
Abstract
Precise separation of ions of the same polarity and similar valence and size remains a critical need in resource recovery from waste streams. Here, we report the rational design and scalable fabrication of a thin film nanocomposite (TFN) cation exchange membrane to achieve precise selectivity for lithium over competing cations. The precise selectivity is realized by an ultrathin polyamide (PA) layer incorporated with amine functionalized β-monoclinic lithium titanium oxide (N-LTO) nanoparticles using a scalable interfacial polymerization process that allows high N-LTO loading while minimizing interfacial defects. The TFN membrane demonstrates superior Li+ permeability, with Li+/Ca2+ and Li+/Na+ selectivity reaching 173.90 and 13.58, respectively. The Li+/Na+ selectivity is attributed to the Li+-exclusive transport pathway in the layered structure of the N-LTO, while size exclusion by the highly cross-linked N-LTO-PA also contrubutes to the Li+/Ca2+ selectivity. Molecular dynamics simulation shows that the electrical field drives Li+ dehydration and accelerates the migration of the dehydrated Li+ while Na+ is blocked due to its larger size than the Li+ cavity. The high Li+ selectivity and permeability enable energy-efficient, precise, and chemical-free lithium extraction using the electrodialysis process. The TFN membrane architecture also allows simple and scalable fabrication of a multi-functional polymer-inorganic nanocomposite membrane.
Similar content being viewed by others

Introduction
The urgent need to achieve net-zero carbon emissions has brought attention to the recovery and recycling of minerals critical to energy transition1,2. Used in large quantities in batteries for electric vehicles and industrial-scale energy storage, lithium is a critical mineral of high demand3,4. In addition to hard-rock ores, there is abundant lithium in surface and underground brine sources5,6, which currently supplies 50 to 75% of the global lithium demand7. However, the conventional method for lithium extraction from brines relies on the use of massive evaporation ponds and suffers from high water intensity, low lithium recovery, and intensive chemical use8,9,10. Direct lithium extraction (DLE) technologies extract lithium from a brine source without evaporation. DLE technologies, including adsorption10,11, ion exchange8,12, solvent extraction13,14, and membrane processes5, have demonstrated lithium recovery of over 80% but face significant challenges ranging from poor long-term stability of adsorbents to extensive chemical usage and high operating costs15,16,17.
Electrodialysis (ED) is an electric potential-driven membrane process. It can extract and concentrate charged species from a brine under ambient conditions with no chemical usage, minimum water consumption and zero pressure, making it highly attractive for DLE18. However, existing cation exchange membranes (CEMs) used for ED cannot separate Li+ from other cations in the brine source, e.g., Ca2+, Mg2+, and Na+, the concentrations of which are usually orders of magnitude higher than that of Li+ 6. Divalent cations, especially Ca2+ and Mg2+, affect lithium product purity due to co-precipitation during chemical precipitation to form Li2CO3, LiOH, or other lithium products. Removal of Ca²⁺ and Mg²⁺ by chemical precipitation pretreatment, on the other hand, reduces lithium recovery due to co-precipitation of Li+ with the Ca2+ and Mg2+ precipitates. Additionally, co-transport of other cations such as Na⁺ and K⁺ increases energy consumption and hence the operating cost19,20,21.
Progress has been made in developing monovalent selective membranes that limit divalent cation transport by size exclusion and/or Donnan exclusion and high selectivity for Li+ over Mg2+ and Ca2+ has been reported22,23,24. However, achieving precise Li⁺ selectivity over competing ions, remains a significant challenge, especially for monovalent cations (e.g., Na+ and K+), which have the same polarity and valences as well as very similar sizes as Li+18,25. Due to its smaller hydrated ion size and lower hydration energy, Na+ and K+ have higher ionic mobility than Li+ and transports faster than Li+ through conventional membranes6,18. Two approaches have been used to reverse this permeability order. Ligand-grafted membranes enhance the permeability of Li+ relative to other monovalent cations by increasing Li+ solubility and diffusivity via specific interactions with the ligand (e.g., crown ethers20,26, sulfonated polymers27 and dipodandic phosphoryl-ligands28). However, the selectivity achieved was low as the non-selective ion transport through the water-swelled polymer matrix is a major contributor to the overall ion transport flux. A more commonly used approach utilizes lithium-ion sieves (LIS) with Angstrom-sized pores to transport partially or fully dehydrated Li+ while excluding the larger Na+ and K+29,30. LISs used include lithium superionic conductors (LiSiCONs) such as Li1.5Al0.5Ge1.5(PO4)3, Li1.3Al0.3Ti1.7(PO4)331, LiMn2O415, Li2TiO332, and Li4Ti5O1233, sodium superionic conductor-like solid state electrolyte34, 2-D nanosheets with modified interlayer spacing35, and chemically modified metal-organic frameworks (MOFs) and covalent organic frameworks (COFs). With their highly crystalline structure, LiSiCONs exhibit excellent adsorption selectivity for Li+. Membranes prepared with only LiSiCONs have demonstrated extremely high Li+ permeation selectivity against mono- and di-valent competing cations36. However, scaling up the fabrication of defect-free LiSiCON membranes is extremely challenging if possible at all. Alternatively, LiSiCONs can be incorporated in a polymer matrix, although the Li+ selectivity reported using this approach was orders of magnitude lower than the adsorption selectivity of the LiSiCONs used37,38. This can be attributed to two main reasons. First, the incompatibility between the inorganic LiSiCON and the membrane polymer limits the LiSiCON loading in the membrane. Because ions preferentially transport through the non-selective polymer matrix, the overall selectivity of the composite membranes remained low39,40,41. Secondly, large defects at the LiSiCON-polymer interface can lead to leakage of ions through the membrane, which reduces the permselectivity between cations and anions as well as the selectivity for Li+ over other cations42,43,44. Moreover, although some research has developed materials with very high Li selectivity, these materials usually lack other necessary properties such as adequate cation/anion separation, mechanical strength or manufacturing scalability45,46,47. Finally, due to lattice constraint and interfacial resistance48,49, the overall low conductivity of LiSiCON contained membrane hinders a rapid separation of Li+ from the complicated water matrix. Therefore, a facile and scalable fabrication method that enables a membrane architecture with high and uniform loading of LISs to maximize Li selectivity is greatly needed.
Here, we report the development of a novel lithium-selective thin film nanocomposite cation exchange membrane (TFN-CEM) using a simple and scalable fabrication method and demonstrate its performance in separating Li+ from both monovalent and divalent competing cations in an ED process in a wide range of solution conditions. Force field molecular dynamic simulation elucidates the transport mechanisms leading to the high selectivity as well as the critical role of the electric field.
Results
Rational design of Li+ selective cation exchange membranes
To achieve the desired properties of a Li+ selective CEM, we designed a TFN-CEM that contains a cation exchange polymer (CEP) layer, a porous polysulfone (PSf) layer, and a PA skin layer embedded with nanosized monoclinic β-LTO (Fig. 1A and Supplementary Fig. 1). The CEP layer separates cations from anions, the PSf layer provides mechanical strength and a suitable interface for the formation of the LTO-embedded PA layer, and the LTO-embedded PA layer provides separation between Li+ and competing monovalent and divalent cations. Because the formation of the PA layer can only happen on the skin layer (i.e., the dense side) of the asymmetric PSf layer, the CEP layer was casted on the opposite side of the PSf layer, where the large pores allow strong adhesion of the CEP on the PSf layer surface. The monoclinic β-LTO features alternating layers of stable (LiTi₂) and exchangeable (Li₃), which form straight, slit-shaped pores to allow highly selective Li+ transport between the (LiTi₂) layers50,51. In order to create defect-free LTO-PA interfaces, the lab-synthesized N-LTO allowing a high degree of cross-linking with acyl chloride during the interfacial polymerization (IP) process that forms the N-LTO embedded PA layer (N-LTO-PA, Fig. 1A and Supplementary Fig. 1). In this design, the dense PA hinders the transport of divalent cations, while the LTO allows only the transport of Li+, resulting in overall high selectivity between Li+ and competing cations.

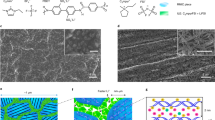
A Schematic illustration of N-LTO synthesis and N-LTO-PA layer formation. In the schematic of the N-LTO-PA layer, the pink area with scattered yellow dots represents the PA network consisting of MPD and TMC. B SEM images of LTO (B1) and N-LTO nanoparticles (B2). C XRD spectra of LTO and N-LTO nanoparticles; peaks that match the standard Li2TiO3 XRD pattern from the Joint Committee on Powder Diffraction Standards (JCPDS) #33-0831 are marked by triangles. D FTIR spectra of LTO and N-LTO nanoparticles. The insert shows the anticipated surface chemistry of N-LTO. The peaks shared by LTO and N-LTO nanoparticles are marked by colored bars. Unique peaks that appeared in N-LTO are marked by arrows. E Photos of the N-LTO-PA side (E1) and the CEP side (E2) of the membrane prepared with 0.5 g L−1 N-LTO (N-LTO-PA-0.5). F Cross-sectional SEM image of the N-LTO-PA-0.5 membrane showing a TFN structure consisting of CEP, PSf and N-LTO-PA layers. The N-LTO-PA layer is too thin to be observed clearly at the top of the PSf layer at this scale, but the surface clearly exhibits characteristic features of the N-LTO-PA layer. G SEM image of the N-LTO-PA layer surface of the N-LTO-PA-0.5 membrane. H Surface SEM images of the commercial CEM (COMM), lab-made PA membrane (PA), unmodified LTO incorpated PA membrnae (LTO-PA), and a membrane formed by reacting N-LTO only with TMC without MPD on the PSf surface (N-LTO-TMC) I Cross-sectional TEM image of the N-LTO-PA-0.5 layer formed on the PSf layer. The bright areas are LTO nanoparticles. J XPS spectra of the N-LTO-PA-0.5 membrane. The insert shows the C1s spectrum.
Achieving uniform and high N-LTO loading in the PA layer
To achieve high Li+ selectivity while maintaining high permeability, it is critical to form an ultra-thin N-LTO-PA layer with uniform and high loading of N-LTO (Fig. 1A). First, the LTO synthesis conditions were controlled to produce nano-sized LTO particles (70.2 ± 8.4 nm in diameter). The scanning electron microscopy (SEM) images (Fig. 1B1) of the LTO showed fairly uniformly sized cubic crystals, similar to what was previously reported52,53. After amine functionalization using 3-aminopropyltriethoxysilane (APTES), the resulting N-LTO showed no significant morphological changes compared to the pristine LTO (Fig. 1B2). Additionally, strong peaks at 18.3°, 35.6°, and 43.3° in the X-ray diffraction (XRD) spectrum of N-LTO match the spectra of standard Li2TiO3 and the unmodified LTO (Fig. 1C), suggesting that the amine functionalization did not change the LTO crystalline structure. Fourier-transform infrared spectroscopy (FT-IR) confirmed successful -NH2 functionalization (Fig. 1D). Both LTO and N-LTO exhibited peaks at 2350, 1850, and 1480 cm⁻¹, corresponding to C=O stretching, Ti-O bond vibration, and O-H bending, respectively54,55. The spectrum of N-LTO, however, featured peaks at 1640 and 3400 cm⁻¹, which are absent in the LTO spectrum and are attributed to N-H bending and N-H stretching, respectively. Distinct peaks at 1060 and 2120 cm⁻¹ were also found on the N-LTO spectrum, corresponding to Si-O and Si-OH bonds originating from APTES56.
The N-LTO nanoparticles were incorporated into the PA matrix to form the N-LTO-PA layer on the skin layer (i.e., the dense side) of the polysulfone support layer (Supplementary Fig. 2). As shown in Fig. 1E1 and E2, the N-LTO-PA layer fabricated with 0.5 g L−1 N-LTO (N-LTO-PA-0.5) was white and shiny, while the CEP layer was brown. The cross-sectional SEM image confirmed the three-layer TFN structure as designed, with a total thickness of 122.3 ± 6.2 μm (Fig. 1F). The dense CEP layer is 64.6 ± 2.1 μm in thickness, and the porous PSf layer is 57.8 ± 3.9 μm. The N-LTO-PA layer exhibits a rugged surface with numerous N-LTO nanoparticles uniformly distributed on the surface (volume fraction DN-LTO,V = 21.4%, Fig. 1G). This is in contrast with the surface of the PA membrane prepared without LTO, which exhibited the ridge and valley surface morphology typical for PA membranes (Fig. 1H and Supplementary Fig. 3). Unlike the as-synthesized N-LTO, the particles on the membrane surface appeared irregular, suggesting that the N-LTO nanoparticles were covered by PA. Unmodified LTO, however, was not successfully incorporated in the PA: few LTO particles were found in the LTO-PA layer with a very low LTO volume fraction of 0.3% (Fig. 1H). The low LTO loading was attributed to the poor chemical compatibility between LTO and PA. Amine functionalization is necessary to create adequate binding between LTO and PA. Furthermore, crosslinking between N-LTO and trimesoyl chloride (TMC) without m-phenylenediamine (MPD) was not able to form a congruent coating (denoted as N-LTO-TMC, DN-LTO,V = 1.4%, Fig. 1H). The formation of a separate PA phase is necessary to ensure proper dispersion of N-LTO in the membrane.
Transmission Electron Microscopy (TEM) characterization of the N-LTO-PA-0.5 layer cross-section showed an ultrathin nanocomposite film of 140.6 ± 12.4 nm in thickness (Fig. 1I). The image also showed high density and uniform distribution of N-LTO throughout the PA layer with a DLTO,V of 21.4%. The Energy Dispersive Spectrometry (EDS) revealed a uniform distribution of Ti, Si and N, which suggests complete coverage of PA and uniform distribution of N-LTO (Supplementary Fig. 4). X-ray photoelectron spectroscopy (XPS) of Si and Ti also confirms the successful incorporation of N-LTO (Fig. 1J). Moreover, the XPS and FT-IR results corroborate the membrane structure as designed (Supplementary Figs. 5 and 6).
Electrochemical impedance of the N-LTO-PA-0.5 membrane was well described via a revised Randles Circuit, where constant phase elements (CPE) and Warburg impedance Ws were used to account for the response caused by the dispersion effect and concentration polarization effect, respectively (Supplementary Information Section 1.3). The Electrochemical impedance spectroscopy (EIS) analyses showed that the membrane has an electrical resistance (5.48 Ω cm² in 0.1 mol L−1 NaCl) similar to commercial CEMs (usually <15 Ω cm²) (Supplementary Fig. 7 and Supplementary Table 1)57,58. A limiting current density of 0.5 mA cm−² was observed in 20 mM NaCl and LiCl solution by chronopotentiometry analyses (Supplementary Fig. 7C). Thus, the current density in subsequent experiments was set at 0.2 mA cm−2.
N-LTO-PA membrane preferentially transports Li+
Adsorption selectivity of LTO
Cation adsorption by LTO was assessed in both single-salt and mixed-salt solutions. LTO was first activated in 0.5 M hydrochloric acid (HCl). The resulting HCl-activated LTO (Supplementary Fig. 8) was then tested for Na+ and Li+ adsorption. Li+ adsorption by LTO follows the Langmuir model, with a saturation adsorption capacity of 34.23 mg g−¹ (Supplementary Fig. 9A), and reached 26.69 mg g−1 Li+ adsorption in a 250 ppm-Li+ solution (Fig. 2A). In contrast, HCl-activated LTO exhibited poor Na+ adsorption, only 0.36 to 0.99 mg g−1 over an initial Na+ concentration range of 250 to 6000 mg L−1 (Supplementary Fig. 9B and Fig. 2A). Similar results were obtained in a mixed-salt solution containing 20 mM each of NaCl and LiCl: 24.73 and 0.33 mg g−1 of Li+ and Na+, respectively, were adsorbed at a LTO dosage of 1 g L−1, a Li+/Na+ adsorption separation factor of 83.26 (Eq. (1)) (Supplementary Fig. 9C). Despite the high adsorption capacity, Li+ adsorption is relatively slow, reaching equilibrium after 48 h (Supplementary Fig. S9). The slow kinetics is due to the small size of the space between the (LiTi2) layers and is a major limitation of most adsorption-based DLE technologies59. The electrical potential in the ED process is expected to accelerate Li+ movement.

A Li+ and Na+ adsorption on bare LTO and N-LTO activated by HCl. B Li+ and Na+ ion flux and Li+/Na+ selectivity obtained by different membranes in ED experiments using a binary (Li+, Na+) solution. The cross-section TEM images (C), Ion flux and Li+/Na+ selectivity (D) and the surface SEM images (E) of membranes prepared at different N-LTO loadings. F The ion selectivity in the extractant chamber using different membranes in a binary (Li, Ca) solution. G Li+ adsorption capacity of HCl activated LTO nanoparticles (top) and ion flux and selectivity of the N-LTO-PA-0.5 membrane (bottom) at different pHs. H The ionic conductivity of different membranes calculated from the measured membrane resistance and thickness. I Comparison of Li+/Na+ and Li+/Ca2+ selectivity (PLi+/Na+ and PLi+/Ca2+) of the membrane developed in the current study (red diamond, “This work”) with other CEMs (blue circles, triangles and squares) reported in the literature. Corresponding data can be found in Supplementary Table 9. Each data point representing one published study. Error bars shown in this figure represent the standard deviation from duplicated or triplicated experiments.
After -NH2 modification, the HCl-activated N-LTO showed reduced Li+ adsorption (17.80 mg g−1 at a dosage of 1 g L−1). Nevertheless, the HCl-activated N-LTO maintained a high Li+ adsorption separation factor of 101.3, with only 0.19 mg g−1 Na+ adsorption in 20 mM NaCl+LiCl solution. The apparent decrease in Li+ adsorption was attributed to a lower effective adsorbent dose (i.e., the added mass of APTES does not contribute to adsorption) (Supplementary Fig. 9D) and blockage of some adsorption sites by the APTES coating. When the mass of APTES was excluded, the adsorption density, 25.26 mg g−1, was similar to that of the HCl-activated LTO.
Ion permeability and selectivity of the TFN-CEM
The ion permeability of the TFN-CEM was first evaluated in a bench-scale ED reactor using the N-LTO-PA-0.5 membrane in place of a CEM using mixed salt solutions (Supplementary Fig. 10). A commercial CEM (denoted as COMM, Supplementary Table 2), a membrane fabricated without N-LTO (denoted as PA)60, a membrane fabricated using unmodified LTO (denoted as LTO-PA), and a membrane fabricated without MPD (denoted as N-LTO-TMC) were tested as controls (Supplementary Table 3). It is noted that all results presented below were obtained by installing the membranes with the polyamide layer facing the feed solution.
The four control membranes exhibited much higher Na⁺ flux (26.46 to 46.27 mmol h−¹ m−²) than Li⁺ (9.74 to 15.16 mmol h−¹ m−²), with a Li⁺/Na⁺ selectivity of 0.21 to 0.5. For the COMM and PA membranes, this is attributed to the smaller hydrated ion size and higher ion mobility of Na+ (Supplementary Table 4, Fig. 2B and Supplementary Fig. 11)61,62,63. Despite the high adsorption selectivity of LTO for Li+, the LTO-PA membrane showed poor Li+ flux relative to Na+ (Li⁺/Na⁺ selectivity of 0.33) due to the very low loading of LTO in the PA layer. The higher Na+ permeability of PA, therefore, led to an overall higher Na+ flux than Li+. The N-LTO-TMC membrane showed spotted coverage of the PSf surface by large aggerates of N-LTO (Fig. 1H). Such incomplete coverage allowed ions to transport through the porous PSf layer without going through a selective layer, resulting in a Li+/Na+ selectivity of only 0.57 (Fig. 2B). In contrast, the N-LTO-PA-0.5 membrane exhibited a Li⁺ flux (41.94 mmol h⁻¹ m⁻²) more than three times that of the Na⁺ flux (13.83 mmol h⁻¹ m⁻²) (Fig. 2B). Because the CEP and PSf layers of the N-LTO-PA-0.5 membrane were the same as those in the PA and LTO-PA membranes (Supplementary Fig. 3), the high Li⁺ flux is attributed to the large number of N-LTO nanoparticles embedded in the PA layer, which provides numerous selective pathways for Li⁺ transport. In addition, N-LTO-PA-0.5 showed a good mass balance (>96.7%) and a cation/anion selectivity of 38.98 (Supplementary Figs. 12 and 13), significantly higher than that of the PA membrane (29.18). This is attributed to the Donnan exclusion by the CEP layer and size exclusion by the PA layer. The size of Cl- (1.81 Å)64 is significantly larger than Na+(0.95 Å), which leads to a complete rejection by surface N-LTO.
Effect of N-LTO loading
As Na+ permeability through PA is higher than Li+, the relative abundance of N-LTO and PA in the N-LTO-PA layer is expected to affect the overall selectivity throughout the membrane. Ion transport through N-LTO-PA membranes prepared using different N-LTO dosages (Supplementary Table 3, denoted as N-LTO-PA-X, X is N-LTO concentrations in g L−1 in the TMC solution) supports this hypothesis. XPS data showed that N-LTO content in the N-LTO-PA layer increased with increasing N-LTO dosage, from 0, to 8.2, 23.4 and 36.9 wt% (Supplementary Section 3.2 and Supplementary Table 5). The cross-sectional TEM images of the PA layers also showed increasing N-LTO density, from 0 to 12.8, 21.4 and 26.8 vol%, with N-LTO dosage (Fig. 2C and Supplementary Table 6). Accordingly, the Li+ to Na+ selectivity increased from 0.24 to 1.61, 3.03, and 13.58 (Fig. 2D). A higher N-LTO loading in the PA formed a better-connected network of N-LTO, which selectively transports Li+. This is evident in the evolution of membrane surface morphology as the N-LTO dosage increased. The PA membrane without N-LTO exhibited a ridge and valley surface morphology (Fig. 2E) typically found in PA reverse osmosis membranes. The number of N-LTO particles visible on the membrane surface increased with increasing N-LTO concentration. The N-LTO-PA-0.5 membrane surface became uniformly covered by N-LTO, providing easily accessible transport channels for Li+. The N-LTO-PA-1.5 membrane surface resembled a tessellation of N-LTO particles due to the very high N-LTO loading throughout the PA layer. This large number of N-LTO nanoparticles forms an extensive network of transport channels for Li+ while excluding Na+. XPS analyses of N-LTO-PA membranes revealed an increased degree of PA cross-linking with increasing N-LTO loading (Supplementary Figs. 14–16 and Supplementary Tables 7, 8), which further reduces Na+ permeability.
Real lithium source brines often contain high concentrations of divalent cations, such as Ca²⁺ and Mg²⁺, which can interfere with Li⁺ extraction by forming scale on the membrane and introducing impurities in the solid lithium product65,66,67. We tested the N-LTO-PA-1.5 membrane, which exhibited highest Li+/Na+ selectivity, in a feed solution containing 20 mM of Li+ and Ca²⁺, respectively. As expected, Li⁺ dominated the overall cation transport, with a Li⁺/Ca²⁺ selectivity of 173.90 (Fig. 2F), 490 times higher than COMM. The very low flux of Ca²⁺ was attributed to its large hydrated ion size and, hence, low permeability through both N-LTO and PA61,68,69,70. The membrane’s high selectivity for Li+ against Na+ and Ca2+ is promising for DLE applications: it is expected to produce high-purity Li products at low energy consumption. The low permeability of Ca2+ will also extend the membrane lifetime by reducing scale formation.
Effect of pH
Li+ adsorption on LTO is a strong function of solution pH, and is known to decrease at lower pH (Fig. 2G) due to (1) competition from H⁺ for adsorption sites, (2) irreversible structural collapse upon H⁺ insertion into the crystal lattice, and (3) electrostatic repulsion by the positive surface charges on HCl activated LTO71,72,73,74. Li+ permeability through the N-LTO-PA-0.5 membranes decreased with decreasing pH with the changes more notable at acidic pHs (Fig. 2G). Na+ permeability, however, increased slightly when pH dropped to below 7, leading to a sharp drop in Li+/Na+ selectivity by 80.6%. Additionally, the charge efficiency decreased significantly at pH <7 (Supplementary Fig. 17), suggesting significant H⁺ permeation through the membrane, which suppresses Li+ transport.
Ionic conductivity
As a CEM, N-LTO-PA membranes need to have adequate ionic conductivity. The ion conductivity of the membranes was determined by measuring the voltage required to achieve a current density of 0.2 mA cm-² in a mixed solution containing 20 mM NaCl and 20 mM LiCl (Fig. 2H and Supplementary Fig. 17)75. Compared with COMM (9.35 mS cm−1), the PA, LTO-PA, and N-LTO-PA-1.5 membranes exhibited notably lower ion conductivity, 2.43, 2.39, and 2.14 mS cm−1, respectively. The decrease in membrane conductivity is attributed to the low conductivity of PA and LTO. The reduced conductivity of all membranes containing PA compared to COMM was due to the resistance of the dense PA network, which is also supported by the voltage profile in Supplementary Fig. 18. However, the slightly lower conductivity of the N-LTO-PA-1.5 membrane than other PA membranes suggests that N-LTO enhanced selectivity significantly with acceptable conductivity loss. Despite such a decrease in membrane conductivity, the overall specific energy consumption (SEC) for lithium recovery using the N-LTO-PA-1.5 membrane, 34.83 kWh (kg Li)−1, is approximately 1/3 of that by COMM and the other control membranes used in this study and comparable with those reported in other studies: reported 12.5 to 95.7 kWh (kg Li)−1 (Supplementary Fig. 19 and Supplementary Table 8).
Performance comparison
The uniform and high loading of N-LTO provided excellent Li+ selectivity against both monovalent and divalent cations (Fig. 2I and Supplementary Table 9). N-LTO-PA-1.5 has the highest Li+/Na+ selectivity among all polymeric or composite membranes reported, and its Li+/Ca2+ selectivity is also among the highest. It is noted that some previously reported lithium selective membranes only have selectivity over divalent cations (e.g. Mg2+) and are labeled as monovalent selective in Fig. 2I with detailed data shown in Supplementary Table 9. In addition, several variations of the membrane architecture were also investigated and their performance compared with the TFN-CEM can be found in Supplementary Section 3.5. Results show that the rationally designed TFC-CEM structure is critical to achieving high selectivity for Li+ (Supplementary Fig. 20).
Modeling Li+ transport through the N-LTO-PA layer
Experimental data shown above suggest that the Li+/Na+ selectivity of the N-LTO-PA membranes may be attributed to the highly selective transport of Li+ through N-LTO. Although monoclinic ß-Li2TiO3 is known to be a selective Li+ adsorbent, Li+ transport through its crystalline structure in the presence of an electrical field has not been studied. To elucidate the ion transport mechanisms through LTO and determine the effect of the electrical field on ion transport rate, molecular dynamics (MD) simulations were performed. In the MD model, the LTO separates the LiCl or NaCl solution from the extractant, i.e., pure water (Supplementary Fig. 21)76. An electric field with intensity ranging from 0.78 to 0.95 V Å−1 was applied parallel to the (Li3) and (LiTi2) layers. It is noted that electric field intensities higher than that used in the experiments were studied in the MD simulation in order to accelerate the simulated processes77, allowing observation of relevant effects within the time frame accessible by MD simulations (typically in the nanosecond range). For comparison, additional simulations were conducted without an applied electrical field (Supplementary Fig. 21).
The MD simulations reveal that Li+ permeation through the LTO is driven by the electric field via an enriching-bumping-hopping mechanism. In the absence of an electric field, Li+ ions are solvated and dispersed in the feed solution (Supplementary Figs. 21A, 22A, 23A), undergoing Brownian motion. Because hydrated Li+ (~7.6 Å in diameter, Supplementary Table 4) is too large to enter the crystal lattice of LTO (opening size ~1.5 Å), dehydration is necessary for Li+ to transport through LTO. MD simulations show that the applied electric potential drives Li+ accumulation at the solution-LTO interface (Fig. 3A and Supplementary Fig. 22), which significantly increases the potential energy of Li+ at the interface, including the adsorbed Li+ and surface Li within the LTO crystal lattice, as well as their thermodynamic fluctuations (Supplementary Fig. 23). This increase in potential energy facilitates the dehydration of Li+, which is evident from the drastic decrease in water density from 30–40 nm−3 to 0 at the solution-LTO interface and the complete absence of water within the LTO structure (Fig. 3B and Supplementary Fig. 24). Inside the LTO structure, Li+ moves along the electrical potential gradient by hopping between Li sites in the (Li3) interlayer, with no Li+ transport occurring in the (LiTi2) layer or at any other atomic sites (Supplementary Fig. 25). The hopping along the electric field moves Li+ through the (Li3) interlayer, eventually releasing Li+ into the extractant (Supplementary Figs. 25 and 26). Increasing the electric field intensity leads to more Li+ accumulation at the LTO-feed solution interface and higher Li+ potential energy (Supplementary Fig. 23), enhancing its thermodynamic fluctuations (evident from the increased scatter distribution in the LTO structure shown in Supplementary Fig. 23A–F) and increasing the probability of hopping and bumping events.

The cross-section number density of A Li+ and B water in the simulated scenario. Different phases are divided by background colors and captions at the top. C Trajectories of Li+ (light blue) and Na+ (purple) transport through an LTO crystal at an electric field intensity of 0.87 V Å−1. The electric field is applied along the +x direction. LTO size: ~15 Å in the x direction; ~26 Å in the z direction. D The number of Li+ and Na+ ions transported through the LTO structure as a function of simulation time and electric field intensity. The steady-state transport rates, determined by linear regression of the data, are also shown (see Supplementary Table 5 for the goodness of fit). For Na+, no transport was observed at all electrical field intensities; data are only shown for the highest intensity (0.95 V Å−1). E The enlarged LTO structure highlights a (Li3) interlayer sandwiched between two (LiTi2) layers. The distance between (LiTi2) layer centers is ~4.4 Å and the width of the vacancies in (Li3) layers is ~1.5 Å. F Schematic of the transport model within the N-LTO-PA layer. The PA matrix (gray-stripped background) and N-LTO (light-blue diamonds) were simplified for clarity. Molecular dynamics trajectory data at the initial and final configurations can be found in Supplementary Data file.
The electric field not only facilitates the dehydration of Li+ but also serves as the primary driving force for its transport. With detailed molecular dynamics trajectory data at the initial and final configurations provided in Supplementary Data 1 file, Fig. 3C presents the trajectories of Li+ transport across the LTO structure, which exhibits two stages: an initial stage with a higher transport rate transitions to a lower, steady-state transport rate. The initial stage corresponds to the fast accumulation of Li+ ions at the LTO-feed solution interface. This leads to a significant momentum transfer from Li+ to LTO, triggering a high Li flux at the beginning. After the accumulation reaches equilibrium, a steady-state flux is reached. The steady-state Li+ flux increases with the electric field intensity (Supplementary Table 10), approaching a plateau at 0.95 V·Å−1 (Supplementary Fig. 27), when Li+ adsorption on the LTO surface reaches saturation, as evidenced by the plateau of Li+ accumulation and potential energy (Supplementary Figs. 22 and 23).
When the NaCl solution is used, the accumulation of Na+ at the solution-LTO interface resulting from the applied electrical field also led to a significant increase in the potential energy of Na+ and Li sites at the interface. This facilitated Na+ dehydration is evidenced by the decrease in the water/Na+ number ratio near the interface upon applying the electric field (Supplementary Fig. 28). However, no Na+ permeation was observed under any conditions simulated (Fig. 3D and Supplementary Figs. 28–30). This is due to the larger size of dehydrated Na+ (1.90-2.32 Å in diameter) that prevents it from entering the vacancies (1.50 Å in channel width)78,79 created by the release of Li in the (Li3) interlayer (Fig. 3E).
Given the structure of the N-LTO-PA layer, ion transport through it can be described using a resistance-in-parallel model (Fig. 3F), where both ions (Li+ and Na+) can permeate through the pristine PA matrix (F1), but only Li+ can transport through N-LTO. With an increased N-LTO loading, the Na+ trajectory becomes more tortuous and lengthier, while Li+ gains a larger network of transport channels (F2).
Stability of TFN-CEM
Structural instability of inorganic LISs due to metal leaching during ion exchange is a well-known issue74,80,81. To evaluate the stability of the TFN-CEM, we conducted both experimental tests and extended MD simulations. After 10 h of consecutive testing, a time frame during which detectable metal leaching and structural collapse have been reported for LTO72,82,83, all membranes maintained their physical integrity, with no visible changes in membrane morphology (Fig. 4A). In addition, no Ti4+ was detected in either the feed or the extractant chambers (<1 μmol L−1), confirming the structural integrity of the LTO (Fig. 4B). The test continued for a total of 24 consecutive experiments, each lasting 2 h. As shown in Fig. 4C, the membrane exhibited highly stable performance with a high Li+ flux of 43.77 ± 2.07 mmol L−1 h−1 and a low Na+ flux of 3.28 ± 0.86 mmol L−1 h−1, maintaining a Li+/Na+ selectivity of 13.42 ± 0.84 throughout the experiments. In addition, the used TFN-CEM showed excellent stability in water. After soaking in DI water for 14 days, its Li+/Na+ and Li+/Ca2+ selectivity remained unchanged (Fig. 4D), and there were no detectable changes in membrane structure and morphology. The stability of the LTO was also demonstrated by MD simulation results. The crystalline structure of the LTO remained stable over an extended simulation time of 4 ns in an electric field (0.78 V Å−1) 106–107 times higher than that typically used in ED processes (Fig. 4E). Since this study primarily reports the initial development of the N-LTO-PA membrane, extended evaluation of membrane stability under realistic water matrices, including systematic monitoring of membrane scaling and fouling, will be the subject of future investigations.

A The SEM images of membranes before and after 5 consecutive 2-h experiments. B Ion concentrations in feed and extractant chambers after 10 h of testing. C The cation flux and Li+/Na+ selectivity of the N-LTO-PA-1.5 membrane in 24 consecutive 2-h experiments. D The Li+/Na+ and Li+/Ca2+ selectivity of the N-LTO-PA-1.5 membrane before and after 14 days of soaking in DI water. E The number of Li+ ions transported through the LTO over an extended simulation time of 4 ns under 0.78 V Å−1 field intensity. Error bars shown in this figure represent the standard deviation value from duplicated or triplicated experiments.
Discussions
In this study, we designed and fabricated a lithium selective cation exchange membrane with a novel TFN structure containing monoclinic β-LTO as the lithium-ion sieve. Experimental results show that the TFN structure allows independent control and tuning of the cation/anion permselectivity, mechanical strength, and Li+ selectivity. Unprecedented selectivity for Li+ over co-existing representative monovalent (Na+) and divalent cations (Ca2+) was achieved in an ED process by the ultra-thin N-LTO-PA layer formed using a modified IP method that overcame key limitations of previously reported fabrication methods for LIS-embedded polymeric membranes. Specifically, surface functionalization of the monoclinic β-LTO and the modified IP protocol improved the compatibility of LTO with the PA polymer matrix and allowed dense cross-linking at the LTO-PA interface, achieving uniform and high LTO loading without forming defects. The polymer matrix also helped maintain LTO structural integrity in long-term operation. Reactive force field molecular dynamics simulations revealed the critical role of the monoclinic β-LTO crystalline structure and the electric field. It showed that the narrow (Li3) interlayers provide an effective transport pathway for dehydrated Li+ but exclude the larger Na+. In addition, the electrical field greatly accelerates Li+ adsorption and transport through the LTO. The excellent properties of the N-LTO-PA membranes would enable highly efficient Li+ extraction using ED processes. More importantly, the rational design of the membrane structure serves as an excellent template for the fabrication of selective ion exchange membranes targeting different ions, a critical need for resource recovery.
Methods
N-LTO nanoparticle synthesis and adsorption experiments
The N-LTO synthesis method was modified from previous reports33,84. LTO was first synthesized using a hydrothermal process and then functionalized with -NH2 functional groups. Briefly, 11.95 g of LiOH (battery grade, Alfa Asear) was dissolved in 100 mL ethanol-water solution (volume ratio of 1:4). Then 1.429 g of TiO2 nanoparticles (~20 nm in diameter, ACS grade, >99%, Thermo Fisher) were added into the LiOH solution and ultrasonicated for 1 h. The suspension was transferred into a Teflon-lined stainless-steel autoclave and heated at 180 °C for 12 h. The white powder obtained was washed with 25 vol% ethanol in DI water 3 times, dried in a vacuum oven at 70 °C for 24 h, and calcined at 700 °C in air for 1 h. Subsequently, 100 mg of the LTO powder harvested was added to 10 mL of 2 vol% (3-aminopropyl) triethoxysilane (APTES, 99%, Alfa Aesar) in 200 proof ethanol (Fisher Scientific). After 2 h of intensive stirring at 50 °C, the modified N-LTO was retrieved by centrifugation at 2400 × g for 10 min, washed with 25 vol% ethanol water solution 3 times, and dried at 40 °C.
Adsorption experiments were conducted by HCl-activated LTO (Supplementary Section 2). For single salt adsorption tests, the electrolyte containing 5 mg of target cations (LiCl: 30.72 mg, >99%, Thermo Fisher; NaCl: 12.71 mg, >99%, Thermo Fisher) was dissolved in 20 mL DI water. Aliquots of 20 mg HCl-activated LTO were added to 20 mL LiCl or NaCl solutions in 40 mL glass vials, each containing 250 mg/L of Li+ or Na+. For the mixture adsorption test, 20 mM of LiCl and 20 mM NaCl mixture solution was used with 1 g L−1 adsorbent dosage and pH was adjusted to 2, 5, 7, 9 or 12 using HCl (>99%, Sigma Aldrich) or KOH (>99%, Sigma Aldrich). The mixtures were agitated for 72 h; samples were taken at predetermined time intervals and analyzed for Li+ or Na+ concentration using inductively coupled plasma optical emission spectroscopy (ICP-OES, detection limit 0.2 ppm) and ion chromatography (IC, detection limit 0.01 ppm). Adsorption separation factor αi/j was calculated as below:
where Qi/j is the adsorption density of ion i/j (mg g−1), Cequilibrium,i/j is the concentration of ion i/j in the solution when adsorption reaches equilibrium (mg L−1).
Fabrication of TFN-CEM membranes
The PSf layer was fabricated first using the non-solvent-induced phase separation (NIPS) technique. Briefly, 15 wt% PSf (average MW ~ 22 kDa, Sigma-Aldrich), 8 wt% polyvinylpyrrolidone (PVP, average MW ~ 10 kDa, Sigma-Aldrich) and 77 wt% N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich) was mixed at 50 °C for 24 h. Then the mixture was casted on a glass plate to form a liquid film and set in a 60 °C water bath for 20 min. The big pore side of PSf was then coated with a layer of CEP (MD9200, MW 112,500 g mol−1, Kraton) using a flow coater with a solution loading of 200 μL cm−2 and a casting speed of 1 cm s−1. The thin N-LTO-PA layer was formed on the opposite side (small pore side) of the PSf layer by interfacial polymerization85. Briefly, 10 mg N-LTO was mixed with 20 mL 0.4 wt% trimesoyl chloride (TMC, 98%, Thermo Fisher) in n-hexane (99%, Thermo Fisher) and sonicated for 30 min. The PSf surface was first contacted with 2.0 wt% MPD aqueous solution for 3 min. After removing the excess MPD solution, the surface was exposed to the N-LTO/TMC mixture for 30 s. This process was repeated 3 times followed by a 10 min drying step in a 40 °C oven after each time for a complete coverage of N-LTO-PA. The sample was then rinsed with n-hexane, dried, and cured at 60 °C for 10 min. The detailed fabrication process and N-LTO-PA membranes with different N-LTO dosage can be found in Supplementary Section 1.
Materials characterization
LTO, N-LTO, and the membranes were characterized by scanning electron microscopy (SEM, FEI Quanta 400, Thermo Fisher Scientific, Waltham, Massachusetts, USA) with energy dispersive spectrometry (EDS) analysis and transmittance electron microscopy (TEM, JEOL JEM-1400 Flash). The chemical composition of the membranes was characterized using X-ray Photoelectron Spectroscopy (XPS, PHI Quentera, ULVAC-PHI, Japan), recorded from 50 to 650 eV in binding energy. The crystallinity of LTO and N-LTO particles was examined by X-Ray Diffraction Crystallography (XRD, Rigaku DMAX West, USA) with a 2θ value ranging from 20° to 80°. The characteristic functional groups of the N-LTO and the membranes were determined by Fourier-Transform Infrared Spectroscopy (FTIR, Nicolet, Thermo fisher Scientific), recorded from 400 to 4000 cm−1 in wave number.
Membrane resistance was measured by Electrochemical Impedance Spectroscopy (EIS) in a 0.1 mol L−1 NaCl solution using a two-compartment cell setup. All measurements were performed using an electrochemical workstation (CHI 760, CH Instrument, TX, US) operated at frequencies from 10−2 Hz to 106 Hz and an amplitude of 2 mV. The limited current density (LCD) of N-LTO-PA membrane was measured in the voltage range of 0 to 10 V using 5 cm2 membrane samples in a solution containing 20 mM NaCl and 20 mM LiCl.
Ion transport in ED
The ion transport properties of the membranes were characterized in a bench-scale three-chamber ED cell (Supplementary Fig. 9) with an effective membrane area of 5 cm2 per membrane. All TFN-CEMs were installed with the polyamide layer facing the feed solution. Mixed solutions containing 20 mM LiCl and 20 mM NaCl at pH 2, 4, 7, 10 or 12 were used as feed solutions and DI water as the extractant. Both feed and extractant solutions were circulated at a flow rate of 20 mL min−1. One mM KCl solution was used as the anolyte. A constant current density of 0.2 mA cm−2 was applied. Each test was run for 120 min. Samples were taken at predetermined intervals from both the feed and extractant chambers and analyzed by ion chromatography (Dionex Aquion). IC calibration was conducted for each solution condition used. Ion flux fi (mol h−1 m−2) was calculated from the concentration change in the receiving chamber:
Here, Cr,i is the measured concentration of ion i in the receiving chamber (mol m−3), t is time (h), Vr is the receiving chamber volume (m3), and A is the effective membrane area (m2). Ion flux calculated from concentration decrease in the feed chamber agreed well (101.6–104.8% of Ji) with that calculated from concentration increase in the receiving chamber. The slightly higher flux calculated from the feed concentration may be ascribed to adsorption by the membrane.
The permeation selectivity of ion i over ion j, Pi/j was calculated as below:
where f i/j is the flux of ion i/j in the receiving chamber (mol m−3), and Cf,i/j is the initial concentration of ion i/j in the feed chamber (mol m−3).
MD simulation of ion transport through LTO
The Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) was used for molecular dynamics simulation, and the Open Visualization86 Tool (OVITO) was used for visualization87,88. The timestep was set as 0.5 fs for all simulations. First, energy minimization was run using the Polak-Ribiere version of the conjugate gradient algorithm. Next, a relaxation stage of 1 ns was run in the NPT ensemble to allow equilibrium at target temperature and pressure (300 K and 1 atm). Finally, a production stage lasting for 1 ns was run in a canonical ensemble (NVT) by applying an external electrical field in the x direction to generate an ionic current. Given the limited time scale of MD simulation, a high electric field with a strength of 0.78–0.95 V Å−1 was used. The atomic trajectory was sampled every 0.5 ps for analysis. Both NPT and NVT ensembles used the Nose-Hoover thermostat and barostat with a temperature damping of 50 fs and pressure damping of 500 fs. ReaxFF was used to describe the interatomic interaction within the built MD system76,88. During the application of the external electric field, the titanium framework was tethered by springs (spring constant = 5000 kJ mol−1 nm−2) to avoid the drift of the LTO structure due to the induced ionic current. An implicit wall with a repulsive-only harmonic spring potential was used on the left edge of the simulation box to enclose the chloride ions and prevent their migration to the right reservoir.
Data availability
All data generated or analyzed during this study are reported in this published article and its Supplementary Information files, and are available from the author upon request.
References
Paranthaman, M. P. et al. Recovery of lithium from geothermal brine with lithium–aluminum layered double hydroxide chloride sorbents. Environ. Sci. Technol. 51, 13481–13486 (2017).
Grosjean, C., Miranda, P. H., Perrin, M. & Poggi, P. Assessment of world lithium resources and consequences of their geographic distribution on the expected development of the electric vehicle industry. Renew. Sustain. Energy Rev. 16, 1735–1744 (2012).
Sbordone, D. A., Di Pietra, B. & Bocci, E. Energy analysis of a real grid connected lithium battery energy storage system. Energy Procedia 75, 1881–1887 (2015).
Kavanagh, L., Keohane, J., Garcia Cabellos, G., Lloyd, A. & Cleary, J. Global lithium sources—industrial use and future in the electric vehicle industry: a review. Resources 7, 57 (2018).
Xiong, J., Zhao, Z., Liu, D. & He, L. Direct lithium extraction from raw brine by chemical redox method with LiFePO4/FePO4 materials. Sep. Purif. Technol. 290, 120789 (2022).
Disu, B. et al. Review of recent advances in lithium extraction from subsurface brines. Geoenergy Sci. Eng. 241, 213189 (2024).
Stringfellow, W. T. & Dobson, P. F. Technology for the recovery of lithium from geothermal brines. Energies 14, 6805 (2021).
Samadiy, M. & Deng, T. Lithium recovery from water resources by ion exchange and sorption method. J. Chem. Soc. Pak. 43, 406–406 (2021).
Zandvakili, S. & Ranjbar, M. Preparation and characterisation of lithium ion exchange composite for the recovery of lithium from brine. Miner. Process. Extr. Metall. 127, 176–181 (2018).
Zhang, L., Zhou, D., Yao, Q. & Zhou, J. Preparation of H2TiO3-lithium adsorbent by the sol–gel process and its adsorption performance. Appl. Surf. Sci. 368, 82–87 (2016).
Ma, L.-W., Chen, B.-Z., Chen, Y. & Shi, X.-C. Preparation, characterization and adsorptive properties of foam-type lithium adsorbent. Microporous Mesoporous Mater. 142, 147–153 (2011).
Nishihama, S., Onishi, K. & Yoshizuka, K. Selective recovery process of lithium from seawater using integrated ion exchange methods. Solvent Extr. Ion Exch. 29, 421–431 (2011).
Lee, D. A., Taylor, W. L., McDowell, W. J. & Drury, J. S. Solvent extraction of lithium. J. Inorg. Nucl. Chem. 30, 2807–2821 (1968).
Nguyen, T. H. & Lee, M. S. A review on the separation of lithium ion from leach liquors of primary and secondary resources by solvent extraction with commercial extractants. Processes 6, 55 (2018).
An, X. et al. Co-doping induced Mn-vacancy LiMn2O4 based membrane electrode for lithium extraction by electrochemically switched ion permselective process. Desalination 591, 118016 (2024).
Flexer, V., Baspineiro, C. F. & Galli, C. I. Lithium recovery from brines: a vital raw material for green energies with a potential environmental impact in its mining and processing. Sci. Total Environ. 639, 1188–1204 (2018).
Swain, B. Recovery and recycling of lithium: A review, Sep. Purif. Technol, 172, 388–403 (2017).
Wu, L. et al. Lithium recovery using electrochemical technologies: advances and challenges. Water Res. 221, 118822 (2022).
Ounissi, T., Dammak, L., Fauvarque, J.-F. & Selmane Bel Hadj Hmida, E. Ecofriendly lithium-sodium separation by diffusion processes using lithium composite membrane. Sep. Purif. Technol. 275, 119134 (2021).
Warnock, S. J. et al. Engineering Li/Na selectivity in 12-crown-4–functionalized polymer membranes. Proc. Natl. Acad. Sci. USA 118, e2022197118 (2021).
Pramanik, B. K., Asif, M. B., Kentish, S., Nghiem, L. D. & Hai, F. I. Lithium enrichment from a simulated salt lake brine using an integrated nanofiltration-membrane distillation process. J. Environ. Chem. Eng. 7, 103395 (2019).
Li, J. et al. Mono-valent cation selective membranes for electrodialysis by introducing polyquaternium-7 in a commercial cation exchange membrane. J. Membr. Sci. 486, 89–96 (2015).
Pang, X. et al. Enhanced monovalent selectivity of cation exchange membranes via adjustable charge density on functional layers. J. Membr. Sci. 595, 117544 (2020).
Ying, J. et al. Layer-by-layer assembly of cation exchange membrane for highly efficient monovalent ion selectivity. Chem. Eng. J. 446, 137076 (2022).
Sun, Y., Wang, Y., Liu, Y. & Xiang, X. Highly efficient lithium extraction from brine with a high sodium content by adsorption-coupled electrochemical technology. ACS Sustain. Chem. Eng. 9, 11022–11031 (2021).
Yin, X., Xu, P. & Wang, H. Modification of cation exchange membranes for enhanced extraction of lithium from magnesium and sodium brine solutions via selective electrodialysis. J. Membr. Sci. 701, 122705 (2024).
Sharma, P. P. et al. Sulfonated poly (ether ether ketone) composite cation exchange membrane for selective recovery of lithium by electrodialysis. Desalination 496, 114755 (2020).
Kireeva, N., Baulin, V. E. & Tsivadze, A. Y. A machine learning-based study of Li+ and Na+ metal complexation with phosphoryl-containing ligands for the selective extraction of Li+ from brine. ChemEngineering 7, 41 (2023).
Weng, D. et al. Introduction of manganese based lithium-ion sieve—a review. Prog. Nat. Sci. Mater. Int. 30, 139–152 (2020).
Xu, X. et al. Extraction of lithium with functionalized lithium ion-sieves. Prog. Mater. Sci. 84, 276–313 (2016).
Seo, D., Lee, J., Kong, S. R., Sim, G. & Park, Y. Selective lithium extraction from salt-lake brine using LATP-incorporated cellulose membranes in electrically driven systems. J. Membr. Sci. 718, 123676 (2025).
Yao, J. et al. Composite flat-sheet membrane adsorbent of Li2TiO3-ethylene-co-vinyl alcohol (LTO-EVAL) for lithium extraction. Chem. Eng. J. 496, 154122 (2024).
Liu, W. et al. Mesoporous spinel Li4Ti5O12 nanoparticles for high rate lithium-ion battery anodes. Electrochim. Acta 133, 578–582 (2014).
Patel, S. K., Iddya, A., Pan, W., Qian, J. & Elimelech, M. Approaching infinite selectivity in membrane-based aqueous lithium extraction via solid-state ion transport. Sci. Adv. 11, eadq9823 (2025).
Lu, Z., Wu, Y., Ding, L., Wei, Y. & Wang, H. A lamellar MXene (Ti3C2T)/PSS composite membrane for fast and selective lithium-ion separation. Angew. Chem. Int. Ed. 60, 22265–22269 (2021).
Fan, F. et al. A bioinspired membrane with ultrahigh Li+/Na+ and Li+/K+ separations enables direct lithium extraction from brine. Adv. Sci. 11, 2402898 (2024).
Yang, H. & Wu, N. Ionic conductivity and ion transport mechanisms of solid-state lithium-ion battery electrolytes: a review. Energy Sci. Eng. 10, 1643–1671 (2022).
Yu, X. & Manthiram, A. A review of composite polymer-ceramic electrolytes for lithium batteries. Energy Storage Mater. 34, 282–300 (2021).
Razmjou, A., Asadnia, M., Hosseini, E., Habibnejad Korayem, A. & Chen, V. Design principles of ion selective nanostructured membranes for the extraction of lithium ions. Nat. Commun. 10, 5793 (2019).
Chung, K.-S., Lee, J.-C., Kim, W.-K., Kim, S. B. & Cho, K. Y. Inorganic adsorbent containing polymeric membrane reservoir for the recovery of lithium from seawater. J. Membr. Sci. 325, 503–508 (2008).
Sun, D. et al. Synthesis of ion imprinted nanocomposite membranes for selective adsorption of lithium. Sep. Purif. Technol. 194, 64–72 (2018).
Sahin, S., Dykstra, J. E., Zuilhof, H., Zornitta, R. L. & de Smet, L. C. P. M. Modification of cation-exchange membranes with polyelectrolyte multilayers to tune ion selectivity in capacitive deionization. ACS Appl. Mater. Interfaces 12, 34746–34754 (2020).
Yang, S. et al. Cation exchange membranes coated with polyethyleneimine and crown ether to improve monovalent cation electrodialytic selectivity. Membranes11, 351 (2021).
Lind, M. L., Eumine Suk, D., Nguyen, T.-V. & Hoek, E. M. V. Tailoring the structure of thin film nanocomposite membranes to achieve seawater RO membrane performance. Environ. Sci. Technol. 44, 8230–8235 (2010).
Pang, X. et al. Preparation of monovalent cation perm-selective membranes by controlling surface hydration energy barrier. Sep. Purif. Technol. 270, 118768 (2021).
Ren, C. E. et al. Charge- and size-selective ion sieving through Ti3C2Tx MXene membranes. J. Phys. Chem. Lett. 6, 4026–4031 (2015).
Nie, X.-Y., Sun, S.-Y., Song, X. & Yu, J.-G. Further investigation into lithium recovery from salt lake brines with different feed characteristics by electrodialysis. J. Membr. Sci. 530, 185–191 (2017).
Bachman, J. C. et al. Inorganic Solid-State Electrolytes for Lithium Batteries: Mechanisms and Properties Governing Ion Conduction. Chem. Rev. 116, 140–162 (2016).
Thangadurai, V. & Weppner, W. Recent progress in solid oxide and lithium ion conducting electrolytes research. Ionics 12, 81–92 (2006).
He, G., Zhang, L., Zhou, D., Zou, Y. & Wang, F. The optimal condition for H2TiO3–lithium adsorbent preparation and Li+ adsorption confirmed by an orthogonal test design. Ionics 21, 2219–2226 (2015).
Sanjeev, M., Gilbert, M. R. & Murphy, S. T. Anisotropic thermal conductivity in Li2TiO3 ceramic breeder materials. Fusion Eng. Des. 170, 112710 (2021).
Du, Y., Du, D., Feng, Q. & Yang, X. Delithation, exfoliation, and transformation of rock-salt-structured Li2TiO3 to highly exposed {010}-faceted anatase. ACS Appl. Mater. Interfaces 7, 7995–8004 (2015).
Guo, H. et al. Synthesis, characterization and sintering of Li2TiO3 nanoparticles via low temperature solid-state reaction. Ceram. Int. 46, 1816–1823 (2020).
Ramaraghavulu, R., Buddhudu, S. & Bhaskar Kumar, G. Analysis of structural and thermal properties of Li2TiO3 ceramic powders. Ceram. Int. 37, 1245–1249 (2011).
Zhang, L.-Y. et al. Preparation of Ni-doped Li2TiO3 using an inorganic precipitation–peptization method. Coatings 9, 701 (2019).
CHUNLIWANG991. FTIR Functional Group Database Table with Search - InstaNANO. https://instanano.com/all/characterization/ftir/ftir-functional-group-search/ (2024).
Honarparvar, S. et al. Frontiers of membrane desalination processes for brackish water treatment: a review. Membranes 11, 246 (2021).
Zhu, S., Kingsbury, R. S., Call, D. F. & Coronell, O. Impact of solution composition on the resistance of ion exchange membranes. J. Membr. Sci. 554, 39–47 (2018).
Yang, S., Wang, Y., Pan, H., He, P. & Zhou, H. Lithium extraction from low-quality brines. Nature 636, 309–321 (2024).
Zhang, F., Fan, J. & Wang, S. Interfacial polymerization: from chemistry to functional materials. Angew. Chem. Int. Ed. 59, 21840–21856 (2020).
Wang, P. et al. Ultrafast ion sieving using nanoporous polymeric membranes. Nat. Commun. 9, 569 (2018).
Volkov, V. I. et al. Hydration and diffusion of H+, Li+, Na+, Cs+ ions in cation-exchange membranes based on polyethylene- and sulfonated-grafted polystyrene studied by NMR technique and ionic conductivity measurements. Membranes 10, 272 (2020).
Ersöz, M. Diffusion and selective transport of alkali cations on cation-exchange membrane. Sep. Sci. Technol. 30, 3523–3533 (1995).
AL-Bazali, T. Insight on the inhibitive property of potassium ion on the stability of shale: a diffuse double-layer thickness (Κ−1) perspective. J. Pet. Explor Prod. Technol. 11, 2709–2723 (2021).
Dobson, P. et al. Characterizing the geothermal lithium resource at the salton sea. https://doi.org/10.2172/2222403 (2023).
Firdiyono, F. et al. The degree of lithium (Li) stability compared to calcium (Ca) and magnesium (Mg) from low lithium grade brine water with addition of limestone and oxalic acid. IOP Conf. Ser. Mater. Sci. Eng. 858, 012044 (2020).
Khalil, A., Mohammed, S., Hashaikeh, R. & Hilal, N. Lithium recovery from brine: recent developments and challenges. Desalination 528, 115611 (2022).
Zhao, Z. et al. Exploring ions selectivity of nanofiltration membranes for rare earth wastewater treatment. Sep. Purif. Technol. 289, 120748 (2022).
Sarkar, P., Modak, S. & Karan, S. Ultraselective and highly permeable polyamide nanofilms for ionic and molecular nanofiltration. Adv. Funct. Mater. 31, 2007054 (2021).
Shen, L. et al. Polyamide-based membranes with structural homogeneity for ultrafast molecular sieving. Nat. Commun. 13, 500 (2022).
Yuan, H., Besselink, R., Liao, Z. & ten Elshof, J. E. The swelling transition of lepidocrocite-type protonated layered titanates into anatase under hydrothermal treatment. Sci. Rep. 4, 4584 (2014).
Dai, X. et al. Al-doped H2TiO3 ion sieve with enhanced Li+ adsorption performance. RSC Adv. 11, 34988–34995 (2021).
Zhao, B. et al. The adsorption behavior of lithium on spinel titanium oxide nanosheets with exposed (1−14) high-index facets. Dalton Trans. 49, 14180–14190 (2020).
Tangkas, I. W. C. W. H. et al. Synthesis of titanium ion sieves and its application for lithium recovery from artificial indonesian geothermal brine. J. Sustain. Metall. 9, 613–624 (2023).
Díaz, J. C. & Kamcev, J. Ionic conductivity of ion-exchange membranes: measurement techniques and salt concentration dependence. J. Membr. Sci. 618, 118718 (2021).
van Duin, A. C. T., Dasgupta, S., Lorant, F. & Goddard, W. A. ReaxFF: a reactive force field for hydrocarbons. J. Phys. Chem. A 105, 9396–9409 (2001).
Makowska-Janusik, M., Reis, H., Papadopoulos, M. G., Economou, I. G. & Zacharopoulos, N. Molecular dynamics simulations of electric field poled nonlinear optical chromophores incorporated in a polymer matrix. J. Phys. Chem. B 108, 588–596 (2004).
Jiang, Y. et al. Thermodynamic stabilities of perfect and vacancy-defected Li 2 Ti O 3 (001) surfaces from first-principles analyses. Phys. Rev. Appl. 11, 054088 (2019).
Nightingale, E. R. Jr Phenomenological theory of ion solvation. Effective radii of hydrated ions. J. Phys. Chem. 63, 1381–1387 (1959).
Saravaia, H., Gupta, H. & Kulshrestha, V. Single step synthesis of a magnesium doped lithium manganese oxide ion sieve nanomaterial and a SPES/ion sieve composite membrane for the separation of lithium. RSC Adv. 6, 106980–106989 (2016).
Li, X. et al. Membrane-based technologies for lithium recovery from water lithium resources: a review. J. Membr. Sci. 591, 117317 (2019).
Wang, Q. et al. Titanium-based lithium ion sieve adsorbent H2TiO3 with enhanced Li+ adsorption properties by magnetic Fe doping. Sep. Purif. Technol. 346, 127455 (2024).
Lawagon, C. P. et al. Adsorptive Li+ mining from liquid resources by H2TiO3: equilibrium, kinetics, thermodynamics, and mechanisms. J. Ind. Eng. Chem. 35, 347–356 (2016).
Wang, L. et al. Understanding the effect of preparative approaches in the formation of “Flower-like” Li4Ti5O12—multiwalled carbon nanotube composite motifs with performance as high-rate anode materials for Li-ion battery applications. J. Electrochem. Soc. 164, A524 (2017).
Nulens, I. et al. MPD and TMC supply as parameters to describe synthesis-morphology-performance relationships of polyamide thin film composite membranes. J. Membr. Sci. 667, 121155 (2023).
Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the open visualization tool. Model. Simul. Mater. Sci. Eng. 18, 015012 (2009).
Thompson, A. P. et al. LAMMPS—a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Ganeshan, K. et al. Structure and dynamics of aqueous electrolytes confined in 2D-TiO2/Ti3C2T2 MXene heterostructures. ACS Appl Mater. Interfaces 12, 58378–58389 (2020).
Acknowledgements
This work was supported by the NSF Nanosystems Engineering Research Center for Nanotechnology-Enabled Water Treatment (EEC-1449500, Q.L., J.L.), the US Department of Interior (R23AC00442, Q.L.), the NSF Big-Data Private-Cloud Research Cyberinfrastructure MRI (CNS-1338099, K.G.). We thank the Shared Equipment Authority at Rice University, which provided access to research equipment at the Electron Microscopy Center (EMC) and other major instrumentation at Rice and the Rice University Center for Research Computing (CRC).
Author information
Authors and Affiliations
Contributions
Conceptualization: Y.F., Y.Z., J.L., Q.L. Methodology: Y.F., Y.Z., W.C., Q.L. Investigation: Y.F., Y.Z., X.H., W.C., X.W., Y.L., N.Y., M.M., T.C., Z.H., C.H., J.L., Q.L. Visualization: Y.F., X.H., K.Z., N.Y., Y.L. Supervision: K.Z., N.Y., K.G., J.L., Q.L. Writing—original draft: Y.F., Y.Z., W.C. Writing—review & editing: Y.F., Y.Z., W.C., X.H., K.G., N.Y., J.L., Q.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Soryong Chae, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Feng, Y., Zhu, Y., Chen, W. et al. A rationally designed scalable thin film nanocomposite cation exchange membrane for precise lithium extraction. Nat Commun 16, 8618 (2025). https://doi.org/10.1038/s41467-025-63660-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63660-3
