
Abstract
N-type sulfide semiconductors are promising photocatalysts due to their broad visible-light absorption, facile synthesis and chemical diversity. However, photocorrosion and limited electron transport in one-step excitation and solid-state Z-scheme systems hinder efficient overall water splitting. Liquid-phase Z-schemes offer a viable alternative, but sluggish mediator kinetics and interfacial side reactions impede their construction. Here we report a stable Z-scheme system integrating n-type CdS and BiVO₄ with a [Fe(CN)₆]³⁻/[Fe(CN)₆]⁴⁻ mediator, achieving 10.2% apparent quantum yield at 450 nm with stoichiometric H₂/O₂ evolution. High activity reflects synergies between Pt@CrOx and Co3O4 cocatalysts on CdS, and cobalt-directed facet asymmetry in BiVO₄, resulting in matched kinetics for hydrogen and oxygen evolution in a reversible mediator solution. Stability is dramatically improved through coating CdS and BiVO4 with different oxides to inhibit Fe4[Fe(CN)6]3 precipitation and deactivation by a hitherto unrecognized mechanism. Separate hydrogen and oxygen production is also demonstrated in a two-compartment reactor under visible light and ambient conditions. This work unlocks the long-sought potential of n-type sulfides for efficient, durable and safe solar-driven hydrogen production.
Similar content being viewed by others

Introduction
Photocatalytic overall water splitting (OWS) is a promising approach to generate hydrogen from sunlight and water, offering a more direct route to solar fuels than PV-electrolysis systems1,2. However, the relatively low efficiency of photocatalytic systems under visible-light illumination remains a major challenge. Sulfide semiconductors are widely regarded as a highly promising class of photocatalysts for solar hydrogen production, owing to their broad visible-light absorption and the rich variety of highly active candidates for the water reduction half-reaction (which forms half of the OWS process)3. Despite five decades of research, single-particle sulfide photocatalysts, such as the prototypical n-type semiconductor CdS, still struggle to achieve OWS, requiring complex design strategies to enable even minimal oxygen evolution resulting in H2:O2 ratios much higher than the theoretical 2:14. Mimicking the Z-scheme systems evolved in natural photosynthesis offers a promising alternative strategy, in which the properties of catalysts for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) can be independently tuned to overcome both thermodynamic and kinetic barriers5. Nonetheless, the development of sulfide Z-scheme systems has remained challenging, with the few successful reports using p-type sulfides6,7 for which solid-state configurations are slightly more active than their liquid-phase counterparts, but still suffer from relatively low efficiencies. Theoretically, n-type metal sulfides are more promising candidates for Z-scheme photocatalysts due to their higher electron mobility, stronger visible-light absorption (vs. metal oxides8), broad compositional diversity, and simpler syntheses (vs. oxynitrides9, nitrides10,11, oxysulfides12, and p-type sulfides7,13). However, constructing a solid-state Z-scheme system using n-type sulfides as the HER photocatalyst is almost impossible, given that the OER also requires n-type semiconductors. Liquid-phase Z-scheme configurations can circumvent the preceding constraint in material selection for solid-state configurations, with reversible ion pairs acting as electron mediators to direct electron flow from the oxygen evolution photocatalyst (OEP) to the hydrogen evolution photocatalyst (HEP). Such configurations also allow spatial decoupling of HER and OER, allowing H₂ and O₂ to evolve at separate locations and thereby eliminating the problematic gas separation inherent to solid-state Z-scheme or one-step OWS systems.
The only example of a liquid-phase Z-scheme system using a p-type sulfide was reported in 201514. Although n-type sulfides offer a broader selection of materials, they have rarely been used to construct Z-scheme systems15, and, as far as is known, have never achieved high efficiency and stability. Designing active HER photocatalysts, necessary for efficient Z-scheme OWS16, is non-trivial as their performance under redox-mediated conditions remains largely unexplored3. Although sulfides exhibit excellent HER activity in the presence of strong electron donors17 (e.g., lactic acid, Na2S, and Na2SO3), CdS, a representative n-type sulfide, is remarkably inactive in solutions containing IO3−/I−, Fe3+/Fe2+, VO2+/VO2+, or [Co(bpy)3]3+/[Co(bpy)3]2+ redox couples (Supplementary Fig. 1). Innovation is thus necessary to unlock the potential of sulfide photocatalysts for liquid-phase Z-scheme OWS. The four-electron water oxidation process is kinetically more demanding, presenting greater challenges in designing OEPs5 compared to those encountered in the development of HEPs4; cocatalysts can promote OER18,19, but effective photocatalyst operation in the presence of competing redox ion pairs ultimately hinges on breakthroughs in photocatalyst materials20. Crystal facet engineering has emerged as a promising approach to expose active surfaces and enable spatially separated redox reactions on single photocatalyst particles by leveraging their structural asymmetry, achieving ~100% apparent quantum yield (AQY) for Al-doped SrTiO3 under UV illumination8. Although facet engineering can spatially separate photogenerated carriers, additional strategies are required to accelerate hole extraction—critical for alleviating sluggish OER kinetics and establishing a well-balanced Z-scheme system. Photocatalyst stability remains a persistent challenge in OWS, notably for sulfide photocatalysts wherein stable operation has eluded researchers for five decades owing to their susceptibility to hole- and oxygen-induced photocorrosion21,22. Although the use of redox pairs can facilitate hole consumption, it may also introduce additional stability challenges, as the interfacial transport, reaction kinetics, and mechanisms of ion pairs on photocatalyst surfaces—critical to Z-scheme OWS—remain poorly understood.
Here, we report an efficient and stable Z-scheme photocatalyst system for OWS, which integrates n-type CdS for HER and n-type BiVO4 for OER with a [Fe(CN)6]3−/[Fe(CN)6]4− redox mediator, enabling a high AQY of 10.2% at 450 nm and stable co-production of nearly stoichiometric hydrogen and oxygen. This liquid-phase Z-scheme system also permits the separate production of hydrogen without sacrificial agents under ambient conditions and visible light, addressing the challenge of H2/O2 separation. The high activity and stability arise from three key breakthroughs. First, the discovery of the inherent potential of CdS for HER in [Fe(CN)6]4− solution, amplified by a core-shell Pt@CrOx cocatalyst that promotes [Fe(CN)6]4− oxidation while simultaneously suppressing [Fe(CN)6]3− reduction and the undesired oxygen reduction reaction (ORR). Second, the development of a cobalt-mediated method to grow decahedral BiVO4 with additional surface-bulk asymmetry while retaining its intrinsic anisotropic and photophysical properties, delivering OER activity 5-fold higher than benchmark BiVO420. Third, the identification of a previously unrecognized catalyst deactivation mechanism through redox-pair degradation at the electrolyte/photocatalyst interface, and the implementation of a dual oxide coating strategy (i.e., TiO2 on CdS and SiO2 on BiVO4-Co) that effectively suppresses such deactivation over multiple reaction cycles. TiO2 coating of CdS also suppresses ORR, previously neglected and partly responsible for the limited progress of sulfides in OWS, and photocorrosion, owing to the stronger affinity of photogenerated holes for sulfur than for water oxidation4. This work provides a roadmap for the design of efficient, stable, and safe photocatalytic OWS systems with integrated H2/O2 separation and highlights the complex interplay between competing reaction networks in liquid-phase Z-scheme systems.
Results
High-activity sulfide photocatalyst for hydrogen evolution
We first explored whether CdS could serve as a high-performance HEP within a liquid-phase Z-scheme OWS system. Single-crystal hexagonal CdS nanoparticles (100–500 nm) with smooth surfaces were synthesized hydrothermally from Na2S and Cd(NO3)2, exhibiting an optical absorption edge of 518 nm and corresponding to a direct band gap of ~2.4 eV (Supplementary Figs. 2 and 3). These unmodified CdS nanoparticles were inactive for photocatalytic HER (<2 µmol·h−1), despite reports of activity in the presence of Na₂S/Na₂SO₃ sacrificial agents23. Surface modification of CdS with nanoparticulate Pt (0.4 wt.%), introduced through H2PtCl6 photodeposition, induced HER activity under redox-mediated conditions (Supplementary Fig. 1). Among the tested mediators, K4[Fe(CN)6] enabled a hydrogen evolution rate of 219 µmol·h−1 (Fig. 1a), far exceeding those obtained with IO3−/I−, Fe3+/Fe2+, or VO2+/VO2+ redox couples.

a Hydrogen evolution rates of CdS with different cocatalysts. The inset shows the spatial distribution of cocatalysts. Error bars indicate the standard deviations. The optimal loadings of Pt, Cr, and Co3O4 were 0.4 wt.%, 0.4 wt.%, and 0.2 wt.%, respectively (Supplementary Figs. 12–14). b Bright-field, dark-field, and HAADF-STEM images of Pt@CrOx/Co3O4 cocatalysts on the CdS surface. DF4 shows weaker Cr signals than DF2 as it collects electrons scattered at higher angles, making it more sensitive to heavier elements. c Photocurrent curves of CdS photoelectrodes with different cocatalysts under intermittent visible-light irradiation (λ ≥ 420 nm). Cathodic currents for d H+ and e [Fe(CN)6]3− reduction on CdS photoelectrodes with different cocatalysts. f Open-circuit potentials (OCPs) of Pt@CrOx/CdS photoelectrodes with and without Co3O4. Source data are provided as a Source data file.
CrOx coating of noble-metal nanoparticles supported on semiconductors has been recognized as an efficient strategy to suppress undesired back reactions in OWS by physically blocking O2 access to the noble-metal24. We explored the potential of chromia photodeposition from K2CrO4 on the Pt/CdS catalyst (Pt:CrOx mass ratio of 1:1) to tackle [Fe(CN)6]3− reduction. Bright-field and dark-field scanning transmission electron microscopy (STEM) imaging confirmed the formation of Pt@CrOx core-shell particles decorating the CdS nanocrystals (Fig. 1b and Supplementary Fig. 4). X-ray photoelectron spectroscopy (XPS) revealed that Pt was predominantly in metallic form (71.2% Pt0; 28.8% Pt2+) and chromium as Cr3+ (with only 22.6% Cr6+; Supplementary Fig. 5). Addition of the CrOx shell more than doubled the H2 productivity to 491 µmol·h−1. Considering Co3O4 is widely employed as a p-type semiconductor for photocatalytic OER25, we hypothesized that its combination with CdS might form a p–n junction, facilitating charge separation and thereby enhancing HER. Hydrothermal treatment of CdS with cobalt acetate yielded CdS decorated with Co₃O₄ nanoparticles. (Supplementary Fig. 5). Subsequent photodeposition of Pt core and the CrOx shell produced a ternary Pt@CrOx/Co3O4/CdS photocatalyst delivering H2 at 568 µmol·h−1 (Supplementary Movie 1). Note that CrOx was deposited by a chemical reduction and thus preferentially coated the electron-rich Pt nanoparticles rather than Co₃O₄, which is generally considered an oxidation catalyst. Attempts to directly deposit CrOx on CdS (using K2CrO4) were unsuccessful (Supplementary Fig. 4d) and did not alter HER activity, consistent with a previous report26, indicating that CrOx was only deposited over Pt cores (Pt@CrOx). Enhanced HER activity following Pt deposition to CdS is likely due to the formation of a metal-semiconductor heterojunction, as evidenced by the enhanced photocurrent response (Fig. 1c). However, subsequent addition of the CrOx shell lowered the photocurrent response, suggesting that the higher HER activity of Pt@CrOx/CdS was not ascribed to improved charge separation. In contrast, Co3O4 incorporation maximized the photocurrent response, attributed to the formation of a p–n junction, as confirmed by Mott-Schottky analysis showing the p-type nature of hydrothermally synthesized Co3O4 (Supplementary Fig. 6).
Unmodified CdS was essentially inert for electrocatalytic HER performed in the dark (Fig. 1d). The addition of Pt induced significant electrocatalytic activity, as evidenced by a large increase in current density, while subsequent CrOx coating further doubled the current density, akin to the synergy observed in photocatalytic HER (Fig. 1a). Although CrOx slightly downgraded the photoelectric properties of Pt/CdS (Fig. 1c), it markedly improved its electrochemical properties (Fig. 1d). The observed increase in HER activity of Pt@CrOx/CdS is therefore attributed to the promoting effect of CrOx. Pt@CrOx/CdS also exhibited a smaller Tafel slope, indicating faster HER kinetics than Pt/CdS (Supplementary Fig. 7), and suppressed [Fe(CN)6]3− reduction (Fig. 1e), which would otherwise compete with proton reduction and consume [Fe(CN)6]3− ions, thereby compromising OWS. Notably, this faster kinetics cannot be attributed to changes in catalyst morphology (surface area or porosity), metal active site density, or liquid-phase diffusion, and must instead reflect improved intrinsic catalytic activity. Encapsulation of Pt by CrOx suppressed the undesired ORR and associated water formation, as previously reported for (Rh@Cr2O3/Ga1−xZnx)(N1−xOx) for which CdS and Pt/CdS both exhibited high ORR activity under nearly neutral conditions (Supplementary Fig. 8). The open-circuit voltage of Pt@CrOx/CdS increased upon Co3O4 addition (Fig. 1f), consistent with enhanced photocurrent responses (Fig. 1c) and reflecting improved charge separation and prolonged carrier lifetime (Supplementary Fig. 9). Electrochemical impedance spectroscopy (EIS) revealed that Co3O4 was inert for [Fe(CN)6]4− oxidation to [Fe(CN)6]3-, which primarily occurred on the CdS surface (Supplementary Figs. 10 and 11). Photo- and electrocatalytic measurements indicate that the excellent HER activity of Pt@CrOx/Co3O4/CdS arises from efficient charge separation across the Pt/CdS and Co3O4/CdS interfaces, unexpected yet remarkable enhancement of HER kinetics by the Pt@CrOx core-shell structure, and suppressed ORR and [Fe(CN)6]3− reduction reactions at the Pt@CrOx interface—factors that are all critical for efficient water reduction under Z-scheme conditions.
High-activity BiVO4 photocatalyst for oxygen evolution
OER is typically the rate-determining step in photocatalytic OWS, whether driven by one-step excitation or Z-scheme systems, as it involves a four-electron process which is kinetically slower than the corresponding two-electron transfer in HER. Achieving high OEP performance in redox-mediator systems is therefore another major challenge to unlocking the potential of CdS-based Z-scheme systems, which is also a common critical challenge for photocatalytic water splitting. Of possible visible-light-responsive OEPs, BiVO4 and WO3 are the most promising with band gaps of 2.4 eV and 2.7 eV, respectively. The narrower-band gap BiVO4 was selected to construct our Z-scheme OWS system. Decahedral BiVO4 nanoparticle modified with Ir/IrO2 cocatalyst represents the most active OEPs in K3[Fe(CN)6] reported to date, owing to the anisotropic exposure of (010) and (110) facets that facilitate spatial charge separation (Fig. 2c)19,27. However, despite improving HER activity by 2.6-fold, the combination of highly active HEP (Pt@CrOx/Co3O4/CdS) and OEP (Ir/IrO2/BiVO4) to construct a liquid-phase Z-scheme OWS system still failed, as evidenced by rapid activity decay with in two cycles (68% for HER, 73% for OER) and a severely off-stoichiometric H2/O2 ratio of 4.3 (Fig. 2a and Supplementary Fig. 15). The latter indicates the insufficient OER kinetics and a significant mismatch between reduction and oxidation rate of [Fe(CN)6]3−/4−, as further supported by photocatalytic studies of [Fe(CN)6]4−/[Fe(CN)6]3− redox chemistry over the two photocatalysts (Supplementary Figs. 16–18). Therefore, to achieve more balanced HER and OER rates, an alternative strategy is urgently required to enhance the OER activity of benchmark decahedral BiVO4 by at least threefold (Supplementary Fig. 16). Doping heteroatoms into facet-engineered BiVO4 could potentially further promote its OER activity, but may adversely affect thermodynamic and kinetic properties of the photocatalyst (by disrupting the decahedral configuration, altering the band structure, or introducing recombination centers) and thereby compromise performance. We thus explored alternative strategies to promote OER activity in decahedral BiVO₄ nanoparticles.

a Impact of HEP and OEP half-reaction performance on activity loss and the H2 to O2 ratio in the Z-scheme reaction. Upward arrows indicate superior HER or OER activity. b Crystal structure of monoclinic scheelite BiVO4. c Modeled structures of decahedral BiVO4 (110) surfaces with and without adsorbed Co. Under illumination, photogenerated electrons and holes will flow towards the (010) and (110) facets, respectively. d Oxygen evolution rates of BiVO4 and BiVO4-Co in Fe(NO3)3 and K3[Fe(CN)6] solutions. Error bars indicate the standard deviations. For K3[Fe(CN)6], an Ir/IrO2 cocatalyst was required to achieve appreciable activity. Cocatalyst optimization and spatial distribution over BiVO4 are shown in Supplementary Figs. 35 and 36. e Refined XRD patterns of BiVO4 and BiVO4-Co. Soft X-ray absorption spectra at f the O K-edge and g V L-edge of BiVO4 and BiVO4-Co in TEY and TFY modes. TAS spectra showing decay kinetics of photogenerated h holes and i electrons in BiVO4 and BiVO4-Co after band gap excitation (fluence: 0.3 mJ·pulse−1, repetition rate: 1 Hz). Source data are provided as a Source data file.
Decahedral monoclinic BiVO4 nanoparticles were first modeled using density functional theory (DFT) calculations. Surfaces were constructed with Bi and V coordinated to eight and four O atoms, respectively, forming distorted BiO8 dodecahedra and VO4 tetrahedra (Fig. 2b). The influence of Co (a common cocatalyst in OWS) substitution or adsorption was subsequently examined. Isomorphic substitution of Co for either Bi or V in the (010) or (110) surfaces of monoclinic BiVO4 was found to be thermodynamically unfavorable (Supplementary Fig. 19 and Supplementary Table 1). However, Co adsorption on the (110) surface of BiVO4 crystals (during hydrothermal synthesis) is thermodynamically favored (−4.58 eV) and induced significant elongation of selected Bi–O bond lengths (Fig. 2c and Supplementary Table 2). Such elongation (surface strain) is anticipated to enhance the OER reactivity of BiVO₄ nanoparticles by amplifying the anisotropic electric field between single-crystal facets, through induced asymmetry between surface and bulk structures. To test this hypothesis, the hydrothermal synthesis of decahedral BiVO4 was performed with and without Co(NO3)2 (2 at.% equivalent, yielding BiVO4-Co), and the resulting OER activities were assessed using K3[Fe(CN)6] and Fe(NO3)3 redox mediators (Fig. 2d). In both cases, decahedral BiVO4 single crystals with lengths of 0.5–2 µm were obtained (Supplementary Fig. 20) in a monoclinic scheelite phase. Rietveld refinement of powder X-ray diffraction (XRD) patterns revealed very similar (but not identical) lattice constants and Bi–O and V–O bond lengths for BiVO4 and BiVO4-Co (Fig. 2e and Supplementary Fig. 21). Despite these bulk structural similarities, the BiVO4-Co photocatalyst exhibited a 4- and 5-fold increase in OER activity compared with benchmark BiVO420 when using Fe(NO3)3 and K3[Fe(CN)6] as electron acceptor, respectively (Fig. 2d). Oxygen productivity exhibited a volcano relationship with Co addition during synthesis, with 2 at.% Co proving optimal (~170 µmol·h−1, Supplementary Fig. 22).
Extensive bulk and surface characterization were subsequently conducted to elucidate the role of Co. Elemental analysis by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (Supplementary Table 3) and XPS (Supplementary Fig. 23) failed to detect any Co. Synchrotron resonant Auger electron spectroscopy (RAS)—a highly surface-sensitive and element-specific technique—further confirmed the absence of Co in the BiVO4-Co selvedge (Supplementary Fig. 24). This observation is consistent with XPS analysis of Bi, V and O core-levels, whose spectra are essentially identical for BiVO4 and BiVO4-Co (Supplementary Fig. 23). The preceding analyses were repeated multiple times to eliminate possible artefacts. Within experimental error, negligible Co existed in either the bulk (<1 ppb) or surface (<0.01 wt.%) of BiVO4-Co, indicating that Co was neither doped into nor adsorbed on BiVO4. Having excluded these alternatives, we conclude that Co can serve as a structure-directing agent to regulate the atomic arrangement of nascent BiVO4 crystals through reversible adsorption and subsequent displacement by Bi and V cations as the crystals grow, without remaining in the final OEP.
Insight into the influence of Co on BiVO4 synthesis was derived from soft X-ray absorption spectroscopy (sXAS) at the O K-edge of BiVO4 for photon energies between 532 eV and 536 eV, which excite electronic transitions into the Bi 6p orbital (Fig. 2f)28. The total fluorescence yield (TFY) spectra of BiVO4 and BiVO4-Co were almost identical; however, the more surface-sensitive total electron yield (TEY) spectra highlighted significant differences in the surface geometric/electronic structure of BiO8 dodecahedra, aligning well with the RAS spectra (Supplementary Fig. 25). In contrast, V L-edge TFY and TEY spectra of BiVO4 and BiVO4-Co were identical (Fig. 2g), indicating that the V local environment was the same in both photocatalysts. Raman spectroscopy, performed under 532 nm and 785 nm excitation to obtain local structural information from varying sample depths, and Fourier transform infrared spectroscopy (FTIR), neither of which are surface sensitive, could not distinguish between BiVO4 and BiVO4-Co, with both exhibiting identical vanadium species (Supplementary Fig. 26). In Raman spectra, bands at 324 cm−1 and 366 cm−1 are ascribed to VO43− tetrahedron while those at 640 cm−1, 710 cm−1, and 826 cm−1 correspond to V–O bonds. Oxygen vacancies are often invoked based on XPS data to explain changes in photo- (and electro-) catalytic activity, despite the difficulty in their reliable assignment from binding energies29,30. To assess the potential influence of Co on oxygen vacancy formation, we therefore analyzed BiVO4 and BiVO4-Co by XPS and the more reliable electron spin resonance (ESR). Neither analysis identified any significant differences in oxygen (or bismuth or vanadium) chemical environment between the samples (Supplementary Fig. 23a–c), or the presence of unpaired electrons in either sample (Supplementary Fig. 27), and hence discounts any contribution from oxygen vacancies to the enhanced OER performance of BiVO4-Co versus BiVO4.
The only difference between BiVO4 and BiVO4-Co lies in the presence of a low concentration (0.1 mmol) of Co(NO3)2·6H2O during the hydrothermal synthesis; therefore, the improved electrochemical properties of BiVO4-Co must arise from the influence of cobalt acting as a structure-directing agent during crystal growth. To better understand this process, the nature of precursors formed prior to the hydrothermal step was investigated. Mixing Bi(NO3)3, Co(NO3)2, NH4VO3, HNO3, NaOH, and acetic acid resulted in spherical tetragonal BiVO4 nanoparticles, which transformed into the decahedral monoclinic BiVO4 during hydrothermal treatment (Supplementary Fig. 28). Elemental analysis by ICP-AES of the precipitate and supernatant from the precursor suspension revealed that cobalt was abundant in the supernatant but present only at trace levels in the tetragonal BiVO₄ precipitate (Supplementary Table 4). Together, these results indicate that cobalt perturbs the tetragonal→monoclinic phase transition during hydrothermal synthesis rather than being incorporated into the BiVO4 precursor. Ion-regulated crystal growth has been reported previously—for example, F− ions regulate the exposed facets of TiO₂ photocatalysts through a similar adsorption-mediated process31.
Further insights into the band structures of BiVO4 and BiVO4-Co were obtained from UV-visible diffuse reflectance spectroscopy (DRS) and ultraviolet photoelectron spectroscopy (UPS). The absorption characteristics and corresponding Tauc plots of both OEPs were almost identical (Supplementary Fig. 29), with band gaps of ~2.48 eV. Although the conduction band minimum and valence band maximum of BiVO4-Co were shifted by −0.03 eV relative to BiVO4, this small difference (compared to the large overpotential for water photooxidation) is unlikely to explain their differing OER activity32. Transient absorption spectroscopy (TAS) of photocarriers in BiVO4 and BiVO4-Co, measured across a range of 500–1667 nm (2.48–0.74 eV, Supplementary Fig. 30), exhibited broad absorption bands at 2.48–1.90 eV and 1.90–0.74 eV, consistent with charge carriers trapped at intrinsic defect states, albeit with different relative intensities. Band assignments can be made based on additional insight from the lifetime of photoexcited charge carriers through their decay kinetics (Fig. 2h, i). Absorption signals at 526 nm and 1667 nm were attributed to holes and electrons, respectively, based on their decay behavior following CoOx decoration, which has been reported to promote hole capture and enhance charge separation33,34. The TA signal at 526 nm decayed faster upon CoOx addition, indicating efficient hole transfer to CoOx. By contrast, the signals at 769 nm and 1667 nm exhibited slower decay in the presence of CoOx, confirming their assignment to photoexcited electrons. Normalized decay profiles of photoinduced charge carriers in BiVO4-Co with and without CoOx nanoparticles (known to efficiently capture photogenerated holes) are also shown in Supplementary Fig. 31. The decay profiles of holes and electrons were fitted by triple- and double-exponential functions according to the following Eq. (1):
where i = 2 or 3 for double- or triple-exponential functions, τi represents the lifetimes of the photoexcited carriers during complex and competing relaxation processes such as electron-hole recombination and charge carrier trapping (Supplementary Table 5). As previously reported for BiVO433,35, photoexcited electrons decayed faster than holes, indicating that electrons were rapidly trapped at intrinsic defects in BiVO4, thereby extending the lifetime of photoexcited holes. Characteristic lifetimes (τi) for holes were τ1 = 2.84 μs, τ2 = 28.7 μs, and τ3 = 347.5 μs in BiVO4, and τ1 = 2.81 μs, τ2 = 29.6 μs, and τ3 = 242.2 μs in BiVO4-Co. Holes in BiVO4-Co decayed faster than in BiVO4 (Fig. 2h), which is attributed to faster hole transfer from the bulk to (more distorted BiO8) surface, where photoexcited holes accumulate. In contrast, the decay of photoexcited electrons in BiVO4-Co was slower than in BiVO4 (Fig. 2i), indicating spatial separation of electrons and holes. However, due to the competing electron trapping process33,35, the effect of Co on electron lifetime was less significant (Supplementary Fig. 32): τ1 = 0.64 μs, τ2 = 15.4 μs for BiVO4-Co versus τ1 = 0.56 μs, τ2 = 13.4 μs for BiVO4. BiVO4-Co also displayed a larger photocurrent response and lower charge transfer resistance (Rct) than BiVO4 (Supplementary Fig. 33), evidencing superior charge separation and transfer in the former that likely contribute to the improved OER performance. Kelvin probe force microscopy (KPFM) further revealed a larger potential difference (~3×) between the (110) and (010) facets of BiVO4-Co compared with BiVO₄, a potential gradient that facilitates directional migration of electrons and holes to distinct facets and thereby enhances OER activity (Supplementary Fig. 34). Distorted surface Bi–O bonds in BiO8 dodecahedra, induced by Co ions transiently adsorbed during BiVO4 synthesis, perturb the unoccupied surface electronic states and thus promote OER, without altering the intrinsic anisotropic and photophysical properties (composition, bulk structure, or crystallinity). The advance of prediction and demonstration of surface-bulk additional asymmetry in anisotropic crystals presented here provides a widely applicable and simple strategy to further promote charge separation and transfer, crucial not only for solar fuels but also for applications in photovoltaics and photodetectors.
Stability enhancement by oxide encapsulation
Combining the optimized HEP and OEP with an equimolar concentration of [Fe(CN)6]4−/[Fe(CN)6]3− mediators (Supplementary Figs. 16 and 37) and appropriate sodium phosphate buffer resulted in approximately two- to threefold enhancement in H2 and O2 productivities (18.0–48.7 µmol·h−1, 4.2–17.3 µmol·h−1), an improved H2:O2 molar ratio much closer to the theoretical value of 2 (4.3–2.8), and mitigated deactivation of both H2 and O2 evolution reactions (74%–47%, two cycles) (Fig. 2a and Supplementary Fig. 38). Phosphate buffers, widely employed in Z-scheme water-splitting systems using [Fe(CN)6]4−/[Fe(CN)6]3− as mediators19,36, proved essential for sustaining high and stable rates of H₂ evolution (Supplementary Fig. 39a), while also yielding trace O₂ from the limited conversion of [Fe(CN)₆]⁴⁻ to [Fe(CN)₆]³⁻ at the HEP. Control experiments at different pH identified the consumption of protons during HER as responsible for a loss in HER activity, with rates declining from 28.7 μmol·h−1 at pH 6.8 to 0.1 μmol·h−1 at pH 11 (Supplementary Fig. 39b). Note that in a Z-scheme reaction, sustained OWS requires matched H2 and O2 evolution rates to maintain a stable concentration of redox mediator. In the present system, this necessitated an HEP:OEP mass ratio of 1:5 to balance the higher HER and slower OER rates (Supplementary Fig. 37). Despite the preceding performance improvements obtained during OWS, both HER and OER still exhibited poor stability, with H2 and O2 productivity decreasing by ~58% and 60%, respectively, after three reaction cycles (Fig. 3a, d). Such instability is commonly observed for sulfide photocatalysts37, which are prone to photocorrosion via reactions with photoexcited charge carriers38,39. We speculated that photocorrosion might be suppressed by coating the sulfide HEP with oxides21. Provided such oxide layers are sufficiently thin, they might still permit charge transfer to the aqueous solution while preventing the direct chemical reaction of oxygen or oxygen-containing radicals with the underlying CdS.

Stability of H2 and O2 production over a uncoated HEP (Pt@CrOx/Co3O4/CdS) and OEP (Ir/IrO2/BiVO4-Co), b TiO2-coated HEP with uncoated OEP, and c TiO2-coated HEP with SiO2-coated OEP. d Catalyst deactivation over four reaction cycles. e Long-term cycling stability over TiO2-coated HEP and SiO2-coated OEP. f Apparent quantum yields of Z-scheme system using TiO2-coated HEP and SiO2-coated OEP, together with DRS curves of uncoated CdS and BiVO4-Co. Error bars indicate the standard deviations. Source data are provided as a Source data file.
Wet chemical syntheses were employed to deposit oxide layers onto the optimal Pt@CrOx/Co3O4/CdS. TiO2 and Ta2O5 were coated by photodeposition, while SiO2 was introduced through hydrolysis. Recycling studies revealed that the coating of TiO2 substantially mitigated deactivation of the HEP, with only a 12% loss of H2 production versus 25% for the uncoated system (Supplementary Fig. 40). In contrast, Ta2O5 and SiO2 coatings were less effective at stabilizing the HEP, each showing ~20% activity loss. Similar trends were observed for O2 production, with only a 5% decrease for the TiO2-coated Pt@CrOx/Co3O4/CdS (versus 19% for the uncoated system), and Ta2O5 and SiO2 coatings were less effective. Notably, coating of the HEP not only improved stability but also enhanced OWS, with TiO2 imparting a 27% increase in H2 production and 67% increase in O2 production. (Supplementary Fig. 40). Control experiments confirmed that improved HER activity was only observed in the Z-scheme OWS system, highlighting the synergy between HEP and OEP, and confirming that evolved O2 did not arise from decomposition of the oxide coating (Supplementary Fig. 41). Optimization of the TiO2 loading further improved OWS stability and activity (Supplementary Fig. 42). A 3 wt.% TiO2 coating on Pt@CrOx/Co3O4/CdS more than halved deactivation over two consecutive reaction cycles, and increased HER and OER activity in the first cycle by 52% and 98%, respectively. Recycling tests still showed a net 30% and 25% decline in H2 and O2 productivity, respectively (Fig. 3b).
Although BiVO4 is generally regarded as a stable OER photocatalyst39, the multiple benefits observed from coating the HEP prompted us to apply the same methodology to the optimized Ir/IrO2/BiVO4-Co (Fig. 3). Of the oxide coatings above, a 5 wt.% SiO2 layer proved the most effective in mitigating OWS deactivation when combined with 3 wt.% TiO2-coated Pt@CrOx/Co3O4/CdS (Supplementary Fig. 43). Over four reaction cycles, the dual-coated OWS system exhibited only a 7.5% fall in H2 production and a 3.3% fall in O2 production, representing ~8- and 18-fold improvements in stability compared to the uncoated system (Fig. 3c, d). Note that the SiO2 coating of Ir/IrO2/BiVO4-Co did not enhance its O2 productivity (Supplementary Fig. 44). The stability of this dual-coated system was further validated through long-term cyclic testing. As shown in Fig. 3e, the overall activity remained essentially stable over 40 h of cyclic testing, with only a 15% decline in H2 and O2 productivity after 112 h (Supplementary Fig. 45a). This durability was corroborated by ICP-AES results that showed an initial period of Cd and S leaching after which a stable system was attained (Supplementary Fig. 45b). Partial activity loss may be due to the photocatalyst powder adhering to the reactor walls and the stirrer (Supplementary Fig. 46), which can be partially recovered by ultrasonication (the fourth cycle in Fig. 3e), indicating the importance of minimizing catalyst loss especially for long-term cycling. The solar-to-hydrogen (STH) conversion efficiency of the dual-coated OWS system reached 0.47% (Supplementary Table 6). The corresponding AQYs were 9.5% at 420 nm and 10.2% at 450 nm (corresponding to maximal absorption from the DRS curves), respectively (Fig. 3f), which compare favorably with reports for OWS using metal chalcogenide photocatalysts (Supplementary Table 7). The AQY remained 3.9% at 500 nm, confirming a broad visible-light response. Control experiments discounted possible reactions of H2 or O2 with the redox mediator, or their recombination in the dark in the presence of oxide-coated HEP and OEP (Supplementary Fig. 47).
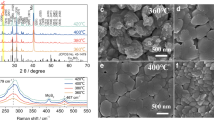
The nature of the oxide coatings and their possible modes of action were further investigated. TiO2 coating of Pt@CrOx/Co3O4/CdS nanocrystals produced an amorphous overlayer of uneven thickness (Fig. 4a and Supplementary Fig. 48) that increased the specific surface area from 5.4 to 8.6 m2·g−1 (Supplementary Fig. 49). The higher surface area of TiO2-coated Pt@CrOx/Co3O4/CdS is expected to enhance HER activity. sXAS indicated that the TiO2 overlayer was amorphous (Supplementary Fig. 50) and sufficiently thick to strongly attenuate core-level X-ray photoemissions from the Pt, CrOx, and particularly Co3O4 cocatalysts (Supplementary Fig. 51). The mean thickness of the TiO2 layer was estimated at ~1 nm based on attenuation of the underlying CdS signal (Supplementary Fig. 52). HRTEM and elemental mapping of the SiO2-coated Ir/IrO2/BiVO4-Co revealed a uniform SiO2 overlayer of ~2.8 nm thickness (Fig. 4a, Supplementary Figs. 53 and 54). Both coated HEP and OEP retained their crystallinity after Z-scheme reaction (Fig. 4b and Supplementary Fig. 55), consistent with their catalytic stability (Fig. 3e). However, a new reflection emerged at 17.5°, the intensity of which was suppressed by oxide coatings. This reflection is attributed to Fe4[Fe(CN)6]3 (Supplementary Fig. 56), formed during Z-scheme reaction, and accounts for the appearance of small cubic particles on uncoated HEP and OEP (Fig. 4c). Elemental mapping confirmed that these smaller particles were primarily comprised of Fe, C, and N (Supplementary Figs. 57 and 58) and the characteristic vibrational bands of C≡N bonds from Fe4[Fe(CN)6]3 were also detected in the Raman spectra of post-reaction samples (Supplementary Fig. 59)40. Prussian blue Fe4[Fe(CN)6]3 and Turnbull’s blue Fe3[Fe(CN)6]2 possess similar crystal structures, rendering them difficult to distinguish by XRD alone, and similar chemical compositions. Their principal difference lies in the FeII:FeIII atomic ratio, which is 0.75 for Fe4[Fe(CN)6]3 and 1.5 for Fe3[Fe(CN)6]2 (Supplementary Table 8). Additional XPS analysis of the FeII:FeIII atomic ratio in these nanoparticles revealed a value of ~0.75, characteristic of Fe4[Fe(CN)6]3 and hence consistent with Prussian blue (Supplementary Fig. 56b). We hypothesize that Prussian blue nanoparticles form from Fe2+/Fe3+ ions liberated at strong acid or base sites41, which dissociate [Fe(CN)6]4−/[Fe(CN)6]3− complexes on the catalyst surface during HER and OER. Such reaction pathways are summarized below from previous literature42:

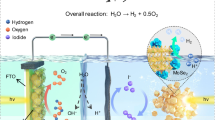
a TEM images of TiO2-coated Pt@CrOx/Co3O4/CdS and SiO2-coated Ir/IrO2/BiVO4-Co. b Post-reaction XRD patterns of different HEPs used in the Z-scheme reaction. c Post-reaction SEM images of HEP and OEP with and without oxide coating used in half-reactions. d Effect of different oxide coatings on sulfur leaching during OWS determined by ICP-AES. e Photocatalytic oxygen reduction over HEPs with and without TiO2 coatings. f Schematic of Z-scheme overall water splitting using optimized HEP (TiO2-coated Pt@CrOx/Co3O4/CdS) and OEP (SiO2-coated Ir/IrO2/BiVO4-Co), and the corresponding setup for H2 and O2 separation. g Separated H2 and O2 production in a two-compartment OWS reactor (inset) under visible light. Source data are provided as a Source data file.
A key role of oxide coatings thus appears to be the suppression of Fe4[Fe(CN)6]3 precipitates that would otherwise lower the concentration of redox mediator required for OWS (Supplementary Figs. 60 and 61). TiO2 coating of the optimized HEP proved the most effective method to suppress Fe4[Fe(CN)6]3 precipitation, in accordance with XRD analysis (Supplementary Fig. 62), and also effectively prevented CdS photocorrosion that would otherwise lead to sulfur leaching (Fig. 4d). Fe4[Fe(CN)6]3 precipitation is likely the major deactivation pathway in our Z-scheme system, as this was also observed for the uncoated OEP (which is not prone to photocorrosion); SiO2 coating on the OEP similarly suppressed this precipitation (Fig. 4c) and markedly improved the stability of Z-scheme OWS (Fig. 4d). The high initial activities of uncoated HEP and OEP reflect faster reaction kinetics at active sites on their pristine surfaces, which become progressively decorated with Fe4[Fe(CN)6]3 as Z-scheme reaction proceeds. The TiO2 coating plays an additional role in suppressing photocatalytic and electrocatalytic ORR (Fig. 4e and Supplementary Fig. 63) over CdS under illuminated and dark conditions, respectively, where ORR is otherwise efficient (Supplementary Fig. 8). This accounts for the improvement in OER activity following coating of the HEP with TiO2 (Fig. 3b), with the additional photoexcited electrons thereby available to drive HER. Therefore, dual-coating effectively suppresses multiple side reactions at HER and OER photocatalyst-electrolyte interfaces, including traditional-recognized photocorrosion, ORR, and a previously unrecognized but crucial redox-shuttle degradation process that may occur universally in liquid Z-scheme systems employing metal-complex mediators, highlighting the broad applicability of the dual-coating strategy (Fig. 4f).
The preceding advances open the possibility of integrating the optimized HEP and OEP into a two-compartment OWS system to circumvent safety and separation issues associated with mixed oxyhydrogen production in single-particle, solid-state Z-scheme systems (Fig. 4g). A two-compartment reactor was fabricated to isolate HEP and OEP in separate chambers connected by a nylon membrane filter (Pore size:10 μm, Delvstlab) permeable to the redox mediator (Fig. 4g inset). A pure stream of H2 was obtained from the HEP compartment with a notable steady state productivity of ~10 μmol·h−1 under visible light and ambient pressure (Fig. 4g), accompanied by a corresponding pure O2 stream from the OEP compartment. Oxygen and hydrogen productivities were approximately in a 1:2 molar ratio. We believe that this represents a leading performance for OWS photocatalysts, achieving spatially separated H2 and O2 production using redox shuttles in a flow-through configuration43.
Discussion
An n-type sulfide (CdS), which is generally considered unstable and rarely used for OWS, is demonstrated as a promising candidate for constructing a redox-driven Z-scheme OWS system with high efficiency and stability under visible light. High efficiency was achieved by promoting both the HEP (CdS) and the OEP (BiVO4) half- reactions in conjunction with liquid-phase redox mediators ([Fe(CN)6]3−/[Fe(CN)6]4−). For CdS, CrOx-coated Pt and Co3O4 cocatalysts accelerate HER kinetics, suppress undesired reactions, and facilitate efficient charge carrier separation. For BiVO4, hydrothermal synthesis in the presence of cobalt cations perturbs Bi–O bonding on the (110) facets of the decahedron while retaining both anisotropic and photophysical properties, thereby enhancing photoexcited hole transfer from the bulk to surface and consequent OER of benchmark BiVO4 by fivefold. High stability was achieved through a dual-coating strategy to inhibit the precipitation of Fe4[Fe(CN)6]3 over both the HEP and OEP—a previously unreported phenomenon identified as a major cause of deactivation in photocatalytic OWS. The synergies between photocatalysts, cocatalysts, and coatings resulted in an excellent AQY of 10.2% at 450 nm for OWS using metal chalcogenide photocatalysts, as well as separated hydrogen production under visible light and ambient pressure in a two-compartment reactor. The latter circumvents downstream separation of H2/O2 mixtures, a critical bottleneck hindering the commercialization of conventional photocatalytic OWS systems. This work unlocks the inherent potential of n-type sulfides as active and stable photocatalysts for OWS. The combination of our active and stable photocatalysts in a Z-scheme configuration provides opportunities to overcome oxyhydrogen safety concerns and gas-separation challenges by directly separating hydrogen and oxygen production at their source.
Methods
Synthesis of CdS single crystals and BiVO4-Co
CdS single crystals were synthesized by a conventional hydrothermal method23. First, 50 mmol Na2S·9H2O (≥98.0%, Aladdin) was completely dissolved in 25 mL pure water, followed by the addition of 10 mmol Cd(NO3)2·4H2O (99.0%, Aladdin) to form an orange slurry under continuous stirring at room temperature. Subsequently, the slurry was transferred into a 50 mL Teflon-lined stainless-steel autoclave and maintained at 473 K for 20 h in an oven after stirring for 1 h. Finally, after cooling the stainless-steel autoclave to room temperature naturally, the resulting orange precipitate was collected by centrifugation, washed several times with ethanol and pure water, and then dried at 333 K under vacuum overnight before use.
BiVO4-Co was synthesized through a one-step hydrothermal method. Typically, 5 mmol Bi(NO3)3·5H2O (≥99.0%, Adamas) and 0.1 mmol Co(NO3)2·6H2O (≥99.9%, Aladdin) were dissolved in 20 mL 2 M HNO3 (65–68%, Sinopharm) solution, labeled as solution A; then 5 mmol NH4VO3 (99.9%; Macklin) was dissolved in 20 mL 2 M NaOH (99.9%, Aladdin) solution, labeled as solution B. Next, Solution B was slowly added to solution A and stirred for 0.5 h to form a yellow suspension. After that, 3 mL of acetic acid was added to the above suspension, followed by 1 h of continuous stirring. Finally, the suspension was transferred into a 50 mL Teflon-lined stainless-steel autoclave and maintained at 473 K for 24 h in an oven. After the stainless-steel autoclave was cooled to room temperature naturally, the resulting yellow precipitate was collected by filtration, washed with pure water several times, and then dried at 333 K in air overnight. The synthesis procedure for BiVO4 was identical to that for BiVO4-Co except for solution A without the addition of Co(NO3)2·6H2O.
Deposition of cocatalysts
Before the photocatalytic water splitting reaction, Pt@CrOx/Co3O4 and Ir/IrO2 cocatalysts were deposited onto CdS as the HEP and BiVO4-Co as the OEP, respectively. For the HEP, Co3O4 was first loaded on CdS through a facile hydrothermal method. Typically, 200 mg of CdS was added into 25 mL of ethanol solution and sonicated for 15 min. Then, 100 μL of 50 mM cobalt acetate (99.9%, Aladdin) aqueous solution and 0.4 mL of ammonium solution were sequentially added into the above suspension with each stirred for 15 min. Finally, the suspension was transferred into a 50 mL Teflon-lined stainless-steel autoclave and maintained at 398 K for 1 h. After the stainless-steel autoclave was cooled naturally to room temperature, the products were collected by filtration, washed several times with ethanol and pure water, and dried at 333 K under vacuum overnight, denoted as Co3O4/CdS. Subsequently, the Pt@CrOx core/shell cocatalyst was further loaded on Co3O4/CdS by a two-step photodeposition process44. The core of Pt was first photodeposited on Co3O4/CdS using H2PtCl6·6H2O (Pt: ≥37.5%, Sinopharm) as the metal precursor. The suspension was evacuated to completely remove dissolved air and then exposed to visible light (λ ≥ 420 nm) under continuous stirring for 3 h. In the second step, the shell of CrOx was formed on the surface of the Pt core. The mass ratio of the CrOx shell to the Pt core was maintained at 1:1. K2CrO4 (99.9%, Macklin) was added to the above solution as a Cr6+ precursor without pH adjustment. After complete degassing, the suspension was further irradiated with visible light (λ ≥ 420 nm). After 3 h of irradiation, the photocatalyst was collected by filtration, washed several times with pure water, and dried at 333 K under vacuum overnight, denoted as Pt@CrOx/Co3O4/CdS. The deposition of Pt and Pt@CrOx on CdS was similar to that of Pt@CrOx/Co3O4.
For the deposition of Ir/IrO2 on BiVO4-Co photocatalyst19, 0.2 g of BiVO4-Co was immersed in a calculated K2IrCl6 (Ir: >39%, Adamas) aqueous solution with ultrasonic agitation for 5 min. After athe bove suspension was completely evaporated in a water bath at 353 K, the resulting powder was thoroughly ground and then reduced at 573 K for 1.5 h under a flow of 3% H2/N2 (200 mL·min−1). After dispersing all above powder in sodium phosphate buffer solution (30 mM, pH = 6.3 ± 0.2) containing K3[Fe(CN)6] (0.2 mM; ≥99.5%, Aladdin), the suspension was evacuated to completely remove dissolved air and then irradiated with full-spectrum light (λ ≥ 300 nm) under continuous stirring to convert deposited Ir species into metallic Ir on the (010) facet and IrO2 on the (110) facet. After 2 h of irradiation, the photocatalyst was collected by filtration, washed several times with pure water, and dried at 333 K under vacuum overnight, denoted as Ir/IrO2/BiVO4-Co.
Oxide coating of HEP and OEP
Oxides were coated on photocatalysts through the photodeposition (or hydrolysis) of the desired metal precursor45,46. Titania was deposited by ultrasonic mixing 20 μL of titanium tetraisopropoxide (95%, Aladdin) and 70 μL of H2O2 (≥30%, Sinopharm) in 1 mL of deionized water for several minutes until a transparent yellow solution was obtained. The resulting Ti-containing peroxide solution was then added to 100 mg of the desired HEP, and the suspension was degassed and irradiated with visible light (λ ≥ 420 nm) for 3 h. The slurry was subsequently filtered, washed repeatedly with ethanol and deionized water, and dried at 333 K under vacuum overnight to obtain an orange-yellow powder. Tantalum pentoxide was similarly deposited on the HEPs from a (in-situ generated) peroxide precursor. Specifically, 4 mg of TaCl₅ (>95%, Aladdin) and 250 μL of H₂O₂ were mixed, to which 56.5 μL of 1 M NaOH (≥98%, Aladdin) solution was added, and the mixture was briefly sonicated to yield a colorless transparent solution. All this solution was added to 50 mg of the photocatalyst, and the suspension was degassed, irradiated, filtered, and dried as described above. Silica deposition was performed by a hydrolysis method: 100 mg of the HEP or OEP and 100 μL of 0.1 M NaOH solution were added to 20 mL of ethanol and sonicated for 5 min, to which 19 μL of tetraethyl orthosilicate (TEOS, >99%, Aladdin) was added. The suspension was sonicated for 30 min and then stirred at room temperature for 24 h to hydrolyze the TEOS. The resulting solid product was filtered and dried at 333 K under vacuum overnight.
Characterization of catalysts
Powder XRD patterns of the catalyst were measured with an X-ray diffractometer (Mini Flex 600, Rigaku, Japan; D8 Advance Da Vinci, Bruker, Germany) using a Cu Kα source. XRD Rietveld refinement analysis was performed using the GSAS-Ⅱ program to further determine the crystal structure variation. Diffuse reflectance spectra (DRS) were obtained using an UV-visible spectrophotometer (UV-2450, Shimadzu, Japan) and were converted from reflectance to absorbance by the Kubelka-Munk function. XPS and UPS were conducted on an X-ray photoelectron spectrometer (AXIS UltraDLD, Kratos, UK). All binding energies were referenced to the C 1s peak at 284.6 eV. Transmission electron microscopy (TEM) and STEM images, as well as energy-dispersive X-ray spectroscopy mapping, were recorded using a field-emission transmission electron microscope (TALOS F200X, FEI, USA). The morphology of catalysts was observed with a field-emission scanning electron microscope (RISE-MAGNA, Tescan, Czech Republic). KPFM images were obtained using an atomic force microscope (MFP-3D, Asylum Research, USA). Nitrogen adsorption-desorption isotherms were conducted using a surface area and porosity analyzer (ASAP 2046, Micromeritics, USA) at 77 K after degassing the catalysts at 423 K for 4 h. The specific surface area was determined by the Brunauer-Emmett-Teller method. Raman spectra were collected on a confocal micro-Raman spectrometer (inVia Qontor, Renishaw, UK). FTIR was conducted using a FTIR spectrometer (Nicolet 6700, Thermo Fisher Scientific, Massachusetts, USA) equipped with an integrating sphere for the mid-infrared region. ESR measurements were performed on a Bruker EMX-8 spectrometer at room temperature. Reaction solutions were analyzed by ICP-AES (Avio 500, PerkinElmer, Singapore). sXAS and mapping of resonant Auger electron spectroscopy were carried out at BL02B of the Shanghai Synchrotron Radiation Facility.
Transient absorption spectroscopy
TAS was conducted to examine the dynamics of photogenerated carriers in BiVO₄ photocatalysts47,48,49. A custom-designed pump-probe setup, featuring an Nd:YAG laser system (Continuum, Surelite I) and home-built spectrometers, was employed. Photocarrier excitation was triggered by 470 nm laser pulses. The resulting transient absorption spectra, ranging from 500 to 1667 nm (2.48–0.74 eV), were recorded using probe beams sourced from a halogen lamp and a MoSi2 heating coil. For broadband visible-NIR detection, the halogen lamp served as the probe source. Specifically, photoexcited electrons were monitored at 1667 nm using infrared light from the MoSi2 coil. This beam was focused onto the sample, and the transmitted IR light was analyzed using a grating spectrometer coupled with a mercury cadmium telluride detector (Kolmar). To probe hole dynamics, decay signals at 526 nm were collected from the reflected visible-NIR light. The probe beam was directed onto the sample, and the reflected light was analyzed via a grating spectrometer and detected by Si photodiodes. The electrical output was amplified using an AC-coupled amplifier (Stanford Research Systems, SR560, 1 MHz), enabling signal acquisition over the microsecond to millisecond timescale. Each decay curve was obtained by averaging 600 successive measurements, with the system’s temporal resolution constrained to 1 μs due to amplifier bandwidth limitations.
Electrochemical measurement
All electrochemical measurements were carried out on an electrochemical workstation (PARSTAT 4000, Princeton Applied Research, USA) using a three-electrode configuration. A Pt coil and an Ag/AgCl (3.5 M KCl) electrode were used as the counter and reference electrodes, respectively. The working electrode was prepared by the drop-casting method. The catalyst powders were suspended in ethanol (4 mg·mL−1), and the suspension was drop-cast onto a fluorine-doped tin oxide (FTO) transparent electrode, accompanied by heating at 333 K until completely dry. Then, the catalyst-loaded FTO substrate was calcined at 573 K for 2 h under Ar. Current–potential curves were measured in a 0.1 M Na2SO4 solution (pH = 6.3 ± 0.2; ≥99.0%, Sinopharm) containing 30 mM sodium phosphate under darkness. Photocurrent measurements were conducted at 0.2 V (vs. Ag/AgCl) in a 0.1 M Na2SO4 solution (pH = 6.8 ± 0.2) under intermittent visible-light irradiation. EIS was recorded in a frequency range of 0.1–105 Hz with an amplitude of 5 mV at the open-circuit potential (OCP) under darkness. OCP curves were collected under light illumination on/off conditions after the OCP had stabilized.
Photocatalytic water splitting reactions
All photocatalytic activity tests were carried out in a Pyrex top-irradiation reaction vessel, which was connected to a closed gas circulation system (Supplementary Fig. 63). For Z-scheme OWS reaction, a measured amount of HEP and OEP was dispersed in 100 mL sodium phosphate buffer solution (30 mM, pH = 6.3 ± 0.2) containing K4[Fe(CN)6] (2 mM; 99.0%, Aladdin) under continuous stirring. The buffer solution was prepared by dissolving 0.3686 g of NaH₂PO₄·H₂O (98%, Aladdin) and 0.0585 g of Na₂HPO₄·2H₂O (≥98%, Aladdin) in 100 mL of deionized water, followed by deaeration and storage in a sealed volumetric flask before use. After complete degassing, the suspension was irradiated with a 300 W Xe lamp equipped with a cold mirror and a cutoff filter (λ ≥ 420 nm). The spectrum of the Xe lamp is shown in Supplementary Fig. 64. The evolved gas products were analyzed by an online gas chromatograph (GC-9160, OuHua) equipped with a thermal conductivity detector and an MS-5A column, using Ar as the carrier gas. H2 and O2 evolution half-reactions were conducted under the same reaction conditions, with K4[Fe(CN)6] and K3[Fe(CN)6] serving as the hole and electron acceptors, respectively. The oxygen photoreduction reaction was performed similarly to the H₂ evolution half-reaction except for the additional introduction of 200 μmol oxygen before the reaction. Photocorrosion of CdS catalysts was evaluated post-reaction by quantifying the S content of the solution by ICP-AES after the filtration of the catalysts. The formation of SOxy− anions was further examined by adding the filtrate from a Z-scheme reaction conducted in an unbuffered aqueous solution of K4[Fe(CN)6] (to avoid interference from PO43− anions) to a BaCl2 (99%, Macklin) solution. The presence of trace SOxy− ions was indicated by the formation of trace BaSO3/BaSO4 white precipitates after standing overnight.
Apparent quantum yield measurement
The AQY was measured in the same experimental setup but with various band-pass filters. All incident photons were assumed to be absorbed by the photocatalyst, and the total incident number of protons was measured by a calibrated Si photodiode. The AQY is determined by the following Eq. (2):
Where R and I represent the rate of gas evolution and incident photons, respectively. A is the number of electrons consumed to generate one molecule of H2 (H2 evolution half-reaction: 2; Z-scheme OWS reaction: 4). The flux of incident photons at wavelengths of 420, 450, and 500 nm was 5.1 × 1020, 8.0 × 1020, and 1.0 × 1021 photons·h−1, respectively. The evolution rates of H2 in the system containing Pt@CrOx/Co3O4/CdS@TiO2 and Ir/IrO2/BiVO4-Co@SiO2 photocatalysts at wavelengths of 420, 450, and 500 nm were 20.2, 33.8, and 16.4 μmol·h−1, respectively.
Solar-to-hydrogen conversion efficiency measurements
The water-splitting reaction was performed under simulated solar irradiation. The STH conversion efficiency is calculated using the following Eq. (3):
where R(H2), ΔG, P, and S denote the rate of H2 evolution during the Z-scheme OWS reaction, the change in Gibbs free energy for the reaction H2O(l) → H2(g) + 1/2O2(g), the energy intensity of the AM 1.5 G solar irradiation, and the effective irradiation area (4 cm2), respectively.
Computational details
Quantum-mechanical DFT calculations were performed as implemented in the Vienna Ab initio Simulation Package code50,51. The projector augmented wave method was used to describe the interaction between core and valence electrons; exchange and correlation potentials were used in the generalized gradient approximation in the Perdew, Burke and Ernzerhof scheme52. A periodically repeating four-layer (Bi4V4O16) slab was chosen to model (010) and (110) surfaces of monoclinic scheelite, with a vacuum thickness of at least 20 Å to separate the slab from its periodic images. The two bottom layers of the slab were fixed at the optimized bulk lattice constants, with the two top layers fully relaxed. Self-consistent total-energy calculations were performed on conventional unit cells consisting of 24 atoms to optimize the BiVO4 structures. Self-consistent DFT energies were converged to 10−4 eV, and geometry optimizations were considered converged when the residual forces on the atoms were smaller than 0.01 eV·Å−1. The atomic coordinates of the optimized crystal structures can be found in Supplementary Data 1. All calculations were performed using a plane-wave basis set with a cutoff energy of 520 eV and a 12 × 12 × 6 Γ-centered k-point mesh for integrations over the Brillouin zone.
Data availability
The data supporting the findings of this study are reported in the main text or the Supplementary Information. Raw data are provided as a Source data file. Source data are provided with this paper.
References
Kudo, A. & Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 38, 253–278 (2009).
Wang, Y. et al. Mimicking natural photosynthesis: Solar to renewable H2 fuel synthesis by Z-Scheme water splitting systems. Chem. Rev. 118, 5201–5241 (2018).
Zhu, Q. H. et al. Recent progress of metal sulfide photocatalysts for solar energy conversion. Adv. Mater. 34, e2202929 (2022).
Wolff, C. M. et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 3, 862–869 (2018).
Maeda, K. Z-scheme water splitting using two different semiconductor photocatalysts. ACS Catal. 3, 1486–1503 (2013).
Iwashina, K., Iwase, A., Ng, Y. H., Amal, R. & Kudo, A. Z-schematic water splitting into H2 and O2 using metal sulfide as a hydrogen-evolving photocatalyst and reduced graphene oxide as a solid-state electron mediator. J. Am. Chem. Soc. 137, 604–607 (2015).
Iwase, A. et al. Water splitting and CO2 reduction under visible light irradiation using Z-scheme systems consisting of metal sulfides, CoOx-loaded BiVO4, and a reduced graphene oxide electron mediator. J. Am. Chem. Soc. 138, 10260–10264 (2016).
Takata, T. et al. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 581, 411–414 (2020).
Maeda, K. et al. Photocatalyst releasing hydrogen from water. Nature 440, 295–295 (2006).
Zhou, P. et al. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 613, 66–70 (2023).
Wang, Z. et al. Overall water splitting by Ta3N5 nanorod single crystals grown on the edges of KTaO3 particles. Nat. Catal. 1, 756–763 (2018).
Wang, Q. et al. Oxysulfide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 18, 827–832 (2019).
Wang, Q. & Domen, K. Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies. Chem. Rev. 120, 919–985 (2020).
Kato, T. et al. Utilization of metal sulfide material of (CuGa)1-xZn2xS2 solid solution with visible light response in photocatalytic and photoelectrochemical solar water splitting systems. J. Phys. Chem. Lett. 6, 1042–1047 (2015).
Chen, S. S., Li, C. Y., Domen, K. & Zhang, F. X. Particulate metal chalcogenides for photocatalytic Z-scheme overall water. Joule 7, 2445–2467 (2023).
Wang, Z. et al. Sequential cocatalyst decoration on BaTaO2N towards highly-active Z-scheme water splitting. Nat. Commun. 12, 1005 (2021).
Simon, T. et al. Redox shuttle mechanism enhances photocatalytic H2 generation on Ni-decorated CdS nanorods. Nat. Mater. 13, 1013–1018 (2014).
Qi, Y. et al. Unraveling of cocatalysts photodeposited selectively on facets of BiVO4 to boost solar water splitting. Nat. Commun. 13, 484 (2022).
Qi, Y. et al. Efficient overall water splitting of a suspended photocatalyst boosted by metal-support interaction. Joule 8, 193–203 (2024).
Lin, S., Huang, H. W., Ma, T. Y. & Zhang, Y. H. Photocatalytic oxygen evolution from water splitting. Adv. Sci. 8, 2002458 (2021).
Weng, B., Qi, M. Y., Han, C., Tang, Z. R. & Xu, Y. J. Photocorrosion inhibition of semiconductor-based photocatalysts: basic principle, current development, and future perspective. ACS Catal. 9, 4642–4687 (2019).
Chen, S. et al. Semiconductor-based photocatalysts for photocatalytic and photoelectrochemical water splitting: Will we stop with photocorrosion?. J. Mater. Chem. A 8, 2286–2322 (2020).
Hao, X. Q. et al. Self-constructed facet junctions on hexagonal CdS single crystals with high photoactivity and photostability for water splitting. Appl. Catal. B 244, 694–703 (2019).
Maeda, K. et al. Noble-metal/Cr2O3 core/shell nanoparticles as a cocatalyst for photocatalytic overall water splitting. Angew. Chem. Int. Ed. 45, 7806–7809 (2006).
Guo, W. Q. et al. Ge-doped cobalt oxide for electrocatalytic and photocatalytic water splitting. ACS Catal. 12, 12000–12013 (2022).
Maeda, K., Teramura, K., Saito, N., Inoue, Y. & Domen, K. Improvement of photocatalytic activity of (Ga1-xZnx)(N1-xOx) solid solution for overall water splitting by co-loading Cr and another transition metal. J. Catal. 243, 303–308 (2006).
Li, R. G. et al. Spatial separation of photogenerated electrons and holes among {010} and {110} crystal facets of BiVO4. Nat. Commun. 4, 1432 (2013).
Cooper, J. K. et al. Electronic structure of monoclinic BiVO4. Chem. Mater. 26, 5365–5373 (2014).
Idriss, H. On the wrong assignment of the XPS O 1s signal at 531-532 eV attributed to oxygen vacancies in photo- and electro-catalysts for water splitting and other materials applications. Surf. Sci. 712, 121894 (2021).
Frankcombe, T. J. & Liu, Y. Interpretation of oxygen 1s X-ray photoelectron spectroscopy of ZnO. Chem. Mater. 35, 5468–5474 (2023).
Yang, H. G. et al. Anatase TiO2 single crystals with a large percentage of reactive facets. Nature 453, 638–U634 (2008).
Valdes, A., Qu, Z. W., Kroes, G. J., Rossmeisl, J. & Norskov, J. K. Oxidation and photo-oxidation of water on TiO2 surface. J. Phys. Chem. C112, 9872–9879 (2008).
Liu, T. et al. Overall photosynthesis of H2O2 by an inorganic semiconductor. Nat. Commun. 13, 1034 (2022).
Liu, T. et al. A general interfacial-energetics-tuning strategy for enhanced artificial photosynthesis. Nat. Commun. 13, 7783 (2022).
Yamakata, A., Ranasinghe, C. S. K., Hayashi, N., Kato, K. & Vequizo, J. J. M. Identification of individual electron- and hole-transfer kinetics at CoOx/BiVO4/SnO2 double heterojunctions. ACS Appl. Energy Mater. 3, 1207–1214 (2020).
Xiao, J. D. et al. Sub-50 nm perovskite-type tantalum-based oxynitride single crystals with enhanced photoactivity for water splitting. Nat. Commun. 14, 8030 (2023).
Ning, X. F. & Lu, G. X. Photocorrosion inhibition of CdS-based catalysts for photocatalytic overall water splitting. Nanoscale 12, 1213–1223 (2020).
Meissner, D., Memming, R., Kastening, B. & Bahnemann, D. Fundamental problems of water splitting at cadmium sulfide. Chem. Phys. Lett. 127, 419–423 (1986).
Chen, S. Y. & Wang, L. W. Thermodynamic oxidation and reduction potentials of photocatalytic semiconductors in aqueous solution. Chem. Mater. 24, 3659–3666 (2012).
Han, L. J. et al. Enhanced activity and acid pH stability of prussian blue-type oxygen evolution electrocatalysts processed by chemical etching. J. Am. Chem. Soc. 138, 16037–16045 (2016).
Kuhn, A. T. & Chan, C. Y. pH changes at near-electrode surfaces. J. Appl. Electrochem. 13, 189–207 (1983).
Luo, J. et al. Unraveling pH dependent cycling stability of ferricyanide/ferrocyanide in redox flow batteries. Nano Energy 42, 215–221 (2017).
Sasaki, Y., Kato, H. & Kudo, A. [Co(bpy)3]3+/2+ and [Co(phen)3]3+/2+ electron mediators for overall water splitting under sunlight irradiation using Z-scheme photocatalyst system. J. Am. Chem. Soc. 135, 5441–5449 (2013).
Maeda, K. et al. Preparation of core-shell-structured nanoparticles (with a noble-metal or metal oxide core and a chromia shell) and their application in water splitting by means of visible light. Chem. Eur. J. 16, 7750–7759 (2010).
Takata, T., Pan, C. S., Nakabayashi, M., Shibata, N. & Domen, K. Fabrication of a core-shell-type photocatalyst via photodeposition of group IV and V transition metal oxyhydroxides: an effective surface modification method for overall water splitting. J. Am. Chem. Soc. 137, 9627–9634 (2015).
Pan, C. S. et al. A complex perovskite-type oxynitride: the first photocatalyst for water splitting operable at up to 600 nm. Angew. Chem. Int. Ed. 54, 2955–2959 (2015).
Yamakata, A., Vequizo, J. J. M. & Kawaguchi, M. Behavior and energy state of photogenerated charge carriers in single-crystalline and polycrystalline powder SrTiO3 studied by time-resolved absorption spectroscopy in the visible to mid-infrared region. J. Phys. Chem. C119, 1880–1885 (2015).
Vequizo, J. J. M. et al. Trapping-induced enhancement of photocatalytic activity on brookite TiO2 powders: comparison with anatase and rutile TiO2 powders. ACS Catal. 7, 2644–2651 (2017).
Lin, L. H. et al. Efficient and stable visible-light-driven Z-scheme overall water splitting using an oxysulfide H2 evolution photocatalyst. Nat. Commun. 15, 397 (2024).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
The research is supported by the National Natural Science Foundation of China (21872093 to Z.J. and 22225301 to X.W.), Shanghai Science and Technology Innovation Action Plan (24520713400 to Z.J.), Leading Innovative and Entrepreneurial Projects in Zhejiang Province (2023R01007 to J.L.), the Fundamental Research Funds for the Central Universities (20720220009 to X.W.), the CAS Project for Young Scientists in Basic Research (YSBR-004 to X.W.), and support from Super Computer Centre of USTCSCC and SCCAS. The authors thank beamline BL02B of the Shanghai Synchrotron Radiation Facility for providing the beamtime. A.F.L. acknowledges funding from the ARC Centre of Excellence for Green Electrochemical Transformation of Carbon Dioxide (CE230100017 to A.F.L.) and the Australian Government.
Author information
Authors and Affiliations
Contributions
Z.J. conceived and designed the research project. Z.J. and J.L. supervised the research project. H. Luo synthesized the catalysts, conducted the catalytic tests and the related data processing, and performed materials characterization and analysis with the help of Z.L. (OEP synthesis, OER data collection), F.H. (OER data collection), and Z.Y. (characterization and data collection). H. Lv and X.W. performed theoretical calculations. J.J.M.V. and A.Y. performed TAS experiments and data analysis. K.D. provided valuable support for manuscript revision. W.S. provided support for data interpretation and research materials. A.F.L. provided valuable discussion and data interpretation. Z.J. and H. Luo analyzed the data. Z.J., H. Luo, A.F.L., M.Z., and J.L. wrote the manuscript with help from all the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Sandeep Lakhera, Xiankui Wei, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, H., Liu, Z., Lv, H. et al. Efficient and stable n-type sulfide overall water splitting with separated hydrogen production. Nat Commun 16, 8786 (2025). https://doi.org/10.1038/s41467-025-63840-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63840-1
