium exchange mass spectrometry
Recombinant BCL-2ΔC proteins (WT and point mutants) were incubated individually (30 μM) or with BAD SAHB 4.2 (protein:peptide, 1:5) for at least 1 h at 23 °C in buffer (150 mM NaCl, 20 mM Tris, pH 7.4). Deuterium labeling of the mixture (2 μL sample) was initiated with an 18-fold dilution into D2O labeling buffer (36 μL, 150 mM NaCl, 20 mM Tris base, pD 7.4, 99.9% D2O). The labeling reaction was quenched over time (10 s, 1 min, or 10 min) by the addition of 38 μL of ice-cold quenching buffer (200 mM sodium phosphate, 4 M guanidinium chloride, 0.72 M TCEP pH 2.3, H2O) and analyzed immediately. Deuterated and control samples were digested online at 15 °C using an AffiPro Nep-2 column (AffiPro, AP-PC-004). The cooling chamber of the HDX system, which housed all chromatographic elements, was held at 0.0 ± 0.1 °C for the duration of measurements. Peptides were trapped and desalted on a VanGuard Pre-Column trap (2.1 mm × 5 mm, ACQUITY UPLC BEH C18, 1.7 µM [Waters, 186002346]) for 3 min at 100 μL/min. Peptides were eluted from the trap using a 5%-35% gradient of acetonitrile with 0.1% formic acid over 6 min at a flow rate of 100 μL/min, and separated using an ACQUITY UPLC HSS T3, 1.8 µm, 1.0 mm × 50 mm column (Waters, 186003535). The back pressure averaged ~12,950 psi at 0 °C in 5% acetonitrile, 95% water, 0.1% formic acid buffer. To eliminate peptide carryover, a wash solution (1.5 M guanidinium chloride, 0.8% formic acid, and 4% acetonitrile) was injected over the protease column during each analytical run. Mass spectra were acquired using a Waters Synapt G2-Si HDMSE mass spectrometer in ion mobility mode. The mass spectrometer was calibrated with direct infusion of a glu-fibrinopeptide (Sigma, F3261) solution at 200 femtomole/μL at a flow rate of 5 μL/min prior to data collection. A conventional electrospray source was used and the instrument was scanned over a 50 to 2000 m/z range. The instrument was configured using the following parameters: capillary 3.2 kV, trap collision energy 4 V, sampling cone 40 V, source temperature 80 °C, and desolvation temperature 175 °C.
The error of determining the average deuterium incorporation for each peptide was at or below ±0.25 Da based on deuterated peptide standards. Peptides were identified from replicate HDMSE analyses of undeuterated control samples using PLGS 3.0.1 (Waters Corporation). Peptide masses were identified from searches using non-specific cleavage of a custom database containing the sequence of BCL-2 (UniProt: P10415), no missed cleavages, no post-translational modifications, low energy threshold of 135, elevated energy threshold of 35, and an intensity threshold of 500. No false discovery rate (FDR) control was performed. The peptides identified in PLGS (excluding all neutral loss and in-source fragmentation identifications) were then filtered in DynamX 3.0 (Waters Corporation), implementing a minimum products per amino acid cut-off of 0.25 and at least 2 consecutive product ions. Peptides that met the filtering criteria were further processed by DynamX 3.0 (Waters Corporation). The relative amount of deuterium in each peptide was determined by the software, which subtracted the centroid mass of the undeuterated form of each peptide from the deuterated form at each time point and for each condition. Deuterium levels were not corrected for back exchange and thus reported as relative56.
Statistical analysis
The number of technical and biological replicates for each experiment are indicated in the corresponding figure legends. Raw data were processed in Microsoft Excel 2024 and curve fitting and mean ± S.D. values were calculated using Prism 10 software (GraphPad).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings of this study are available within the paper, its Supplementary Information, and Source Data, including Supplementary Data 1. Crystallographic data for the structures reported in this article have been deposited to the Protein Data Base with accession numbers PDB 9O14, 9O15, and 9O16. HDX MS datasets have been submitted to the ProteomeXchange Consortium via PRIDE57 with identifier PXD062817. Source data are provided with this paper.
Code availability
This study did not generate new code.
References
Walensky, L. D. BCL-2 in the crosshairs: Tipping the balance of life and death. Cell Death Differ. 13, 1339–1350 (2006).
Sattler, M. et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 275, 983–986 (1997).
Chittenden, T. et al. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. EMBO J. 14, 5589–5596 (1995).
Cheng, E. H. et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711 (2001).
Letai, A. et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192 (2002).
Gavathiotis, E. et al. BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 (2008).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Walensky, L. D. et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470 (2004).
Cleary, M. L., Smith, S. D. & Sklar, J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47, 19–28 (1986).
Tsujimoto, Y., Finger, L. R., Yunis, J., Nowell, P. C. & Croce, C. M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099 (1984).
Vaux, D. L., Cory, S. & Adams, J. M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442 (1988).
Tse, C. et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68, 3421–3428 (2008).
Muchmore, S. W. et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381, 335–341 (1996).
Mason, K. D. et al. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007).
Roberts, A. W. & Huang, D. Targeting BCL2 with BH3 mimetics: Basic science and clinical application of venetoclax in chronic lymphocytic leukemia and related B cell malignancies. Clin. Pharm. Ther. 101, 89–98 (2017).
Souers, A. J. et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 19, 202–208 (2013).
Fischer, K. et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N. Engl. J. Med. 380, 2225–2236 (2019).
Seymour, J. F. et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N. Engl. J. Med 378, 1107–1120 (2018).
Lin V. S., Xu Z. F. & Huang D. C. S. Thijssen R. BH3 mimetics for the treatment of B-cell malignancies-insights and lessons from the clinic. Cancers (Basel) 12, 3353 (2020).
Roberts, A. W., Wei, A. H. & Huang, D. C. S. BCL2 and MCL1 inhibitors for hematologic malignancies. Blood 138, 1120–1136 (2021).
Blombery, P. et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 9, 342–353 (2019).
Blombery, P. et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br. J. Haematol. 186, e188–e191 (2019).
Fresquet, V., Rieger, M., Carolis, C., Garcia-Barchino, M. J. & Martinez-Climent, J. A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 123, 4111–4119 (2014).
Blombery, P. et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 135, 773–777 (2020).
Lucas, F. et al. Novel BCL2 mutations in venetoclax-resistant, ibrutinib-resistant CLL patients with BTK/PLCG2 mutations. Blood 135, 2192–2195 (2020).
Sullivan, G. P., Flanagan, L., Rodrigues, D. A. & Ni Chonghaile, T. The path to venetoclax resistance is paved with mutations, metabolism, and more. Sci. Transl. Med 14, eabo6891 (2022).
Tausch, E. et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 104, e434–e437 (2019).
Wei, A. H. & Roberts, A. W. Polyclonal heterogeneity: The new norm for secondary clinical resistance to targeted monotherapy in relapsed leukemia?. Cancer Discov. 9, 998–1000 (2019).
Birkinshaw, R. W. et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 10, 2385 (2019).
Guo, Y. et al. Discovery of the clinical candidate sonrotoclax (BGB-11417), a highly potent and selective inhibitor for both WT and G101V mutant Bcl-2. J. Med Chem. 67, 7836–7858 (2024).
Ravikrishnan, J. et al. LP-118 is a novel B-cell lymphoma 2 / extra-large inhibitor that demonstrates efficacy in models of venetoclaxresistant chronic lymphocytic leukemia. Haematologica 110, 78–91 (2025).
Walensky, L. D. & Bird, G. H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med Chem. 57, 6275–6288 (2014).
Bird, G. H. et al. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc. Natl. Acad. Sci. USA 107, 14093–14098 (2010).
Walensky, L. D. et al. A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell 24, 199–210 (2006).
LaBelle, J. L. et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J. Clin. Invest 122, 2018–2031 (2012).
Stewart, M. L., Fire, E., Keating, A. E. & Walensky, L. D. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 6, 595–601 (2010).
Huhn, A. J., Guerra, R. M., Harvey, E. P., Bird, G. H. & Walensky, L. D. Selective covalent targeting of anti-apoptotic BFL-1 by cysteine-reactive stapled peptide inhibitors. Cell Chem. Biol. 23, 1123–1134 (2016).
Hauseman, Z. J. et al. Homogeneous oligomers of pro-apoptotic BAX reveal structural determinants of mitochondrial membrane permeabilization. Mol. Cell 79, 68–83 e67 (2020).
Bloch, N. B. et al. The conformational stability of pro-apoptotic BAX is dictated by discrete residues of the protein core. Nat. Commun. 12, 4932 (2021).
Kotmayer L. et al. Landscape of BCL2 resistance mutations in a real-world cohort of patients with relapsed/refractory chronic lymphocytic leukemia treated with venetoclax. Int. J. Mol. Sci 24, 5802 (2023).
Shah, N. P. et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002).
Sun, Y. et al. Hydrogen/deuterium exchange and protein oxidative footprinting with mass spectrometry collectively discriminate the binding of small-molecule therapeutics to Bcl-2. Anal. Chem. 97, 4329–4340 (2025).
Brown J. R. et al. Acquired mutations in patients with relapsed/refractory CLL who progressed in the alpine study. Blood Adv. 9, 1918–1926 (2025).
Bird, G. H. et al. A stapled lipopeptide platform for preventing and treating highly pathogenic viruses of pandemic potential. Nat. Commun. 15, 274 (2024).
Bernal, F. et al. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18, 411–422 (2010).
Guerlavais, V. et al. Discovery of sulanemadlin (ALRN-6924), the first cell-permeating, stabilized alpha-helical peptide in clinical development. J. Med Chem. 66, 9401–9417 (2023).
Saleh, M. N. et al. Phase 1 trial of ALRN-6924, a dual inhibitor of MDMX and MDM2, in patients with solid tumors and lymphomas bearing wild-type TP53. Clin. Cancer Res 27, 5236–5247 (2021).
Bird, G. H., Bernal, F., Pitter, K. & Walensky, L. D. Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 446, 369–386 (2008).
Pitter, K., Bernal, F., Labelle, J. & Walensky, L. D. Dissection of the BCL-2 family signaling network with stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 446, 387–408 (2008).
Winter, G. et al. DIALS as a toolkit. Protein Sci. 31, 232–250 (2022).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl Crystallogr 40, 658–674 (2007).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr 66, 213–221 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. Biol. Crystallogr 60, 2126–2132 (2004).
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Hunter, J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 9, 90–95 (2007).
Wales, T. E. & Engen, J. R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 (2006).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank E. Smith for figure preparation, B. Tesar for editorial assistance, K. Song, L. Sebastian, and C. Oyoung for crystallography technical support, and P. Cooper and R. Rusty for helpful discussions. This work was supported by NIH grant R35CA197583 and a William Lawrence and Blanche Hughes Foundation award to L.D.W.; Alex’s Lemonade Stand Foundation Pediatric Oncology Student Training (POST) award and Colby College Linde-Packman Fellowship to T.M.D.; NIH grant R50CA211399 to G.H.B; and NIH grant P30GM133893 for the Advanced Photon Source of the Northeastern Collaborative Access Team beamlines. Additional support was provided by the Wolpoff Family Foundation, the Todd J. Schwartz Memorial Fund, and a research collaboration between T.E.W. and the Waters Corporation.
Author information
Authors and Affiliations
Contributions
L.D.W., T.M.D., U.A., and K.J.K. conceived of the study. G.H.B. synthesized BAD SAHBs. K.J.K. and T.M.D. generated the bacterial and mammalian expression vectors. T.M.D., U.A., and K.J.K. performed pull-down experiments. C.R.B-S. and T.M.D. generated recombinant proteins and performed FP experiments. T.M.D. performed liposomal release assays. X-ray crystallography was conducted by T.M.D. and H.S.S. under the supervision of S.D.P. C.M.C. generated difference distance matrix plots. HDX MS experiments were performed by T.M.D. under the supervision of T.E.W. The paper was written by L.D.W. and T.M.D., and reviewed by all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ganesh Srinivasan Anand, Tudor Moldoveanu and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
DeAngelo, T.M., Adhikary, U., Korshavn, K.J. et al. Structural insights into chemoresistance mutants of BCL-2 and their targeting by stapled BAD BH3 helices. Nat Commun 16, 8623 (2025). https://doi.org/10.1038/s41467-025-63657-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63657-y
