
Abstract
Aberrant activation of Wnt/β-catenin signaling is proposed as a major molecular mechanism underlying the occurrence and progression of colorectal cancer (CRC). However, the precise mechanisms controlling the accumulation of β-catenin protein in CRC cells remain incompletely understood. Here, we show that TRIM24 is elevated in CRC tissues and partially distributed in the cytoplasm. TRIM24 is phosphorylated at serine 1042 by Aurora kinase B (AURKB), which promotes its cytoplasmic distribution. Subsequently, TRIM24 activates Wnt/β-catenin signaling by facilitating AKT activation through interaction with and ubiquitination of its negative regulator von Hippel-Lindau (VHL), resulting in β-catenin accumulation and enhanced proliferation of CRC cells. Moreover, chemical inhibition of AURKB suppresses tumor growth in subcutaneous mouse model and exhibits particular effectiveness against tumors derived from CRC cells characterized by prominent cytoplasmic TRIM24 distribution. Together, these findings reveal a critical role of TRIM24 in CRC cell proliferation, particularly through activating Wnt/β-catenin signaling.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most common cancer and the fourth leading cause of cancer-related death worldwide, with both incidence and mortality expected to rise in the coming decades1,2. Challenges in CRC treatment include metastasis and resistance to conventional chemo- and radiotherapies, with approximately 40% of cases progressing to metastasis and a survival rate for metastatic CRC below 10%3. Therefore, there are clearly unmet clinical needs to unravel the molecular mechanisms underpinning CRC pathogenesis and to pinpoint molecular checkpoints governing its initiation, progression, and metastasis.
The Wnt/β-catenin signaling pathway, crucial for regulating development, proliferation, stemness, cell-cell adhesion, and epithelial-mesenchymal transition, is among the most frequently altered signaling pathways in clinical CRC specimens4. Under physiological conditions, β-catenin, the key component of the Wnt signaling pathway, is tightly regulated by a destruction complex composed of adenomatous polyposis coli (APC), AXIN, and glycogen synthase kinase 3β (GSK3β)5, which promotes the phosphorylation of β-catenin. Subsequently, phosphorylated β-catenin is recognized and degraded by E3 ubiquitin ligase β-TrCP. Dysfunction of the destruction complex leads to the stabilization of β-catenin; the accumulated β-catenin translocates into the nucleus, thereby constitutively activating Wnt/β-catenin signaling in the absence of stimuli from the Wnt ligand6. A close interplay between Wnt/β-catenin and AKT signaling pathways has been reported in inflammatory responses, cell proliferation, and differentiation7,8. The inactivation of GSK3β by AKT-mediated phosphorylation impacts the protein stability and nuclear translocation of β-catenin9. APC functions as a tumor suppressor, and mutations in APC are detected in approximately 80% of sporadic CRC cases10. Inactivation of APC is believed to disrupt the destruction complex, leading to β-catenin stabilization, its accumulation in the cytoplasm, nuclear translocation, and activation of TCF/LEF transcription factors to initiate Wnt/β-catenin target gene expression11. Of note, emerging evidence indicates a lack of correlation between APC mutations and β-catenin regulation in CRC patient samples12, which supports a previous finding that aminopeptidase P is a key factor in the control of the abundance of β-catenin13. Hence, further investigation into the regulatory mechanisms that govern β-catenin protein stability in CRC is needed.
Tripartite motif containing 24 (TRIM24), also known as transcription intermediary factor 1-alpha (TIF1α), is an important member of the transcription intermediary factor family14. It regulates gene expression by interacting with DNA-bound proteins, such as nuclear receptors, and by directly binding to chromatin through its tandem PHD-bromodomain15,16,17. TRIM24 also contains a RING domain, two B-boxes, and a coiled-coil region18. Although TRIM24 has been shown to function as a RING-type E3 ubiquitin ligase, only several substrates have been identified to date, including p53, TRAF3 and components of P-bodies19,20,21. Nevertheless, the physiological importance and specific molecular targets of TRIM24 E3 ligase activity remain elusive.
Here we report that TRIM24 is aberrantly expressed in human CRC tissues, and its cytoplasmic distribution is augmented by phosphorylation at serine 1042 by aurora kinase B (AURKB). Moreover, TRIM24 interacts directly with tumor suppressor von Hippel-Lindau (VHL), a negative regulator of AKT, and functions as an E3 ubiquitin ligase to facilitate VHL degradation, which in turn activates the Wnt/β-catenin signaling pathway. Indeed, AURKB inhibition shows therapeutic potential for CRC in mouse models, especially for tumors with significant subcellular localization of TRIM24. These findings demonstrate a regulatory role for TRIM24 as an E3 ligase in controlling the activation of Wnt/β-catenin signaling and provide a potential therapeutic strategy for the treatment of CRC.
Results
TRIM24 is critical for the proliferation of CRC cells
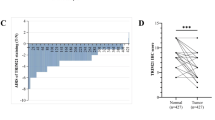
To evaluate the role of TRIM24 in CRC, we first assessed the expression levels and prognostic value of TRIM24 in CRC patients mining the data in The Cancer Genome Atlas (TCGA) dataset and the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database. Indeed, the mRNA levels of TRIM24 were significantly elevated in the tumor tissues compared to the adjacent normal tissues (Fig. 1A), which is consistent with the protein abundance trend (Fig. 1C). Kaplan–Meier analysis revealed that CRC patients with high TRIM24 expression had significantly worse disease-free survival compared to those with low expression of TRIM24 (Fig. 1B). In the tissue samples derived from CRC patients, the protein levels of TRIM24 were notably elevated in 10 out of 12 (83%) cases compared to the corresponding adjacent normal tissues (Fig. 1D, Supplementary Fig. S1, and Supplementary Table 1). Furthermore, the expression levels of TRIM24 were significantly higher in various human CRC cell lines compared to those in the normal human colonic epithelial cell line FHC (Fig. 1E). To assess the biological role of TRIM24 in CRC cells, we primarily employed two CRC cell lines, DLD1 and SW620, which exhibited the highest levels of TRIM24 expression (Fig. 1E). Silencing TRIM24 using two different small interfering RNAs (siTRIM24-1 and siTRIM24-2) or CRISPR-Cas9-mediated knockout cell clones (KO-1 and KO-2) significantly suppressed colony formation (Fig. 1F and Supplementary Fig. S2A–C) and induced G2/M phase arrest in CRC cells (Fig. 1G), whereas overexpression of TRIM24 significantly restored both colony formation and G2/M arrest in DLD1-KO cells (Supplementary Fig. S3A–E). CD44, a compelling marker of CRC stem cells, was previously linked to the proliferation of CRC cells22. The depletion of TRIM24 resulted in a significant reduction in the proportion of CD44-positive cells (Fig. 1H). Moreover, knockout of TRIM24 in DLD1 cells retarded tumor growth in a subcutaneous xenograft tumor mouse model (Fig. 1I, J). Proliferating cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining showed that TRIM24 depletion inhibited CRC cell proliferation (Fig. 1K) in tumor tissues without impacting cell death (Supplementary Fig. S4B). Consistently, TRIM24 knockdown impeded the growth of CRC patients-derived organoids (Fig. 1L). Therefore, these results suggest that TRIM24 could execute a critical function in the regulation of CRC cell proliferation.

A The mRNA levels of TRIM24 in normal colon tissues (n = 51) and CRC tissues (n = 622) from TCGA-COAD. Two-tailed Student’s t-test was used. The mRNA analysis of TRIM24 was carried out by BEST. B The Kaplan–Meier survival curves based on the expression of TRIM24 showed CRC patients with high expression of TRIM24 had a shorter disease-free survival. The Kaplan–Meier survival analysis was performed by BEST and the cut-off value was set to “median”. “Number at risk” means the number of patients exposed to outcome risk at each point on the curve. C The protein abundance of TRIM24 in normal colon tissues (n = 93) and CRC tissues (n = 95) derived from CPTAC database. Two-tailed Student’s t-test was used. (https://proteomics.cancer.gov/programs/cptac). Boxplots in (A) and (C) showing 0th, 25th, 50th,75th, and 100th centiles. Outliers were defined as values larger than 1.5 × lQR (Interquartile Range) + 75th centile. D Left: Representative immunoblots of TRIM24 in colonic epithelial cells isolated from normal (N) or tumor (T) tissues derived from patients with colorectal adenocarcinoma (n = 12), with Actin as a loading control. Right: The comparison between the expression levels of TRIM24 in tumor tissues and paired adjacent normal tissue from CRC patients in our facility, with relative TRIM24 protein levels normalized to Actin. Data were expressed as two-tailed Student’s t-test. E Normal human colonic epithelial cell line FHC and human CRC cell lines were harvested and immunoblotted for TRIM24. Shown are the representative results from three independent experiments. F After 24 h transfection with siNC or two small interfering RNAs targeting distinct sequences of TRIM24 (siTRIM24-1/2), DLD1 or SW620 cells from each group were evenly spread in dishes. After 1–2 weeks, the colonies were stained and photographed for colony formation assays. Experiments were conducted independently three times, consistently producing similar results. G Left: After 72 h transfection with siNC or siTRIM24-1/2, DLD1 cells were collected and incubated with PI. The cell cycle analysis was performed by flow cytometry. Right: Graph representing percentage cells in each phase of cell cycle. Experiments were conducted independently three times, consistently producing similar results. H Left: After 72 h transfection with siNC or siTRIM24-1/2, SW620 cells were collected and incubated with FITC-CD44. The proportion of CD44-positive cells was analyzed by flow cytometry. Right: Quantification of CD44 positive cells in the indicated groups. n = 3 biologically independent samples per group. Data were expressed as means ± SEM, two-tailed Student’s t-test. I In subcutaneous model, DLD1-WT (wild-type DLD1) cells were subcutaneously inoculated into the left flank area of five-week-old male BALB/c nude mice with the same amount of DLD1-KO-1 (TRIM24 knockout DLD1 clone-1) cells into the right flank (n = 11 mice per group). Tumor size was measured every 3 days from 8 days post-inoculation until day 25. Data were expressed as means ± SEM, two-tailed Student’s t-test. J Representative pictures of tumors dissected from mice in (I) on day 25 post-inoculation. K Left: Tumor tissues collected from mice in (I) were subjected to PCNA staining, with nuclei counterstained by hematoxylin. Scale bar, 50 µm. Right: PCNA staining intensity was quantified in randomly selected fields (WT group n = 6; KO-1 group n = 5) using ImageJ with the H-DAB color deconvolution vector, and relative intensity was calculated after normalization to the WT group. Data were expressed as means ± SEM, two-tailed Student’s t-test. L Left: The representative images of CRC patient-derived organoids at 7 days post transfect with siNC or siTRIM24-1. Scale bar, 100 µm. Right: Quantification of the size of organoids in each indicated group. n = 10 biologically independent samples per group. Data were expressed as means ± SEM, two-tailed Student’s t-test.
TRIM24 determines the activation of the Wnt/β-catenin signaling pathway in CRC cells
To explore the TRIM24-mediated signaling pathway involved in regulating CRC cell proliferation, we divided the TCGA-COAD dataset into two groups based on the median TRIM24 expression level and performed GSEA using an online platform (https://rookieutopia.hiplot.com.cn/app_direct/BEST/), revealing the top fifteen positively correlated gene sets (Fig. 2A). We observed that Wnt/β-catenin signaling, PI3K/AKT/mTOR signaling, and G2/M checkpoint regulation were substantially enriched (Fig. 2A, B and Supplementary Fig. S5). In support, depletion of TRIM24 resulted in G2/M phase cell cycle arrest in CRC cells (Fig. 1G and Supplementary Fig. S3B–E), aligning with previous studies showing that the blockade of Wnt/β-catenin signaling caused cell-cycle arrest at G2/M phase in various cancers23,24. Importantly, AKT and Wnt/β-catenin signaling activation exhibit crosstalk in controlling the cell cycle. In particular, inhibition of AKT, which failed to promote GSK3β deactivation phosphorylation, led to β-catenin degradation and G2/M phase arrest in cancer cells25. To examine the possible association between TRIM24 and activation of Wnt/β-catenin signaling pathway, TOPFlash/FOPFlash luciferase reporter assays, widely used to evaluate β-catenin-dependent transcriptional activity26, were conducted using SW620 and DLD1 cells. The TOPFlash, but not FOPFlash, activity is significantly reduced in TRIM24-deficient CRC cells (Fig. 2C). TRIM24 knockout resulted in a dramatic reduction in both the mRNA (Fig. 2D) and protein levels (Fig. 2E) of multiple Wnt/β-catenin downstream target genes, which are associated with cell cycle regulation and cell proliferation in CRC cells, including c-MYC. To rule out potential off-target effects from the CRISPR-Cas9 system, we utilized different small RNAs to interfere with TRIM24 expression in CRC cells, yielding similar results (Supplementary Fig. S6A). We noticed that MYC targets v1 pathway and MYC targets v2 pathway were also enriched by GSEA analysis (Fig. 2A and Supplementary Fig. S5). Of note, TRIM24 deficiency resulted in significantly reduced levels of p-AKT (phosphor-AKT, indicator of AKT activation) and p-GSK3β (phosphor-GSK3β, indicator of GSK3β inactivation) in CRC cells (Fig. 2E and Supplementary Fig. S6A, B) and CRC patients-derived organoids (Supplementary Fig. S6C), without affecting levels of total AKT and GSK3β. Moreover, ectopic expression of TRIM24 in TRIM24-deficient CRC cells restored the levels of p-AKT and p-GSK3β, the accumulation of β-catenin, and the protein levels of downstream targets of Wnt/β-catenin signaling (Supplementary Fig. S6D). Hence, our results suggest that TRIM24 executes an important function in Wnt/β-catenin signaling regulation through controlling AKT activation.

A The Gene set enrichment analysis (GSEA) based on the expression of TRIM24. CRC samples from TCGA were divided into two groups based on the median value of TRIM24 expression and then subjected to GSEA (https://rookieutopia.hiplot.com.cn/app_direct/BEST/) to investigate whether genes in the two groups were rich in meaningful biological processes. B GSEA showed that Wnt/β-catenin signaling and PI3K/AKT/mTOR signaling were hyper-activated in samples with high expression of TRIM24. NES represents normalized enrichment scores. C DLD1-WT/DLD1-KO-1 and SW620-WT/SW620-KO-1 cells were co-transfected with Renilla luciferase and TOPFlash plasmids (or FOPFlash plasmids, negative control). The cells were harvested 48 h after transfection, and firefly and Renilla luciferase substrates were added successively. The ratio of the detection value of firefly luciferase to the detection value of Renilla luciferase is taken to compare the difference in the results between the two groups of cells. Data were expressed as means ± SEM, two-tailed Student’s t-test. n = 3 biological replicates per group. D Total RNA was extracted from DLD1-WT-1 and DLD1-KO-1 cells and mRNA profiles of MYC, CD44, CDC25A, EFNB2, HNF1A, and ACTB were analyzed by qRT-PCR. Data were expressed as means ± SEM, two-tailed Student’s t-test. n = 3 biological replicates per group. E Whole-cell lysates from DLD1-WT/DLD1-KO-1 and SW620-WT/SW620-KO-1 cells were immunoblotted for indicated proteins, with Tubulin as a loading control. Shown are the representative results from three independent experiments.
TRIM24 interacts with VHL and promotes its ubiquitination
TRIM24 primarily serves as a transcriptional regulator and E3 ubiquitin ligase27. TRIM24 depletion notably decreased p-AKT levels without altering total AKT proteins, indicating that TRIM24 possibly functions as an E3 ubiquitin ligase that influences AKT activation by modulating its negative regulator(s). It was reported that the PH domain leucine-rich repeat protein phosphatases (PHLPP1 and PHLPP2) and von VHL bound directly to AKT and inhibited AKT activity28,29,30. Therefore, we first determined whether there is a functional link between TRIM24 and the polyubiquitination of AKT negative regulator(s). The co-immunoprecipitation assays showed that TRIM24 markedly facilitated the polyubiquitination of VHL, but not PHLPP1 and PHLPP2, whereas the TRIM24 mutant lacking the RING domain (△RING) did not (Fig. 3A). Consistently, TRIM24 effectively catalysed the polyubiquitination of VHL in vitro, whereas the △RING mutant failed to do so (Fig. 3B). Next, we observed an endogenous interaction between TRIM24 and VHL in CRC cells (Fig. 3C). Moreover, GST pull-down assay and co-immunoprecipitation assays using full-length as well as a series of truncated mutants of TRIM24 showed that TRIM24 directly interacted with VHL in vitro (Fig. 3D) and in DLD1 cells (Supplementary Fig. S7), both of which require the B-boxes and the coiled-coil region (residues 141-400) of TRIM24. In line with the findings that the VHL protein undergoes degradation via the proteasome pathway31, the proteasome inhibitor N-carbobenzoxy-l-leucinyl-lleucinyl-l-norleucinal (MG132) treatment markedly augmented VHL protein levels in CRC cells (Fig. 3E). Similarly, deletion of TRIM24 resulted in the elevated VHL protein levels in these cells (Fig. 3E); VHL protein stability was also significantly increased in the TRIM24 depleted CRC cells (Fig. 3F). Interestingly, TRIM24 protein levels were elevated in colorectal adenocarcinoma tissues compared to adjacent normal tissues (Fig. 1D), such increase in TRIM24 showed a negative correlation with VHL levels in the tissues from CRC patients (Fig. 3G, H), while remaining positively correlated with β-catenin levels (Fig. 3G, I). These results suggest that TRIM24, as an E3 ubiquitin ligase, binds to and ubiquitinates VHL in CRC cells.

A HEK293T cells were co-transfected with HA-PHLPP1/HA-PHLPP2/HA-VHL and Flag-TRIM24/Flag-ΔRING (truncated TRIM24 mutant, lacks RING domain) plasmids and treated with or without proteasome inhibitor MG132 (10 μM) for 8 h. Whole cell lysates (Input) were derived and immunoblotted directly or after immunoprecipitation with anti-HA antibody for indicated proteins. Data are representative of three independent experiments. B Upper: In vitro ubiquitination assays. HEK293T cells transfected with GFP-TRIM24 or GFP-ΔRING for 36 h and cell lysates were immunoprecipitated using an anti-GFP antibody. The immunoprecipitants and recombinant VHL were used for in vitro ubiquitination assays. The reaction products were immunoblotted using indicated antibodies. Bottom: The levels of indicated proteins, normalized to vehicle control, were quantified (data are representative of three independent experiments). AU, arbitrary unit. Data were expressed as means ± SEM, two-tailed Student’s t-test. Data are representative of three independent experiments. C DLD1 cell lysates (input) were immunoprecipitated by anti-TRIM24 antibody, IgG control antibody, or anti-VHL antibody, respectively. Data are representative of three independent experiments. The input and the immunoprecipitants were immunoblotted for indicated proteins. D Upper: Schematic diagram of full-length human TRIM24. RING domain; B1 and B2, B-box 1 and 2; CC, coiled-coil domain; PHD, plant homeodomain; BROMO, bromodomain. Bottom: GST pull-down assays with GST, GST-fusion proteins (TRIM24 and truncated TRIM24 mutants), and His-VHL. The input and the bound proteins were immunoblotted with indicated antibodies. Data are representative of three independent experiments. E DLD1-WT/DLD1-KO-1 and SW620-WT/SW620-KO-1 cells were treated with 10 μM MG132 for 8 h. Whole cell lysates were immunoblotted for indicated proteins, with Tubulin as a loading control. Data are representative of three independent experiments. F DLD1-WT/DLD1-KO-1 and SW620-WT/SW620-KO-1 cells were treated with 100 μg/mL cycloheximide (CHX) for the indicated time periods. Whole cell lysates were derived and immunoblotted for indicated proteins. Data are representative of three independent experiments. G Representative immunoblots of indicated proteins in adjacent normal tissue and tumor tissue in patients with CRC. Shown are the representative results from three independent experiments. H, I Linear regression analysis of the levels of TRIM24 protein versus VHL (H) and β-catenin (I) in colonic epithelial cells isolated from normal and tumor tissues of patients with colorectal adenocarcinoma (n = 10). AU, arbitrary unit. Linear regression model with two-sided test was used for the statistical analysis, and multiple R-squared was used. Relationships are denoted with solid lines and fit statistics (R2 and p values).
Phosphorylation of serine 1042 is critical for cytoplasmic localization of TRIM24
VHL predominantly localizes in the cytoplasm and/or the cell membrane (Supplementary Fig. S8)32,33, whereas TRIM24 was primarily found in the nucleus21,34. To elucidate how TRIM24 interacts with and regulates the degradation of VHL in CRC cells, we conducted immunohistochemical staining of TRIM24 on CRC tissue specimens. Intriguingly, TRIM24 proteins displayed partially cytoplasmic distribution in approximately 60% (7/12) of CRC tissues (Fig. 4A and Supplementary Table 2). The amino acid sequence analysis35 revealed two potential nuclear localization sequences (NLS-1, residues 902–911; NLS-2, residue 1033–1045) in TRIM24 (Fig. 4B). Indeed, we experimentally ascertained that the carboxyl-terminal NLS-2, rather than NLS-1, is the bona fide functional NLS of TRIM24, as evidenced by the finding that the NLS-2 mutant exhibited significant localization in the cytoplasm (Fig. 4E). AlphaFold336 was further employed to characterize the molecular structure of TRIM24, revealing that the carboxyl-terminal residues 1036 to 1048 form an α-helical structure within the protein (Fig. 4C). Notably, it is well known that phosphorylation of residue(s) within the α-helix typically disrupts the helical structure37; serine 1042 (S1042), located in the core of the α-helix, is highly conserved across different species (Fig. 4D). We hypothesized that phosphorylation of S1042 could impact the α-helix conformation and subsequent NLS function in human TRIM24 protein, thereby altering its subcellular localization. Indeed, our data showed that the phospho-mimicking S1042E mutant TRIM24 exhibited pronounced cytoplasmic distribution, whereas the phospho-dead S1042A mutant TRIM24 remained nuclear localization similar to wild-type TRIM24, when ectopically expressed in HEK293T cells (Fig. 4B, E, F). To confirm the presence of S1042 phosphorylation on TRIM24 in CRC cells, we generated a specific polyclonal antibody p-TRIM24, targeting the S1042-phosphorylated TRIM24 (Supplementary Fig. S9A, B). While the total TRIM24 protein levels appeared notably high across various CRC cells, the levels of p-TRIM24 were most prominent in DLD1 and SW620 cells, with relatively lower abundance observed in cells such as SW1643, LS174T, and HT29 (Fig. 4G). Subcellular fraction assays revealed that the cytoplasmic distribution of TRIM24 generally corresponds to p-TRIM24 levels in various CRC cells (Fig. 4H). Consistent with these results, overexpression of wild-type TRIM24 and S1042E mutant rescued VHL-AKT axis-mediated activation of Wnt/β-catenin signaling in cell lines with low p-TRIM24 levels, whereas overexpression of S1042A failed to do so (Fig. 4I). Moreover, mass spectrometry analysis further ascertained the presence of the endogenous phosphorylation on S1042 of TRIM24 in DLD1 cells (Fig. 4J). These results suggest the importance of S1042 phosphorylation in conferring the subcellular localization of TRIM24 in CRC cells.

A Immunohistochemical staining of TRIM24 in CRC tissues (n = 12) obtained from the Human Protein Atlas database (https://www.proteinatlas.org/). Shown are representative images of strict nuclear localization (N: 5/12) and partially cytoplasmic distribution (C + N: 7/12) of TRIM24, respectively. B Schematic diagram of human TRIM24, predicted nuclear localization sequences (NLS1 and NLS2) using cNLS Mapper, the mutant amino acid sites of NLSs (NLS1-M and NLS2-M), and the mutants of TRIM24 at serine 1042 (S1042). C The structure of human TRIM24 protein (left) and its carboxyl terminus (right) were obtained from AlphaFold3 (UniProt ID: O15164). The S1042 residue is illustrated in orange (right). D The amino acid sequences of TRIM24 from human, mouse, rat, and monkey were aligned, with the potential phosphorylation site S1042 (or S1043) highlighted in bold. E Immunofluorescence micrographs of HEK293T cells transfected with GFP-TRIM24 plasmid or TRIM24 mutant plasmid as shown in (B), with the nucleus counterstrained with DAPI. Scale bar, 10 µm. F The statistical analysis based on TRIM24 distribution shows localization in the nucleus (N) or in both the cytoplasm and nucleus (C + N) within HEK293T cells in (E). The subcellular localization of GFP and GFP-fusion proteins was quantified separately in five randomly selected fields for each group. Experiments were conducted independently three times, consistently producing similar results. G Normal human colonic epithelial cell line FHC and human CRC cell lines were harvested and immunoblotted for indicated proteins, with Actin as a loading control. Data are representative of at least three independent experiments. H The cytosolic and nuclear fractions of CRC cell lines were extracted and immunoblotted for indicated proteins. GAPDH and Lamin-A were employed as loading controls of cytosol fraction and nucleus fraction, respectively, and indicated the purity of each fraction. Data are representative of at least three independent experiments. I Left: HT29 cell were transfected with GFP, GFP-TRIM24, or TRIM24 mutant plasmids. After 36 h, whole cell lysates were derived and immunoblotted for indicated proteins. Right: The levels of indicated proteins, normalized to vehicle controls, were quantified from three independent experiments. Data were expressed as means ± SEM, two-tailed Student’s t-test. J TRIM24 S1042 phosphorylation in DLD1 cells was detected by label-free relative quantitative mass spectrum analysis.
S1042 phosphorylation of TRIM24 by AURKB kinase is critical for the activation of Wnt/β-catenin signaling
Protein phosphorylation, catalysed by kinases, serves as a crucial cellular regulatory mechanism to activate or deactivate numerous signaling proteins38. Bioinformatic analyses using multiple phosphorylation site prediction tools (KinasePhos 2.0, GPS 3.0, and iGPS 1.0) revealed that several candidate kinases could phosphorylate TRIM24 at S1042 (Fig. 5A). To validate them, we initially utilized small RNA to silence the top-ranked kinases in DLD1 and SW620 cells, in which p-TRIM24 levels are substantially elevated. Interestingly, we observed a significant reduction in the level of p-TRIM24 (S1042) upon knockdown of AURKB, one of the primary forms of AURORA kinase, but not AURKA, CK2, or AKT (Fig. 5B). Consistently, the AURKB inhibitor hesperadin markedly reduced p-TRIM24 levels, whereas the CK2 and AKT inhibitors had no such effect (Fig. 5C). The In vitro phosphorylation assays showed that TRIM24, but not the S1042A mutant, was phosphorylated by AURKB and such phosphorylation was completely inhibited by hesperadin (Fig. 5D). Of note, treatment with AURKB inhibitors, including hesperadin and barasertib, led to the inactivation of Wnt/β-catenin signaling in CRC cells, accompanied by elevated VHL protein levels (Fig. 5E and Supplementary Fig. S10). In addition, hesperadin treatment accelerated TRIM24 translocation to the nucleus and attenuated β-catenin accumulation in the cytosol and its nuclear distribution (Fig. 5F). Furthermore, AURKB inhibition notably attenuated TOPFlash luciferase activity in parental CRC cells but had no effect on this activity in TRIM24-deficient cells (Fig. 5G), suggesting that the inhibitory effect of hesperadin on Wnt/β-catenin signaling is TRIM24-dependent. These results suggest that the phosphorylation of TRIM24 by AURKB is critical for the activation of Wnt/β-catenin signaling in CRC cells.

A Kinase prediction tools (KinasePhos 2.0, GPS 3.0, and iGPS 1.0) were used to identify potential kinases targeting TRIM24 at Ser1042. “+” indicates a positive prediction by the respective tool; “−” indicates no prediction. B DLD1 and SW620 cells were transferred with si-NC, si-AURKA, si-AURKB, si-CK2α, and si-AKT. After 72 h, whole cell lysates were derived and immunoblotted for indicated proteins, with Tubulin as a loading control. Experiments were conducted independently three times, consistently producing similar results. C DLD1 and SW620 cells were treated with hesperadin (AURK inhibitor, which has a particularly significant inhibitory effect on AURKB, 20 μM), CX-4945 (CK2 inhibitor, 20 μM), and AKT Inhibitor VIII (AKT1/AKT2 inhibitor, 20 μM) for 4 h and 8 h, respectively. Whole cell lysates were derived and immunoblotted for indicated proteins. Experiments were conducted independently three times, consistently producing similar results. D In vitro phosphorylation assays utilized the purified GST-fusion proteins and AURKB, incubated with hesperadin or DMSO vehicle control. The reaction products were immunoblotted using indicated antibodies. AKT was employed as a positive control. Data are representative of three independent experiments. E SW620 cells were treated with hesperadin for 72 h, whole cell lysates were derived and immunoblotted for indicated proteins. Data are representative of three independent experiments. F Cytosolic (Cyto) and nuclear (Nuc) fractions derived from DLD1 and SW620 cells stimulated with 20 μM of hesperadin for 8 h were immunoblotted for indicated proteins. PARP1 and Caspase-3 served as loading controls and nuclear/cytosolic markers, respectively. Shown are the representative results from three independent experiments. G DLD1-WT/DLD1-KO-1 and SW620-WT/SW620-KO-1 cells were co-transfected with Renilla luciferase and TOPFlash plasmids. The cells were harvested 48 h after transfection, and firefly and Renilla luciferase substrates were added successively. The ratio of the detection value of firefly luciferase to the detection value of Renilla luciferase is taken to compare the difference in the results between the two groups of cells. Data were expressed as means ± SEM, two-tailed Student’s t-test. n = 3 biological replicates per group.
AURKB inhibition retards development of CRC in mouse models
AURKB inhibitors have emerged as a promising alternative for cancer treatment39,40, we therefore examined the effect of hesperadin on CRC cell lines. The colony formation assays revealed that hesperadin markedly suppressed the proliferation of DLD1 and SW620 cells (Fig. 6A, B), which exhibited remarkable cytoplasmic distribution of TRIM24 and high p-TRIM24 levels (Fig. 4G, H and Supplementary Fig. S11A, B), upon treatment with hesperadin, whereas hesperadin had less profound impact on the proliferation of RKO and SW1643 cells (Fig. 6A, B), which exhibited minimal cytoplasmic distribution of TRIM24 and low p-TRIM24 levels (Fig. 4G, H and Supplementary Fig. S11A, B). Knockdown or pharmacological inhibition of AURKB markedly reduced the cytoplasmic localization of TRIM24 in DLD1 cells (Supplementary Fig. S11C–F). Then we performed subcutaneous xenograft assay in male BALB/c nude mice using DLD1 and SW1643 cells (Fig. 6C–E). While hesperadin (Fig. 6C, D) or barasertib (Supplementary Fig. S12A–C) administration showed no impact on tumor growth in mice inoculated with SW1643 cells, it significantly suppressed tumor growth in mice inoculated with DLD1 cells. Of note, immunohistochemical staining of PCNA indicated that hesperadin administration inhibited cell proliferation in tumors formed by DLD1 cells but not in those by SW1643 cells (Fig. 6E), and did not result in significant cell death in tumor tissues (Supplementary Fig. S13). Immunohistochemical staining revealed that TRIM24 was distributed in both the nucleus and cytoplasm in tumors derived from DLD1 cells, but shifted predominantly to the nucleus following continuous hesperadin injection; in contrast, TRIM24 remained nuclear in SW1643-derived tumors regardless of treatment (Fig. 6F). Moreover, substantial p-TRIM24 staining was detected only in DLD1-derived tumors, with no detectable signal in the other groups (Fig. 6G). Collectively, our results showed that hesperadin treatment harbors therapeutic potential for CRC, particularly in inhibiting the proliferation of CRC cells with significant cytoplasmic TRIM24 distribution.

A DLD1, SW620, RKO, and SW1643 cells were grown in the absence or presence of hesperadin at the indicated concentrations for 7 days, then fixed and stained for colony-formation assessment. Experiments were conducted independently three times, consistently producing similar results. B The number of colonies in (A) was counted by ImageJ, and normalized to those under 0 nM hesperadin treatment. C Upper: A schematic diagram of the experimental timeline for the subcutaneous xenograft assay (n = 4 mice per group). Bottom: The tumor growth curve of the mice subcutaneously injected with DLD1 or SW1643 cells after intraperitoneal administration of hesperadin (2.5 mg/kg) or PBS. Data were expressed as means ± SEM, two-tailed Student’s t-test. D Representative pictures of tumors dissected from mice at day 24 post-implantation. Scale bar, 1 cm. E Left: Tumor tissues collected from mice in (D) were subjected to PCNA staining, with nuclei counterstained by hematoxylin. Scale bar, 50 µm. Right: PCNA staining intensity was quantified in randomly selected fields (n = 5, 6, 8, and 6 for the four histograms, respectively) using ImageJ with the H-DAB color deconvolution vector, and relative intensity was calculated after normalization to the PBS control group. F and G Tumor tissues collected from mice in (D) were subjected to TRIM24 (F) or p-TRIM24 (G) staining, with nuclei counterstained by hematoxylin. Scale bar, 20 µm.
Discussion
Despite the hyperactivation of Wnt/β-catenin signaling being considered the initiating and driving event of CRC41,42, the precise mechanisms that regulate β-catenin homeostasis in CRC cells are still not well defined. Our results demonstrate that TRIM24 is a previously unappreciated regulator of Wnt/β-catenin signaling through regulating VHL-AKT-GSK3β axis-mediated β-catenin degradation, thus promoting the proliferation of CRC cells.
TRIM24 has been reported to be aberrantly activated in various cancers, including breast cancer, glioma, and prostate cancer, and others16,17,43. We reveal that TRIM24 mRNA and protein levels are significantly elevated in CRC tissues compared to adjacent normal tissues (Fig. 1A, C, D). Furthermore, Kaplan-Meier survival analysis illustrates that TRIM24 overexpression in CRC patients is associated with worse disease-free survival (Fig. 1B), in line with previous studies44. Interestingly, TRIM24 is associated with activation of Wnt/β-catenin in CRC based on bioinformatics analysis of the TCGA-COAD data (Fig. 2A, B). Deletion of TRIM24 in CRC cells with high TRIM24 expression results in inactivation of the AKT-GSK3β axis and degradation of β-catenin, and ultimately inactivation of Wnt/β-catenin signaling (Fig. 2C–E). Moreover, TRIM24 depletion retards the proliferation of CRC cells in vitro and in a subcutaneous xenograft tumor mouse model (Fig. 1F–L and Supplementary Fig. S2A–C). Interestingly, although TRIM24 has also been reported to regulate cell proliferation in colorectal cancer via YAP signaling44, dual-luciferase reporter assays revealed that TRIM24 depletion had a more pronounced effect on Wnt/β-catenin activity than YAP/TAZ activity (Supplementary Fig. S14A, B). This suggests that Wnt/β-catenin signaling could be more dominant in driving proliferation of CRC cells.
TRIM24 is a multifunctional protein that acts as a co-regulator of nuclear receptors and a histone reader, typically localized in the nucleus. In addition to the aforementioned functions, TRIM24 could act as an E3 ubiquitin ligase, particularly with the characteristic RING domain27. Although evidence demonstrates that TRIM24 functions as an E3 ligase promoting protein ubiquitination and degradation, only a few substrate proteins have been identified so far, including p53 and components of P-bodies21,45. Here, we identified the tumor suppressor protein VHL as a TRIM24-binding protein (Fig. 3C, D and Supplementary Fig. S7). TRIM24 confers the RING domain-dependent polyubiquitination and degradation of VHL (Fig. 3A, B). VHL has been recognized as a suppressor of AKT30, which phosphorylates GSK3β at serine 9, thereby promoting the degradation of β-catenin. Previous studies have suggested that TRIM24 is involved in the Wnt/β-catenin signaling pathway in gastric cancer and colorectal cancer46,47, however, the precise underlying mechanism remains a mystery. Of note, the negative correlation between TRIM24 and VHL levels in CRC patient tissues, along with the positive correlation between TRIM24 and β-catenin abundance (Fig. 3G–I), suggests that TRIM24 could be essential for CRC development through regulating Wnt/β-catenin signaling. In support of this notion, ectopic expression of TRIM24 in CRC cells with low basal TRIM24 levels resulted in reduced VHL levels and elevated p-AKT, p-GSK3β, and β-catenin levels (Fig. 4I), highlighting its pivotal role in CRC progression.
To elucidate the causal relationship between TRIM24 and Wnt/β-catenin signaling in CRC cells, a series of mechanistic experiments were performed. First, overexpression of a constitutively active AKT mutant (AKTS473D) markedly increased the levels of p-GSK3β and β-catenin. TRIM24 knockdown significantly reduced p-GSK3β and β-catenin levels in control cells expressing an empty vector; however, this effect was abolished in cells overexpressing AKTS473D (Supplementary Fig. S15A–C), suggesting that TRIM24 acts upstream of AKT in regulating β-catenin signaling. Second, silencing of VHL led to enhanced phosphorylation of AKT, GSK3β, and accumulation of β-catenin. TRIM24 knockdown reduced the levels of p-AKT, p-GSK3β, and β-catenin in control cells, but not in VHL-deficient cells (Supplementary Fig. S16A–C), further indicating that TRIM24 modulates this pathway via VHL-AKT signaling. Similarly, inhibition of AURKB reduced p-TRIM24, p-AKT, p-GSK3β, and β-catenin levels in control cells. In contrast, AURKB inhibition in VHL-knockdown cells still suppressed p-TRIM24 but had no significant effect on downstream p-AKT, p-GSK3β, or β-catenin levels (Supplementary Fig. S17A, B). Moreover, in cells overexpressing GFP, AURKB inhibition led to decreased levels of p-AKT, p-GSK3β, and β-catenin, whereas these effects were abolished in cells overexpressing the S1042E mutant (Supplementary Fig. S18). Third, deletion of TRIM24 in both wild-type β-catenin (DLD1 and SW620) and mutant β-catenin (HCT116 and L174T) CRC cells significantly decreased β-catenin levels and inhibited tumor cell proliferation (Supplementary Fig. S19A–D), suggesting that TRIM24-mediated regulation of β-catenin may occur independently of APC-mediated degradation. Although TRIM24 deficiency significantly decreased β-catenin protein accumulation, it did not alter its phosphorylation at S675 or nuclear localization (Supplementary Fig. S20), suggesting that TRIM24 primarily affects β-catenin stability rather than nuclear translocation. Collectively, our findings identify the AURKB-TRIM24-VHL-AKT-GSK3β-β-catenin signaling axis as a critical pathway through which TRIM24 modulates Wnt/β-catenin signaling activity in colorectal cancer.
It has been reported that VHL is primarily distributed in the cytoplasm and/or the cell membrane32,33, whereas TRIM24, as mentioned above, is distributed in the nucleus21,34. Of note, our study observed a significant proportion of TRIM24 in CRC tumor specimens (7/12) with a clear cytoplasmic distribution (Fig. 4A). TRIM24 has been reported to localize in the cytoplasm under specific conditions. It functions as an insulin-responsive regulator of P-bodies, with AKT-mediated phosphorylation triggering its translocation from the nucleus to the cytoplasm upon insulin stimulation21. Additionally, vesicular stomatitis virus infection induces a substantial shift of TRIM24 to mitochondria, where it plays a crucial role in activating antiviral signaling20. In breast cancer, TRIM24 was detected in the cytoplasm of some tumor specimens, and its presence was associated with poorer overall survival16. Cytoplasmic TRIM24 expression in HNSCC is associated with poor overall survival and progression-free survival, as well as the development of local recurrence, suggesting its potential role in tumor biology, possibly as an E3 ubiquitin ligase48. Intriguingly, while the cytoplasmic distribution of TRIM24 has been observed in various tumor cells48,49, the mechanism driving its cytoplasmic distribution in these cancer cells remains unclear. Our findings reveal that TRIM24 possesses a carboxyl-terminal NLS (1033 to 1045) (Fig. 4B). Additionally, we analyzed the amino acid sequence of TRIM24 and identified an α-helical structure formed by amino acids 1036 to 1048 at the carboxyl terminus (Fig. 4C). Within this sequence, a serine (Ser1042) residue is located in the core region of human TRIM24, and its phosphorylation has been detected proteomically in other types of cancers including breast cancer and non-small cell lung cancer50,51. We validated the phosphorylation of Ser1042 in CRC cells, in particular, the phosphorylation of TRIM24 Ser1042 in DLD1 cells was detected by mass spectrometry (Fig. 4J). In line with the reports suggesting that phosphorylation disrupts the α-helical structure37, our study revealed that phosphorylation of Ser1042 influences the subcellular localization of TRIM24, with the S1042A mutant mainly distributed in the nucleus and the S1042E mutant demonstrating a clear cytoplasmic distribution (Fig. 4E, F). TRIM24 and the S1042E mutant exhibit comparable binding affinity to VHL in vitro (Supplementary Fig. S21). A recent study indicates that insulin activates AKT, which phosphorylates TRIM24, altering its cellular localization21. Both AKT and AURKB were able to phosphorylate TRIM24 in vitro (Fig. 5D). However, we found that TRIM24 is phosphorylated at Ser1042 by the kinase AURKB, but not by AKT in CRCs (Fig. 5B, C). This discrepancy may arise because our study focused on CRC cells, whereas the previous research used non-tumor cells such as HEK293 cells. Of note, a significant positive correlation was observed between AURKB expression levels and the abundance of cytoplasmic TRIM24 (Supplementary Fig. S22B). The inhibitor hesperadin significantly facilitates the nuclear distribution of TRIM24 and the protein amount of β-catenin, thereby inhibiting Wnt/β-catenin activity (Fig. 5F, G). Pharmacological inhibition of AURKB effectively inhibits the proliferation of CRC cells with high Ser1042 phosphorylation level in vitro and in mouse models, but is less effective for CRC cells with low Ser1042 phosphorylation (Fig. 6A–D and Supplementary Fig. S12A–C).
In summary, our study demonstrates that TRIM24 is a pivotal protein that controls the VHL-AKT-GSK3β axis-mediated β-catenin degradation, which activates Wnt/β-catenin signaling and promotes the proliferation of CRC cells. These findings provide insights into the pathogenic mechanisms of CRC and a potential therapeutic strategy for CRC treatment, particularly for the patients with cytoplasmic TRIM24 distribution in the tumor cells.
Methods
Ethics statement
All procedures involving human participants were conducted in compliance with the ethical standards set by the institutional and national research committees, in alignment with the 1964 Helsinki Declaration and its subsequent amendments. The project was approved by the institutional ethics committee of the First Affiliated Hospital of the University of South China (approval No: 2023LL0810002). Informed consent was obtained from all participants involved in the research. The Animal Ethics Committee of the University of South China approved all animal experiments (approval No: USC2024XS247). Tumor burden was strictly monitored and did not exceed the maximum permitted size (1.5 cm in diameter or 2000 mm³ in volume for mice) as per institutional guidelines. All participants gave informed consent to be included in the study. We fully adhere to SAGER guidelines and our study design was not related to gender.
Database and bioinformatics analysis
The TCGA CRC dataset was used in this study, which containing 632 CRC samples. The bioinformatics analysis was performed using BEST. The Kaplan–Meier curves of disease-free survival (DFS) was performed with log-rank test by a cutoff “median”. The Hallmark gene set enrichment analysis (GSEA) was obtained from Molecular Signatures Database (MSigDB).
Cell culture, reagents, and antibodies
All cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences (China) and BDbio (China). TRIM24 KO DLD1 cells and TRIM24 KO SW620 cells were obtained through the CRISPR-Cas9 system. FHC, DLD1, SW620, HCT116, RKO, SW480, HT29, LS174T, SW1643, LOVO, and HEK293T cells were cultured in DMEM medium containing 10% fetal calf serum, 100 U/mL penicillin and streptomycin. In addition to the above conditions, FHC also requires extra 10 mM HEPES (for a final conc. of 25 mM); 10 ng/mL cholera toxin; 0.005 mg/mL insulin; 0.005 mg/mL transferrin; 100 ng/mL hydrocortisone; 20 ng/mL human recombinant EGF. Hesperadin and cycloheximide were purchased from MCE (USA). MG132 and AKT Inhibitor VIII were purchased from Beyotime (China). Alkaline phosphatase was purchased from Thermo Fisher (USA). 3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di- phenytetrazoliumromide (MTT) was purchased from Coolaber (China). Barasertib and CX-4945 were purchased from TOPSCIENCE (China). Recombinant human AURKB, recombinant human AKT1 protein and crystal violet were purchased from Solarbio (China). Antibodies used were as follows: TRIM24 (14208-1-AP, Proteintech, China), TRIM24 (R27382, ZEN BIO, China), TRIM24 (sc-271266, Santa Cruz Biotechnology, USA), p-AKT (Ser473) (R22961, ZEN BIO), AKT (R23412, ZEN BIO), p-GSK3β (Ser9) (9323, Cell Signaling Technology, USA), GSK3β (22104-1-AP, Proteintech), β-catenin (sc-7963, Santa Cruz Biotechnology), c-MYC (10828-1-AP, Proteintech), CD44 (200840, ZEN BIO), EFNB2 (YN0096, ImmunoWay Biotechnology, China), HNF1A (YN2932, ImmunoWay Biotechnology), Beta Tubulin (66240-1-Ig, Proteintech), ubiquitin (10201-1-Ig, Proteintech), HA (66006-1-Ig, Proteintech), Flag tag (20543-1-AP, Proteintech), Flag tag (A02010, Abbkine, China), VHL (24756-1-AP, Proteintech), GFP tag (50430-2-AP, Proteintech), GFP tag (ABM40124, Abbkine), His (A02050, Abbkine), GST tag (300195, ZEN BIO), Beta Actin (66009-1-Ig, Proteintech), GAPDH (60004-1-Ig, Proteintech), AURKA (200525, ZEN BIO), AURKB (380145, ZEN BIO), CK2α (R26636, ZEN BIO), PARP1 (13371-1-AP, Proteintech), Caspase-3 (sc-7272, Santa Cruz Biotechnology), PCNA (ET1605-38, HUABIO, China), LGR5 (ab75850, Abcam), AXIN2 (db15410, Diagbio), CCND1 (db11659, Diagbio), p-β-catenin (Ser675) (80084-1-RR, Proteintech), AURKB (380145, ZENBIO). Anti-TRIM24 pS1042 (p-TRIM24) antibody was prepared from whole rabbit serum produced by repeated immunizations with the phosphorylated synthetic peptide CKKRLK(phos-S-)IEERQLLK-NH2, corresponding to a c-terminal region with Serine 1042 of human TRIM24 protein (Dia-An, China). All antibodies were used at a dilution of 1:1000.
RNA interference and transfection
Human siRNA was designed and synthesized by GenePharma (China). We performed liposome-mediated transfection for knockdown using Lipofectamine™ 2000 (Life Technologies, USA) according to the instructions. The specific siRNA sequence are as follows: TRIM24-1 siRNA (sense sequence: 5′-GAGCUCAUCAGAGGGUAAATT-3′, antisense sequence: 5′-UUUACCCUCUGAUGAGCUCTT-3′), TRIM24-2 siRNA (sense sequence: 5′-GCCACCAAGUGGUUUAUCATT-3′, antisense sequence: 5′-UGAUAAACCACUUGGUGGCTT-3′), AKT1 siRNA (sense sequence: 5′-AGGAAGUCAUCGUGGCCAATT-3′, antisense sequence: 5′-UUGGCCACGAUGACUUCCUTT-3′), AKT2 siRNA (sense sequence: 5′-GCUCUUGAGUACUUGCACUTT-3′, antisense sequence: 5′-AGUGCAAGUACUCAAGAGCTT-3′), AKT3 siRNA (sense sequence: 5′-GCCUUGGACUAUCUACAUUTT-3′, antisense sequence: 5′-AAUGUAGAUAGUCCAAGGCTT-3′), AURKA siRNA (sense sequence: 5′-GCAAUUUCCUUGUCAGAAUTT-3′, antisense sequence: 5′-AUUCUGACAAGGAAAUUGCTT-3′), AURKB siRNA (sense sequence: 5′-GCAUUGGAGUGCUUUGCUATT-3′, antisense sequence: 5′-UAGCAAAGCACUCCAAUGCTT-3′), CK2α siRNA (sense sequence: 5′-CCUCUCCUUGAUGCAGCUUTT-3′, antisense sequence: 5′-AAGCUGCAUCAAGGAGAGGTT-3′), VHL siRNA (sense sequence: 5′-GAGGUCACCUUUGGCUCUUTT-3′, antisense sequence: 5′-AAGAGCCAAAGGUGACCUCTT-3′).
Three-dimensional organoid culture
The CRC tissue was collected from a CRC patient at the First Affiliated Hospital, University of South China. The human tissue-related experiments were approved by the Medical Ethics Committee of the First Affiliated Hospital, University of South China, and the informed consent was obtained from the patient. The CRC tissue was removed from the resected colon segment and stored in ice-cold RPMI 1640 with 1% Penicillin-Streptomycin. The tissue was washed in ice-cold PBS supplemented with 1% Penicillin-Streptomycin, then cut into 1–3 mm3 cubes. The supernatant was removed after centrifuging at 200 × g for 5 min, and the pellet was then digested using Gentle Cell Dissociation Reagent (StemCell Technologies, Canada) with 10 μM ROCK inhibitor Y-27632 dihydrochloride (MCE) at 37 °C for 1 h. The tissue was then centrifuged at 200 × g for 5 min at 4 °C after being washed with 10 mL of DMEM (Servicebio, China) supplemented with Y-27632. After being resuspended in DMEM with Y-27632, the pellet was filtered through a 70 μm cell strainer. The supernatant was discarded following centrifugation at 200 × g for 5 min at 4 °C, and the pellet was then resuspended in 70% matrigel (Corning, USA). 30 μL of the mixture was plated on the bottom of 24-well plates. 500 μL Organoid Growth Medium (Human) (StemCell Technologies) supplemented with 1% Penicillin-Streptomycin and 15 mg/mL Gentamicin was added to each well generously. The organoid was incubated at 37 °C with 5% CO2 and the organoid growth medium was changed every 3 days, and the organoids were pass after 7–10 days of culture.
Plasmids
TRIM24 DNA was purchased from Addgene (Plasmid #28138, USA). Point and deletion mutants of TRIM24 were generated by subcloning PCR products. VHL DNA was purchased from Addgene (Plasmid #20790). YAP/TAZ luciferase reporter was purchased from Addgene (Plasmid #34615).
TOPFlash/FOPFlash assay
TOPFlash and FOPFlash constructs are commonly used to evaluate β-catenin-dependent transcriptional activity26. TOPFlash contains wild-type TCF binding sites and drives luciferase expression under the influence of Wnt activity, while FOPFlash has mutated, non-functional TCF binding sites and serves as a negative control. These plasmids were each cotransfected with the Renilla luciferase plasmid pRLTK to control the transfection efficiency.
Cell viability assay and colony formation assay
Cell viability assay is a method to detect cell survival and growth, and the final concentration of MTT is usually 5 mg/mL. The target cells were treated according to the experimental requirements and inoculated into 96-well plates. After a certain time, add 10% MTT solution to each well and incubate in the incubator for 4 h. Then an equal amount of triple solution (10 mM HCl, 10% SDS, and 5% isobutanol) was added to each well and incubated for 4–6 h. After all the crystals were dissolved, the absorbance was measured at 570 nm. Clonal formation assay is an effective method to determine the proliferative ability of individual cells. Cells of logarithmic growth stage were inoculated in a culture dish with appropriate cell density and cultured in a cell incubator for 1–2 weeks. When visible clones appear, the culture is terminated. Clean with 1 × PBS and fix with 4% paraformaldehyde (Biosharp, China) for 15 min. Add an appropriate amount of 0.01% crystal violet solution for 10–20 min, then slowly wash away the dyeing solution with running water, and air dry. The plate was inverted and photographed for statistics.
Flow cytometric analysis of cell cycle
The cells were digested with trypsin and the cell suspension was collected into the centrifuge tube. After washing the cells with pre-cooled PBS twice, about 50 μL of PBS was left in the suspended cells. The cells were added to 1 mL of pre-cooled 70% ethanol and fixed at 4 °C for 2 h or overnight. The cells to be tested were added with 500 μL prepared PI staining solution (20 μg/mL) (Yeasen Biotechnology, China) and incubated at room temperature for 30 min away from light. Cell cycle analysis was performed by flow cytometry. Flow cytometry data were acquired using a BD FACSCANTO II and analyzed with FlowJo software. Cell cycle distribution was evaluated using MODFIT. Cells were first gated on forward scatter (FSC) vs. side scatter (SSC) to define the main population (P1), followed by doublet exclusion using FSC-A vs. FSC-H (P2). A total of 10,000 events were collected per sample. The final gated population (P2) was analyzed for cell cycle distribution using propidium iodide (PI) staining and MODFIT software.
Flow cytometric analysis of CD44 in CRC cells
The cells were digested with trypsin and collected (1 × 106) in 1.5 mL EP tubes. At room temperature, the cells were washed twice with an appropriate amount of PBS, and then the cells were re-suspended with 100 μL PBS. Then appropriate amount of FITC-CD44 antibody (BD Pharmingen, USA) was added and incubated at room temperature for 30–40 min. Finally, the CD44 positive proportion of cells was analyzed by flow cytometry. Flow cytometry was performed using a BD FACSCANTO II and analyzed with FlowJo software. Cells were first gated on FSC vs. SSC to define the main population (P1), followed by doublet exclusion using FSC-A vs. FSC-H (P2). The gated population (P2) was then analyzed for CD44 expression using FITC-conjugated anti-CD44 antibody. A total of 10,000 events were collected per sample, and the percentage of CD44-positive cells was quantified based on fluorescence intensity.
Real-time quantitative PCR (RT-qPCR)
First, prepare the RNA sample to be detected, including the extraction of target RNA molecules (Trizol method), purification, and quantification. cDNA was synthesized by reverse transcription using cDNA synthesis kit (Novoprotein, China). Using cDNA as template, qPCR kit (Yeasen Biotechnology) was used for PCR amplification. Real-time PCR instrument can monitor the change of fluorescence intensity in the process of PCR reaction in real time, and carry out quantitative analysis of DNA content according to the change curve of light signal. ACTB is used as an internal parameter for data normalization. The qRT-PCR primer sequences are listed in Supplementary Table 3.
Immunoprecipitation and immunoblot
Immunoprecipitation was performed using a Beads Protein A/G immunoprecipitation Kit (Beaver, China) according to the manufacturer’s instructions. Briefly, certain antibodies were applied to 30 µl of protein A/G magnetic beads in PBS containing 0.1% BSA, and rotated for 2 h at 4 °C. Then, magnetic beads were washed twice with PBS. Cells were collected and lysed on ice in 0.4 mL of lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, and 0.5% sodium deoxycholate, supplemented with a complete mini protease inhibitor cocktail [Roche, USA]) for 30 min. After centrifugation at 13,000 × g for 20 min at 4 °C, the supernatants were incubated with the obtained magnetic beads, and rotated for more than 2 h at 4 °C. Finally, the beads were washed at least four times with cold lysis buffer followed by a separation by SDS-PAGE under reduced and denaturing conditions, the resolved protein bands were transferred onto nitrocellulose membranes, probed with appropriate antibodies, developed by the SuperKine West Femto Maximum Sensitivity Substrate (Abbkine) according to the manufacturer’s instructions, and imaged using a Chemiluminescent Imager (Qinxiang, China).
GST pull-down assays
GST or GST-fused proteins were bacterially expressed. The bacterial lysate was applied to GST purification magbeads (Absin, China), and incubated for 1 h at 4 °C. After washing twice with PBS, His-labelled recombinant proteins were added and incubated for 2 h at 4 °C. The magbeads were washed four times with HNTG buffer (HEPES [pH7.4], NaCl 150 mM, glycerol 10%, Triton ×-100 0.1%, supplemented with a complete mini protease inhibitor cocktail [Roche]), and subjected to immunoblotted.
Subcellular fractionation assay
The subcellular fractionation assay was performed as previously described52. The cells were digested with trypsin (2–5 × 106), transferred to 1.5 mL EP tubes, and washed twice with pre-cooled PBS. The cells were suspended in pre-cooled Buffer A (10 μM HEPES [pH7.9], 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.5 mM DTT, 0.4% NP-40, 0.5 mM PMSF) with a protease inhibitor cocktail (Roche, China) and placed on ice for 5 min. Then cells were centrifuged at 500 × g for 3 min at 4 °C and the supernatant was collected as cytoplasmic extract. The remaining cell granules were treated with Buffer C (20 mM HEPES [pH7.9], 420 mM NaCl, 1.5 mM MgCl2, 25% glycerol, 0.5 mM PMSF, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mm DTT, a protease inhibitor cocktail) was suspended and swirled for 1 min. After incubating on ice for 10 min, vortex for 1 min. Centrifuge at 15,000 × g 10 min at 4 °C and collect supernatant as nuclear extract. Loading buffer was added and the samples were boiled at 95 °C for 6 min for western blot test.
In vitro kinase assays
The GST-TRIM24 and GST-TRIM24-S1042A recombinant proteins were induced and purified as previously described. In a kinase buffer (50 mM Hepes [pH 7.5], 150 mm NaCl, 4 mM MnCl2, 6 mM MgCl2, 10% glycerol, 1 mM dithiositol, and 100 μM NaVO4), We added the above GST purified magnetic beads, recombinant AURKB or AKT, ATP (20 µM), hesperadin (100 mM), or DMSO and incubated at 30 °C for 15 min. The samples were boiled at 95 °C for western blot detection.
Mouse tumor xenografts and treatments
Male BALB/c nude mice (6–8 weeks old) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China) and maintained under specific pathogen-free (SPF) conditions with a 12-h light/dark cycle, ambient temperature of ~22 °C, and relative humidity of 50 ± 5%. For xenograft studies, mice were randomly divided into two groups and subcutaneously injected with 1 × 10⁶ DLD1 or SW1643 cells suspended in 0.1 mL PBS. When tumors became palpable (typically by day 6 or 7), each group was further divided into two subgroups and treated with either hesperadin (2.5 mg/kg), barasertib (2 mg/kg or 10 mg/kg), or PBS (vehicle control) via intraperitoneal injection every 2 days. Tumor size was measured approximately every 3 days using calipers, and tumor volume was calculated using the formula: 0.52 × length × width². In a separate experiment, 1 × 10⁶ DLD1-WT or DLD1-KO-1 cells were subcutaneously injected into male BALB/c nude mice (6–8 weeks old). Tumor growth was monitored every 3 days starting around day 7 post-injection. The experiment was terminated when tumor volume exceeded approximately 300 mm³. At the endpoint, all mice were euthanized, and the subcutaneous tumors were excised, photographed, and fixed in 4% paraformaldehyde for subsequent immunohistochemical analysis.
Immunocytochemistry
Paraffin-embedded samples were cut into 5 μm thick sections. The sections were completely immersed in citric acid antigen retrieval solution (pH = 6.0, Servicebio) and heated to boiling for 15–20 min for antigen retrieval. Endogenous tissue peroxidase was blocked by treatment with 3% hydrogen peroxide for 15–20 min. After 5% BSA blocking, the sections were incubated with PCNA antibody at 4 °C overnight. Immunodetection was performed the next day using DAB staining.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as previously described53. Briefly, HEK293T cells or CRC cells were cultured on poly-L-lysine-coated coverslips and were transiently transfected with plasmids for 36 h, or transfected with siRNAs for 72 h. Then cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.05% Triton ×-100 in PBS for 5 min, and blocked with 5% goat serum in PBS for 30 min. Subsequently, the cells were stained with 2 μg/mL of DAPI (Absin) for 5 min at room temperature. The slides were then rinsed with PBS three times and cover mounted for fluorescence microscopy. Images were acquired under Leica Stellaris confocal microscopes using identical laser settings across each group.
In vitro ubiqutination assay
HEK293T cells were transiently transfected with GFP-TRIM24 or TRIM24 mutant (GFP-△Ring) for 36 h and cell lysates were immunoprecipitated using an anti-GFP antibody. The immunoprecipitants were washed twice with lysis buffer and twice with ubiquitylation buffer (25 mM Tris-HCl [pH 7.6], 5 mM MgCl2, 100 mM NaCl, 1 mM DTT, 2 mM ATP) and resuspended with ubiquitylation buffer. Then the recombinant VHL protein, UbE1 (0.5 μg, Yeasen Biotechnology), UbE2 (0.5 μg, Yeasen Biotechnology) and ubiquitin (5 μg, ProSpec, Israel), were added into ubiquitylation buffer to the final volume 30 μL. After incubation for 2 h at 37 °C, post-reaction mixtures were immunoblotted with indicated antibodies.
Label-free relative quantitative mass spectrum analysis
This analysis was conducted at Institutional Center for Shared Technologies and Facilities of SIMM, Chinese Academy of Sciences. In brief, immunoprecipitation experiments were carried out to enrich TRIM24 following the lysis of DLD1 cells. The sample was added to 100 µL of 100 mM NH4HCO3, followed by trypsin (10 ng/µL) digestion at 37 °C for 17 h. Then, the sample was centrifuged at 12,000 × g for 30 min, and the supernatant was freeze-dried, desalted, and freeze-dried again. Subsequently, 15 µL of 0.1% FA solution was added to dissolve the freeze-dried peptide. After centrifugation at 12,000 × g for 5 min, the supernatant was transferred to the sample vial for mass spectrometry (Q Exactive HFX) analysis. The pFind (3.1.5) database search was performed, with Oxidation (M), Acetyl (Protein N-term), and Phospho (STY) set as variable modifications, and the false discovery rate of protein identification was controlled to be less than 1%.
TUNEL
TUNEL in situ on tumor tissue sections was performed using the TUNEL BrightGreen Apoptosis Detection Kit (Vazyme, China) following the manufacturer’s instructions.
Statistical analysis
All statistical analysis was performed using GraphPad Prism version 8.0. The differences between treated and control groups were examined by unpaired Student’s t-tests, except log-rank tests were used for Kaplan–Meier survival curves. Standard error of mean (SEM) was plotted in graphs, ns means nonsignificant difference.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The kinases potentially responsible for TRIM24 Ser1042 phosphorylation were predicted using online tools including KinasePhos 2.0, GPS 3.0, and iGPS 1.0. The specific website URL is as follows: http://kinasephos2.mbc.nctu.edu.tw/, http://hprd.org/PhosphoMotif_finder, http://gps.biocuckoo.cn/, http://igps.biocuckoo.org/index.php. RNA-seq data and corresponding clinical information of CRC were extracted from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/). Bioinformatics analysis was performed using BEST. The data of protein abundance of CRC patients was downloaded from CPTAC database (https://proteomics.cancer.gov/programs/cptac). Immunohistochemical staining image of TRIM24 in CRC tissues were obtained from the Human Protein Atlas database54,55 (https://www.proteinatlas.org/, DOI: 10.1126/science.aan2507). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD063426. Source data are provided with this paper. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Hossain, M. S. et al. Colorectal cancer: a review of carcinogenesis, global epidemiology, current challenges, risk factors, preventive and treatment strategies. Cancers 14, 1732 (2022).
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Tsikitis, V. L., Larson, D. W., Huebner, M., Lohse, C. M. & Thompson, P. A. Predictors of recurrence free survival for patients with stage II and III colon cancer. BMC Cancer 14, 336 (2014).
Zhao, H. et al. Wnt signaling in colorectal cancer: pathogenic role and therapeutic target. Mol. cancer 21, 144 (2022).
Albrecht, L. V., Tejeda-Muñoz, N. & De Robertis, E. M. Cell biology of canonical Wnt signaling. Annu. Rev. Cell Dev. Biol. 37, 369–389 (2021).
Liu, J. et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 7, 3 (2022).
Prossomariti, A., Piazzi, G., Alquati, C. & Ricciardiello, L. Are Wnt/β-catenin and PI3K/AKT/mTORC1 distinct pathways in colorectal cancer?. Cell. Mol. Gastroenterol. Hepatol. 10, 491–506 (2020).
Daisy Precilla, S., Biswas, I., Kuduvalli, S. S. & Anitha, T. S. Crosstalk between PI3K/AKT/mTOR and WNT/β-Catenin signaling in GBM - Could combination therapy checkmate the collusion?. Cell. Signal. 95, 110350 (2022).
Nagini, S., Sophia, J. & Mishra, R. Glycogen synthase kinases: moonlighting proteins with theranostic potential in cancer. Semin. Cancer Biol. 56, 25–36 (2019).
Rowan, A. J. et al. APC mutations in sporadic colorectal tumors: a mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 97, 3352–3357 (2000).
Aghabozorgi, A. S. et al. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 157, 64–71 (2019).
Bala, P., Kavadipula, P., Sarkar, S. & Bashyam, M. D. To β or not to β: lack of correlation between APC mutation and β-catenin nuclear localization in colorectal cancer. J. Gastrointest. Cancer 54, 1181–1184 (2023).
Blundon, M. A. et al. Proteomic analysis reveals APC-dependent post-translational modifications and identifies a novel regulator of β-catenin. Development 143, 2629–2640 (2016).
McAvera, R. M. & Crawford, L. J. TIF1 proteins in genome stability and cancer. Cancers 12, 2094 (2020).
Khetchoumian, K. et al. Loss of Trim24 (Tif1alpha) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat. Genet. 39, 1500–1506 (2007).
Tsai, W. W. et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932 (2010).
Groner, A. C. et al. TRIM24 is an oncogenic transcriptional activator in prostate cancer. Cancer Cell. 29, 846–858 (2016).
Reymond, A. et al. The tripartite motif family identifies cell compartments. EMBO J. 20, 2140–2151 (2001).
Jain, A. K., Allton, K., Duncan, A. D. & Barton, M. C. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol. Cell. Biol. 34, 2695–2709 (2014).
Zhu, Q. et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med. 217, e20192083 (2020).
Wei, W. et al. TRIM24 is an insulin-responsive regulator of P-bodies. Nat. Commun. 13, 3972 (2022).
Xu, H., Niu, M., Yuan, X., Wu, K. & Liu, A. CD44 as a tumor biomarker and therapeutic target. Exp. Hematol. Oncol. 9, 36 (2020).
Dutta-Simmons, J. et al. Aurora kinase A is a target of Wnt/beta-catenin involved in multiple myeloma disease progression. Blood 114, 2699–2708 (2009).
Leow, P. C., Tian, Q., Ong, Z. Y., Yang, Z. & Ee, P. L. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Invest. New Drugs 28, 766–782 (2010).
Sarkar, S., Mandal, C., Sangwan, R. & Mandal, C. Coupling G2/M arrest to the Wnt/β-catenin pathway restrains pancreatic adenocarcinoma. Endocr. -Relat. Cancer 21, 113–125 (2014).
Ishitani, T. et al. The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature 399, 798–802 (1999).
Yao, Y., Zhou, S., Yan, Y., Fu, K. & Xiao, S. The tripartite motif-containing 24 is a multifunctional player in human cancer. Cell Biosci. 14, 103 (2024).
Gao, T., Furnari, F. & Newton, A. C. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell. 18, 13–24 (2005).
Brognard, J., Sierecki, E., Gao, T. & Newton, A. C. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell. 25, 917–931 (2007).
Guo, J. et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932 (2016).
McClellan, A. J., Scott, M. D. & Frydman, J. Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121, 739–748 (2005).
Lee, S. et al. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc. Natl. Acad. Sci. USA 93, 1770–1775 (1996).
Iliopoulos, O., Kibel, A., Gray, S. & Kaelin, W. G. Jr. Tumour suppression by the human von Hippel-Lindau gene product. Nat. Med. 1, 822–826 (1995).
Wang, Y. et al. TRIM24 is critical for the cellular response to DNA double-strand breaks through regulating the recruitment of MRN complex. Oncogene 42, 586–600 (2023).
Kosugi, S., Hasebe, M., Tomita, M. & Yanagawa, H. Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 106, 10171–10176 (2009).
Varadi, M. et al. AlphaFold protein structure database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–d375 (2024).
Andrew, C. D., Warwicker, J., Jones, G. R. & Doig, A. J. Effect of phosphorylation on alpha-helix stability as a function of position. Biochemistry 41, 1897–1905 (2002).
Ardito, F., Giuliani, M., Perrone, D., Troiano, G. & Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40, 271–280 (2017).
Tanaka, K. et al. Targeting Aurora B kinase prevents and overcomes resistance to EGFR inhibitors in lung cancer by enhancing BIM- and PUMA-mediated apoptosis. Cancer Cell. 39, 1245–1261.e1246 (2021).
Zhang, Y. et al. Hesperadin suppresses pancreatic cancer through ATF4/GADD45A axis at nanomolar concentrations. Oncogene 41, 3394–3408 (2022).
Disoma, C., Zhou, Y., Li, S., Peng, J. & Xia, Z. Wnt/β-catenin signaling in colorectal cancer: is therapeutic targeting even possible?. Biochimie 195, 39–53 (2022).
Schatoff, E. M., Leach, B. I. & Dow, L. E. Wnt signaling and colorectal cancer. Curr. Colorectal Cancer Rep. 13, 101–110 (2017).
Zhang, L. H. et al. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 34, 600–610 (2015).
Xie, W. et al. Tripartite motif containing 24 regulates cell proliferation in colorectal cancer through YAP signaling. Cancer Med. 9, 6367–6376 (2020).
Allton, K. et al. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 106, 11612–11616 (2009).
Fang, Z. et al. TRIM24 promotes the aggression of gastric cancer via the Wnt/β-catenin signaling pathway. Oncol. Lett. 13, 1797–1806 (2017).
Tian, H. et al. TRIM24 promotes colorectal cancer cell progression via the Wnt/β-catenin signaling pathway activation. Am. J. Transl. Res. 14, 831–848 (2022).
Klapper, L. et al. TRIM24 expression as an independent biomarker for prognosis and tumor recurrence in HNSCC. J. Pers. Med. 12, 991 (2022).
Kikuchi, M. et al. TRIM24 mediates ligand-dependent activation of androgen receptor and is repressed by a bromodomain-containing protein, BRD7, in prostate cancer cells. Biochim. Biophys. Acta 1793, 1828–1836 (2009).
Mertins, P. et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016).
Schweppe, D. K., Rigas, J. R. & Gerber, S. A. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteom. 91, 286–296 (2013).
Liu, Y. et al. Bacterial genotoxin accelerates transient infection-driven murine colon tumorigenesis. Cancer Discov. 12, 236–249 (2022).
Fu, K. et al. Sam68/KHDRBS1-dependent NF-kappaB activation confers radioprotection to the colon epithelium in gamma-irradiated mice. Elife 5, e21957 (2016).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Uhlen, M. et al. A pathology atlas of the human cancer transcriptome. Science 357, eaan2507 (2017).
Acknowledgements
We are grateful to the Cancer Genome Atlas Program for the database. We thank the online bioinformatics analysis site BEST, by which Figs. 1A, B and 2A, B and supplementary Fig. S5 were created. This work was supported by National Natural Science Foundation of China [32100580 and 32170726] to Y.W. and K.F.; Ministry of Science and Technology of China [2022YFE0133400] to K.F.; Hunan Provincial Science and Technology Department [2024JJ8147] to Y.W.
Author information
Authors and Affiliations
Contributions
K.F., FW., and S.X. performed study concept and design; Y.W., Y.Y., Z.L., S.L., F.Y., Q.F., D.W., Q.Z., and H.Z. performed experiments and analysed the data; DW provided technical and material support; K.F. and Y.W. provided funding acquisition; K.F., Y.W., and Y.Y. wrote, reviewed, and revised the paper; Y.W. and Y.Y. provided acquisition, analysis and interpretation of data, and statistical analysis. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Joong Sup Shim and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Yao, Y., Liu, Z. et al. Cytoplasmic TRIM24 promotes colorectal cancer cell proliferation by activating Wnt/β-catenin signaling. Nat Commun 16, 8598 (2025). https://doi.org/10.1038/s41467-025-63685-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63685-8
