
Abstract
The metal–oxygen redox behavior governs the performance of transition metal oxides in many electrochemical reactions, especially for RuO2 with the activity–stability paradox in the anode oxygen evolution reaction of proton exchange membrane water electrolyzers. Herein, we modulate the electronic structure of RuO2 near the Fermi level to promote reversible Ru redox while suppressing the oxidative release of lattice oxygen. As a result, the RuO2 integrated with electron-rich p-block metals Sb achieves an overpotential of 220 mV and long-term operational stability of 1200 h at 10 mA cm−2. The assembled proton exchange membrane water electrolyzers can operate steadily over 100 h at 100, 500, and 1000 mA cm−2. Further advanced in-situ characterizations reveal the more reversible and milder Ru redox and the passivated lattice oxygen reactivity, which suppresses drastic structural changes of RuO2 during the oxygen evolution reaction. This work highlights the importance of engineering metal-oxygen redox behavior and provides insights for designing high-performance catalysts for energy conversion and storage devices.
Similar content being viewed by others


Introduction
The metal–oxygen redox behavior affects the performance of transition metal oxides in many electrochemical energy storage and conversion-related fields, such as Li-ion batteries1,2, fuel cells3,4, and rechargeable metal-air batteries5, and electrochemical water splitting6,7. Wherein, the redox of metal cation sites governs the frequent electron transfer process, directly impacting the charge capacity or intrinsic catalytic activity8,9,10,11. The redox of lattice oxygen anion sites is strongly associated with the structural stability of the metal oxides12,13,14. The irreversible oxidative release of lattice oxygen anion sites can result in the formation of oxygen vacancies and subsequent structural corrosive collapse under continuous oxidative potentials6,15. Unfortunately, many transition metal oxides usually fail to maintain sufficient stability despite exhibiting notable activity in electrochemical reactions, such as oxygen evolution reaction (OER). This deactivation phenomenon can be observed across various types of metal oxide electrocatalysts, including rutile oxides (e.g., RuO2)16, spinel oxides (e.g., Co3O4)17,18, and perovskite oxides (e.g., SrRuO3)19,20, limiting the application in proton exchange membrane water electrolyzers (PEMWE). Particularly, ruthenium oxide electrocatalysts featuring the advantages of their high activity (ƞ10 ≈ 260 mV vs. 340 mV, IrO2) and cost-effectiveness (10 times cheaper than Ir), are widely regarded as promising alternatives for commercial IrO221,22. However, they still perform inferior stability and short lifespan (lifespan is usually less than 50 h in PEMWE)23,24,25,26,27,28,29,30,31. Therefore, from fundamental understanding to practical application, the activity and stability of low-cost Ru oxide electrocatalysts need optimization to meet the demands of PEMWE industrial hydrogen production.
The conventional lattice oxygen oxidation mechanism (LOM) and the adsorbate evolution mechanism (AEM) are closely linked to whether metal or oxygen serves as the redox center (Fig. 1a)7,32,33,34,35. From the electronic states around the Fermi level, when the O2p energy is higher than that of the metal d band, lattice oxygen acts as the redox center for losing electrons6. The LOM pathway is thermodynamically favored but damages the surface structure of the electrocatalyst during acidic OER. The O2p band centers have been reported as descriptors to evaluate the reactivity of lattice oxygen for perovskite oxides17,20. Moderately upshifting the metal d-band center while downshifting the O2p band center may shift the redox center to the metal sites. In this case, the electron transfer for water oxidation occurs at the metal sites. In contrast, the oxidation of lattice oxygen may be partially suppressed, which results in a stable AEM pathway36,37. Specifically for RuO2 electrocatalysts, the common optimization approaches, such as introducing electron-donating components (such as Na38, Cr39, and Cu40, etc.) or constructing sacrificial layer heterojunctions (Co3O441, CoMnO342, etc.), aim to compensate charge into Ru sites to prevent further Ru over-oxidation. However, solely suppressing the changes of Ru valence state often constrains the flexibility of Ru redox, which reduces their intrinsic activity8,43,44. In fact, the highly reversible and flexible Ru redox and the suppressed O redox are expected for an ideal Ru–O catalytic unit in acid OER9,32,35,45,46. Optimizing the metal–oxygen redox behavior offers an opportunity for designing highly active and durable OER catalysts. However, it is closely associated with the electronic states near the Fermi level, and its precise regulation remains challenging.

a The illustration of the relationship between the electronic states around the Fermi level, the adsorbate evolution mechanism, lattice oxygen oxidation mechanism, and the metal–oxygen redox center. b The projected density of states (PDOS) of the eg and t2g orbitals in the M-RuO2 (M = Ti, Ce, Bi, Sb). c The O K-edge X-ray absorption spectroscopy of M-RuO2, RuO2, and rutile IrO2. d The quantitative t2g*/eg* peak intensity ratio extracted from plot c. e The variation of the O2p band center and O charge in M-RuO2. f The charge density difference of RuO2 and Sb-RuO2. The yellow part refers to the charge accumulation, and the blue part refers to the charge decrease. g The illustration of electronic states near the Fermi level of Ru–O–Ru and Ru–O–Sb, and the potential effect on Ru–O redox behavior during OER. Source data for Fig. 1 are provided as a Source Data file.
Herein, by combining density functional theory (DFT) with experimental spectroscopy, we modulate the electronic structure of RuO2 near the Fermi level by implantation of different types of heteroatoms (transition metals, lanthanides, main group metals). Typically, the Sb heteroatoms increase the occupancy of π-type orbitals above the Fermi level and downshift the O2p band center. This thermodynamically polarizes the Ru–O redox behavior, promoting reversible Ru redox while suppressing the oxidative release of lattice oxygen. As a result, the SbRuO2 electrocatalysts achieve an overpotential of 220 mV at 10 mA cm−2 and maintain steady operation for 1200, 400, and 350 h at 10, 25, and 50 mA cm−2, respectively. More importantly, the PEMWE assembled with Sb-RuO2 maintains stable operation for over 100 h at 100, 500, and 1000 mA cm−2. Furthermore, Insitu Ru Kedge Xray absorption spectroscopy, electrochemical characterization, and insitu differential electrochemical mass spectroscopy confirm that the more reversible and milder redox behavior at Ru sites and the reduced participation of lattice oxygen and suppressed drastic structural changes of Sb-RuO2. The method of regulating the metal–oxygen redox offers insights into the design of highly active and stable catalysts in energy storage and conversion devices.
Results
Regulation of electronic states near the Fermi level of RuO2
The frequent electron transfer in OER is closely related to the electronic states near the Fermi level in transition metal oxides. We combined DFT calculations and synchrotron soft X-ray absorption spectroscopy to probe the modulation effects of heteroatom dopants on the electronic structure of RuO₂. Firstly, some typical M dopants (Ti of transition metals, Ce of lanthanides, Bi and Sb of main group metals) were selected and integrated into the RuO2 lattice to construct structural models (Supplementary Fig. 1, Supplementary Data 1–11). The projected density of states of the metal–oxygen covalent orbitals suggests large amounts of DOS of t2g orbitals above the Fermi level (EF) are generated with Sb and Bi dopants (Fig. 1b). Meanwhile, the experimental O K-edge X-ray absorption spectroscopy (XAS) also reflects the electron occupancy of the metal–oxygen covalent orbitals above EF. (Fig. 1c and Supplementary Fig. 2, samples seen in synthesis details). The two peaks at 530.1 and 533.2 eV are assigned to electron excitation from the O 1 s orbital to unoccupied states of t2g* and eg* orbitals. The modifications of electron-rich p-block metals Sb and Bi reduce the peak intensity corresponding to t2g* orbitals. Furthermore, the quantitative t2g* /eg* intensity ratio decreases from 1.03 (RuO2), 0.99 (Ti-RuO2), and 0.99 (Ce-RuO2) to 0.89 (Bi-RuO2) and 0.91 (Sb-RuO2) and approached 0.89 (IrO2) (Fig. 1d). The lower ratio indicates increased electron occupancy in the anti-bonding type t2g* orbitals above EF, which is consistent with the theoretical calculations. These results demonstrate that Sb and Bi dopants help to increase the occupancy of t2g* above EF and weaken Ru–O π-type covalency to the level of the Ir–O units47,48.
The valence bond electronic structure below the Fermi level is reflected in the projected density of Ru 4 d and O 2p states (Supplementary Fig. 3). The O2p band center associated with the reactivity of lattice oxygen shows upshifting that of Ti-RuO2 (−3.16 eV), almost unchanged that of Bi-RuO2 (−3.25 eV) and downshifting that of Ce-RuO2 (−3.31 eV) and Sb-RuO2 (−3.31 eV) compared to pure RuO2 (−3.24 eV) (Fig. 1e). Meanwhile, Bader charge analysis shows the more the O2p centers shift downward, the more charge the lattice oxygen increases (Ce-RuO2 + 0.98, Sb-RuO2 + 0.99, Supplementary Fig. 4). The downshifted O2p band center and the increased charge of lattice oxygen contributes to the stabilization of lattice oxygen17. The charge density difference reveals increased charge donation to the Ru–O unit from Sb in the asymmetric Ru–O–Sb structure (Fig. 1f). The modulation of electronic states near the Fermi level potentially affects the Ru–O redox behavior (Fig. 1g). The energetically close 4 d and 2p orbitals in RuO2 enable electron transfer from lattice oxygen to the Ru sites during deep oxidation. However, the increased occupancy of π-type orbitals above the EF benefits the metal sites to extract electrons from higher energy orbitals. The downshifted O2p band center is unfavorable for oxygen anions to lose electrons. This may facilitate metal redox and passivate the reactivity of lattice oxygen to realise the polarised M–O redox.
Synthesis and structural characterization of catalysts
The different types of metal-doped RuO2 (M-RuO2, such as Ti, Ce, Bi, and Sb) were synthesized using the modified sol-gel method with citric acid and tartrate as the coordination agents. The general process involves uniformly dispersing the dopant and Ru species within a three-dimensional sol-gel network composed of citrate and tartrate, followed by drying and annealing to prepare a series of M-RuO₂ with various dopants and doping levels. High-resolution transmission electron microscopy (HRTEM) images reveal that the obtained M-RuO₂ samples exhibit polycrystalline nanosheets composed of numerous fine grains smaller than 10 nm (Fig. 2a, b and Supplementary Figs. 5 and 6). Their good crystallinity with the same characteristic peaks of rutile-phase RuO2 can also be observed in the X-ray diffraction patterns (Fig. 2c). The absence of any diffraction peaks for metallic Ru and doped metal oxides indicates the complete oxidation of Ru and uniform doping. The slight shift towards the higher 2θ of the diffraction peak corresponding to (101) planes in RuO2 samples with Sb dopants suggests a local and slight lattice contraction. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and fast Fourier transform pattern of Sb-RuO2 reveal a characteristic tetragonal system with high crystallinity and interplanar spacing of 0.251 nm and 0.229 nm corresponding to (101) and (020) planes respectively (Fig. 2d). The uniform doping of Sb elements (13.9 wt.%) without aggregation was observed in the energy-dispersive spectroscopic elemental mapping (Supplementary Fig. 7). Furthermore, by overlaying the Sb and Ru mappings combined with the corresponding HAADF-STEM image, higher-precision atomically resolved elemental mapping performs the atomic-level dispersion of the Sb atoms solid solution into the RuO2 lattice (Fig. 2e)49.

a, b HRTEM images of the Sb-RuO2 polycrystalline nanosheets. c XRD pattern of M-RuO2 (M=Ti, Ce, Bi, Sb). d HAADF-STEM image of Sb-RuO2 and corresponding fast Fourier transform pattern. e The corresponding atomically resolved elemental mapping of Sb-RuO2. f Raman spectra of Sb-RuO2 and RuO2. g Ru K-edge and Sb K-edge XANES spectra of Sb-RuO2. h Corresponding Fourier transform of extended Sb K-edge EXAFS in R spaces (R: radial distance). Source data for Fig. 2 are provided as a Source Data file.
Raman spectra of Sb-RuO2 and RuO2 display characteristic Eg, A1g, and B2g peaks (Fig. 2f)14, in which the Eg and A1g peaks appear redshifted and the B2g peaks disappear after Sb doping. This result indicates that Sb locally substitutes Ru atoms rather than surface adsorption50. The reduced Ru–O vibration frequency may be attributed to the weakened covalency of the Ru–O bonds, which reduces Ru–O bond stiffness51. X-ray absorption near edge structure (XANES) was utilized to probe the chemical states of Ru and Sb species and the fine structure of Sb-RuO2 (Fig. 2g). The absorption threshold of Sb-RuO2 in Ru K-edge is close to that of RuO2 but with a slight shift towards lower energy, which indicates the valence state of the Ru species is slightly less than Ru4+ and the average oxidation state of +3.7 is identified by further fitting results (Supplementary Fig. 8). This observation is in agreement with the Ru 3 d XPS results (Supplementary Fig. 9). The fitted peaks of Sb-RuO2 at 281.3 (Ru4+) and 282.3 eV (Ru3+) in the Ru 3 d XPS exhibit a lower integral peaks area ratio of Ru4+/Ru3+ than that of RuO2. Similarly, the Ru 4p XPS shows a decreased Ru⁴⁺/Ru³⁺ ratio upon Sb doping (from 1.81 in RuO₂ to 1.51 in Sb-RuO₂), supporting a partial reduction of Ru. Meanwhile, the absorption threshold of Sb-RuO2 in the Sb K-edge exhibits a slight shift towards higher energy. The further quantitative XANES analysis yields an average Sb oxidation state of approximately +3.3 (Supplementary Fig. 8). The Sb 3 d₃/₂ peak appears at 540.5 eV across all Sb-doped samples in the Sb 3 d XPS spectra (Supplementary Fig. 9). The gradual shift to higher energy of the peak positions with increasing Sb content supports a slightly increasing average valence, which is in agreement with the XAS analysis. Together, both the XANES and XPS data consistently support that Sb dopants play a role in charge compensation within the RuO2 lattice, resulting in charge transfer from Sb to the Ru–O units. Fourier transform extended X-ray absorption fine structure (EXAFS) spectra of Ru K-edge show structural information. The fitting results indicate that the Ru–O bond length remains largely unchanged (R = 1.98 Å), while the Ru–O coordination number (CN = 5.8) slightly decreases (Supplementary Fig. 10 and Table 1). This may be attributed to an unsaturated Ru–O coordination with surface oxygen vacancies52. EXAFS of Sb K-edge shows the first coordination shell peak located at 1.50 Å is assigned to Sb–O and the second coordination shell peak located at 3.25 Å is assigned to Sb–Ru (Fig. 2h). The increased peak intensity of the Sb–O bond in Sb-RuO2 compared to that of Sb2O3 indicates the increase in the coordination number (CN) from theoretical 3 to 6 (Supplementary Fig. 11). This significant change in coordination number also indicates that Sb species occupy the RuO₂ lattice in the form of Sb–O octahedra. The second-shell Sb–Ru bond length in Sb-RuO₂ is shortened compared to the Sb–Sb bond in Sb2O3. The fitting results also show that the Sb–Sb distances of 3.60 Å/3.90 Å decrease to Sb–Ru distances of 3.12 Å/3.64 Å (Supplementary Fig. 12 and Table 2), which matches well with the theoretical Sb–Ru distance in rutile RuO₂ and the Sb–Sb distance in Sb2O3. These results further confirm that Sb is incorporated in the RuO2 lattice by substituting Ru atoms and forming highly complete Sb–O octahedral units.
The performance of acidic OER and the corresponding PEMWE
To evaluate the acidic OER activity and stability of the series of M-RuO₂ electrocatalysts we predicted and synthesized, the specific electrochemical performance of the as-prepared M-RuO₂ electrocatalysts was determined in O2-saturated 0.5 M H2SO4 by a standard three-electrode system. Linear sweep voltammetry (LSV) curves and corresponding Tafel plots of M-RuO2 (M = Sb, Bi, Ce, Ti), homemade RuO2 (HM-RuO2), and commercial RuO2 (Com. RuO2) with the loading of 0.2 mg cm−2 are shown in Fig. 3a, b. The LSV curves without iR correction are presented in Supplementary Fig. 13. The Sb-RuO2, Ti-RuO2, Ce-RuO2, and Bi-RuO2 electrocatalysts exhibit an overpotential of 220 ± 4, 223 ± 5, 213 ± 5, and 218 ± 6 mV at 10 mA cm−2, respectively, which are lower than that of Com. RuO2 (273 ± 6 mV) and HM-RuO2 (259 ± 7 mV). The results indicate these dopants are all advantageous for enhancing activity. The Sb-RuO2 electrocatalyst also demonstrates advantages (overpotential of 313 ± 9 mV) under a high current density of 100 mA cm−2. Moreover, its lower Tafel slope (55.5 ± 0.3 mV dec−1) than that of Com. RuO2 (70 ± 0.4 mV dec−1) and HM-RuO2 (67.6 ± 0.3 mV dec−1) indicate that the Sb-RuO₂ electrocatalyst can drive oxygen evolution with smaller potential steps and faster reaction kinetics. Furthermore, we found that the effect of a low doping level (5 at.%) on the electronic structure is localized, which can be observed in the O K-edge XAS (Supplementary Fig. 2). High doping levels (15 at.%) can reduce the number of active Ru sites and introduce more lattice distortions and defects, leading to decreased activity and stability. In contrast, a moderate doping level (10 at.%) balances effective orbital modification and structural stability, resulting in optimal performance (Supplementary Fig. 14). Therefore, the Sb-RuO2 (10 at.% Sb) electrocatalyst is mainly discussed unless specified otherwise. The electrochemical impedance spectroscopy indicates that the Sb-RuO2 electrocatalyst shows a smaller charge transfer resistance and non-capacitive behavior (Supplementary Fig. 15), which reflects a more direct and faster electron transfer between H2O and the electrocatalyst.

a OER polarization curves and b corresponding Tafel plots of the prepared M-RuO2 (M = Ti, Ce, Bi, Sb), homemade RuO2 (HM-RuO2), and commercial RuO2 (Com. RuO2) in O2-saturated 0.5 M H2SO4 solution. The solution resistance: 5 ± 0.5 Ω. c Dissolved Ru and Sb ion concentration and corresponding mass loss of Sb-RuO2 and RuO2 in 70 mL electrolyte determined via ICP-MS. d Chronopotentiometric stability tests of Sb-RuO2 and RuO2 at an anodic current density of 10, 25, and 50 mA cm−2 without iR correction. e Comparison of OER performance (at 10 mA cm−2) for Sb-RuO2 and benchmark RuO2. f Polarization curve of Sb-RuO2 ǁ Pt/C, IrO2 ǁ Pt/C, and RuO2 ǁ Pt/C in PEMWE operated at 80 °C. g Stability test of Sb-RuO2 ǁ Pt/C in PEMWE at 100, 500, and 1000 mA cm−2. Source data for Fig. 3 are provided as a Source Data file.
The dissolution and mass loss of the catalyst during the acidic OER are evaluated in a membrane-separated H-type electrolyzer. The time-dependent Ru concentration dissolved from Com. RuO2 increases rapidly and reaches 624 ppb (mass loss of 2.18 wt.%) at 40 h. In contrast, the lower Ru and Sb concentrations dissolved from Sb-RuO2 remain stable at 146 ppb and 164 ppb (mass loss of 0.51 and 0.57 wt.%) at 200 h (Fig. 3c). The calculated stability number (S-number) is up to 1.8 × 105. Lower dissolution indicates that the introduction of Sb suppresses the dissolution of Ru and stabilizes the structure of RuO2 during prolonged acidic OER. In addition, the durability test of Sb-RuO2 was evaluated by chronopotentiometry with a loading of 2 mg cm−2 on hydrophilic carbon paper (Fig. 3d). The Sb-RuO2 electrocatalyst achieves continuous and stable 1200 h, 400 h, and 350 h operation at a current density of 10, 25, and 50 mA cm−2. The corresponding potential decay rates are only 56.5, 225, and 128 μV h−1, respectively. However, Com. RuO2 experiences a significant degradation within the initial 40 h, possibly due to its rapid dissolution in acidic OER. After durability testing, the high-resolution TEM image and elemental distribution of Sb-RuO2 show intact grain boundaries and uniform elemental distribution without surface amorphization (Supplementary Fig. 16). The stability evaluation of other Ti-RuO2, Ce-RuO2, and Bi-RuO2 electrocatalysts is shown in Supplementary Fig. 17, Notably, the Ce-RuO2 also exhibits improved operational stability, which is possibly attributed to the passivated reactivity of oxygen anion caused by the downshifted O2p band center. The Sb-RuO2 electrocatalyst demonstrates competitive performance in terms of overpotential, Tafel slope, mass activity (608 ± 91 versus 135 ± 22 A g−1Ru, RuO2), stability, and S-number (Fig. 3e and Supplementary Table 3), particularly achieving an order of magnitude improvement in stability compared to benchmark RuO2. These merits make Sb-RuO2 an inexpensive and promising non-Ir-based electrocatalyst for acidic OER.
PEMWE were employed to evaluate the practical potential of electrocatalysts. A single cell was assembled with Sb-RuO2 or Com. RuO2 as anode electrocatalyst combined with a proton exchange membrane and cathodic commercial 60 wt.% Pt/C for pure water electrolysis (Supplementary Fig. 18, see Methods for details). The polarization curve of membrane electrode assemblies (MEAs) of Sb-RuO2||PEM||Pt/C operated at 80 °C (Fig. 3f) shows performance improvements. Specifically, the current densities of 1 A cm−2 and 2 A cm−2 for hydrogen production are achieved at cell voltages of 1.61 V and 1.74 V, which are lower than those of Com. RuO2 (1.68 V and 1.86 V) and commercial IrO2 (1.77 V and 1.97 V) as an anode. More importantly, the long-term operational stability (Fig. 2g) shows that PEMWE based on Sb-RuO2 performs 300 h, 200 h, and 100 h continuous well-maintained operation at a current density of 100, 500, and 1000 mA cm−2. PEMWE also exhibited cell voltages of 1.68 V and 1.75 V at 1 A cm−2 under 60 °C and 40 °C, respectively, along with continuous operation for 100 h at 60 °C, and 500 mA cm−2 (Supplementary Fig. 19). These results confirm the enhanced resistance to deactivation of the Sb-RuO2 electrocatalyst and its practical application potential for PEMWE53. Meanwhile, the SEM image and element mapping of the cross-section of the MEAs after durability test (100 mA cm−2) indicate that the anode Ru is almost not accumulated in the PEM and cathode. HRTEM images show that the Sb-RuO₂ catalysts in MEA after durability test (100 mA cm−2) retain the polycrystalline structure (Supplementary Fig. 20). The Sb-RuO₂ electrocatalyst-based PEMWE achieves high energy efficiencies of 46.0 and 49.7 \({{\rm{kWh}}}\, {{\rm{kg}}}^{-1}_{{{\rm{H}}_{2}}}\) at 1 and 2 A cm−2, respectively, which is comparable to that of previously reported Ru-based electrocatalysts (Table S4). The calculated anode noble metal cost of 0.057 $ cm−2 is competitive with that of reported Ir-based PEMWE devices previously (Supplementary Fig. 21 and Table S5), which contributes to the widespread application of PEMWE.
In situ structural changes and investigation of Ru–O redox behavior
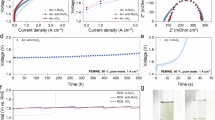
To further investigate the relationship between the improved stability of Sb-RuO2 and Ru–O redox during acidic OER, in situ XAS measurements were carried out to probe the dynamic change in oxidation states and the local structure of Ru. The Fourier transform EXAFS reveals in situ structural changes, including Ru–O bond lengths (corresponding peak at 1.56 Å) and average coordination numbers (Fig. 4a). The position and intensity of the peak in RuO2 and Sb-RuO2 remain relatively stable in the pre-catalysis stages (OCP to 1.4 V vs. RHE). However, in the on-site and high overpotential stages (1.4 V to 1.8 V vs. RHE), the peak shifts towards shrinkage of the Ru–O bond, and the peak intensity decreases in the pure RuO2 sample. The Ru–O bond length shortens and the average coordination number of Ru–O decreases in the pure RuO2 sample during the OER process (Supplementary Fig. 22 and Table 1), which can be understood as the breaking of surface Ru–O bonds and the generation of oxygen vacancies, which reflects the structural instability of pure RuO2 under high oxidation potentials9,12,24. In contrast, A slight change in peak intensity and almost no shift in peak position are observed in the Sb-RuO2 sample with a continuous increase in applied potential, indicating its smaller in situ structural changes. As summarized in Fig. 4b, the changes in the valence state are revealed via Ru K edge XANES of Sb-RuO2 and RuO2 at different applied potentials (Supplementary Fig. 23). The absorption edge position of the RuO2 sample shifts positively and changes dramatically. In contrast, that of the Sb-RuO2 sample undergoes a slight positive shift but remains relatively stable. The specific fitting results show the oxidation state of Ru increases from +3.96 to +4.76 in pure RuO2 but changes negligibly from 3.61 to 3.69 in Sb-RuO2. Notably, relatively minor changes in valence observed in the XAS measurements with limited temporal resolution do not imply the absence of high-valence Ru species. Redox-active high-valence Ru species are essential for catalyzing water oxidation. Transient high-valence Ru species with reversible redox do not accumulate on the stable catalyst surface, whereas those with irreversible redox can further diffuse through the corroded surface into the hydrated subsurface and bulk, which is detectable by XAS. In situ Raman spectroscopy also reflects the same in situ structural changes (Fig. 4c). The peak at 519 cm−1 (Eg vibration modes) shows no significant change in Sb-RuO2 but undergoes a blue shift in RuO2 at higher overpotentials, which suggests the surface Ru–O bonding structure of Sb-RuO2 samples maintains consistency, but that of RuO2 shrinks during OER12. This result is consistent with the analysis from the Fourier transform EXAFS. Moreover, XAS analysis of Sb-RuO2 after long-term stability testing indicates that the oxidation state of Ru remains relatively stable without a significant increase, and the changes in the first-shell Ru–O bond are negligible (Supplementary Fig. 24 and Table S1).

a In situ Fourier transform EXAFS of Sb-RuO2 and RuO2 during acidic OER. b The variation trend in the fitting Ru oxidation state from Ru K-edge absorption energy and the fitting Ru–O bond length from EXAFS of Sb-RuO2 and RuO2. c In situ Raman spectroscopy of Sb-RuO2 and RuO2 during acidic OER. d The time-dependent cyclic voltammetry curve of Sb-RuO2 and RuO2 at a scan rate of 0.1 V · s−1 in 0.5 M H2SO4 with 0, 10, 20, and 30 min OER at 10 mA cm−2 without iR correction. e Square-wave voltammetry (SWV) profiles of Sb-RuO2 and RuO2 with capacitive current correction without iR correction. The DEMS signals of 32O2, 34O2, and 36O2 from the produced oxygen of f Sb-RuO2 and g RuO2 during OER. h Illustration of adjusting Ru–O redox and corresponding structural changes during OER. Source data for Fig. 4 are provided as a Source Data file.
The redox peaks of Ru3+/4+ (0.6 V), Ru4+/6+ (1.25 V), and Ru6+/8+ (1.48 V) can be detected in the time-dependent cyclic voltammetry curve (Fig. 4d)29,54,55, which corresponds to H₂O/OH, OH/O, and O/OOH transitions of the deprotonation and O–O coupling processes during catalytic water oxidation56,57. Notably, the reduction peak from Ru8+ to Ru6+ for pure RuO2 disappeared in the reverse scanning at the initial 0 min. However, as the water oxidation time increased, this peak gradually emerged, and the capacitive behavior in the CV curves also increased. This may be attributed to the formation of a loose structure of amorphous RuOₓ on the surface. In contrast, the CV curves of Sb-RuO₂ showed no significant changes during 30 min of water oxidation, indicating that the Ru redox and the catalytic water oxidation proceeded on the stable catalyst surface. Square-wave voltammetry (SWV) method exhibits higher sensitivity for redox behavior by diminishing the contribution of the charging current (Fig. 4e). All the redox peaks of Sb-RuO₂ are negatively shifted, suggesting the promoting redox of Ru under a smaller driving potential. The generation of high-valence Ru at low potentials is mainly responsible for the enhanced OER activity of Sb-RuO₂22. More importantly, the oxidation/reduction peak intensity ratio of Ru3+/4+, Ru4+/6+, and Ru6+/8+ for Sb-RuO2 is closer to 1, indicating the highly reversible redox behavior of surface Ru species in the Sb-RuO2 sample. However, the non-coincidence of oxidation and reduction behaviors indicates an additional irreversible lattice oxygen oxidation and irreversible destruction process on the RuO2 surface56,58. The time-dependent SWV plots further reflect the changes in Faradaic currents associated with various redox transitions as the water oxidation time increases (Supplementary Fig. 25). Different from the increase in non-Faradaic currents associated with capacitive behavior in the CV curves of RuO2, the Faradaic current of the redox peak at 0.6 V decreased over time. This suggests that the transition involving the lower oxidation states of Ru³⁺/Ru⁴⁺ diminishes over time, which may be attributed to the formation of an amorphous surface structure and the increased presence of Ru species that cannot undergo reversible redox transitions, leading to a decline in their activity for the deprotonation step (H₂O/OH). In contrast, the Faradaic current of the Sb-RuO₂ sample showed no significant change over time, suggesting the stability of the surface structure and the reversibility of the Ru redox processes.
In situ differential electrochemical mass spectroscopy (DEMS) measurements were used to investigate the participation of lattice oxygen during OER (Fig. 4f, g). Before evaluation, the catalyst surface was labeled with 18O by exchanging with H218O (see Methods)13,44. The evolved 18O16O originates from the coupling of surface-labeled 18O and 16O from the electrolyte through the LOM mechanism, reflecting the lattice oxygen participation. The results showed a suppressed 18O16O signal for Sb-RuO2 compared to RuO2, which indicates that the over-oxidation of lattice oxygen is somewhat suppressed. Thus, surface amorphization induced by the formation of oxygen vacancies is also suppressed, which results in the enhanced stability of Sb-RuO2.
According to the collective results, it can be concluded that the enhanced OER activity is attributed to the facilitated Ru redox process, while the enhanced stability results from the suppressed lattice oxygen oxidation and the stabilized surface structure (Fig. 4h). On the one hand, the weakened Ru–O bond covalency prevents electron flow from lattice oxygen to the Ru, thereby enhancing the reversibility of Ru redox. The mild oxidation of Ru further stabilizes the surface structure, preventing surface corrosion and deep oxidation from propagating into the subsurface and bulk regions. On the other hand, the energy increase of the t2g above EF with metal character boosts Ru redox, and the energy decrease of the O2p state with O character passivates the reactivity of lattice oxygen. Thus, the coupled Ru–O oxidation is tuned into a reversible Ru redox process. The drastic structural changes during the OER process are suppressed.
Detection of oxygen intermediates and understanding of the OER mechanism
In situ attenuated total reflection surface enhanced infrared absorption spectroscopy (ATR-SEIRAS) was conducted to investigate the reaction intermediates during the OER process (Fig. 5a). According to previous reports, the peak at 1230 cm−1 could be attributed to the Si–O–Si prism59. The peak at 1145 cm−1 and 902 cm−1 corresponds to the linearly bonded *OOH and *OO intermediate on the coordinatively unsaturated Ru sites53,57,60. Above 1.4 V, these peaks appear and continue to increase in the Sb-RuO2 samples, whereas these peaks only appear above 1.7 V in RuO2 (Supplementary Fig. 26). These results indicate greater adherence to the AEM mechanism for the Sb-RuO2 samples. The *OH, as a typical intermediate for OER, can be detected by methanol probes (Fig. 5b and Supplementary Fig. 27)61. Upon the addition of methanol, the current density of the CV curve increases during the on-site stage, which is due to the methanol oxidation facilitated by *OH intermediates. At high overpotential stages, the current density decreases due to the consumption of generated *OH, which hinders subsequent oxygen evolution. The kinetic isotope effect was used to evaluate whether the deprotonation step is the potential-determining step (PDS) of the OER62. The deprotonation step in the LOM pathway requires a higher energy barrier and is more influenced by the isotope effect63,64. The introduction of D2O caused a decrease in OER activity and the Tafel slope (Fig. 5c). The corresponding LSV curves without iR correction are presented in Supplementary Fig. 28. In comparison, the Sb-RuO2 sample exhibited a smaller change, suggesting the deprotonation rate is not the PDS and that the LOM pathway is less involved53.

a Operando ATR-SEIRAS of Sb-RuO2 at the applied potential of OER. b The cyclic voltammetry curve of Sb-RuO2 recorded with and without 1 M methanol in 0.5 M H2SO4 at a scan rate of 10 mV s−1 without iR correction. c Linear sweep voltammetry curve and the corresponding Tafel slope of Sb-RuO2 and RuO2 in 0.5 M H2SO4 dissolved in H2O (solution resistance: 5.5 ± 0.5 Ω) and D2O (solution resistance: 12 ± 1 Ω) with iR correction. d The Bader charge analysis of the RuO2 and Sb-RuO2 with adsorbed O (*O) intermediates. e Free energy diagram for OER on Ru sites of Sb-RuO2 and RuO2 at 0 and 1.23 V versus RHE, respectively. Source data for Fig. 5 are provided as a Source Data file.
The Bader charge analysis of key steps (*O) at the watershed between LOM and AEM still reflects that more negative charge accumulates on the bridging oxygen between Ru and Sb, meanwhile, the less negative charge remains on the adsorbed oxygen intermediates (*O) in Sb-RuO2 (Fig. 5d), which suggests that the introduction of Sb enhances the oxidative capacity of neighboring Ru sites for adsorbed oxygen. In addition, more negatively charged bridging oxygen increases the difficulty of coupling *O with lattice O in LOM (Supplementary Fig. 29). According to the analysis above, the OER barrier based on the AEM mechanism was further calculated. The reaction intermediate configurations and the free energy diagram on Sb-RuO2 (110) are shown in Fig. 5e. After applying a voltage (U = 1.23 V) in DFT calculation, the energy barrier for the step where *H2O is split into *OH and subsequently deprotonated to form *O decreases to a negligible level, while the step from *O to *OOH still maintain a high energy barrier. Therefore, this step becomes the PDS and determines the OER activity65,66. The calculated PDS energy barrier of Sb-RuO2 decreases compared to RuO2 at U = 0 V (from 1.83 eV to 1.62 eV) and U = 1.23 V (from 0.60 eV to 0.39 eV), which supports a lower overpotential of Sb-RuO2. We conclude that the Sb dopants compensate for the negative charge on the bridging oxygen and result in a lower PDS energy barrier of AEM in acidic OER.
In summary, we regulate the metal–oxygen redox behavior in RuO2 by modifying the electronic states near the Fermi level. The increased occupancy of π-type orbitals above the Fermi level and the downshifted O2p band center thermodynamically promote the highly reversible metal redox, while suppressing the oxidative release of lattice oxygen. Thereby, the Sb-RuO2 electrocatalysts deliver enhanced stability and activity. Moreover, the Sb-RuO2-based PEMWE requires a voltage of 1.61 V to achieve 1000 mA∙cm−2 for electrolysis of pure water and steadily operates for more than 100 h at 100, 500, and 1000 mA cm−2. Further electrochemical characterization, in situ XAS, in situ DEMS, and DFT calculation results demonstrate the optimized activity and stability benefit from mild and reversible Ru redox behavior and passivated reactivity of lattice oxygen. This strategy of polarizing the metal–oxygen redox center not only contributes to engineering highly active and durable OER catalysts but also has potential applications in electrode materials of more energy devices.
Methods
Chemicals
All chemicals were used as received without further purification. The Citric acid monohydrate (C₆H₈O₇·H2O, 99.0%), Sodium tartrate dibasic dihydrate (Na₂C₄H₄O₆·2H2O, 99.0%), Titanium(IV) sulfate (Ti(SO4)2, ≥98.0%), Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99.0%), Bismuth(III) nitrate pentahydrate (Bi(NO3)3·5H2O, ≥99.0%), Nitric acid (HNO3, 65.0–68.0%), Hydrochloric acid (HCl, 36.0–38.0%) and Sulfuric acid (H2SO4, 98.0–98.5%) were purchased from Shanghai Chemical Reagents. Ruthenium(III) chloride trihydrate (RuCl3·3H2O, ≥98.0%), Antimony(III) chloride (SbCl3, ≥99.0%), deuteroxide (D2O, 99.9%), Water-18O (H18O, ≥97 atom% 18O), and Nafion solution (D521CS) were purchased from Sigma-Aldrich. Deionized water was used throughout this study. 60% Pt/carbon (Pt/C), Ruthenium dioxide anhydrous (RuO2, 99.95%) were obtained from Alfa Aesar.
Preparation of M-RuO2 catalysts
All samples were prepared using the sol-gel method. Typically, 228 mg (1 mmol) SbCl3 and 210 mg (1 mmol) Citric acid monohydrate were dissolved in 9 mL of deionized water with 1 mL of HCl to form a uniform SbCl3 solution. Next, 235 mg (0.9 mmol) RuCl3·3H2O, and 420 mg (2 mmol) Citric acid monohydrate, and 468 mg (2 mmol) Sodium tartrate dibasic dihydrate and 5 mL HNO3 were dissolved in ~20 mL deionized water. 1 mL SbCl3 solution was added to the above solution dropwise with vigorous stirring for 1 h. Then, the above solution is transferred to a ceramic crucible and heated to 80 °C overnight to evaporate all the water. The obtained precursor powder was ground and transferred to a muffle furnace and annealed at 400 °C in an air atmosphere for 6 h. The black powder was collected and further ground into more uniform and finer samples (donated as Sb-RuO2).
Similarly, other samples, including Ti, Ce, and Bi-doped RuO₂, were synthesized using the above method with Ti (SO4)2, Ce (NO3)3·6H2O, and Bi (NO3)3·5H2O instead of SbCl3. Notably, Ti (SO4)2 was dissolved in 1 M Citric acid monohydrate and Sodium tartrate dibasic dihydrate solution with 1 mL of HNO3. The doping amount is adjustable by varying the proportions of the precursor solution.
Characterization
The scanning electron microscope (SEM) images were recorded on a GeminiSEM 360. The high-resolution TEM and HAADF-STEM and EELS mapping images were recorded on a JEOL-2100Plus high-resolution transmission electron microscope working at 200 kV and a ThermoFisher Themis Z with a spherical aberration corrector worked at 300 kV. Powder X-ray diffraction (XRD) measurements were recorded on a Rigaku Miniflex-600 operated at 40 kV voltage and 15 mA current using a Cu Kα radiation (λ = 0.15406 nm) at a step width of 8° min−1. The ICP measurement was performed on Optima 7300 DV inductively coupled plasma atomic emission spectrometer. X-ray photoelectron spectroscopy (XPS) was recorded on ESCALAB 250Xi. Raman scattering spectra were recorded with a Renishaw System 2000 spectrometer using the 514.5 nm line of Ar+ for excitation.
The X-ray absorption spectra of O K-edges were obtained from the BL10B beamline station of the National Synchrotron Radiation Laboratory in Hefei (China). Hard XAFS measurement and data analysis: XAFS spectra at the Ru K-edge and Sb K-edge were recorded at the XAS station (BL14W1) of the Shanghai Synchrotron Radiation Facility (SSRF), China. The storage ring worked at an energy of 3.5 GeV. The hard X-ray was monochromatized with Si (111) double-crystals. The photograph of custom-built electrochemical cells for in situ X-ray absorption spectra is provided in Supplementary Fig. 30. The acquired EXAFS data were extracted and processed according to the standard procedures using the ATHENA module implemented in the IFEFFIT software packages. To obtain detailed structural parameters around the Ru and Sb atoms, quantitative curve fittings were carried out for R-space using the ARTEMIS module of IFEFFIT.
Electrochemical measurements
A three-electrode system was used to conduct the electrochemical measurements on an electrochemical workstation (CHI 760E). An Ag/AgCl was used as the reference electrode. A Pt wire was used as the counter electrode. The 0.5 M H2SO4 electrolyte (pH=0.3 ± 0.05) was prepared by adding 27.8 mL of sulfuric acid (95%) into a container with 500 mL of deionized water, followed by adjusting the volume to 1000 mL in a volumetric flask. The electrolyte was used immediately after preparation and stored under standard laboratory conditions. To prepare the working electrode, the catalyst ink was prepared by mixing catalyst powder (2 mg) and carbon powder (XC-72, 1 mg) with a mixture of 500 μL isopropyl alcohol, 250 μL ethanol, 220 μL H2O, and 30 μL 5% Nafion solution under ultrasonic conditions for 20 min. A glassy carbon (GC) RDE (geometric area: 0.196 cm2) of 5 mm in diameter coated with the catalyst ink served as the working electrode. 20 μL of catalyst ink was spin-coated on a GC RotatingRing-Disk Electrode. Catalyst loading (mloading) is ~0.2 mg cm−2, which was calculated according to the following equation:
where Vink is the volume of the catalyst ink. Ccatalyst is the concentration of catalyst in the ink. Aelectrode is the geometric area of the electrode.
Prior to the electrochemical tests, 10 cycles of cyclic voltammetry at 0.05 V s−1 were conducted to stabilize the performance of the catalysts. LSV was performed from 1 to 1.6 V versus Ag/AgCl at a scan rate of 0.01 V s−1 at 1600 rpm after purging O2 in the 0.5 M H2SO4 electrolyte for 20 min. LSV is measured independently three times. The solution resistance (R) of the electrochemical cell was determined by electrochemical impedance spectroscopy (EIS) at open-circuit potential. All of the potentials in LSV are corrected with 90% iR-compensation. EIS was conducted in the frequency range from 100 kHz to 0.01 Hz. The redox peaks of the catalysts were analyzed by cyclic voltammetry curve at a scan rate of 0.1 V s−1 without iR correction. The SWV profiles were recorded with a step potential of 5 mV, an amplitude of 50 mV, and a scan frequency of 50 Hz.
All potentials are referenced to the RHE by the Nernst equation:
where ERHE is the potential versus the reversible hydrogen electrode (RHE). EAg/AgCl is experimentally measured potential versus the Ag/AgCl reference electrode. Ecalibration (−0.005 V) is an additional correction determined from experimental calibration of the reference electrode (Supplementary Fig. 31).
The stability tests and the metal dissolution in the electrolyte were evaluated in an H-type electrolytic cell separated by a proton exchange membrane (Nafion 212). The volume of electrolyte in each compartment is 70 mL. An Ag/AgCl was used as the reference electrode. A Pt wire was used as the counter electrode. An amperometric i-t curve was conducted and recorded in 0.5 M H2SO4 with 1 cm2 hydrophobic carbon paper (catalyst loading: 2 mg cm−2).
Mass activity (jmass) was calculated according to the following equation:
where igeo is the geometric current (A) obtained from LSV at 1.53 V. The mloading is the loading of catalysts on the electrode, and Ru wt.% is the mass ratio of Ru in M-RuO2 catalysts.
S-number was calculated according to the following equation:
where nO2 is the number of moles of oxygen evolved during the electrolysis, and nRu, dissolved is the number of moles of Ru dissolved into the electrolyte, which is determined by ICP-MS.
Proton exchange membrane water electrolyzers test
PEMWE with anodic grid flow channel (effective area: 2 × 2 cm2) were used to test the practical applications of catalysts. 60% Pt/C as a cathode catalyst (60 mg) and Sb-RuO2 and RuO2 samples as an anode catalyst (300 mg) were first prepared as 20 mL of catalyst ink with 5 wt.% Nafion solution (750 μL for anode and 120 μL for cathode), 10 mL isopropanol, 9 mL ethanol, and 250 μL deionized water. After 30 min of ultrasonic dispersion, the mixture was further subjected to intermittent ball milling at 400 rpm for 2 h to obtain a well-dispersed catalyst ink. Then, the catalyst ink was uniformly sprayed onto both sides of the proton exchange membrane (Nafion 115) using an automated spray coater. Nafion 115 membranes (4 × 4 cm², ca. 127 µm thickness) were pretreated by boiling sequentially in 3% H₂O₂ solution for 1 h, deionized water, and 0.5 M H₂SO₄ for 3 h, followed by rinsing in deionized water. The loading amount of anodic Sb-RuO2 and RuO2 catalysts was ~3.0 mg cm−2 and cathodic 60% Pt/C was ~0.5 mg cm−2. Platinum-coated titanium felt and carbon paper were used as anode and cathode porous current collectors. The membrane electrode assembly was then hot-pressed at 4 MPa and 80 °C for 3 min. The PEMWE test was operated at 80 °C with a peristaltic pump pumping deionized water at a flow rate of 70 ml min−1. Polarization curves were obtained from 0 to 3 A cm−2 at a stepwise rate of 0.025, 0.05, and 0.1 A cm−2 by an electrochemical workstation (Henghui HLR-12H30D). A long-term stability test was conducted at 80 °C with an activated Nafion 115 proton exchange membrane.
DEMS measurements
Mass signals were measured using a Linglu mass spectrometer equipped with a Balzers QMA 200 M quadrupole (Pfeiffer). The working electrodes were prepared by sputtering Au onto 50 μm-thick porous PTFE films. The catalyst ink is drop-cast onto it. An Ag/AgCl as the reference electrode and Pt wire as the counter electrode constitute a three-electrode system. Firstly, the catalysts were labelled with 18O isotopes by performing 5 CV cycles at 1–1.35 V (versus Ag/AgCl) in 0.5 M H2SO4 dissolved in H218O. After the reaction, the electrodes are removed and rinsed with deionised water and then dried. The dried electrodes are tested in 0.5 M H₂SO₄ dissolved in H216O. Five cycles of LSV from 1.0 V to 1.4 V at a scan rate of 0.01 V s−1 are performed to measure the mass-to-charge ratio (m/z) of the five gas pulses produced. The photograph of custom-built electrochemical cells for in situ DEMS is provided in Supplementary Fig. 30.
In situ ATR-SEIRAS measurements
In situ ATR-SEIRAS was carried out with on Bruker 70 V Fourier transform infrared spectrometer with an HgCdTe (MCT) detector cooled with liquid N2. The working electrodes were prepared by magnetron sputtering Au onto the Si prism. The catalyst ink is drop-cast onto the gold ultrathin layer. The Ag/AgCl electrode and Pt wire served as the reference and counter electrodes, respectively. The 0.5 M H2SO4 solution was used as the electrolyte. Amperometric i-t is testing was used to collect spectra at different applied potentials. ATR-SEIRAS measurement was conducted at a spectral resolution of 4 cm−1 for each spectrum for 32 scans. The photograph of custom-built electrochemical cells for in situ ATR-SEIRAS measurements is provided in Supplementary Fig. 30.
Computational methods
All calculations were executed via periodic DFT utilizing the Vienna Ab-initio Simulation Package code. The projector augmented wave method was used to describe the interaction between the atomic cores and electrons. The kinetic energy cut-off for the planewave expansion was set to 400 eV. Exchange-correlation effects were approximated using the generalized gradient approximation with the Perdew-Burke-Ernzerhof functional. The convergence thresholds of the energy change and the maximum force for the geometry optimizations were set to 10-5 eV and 0.05 eV/Å, respectively. Spin polarization was considered in all calculations to account for potential magnetic effects. Van der Waals corrections were included using the DFT-D3 method as proposed by Grimme et al. Dipole corrections were applied along the z-direction to account for the asymmetry of the slab model. In this work, a rutile-phase RuO₂(110) slab model with dimensions 9.31 Å × 12.72 Å × 23.84 Å was constructed, in which the bottom three layers of atoms were fixed to simulate the bulk environment. The vacuum distance was ~15 Å between slabs to avoid undesired interactions. The Brillouin-zone integrations were sampled using a (3 × 2 × 1) Monkhorst–Pack mesh.
Data availability
The data generated in this study are provided in the Supplementary Information/Source Data file. All data supporting the findings of this study are available within the paper and its Supplementary files. Any additional information related to the study is available from the corresponding author upon request. Source data are provided with this paper.
References
Seo, D.-H. et al. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016).
McCalla, E. et al. Visualization of O-O peroxo-like dimers in high-capacity layered oxides for Li-ion batteries. Science 350, 1516–1521 (2015).
Risch, M. et al. La0.8Sr0.2MnO3−δ decorated with Ba0.5Sr0.5Co0.8Fe0.2O3−δ: a bifunctional surface for oxygen electrocatalysis with enhanced stability and activity. J. Am. Chem. Soc. 136, 5229–5232 (2014).
Jung, J.-I. et al. Optimizing nanoparticle perovskite for bifunctional oxygen electrocatalysis. Energy Environ. Sci. 9, 176–183 (2016).
Takeguchi, T. et al. Layered perovskite oxide: a reversible air electrode for oxygen evolution/reduction in rechargeable metal-air batteries. J. Am. Chem. Soc. 135, 11125–11130 (2013).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Wang, X., Zhong, H., Xi, S., Lee, W. S. V. & Xue, J. Understanding of oxygen redox in the oxygen evolution reaction. Adv. Mater. 34, 2107956 (2022).
Stoerzinger, K. A. et al. The role of Ru redox in pH-dependent oxygen evolution on rutile ruthenium dioxide surfaces. Chem. 2, 668–675 (2017).
Hao, Y. et al. Polarized ultrathin BN induced dynamic electron interactions for enhancing acidic oxygen evolution. Angew. Chem. Int. Ed. 63, e202402018 (2024).
Enman, L. J., Burke, M. S., Batchellor, A. S. & Boettcher, S. W. Effects of intentionally incorporated metal cations on the oxygen evolution electrocatalytic activity of nickel (Oxy)hydroxide in alkaline media. ACS Catal. 6, 2416–2423 (2016).
Trotochaud, L., Young, S. L., Ranney, J. K. & Boettcher, S. W. Nickel–iron oxyhydroxide oxygen-evolution electrocatalysts: the role of intentional and incidental iron incorporation. J. Am. Chem. Soc. 136, 6744–6753 (2014).
Shi, Z. et al. Customized reaction route for ruthenium oxide towards stabilized water oxidation in high-performance PEM electrolyzers. Nat. Commun. 14, 843 (2023).
Ping, X. et al. Locking the lattice oxygen in RuO2 to stabilize highly active Ru sites in acidic water oxidation. Nat. Commun. 15, 2501 (2024).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Gao, J. et al. Breaking long-range order in iridium oxide by alkali ion for efficient water oxidation. J. Am. Chem. Soc. 141, 3014–3023 (2019).
Sun, Y. et al. Covalency competition dominates the water oxidation structure–activity relationship on spinel oxides. Nat. Catal. 3, 554–563 (2020).
Wu, T. et al. Iron-facilitated dynamic active-site generation on spinel CoAl2O4 with self-termination of surface reconstruction for water oxidation. Nat. Catal. 2, 763–772 (2019).
Pan, Y. et al. Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation. Nat. Commun. 11, 2002 (2020).
Grimaud, A. et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 4, 2439 (2013).
Gao, J., Tao, H. & Liu, B. Progress of nonprecious-metal-based electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 33, 2003786 (2021).
Zhu, W. et al. Stable and oxidative charged Ru enhance the acidic oxygen evolution reaction activity in two-dimensional ruthenium-iridium oxide. Nat. Commun. 14, 5365 (2023).
Song, H. et al. RuO2–CeO2 lattice matching strategy enables robust water oxidation electrocatalysis in acidic media via two distinct oxygen evolution mechanisms. ACS Catal. 14, 3298–3307 (2024).
Deng, L. et al. Valence oscillation of ru active sites for efficient and robust acidic water oxidation. Adv. Mater. 35, 2305939 (2023).
Liu, T. et al. Rare-earth oxychlorides as promoters of ruthenium toward high-performance hydrogen evolution electrocatalysts for alkaline electrolyzers. Adv. Mater. 37, 2417621 (2025).
Wang, Y. et al. Breaking the Ru−O−Ru symmetry of a RuO2 catalyst for sustainable acidic water oxidation. Angew. Chem. Int. Ed. 63, e202316903 (2024).
Hao, Y. et al. Switching the oxygen evolution mechanism on atomically dispersed Ru for enhanced acidic reaction kinetics. J. Am. Chem. Soc. 145, 23659–23669 (2023).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium–oxygen–cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995–18006 (2023).
Liu, H. et al. Eliminating over-oxidation of ruthenium oxides by niobium for highly stable electrocatalytic oxygen evolution in acidic media. Joule 7, 558–573 (2023).
Han, N. et al. Lowering the kinetic barrier via enhancing electrophilicity of surface oxygen to boost acidic oxygen evolution reaction. Matter 7, 1330–1343 (2024).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Kuznetsov, D. A. et al. Tuning redox transitions via inductive effect in metal oxides and complexes, and implications in oxygen electrocatalysis. Joule 2, 225–244 (2018).
Laha, S. et al. Ruthenium oxide nanosheets for enhanced oxygen evolution catalysis in acidic medium. Adv. Energy Mater. 9, 1803795 (2019).
Wang, L. et al. Modulating the covalency of Ru-O bonds by dynamic reconstruction for efficient acidic oxygen evolution. Nat. Commun. 16, 3502 (2025).
Gao, T. et al. Switchable acidic oxygen evolution mechanisms on atomic skin of ruthenium metallene oxides. J. Am. Chem. Soc. 147, 4159–4166 (2025).
Miao, X. et al. Quadruple perovskite ruthenate as a highly efficient catalyst for acidic water oxidation. Nat. Commun. 10, 3809 (2019).
Hao, S. et al. Dopants fixation of Ruthenium for boosting acidic oxygen evolution stability and activity. Nat. Commun. 11, 5368 (2020).
Zhang, L. et al. Sodium-decorated amorphous/crystalline RuO2 with rich oxygen vacancies: a robust ph-universal oxygen evolution electrocatalyst. Angew. Chem. Int. Ed. 60, 18821–18829 (2021).
Lin, Y. et al. Chromium-ruthenium oxide solid solution electrocatalyst for highly efficient oxygen evolution reaction in acidic media. Nat. Commun. 10, 162 (2019).
Su, J. et al. Assembling ultrasmall copper-doped ruthenium oxide nanocrystals into hollow porous polyhedra: highly robust electrocatalysts for oxygen evolution in acidic media. Adv. Mater. 30, 1801351 (2018).
Du, K. et al. Interface engineering breaks both stability and activity limits of RuO2 for sustainable water oxidation. Nat. Commun. 13, 5448 (2022).
Zhang, J. et al. Constructing adjustable heterointerface for enhancing acidic oxygen evolution performances of RuO2@CoMnO3 nanosheets electrocatalysts. ACS Mater. Lett. 6, 3016–3024 (2024).
Jin, H. et al. Dynamic rhenium dopant boosts ruthenium oxide for durable oxygen evolution. Nat. Commun. 14, 354 (2023).
Wen, Y. et al. Stabilizing highly active Ru sites by suppressing lattice oxygen participation in acidic water oxidation. J. Am. Chem. Soc. 143, 6482–6490 (2021).
Kornienko, N. Enhancing catalysis through substitute-driven redox tuning. Joule 2, 207–209 (2018).
Mu, Y. et al. Tuning Ru–O coordination for switching redox centers in acidic oxygen evolution electrocatalysis. Angew. Chem. Int. Ed. 64, e202504876 (2025).
Suntivich, J. et al. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 3, 546–550 (2011).
Yu, Z.-Y. et al. General synthesis of tube-like nanostructured perovskite oxides with tunable transition metal–oxygen covalency for efficient water electrooxidation in neutral media. J. Am. Chem. Soc. 144, 13163–13173 (2022).
Li, J. et al. Surface evolution of a Pt–Pd–Au electrocatalyst for stable oxygen reduction. Nat. Energy 2, 17111 (2017).
Shan, J. et al. Short-range ordered iridium single atoms integrated into cobalt oxide spinel structure for highly efficient electrocatalytic water oxidation. J. Am. Chem. Soc. 143, 5201–5211 (2021).
Hu, T. et al. Covalency-dependent vibrational dynamics in two-dimensional titanium carbides. J. Phys. Chem. A 119, 12977–12984 (2015).
Li, Y. et al. Arming Ru with oxygen-vacancy-enriched RuO2 sub-nanometer skin activates superior bifunctionality for ph-universal overall water splitting. Adv. Mater. 35, 2206351 (2023).
Zhao, G. et al. Metallic Ru─Ru interaction in ruthenium oxide enabling durable proton exchange membrane water electrolysis. Adv. Mater. n/a, 2404213 (2024).
Lee, C.-Y. & Bond, A. M. Revelation of multiple underlying RuO2 redox processes associated with pseudocapacitance and electrocatalysis. Langmuir 26, 16155–16162 (2010).
Wen, Y. et al. Introducing Brønsted acid sites to accelerate the bridging-oxygen-assisted deprotonation in acidic water oxidation. Nat. Commun. 13, 4871 (2022).
Rao, R. R. et al. Towards identifying the active sites on RuO2(110) in catalyzing oxygen evolution. Energy Environ. Sci. 10, 2626–2637 (2017).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516–525 (2020).
He, W. et al. Grain-boundary-rich RuO2 porous nanosheet for efficient and stable acidic water oxidation. Angew. Chem. Int. Ed. 63, e202405798 (2024).
Wu, H. et al. Atomically engineered interfaces inducing bridging oxygen-mediated deprotonation for enhanced oxygen evolution in acidic conditions. Nat. Commun. 15, 10315 (2024).
Lin, C. et al. In-situ reconstructed Ru atom array on α-MnO2 with enhanced performance for acidic water oxidation. Nat. Catal. 4, 1012–1023 (2021).
Tao, H. B. et al. A general method to probe oxygen evolution intermediates at operating conditions. Joule 3, 1498–1509 (2019).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Fabbri, E. & Schmidt, T. J. Oxygen evolution reaction—the enigma in water electrolysis. ACS Catal. 8, 9765–9774 (2018).
Yoo, J. S., Rong, X., Liu, Y. & Kolpak, A. M. Role of lattice oxygen participation in understanding trends in the oxygen evolution reaction on perovskites. ACS Catal. 8, 4628–4636 (2018).
Hubert, M. A. et al. Acidic oxygen evolution reaction activity–stability relationships in ru-based pyrochlores. ACS Catal. 10, 12182–12196 (2020).
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J. & Nørskov, J. K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89 (2007).
Acknowledgements
This work was supported by National Key R&D Program of China (2021YFA1600800 [H.Z.], 2022YFB4002001 [Y.W.]), the National Natural Science Foundation of China (22422506 [H.Z.], 22201271 [H.Z.], 22221003 [Y.W.], 22372153 [S.H.]), the Joint Funds of the National Natural Science Foundation of China (U23A2081 [Y.W.]), the Anhui Provincial Key Research and Development Project (2023z04020010 [Y.W.]), the Anhui Provincial Natural Science Foundation (2108085UD06 [Y.W.], 2208085UD04 [H.Z.], 2208085J27 [S.H.]), the USTC Research Funds of the Double First-Class Initiative (YD2060006005 [Y.W.]), and the Joint Funds from Hefei National Synchrotron Radiation Laboratory (KY2060000180 [Y.W.]), National Program for Support of Top-notch Young Professional, Chinese Academy of Sciences Youth Innovation Promotion Association [S.H.]. We acknowledge the Experimental Centre of Engineering and Material Science at the University of Science and Technology of China. The simulations and model training were performed on the robotic AI-Scientist platform of the Chinese Academy of Sciences. We thank the photoemission end-stations BL1W1B in Beijing Synchrotron Radiation Facility (BSRF), BL14W1 in SSRF, BL10B and BL11U in the National Synchrotron Radiation Laboratory (NSRL) for the help in characterizations, and the Supercomputing Centre of the University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
Y.W., H.Z. conceived and designed the experiments. X.L., Z.W. performed the experiments. Z.C., Z.L. conducted in situ DEMS measurements. X.L., Z.W., X.M., D.H., X.W., X.Z., J.L., and G.Y. analyzed the data. P.L., X.Z. analyzed and fitted the XAFS data. J.Z., S.H., and W.L. performed DFT calculations. J.X. performed the electron microscopy characterization.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yan-Gu Lin who co-reviewed with Chun-Kuo Peng and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, X., Liu, P., Zheng, J. et al. Electronic tuning of RuO₂ polarizes metal–oxygen redox for proton exchange membrane water electrolysis. Nat Commun 16, 8709 (2025). https://doi.org/10.1038/s41467-025-63721-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63721-7
