
Abstract
Disentangling the drivers of global biodiversity patterns is a cornerstone of biogeography that remains elusive for many diverse biological groups. Here we present a complete species-level phylogeny of the ant subfamily Ponerinae based on new genomic sequencing and taxonomic grafting. We combine results with a large-scale geographic database to explore the contribution of three main mechanisms in shaping global ponerine biodiversity patterns: time for accumulation, differences in diversification rate, and asymmetric dispersal. We show that extant ponerine ants originated in Gondwana, spread eastward across tropical bioregions, and more recently colonized temperate areas. The relative timing of colonization events was identified as the prominent driver of present-day biodiversity patterns, supporting the time for accumulation hypothesis. Conversely, differences in diversification rates and asymmetrical dispersal histories mitigated the heterogeneity in biodiversity by fueling accumulation of lineages in the least diverse bioregions. These findings suggest that tropical niche conservatism played a major role in shaping the biogeographic and evolutionary history of Ponerinae. Overall, we emphasize the importance of considering the relative timing of past dispersal events and variations in diversification rates over evolutionary time to gain a deeper understanding of Earth’s biodiversity patterns.
Similar content being viewed by others

Introduction
Unraveling the drivers of the distribution of biodiversity patterns worldwide has been a core task of biogeographers since the dawn of the discipline. Multiple non-exclusive mechanisms have been proposed to explain the heterogeneous patterns of biodiversity at a large spatial scale1. Three overarching hypotheses offer predictions that can be put to scrutiny by using phylogenetic and historical biogeographic approaches to reconstruct diversification dynamics across space and time. The Diversification Rate Hypothesis (DRH) states that differences in net diversification rates across bioregions are responsible for the current imbalance in diversity patterns2,3. The Time-for-Accumulation Hypothesis (TfAH) posits that the timing of colonization of bioregions is crucial to explain biodiversity patterns, as more time to diversify leads to the accumulation of more local richness over time4. Finally, the Asymmetric Dispersal Hypothesis (ADH) relies on a pervasive imbalance in lineage exchanges across bioregions over time to generate macroevolutionary sources and sinks of lineages and explain biodiversity heterogeneity5,6.
The task of disentangling the importance of these three mechanisms for generating present-day biodiversity patterns has been reinvigorated by the emergence of global species-level occurrence databases and genome-scale sequencing data used to reconstruct comprehensive phylogenies for macroevolutionary and historical biogeographic analyses7. However, that new power has been applied primarily to large organisms: vertebrates and plants8,9,10. Invertebrates, especially terrestrial arthropods, are a major biodiversity component that has been historically overlooked relative to its importance11,12. In terrestrial systems, ants are dominant invertebrates and ecosystem engineers whose diversification dynamics play a major role in the evolution of terrestrial life in general13,14. In this study, we aim to disentangle the importance of the different mechanisms at play for generating present-day biodiversity patterns in a cosmopolitan and ecologically significant insect group, the ponerine ants.
Ponerinae rank as the third most diverse subfamily of ants with 50 described genera and ca. 1400 extant species, including the largest known ant species15. The subfamily is superseded in diversity only by Myrmicinae (ca. 7200 sp.) and Formicinae (ca. 3300 sp.), and dwarfs all other closely related subfamilies in the poneroid clade that each record fewer than 200 described extant species16. As ubiquitous and abundant predators of small invertebrates, ponerine ants are a key component of trophic webs, especially in tropical ecosystems17. Ponerines are also widely distributed across all biogeographic regions except Antarctica and constitute an important presence in the fossil record18. Therefore, the group is well-suited to exploring diversity dynamics across large scales of space and time.
Ant species richness is centered in the tropics with hotspots located in the Amazon basin, Mesoamerica, the Brazilian Atlantic Forest, the African Equatorial forest, and Southeast Asia. Outside of the tropical belt, the Mediterranean basin, Eastern Australia, and Madagascar appear as secondary hotspots19. Economo et al.20 examined two hypotheses to explain latitudinal diversity patterns across ants, the aforementioned DRH and the Tropical Conservatism Hypothesis (TCH), which assumes that tropical ecosystems have remained stable over longer evolutionary timeframes, allowing for more lineages to accumulate due to longer diversification times. This study included phylogenetic data from 90 ponerine taxa, while the remaining species were added to the phylogeny by random grafting. The inferred diversification dynamics indicated an absence of signal for the DRH but are consistent with TCH and time for accumulation being the most important factor in driving latitudinal diversity in ants20. Whether these patterns will also hold true on a finer scale, such as the subfamily level, and within a more robust phylogenetic framework, remains to be tested.
This strong latitudinal gradient observed in ants hints at a tropical origin for most ant subfamilies, including the Ponerinae. A previous attempt to reconstruct the ancestral ranges of ants led to strong support for a Neotropical origin of ponerine ants21. This result, based on a reduced sample of 27 ponerine species, predicted an emergence of Ponerinae in the Neotropics during the Paleocene (65-55 Mya). However, this age estimate, among the youngest for Ponerinae origin22, was later contradicted by the identification of older fossils found in Kachin amber from Myanmar (Late Cretaceous: 98.79 ± 0.62 My)23. Beyond this uncertainty in the origin of Ponerinae, the details of their current diversity patterns, diversification dynamics, and dispersal history across the world are poorly known. While Moreau and Bell21 identified the Neotropics as an important center for early diversification of ants as a whole, acting as both a museum and a cradle for ant diversity, it is unclear whether this pattern holds for ponerine ants.
Here, we produce a complete species-level phylogeny of ponerine ants, based on new genomic sequencing and taxonomic grafting, and leverage comprehensive online occurrence databases to reconstruct the diversification dynamics and dispersal history of the extant taxa in the subfamily to investigate the contribution of the three hypotheses (DRH, TfAH, and ADH) as drivers of biogeographic and diversification patterns in Ponerinae. We model ancestral range estimates to trace the origins of the clade and its major lineages. We map current species richness and compare net diversification rates across bioregions along geological time. Finally, we investigate the main drivers of the richness of bioregional radiations and quantify the immigration age of lineages. Altogether, our study paints a complex biogeographic and evolutionary history for an ecologically significant group of insects, contributing to the much-needed invertebrate perspective on global biodiversity dynamics11,12.
Results
A new comprehensive species-level time-calibrated phylogeny
We produced the most comprehensive species-level time-calibrated phylogeny of Ponerinae, including 789 phylogenetically-informed taxa (Supplementary Figs. S3 and S5), complemented with 745 taxonomically-informed taxa to reach an exhaustive coverage of described ponerine species to date (Fig. 1 and Supplementary Fig. S5). We obtained a median root age estimate of 122.8 My with a 95% Highest Posterior Density (HPD) interval ranging from 96.6 My to 150.8 My across the 1000 backbone posterior trees processed with TreePL (Supplementary Tables S5–S8). This estimate falls within the upper range of previous estimates and dates the emergence of the extant ponerine ant clade during the Early Cretaceous, coinciding with the timing of origins of other ant subfamilies22. We retrieved monophyly for all previously described tribes and genus-groups24. Within genus groups, a few cases of paraphyletic genera were discovered, providing a phylogenetic framework for future taxonomic revisions (Supplementary Figs. S3 and S5, S6).

Ancestral range estimates (ARE) are mapped on the grafted Maximum Clade Credibility (MCC) tree using pies to represent posterior marginal likelihood of bioregions estimated from 1000 Biogeographic Stochastic Maps simulated with the DEC + J model. ARE of the 7 genus-groups are shown with pies at respective Most Recent Common Ancestor (MRCA) nodes. Regional In Situ Radiations (RISR) used in analyses are displayed with rectangles on their crown nodes. Capitalized names correspond to genus-groups from Schmidt & Shattuck24: PLAT. Platythyrea group; PLEC. Plectroctena group; H. Harpegnathos group. Ant images are modified from original pictures by F. Brassard. Results for alternative divergence dating hypotheses are shown in Supplementary Fig. S9.
Gondwanan origin of Ponerinae
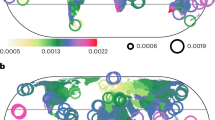
The distribution of ponerine diversity across the globe presents a striking latitudinal gradient with only 95 (6.2%) extant lineages found in the entire Holarctic (Fig. 2A and Supplementary Fig. S8A). However, the bulk of ponerine diversity shows a balanced distribution across the tropics with 397 taxa found in the Neotropics, 452 in the Afrotropics, 380 in Indomalaya, and a relatively lower record of 236 taxa in Australasia (Fig. 2A and Supplementary Fig. S8B).

A Species richness; B Overall dispersal network; C Current net diversification rates; D Mean biogeographic immigration ages. A Species richness based on stacked spatial ranges estimated as alpha-hulls from geolocalized occurrences. B Overall network of dispersal events across bioregions averaged across 1000 Biogeographic Stochastic Mapping (BSM) simulations of biogeographic histories based on a DEC + J model. Arrow width scaled to overall counts of dispersal events displayed on arrow tips. Node size scaled to bioregional richness displayed in each node. Counts of taxa encompassing multiple bioregions are split equally among them. See Supplementary Fig. S10 for details of dispersal network per geological epoch. C Mean net diversification rates as mean tip rates obtained from BAMM rate estimates combined with taxon ranges. Areas with more than two taxa are shown. D Mean biogeographic immigration ages as mean taxon biogeographic immigration ages combined with taxon ranges. Biogeographic immigration ages depict the timing of the first arrival of each taxon and its ancestral lineages in its current bioregion, as estimated from 1000 BSM simulations. Areas with more than two taxa are shown. Results for alternative divergence dating hypotheses are shown in Supplementary Fig. S23.
We found very high agreement for ancestral range estimates between the different divergence dating hypotheses, with Cohen’s Kappa indices higher than 0.90 (MCC vs. Youngest = 0.971; MCC vs. Oldest = 0.907), reflected by the high similarity in ancestral ranges mapped on the respective phylogenies (Fig. 1 and Supplementary Fig. S9). The most likely ancestral range for Ponerinae estimated on the grafted Maximum Clade Credibility (MCC) tree with the best fitted DEC + J model (mean Akaike’s weight = 99.95%; Supplementary Tables S1 and S2) encompassed both the Afrotropics and the Neotropics (scaled marginal likelihood (ML) = 0.482). Lower support values for Afrotropics-Indomalaya (ML = 0.235) and Afrotropics alone (ML = 0.221) were also found. Alternative time-calibrated grafted trees, including the most extreme youngest/oldest hypotheses, led to congruent results (Supplementary Tables S4 and S6–S8). Altogether, those ancestral ranges point toward an origin of ponerine ants in Gondwana, at a time when the African, South American, and Indian plates were part of the Gondwanan supercontinent. By contrast, the seven genus-groups, which emerged after the split of Gondwana ca. 120–100 Mya25 in the MCC tree, are estimated to originate from unique areas distributed across three bioregions in the Tropics: Odontomachus group and Pachycondyla group in the Neotropics; Plectroctena group, Hypoponera group, and Platythyrea group in the Afrotropics; and Ponera group and Harpegnathos group in Indomalaya (Supplementary Tables S5–S8). Most lineages were found within these three tropical bioregions (i.e., Afrotropics, Neotropics, and Indomalaya) across the evolutionary history of ponerine ants (Fig. 1 and Supplementary Table S9).
Asymmetry in dispersal dynamics
We reconstructed the dispersal network of ponerine ants across bioregions based on the aggregation of 1000 Biogeographic Stochastic Mapping (BSM) simulations (Fig. 2B; An animated figure showing the overall dispersal and diversification dynamics of ponerine ants through time is available in the online version). Overall, the most important dispersal routes occurred between tropical regions from the Neotropics to Australasia, going through the Afrotropics and Indomalaya. Extensions towards temperate zones occurred mostly from the Neotropics to the Nearctic, from the Afrotropics to the Western Palearctic, and from Indomalaya to the Eastern Palearctic, strikingly with almost no dispersal recorded between temperate bioregions (Fig. 2B). The Afrotropics were the most important source of dispersal events in early times (Late Cretaceous to Oligocene), while Indomalaya emerged as the most important source since the Miocene. The Neotropics remained an important source throughout the history of Ponerinae but received relatively few new lineages since the Miocene (Fig. 3A, B and Supplementary Fig. S10). Ponerinae likely reached Australasia for the first time around 60 Mya with a first wave of colonization from the Afrotropics, followed by a resurgence during the Miocene when the Australian plate collided with Southeast Asia creating temporary land bridges with Indomalaya26 (Fig. 3C, D and Supplementary Fig. S10). The Eastern Palearctic was colonized ca. 40–30 Mya from Indomalaya. The Nearctic was reached for the first time 30–20 Mya from South America through the Caribbean plate. Finally, our analyses indicate arrival of extant ponerines in the western Palearctic ca. 20–15 Mya, after the African plate closed on the European plate27. Across all Ponerinae history, the Neotropics (N) and Afrotropics (A) appeared as macroevolutionary sources of lineages, recording on average more departing lineages (Fig. 4A; N: 53.8 ± 3.4 taxa out; A: 67.0 ± 4.8 taxa out) than newcomers (Fig. 4A; N: 22.4 ± 2.6 taxa in; A: 31.6 ± 3.1 taxa in). Indomalaya (100.7 ± 5.2 taxa in vs. 168.1 ± 7.2 taxa out) displayed a more neutral balance, while Australasia (115.4 ± 3.9 taxa in vs. 54.2 ± 5.7 taxa out), and even more clearly the temperate bioregions (Nearctic: 18.5 ± 0.7 taxa in vs. 5.0 ± 1.6 taxa out; Western Palearctic: 17.0 ± 0.4 taxa in vs. 3.1 ± 1.6 taxa out; Eastern Palearctic: 58.5 ± 1.5 taxa in vs. 12.9 ± 3.9 taxa out) acted as macroevolutionary sinks (Fig. 4A). Overall, we reconstructed a longitudinal spatiotemporal asymmetry of dispersal from a Western origin toward the Eastern continental areas with a hierarchical order of source bioregions providing on average more lineages than they received from others such as the Neotropics (N) > Afrotropics (A) > Indomalaya (I) > Australasia (U) (Fig. 2B; N → A = 11 > A → N = 8; N → I = 16 > I → N = 8; N → U = 8 > U → N = 1; A → I = 37 > I → A = 13; A → U = 14 > U → A = 5; I → U = 89 > U → I = 41). Similarly, we found a clear latitudinal asymmetry from a tropical origin towards the temperate areas, with all shared dispersal routes recording more dispersal towards higher latitudes than towards tropical areas (Fig. 2B). These asymmetrical dispersal dynamics were also found under the two alternative dating hypotheses (Supplementary Fig. S23A, B). Even though the absolute timing of events estimated differed, the conclusions regarding the primary direction of dispersal flux and the status of bioregions as macroevolutionary sources and sinks were invariant to the divergence ages selected (Supplementary Figs. S11–S13).

A, B Sources of dispersal events. C, D Destinations of dispersal events. E, F Locations of in situ speciation events. Mean counts on the left-side. Percentages of events on the right-side. All events are averaged across 1000 Biogeographic Stochastic Mapping (BSM) simulations within sliding windows of 10 My, e.g., counts/proportions reported for 5 Mya (the most recent time shown) represent the average counts/proportions of events recorded between 0 to 10 Mya for a single BSM simulation. Results for alternative divergence dating hypotheses are shown in Supplementary Figs. S11 and S12.

A Dispersal events; B In situ diversification events. A Mean counts of dispersal events aggregated over evolutionary time (123.6 My–0 My) as detected from 1000 Biogeographic Stochastic Mapping (BSM) simulations using DEC + J model. Upper bar plots represent the mean counts of arrivals to a bioregion acting as a destination. Lower bar plots represent the mean counts of departures from a bioregion acting as a source. B Mean counts of in situ cladogenetic events within bioregions aggregated over evolutionary time (123.6 My–0 My) as detected from 1000 BSM simulations using the DEC + J model. Error bars on both plots are standard deviation recorded across the 1000 BSM simulations. Results for alternative divergence dating hypotheses are shown in Supplementary Fig. S13.
Diversification dynamics across space and time
We recorded the location of in situ diversification events observed across the 1000 BSM simulations. The Neotropics arose as the primary location for in situ diversification during the Late Cretaceous comprising the majority of the few in situ speciation events recorded during this epoch (Fig. 3E, F). During the Paleocene and Eocene (66–34 My), the Afrotropics emerged progressively as the new major area of in situ speciation until the present-day balanced picture between Neotropics, Afrotropics, and Indomalaya set up during the Oligocene (Fig. 3E, F). Across Ponerinae history, the Afrotropics (420.9 ± 3.3 events = 34.9%) were the major source of in situ diversification, followed by the Neotropics (377.7 events ±2.9 events = 31.3%), Indomalaya (281.8 ± 7.4 events = 23.4%), and Australasia (120.0 ± 5.0 events = 9.9%) (Fig. 4B). Strikingly, the temperate bioregions recorded very few in situ diversifications (6.3 ± 2.9 events = 0.5%), hinting that temperate biodiversity of ponerine ants originated mostly from dispersal events from the Tropics rather than regional diversification. This result was also found when assuming different divergence dating hypotheses (Supplementary Figs. S11 and S13).
Complementary to empirical counts of speciation events, we modeled changes in diversification rates along branches in the BAMM framework. Next, we mapped current net diversification rates in space computing mean tip rates among local taxa. We observed a longitudinal gradient of current diversification rates in ponerine ants increasing from West to East, with higher rates found in Indomalaya and Australasia (Fig. 2C and Supplementary Fig. 23C, D). Mean rates in the New World (Nearctic and Neotropics) and Afrotropics were found to be marginally lower than what can be expected from a random spatial distribution of taxa (Supplementary Fig. S20A). Rates in the northern temperate regions (Latitude > 30° N) appeared lower than average, yet, due to the relatively low taxon richness were not significant compared to a random spatial distribution of taxa (Supplementary Fig. S20B).
Diversification rates hypothesis is not supported at any time point in the evolution of Ponerinae
We recovered on average 12.1 ± 4.1 regime shifts in diversification dynamics along ponerine ant history from BAMM applied on the MCC tree. Those regime shifts corresponded to seven major taxonomic groups (Supplementary Fig. S14 and Supplementary Table S10). While we found differences in absolute net diversification rates estimated from the MCC tree and the youngest and oldest divergence dating hypotheses due to differences in the timing of speciation events, the relative distribution of rates across shared clades are significantly correlated between the hypotheses (Spearman’s rank correlation: MCC vs. Youngest: t303 = 13.7, p < 0.001, ρ = 0.619, 95% CI = 0.544–0.684; MCC vs. Oldest: t313 = 37.4, p < 0.001, ρ = 0.904, 95% CI = 0.882– 0.922; Supplementary Figs. S14 and S15). We formally tested for differences in diversification rates along a latitudinal gradient in the Tropics and Temperate regions and a longitudinal gradient between the New World and the Old World with two-sided STRAPP tests28. In current time, Old World taxa did not present significantly higher mean net diversification rates than New World taxa (STRAPP test: Δ Mann–Whitney–Wilcoxon’s U 5% = −26230, p = 0.376, ROW = 0.1070, RNW = 0.0982). Notably, when testing for differences at earlier time steps, we found no period during Ponerinae evolutionary history with a significant difference in diversification rates between the New World and the Old World (Fig. 5A and Supplementary Fig. S18A). Similarly, we found no significant differences in net diversification rates between temperate and tropical taxa either in current time (STRAPP test: Δ Mann–Whitney–Wilcoxon’s U 5% = −9431, p = 0.474, RTrop = 0.1071, RTemp = 0.1077), or along any evolutionary time-step (Fig. 5B and Supplementary Fig. S18B). Furthermore, no overall difference of diversification rates across the seven bioregions used for historical biogeographic inferences was found in the present-day (STRAPP test: Δ Kruskal−Wallis’s Khi² 5% = −53.58, p = 0.594), nor along evolutionary times (Supplementary Fig. S18C). The youngest and oldest divergence dating hypotheses, while yielding different absolute net diversification rates in line with their different timeframes (Supplementary Fig. S23C, D), led to similar conclusions, with no differences in net diversification rates detected between bioregions, Old World vs. New World, and Tropics vs. Temperate areas along evolutionary times (Supplementary Figs. S17 and S19).

A Old World vs. New World; B Tropics vs. Temperate bioregions. Net diversification rates through time are extracted from 1000 posterior samples of a BAMM run. Lineages are attributed to a group of bioregions (Old World vs. New World, Tropics vs. Temperate) according to their posterior probability of presence in a bioregion as estimated from 1000 Biogeographic Stochastic Mapping (BSM) simulations using DEC + J model. Each semi-transparent line corresponds to a BAMM posterior sample and reflects the confidence intervals. The opaque plain line shows the average rates. Two-sided STRAPP tests were carried out at different time steps to test for rate differences along evolutionary time (see Supplementary Fig. S18 for significance plots). Results for alternative divergence dating hypotheses are shown in Supplementary Fig. S17.
The New World is older than the Old World for Ponerinae
To explore more thoroughly the main factors shaping differences in diversification rates across bioregions, we investigated the diversification dynamics of Regional In Situ Radiations (RISR). Neotropical and Afrotropical radiations were revealed to be on average more diverse and older but showed no significant differences with Indomalaya in diversification dynamics in terms of crown rates, current rates, or time-variation in rates (Fig. 6). On the other end of the spectrum, Australasian radiations appeared younger and are experiencing currently higher rates of diversification induced by significant rate increases over time (Fig. 6).

A Current richness. B Crown age. C Linear regression: current richness ~ crown age. D Current net diversification rates. E Net diversification rates at crown age. F Time-variation parameters. Rate parameters are obtained from time-dependent birth-death models fit on each RISR. Current net diversification rates are speciation minus extinction rates as estimated from the present-days. Crown net diversification rates are initial rates estimated from the crown age. Time-variation parameters are controlling the shape of the exponential relationship between speciation rates and time: positive = increase, negative = decrease. Box elements represent median (plain lines), upper and lower quartiles (minima and maxima), 1.5x interquartile ranges (whiskers), and outliers (points) as the default option in R package ggplot2. Overall differences tested with non-parametric Kruskal–Wallis rank tests. Pairwise differences tested with post hoc two-sided Wilcoxon tests. Effect of bioregions on the linear regression of current richness per crown age was tested with likelihood ratio test (LRT) against a nested model with no bioregion effect. Lower case letters group bioregions with no significant differences. Sample size: Afrotropics = 9, Australasia = 5, Indomalaya = 12, Neotropics = 5.
The older ages of Neotropical radiations and younger ages of Australian radiations were also reflected in the spatial distribution of taxon immigration ages. We found striking latitudinal and longitudinal gradients of immigration ages, with the temperate regions displaying significantly younger taxa (Fig. 2D and Supplementary Fig. S21A). Meanwhile, within the tropical belt, immigration ages decreased from the New World in the Western hemisphere toward the Old World in the Eastern hemisphere, with the oldest communities found in the Neotropics, intermediate-aged communities in the Afrotropics, and younger tropical communities in Indomalaya and Australasia (Fig. 2D and Supplementary Fig. S21B). These striking gradients of taxon immigration ages were also found under the alternative divergence dating hypotheses. While the absolute ages varied in accordance with the timeframe of each hypothesis, the relative patterns remained robust. Especially the Neotropics and Afrotropics appeared to host taxa with the oldest immigration ages, while Indomalaya, Australasia, and the temperate bioregions exhibited younger mean immigration ages (Supplementary Fig. S23E, F).
Lastly, we found contrasting origins for taxa between bioregions. Neotropical and Afrotropical ponerine diversity is currently composed of 97% and 95% of taxa originating from in situ speciation events as opposed to dispersal, illustrating their constant high proportion of autochthonous lineages in the regional ponerine fauna through evolutionary time (Fig. 7). Other tropical bioregions currently display high levels of autochthonous fauna (82–84%) that have increased since the Cretaceous origins for Indomalaya, and more recently during the Cenozoic in Australasia (Fig. 7). Temperate regions have also witnessed increases in their proportions of autochthonous fauna since their respective colonizations. However, in sharp contrast with the tropics, they still currently record low levels of autochthonous taxa with 44% to 69% of their current diversity being of exotic origin (Fig. 7). These contrasting origins were retrieved similarly for the two alternative dating hypotheses, albeit with different absolute timing along evolutionary times (Supplementary Fig. S22).

A Current; B Through time. Proportions of lineages originating from in situ speciation events (i.e., autochthonous lineages) within each bioregion. Origins were examined by comparing the lineage ranges at the focal time step with the range of their most direct parental node. Similar ranges were counted as an in situ origin; different ranges reflected an origin from dispersal events. Estimates through time were extracted from 1000 Biogeographic Stochastic Mapping (BSM) simulations. Semi-transparent intervals correspond to each quantile among the simulations and reflect the confidence intervals. The opaque plain lines show the average proportions of autochthonous lineages recorded across simulations. Results for alternative divergence dating hypotheses are shown in Supplementary Fig. S22.
Discussion
In this study, we built upon a new species-level phylogeny of ponerine ants to reconstruct the comprehensive dispersal and diversification history of the extant taxa and their ancestors in the subfamily. Our results supported the Time-for-Accumulation Hypothesis (TfAH), revealing that differences in the timing of colonization events, and not differences in diversification rates (DRH) or asymmetrical dispersal histories (ADH), are the key mechanisms explaining current diversity patterns in Ponerinae (Table 1). The most striking pattern of the distribution of Ponerinae diversity is their strong latitudinal gradient bias toward the tropics. Indeed, temperate bioregions have been colonized considerably more recently than tropical areas and still owe an important proportion of their current diversity from the steady flow of lineages arriving from tropical areas. Meanwhile, no significant differences in net diversification rates were revealed across time between temperate and tropical areas. Within tropical latitudes, diversity is relatively balanced across the Neotropics, the Afrotropics, and Indomalaya, with Australasia harboring fewer lineages. The origin of ponerine ants in Gondwana, followed by a pervasive eastward colonization of continental masses, mostly explains the relatively lower richness of the more recent radiations in Australasia. Altogether, this study highlights the importance of reconstructing the timing of past biogeographic and evolutionary events and considering diversification rates over time in complement to current diversification rates, to better understand patterns of biodiversity on Earth.
Prominence of tropical niche conservatism in shaping the latitudinal diversity gradient has been reported for many taxa, including New World trees29, birds30, nymphalid butterflies31, and even the whole family of ants20. In this study, we found the origin of ponerine ants to lie in the tropical supercontinent Gondwana during the Cretaceous. Such a tropical origin associated with broad-scale tropical niche conservatism is likely to have favored dispersal exchanges between the tropical areas while preventing latitudinal expansion towards temperate regions, delaying colonization of the Nearctic and Palearctic, and strongly shaping the current patterns of ponerine ant diversity via the effect of the TfAH. This key finding, similar to what was recently uncovered for squamates32, advocates for the importance of geographic origin and timing of colonizations as an overarching mechanism for the distribution patterns of terrestrial life, particularly in shaping the modern-day latitudinal biodiversity gradient observed throughout the tree of life33. Our sensitivity analyses using two extreme alternative divergence dating hypotheses strongly supported this conclusion. They also emphasize that the relative timing of colonization events across bioregions is the key factor, independently from the absolute estimated timing of events.
In parallel, our results showed that the delayed colonization of temperate areas is accompanied by a recent history of asymmetrical dispersal events between the tropical and temperate regions compatible with an “Out-of-the-Tropics” scenario6. Within the tropical belt, we detected an eastward shift in time of the locations of the main dispersal routes from the early Neotropical and Afrotropical sources to the more recent Australasian sink. Such asymmetries may be due to a diversity-dependence effect where bioregions colonized early already have high diversity and present fewer available niche opportunities that in turn prevent further successful emigration from occurring34 (but see Wiens35). Consequently, both the latitudinal and longitudinal gradients of source-sink dynamics (i.e., ADH) have acted as buffers against the effects of differences in colonization times (i.e., TfAH) and limited to some extent the heterogeneity in diversity across bioregions.
Additionally, the DRH was not supported by our results for either latitudinal or longitudinal trends, adding to a growing body of literature on, e.g., ants, butterflies, birds, and mammals, indicating that differences in diversification rates may not be a prominent factor in shaping large-scale diversity patterns20,31,36. Yet, we found that the more recent Australasian radiations currently have higher rates of diversification than in other tropical bioregions, hinting that differences in diversification rates tend to buffer differences in bioregional diversity between tropical bioregions rather than generating this pattern. This increase in diversification dynamics associated with different independent colonizations of Australasia may reflect the effect of ecological opportunity37. Indeed, Australia has witnessed the ongoing expansion of subtropical and dry environments during its drift toward lower latitudes throughout the Cenozoic38. Access to these new habitats suitable for tropical ponerine ants could have driven the current rapid radiations (i.e., DRH), thus buffering the effects on diversity of more recent colonization (i.e., TfAH). Such a buffering effect of ecological opportunity in recently colonized areas is not supported in temperate bioregions despite recent evidence indicating multiple clades are currently experiencing higher rates of diversification in temperate regions39,40. As most, if not all, ponerine lineages dispersing toward temperate bioregions come from the tropics, they may lack the pre-adaptations that would enable them to rapidly thrive in the newly colonized temperate environments, thus limiting the occurrence of in situ radiations41. Ponerines typically have small colonies, rarely hosting more than a few hundred individuals, and low caste differentiation between queens and workers24. Meanwhile, temperate climates seem to favor species with larger colonies and complex social structures, allowing them to cope better with seasonal environments42. However, this picture may soon change as temperate bioregions are expected to witness an extension of subtropical climatic conditions due to anthropogenic climate change43, and they are already experiencing critical numbers of arrivals of tropical lineages due to anthropogenic dispersals44.
Overall, while differences in relative timing of colonizations (i.e., TfAH) appear to be the main driver of current biodiversity in ponerine ants, we found that asymmetries in dispersal events (i.e., ADH) and higher diversification rates (i.e., DRH) in the more recently colonized and less diverse bioregions may help shrink the gaps in biodiversity over evolutionary time. Chronologically, the source-sink dynamic providing higher numbers of arrivals than departures (i.e., ADH) is likely the main buffering mechanism in early colonization, as shown by the high proportion of lineages in temperate regions originated from dispersal rather than in situ radiation. In the longer run, higher diversification rates (i.e., DRH) are likely the main buffering mechanism: as the regional pool of lineages in the newly colonized regions increases, so does the proportion of lineages originating from in situ speciation, a phenomenon observed in Australasia.
Our study presents a complex biogeographic and evolutionary history based on an exhaustive sampling of extant described species of Ponerinae. We used fossils to time-calibrate our phylogenies. However, we did not include the fossil record in our biogeographic and diversification framework due to the difficulty in obtaining a representative sampling beyond taphonomic and taxonomic biases (but see Jouault et al.18), and ensuring proper placement and dating of fossils within our species-level time-calibrated phylogenies. Such constraints have likely led to the underestimation of speciation and extinction rates. To limit bias, we restricted our analysis and interpretation to net diversification rates31. We acknowledge that the identity and location of an early Ponerini fossil in Kachin amber in Myanmar (98.79 ± 0.62 My)23, and Ponera and Hypoponera fossils in the Baltic amber (34–48 My)45 suggest earlier and larger expansions of extinct ponerine lineages than recovered in our study focusing on extant taxa. Indeed, the temperate zone was occupied partly by megathermal biomes during the Paleocene and Eocene (65–34 My)46 allowing range expansion of tropical lineages, which mostly went extinct47, or retracted to lower latitudes following the global cooling of the Eocene-Oligocene climate transition48,49. Therefore, our study does not cover extensions of extinct lineages that may have ventured into the temperate area and did not persist to the present day. Neither does it account for scenarios of range extension and contraction, as those cannot be supported by our current likelihood framework for historical biogeographic models favoring the most likely scenarios.
Nonetheless, our study provides a crucial invertebrate perspective on the contribution of three main mechanisms shaping global biodiversity patterns and dynamics over time (i.e., TfAH, DRH, and ADH). Our key finding reveals that differences in the relative timing of colonization events are the main mechanism explaining current diversity patterns in Ponerinae, consistent with the Time-for-Accumulation Hypothesis (TfAH). This advocates for the importance of the timing of colonization as a general rule for the distribution patterns of terrestrial life32 and calls for further investigations across other diverse groups. We also produced a new species-level phylogeny that provides a broad synthesis of the state of knowledge in the systematics of Ponerinae. This phylogeny opens the door to refinements of current taxonomic classifications and more in-depth explorations of macroevolutionary patterns within Ponerinae that can function as models for studies on the diversification of other insect taxa. In particular, this work paves the road for identifying the critical ecological and phenotypic drivers that may be associated with insect diversification dynamics, such as the evolution of mandibular shapes in relation to prey diets50, or the dispersal and diversification of angiosperms13.
Methods
Taxon sampling
No new specimens were collected specifically for this study. All specimens used were obtained during prior research projects and were collected in full compliance with the relevant permits and regulations. Access and sampling of museum specimens was granted from the respective curators of the collections, including the authors, as listed under “contact persons” in Supplementary Data S1. We selected a total of 789 ponerine taxa plus 3 outgroups for phylogenetic analysis. Among these samples, 136 were sequenced previously51,52,53,54,55; 656 were newly sequenced for this study. We aimed to include UltraConserved Elements (UCE) data for as many unique species-level taxa as possible. In total we were able to include 528 (41.5 %) of the 1273 described species, and an additional batch of 261 expert-identified morphospecies suspected to represent independent taxa. Worker specimens have been favored for sampling, but queen and males have been sporadically used if no worker was available. Specimens have collecting year ranges from 1999 to 2023. Information on taxon sampling with NCBI accession numbers and references is presented in Supplementary Data S1 and S2. Our taxon sampling covers all geographic regions where the subfamily occurs, with taxonomic coverage ranging from 30.9% in the Eastern Palearctic to 87.2% in the Nearctic. This sampling scale is considerably greater than in previous studies with a similar or larger ant focus, and multiplies the taxonomic coverage of sequenced species several fold20,21,56. Such resources open the door to reliable investigations of diversification dynamics at large taxonomic and temporal scales.
UCE data generation and processing
Samples were extracted using Qiagen DNeasy Blood and Tissue kits following manufacturer’s protocols, with the following modifications: samples were incubated overnight for up to 16 hours in digestion buffer; AE buffer was heated to 60 C prior to DNA elution; and two rounds of elution were performed whereby 50–100 μL of AE buffer was added to each column and incubated at room temperature for 3 min prior to centrifugation. Extracted DNAs were then quantified using a Qubit fluorometer. Up to 50 ng of extracted DNA was then sheared to an approximate fragment distribution of 400–600 bp using a Qsonica Q800R sonicator. The sonicated DNA was then processed in-house, or sent to either Rapid Genomics (now LGC Bioresearch Technologies) in Gainesville, FL, or the genomics core facility at the University of Utah for library preparation, UCE enrichment, pooling, and sequencing. For the in-house work, libraries were prepared following the methods reported in Branstetter et al.51. In brief, libraries were prepared using Kapa HyperPrep kits and custom, dual-indexing adapters described in Glenn et al.57. Libraries were pooled into batches of up to 12 samples and then enriched using the HymV2 ant-specific probe set, synthesized by Arbor Biosciences. Enrichment was performed following Arbor Biosciences v4 protocol for day 1 and an older UCE enrichment protocol for day 2 (enrichment protocol v1.5 available at ultraconserved.org; based on Blumenstiel et al.58). Following enrichment, pools were quantified using Qubit and qPCR and then pooled together into a final pool containing up to 120 samples. The final pool was then sent to either Novogene or the University of Utah for sequencing on Illumina HiSeq X or NovaSeq. Rapid Genomics and the University of Utah followed similar procedures, except that they sequenced 2–3 million PE reads for UCE-enriched libraries and also ran additional sequencing on unenriched libraries for 1–2 million reads (whole genome sequence data).
We combined UCE and Whole-Genome Sequencing (WGS) reads produced for this study with reads available from the NCBI Sequence Read Archive (See “Data availability” in the main text for accession codes). Most data processing steps were performed using Phyluce v1.759. Reads were trimmed using Illumiprocessor v2.060 and assembled using SPAdes v3.14.161. UCEs were extracted using the HymV2 ant-specific probe file and default settings51. UCE contigs were assembled using MAFFT v7.130b, as implemented in Phyluce. Alignments were trimmed with (1) TrimAl v1.4.rev1562, as implemented in Phyluce, and (2) Spruceup v2022.2.463. Several iterative rounds of Spruceup were performed, with the final analysis implementing the lognormal criterion, a global cutoff of 0.93, and several manual cutoffs for taxa with noticeably longer branch lengths (Hypoponera_MG024_MAMI0635_CASENT0175229: 0.05; Leptogenys_diminuta_D0273: 0.05; Neoponera_magnifica2_EX2401: 0.04; Odontomachus_rufithorax_EX2952: 0.05; Ponera_selenophora_EX2971: 0.03; Pseudoneoponera_AU_A_EX2579: 0.07). The trimmed alignments were then split using AMAS64 and filtered to remove gap-only columns using a custom script (https://github.com/marekborowiec/remove_empty_columns). The alignments were then processed to retain only those that included at least 75% of all samples. This resulted in a final dataset that comprised 2365 UCE loci and 1,055,001 bp of sequence data, of which 542,612 bp were informative. The dataset included only 17.2% missing data.
Phylogenetic inferences
We concatenated the filtered loci and inferred phylogenetic trees in a Maximum Likelihood (ML) framework using IQ-Tree v2.265. Three different partitioning schemes were evaluated: unpartitioned, by locus with merging, and Sliding-Window Site Characteristics based on Entropy (SWSC-EN) with merging. For the unpartitioned analysis, we followed recommendations from Abadi et al.66 and selected a unique parameter-rich substitution model, which encompasses simpler models as special cases, and yields similar performances for phylogenetic inferences, while avoiding including a time-consuming model selection step in our computationally intensive workflow. Therefore, we used a General Time Reversible (GTR) model of sequence evolution with Gamma rate variation across sites (G4) and empirical base frequencies (F) as a substitution model. For the by locus analysis and SWSC-EN analyses, we set up analyses that combined partition merging, full model selection, and tree inference. For the by-locus analysis, we used as input the concatenated alignment and a by locus partition file into IQ-Tree v2. The best partitioning scheme was inferred using ModelFinder267. We used the rclusterf algorithm limited to testing 10% of possible dataset combinations and using only the GTR + F + G4 model of sequence evolution. We used the corrected Akaike Information Criterion (AICc) as criterion for selection. After merging, best-fit substitution models for each partition were inferred using the “-m MFP” command in IQ-Tree. For the SWSC-EN analysis, we used the program CURE68 to separate loci into right and left flanks and a core region using the Sliding Window Site Characteristics based on Entropy69. We then used the same merging, model selection, and tree inference approach as done for the by locus analysis going from 6715 partitions down to 2565 after merging. In all cases, support values were assessed using 1000 replicates of ultrafast bootstraps (UFBS)70 and SH-like approximate likelihood ratio test71. The three partitioning schemes led to identical topologies, except for five clades (unpartitioned: 99.4% identity) and one clade (by locus: 99.9% identity), respectively, differing from the SWSC-EN topology due to uncertain placement of single isolated rogue taxa (Supplementary Fig. S3 and S4). The SWSC-EN with merging scheme had the highest likelihood (unpartitioned: −37.82 × 106, by locus with merging: −37.30 × 106, SWSC-EN with merging: −37.08 × 106) and the best overall support values (mean UFBS: unpartitioned = 97.48%; by locus with merging = 97.84%; SWSC-EN with merging = 97.95%). Moreover, the complexity of the SWSC-EN scheme encompasses the unpartitioned and by locus schemes in allowing different evolutionary dynamics to occur in the ultra-conserved and variation section of each locus. Therefore, the topology from the SWSC-EN analysis was selected for downstream analyses. The ML phylogram with 789 ponerine ants is shown in Supplementary Fig. S3 and available in Supplementary Data 8.1. Configuration files, analysis output, and custom scripts used for phylogenetic inferences can be found in the dedicated part of the Zenodo archive at https://doi.org/10.5281/zenodo.14216556.
Divergence dating
We used a two-step approach to make the generation of time-calibrated phylogenies computationally feasible. First, we applied Bayesian time tree inference in MCMCTree implemented in PAML v4.10.724,25 using node calibrations on a backbone tree with reduced taxon sampling. We then ran a penalized likelihood approach26 in TreePL v1.027 using secondary calibrations from the backbone tree to infer ages of all nodes.
We selected fossil calibrations following three guiding principles: (1) fossils had to be confidently assignable to total groups; (2) in the absence of total-evidence phylogenetic placement, we conservatively assumed that fossils could belong to either stem or crown groups; and (3) when multiple fossils of similar age could inform a set of nested nodes, we calibrated only the shallowest such node to avoid redundant constraints. These principles were applied in conjunction with best practices outlined by Parham et al.72 for fossil selection and have been applied successfully in divergence dating of ants and other Hymenoptera73,74,75. Following this approach, we identified seven fossil calibrations appropriate for our analyses, prioritizing those with clear morphological justification, sufficient preservation, and unambiguous taxonomic placement.
For the first step, we produced a reduced dataset for MCMCTree analyses by trimming taxon sampling to 69 taxa spanning all clades that could be dated using our seven fossil calibrations. We set Cauchy distribution priors with a 0.1 offset, scale parameter of 1, and soft minimum bounds on nodes calibrated by fossils. Details about fossils, nodes, and age constraints used can be found in Supplementary Data S3. To explicitly account for the large uncertainty surrounding divergence estimates in ants, we set a normal distribution for the required root calibration prior based on ranges inferred for this node in seven recent divergence dating studies of ants reviewed by Borowiec et al.22. This normal distribution was centered around 113 Ma, which is the average of mean Highest Posterior Densities (HPD) or confidence intervals from these studies, and with 95% of density falling between 69 and 149 Ma, corresponding to the lowest and highest-end estimates from those dating studies.
In addition to reducing taxon sampling, we reduced the molecular dataset to 100 UCE loci selected based on the properties of their individual locus trees. We sorted the loci based on the weighted average of three properties that have been shown to identify loci suitable for divergence dating analyses76. The most important property considered was “clocklikeness” (weight 0.5), here defined as the minimum coefficient of variation of root to tip distances, followed by average locus tree branch length (weight 0.3), and Robinson-Foulds distances to the 792-taxon ML tree inferred by concatenated analysis (weight 0.2), evaluated using kinda_date74 (https://github.com/marekborowiec/kinda_date). These properties were computed on locus trees with complete taxon sampling. We ran the MCMCTree analyses using fixed concatenated topology and likelihood approximation77 for four independent chains for 10,000,000 generations each, sampling every 1000th generation and discarding samples from the first 1,000,000 (10%) generations as burn-in. We combined the output of the four chains and confirmed that the analyses converged by visually examining the posterior traces of each parameter in Tracer v1.7.178 and ensuring effective sample size (ESS) for each parameter was above 300. We also compared the results to a prior-only run to confirm that the posterior was informed by the data.
To account for and ensure the robustness of our downstream analyses to dating uncertainty, we randomly selected 1000 reduced 69-taxon trees from the 40,004 total posterior samples from MCMCTree. We extracted node ages and created a TreePL config file with constraints extracted from each of those reduced 69-taxon trees. This ensures each calibrated node has min and max calibration ages equal to the age of the corresponding node in each input tree.
We used the 792-taxon ML tree as input, set the number of sites to 1,055,001, as in the full alignment that tree was inferred under, and turned on the “thorough” setting. We used random-sampled cross-validation to find the optimal smoothing value for one of the randomly selected trees from a range of 1000 down to 0.0000001. The best value turned out to be 0.01. We used this value for all the configuration files and inferred 1000 dated backbone trees using TreePL.
We obtained a single backbone tree with node age confidence intervals using TreeAnnotator v1.1079 to summarize the 1000 trees into a Maximum Clade Credibility tree (subsequently, MCC tree) with mean node heights. We also extracted the two most extreme divergence dating hypotheses with the lowest and highest sum of estimated node ages to use for sensitivity analyses (subsequently, youngest and oldest hypotheses). The time-calibrated MCC tree with 789 ponerine ants is shown in Supplementary Fig. S5 and available in Supplementary Data 8.2. Posterior time-calibrated trees are available in Supplementary Data 8.3. Configuration files, analysis output, and custom scripts used for divergence dating analyses can be found in GitHub at https://github.com/marekborowiec/ponerine_phylogeny.
Tree grafting
Analyses of diversity dynamics have increased power and lower estimation bias on imputed exhaustive phylogenies relative to analyses that use sampling fractions to account for missing taxa80. Moreover, a grafting approach accounts for the location of missing taxa across clades, while avoiding potentially incorrect likelihood behaviors and identifiability issues associated with clade-specific sampling fractions81,82,83. Therefore, we followed the recommended approach for accounting for non-random incomplete taxon sampling in diversification studies (http://bamm-project.org/advanced.html; Section 9.1; accessed on April 16th, 2025). We generated a distribution of fully sampled phylogenies encompassing all known ponerine ant species by using phylogenetically informed imputations of missing taxa on the UCE-based time-calibrated backbone phylogenies obtained from TreePL.
We used all taxonomic and biogeographic information available in the literature to define, for each of the 745 missing taxa, a target clade within which to graft the missing taxa, including its stem branch (Supplementary Data S4). When relevant, we also identified “excluded subclades” within the target clade to exclude from potential grafting locations. For instance, we prevented missing taxa distributed in one bioregion from being grafted in a subclade known to be restricted to another distinct bioregion, limiting overestimation of dispersal events that would arise from a purely random grafting in widely spread target clade. Within each identified target clade, we fitted a birth-death model with the sampling fraction adjusted to the number of missing taxa to graft within. Then, we simulated ages of missing taxa with the corsim function from the TreeSim R package84, extracted the set of branches present at the given age, and randomly selected the grafting branch while excluding the branches belonging to the excluded subclade(s) from the set of options. This imputation procedure is conservative, because it minimizes among-lineage diversification rate heterogeneity within any target clades85. Imputations were run in a stepwise nested fashion, starting with nested target clades. We ran an independent random imputation of all missing taxa on each of the 1000 backbone time-calibrated phylogenies to generate a set of exhaustive ponerine phylogenies for sensitivity analyses; these account simultaneously for uncertainty in divergence age estimates and informed imputations of missing taxa. This distribution includes the youngest and oldest divergence dating hypotheses used for sensitivity analyses downstream. We also ran a random imputation on the MCC tree obtained from summarizing TreePL outputs across all posterior time-calibrated trees to produce a grafted MCC tree used for reporting results of downstream analyses. The grafted MCC tree with 1534 ponerine ants is shown in Supplementary Fig. S6 and available in Supplementary Data 8.4. Posterior grafted trees are available in Supplementary Data 8.5. The overall workflow used to produce phylogenies is visually summarized in Supplementary Fig. S1.
Biogeographic data
Data sources and curation
All available georeferenced occurrence data for ponerine ants were retrieved from AntWeb (62,301 records; https://www.antweb.org/, accessed on 30/05/2024) and AntMaps86 (93,557 records; GABI v1.0). Coordinates were screened with CoordinateCleaner87 to detect common errors in biodiversity datasets, including locations in bodies of water, within biodiversity institutions, in centroids of political entities, and outliers based on distance to other occurrences of the same taxa. Four thousand one hundred and forty-three records were flagged and manually curated in the process. We also detected mismatches between coordinates and assigned geographic administrative entities at several hierarchical levels (adm2, adm1, and countries from geoBoundaries88, http://www.geoboundaries.org, accessed on 15/02/2024; bentity2 from AntMaps86, https://antmaps.org/) and solved conflicts for those 15,244 flagged records. Additionally, we designed a custom screening procedure to detect bioregional outliers. For each taxon, we flagged all records falling in a bioregion represented by less than 20% of records, highlighting potential cases of erroneous outliers likely to affect our ancestral range estimates. Two hundred and fifty-six dubious records belonging to 101 taxa were removed, while documented cases of range expansion across bioregions were conserved. Finally, we retrieved coordinates for 50,114 records missing GPS data based on their assignment to administrative entities. For each record, we randomly drew coordinates within the smallest assigned entity and recorded uncertainty as the area of the assigned entity. Such uncertainty has no effect on our downstream analyses as they are carried out at a bioregional level encompassing all geographic administrative entities used in this process. Finally, we cleaned records for duplicates to obtain a final dataset of 38,128 curated unique taxon-location georeferenced records based on one degree-grid cells, covering all 1534 ponerine taxa included in our study (all 1273 described species and 261 UCE-sequenced morphospecies). The map of curated occurrences is shown in Supplementary Fig. S7. The overall workflow for curation of biogeographic data is visually summarized in Supplementary Fig. S2. The exhaustive database of 149,541 curated geolocalized occurrences of ponerine ants is available in Supplementary Data S5.
Mapping current species richness
We estimated the range of taxa based on georeferenced occurrences, excluding those retrieved from administrative entities. First, we drew alpha-hull shapes around occurrences with an alpha radius = 1000 km, allowing us to retrieve disjunct ranges in case occurrences formed distant clusters. Next, we applied a taxon-specific buffer around alpha-hulls based on the density of sampling for each taxon using a minimal buffer of one arc-degree, and a maximum buffer estimated as the 80% quantile of minimal pairwise distances between occurrences as a way to account for uncertainty in the sampling of the actual species range89. Finally, we rasterized and stacked all buffered alpha-hulls representing taxon ranges and computed local species richness as the sum of taxon presence recorded in each quarter-degree cell to obtain a map of worldwide ponerine species richness.
Historical biogeography
Bioregional scheme
Each occurrence was attributed to a bioregion based on the bioregion scheme used in AntWeb (https://www.antweb.org/). We lumped Madagascar with the Afrotropics and split the Palearctic between Western and Eastern Palearctic to ensure a more homogeneous split of terrestrial areas. The latter also allowed discrimination of Nearctic/Palearctic dispersal through the Bering land bridge in the Northern Pacific or the Thule land bridge in the Northern Atlantic. We also classified India and the Burmese terrane under a unique IndoMalayan bioregion, even though those territories have been allegedly separated over geological time90. The vast majority of ponerines found in the present-day Burmese area and in the Indian subcontinent are also encroaching on the surrounding South-Asian regions, limiting vastly the informative power of historical biogeographic inferences treating those areas as separate entities. This treatment also ensures a more balanced area division of terrestrial lands. Recent investigations of current native ant bioregionalization have identified sets of different bioregions91,92 that remain largely concomitant with the broad delineation we used. Such delineations are based on the current distribution of taxa but are less suitable for use in historical biogeographic analyses accounting for continental drift and investing taxon distributions over evolutionary time. Thus, we favored the use of our larger bioregion scheme in line with past and recent endeavors in zooregionalization and historical biogeography31,93,94,95,96,97. Binary tables of presence/absence of the 1534 ponerine ant taxa across bioregions used in this study are available in Supplementary Data S6.
Ancestral range estimates with BioGeoBEARS
To study support for the Time-for-Accumulation Hypothesis (TfAH), we estimated the ancestral ranges of ponerine ants in our exhaustive phylogeny. We ran a set of time-stratified historical biogeographic models within the BioGeoBEARS framework accounting for different combinations of biogeographic events such as range expansion and contraction, vicariance, subset speciation, and jump-dispersal, with DIVALIKE, DIVALIKE + J, DEC, and DEC + J models98. Such a time-stratified approach accounts for changes in bioregion connectivity by providing adjacency matrices (Supplementary Data S7) associated with each geological epoch and preventing lineages from harboring ranges encompassing non-adjacent bioregions at any given point in time. Although we acknowledge that +J models may yield unrealistically low regional extinction rates and tend to limit the residence times of widespread ranges99, they also allow long-distance jump-dispersal events concomitant with speciation (i.e., founder effect) to occur, even between non-adjacent areas. This releases an important, and likely unrealistic, constraint of other models with the j parameter set to 0 that prevents any dispersal to occur between non-adjacent areas. Therefore, we chose to rely on likelihood-based statistical comparison to select the best-fitting model for our analyses100 and ran a whole set of models as listed above, without a priori deciding which one should be favored.
The DEC + J model was selected as the best model across both the grafted MCC tree and all 1000 grafted time-calibrated phylogenies, accounting for uncertainty in both divergence dating and taxon grafting, including the youngest and oldest divergence dating hypotheses (Mean Akaike’s weight = 99.95%; Supplementary Tables S1–S4). Similarly, ancestral range estimates based on posterior marginal likelihoods using those fitted DEC + J models appeared congruent across all phylogenies (Supplementary Tables S5–S8). Therefore, we conducted all downstream analyses on the grafted MCC tree under the DEC + J model. In parallel, we performed sensitivity analyses by conducting the same analytical pipeline on both the youngest and oldest divergence dating hypotheses in order to fully assess the robustness of our conclusions to alternative divergence age estimates. We also employed the Cohen’s Kappa index for classification similarity (Cohen, 1960) to quantify agreement in the most likely ancestral range estimated for shared nodes between the different divergence dating hypotheses (i.e., MCC, Youngest, and Oldest).
Biogeographic stochastic mapping (BSM)
We applied Biogeographic Stochastic Mapping101 (BSM) using the best-fitted model for historical biogeography (DEC + J) to simulate 1000 possible biogeographic histories for ponerine ants on the grafted MCC tree. BSM draws random ranges at each internal node based on the estimated posterior marginal probabilities, then simulates range evolution on each branch using estimated state transition parameters until it finds a biogeographic history compatible with allowed ranges. From BSM simulations, we retrieved the posterior frequencies of anagenetic and cladogenetic events and recorded the sources and destinations of dispersal events in time. In particular, we quantified the counts and relative frequencies of events along 10 My sliding windows to compare dispersal dynamics in time across bioregions and investigate support for the Asymmetrical Dispersal Hypothesis (ADH). In the case of ranges encompassing multiple bioregions, the lineages were equally divided among the bioregions to contribute to respective bioregional counts.
Mapping biogeographic and diversification dynamics through time
To visualize the overall dynamic of ponerine ants in space and time, we reconstructed paleomaps of bioregions based on a geological model of continental plate drift102 implemented in the rgplates R package103. We then overlaid the network of dispersal events across moving bioregions at each time step (1 My), with edge width scaled by event counts recorded within time-windows of 5 My, and vertex size scaled by regional species richness quantified as number of lineages present in each region at each time step, aggregated across the 1000 BSM simulations. The final animation of ponerine ant biogeographic and diversification dynamics is available in Supplementary Movie 1.
Diversification dynamics
Empirical in situ speciation rates
Complementary to frequencies of dispersal events, we also recorded the source of cladogenetic events and quantified the frequencies and rates of in situ speciation as the number of range inheritance events within time-sliding windows. As with dispersal events, ranges encompassing multiple bioregions contributed equally to respective bioregional counts.
BAMM diversification rates
Besides empirical speciation rates estimated from in situ speciation event counts on time-calibrated phylogenies, we also used BAMM v2.5.0 to estimate diversification rates among lineages using a mechanistic approach modeling rate shifts104. Several independent runs with four reversible-jump MCMC on 107 generations were carried out for 1, 5, 10, and 20 expected number of shifts, with all other priors set to default using the R package BAMMtools v2.1.1228. We combined the output of the four chains in each run and confirmed that all analyses converged by visually examining the posterior traces of parameters (Supplementary Fig. S16) and ensuring effective sample size (ESS) for each parameter was above 200. We sampled 1000 posterior generations after discarding burn-in and selected the run with 10 expected shifts for downstream analyses since its prior distribution was the most similar to the posterior. From this, we obtained 1000 evolutionary scenarios from which we retrieved median diversification rates along branches (Supplementary Figs. S14 and S15). We compared median rates estimated for the grafted MCC tree and the alternative divergence dating hypotheses using Spearman’s sum-rank correlation tests across branches associated with shared clades. Estimated rates were aggregated per bioregion membership, as estimated from the BSM simulations, to compute rates through time evolution in relation to bioregions31. For the MCC tree, we recovered on average 12.1 ± 4.1 regime shifts in diversification dynamics along ponerine ant history, of which seven concerned major radiations (see Supplementary Table S10 for regime shifts metadata from the Maximum Shift Credibility (MSC) configuration).
BAMM, and other similar methods used to estimate time-varying diversification rates and identify regime shifts suffer from identifiability issues105. To limit potential issues, we focused our analyses on net diversification rates rather than on the absolute estimation of speciation and extinction rates individually. As a Bayesian approach, BAMM samples a large number of alternative diversification histories that can lead to similar likelihood, effectively accounting for a portion of the uncertainty regarding the identification of the “real” diversification history. Such uncertainty was carried out down to our tests for diversification rate differences as we performed STRAPP tests across posterior BAMM samples (see Section 9.1 below). Additionally, we combined BAMM analyses with a different approach based on Regional In-Situ Radiations (see Section 9.2 below) in order to cross-validate our results and address identifiability issues to the best possible extent.
Mapping current diversification rates
To visualize the spatial distribution of current diversification rates, we assigned taxon-specific rates estimated from BAMM to each respective taxon range, as estimated from buffered alpha-hulls, and extracted the mean current rates recorded among taxa found in each quarter-degree grid cell. To limit artifactual outlier values, we recorded mean rates only for grid cells with more than two taxa. Additionally, we extracted mean rates along latitudinal and longitudinal bands of one arc-degree based on taxa with a range overlapping each band. We extracted such geographic gradients across all 1000 BAMM posterior samples and generated a null distribution of mean rates along gradients by permuting species identities among range data. We identified latitudinal and longitudinal bands where the mean current diversification rates are lower/higher than expected if species were distributed randomly.
Diversification-dispersal relationships
BAMM rates across bioregions: STRAPP tests
We carried out STRAPP tests using the traitDependentBAMM function in the R package BAMMtools v2.1.1228 to investigate differences in current diversification rates across bioregions and formally test for support for the Diversification Rate Hypothesis (DRH). For greater statistical power, we focused on differences between temperate and tropical bioregions along a latitudinal gradient and between the Old World and the New World along a longitudinal gradient (Fig. 5), but also report results considering the seven bioregions altogether (Supplementary Information Fig. S10). Such a test permutates tip rates across taxa in the posterior samples from a BAMM analysis using a block-design that accounts for the non-independence of tip rates data via permutations restricted among macroevolutionary regimes106. Then, statistics from two-sided Wilcoxon tests (for pairwise gradient tests) and Kruskal-Wallis tests (across all bioregions) are compared to the value obtained from the observed data to assess significance of rate differences against random rate distribution across regimes.
Additionally, we designed a modified version of the STRAPP test that allows us to test for diversification rate differences through evolutionary time, and not only between present-day rates, as those are unlikely to be informative about long-term past dynamics shaping biodiversity32,107. We used bioregion membership estimates from the BSM simulations and cut off the phylogenies mapped with biogeographic histories every 0.1 My time step to be able to carry out STRAPP tests across lineages present at any given point in time. As a result, we obtained p-values for rate differences along evolutionary time and could identify timeframes of significance.
RISR analyses
To identify the main factors explaining differences in diversification rates across bioregions, we investigated the diversification dynamics of Regional In Situ Radiations (RISR). RISR are independent clades that diversify almost exclusively within a unique bioregion, allowing for statistical tests between evolutionary independent units31. First, we identified RISR as clades whose lineages spent more than 75% of cumulated time along branches in a single bioregion, with a minimum of 10 extant taxa. As such, we identified 9 RISR in Afrotropics, 5 in Australasia, 12 in Indomalaya, and 5 in Neotropics. For each clade, we fitted a time-dependent birth-death model with speciation rates (\({\lambda }\)) being an exponential function of time since the crown age (t = 0) and extinction rates (\({\mu }\)) being constants similar to macroevolutionary regimes in BAMM104 (Eqs. 1, 2 and 3). Models were fitted with the R package diversitree v0.10-1108.
We extracted the crown age, the crown net diversification rate (Rcrown), the current net diversification rate (Rcurrent), and the time-variation parameter (\({\alpha }\)) associated with each RISR. We then tested for differences in parameter values across bioregions using two-sided Kruskal–Wallis and pairwise post hoc Wilcoxon tests. Finally, we used a Poisson regression to predict present-day taxonomic richness of RISR across bioregions based on crown ages.
Quantifying lineage origins through time
We designed a new method to estimate and map the age of immigration for lineages within a new bioregion and gather additional support for the Time-for-Accumulation Hypothesis (TfAH). We browsed our 1000 BSM simulations from tips-to-root to retrieve for each present-day taxon, the timing of the range shift when the lineage reached its current bioregion for the first time. For taxa harboring a range encompassing multiple bioregions, the “biogeographic immigration age” is averaged between areas. Taxon immigration ages were then averaged across all simulations. As with current diversification rates, we mapped the spatial distribution of biogeographic immigration ages using taxon ranges as estimated from buffered alpha-hulls and extracted the mean immigration ages of taxa recorded in each quarter-degree grid cell. To limit artifactual outlier values, we recorded mean ages only for grid cells with more than two taxa. Additionally, we computed geographic gradients of mean ages along latitudinal and longitudinal bands of one arc-degree based on taxa with a range overlapping each band. For statistical comparison, we generated a null distribution of mean immigration ages along geographic gradients by permuting species identities among range data.
Finally, we investigated changes through time in the origin of lineages across bioregions. Using the BSM simulations, we recorded at each time step (1 My) the origin of lineages found in each bioregion. The origins were examined by comparing the lineage ranges at the focal time step with the range of their most direct parental node. Autochthonous scores of each lineage were computed as the proportion of unique areas in the lineage range found among the unique areas of its parental range. Then we computed the mean autochthonous score per bioregion by aggregating lineages across their focal time ranges, splitting lineages equally among bioregions in the case of ranges encompassing multiple areas. As such, for each time step and each bioregion, we obtained mean autochthonous scores representing the counts and proportions of lineages present at a given time step whose presence is due to in situ diversification as opposed to dispersal.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence reads newly generated in this study have been deposited in the NCBI Sequence Read Archive under BioProject accession code PRJNA1196817. Newly generated COI barcode data are available under accession codes PV930584 to PV931323. The UCE sequence data from prior publications used in this study are available under BioProject accession codes PRJNA360290, PRJNA513200, PRJNA563172, PRJNA778536, and PRJNA1125038. The raw sequence data can also be accessed using individual accession numbers given in Supplementary Data S2. Information on the location of voucher specimens is provided in Supplementary Data S1. Other data sources for this study (fossil calibrations, grafting information, occurrence database, bioregion membership, and adjacency tables) and ready-to-use phylogenies produced for this study can be found in Supplementary Data S3–S8 and in the associated Zenodo repository at https://doi.org/10.5281/zenodo.14216556. Specimen requests can be addressed to collection curators indicated in Supplementary Data S1 according to the origin of the material.
Code availability
All scripts and files used to carry out analyses are available in the associated Zenodo archive (https://doi.org/10.5281/zenodo.14216556). In parallel, scripts and files are also available on GitHub: for divergence dating analyses (https://github.com/marekborowiec/ponerine_phylogeny), and for curation of the taxonomic and biogeographic databases, and for carrying out tree grafting and all diversification and biogeographic analyses (https://github.com/MaelDore/Ponerinae_Historical_Biogeography; Release v1.0.0).
References
Mittelbach, G. G. et al. Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecol. Lett. 10, 315–331 (2007).
Scholl, J. P. & Wiens, J. J. Diversification rates and species richness across the Tree of Life. Proc. R. Soc. B Biol. Sci. 283, 1838 (2016).
Rohde, K. Latitudinal gradients in species diversity: the search for the primary cause. Oikos 65, 514 (1992).
Stephens, P. R. & Wiens, J. J. Explaining species richness from continents to communities: the time-for-speciation effect in emydid turtles. Am. Nat. 161, 112–128 (2003).
Roy, K. & Goldberg, E. E. Origination, extinction, and dispersal: integrative models for understanding present-day diversity gradients. Am. Nat. 170, S71–85 (2007).
Jablonski, D., Kaustuv, R. & Valentine, J. W. Out of the tropics: evolutionary diversity gradient. Science 314, 102–106 (2006).
Fine, P. V. A. Ecological and evolutionary drivers of geographic variation in species diversity. Annu. Rev. Ecol. Evol. Syst. 46, 369–392 (2015).
Rolland, J., Condamine, F. L., Jiguet, F. & Morlon, H. Faster speciation and reduced extinction in the tropics contribute to the mammalian latitudinal diversity gradient. PLoS Biol. 12, e1001775 (2014).
Jetz, W., Thomas, G. H., Joy, J. B., Hartmann, K. & Mooers, A. O. The global diversity of birds in space and time. Nature 491, 444–448 (2012).
Tietje, M. et al. Global variation in diversification rate and species richness are unlinked in plants. Proc. Natl. Acad. Sci. USA 119, 1–9 (2022).
Titley, M. A., Snaddon, J. L. & Turner, E. C. Scientific research on animal biodiversity is systematically biased towards vertebrates and temperate regions. PLoS ONE 12, 1–14 (2017).
Eisenhauer, N. & Hines, J. Invertebrate biodiversity and conservation. Curr. Biol. 31, R1214–R1218 (2021).
Moreau, C. S., Bell, C. D., Vila, R., Archibald, S. B. & Pierce, N. E. Phylogeny of the ants: diversification in the age of angiosperms. Science 312, 101–104 (2006).
Rico-Gray, V. & Oliveira, P. S. The Ecology And Evolution Of Ant-plant Interactions (University of Chicago Press, 2008).
Kempf, W. W. A preliminary review of the ponerine ant genus Dinoponera Roger (Hymenoptera: Formicidae). Stud. Entomol. 14, 369–394 (1971).
Bolton, B. AntCat. An online catalog of the ants of the world. https://antcat.org/ (2025).
Hanisch, P. E., Drager, K., Yang, W. H., Tubaro, P. L. & Suarez, A. V. Intra- and interspecific variation in trophic ecology of ‘predatory’ ants in the subfamily Ponerinae. Ecol. Entomol. 45, 444–455 (2020).
Jouault, C., Condamine, F. L., Legendre, F. & Perrichot, V. The angiosperm terrestrial revolution buffered ants against extinction. Proc. Natl. Acad. Sci. USA 121, 2017 (2024).
Kass, J. M. et al. The global distribution of known and undiscovered ant biodiversity. Sci. Adv. 8, eabp9908 (2022).
Economo, E. P., Narula, N., Friedman, N. R., Weiser, M. D. & Guénard, B. Macroecology and macroevolution of the latitudinal diversity gradient in ants. Nat. Commun. 9, 1–8 (2018).
Moreau, C. S. & Bell, C. D. Testing the museum versus cradle tropical biological diversity hypothesis: phylogeny, diversification, and ancestral biogeographic range evolution of the ants. Evolution 67, 2240–2257 (2013).
Borowiec, M. L., Moreau, C. S. & Rabeling, C. Ants: phylogeny and classification. in Encycl. Soc. Insects (ed. Starr, C. K.) 1–18 (Springer, 2020).
Shi, G. et al. Age constraint on Burmese amber based on U-Pb dating of zircons. Cretac. Res. 37, 155–163 (2012).
Schmidt, C. A. & Shattuck, S. O. The higher classification of the ant subfamily Ponerinae (Hymenoptera: Formicidae), with a review of Ponerine ecology and behavior. Zootaxa 3817, 1–242 (2014).
Blakey, R. C. Gondwana paleogeography from assembly to breakup—A 500 my odyssey. in Resolving the Late Paleozoic ice age in time and space (eds. Fielding, C. R., Frank, T. D. & Isbell, J. L.) 1–28 (Geological Society of America, 2008).
Hall, R. Sundaland and Wallacea: geology, plate tectonics and palaeogeography. in Biotic Evolution and Environmental Change in Southeast Asia (eds. Gower, D. et al.) 32–78 (Cambridge University Press, 2012).
Jolivet, L. & Faccenna, C. Mediterranean extension and the Africa-Eurasia collision. Tectonics 19, 1095–1106 (2000).
Rabosky, D. L. et al. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5, 701–707 (2014).
Kerkhoff, A. J., Moriarty, P. E. & Weiser, M. D. The latitudinal species richness gradient in New World woody angiosperms is consistent with the tropical conservatism hypothesis. Proc. Natl. Acad. Sci. USA 111, 8125–8130 (2014).
Duchêne, D. A. & Cardillo, M. Phylogenetic patterns in the geographic distributions of birds support the tropical conservatism hypothesis. Glob. Ecol. Biogeogr. 24, 1261–1268 (2015).
Chazot, N. et al. Conserved ancestral tropical niche but different continental histories explain the latitudinal diversity gradient in brush-footed butterflies. Nat. Commun. 12, 5717 (2021).
Stephens, P. R. et al. Global diversity patterns are explained by diversification rates and dispersal at ancient, not shallow, timescales. Syst. Biol. 0813, 1–11 (2025).
Krug, A. Z., Jablonski, D. & Valentine, J. W. Contrarian clade confirms the ubiquity of spatial origination patterns in the production of latitudinal diversity gradients. Proc. Natl. Acad. Sci. USA 104, 18129–18134 (2007).
Beaugrand, G., Kléparski, L., Luczak, C., Goberville, E. & Kirby, R. R. A niche-based theory of island biogeography. Ecol. Evol. 14, e11540 (2024).
Wiens, J. J. The niche, biogeography and species interactions. Philos. Trans. R. Soc. B Biol. Sci. 366, 2336–2350 (2011).
Belmaker, J. & Jetz, W. Relative roles of ecological and energetic constraints, diversification rates and region history on global species richness gradients. Ecol. Lett. 18, 563–571 (2015).
Stroud, J. T. & Losos, J. B. Ecological opportunity and adaptive radiation. Annu. Rev. Ecol. Evol. Syst. 47, 507–532 (2016).
Martin, H. A. Cenozoic climatic change and the development of the arid vegetation in Australia. J. Arid Environ. 66, 533–563 (2006).
Cerezer, F. O., Machac, A., Rangel, T. F. & Dambros, C. S. Exceptions to the rule: Relative roles of time, diversification rates and regional energy in shaping the inverse latitudinal diversity gradient. Glob. Ecol. Biogeogr. 31, 1794–1809 (2022).
Dimitrov, D. et al. Diversification of flowering plants in space and time. Nat. Commun. 14, 1–16 (2023).
Gehrke, B. Staying cool: preadaptation to temperate climates required for colonising tropical alpine-like environments. PhytoKeys 125, 111–125 (2018).
Kaspari, M. & Vargo, E. L. Colony size as a buffer against seasonality: Bergmann’s rule in social insects. Am. Nat. 145, 610–632 (1995).
Rajaud, A. & de Noblet-Ducoudré, N. Tropical semi-arid regions expanding over temperate latitudes under climate change. Clim. Change 144, 703–719 (2017).
Wong, M. K. L., Economo, E. P. & Guénard, B. The global spread and invasion capacities of alien ants. Curr. Biol. 33, 566–571.e3 (2023).
Perkovsky, E. E. Tropical and Holarctic ants in late Eocene ambers. Vestn. Zool. 50, 111–122 (2016).
Morley, R. J. Cretaceous and Tertiary climate change and the past distribution of megathermal rainforests. in Tropical Rainforest Responses to Climatic Change (eds. Bush, M., Flenley, J. & Gosling, W.) 1–34 (Springer Praxis Books, 2011).
Meseguer, A. S. & Condamine, F. L. Ancient tropical extinctions at high latitudes contributed to the latitudinal diversity gradient*. Evolution 74, 1966–1987 (2020).
Liu, Z. et al. Global cooling during the Eocene-Oligocene climate transition. Science 323, 1187–1190 (2009).
Branstetter, M. G., Longino, J. T., Reyes-López, J. L., Brady, S. G. & Schultz, T. R. Out of the temperate zone: a phylogenomic test of the biogeographical conservatism hypothesis in a contrarian clade of ants. J. Biogeogr. 49, 1640–1653 (2022).
Booher, D. B. et al. Functional innovation promotes diversification of form in the evolution of an ultrafast trap-jaw mechanism in ants. PLoS Biol. 19, 1–22 (2021).
Branstetter, M. G., Longino, J. T., Ward, P. S. & Faircloth, B. C. Enriching the ant tree of life: enhanced UCE bait set for genome-scale phylogenetics of ants and other Hymenoptera. Methods Ecol. Evol. 8, 768–776 (2017).
Branstetter, M. G. & Longino, J. T. Ultra-conserved element phylogenomics of New World Ponera (Hymenoptera: Formicidae) illuminates the origin and phylogeographic history of the endemic exotic ant Ponera exotica. Insect Syst. Divers. 3, 1–13 (2019).
Longino, J. T. & Branstetter, M. G. Phylogenomic species delimitation, taxonomy, and ‘Bird Guide’ identification for the neotropical ant genus Rasopone (Hymenoptera: Formicidae). Insect Syst. Divers. 4 (2020).
Branstetter, M. G. & Longino, J. T. UCE phylogenomics of New World Cryptopone (Hymenoptera: Formicidae) elucidates genus boundaries, species boundaries, and the vicariant history of a temperate–tropical disjunction. Insect Syst. Divers. 6, 1–23 (2022).
Camacho, G. P., Loss, A. C., Fisher, B. L. & Blaimer, B. A zoogeographic model for the evolution of diversity and endemism in Madagascar. Ecography 2025, e07661 (2025).
Schmidt, C. A. Molecular phylogenetics of ponerine ants (Hymenoptera: Formicidae: Ponerinae). Zootaxa 3647, 201–250 (2013).
Glenn, T. C. et al. Adapterama I: universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ 2019, 1–31 (2019).
Blumenstiel, B. et al. Targeted exon sequencing by in-solution hybrid selection. Curr. Protoc. Hum. Genet. 66, 18.4.1–18.4.24 (2010).
Faircloth, B. C. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 32, 786–788 (2016).
Faircloth, B. C. illumiprocessor: a trimmomatic wrapper for parallel adapter and quality trimming. at https://doi.org/10.6079/J9ILL.tle (2013).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes De novo assembler. Curr. Protoc. Bioinforma. 70, 1–29 (2020).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Borowiec, M. L. Spruceup: fast and flexible identification, visualization, and removal of outliers from large multiple sequence alignments. J. Open Source Softw. 4, 1635 (2019).
Borowiec, M. L. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 2016, e1660 (2016).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Abadi, S., Azouri, D., Pupko, T. & Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 10, 934 (2019).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Freitas, F. V. et al. UCE phylogenomics, biogeography, and classification of long-horned bees (Hymenoptera: Apidae: Eucerini), with insights on using specimens with extremely degraded DNA. Insect Syst. Divers. 7, 1–21 (2023).
Tagliacollo, V. A. & Lanfear, R. Estimating improved partitioning schemes for ultraconserved elements. Mol. Biol. Evol. 35, 1798–1811 (2018).
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Parham, J. F. et al. Best practices for justifying fossil calibrations. Syst. Biol. 61, 346–359 (2012).
Peters, R. S. et al. Evolutionary history of the Hymenoptera. Curr. Biol. 27, 1013–1018 (2017).
Borowiec, M. L. et al. Evaluating UCE data adequacy and integrating uncertainty in a comprehensive phylogeny of ants. Syst. Biol. 7, 1–37 (2025).
Blaimer, B. B. et al. Key innovations and the diversification of Hymenoptera. Nat. Commun. 14, 1212 (2023).
Smith, S. A., Brown, J. W. & Walker, J. F. So many genes, so little time: a practical approach to divergence-time estimation in the genomic era. PLoS ONE 13, e0197433 (2018).
dos Reis, M. & Yang, Z. Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times. Mol. Biol. Evol. 28, 2161–2172 (2011).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Chang, J., Rabosky, D. L. & Alfaro, M. E. Estimating diversification rates on incompletely sampled phylogenies: theoretical concerns and practical solutions. Syst. Biol. 69, 602–611 (2020).
Moore, B. R., Höhna, S., May, M. R., Rannala, B. & Huelsenbeck, J. P. Critically evaluating the theory and performance of Bayesian analysis of macroevolutionary mixtures. Proc. Natl. Acad. Sci. USA 113, 9569–9574 (2016).
Fitzjohn, R. G., Maddison, W. P. & Otto, S. P. Estimating trait-dependent speciation and extinction rates from incompletely resolved phylogenies. Syst. Biol. 58, 595–611 (2009).
Beaulieu, J. M. The problem with clade-specific sampling fractions. at https://cran.r-project.org/web/packages/hisse/vignettes/Clade-specific-sampling.pdf (2020).
Stadler, T. TreeSim: simulating phylogenetic trees. R package version 2.4. at https://cran.r-project.org/package=TreeSim (2019).
Title, P. O. et al. The macroevolutionary singularity of snakes. Science 383, 918–923 (2024).
Guénard, B., Weiser, M. D., Gómez, K., Narula, N. & Economo, E. P. The Global Ant Biodiversity Informatics (GABI) database: synthesizing data on the geographic distribution of ant species (Hymenoptera: Formicidae). Myrmecol. News 24, 83–89 (2017).
Zizka, A. et al. CoordinateCleaner: standardized cleaning of occurrence records from biological collection databases. Methods Ecol. Evol. 10, 744–751 (2019).
Runfola, D. et al. GeoBoundaries: a global database of political administrative boundaries. PLoS ONE 15, 1–9 (2020).
Doré, M. et al. Anthropogenic pressures coincide with Neotropical biodiversity hotspots in a flagship butterfly group. Divers. Distrib. 28, 2912–2930 (2022).
Scotese, C. R. The PALEOMAP Project: paleogeographic atlas and plate tectonic software. Oceanogr. Lit. Rev. 3, 606–607 (1998).
Wang, R., Kass, J. M., Chaudhary, C., Economo, E. P. & Guénard, B. Global biogeographic regions for ants have complex relationships with those for plants and tetrapods. Nat. Commun. 15, 5641 (2024).
Aulus-Giacosa, L., Ollier, S. & Bertelsmeier, C. Non-native ants are breaking down biogeographic boundaries and homogenizing community assemblages. Nat. Commun. 15, 1–11 (2024).
Wallace, A. R. The Geographical Distribution of Animals (Harper & Brothers, 1876).
Rueda, M., Rodríguez, M. Á & Hawkins, B. A. Identifying global zoogeographical regions: Lessons from Wallace. J. Biogeogr. 40, 2215–2225 (2013).
Kawahara, A. Y. et al. A global phylogeny of butterflies reveals their evolutionary history, ancestral hosts and biogeographic origins. Nat. Ecol. Evol. 7, 903–913 (2023).
Reginato, M. et al. Historical biogeography of the Melastomataceae. in Systematics, Evolution, and Ecology of Melastomataceae 87–105 (Springer International Publishing, 2022).
Udvardy, M. D. F. A. A Classification of the Biogeographical Provinces of the World. IUCN Occasional Paper Vol. 18 (International Union for Conservation of Nature and Natural Resources, 1975).
Matzke, N. J. BioGeoBEARS: bioGeography with Bayesian (and likelihood) Evolutionary Analysis with R Scripts. at https://doi.org/10.5281/zenodo.1478250 (2018).
Sanmartín, I. & Meseguer, A. S. Extinction in phylogenetics and biogeography: from timetrees to patterns of biotic assemblage. Front. Genet. 7, 1–17 (2016).
Matzke, N. J. Statistical comparison of DEC and DEC+J is identical to comparison of two ClaSSE submodels, and is therefore valid. J. Biogeogr. 49, 1805–1824 (2022).
Matzke, N. J. Stochastic mapping under biogeographical models. PhyloWiki BioGeoBEARS website. http://phylo.wikidot.com/biogeobears#stochastic_mapping (2016).
Müller, R. D. et al. A tectonic-rules-based mantle reference frame since 1 billion years ago - implications for supercontinent cycles and plate-mantle system evolution. Solid Earth 13, 1127–1159 (2022).
Kocsis, Á. T., Raja, N. B., Williams, S. D. & Dowding, E. M. rgplates: R interface for the GPlates Web Service and Desktop Application. at https://zenodo.org/doi/10.5281/zenodo.8093990 (2024).
Rabosky, D. L. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 9, e89543 (2014).
Louca, S. & Pennell, M. W. Extant timetrees are consistent with a myriad of diversification histories. Nature 580, 502–505 (2020).
Rabosky, D. L. & Huang, H. A robust semi-parametric test for detecting trait-dependent diversification. Syst. Biol. 65, 181–193 (2016).
Schluter, D. & Pennell, M. W. Speciation gradients and the distribution of biodiversity. Nature 546, 48–55 (2017).
Fitzjohn, R. G. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 3, 1084–1092 (2012).
Acknowledgements
This work was supported by National Science Foundation grants under the Ants of the World project: DEB-1932405 (J.T.L. and M.G.B.), DEB-2019431 (BBB), DEB-1932062 (PSW), DEB-1932467 (BLF). No new specimens were collected specifically for this study. All specimens used were obtained during prior research projects and were collected in full compliance with the relevant permits and regulations. Collecting permits in Costa Rica were granted by the “Comisión Institucional de Biodiversidad” (Institutional Biodiversity Committee, University of Costa Rica; Resolution Number 028) and authorized by the owners of each property. In Madagascar, permits to research, collect, and export arthropods were obtained from the Ministry of Environment and Forest as part of an ongoing collaboration between the California Academy of Sciences and the Ministry of Environment and Forest, Madagascar National Parks, and Parc Botanique et Zoologique de Tsimbazaza. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the authors. USDA is an equal opportunity provider and employer. We thank Brendon Boudinot, Amanda Dias, Rodrigo Feitosa, Fernando Fernández, Peter Hawkes, Milan Janda, Bob Johnson, John Lattke, Cong Liu, Laís Mizraim, Adrian Troya, Wendy Wang, and Alex Wild for providing specimens. We thank Rodolfo Probst, Elaine Tan, Zachary Griebenow, Ziv Lieberman, and Jill Oberski for help with sample preparation and extraction. We thank Brian Dalley at the University of Utah for helping with sample processing and sequencing. We thank Michele Esposito for specimen preparation, imaging, and databasing. We are grateful to François Brassard and Alex Wild for providing ant images for illustration.
Author information
Authors and Affiliations
Contributions
M.D., M.L.B., M.G.B., G.P.C., J.T.L., P.S.W., B.L.F. and B.B.B. conceived the study. M.G.B., J.T.L., P.S.W., B.L.F. and B.B.B. provided financial and material resources. M.D., G.P.C., J.T.L., P.S.W., B.L.F. and B.B.B. provided samples and contributed to data curation. M.G.B processed the molecular data and performed phylogenetic inferences. M.L.B. ran divergence dating analyses. M.D. built the scripts and carried out tree grafting and all diversification and biogeographic analyses. M.D. produced figures and wrote the original draft for the manuscript. All authors contributed to revising subsequent drafts of the paper and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Doré, M., Borowiec, M.L., Branstetter, M.G. et al. Evolutionary history of ponerine ants highlights how the timing of dispersal events shapes modern biodiversity. Nat Commun 16, 8297 (2025). https://doi.org/10.1038/s41467-025-63709-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63709-3

{kind=link}